
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DFT investigation of the mechanism and role of N-heterocyclic carbene (NHC) in constructing asymmetric organosilanes using NHC-catalyzed [4+2] cycloaddition reaction†
Batoul Alipour *
*
Department of Chemistry, Tarbiat Modares University, P.O. Box 14115 175, Tehran, Iran. E-mail: a.batoul@modares.ac.ir
First published on 6th November 2024
Abstract
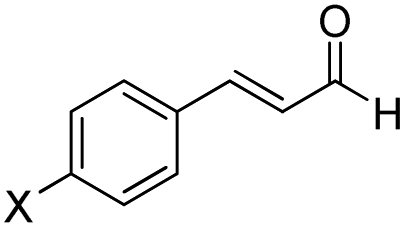
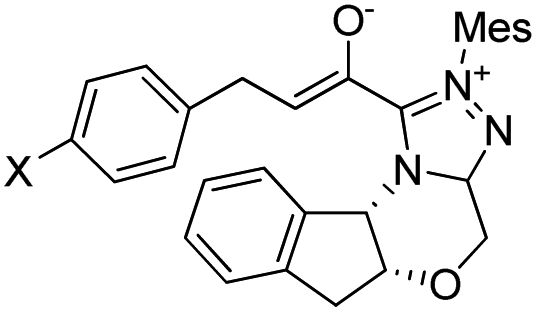
Herein, the mechanism and origin of stereoselectivity for the asymmetric [4+2] cycloaddition between (E)-3-(p-tolyl)acrylaldehyde (R1) and phenyl-3-(trimethylsilyl)prop-2-en-1-one (R2) in the presence of an N-heterocyclic carbene (NHC) were theoretically scrutinized. The desirable catalytic cycle is characterized by five steps: (1) the coupling reaction of the NHC catalyst with R1, the formation of the Breslow and enolate intermediates in the second and third steps, (4) the formal [4+2] cycloaddition reaction to form the stereoselective C–C bond, and (5) the regeneration of NHC to obtain asymmetric organosilanes. In the most energetically favorable pathway, the formation of the enolate intermediate exhibits the highest energy barrier of about 19.48 kcal mol−1 (Re-TS2BA) and is the rate-determining step. The [4+2] cycloaddition reaction is the stereoselectivity-determining step forming the chiral C–C bond with RR, RS, SR and SS configurations, among which RS is the most desirable configuration. The origin of stereoselectivity was investigated using distortion energy analysis. The first and fourth steps helped in investigating the effects of electron-donating (Me) and electron-withdrawing (Cl) groups on cinnamaldehyde. Conceptual DFT (CDFT) analysis was carried out to confirm the critical role of the NHC catalyst as a Lewis base during the reaction processes.
1. Introduction
Asymmetric organosilanes have interesting and useful applications in different fields, including organic synthesis,1 agroscience,2 materials science,3 and medicinal chemistry.4 Owing to the presence of the silicon atom in their structures, they have a particular physical and electronic nature. Several synthetic strategies have been developed to produce asymmetric organosilanes, which rely on transition metals as catalysts.5,6 In contrast, developing organocatalysis-based strategies to construct asymmetric organosilanes has become a great and essential challenge.7 N-Heterocyclic carbenes (NHCs) act as key and powerful tools to construct chiral C–X (X = C, N or O) bonds in various asymmetric organic reactions, such as Stetter, cross-benzoin, Mannich, Michael, and cycloaddition reactions.8–12 Interestingly, many annulation reactions in the formation of carbocyclic and heterocyclic systems using NHC catalysts have been reported. These reactions have been performed with excellent stereo- and regioselectivity. NHC-catalyzed annulation reactions were carried out with the Umpolung of enals and diverse electrophilic substrates, such as aldehydes, imines, and ketones.13 In fact, the complexation of NHCs and enals results in the inversion of natural reactivity (i.e., Umpolung reaction) and forms the Breslow intermediate, which works as a prenucleophile, as shown in Scheme 1.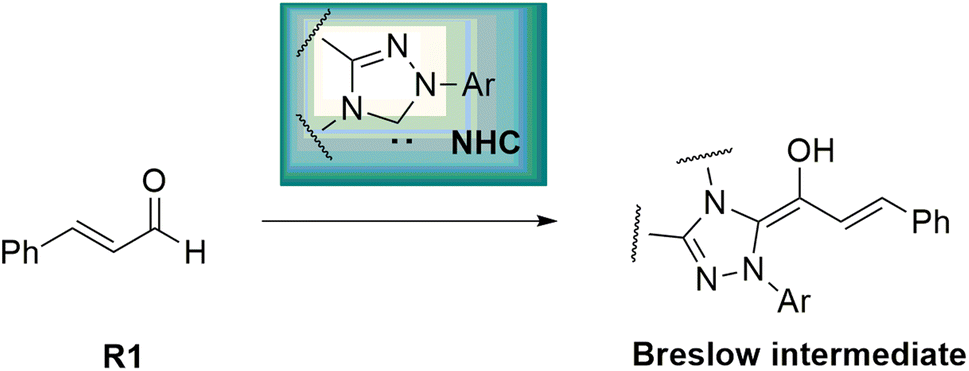 | ||
| Scheme 1 Formation of the Breslow intermediate. | ||
This leads to the formation of various reactive intermediates possessing one, two, three, and four active carbon centers. α-Carbon (enolate intermediate b),14,15 β-carbon (homoenolate intermediate c),16–20 γ-carbon (dienolate intermediate d),21–23 and carbonyl carbon (acyl anion equivalent intermediate a) are among these intermediates (Scheme 2).24–26
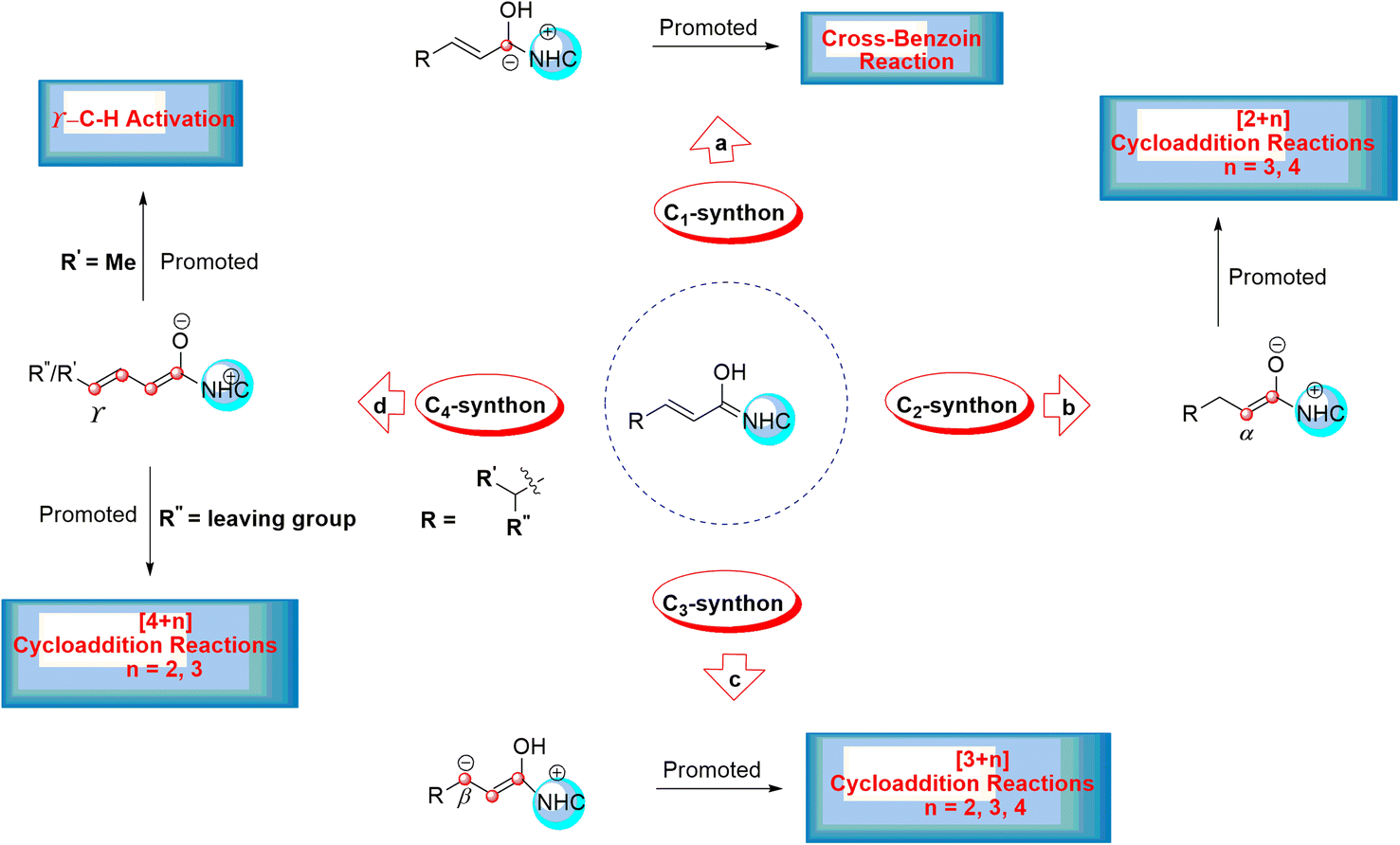 | ||
| Scheme 2 The formation of different reactive intermediates from the Breslow intermediate. | ||
This intermediate serves as a CX (X = 1, 2, 3, and 4) carbon synthon in diverse annulation reactions (Scheme 2). For instance, the cross-benzoin reaction occurs when the Breslow intermediate acts as a C1-synthon.24–26 A C2-synthon (enolate intermediate) performs different types of [2+n] (n = 3, 4) annulation reactions.27,28 A C3-synthon (homoenolate intermediate) shows different types of [3+n] (n = 3, 4) annulation reactions.8 Finally, the NHC-catalyzed [4+n] (n = 2, 3) annulation reaction occurs through a C4-synthon (dienolate intermediate).29,30 Glorius and Bode reported that the NHC-catalyzed reactions between enals and aldehydes resulted in the production of γ-lactones generated by the homoenolate intermediates.16,17 These results lead to much attention for the construction of different kinds of compounds through the activated β-carbon in the homoenolates.31 Also, Bode suggested that the conjugated Breslow intermediate can undergo the protonation process and form the enol or enolate intermediate.32 In addition, Scheidt33 and Chi15 indicated that the proton transfer of the β-carbon of homoenolate led to the generation of the enolate intermediate under certain experimental conditions.
In 2018, Fu and Huang advanced an interesting [4+2] cycloaddition reaction between (E)-3-(p-tolyl)acrylaldehyde (R1) and phenyl-3-(trimethylsilyl)prop-2-en-1-one (R2).34 This reaction was performed in the presence of triazolium salt as a precatalyst and Cs2CO3 and 1,4-dioxane (DO) as the base and solvent, respectively. This reaction proceeded smoothly at ambient temperature to afford the asymmetric organosilanes in good yields and excellent stereoselectivities (Scheme 3).
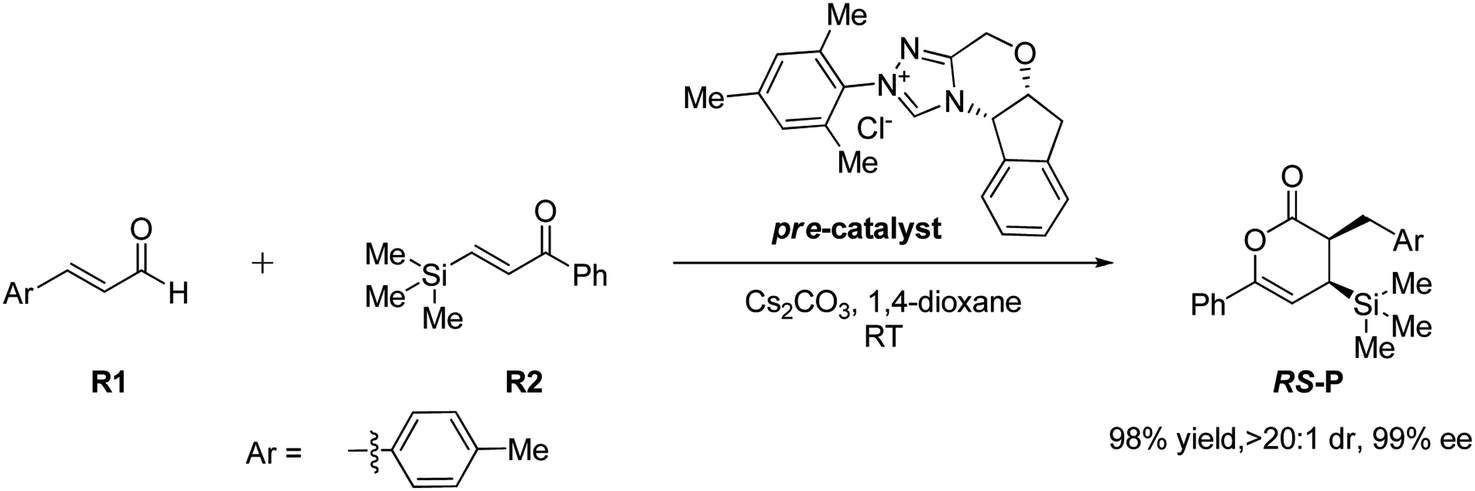 | ||
| Scheme 3 NHC-catalyzed [4+2] annulation reaction of (E)-3-(p-tolyl)acrylaldehyde (R1) and phenyl-3-(trimethylsilyl)prop-2-en-1-one (R2). | ||
Although construction methods of carbon–carbon chiral bonds have been developed, the comprehension of their mechanism continues to be challenging. Over the years, numerous theoretical studies have been published to elucidate the characterization of NHC as a catalyst and the origin of stereoselectivity in annulation reactions.35–39 Sunoj and co-workers explored the detailed mechanism of several reactions in NHC-catalyzed [3+2] and [4+2] annulation reactions using DFT calculations.40 Wei and co-workers revealed the origin of stereoselectivity in different types of NHC-catalyzed cycloaddition reactions through density functional theory (DFT).37,41,42 More theoretical investigations have been reported to elucidate the detailed mechanism of NHC-catalyzed [n+2] (n = 2, 3, 4) and [3+4] annulation reactions by other groups.36,43–46
Herein, DFT computations of the NHC-catalyzed [4+2] cycloaddition reaction between R1 and R2 have been performed, as shown in Scheme 3. This study aims to describe the potential role of N-heterocyclic carbene (NHC) as an organocatalyst in promoting [4+2] cycloaddition reactions with a deep insight and a broad understanding of their important and influential role in organic synthesis, which solved the following questions.
(1) How can the unsaturated sp2 carbon of R1 be activated in this reaction? (2) How can proton transfer be performed in this reaction? (3) What is the role of NHC and Cs2CO3? (4) Which step determines the rate or stereoselectivity? (5) What is the origin of the stereoselectivity?
And (6) what are the effects of substitution on the addition of NHC to R1 and stereoselectivity?
2. Computational method
Gaussian 09 software was employed to investigate all DFT calculations.47 The reported DFT calculations show that the M06-2X method gives an actual outcome of the kinetic and thermodynamic parameters.48–55 The geometries of all structures were investigated using the M06-2X48,56,57 method along with the 6-31G(d,p) basis set to optimize the C, H, N, and O atoms. Also, lanl2dz58,59 was applied to optimize the Cs atom. The solvent environmental effects were considered in all theoretical calculations in 1,4-dioxane solvent through the integral equation formalism polarizable continuum model (IEF-PCM).60,61 The vibrational frequency was computed at the same level to access the corrections in the Gibbs free energies and verify the character of each stationary point. The minima state had no imaginary frequencies (NIMAG = 0), while the transition state (TS) had only one imaginary frequency (NIMAG = 1). Single-point energy for all optimized structures were calculated at the M06-2X/6-311++G(2df,2pd) level in implicit 1,4-dioxane using IEF-PCM. Also, tight convergence criterion and ultrafine integral grid were used to enhance the accuracy of the calculations. In this study, all of the detailed mechanisms were scrutinized using the Gibbs free energies resulting from the addition of the thermal corrections Gibbs free energies at the M06-2X/6-31G(d,p)//(dioxane) IEF/PCM level (L1) to the relevant single point energies at the M06-2X/6-311++G(2df,2pd)//(dioxane) IEF/PCM level (L2). Intrinsic reaction coordinate (IRC)62,63 calculations were done at the M06-2X/6-31G(d,p) level to confirm the transition states that led to the desired reactants and products. Natural bond orbital (NBO) analysis64–66 was used at the M06-2X/6-31G(d,p) level to calculate the atomic charge distribution. In addition, the CDFT-derived reactivity index was described similarly to the global reactivity index (GRI) analysis,67 which was executed to indicate the role of the organocatalyst. In this analysis, the electrophilicity index ω was obtained through ω = (μ2/2η) (eV), in which μ was defined as electronic chemical potential and η was defined as chemical hardness.68,69 With the use of expressions μ ≈ (EH − EL)/2 and η ≈ (EL − EH), the values of μ and η were obtained. The EH and EL are the one-electron energy of the HOMO and LUMO orbitals in the corresponding molecules, respectively. It is worth mentioning that the nucleophilicity index N = EH(Nu) − EH(TCNE), in which tetracyanoethylene (TCNE) is known as the ref. 70–72. The Parr functions (Pk+ and Pk−)73,74 were assessed to compute both the local electrophilicity and local nucleophilicity indexes, which were obtained through expressions ωk (ωk = ωPk+)75 and Nk (Nk = NPk−)76 for some key atoms in the reactants and intermediates, respectively. Three-dimensional (3D) structures in this article were performed using the CYL-view software.77 In addition, Boltzmann distribution calculation was performed to determine the extent of enantioselectivity in terms of enantiomeric excess (ee%). The value of diastereomeric (ee%) transition states were obtained by the following equations.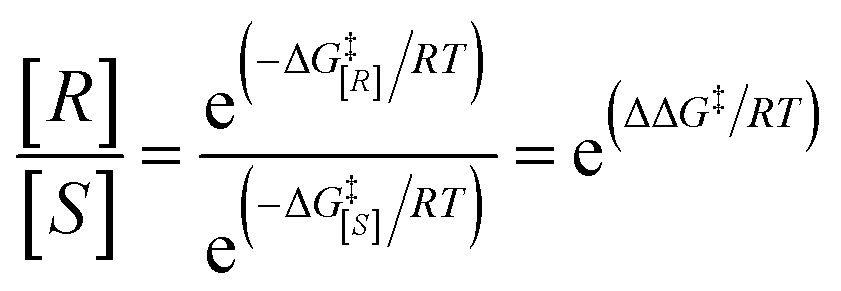
where G‡ is the Gibbs free energy barrier of the competing diastereomeric transition state and ΔG‡ is the Gibbs free energy difference between two competing intended diastereomeric transition states.
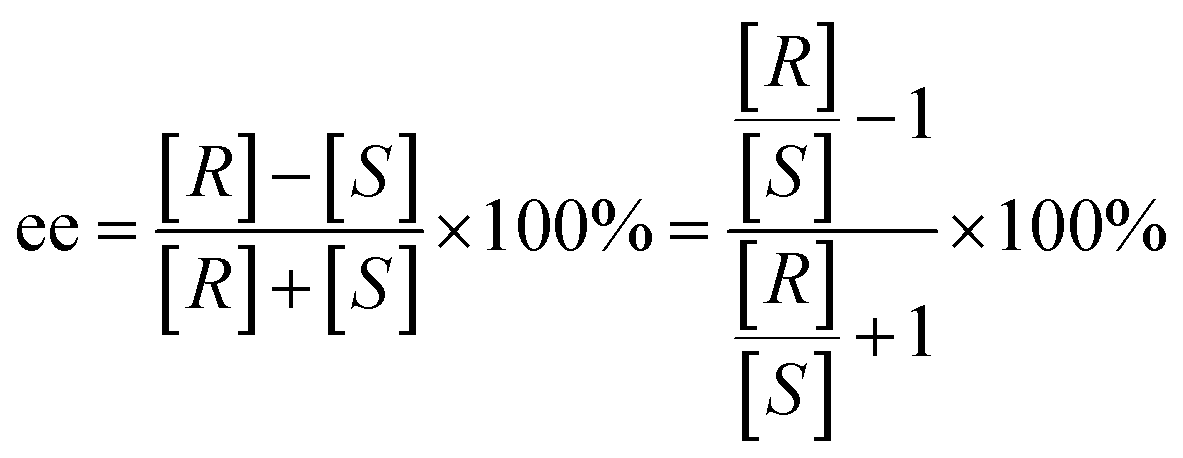
3. Results and discussion
In this study, two possible routes for constructing the asymmetric organosilanes of (E)-3-(p-tolyl)acrylaldehyde (R1) and phenyl-3-(trimethylsilyl)prop-2-en-1-one (R2) are proposed and investigated, which are discussed in detail as follows.3.1. The first reaction route
As shown in Scheme 4, a possible mechanism has been proposed for this reaction by initiating the removal of a proton of the pre-catalyst by Cs2CO3 base to prepare the active NHC, a molecule of CsCl and a molecule of CsHCO3. The catalytic cycle involves five steps: (1) the formation of a zwitterionic intermediate by the addition of NHC to R1; (2) [1,2]-proton transfer processes to form the Breslow intermediate; (3) [1,4]-proton transfer processes to generate the enolate intermediate; (4) formal [4+2] annulation reaction or Diels–alder reaction; (5) regeneration of NHC to afford the expected product.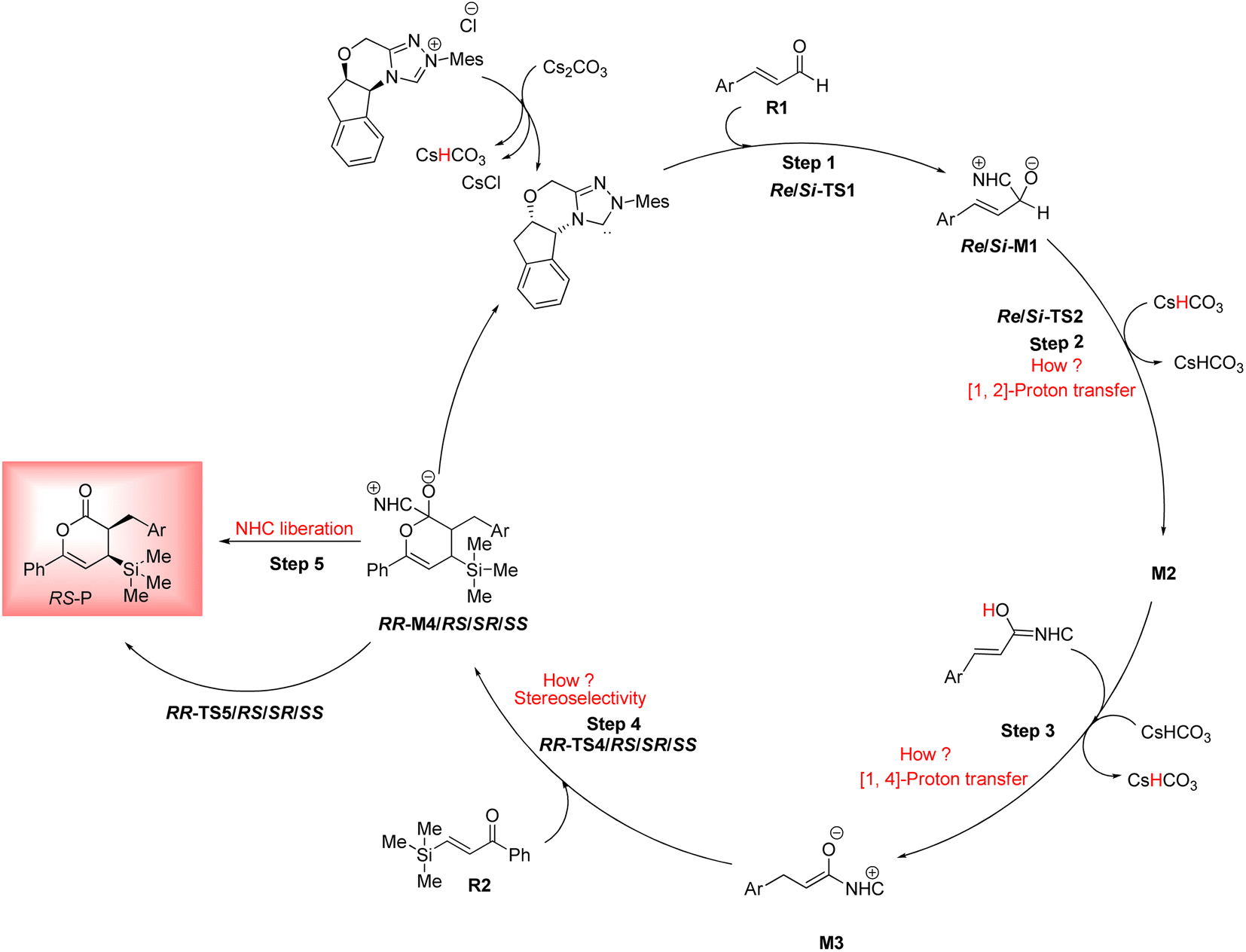 | ||
| Scheme 4 Possible mechanism for the NHC-catalyzed [4+2] annulation reaction of (E)-3-(p-tolyl)acrylaldehyde (R1) and phenyl-3-(trimethylsilyl)prop-2-en-1-one (R2). | ||
 | ||
| Scheme 5 The possible pathways for the formation of the Breslow intermediate. | ||
DPT route: The optimized geometry of Re/Si-TS2D reveals the formation of a three-membered ring between C2, O3, and H4 atoms. The bond length of O3–H4 is reduced from 2.08/2.08 Å in Re/Si-M1 to 1.36/1.35 Å in Re/Si-TS2D, whereas the distance between C2 and H4 is elongated from 1.13/1.12 Å in Re/Si-M1 to 1.17/1.18 Å in Re/Si-TS2D, respectively (ESI†). The free energy barrier was calculated at 37.93/33.49 (kcal mol−1) in Re/Si-TS2D, which is challenging to overcome at room temperature.
BPT route: Using the reported DFT studies on the NHC-catalyzed dimerization,79,82 this route has been proposed. This mechanism describes a bimolecular proton transfer that occurs using another molecule of enals R1. When the enal R1 approaches intermediate Re/Si-M1, the H4′ atom is transferred from the C2′ atom to the O3 atom from Re/Si-M1 and the H4 atom is shifted from the C2 atom to the C2′ atom of R1. This leads to the formation of a five-membered ring (C2, H4, O3, C2′, and H4′) through the Re/Si-TS2B transition state. The energy barrier (37.61/45.35 kcal mol−1 in Re/Si-TS2B) is higher than that of Re/Si-TSD, which is difficult to overcome at room temperature. The optimized geometry of Re/Si-TS2B indicates that these two proton transfers are asynchronous, and the transfer of the H4′ atom occurs faster than that of the H4 atom.
BAPT route: As shown in Fig. 1, this route is scrutinized as an intermolecular proton transfer process in which cesium bicarbonate (CsHCO3) serves as the base and assists in performing proton transfer under the experimental conditions. The investigation of this mechanism was inspired by the reported studies in the literature.82,83 Cesium bicarbonate, for the aid of proton transfer, can reduce the energy barrier through the formation of the seven-membered ring (C2–H4–O9–C10–O7–H8–O3). This mechanism is more energetically favorable than that of the described mechanisms. As shown in Fig. 1, Re/Si-M1 in the presence of CsHCO3 forms Re/Si-M02BA. Next, Re/Si-M02BA performs the proton transfer through the transition state Re/Si-TS2BA and produces Re/Si-M03BA. In this Re/Si-TS2BA, the H4 is shifted from the C2 atom to the O9 of carbonyl, and following the hydrogen, H8 from the O7 atom is transferred to the O3 atom. The calculated free energy barrier was 9.23/14.49 kcal mol−1 in Re/Si-TS2BA, which has the lowest energy barrier of all the routes and is easily performed under experimental conditions. In addition, the energy barriers of Re/Si-M02BA and Re/Si-M03BA were obtained as 9.96/15.87 and −3.95/−4.55 kcal mol−1, respectively. These outcomes show that CsHCO3 can be easily linked with Re/Si-M1. In contrast, the energy barrier Re/Si-M03BA shows that CsHCO3 is quickly dissociated and the Breslow intermediate Re/Si-M2 is formed. The formation of a seven-membered ring leads to decreased strain and energy barriers compared to the other routes. Owing to the ring expansion, the reactivity centers increase in three-, five-, and seven-membered rings, respectively. As discussed above, the desired catalytic cycle was performed and continued using Re-M2 because the complexation of NHC and R1 (Re-face) in both thermodynamics and kinetics is more favorable than the coupling of NHC and R1 (Si-face). Therefore, the mechanism of the nucleophilic attack of NHC on the Si-face of R1 is ignored.
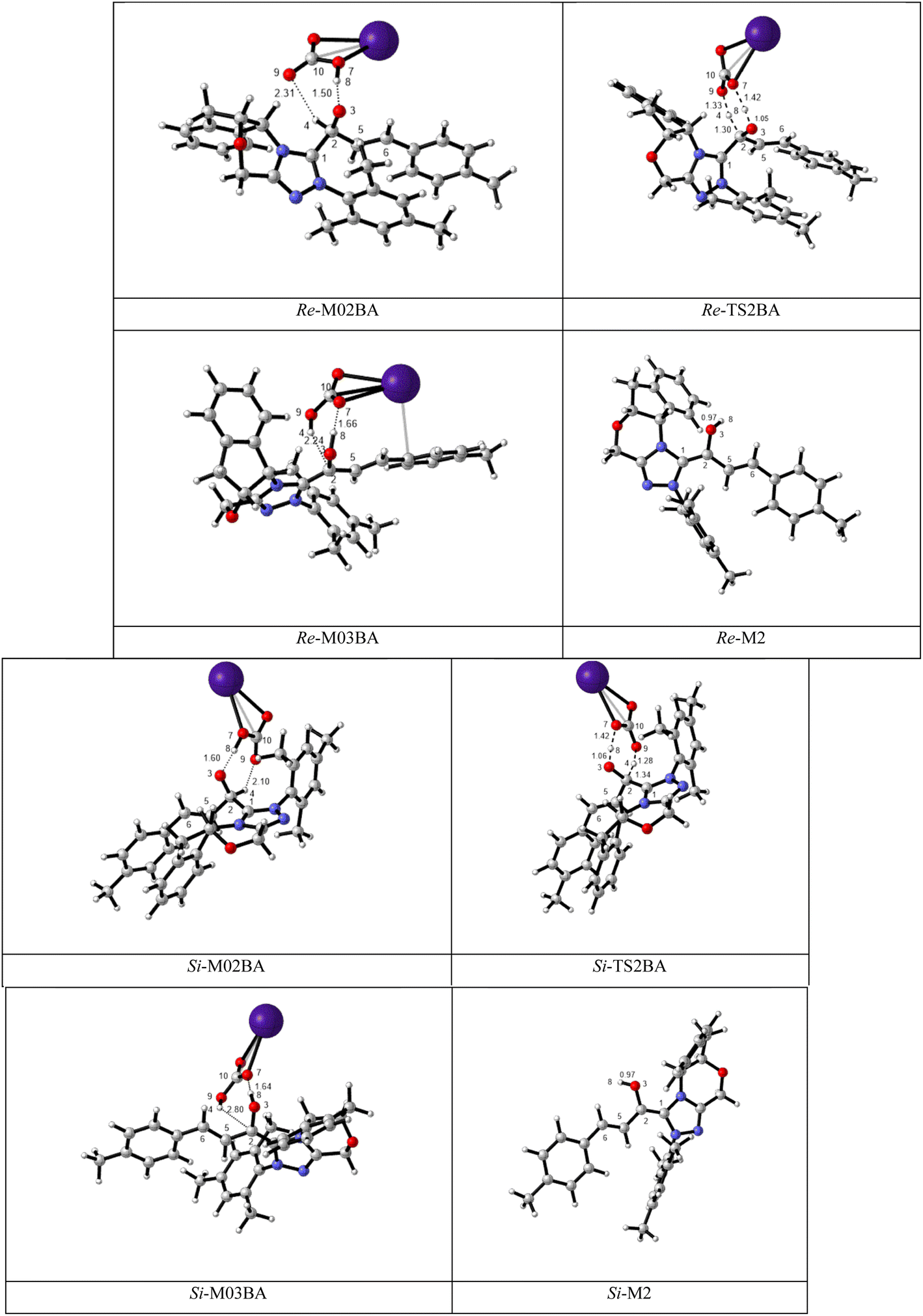 | ||
| Fig. 1 The geometry and optimization of the BAPT route for the formation of the Breslow intermediate. | ||
BAPT1 route: The bicarbonate anion (HCO3−) is also used for assisting proton transfer, which decreased the energy barrier by forming a seven-membered ring (C2–H4–O9–C10–O7–H8–O3). This route is more energetically favorable than that of the DPT and BPT routes. The calculated free energy barrier is 13.30/16.97 kcal mol−1 in Re/Si-TS2BA1, which has a high energy barrier in the BAPT mechanism and can be performed under experimental conditions. Notably, the strong interactions between the cesium and oxygen atoms lead to reduce energy barrier of the BAPT mechanism (BAPT vs. BAPT1) and were obtained simply under the experimental conditions. Scheme 6 illustrates the relative Gibbs free energy to construct the Breslow intermediate in both the first and second steps.
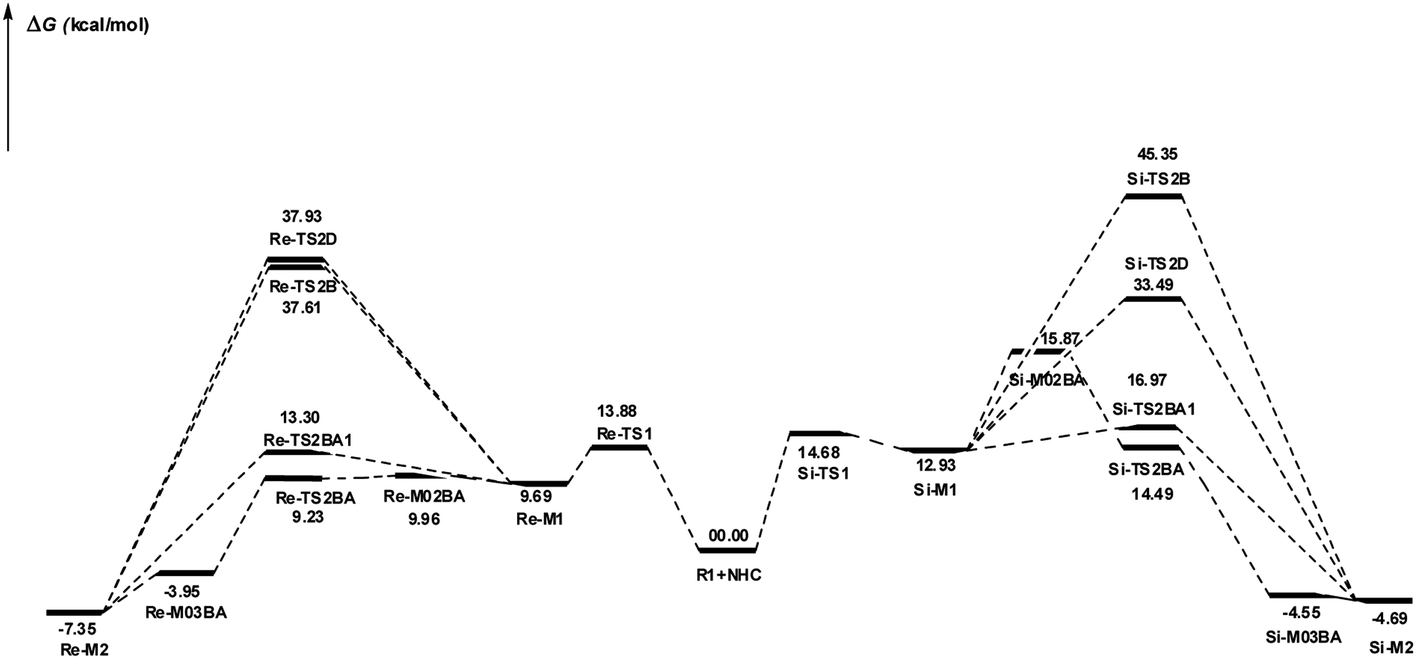 | ||
| Scheme 6 Gibbs free energy profiles of steps 1 and 2. | ||
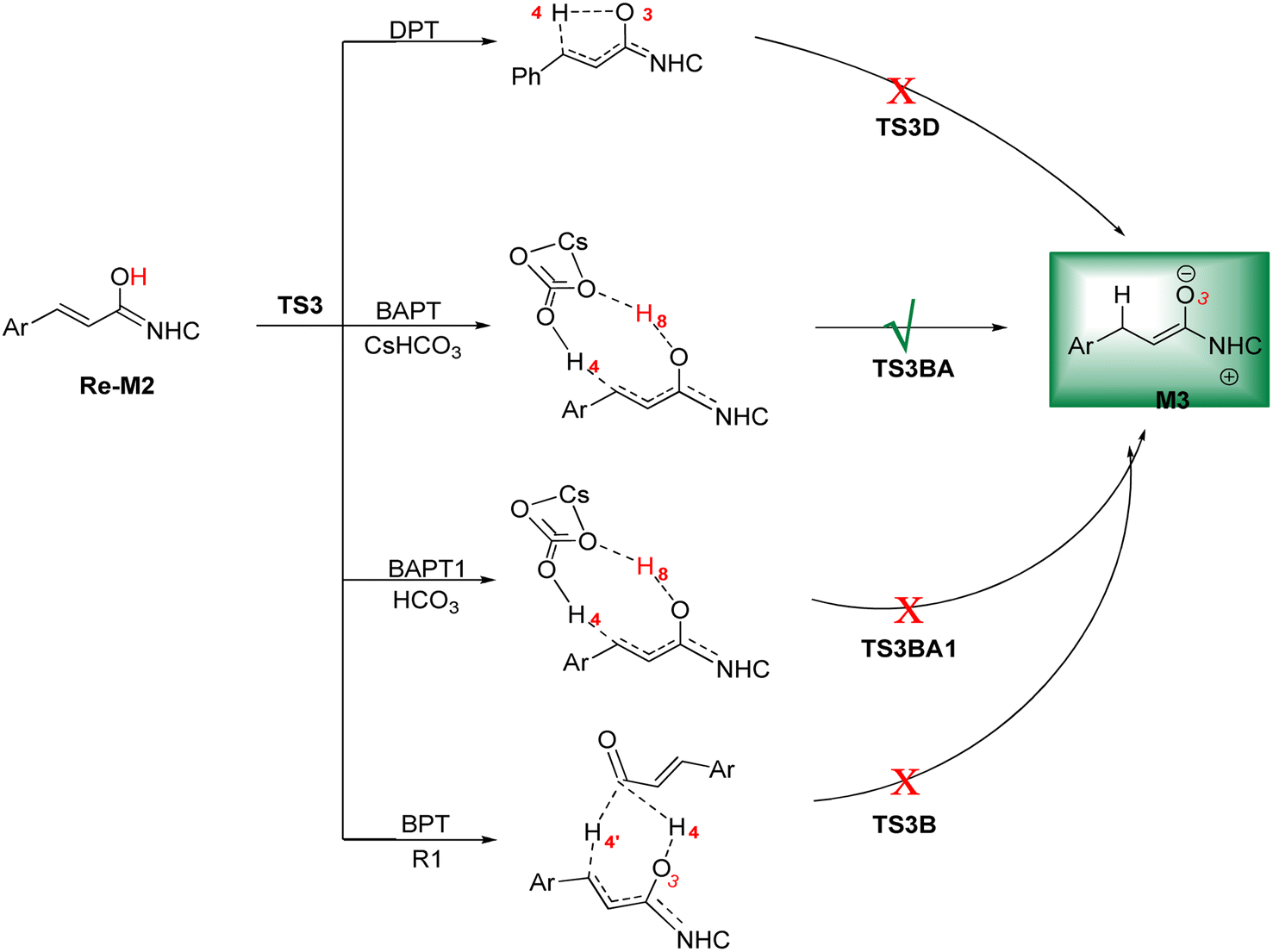 | ||
| Scheme 7 The possible routes for the formation of the enolate intermediate. | ||
DPT route: This process forms a five-membered ring via the [1,4]-proton transfer. The optimized geometry of TS3D shows that a five-membered ring between C2, C5, C6, O3, and H4 atoms was formed. The bond length of O3–H4 was elongated from 0.97 Å in Re-M2 to 1.27 Å in TS3D, whereas the distance between C6 and H4 was reduced from 2.83 Å in Re-M2 to 1.43 Å in TS3D, respectively (ESI†). For this TS, the energy barrier was calculated to be 32.37 kcal mol−1.
BPT route: This mechanism explored a bimolecular proton transfer using another molecule of enals R1, which is done in two steps; firstly, the enal R1 approaches intermediate Re-M2, and the H4′ atom is transferred from the C2′ atom of R1 to the C6 atom of Re-M2. Following this, the H4 atom is shifted from the O3 atom to the C2′ atom of R1. For the transfer of the H4′ atom to the C6 atom in the first step, the energy barrier (61.12 kcal mol−1) is more than that of the DPT route, which leads to neglecting the transfer of H4 to C2′ calculation.
BAPT route: As depicted in Fig. 2, this route is described as an intermolecular proton transfer process in which the cesium bicarbonate base works as the medium for the proton-assisted transfer process. Cesium bicarbonate reduces the energy barrier through the formation of the nine-membered ring (C2, H8, O3, C5, C6, O7, H4, O9, and C10 atoms). This mechanism is more energetically favorable than that of other routes. As depicted in Fig. 2, CsHCO3 and Re-M2 formed intermediate M04BA. Then, M04BA undergoes the proton transfer through the transition state TS3BA and creates intermediate M05BA. In this T3BA, the H8 is shifted from the O3 atom to the O7 of carbonyl, following which the hydrogen H4 from the O9 atom in cesium bicarbonate is transferred to the C6 atom in Re-M2. The calculated free energy barrier is 19.48 kcal mol−1, the lowest barrier in this step and is easily performed under experimental conditions. In addition, the calculated results show that the energy barriers of M04BA and M05BA are −7.53 and −2.01 kcal mol−1, indicating that CsHCO3 is highly prone to react with Re-M2; in contrast, the energy barrier of M05BA shows that CsHCO3 is easily separated and formed enolate intermediate M3.
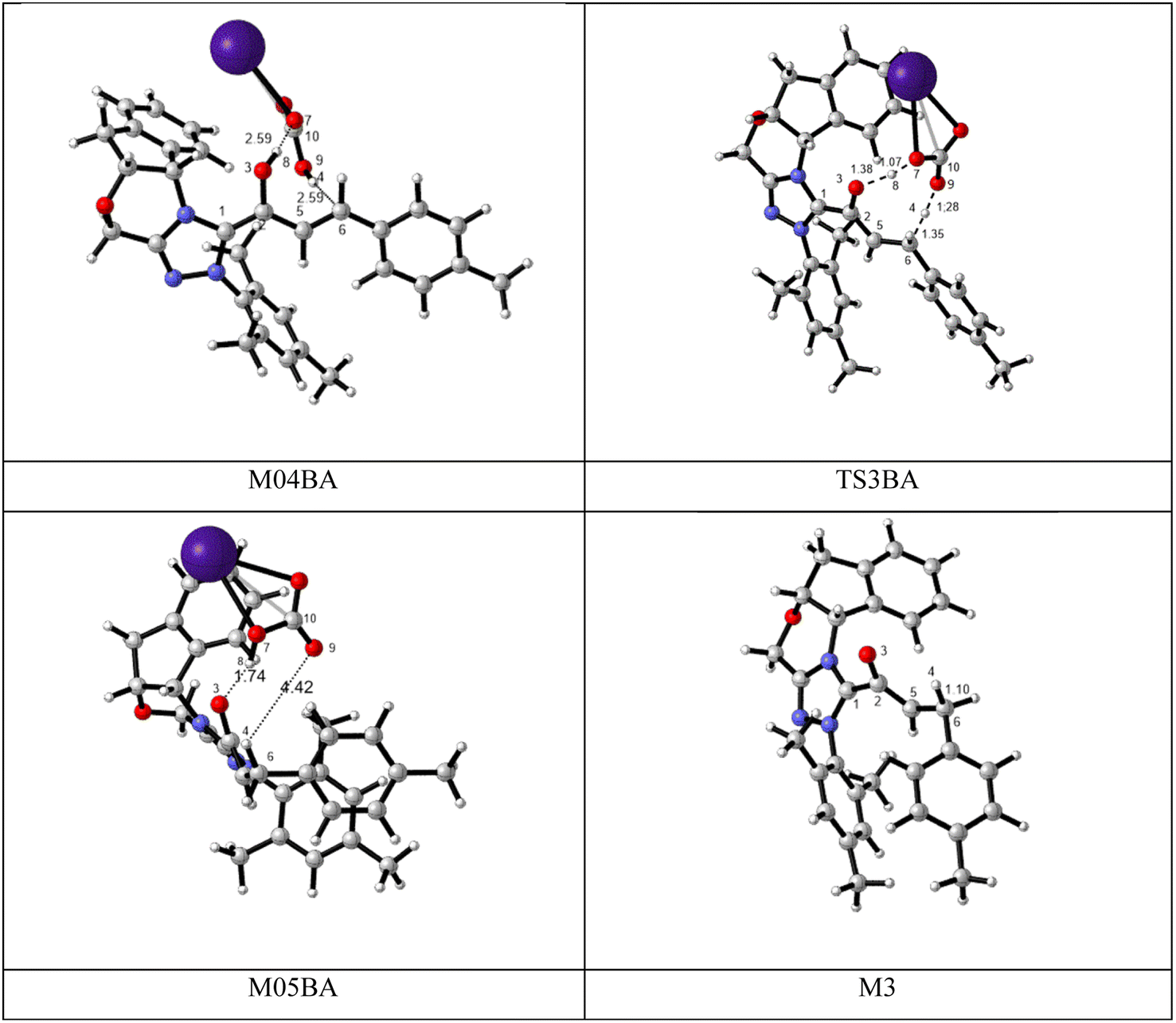 | ||
| Fig. 2 The optimized structures of the BAPT route for the formation of the enolate intermediate. | ||
BAPT1 route: The bicarbonate anion applied for the aid of proton transfer diminished the energy barrier by making a nine-membered ring (C2–H4–O9–C10–O7–H8–O3). This pathway is more thermodynamically favorable than that of the DPT and BPT routes. The calculated free energy barrier is 22.37 kcal mol−1 in TS3BA1, which has a high energy barrier of the BAPT pathway and can be done under experimental conditions. Due to the presence of the Cs atom in the BAPT route, the dative interactions between the O and Cs atoms lead to reduced barrier energy (stability) versus the BAPT1 route.
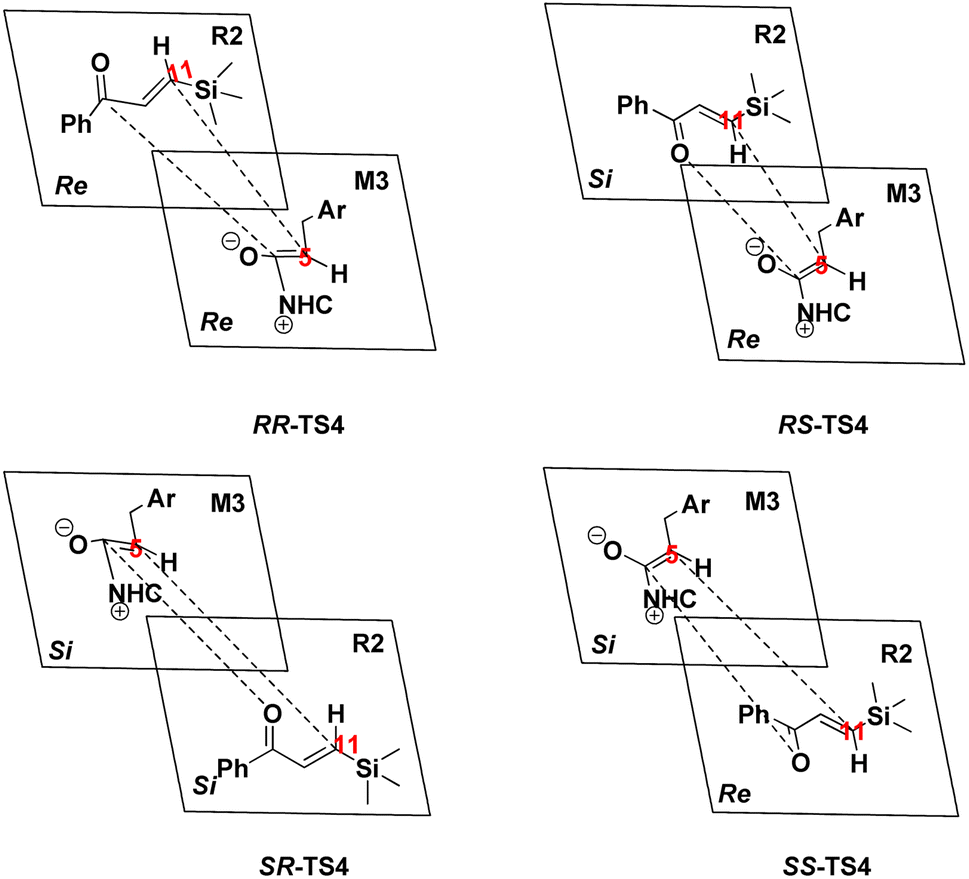 | ||
| Scheme 8 Stereochemistry for the [4+2] annulation reaction in the fourth step. | ||
Generally, the addition of the Re face from M3 to the Re or Si face of R2 is more stable and prominent both kinetically and thermodynamically compared to the addition of the Si face from M3 to the Re or Si face of R2.
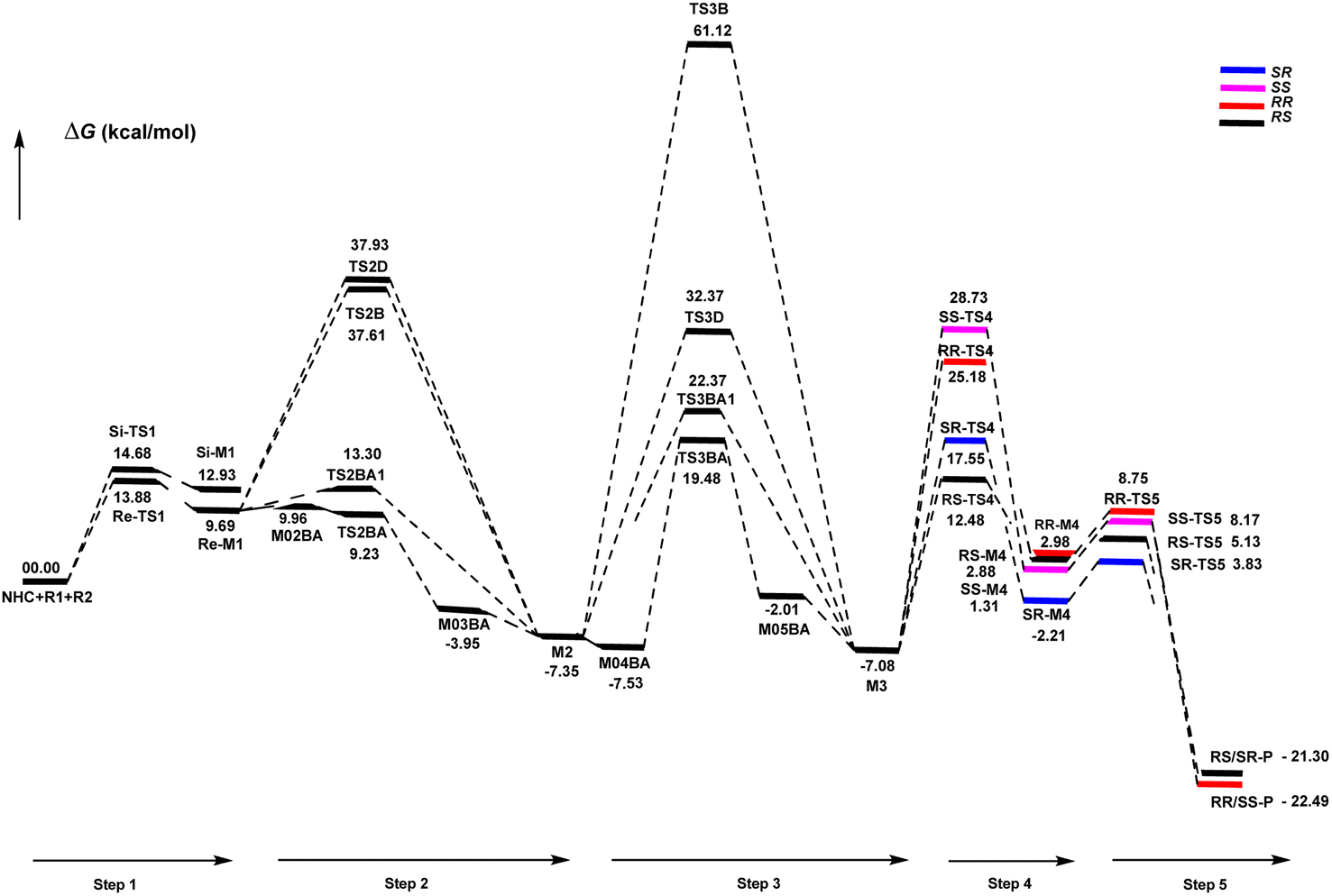 | ||
| Scheme 9 Gibbs free energy profiles of steps 1–5. | ||
3.2. The second reaction route
Based on Wei's research in 2014,41 this route is suggested to further investigate the reaction of R2 and NHC catalyst. As shown in Scheme 10, the catalytic cycle recognizes three steps: (1) the formation of zwitterionic intermediate Re/Si-M1′ by the Michael addition of NHC to Re/Si-R2; (2) the construction of the C–O bond through the addition of Re-M1′ to R1 (keten); (3) the formation of the chiral C–C bond via intermolecular annulation reaction and regeneration of NHC to afford the expected product. For performing this pathway, it essentially converted the (E)-3-(p-tolyl)acrylaldehyde (R1) to the keten molecule (R1′) via [1,3]-proton transfer, where the H4 from C2 atom is transferred to the C6 atom. The conversion of R1 to R1′ was studied through three mechanisms including the direct proton transfer (DPT) through the formation of the four-membered ring, the bimolecular proton transfer (BPT) through the help of another R1 by forming the six-membered ring, and the aid of CsHCO3 base (BAPT) by making the eight-membered ring. The calculated results indicated that the energy barrier for these TSs is 65.34, 75.17, and 51.56 kcal mol−1 for the DPT, BPT, and BAPT pathways, respectively. Owing to the instability of the formed four, six, and eight-membered ring structures in these TSs, the preparation of the keten molecule (R1′) structure from the enal (R1) is impossible under ambient temperatures. Additionally, we investigated the first step of this route that started with the Michael addition of the NHC on the Re/Si-face of C11 from R2 to afford the intermediate Re/Si-M1′, identified as the zwitterionic intermediate via the transition state Re/Si-TS1′. The optimized geometries demonstrate that the distance between C1 and C11 atoms is 2.06/2.01 Å in Re/Si-TS1′, respectively. The DFT outcomes indicated that the energy barrier Re/Si-TS1′ (17.22/18.24 kcal mol−1) is 3.34/3.56 kcal mol−1 higher than that of Re/Si-TS1 (13.88/14.68 kcal mol−1). These outcomes confirm that the nucleophilic addition of NHC to the Re-face of R1 is both thermodynamically and kinetically favorable than the Michael addition of the NHC catalyst on the Re/Si-face of R2. Therefore, we did not continue the other steps and ignored the second reaction route.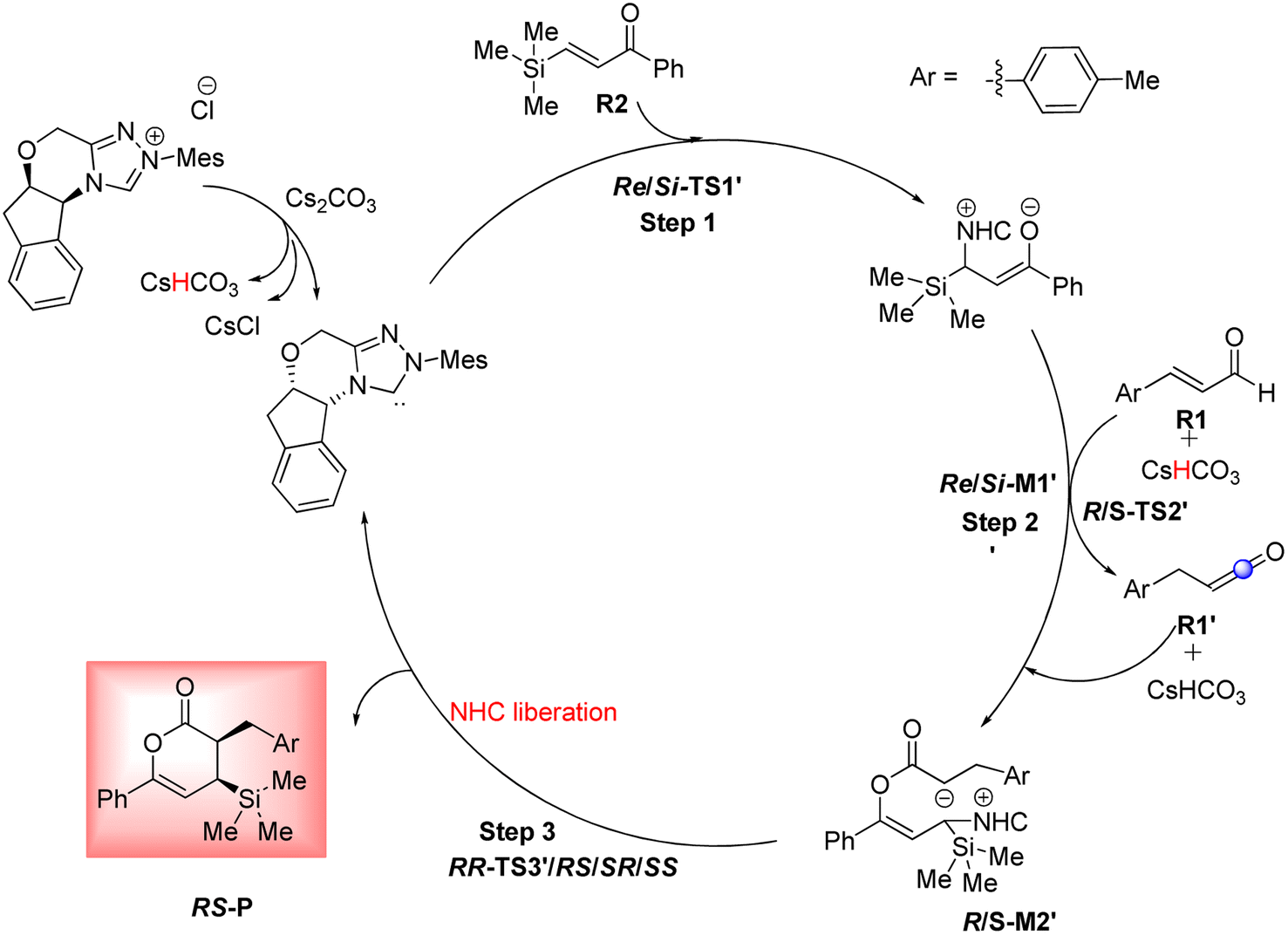 | ||
| Scheme 10 The second reaction mechanism for the NHC-catalyzed annulation reaction of (E)-3-(p-tolyl)acrylaldehyde (R1) and phenyl-3-(trimethylsilyl)prop-2-en-1-one (R2). | ||
3.3. Origin of the stereoselectivity
As discussed above, approaching the intermediate M3 to R2, the two chiral centers (C5 and C11 atoms) formed in the stereoselectivity step. Therefore, the transition states RR-TS4, RS-TS4, SR-TS4, and SS-TS4 are crucial to deeply understanding this mechanism. The energy difference between RS-TS4 and SR-TS4 is 5.08 kcal mol−1, corresponding to an enantiomeric excess of >99% in favor of the RS diastereomer, and the free energy difference between RS-TS4 and RR-TS4 (12.71 kcal mol−1) can be associated with the diastereomeric excess. These results are in good agreement with the experimentally observed ee of 99%. Herein, the origin of the stereoselectivity is described for the NHC-catalyzed [4+2] annulation reaction. The distortion/interaction analysis was performed to comprehensively understand the stereoselectivity of the transition state TS4. Based on Houk's definition,84 each transition state is dissociated into two parts (i.e., M3_part and R2_part). The distortion energy is defined as the energy level difference between the decomposed part and the corresponding optimized structure in the absence of the other part (i.e., ΔE≠dist_M3 and ΔE≠dist_R2), and the total distortion energy (ΔE≠dist_total) is defined as ΔE≠dist_M3 + ΔE≠dist_R2. The interaction energy is obtained through the relationship ΔE≠int = ΔE≠ − ΔE≠dist. The computed distortion and interaction energies are obtained for the reactants in the transition state geometries (Table 1). The distortion/interaction energies were compared for these four transition states. These results show that the distortion energy of RS-TS4 (17.92 kcal mol−1, Table 1) is substantially lower than that of RR-TS4 and SR-TS4 (29.55 and 21.79 kcal mol−1, Table 1). In addition, the most stable interaction energy (−1.68 kcal mol−1, Table 1) of RS-TS4 is presented. The most stable interaction energy and the distortion energy are observed for RS-TS4. These data determine that the RS-configuration product is preferentially formed.| SR | ΔΔG | ΔE≠ | ΔE≠dist (M3) | ΔE≠dist (R2) | ΔE≠dist (total) | ΔEint |
|---|---|---|---|---|---|---|
| RR-TS4 | 12.71 | 30.01 | 15.33 | 14.22 | 29.55 | 0.46 |
| RS-TS4 | 00.00 | 16.24 | 8.58 | 9.34 | 17.92 | −1.68 |
| SR-TS4 | 5.08 | 21.97 | 12.21 | 9.58 | 21.79 | 0.18 |
| SS-TS4 | 16.26 | 33.41 | 16.77 | 10.32 | 27.08 | 6.33 |
The atom in molecules (AIM) analysis was also applied to further gain insight into the stereo-controlling step. and distinguish the nature of interactions of the chiral C5–C11 bond in TS4 using the properties of electron density by indicating the bond critical points (BCPs). These desirable topological parameters, such as electron density, ρ(r), Laplacian of electron density, ∇2ρ(r) and electronic energy density, H(r) interaction for the transition states RR, RS, SR, and SS-TS4, are shown in Table 2. According to this table, the C5–C11 bond should be considered as a partially electrostatic and covalent interaction (1 < |V(r)|/G(r) < 2).
| Compound | ρ(r) | ∇2ρ(r) | H(r) | V(r) | G(r) | |V(r)|/G(r) |
|---|---|---|---|---|---|---|
| RR-TS4 | 0.049 | 0.048 | −0.008 | −0.028 | 0.020 | 1.40 |
| RS-TS4 | 0.059 | 0.049 | −0.011 | −0.034 | 0.023 | 1.48 |
| SR-TS4 | 0.057 | 0.047 | −0.013 | −0.037 | 0.025 | 1.48 |
| SS-TS4 | 0.058 | 0.047 | −0.013 | −0.038 | 0.025 | 1.52 |
The localized orbital locator (LOL) map was calculated (Fig. 3).58,59 The obtained results illustrate that there is usually a strong bond along with a high electron density at the BCP. This parameter shows that the C5–C11 bond (RS-TS4) is stronger than RR, SR, and SS-TS4.
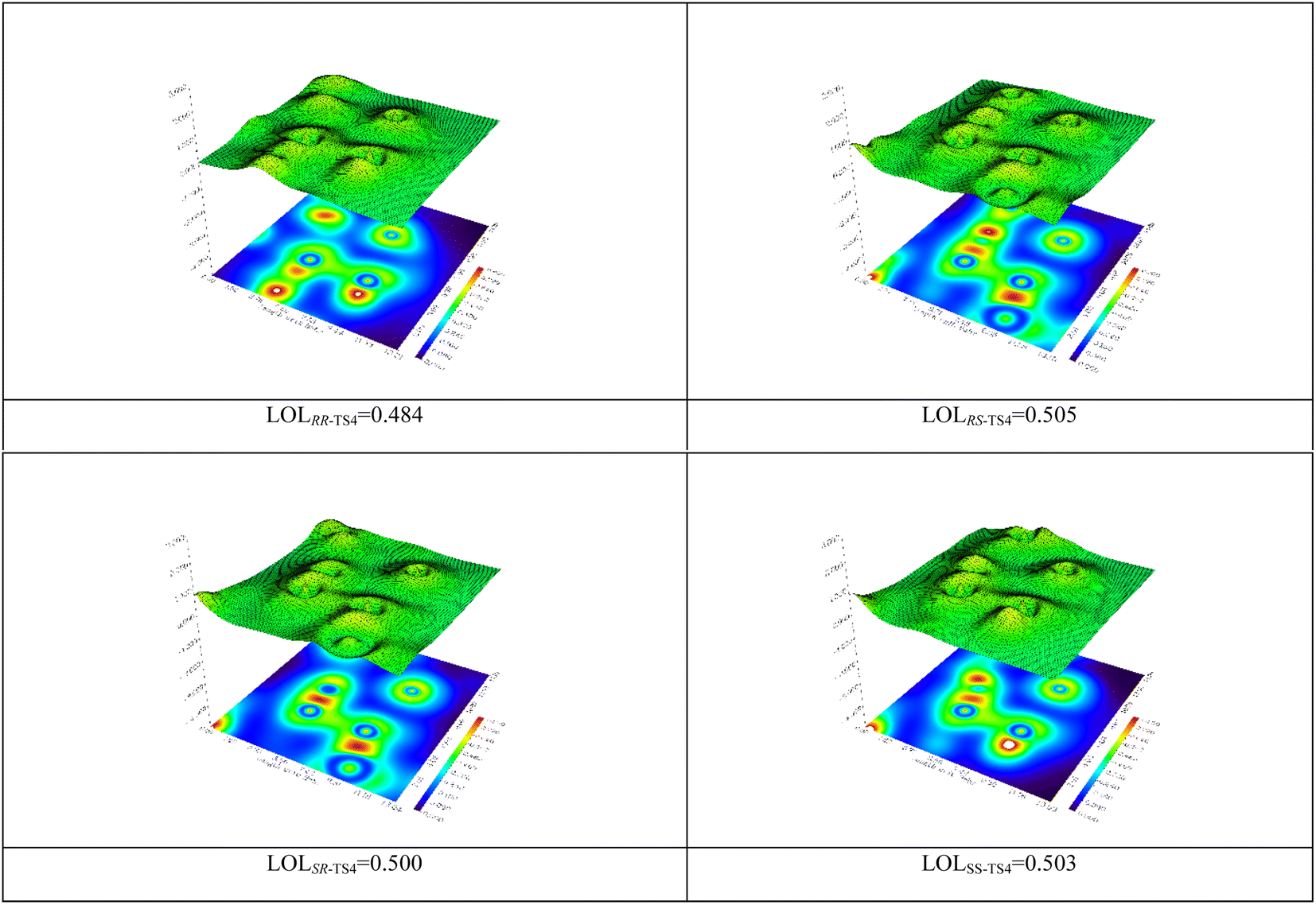 | ||
| Fig. 3 The LOL analysis for the transition states RR-TS4, RS, SR, and SS. | ||
Also, global electron density transfer (GEDT)85–87 is used to disclose the kind of reaction and charge transfer in the transition states TS4. GEDT results are indicated along with the molecular electrostatic potential maps for RR, RS, SR, and SS-TS4 in Fig. 4. For the four transition states, the obtained GEDT indicates that R2 is the negative part and has a low electron density (LUMO), whereas M3 is the positive part with a high electron density (HOMO). The charge transfer of M3 to R2 confirms that the inverse Diels–Alder reaction occurred. It should be mentioned that the GEDT value for the four transition states is more than 0.15 eV. This GEDT value shows that the [4+2] reaction has a polar nature and absorption occurs in all of the transition states TS4.
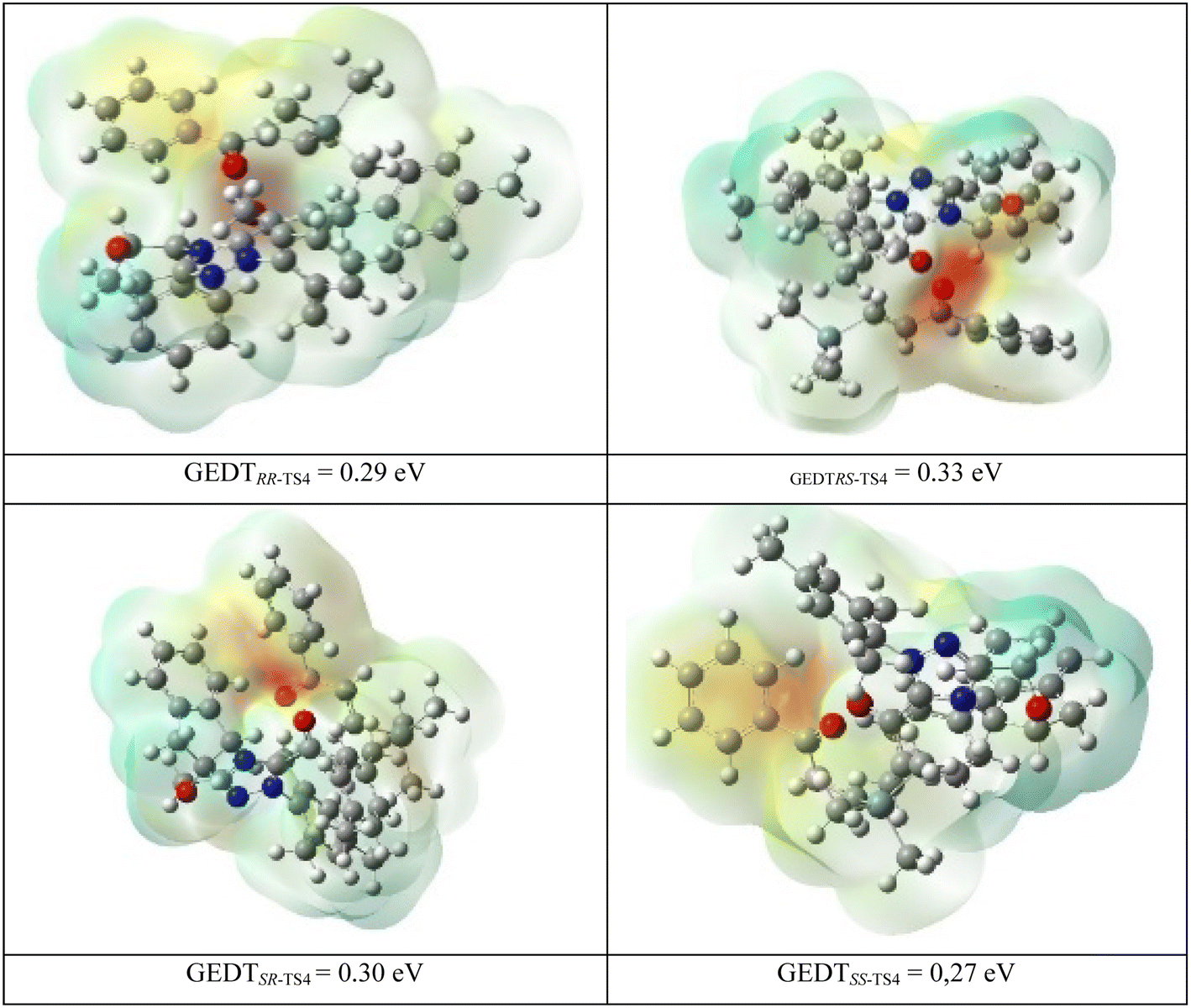 | ||
| Fig. 4 Molecular electrostatic potential (MEP) maps of TSs in the [4+2] annulation reaction step. | ||
3.4. Explication of the effect of substitution
As shown in Table 3, Parr Functions (Pk+ and Pk−) were studied to obtain the effect of the substitution for the R1 and M3 derivatives in the first and fourth steps. We chose chlorine (Cl) and hydrogen (H) atoms in contrast to the methyl group (Me) as the model to investigate the transition states of Re/Si-TS1 and RR, RS, SR, SS-TS4. Table 3 shows that the Cl atom in R1 reduces the barrier energy compared to the hydrogen and methyl derivatives of both the Re and Si-TS1. The local electrophilicity index Pk+ increases the C2 atom of the Cl-R1 through the resonance effect with the phenyl ring. This phenomenon caused the treatment of Cl-R1 and NHC to proceed easily compared to the substituted H and Me-R1. It is observed that the barrier energy for the substituted Re-TS1 is lower than that of the substituted Si-TS1. Also, this table indicates that the substituted Cl atom in M3 decreases the barrier energy compared to the substituted H and Me derivatives in the RR, RS, SR, and SS-TS4. The local nucleophilicity index Pk− value of the C5 atom in Cl-M3 is 0.67 eV, while this value of Pk− for the C5 atom in H and Me-M3 is 0.66 and 0.65 eV, respectively. These results illustrate that the nucleophilicity of the C5 atom in the substituted Cl atom in M3 is increased compared to the substituted H and Me in M3 and leads a reduction in the barrier energy Cl-TS4s. The RS configuration has the lowest barrier energy of all the substituted TS4s.
|
|
|||||
|---|---|---|---|---|---|---|
| R1 | M3 | |||||
| R1 (X) | Pk+ (C2) | Pk− (C2) | Re-TS1 | Si-TS1 | ||
| Cl | 0.20 | −0.10 | 11.88 | 12.83 | ||
| H | 0.16 | −0.11 | 12.91 | 13.88 | ||
| Me | 0.16 | −0.10 | 13.88 | 14.68 | ||
| M3 (X) | Pk+ (C5) | Pk− (C5) | RR-TS4 | RS-TS4 | SR-TS4 | SS-TS4 |
| Cl | 0.09 | 0.67 | 23.61 | 10.71 | 15.38 | 26.38 |
| H | 0.10 | 0.66 | 24.43 | 11.80 | 16.49 | 27.53 |
| Me | 0.10 | 0.65 | 25.18 | 12.47 | 17.55 | 28.73 |
Therefore, in our study, the observed substituent effects were primarily attributed to the electronic factors. The Parr functions employed provide a quantitative measure of local electron density, elucidating the impact of different substituents on the reaction centers. Though steric effects cannot be entirely discounted, especially for significantly bulky substituents, their influence in our system is considered secondary.
3.5. Explication of the nature of the NHC catalyst
To further understand the details of the NHC-catalyzed formation of chiral organosilanes through the [4+2] annulation of R1 and R2, it is essential that the role of the catalyst be revealed in the suggested catalytic cycle. The conceptual DFT (CDFT), FMO, LRI, and NBO analyses were applied to describe the role of NHC in the catalytic mechanism. Global reactivity indices derived from CDFT for the NHC, R1, Re-M1, M3, and R2 are listed in Table 4. Domingo's describes that the molecular electrophilicity feature is determined as the global electrophilicity index (ω), whereas the molecular nucleophilicity feature is quantified as the global nucleophilicity index (N). As depicted in Table 4, the catalyst NHC contains an electrophilicity value of 0.73 eV and a nucleophilicity value of 1.48 eV. On the other hand, the electrophilicity and nucleophilicity values of R1 are 1.508 and 1.56 eV, respectively. Thus, the NHC carbene coordinates successfully with R1. Moreover, it is observed that the interaction of NHC with R1 remarkably decreased the electrophilicity value to 0.79 eV; in contrast, the global nucleophilicity value increased to 2.73 eV in the Re-M1. In addition, the electrophilicity and nucleophilicity values are 0.76 and 3.40 eV for intermediate M3, respectively. Accordingly, the enolate intermediate M3 can now interact successfully with the electrophilic center of R2, which has a global electrophilicity value of 1.56 eV. These outcomes corroborated that the inversion of polarity occurs in R1.| SR | μ | η | ω | N |
|---|---|---|---|---|
| NHC | −0.12 | 0.29 | 0.73 | 1.48 |
| R1 | −0.16 | 0.23 | 1.51 | 1.56 |
| Re-M1 | −0.11 | 0.21 | 0.79 | 2.73 |
| M3 | −0.10 | 0.19 | 0.76 | 3.40 |
| R2 | −0.17 | 0.25 | 1.56 | 0.72 |
The role of NHC catalyst in the catalytic cycle was evaluated by studying the FMO analysis. This theory is defined as the energies of the highest occupied and the lowest unoccupied molecular orbitals, known as the HOMO and LUMO energies, respectively. The FMO diagram for the interaction of R1 with R2 in the presence and absence of the NHC catalyst is shown in Fig. 5. This diagram illustrates that the reaction between R1 and R2 is not possible in the absence of an NHC catalyst at the experimental conditions owing to a higher energy gap (6.55 eV) between the HOMO and LUMO orbitals of R1 and R2, respectively. The incorporation of the NHC catalyst and R1 forms an intermediate M3 in which the HOMO energy level is 5.75 eV, reducing the LUMO–HOMO energy gap to 4.74 eV. This energy gap leads to the reaction with R2 under the experimental conditions. Owing to the existence of the NHC catalyst, the polarity of R1 changes by forming M3 and allows the annulation reaction to take place with R2. Thus, the description of FMO theory is well associated with the CDFT assessment, indicating the role of the NHC catalyst in the catalytic cycle of this reaction.
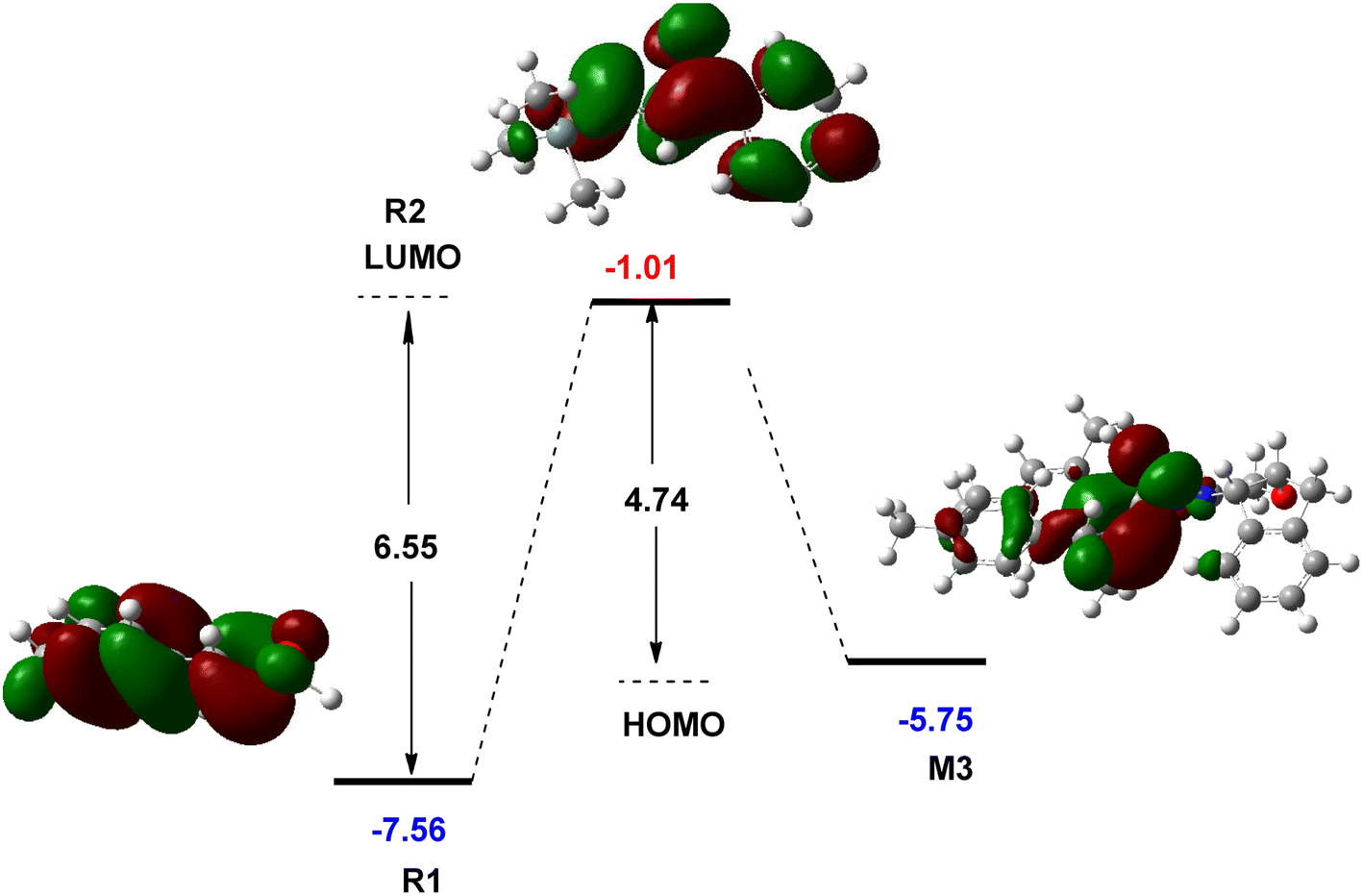 | ||
| Fig. 5 Calculated frontier molecular orbitals of R1, R2, and M3 at the M06-2X/6-31G(d,p) level (unit in eV). | ||
As shown in Table 5, the local reactivity index (LRI) was also assessed to obtain further insights into the nature of NHC at the M06-2X/6-31G(d,p) level of theory. This table shows more details about the Umpolung properties of the NHC catalyst. It is seen that the ωk value of the C2 atom in R1 is 0.24 eV, while the Nk value of the C1 atom in NHC is 1.55 eV. The ωk and Nk values of the O3 atom in R1 are 0.31 eV and 0.21 eV, changing to 0.004 eV and 2.33 eV in Re-M1, respectively. This shows that the polarity of the O3 atom in the generated Re-M1 is changed. The interaction of the C1 and C2 atoms in NHC and R1 changes the nature of the C5 atom in the generated M3. The ωk and Nk values at the C5 atom of M3 were calculated to be 0.07 and 2.24 eV, respectively. The calculated LRI values show that reactant R2 has two electrophilic centers at C11 and C13 with ωk values of 0.43 and 0.35 eV, respectively. Thus, the NHC organocatalyst promoted the annulation reaction between R1 and R2 under the experimental conditions.
| SR | Atom | Pk+ | Pk− | ωk | Nk |
|---|---|---|---|---|---|
| NHC | C1 | 0.11 | 1.05 | 0.08 | 1.55 |
| R1 | C2 | 0.155 | −0.10 | 0.24 | −0.14 |
| O3 | 0.20 | 0.14 | 0.31 | 0.21 | |
| C5 | 0.11 | 0.44 | 0.17 | 0.66 | |
| Re-M1 | C2 | −0.01 | −0.08 | −0.01 | −0.21 |
| O3 | −0.004 | 0.85 | −0.004 | 2.33 | |
| C5 | 0.02 | 0.08 | 0.02 | 0.23 | |
| M3 | O3 | 0.02 | 0.45 | 0.01 | 1.52 |
| C5 | 0.10 | 0.66 | 0.07 | 2.24 | |
| R2 | C13 (CO) | 0.22 | −0.08 | 0.35 | −0.06 |
| C11 (Cβ) | 0.27 | −0.02 | 0.43 | −0.01 |
As shown in Table 6, the NBO charge analysis illustrates that the charge on the C1 atom is enhanced from 0.14 e in NHC to 0.51 e in Re-M1, while the charge value on the C2 atom is reduced from 0.38 e in R1 to 0.10 e in Re-M1. Furthermore, the negative charge on the N atom in NHC reduces (−0.46 e in NHC vs. −0.39 e in Re-M1), whereas the negative charge on the oxygen atom increases (−0.57 e in R1 vs. −0.92 e in Re-M1). Also, the NBO charge of the H4 atom is enhanced slightly from 0.16 e in R1 to 0.17 e in Re-M1. The cooperation of NHC and R1 increases the acidity of the H4 atom, which makes the breakage of the C2–H4 bond easier. NBO results show that the electron is shifted from the NHC catalyst to R1 in the process of NHC + R1 → Re-M1. The increased NBO charge of the O3 and H4 atoms in the conversion of (R1 → Re-M1) would remarkably facilitate the proton transfer of H4 to the O3 atom. Therefore, NHC activates the C2–H4 bond of R1 by enhancing the acidity of the H4 atom and improving the feasibility of bond fracture. In addition, many investigations have indicated that NHC mainly serves as a Lewis base in the annulation reaction.
| SR | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| NHC | R1 | Re-M1 | |||||||
| N | C1 | C2 | O3 | H4 | N | C1 | C2 | O3 | H4 |
| −0.46 | 0.14 | 0.38 | −0.57 | 0.16 | −0.39 | 0.51 | 0.10 | −0.92 | 0.17 |
To predict and assay the catalytic efficiency of the NHC catalyst compared to the achiral NHC (A), we carried out additional calculations on the stereoselectivity-determining step (Table 7). The energy barrier for RS-TS4_NHC (A) is 6.94 kcal mol−1, which is higher than that of the RS-TS4_NHC (B). These results are consistent with the experimental observations, i.e., the NHC provides higher reaction yields than NHC (A).
|
|
||
|---|---|---|---|
| Achiral NHC (A) | Chiral NHC (B) | ||
| SR | ΔG‡ | SR | ΔG‡ |
| RR-TS4 | 17.31 | RR-TS4 | 18.10 |
| RS-TS4 | 6.94 | RS-TS4 | 5.40 |
| SR-TS4 | 6.42 | SR-TS4 | 10.47 |
| SS-TS4 | 20.83 | SS-TS4 | 21.65 |
4. Conclusion
In this article, the detailed mechanisms and origin of stereoselectivity for the asymmetric [4+2] cycloaddition between an (E)-3-(p-tolyl)acrylaldehyde (R1) and phenyl-3-(trimethylsilyl)prop-2-en-1-one (R2) in the presence of NHC catalyst have been theoretically investigated. The most favorable mechanism proceeds through the following steps: the coupling reaction of the NHC catalyst with the (E)-3-(p-tolyl)acrylaldehyde (R1), [1,2]-proton transfer to form the Breslow intermediate, [1,4]-proton transfer to generate the enolate intermediate, [4+2] cycloaddition to form the stereoselective C__C bond, and regeneration of the NHC catalyst to give the final product. The computed results show that the additive CsHCO3 mainly works in proton-assisted transfer to form the Breslow and enolate intermediates via TS2BA and TS3BA routes and plays a key role in the [1,2] and [1,4]-proton transfer process. The [1,2]-proton transfer is a rate-determining step, while the [4+2] cycloaddition reaction is characterized as a stereoselectivity-determining step.Also, the Re/Si pattern leads to the formation of the RS-configurational product, which is consistent with the experimental results.
Distortion energy is the dominant parameter determining stereoselectivity, and the RS-configuration product is preferentially formed. The first and fourth steps investigate the effects of electron-donating (Me) and electron-withdrawing (Cl) groups on the cinnamaldehyde. The electron-withdrawing (Cl) group on R1 and M3 has the lowest barrier energy in the first and fourth steps.
CDFT and FMO analyses have been utilized to deeply understand the role of NHC in the proposed catalytic cycle. NHC serves as a Lewis base and plays a crucial role in the polarity inversion of (E)-3-(p-tolyl)acrylaldehyde (R1).
Data availability
The data that support the findings of this study are available within the ESI† files of this article.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The support from Tarbiat Modares University (TMU) is gratefully acknowledged.References
- T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS.
- G. G. Cash, Pestic. Sci., 1997, 49, 29–34 CrossRef CAS.
- Z. Rappoport, The Chemistry of Functional Groups, 2001, 3 Search PubMed.
- G. K. Min, D. Hernández and T. Skrydstrup, Acc. Chem. Res., 2013, 46, 457–470 CrossRef CAS PubMed.
- M. A. Kacprzynski, S. A. Kazane, T. L. May and A. H. Hoveyda, Org. Lett., 2007, 9, 3187–3190 CrossRef CAS PubMed.
- K.-s. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898–2900 CrossRef CAS PubMed.
- J. M. O'Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712–7715 CrossRef PubMed.
- Y. Zhang, Y. Lu, W. Tang, T. Lu and D. Du, Org. Biomol. Chem., 2014, 12, 3009–3015 RSC.
- M. Wang, Z. Huang, J. Xu and Y. R. Chi, J. Am. Chem. Soc., 2014, 136, 1214–1217 CrossRef CAS PubMed.
- S. Hu, B. Wang, Y. Zhang, W. Tang, M. Fang, T. Lu and D. Du, Org. Biomol. Chem., 2015, 13, 4661–4667 RSC.
- N. Zhou, F. Zhao, L. Wang, X. Gao, X. Zhao and M. Zhang, Org. Lett., 2023, 25, 6072–6076 CrossRef CAS PubMed.
- Y. Li, X.-Y. Duan, C. Yang, Y. Wei, J. Li, X. Ren and J. Qi, J. Org. Chem., 2023, 88, 11299–11309 CrossRef CAS PubMed.
- S.-Y. Zhu, Y. Zhang, W. Wang and X.-P. Hui, Org. Lett., 2017, 19, 5380–5383 CrossRef CAS PubMed.
- Y. Lin, L. Yang, Y. Deng and G. Zhong, Chem. Commun., 2015, 51, 8330–8333 RSC.
- Z. Fu, H. Sun, S. Chen, B. Tiwari, G. Li and Y. R. Chi, Chem. Commun., 2013, 49, 261–263 RSC.
- C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS PubMed.
- S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS PubMed.
- C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232–10236 CrossRef CAS PubMed.
- A. Chan and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 5334–5335 CrossRef CAS PubMed.
- K. Liu, L. Zhang, T. Cao, C. Jin, D. Qiu, Q. Zhou, A. Zettl, P. Yang, S. G. Louie and F. Wang, Nat. Commun., 2014, 5, 4966 CrossRef CAS PubMed.
- X. Y. Chen, F. Xia, J. T. Cheng and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 10644–10647 CrossRef CAS PubMed.
- J. Mo, X. Chen and Y. R. Chi, J. Am. Chem. Soc., 2012, 134, 8810–8813 CrossRef CAS PubMed.
- R. Liu, C. Yu, Z. Xiao, T. Li, X. Wang, Y. Xie and C. Yao, Org. Biomol. Chem., 2014, 12, 1885–1891 RSC.
- L. H. Sun, Z. Q. Liang, W. Q. Jia and S. Ye, Angew. Chem., 2013, 125, 5915–5918 CrossRef.
- X. Fang, X. Chen, H. Lv and Y. R. Chi, Angew. Chem., 2011, 123, 11986–11989 CrossRef.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402–10405 CrossRef CAS PubMed.
- J. Kaeobamrung, M. C. Kozlowski and J. W. Bode, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20661–20665 CrossRef CAS PubMed.
- L. Yang, F. Wang, P. J. Chua, Y. Lv, L.-J. Zhong and G. Zhong, Org. Lett., 2012, 14, 2894–2897 CrossRef CAS PubMed.
- H. Lv, W. Q. Jia, L. H. Sun and S. Ye, Angew. Chem., 2013, 125, 8769–8772 CrossRef.
- Y. Xie, Y. Que, T. Li, L. Zhu, C. Yu and C. Yao, Org. Biomol. Chem., 2015, 13, 1829–1835 RSC.
- R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC.
- M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418–8420 CrossRef CAS PubMed.
- B. E. Maki, A. Chan and K. A. Scheidt, Synthesis, 2008, 1306–1315 CAS.
- Y. Zhang, J. Huang, Y. Guo, L. Li, Z. Fu and W. Huang, Angew. Chem., 2018, 130, 4684–4688 CrossRef.
- N. Munir, N. Gürbüz, M. N. Zafar, E. Evren, B. Şen, M. Aygün and İ. Özdemir, J. Mol. Struct., 2024, 1295, 136679 CrossRef CAS.
- Y. Li, L. Geng, Z. Song and Z. Zhang, New J. Chem., 2023, 47, 21815–21824 RSC.
- Y. Wang, B. Wu, L. Zheng, D. Wei and M. Tang, Org. Chem. Front., 2016, 3, 190–203 RSC.
- J. Zhang, Q. Qiao, Z. Wu, Z. Pang, Q. Shi, Y. Wang, Y. Qiao and D. Wei, Org. Biomol. Chem., 2022, 20, 1662–1670 RSC.
- K. M. Melancon and T. R. Cundari, Org. Biomol. Chem., 2020, 18, 7437–7447 RSC.
- (a) Y. Reddi and R. B. Sunoj, ACS Catal., 2017, 7, 530–537 CrossRef CAS; (b) M. Pareek and R. B. Sunoj, ACS Catal., 2016, 6, 3118–3126 CrossRef CAS.
- Z. Li, D. Wei, Y. Wang, Y. Zhu and M. Tang, J. Org. Chem., 2014, 79, 3069–3078 CrossRef CAS PubMed.
- X. Li, R. Duan, Y. Wang, L.-B. Qu, Z. Li and D. Wei, J. Org. Chem., 2019, 84, 6117–6125 CrossRef CAS PubMed.
- L. Zhang and Y. Wang, Comput. Theor. Chem., 2023, 1220, 114007 CrossRef CAS.
- A. Mavroskoufis, M. Lohani, M. Weber, M. N. Hopkinson and J. P. Götze, Chem. Sci., 2023, 14, 4027–4037 RSC.
- Y. Li, M. Zhang and Z. Zhang, J. Org. Chem., 2023, 88, 12997–13008 CrossRef CAS PubMed.
- Y. Wang, Y. Luo, M. Zhao and Y. Qiao, Mol. Catal., 2023, 551, 113621 CrossRef CAS.
- R. A. Gaussian 09, Inc., Wallingford CT, 2009, 121, 150–166 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- H. Yang and M. W. Wong, J. Org. Chem., 2011, 76, 7399–7405 CrossRef CAS PubMed.
- E. H. Krenske, E. C. Davison, I. T. Forbes, J. A. Warner, A. L. Smith, A. B. Holmes and K. Houk, J. Am. Chem. Soc., 2012, 134, 2434–2441 CrossRef CAS PubMed.
- M. W. Wong and A. M. E. Ng, Aust. J. Chem., 2014, 67, 1100–1109 CrossRef CAS.
- H. Yang and M. W. Wong, J. Am. Chem. Soc., 2013, 135, 5808–5818 CrossRef CAS PubMed.
- H. Xue, D. Jiang, H. Jiang, C. W. Kee, H. Hirao, T. Nishimura, M. W. Wong and C.-H. Tan, J. Org. Chem., 2015, 80, 5745–5752 CrossRef CAS PubMed.
- Y. Li, Z. Zhang and C. Liang, Org. Biomol. Chem., 2018, 16, 9251–9258 RSC.
- Y. Li, Z. Zhang and Z. Li, Mol. Catal., 2020, 492, 111030 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. Zhang, D. G. Truhlar and M. Tang, J. Chem. Theory Comput., 2013, 9, 3965–3977 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- a. J. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- N. Version, Google Scholar There is no corresponding record for this reference, 1988.
- R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS PubMed.
- L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, J. Phys. Chem. A, 2002, 106, 6871–6875 CrossRef CAS.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486–1494 RSC.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2016, 21, 748 CrossRef PubMed.
- P. Pérez, L. R. Domingo, M. Duque-Noreña and E. Chamorro, J. Mol. Struct.: THEOCHEM, 2009, 895, 86–91 CrossRef.
- R. R. Contreras, P. Fuentealba, M. Galván and P. Pérez, Chem. Phys. Lett., 1999, 304, 405–413 CrossRef CAS.
- C. Legault, CYLview User Manual, 2012 Search PubMed.
- Y. Wang, D. Wei, W. Zhang, Y. Wang, Y. Zhu, Y. Jia and M. Tang, Org. Biomol. Chem., 2014, 12, 7503–7514 RSC.
- Y. Li, Y. Zhu, W. Zhang, D. Wei, Y. Ran, Q. Zhao and M. Tang, Phys. Chem. Chem. Phys., 2014, 16, 20001–20008 RSC.
- X.-K. Guo, L.-B. Zhang, D. Wei and J.-L. Niu, Chem. Sci., 2015, 6, 7059–7071 RSC.
- Y. Wang, X. Guo, M. Tang and D. Wei, J. Phys. Chem. A, 2015, 119, 8422–8431 CrossRef CAS PubMed.
- X. Zhang, M. Tang, Y. Wang, Y. Ran, D. Wei, Y. Zhu and W. Zhang, J. Org. Chem., 2016, 81, 868–877 CrossRef CAS PubMed.
- Y. Wang, L. Zheng, D. Wei and M. Tang, Org. Chem. Front., 2015, 2, 874–884 RSC.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10002 CrossRef CAS.
- L. R. Domingo, RSC Adv., 2014, 4, 32415–32428 RSC.
- L. R. Domingo, A. Asensio and P. Arroyo, J. Phys. Org. Chem., 2002, 15, 660–666 CrossRef CAS.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03676j |
| This journal is © The Royal Society of Chemistry 2024 |