
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-based metal–organic framework/graphene oxide composite electrodes for efficient flow-injection amperometric detection of dexamethasone†
Chanida Jakkrawhadab,
Fonthip Makkliangac,
Piyaluk Nurerkab,
Mohamed Siaj d and
Sujittra Poorahong*ab
d and
Sujittra Poorahong*ab
aFunctional Materials and Nanotechnology Center of Excellence, Walailak University, Thasala, Nakhon Si Thammarat 80160, Thailand. E-mail: sujittra.po@mail.wu.ac.th
bDepartment of Chemistry, School of Science, Walailak University, Thasala, Nakhon Si Thammarat 80160, Thailand
cSchool of Languages and General Education, Walailak University, Thasala, Nakhon Si Thammarat 80160, Thailand
dDepartment of Chemistry, Université du Québec à Montréal, Montréal, Québec H3C 3P8, Canada
First published on 31st July 2024
Abstract
A highly stable flow-injection amperometric sensor for dexamethasone (DEX) was developed using a pencil graphite electrode (PGE) modified with Fe-based metal organic frameworks, MIL-100(Fe) and graphene oxide composite materials (MIL-100(Fe)/GO). Scanning electron microscopy and energy-dispersive X-ray spectroscopy, transmission electron microscopy, powder X-ray diffraction, and Fourier-transform infrared spectroscopy were used to characterize the MIL-100(Fe) composites. The MIL-100(Fe)/GO-modified PGE (denoted MIL-100(Fe)/GO/PGE) was further electrochemically characterized using cyclic voltammetry. As an electrode material, MIL-100(Fe) is a sensing element that undergoes oxidation from Fe(II)-MOF to Fe(III)-MOF, and GO possesses high conductivity and a large surface area, which exhibits high absorbability. In the presence of DEX, Fe(III) is reduced, which accelerates electron transfer at the electrode interface. Therefore, DEX can be quantitatively detected by analyzing the anodic current of MIL-100(Fe). When coupled with amperometric flow injection analysis, excellent performance can be obtained even when a low detection potential is applied (+0.10 V vs. Ag/AgCl). The concentration was linear in the range 0.10–5.0 μM and 0.010–5.0 mM with LOD of 0.030 μM based on 3(sd/slope). The modified electrode also exhibited a remarkably stable response under optimized conditions, and up to 55 injections can be used per electrode. The sensor exhibits high repeatability, reproducibility, and anti-interference properties when used for DEX detection. The effective determination of dexamethasone in real pharmaceutical and cosmetic samples demonstrated the feasibility of the electrochemical sensor, and the results were in good agreement with those obtained from the HPLC-DAD analysis. Acceptable percentage recoveries from the spiked pharmaceutical and cosmetic samples were obtained, ranging from 93–111% for this new method compared with 84–107% for the HPLC-DAD standard method.
Introduction
Dexamethasone (DEX) is a corticosteroid primarily used for anti-inflammatory and immunosuppressive properties in medical treatment. In the field of dermatology, dexamethasone may be prescribed by medical professionals to treat specific skin conditions, such as eczema, psoriasis, or dermatitis.1 However, it is important to note that the use of dexamethasone in whitening cosmetics or skincare products is uncommon, and generally not recommended. Owing to a variety of unwanted side effects, including hypertension, diabetes, osteoporosis, skin atrophy, and suppression of the hypothalamic–pituitary–adrenal axis, the long-term administration of corticosteroids must be closely monitored, particularly in pediatric patients.2 Several analytical techniques have been reported for the qualitative and quantitative determination of DEX, including radioimmunoassay,3 liquid chromatography-tandem mass spectrometry,4 high-performance liquid chromatography,5 chemiluminescence,6 and thin-layer chromatography.7 Although these methods have a high accuracy with a low limit of detection, major drawbacks such as cost-effectiveness, time consumption, and complex preconcentration of samples have also been reported.8 Therefore, it is necessary to develop an alternative technique that is simple and inexpensive for detecting DEX in cosmetics.Electrochemical detection has lately gained interest in the monitoring field because of its high sensitivity, simple instrumentation, short time consumption, and low cost.9,10 Various electrochemical methods and modified electrodes have been used to detect DEX. Square wave voltammetry (SWV) with a multiwalled carbon nanotubes (MWCNTs) modified electrode was effective in detecting the oxidation of DEX (detection potential of +0.8 V) with a low detection limit.11 Other studies also used SWV with MWCNTs-modified graphite electrode, which can be observed in both reduction and oxidation peaks at −1.3 V and +0.8 V, respectively.8 Additionally, by using graphene nanoplate-modified glassy carbon electrode was able to shift the reduction peak from −1.33 to −1.29 V due to electron mediator property of a graphene nanoplate when compared with the bare electrode.12 Although a high sensitivity can be obtained, the detection of DEX in both the reduction and oxidation pathways requires a high detection potential. This can be a complication when applied to real cosmetic sample analysis due to the presence of interferences can influence the detection. To address these limitations, novel sensing materials with high sensitivity and selectivity, rapid response time and low detection potential need to be explored.
Among the various sensing materials reported to date, nanomaterials have recently found a wide range of applications in electrochemical sensor construction. Metal–organic frameworks (MOFs) have many advantages such as a large specific surface area, high porosity and loading efficiency, easy functionalization, tunable pore sizes, biocompatibility and biodegradability, flexible networks, and topological diversity.13 Therefore, MOFs have attracted attention for various applications. Among these applications, MOF-based nanomaterials have shown outstanding performance in the development of electrochemical sensors.14 However, single-phase MOFs exhibit limited performance due to their intrinsically weak properties such as electrical conductivity, electroactivity, and stability. Therefore, composite MOFs with other materials have been developed via combining with metallic nanoparticles to improve the performance of sensors through synergistic effects.9,15
In addition, carbon-based materials such as graphene, graphene oxide and carbon nanotubes are characterized by good conductivity, large specific surface area, and diverse functional groups. Graphene oxide (GO) have often been used for the electrochemical detection of organic compounds, because of their effective surface area, satisfactory electrical conductivity, improved charge transportation, outstanding electrochemical stability, and strong adsorption abilities through π–π interactions with analytes.16,17 Considering the characteristics of GO, which is a good for increasing the base electrode surface area.
Therefore, this work aims to present a novel method for detecting dexamethasone (DEX) using a pencil graphite electrode modified with a MIL-100(Fe)–graphene oxide composite (MIL-100(Fe)/GO/PGE). This approach significantly enhances the anodic current response at a low applied potential (+0.1 V vs. Ag/AgCl), resulting in improved sensitivity compared to bare electrodes. When combined with flow injection analysis, the method offers advantages such as simplicity, rapid response, high stability, and selectivity. These improvements suggest potential applications in healthcare, the pharmaceutical industry, and personal wellness.
Experimental
Preparation of nanomaterials for modified electrode
Graphene oxide (GO) and MIL-100(Fe) synthesis procedures are described in the ESI.† Then, 10 mg mL−1 of MIL-100(Fe) and GO; denoted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of MIL-100(Fe):GO were dispersed in 200 μL of DI water and 15 μL of Nafion as the binder. Then 2.0 μL of the dispersed suspension was drop-cast onto the circular surface (diameter 2.0 mm) of a cleaned PGE and dried at room temperature overnight before use. The PGE modified with only GO or MIL-100(Fe) were also prepared using the same procedure.
:1 ratio of MIL-100(Fe):GO were dispersed in 200 μL of DI water and 15 μL of Nafion as the binder. Then 2.0 μL of the dispersed suspension was drop-cast onto the circular surface (diameter 2.0 mm) of a cleaned PGE and dried at room temperature overnight before use. The PGE modified with only GO or MIL-100(Fe) were also prepared using the same procedure.
Flow-based amperometric set-up
Electrochemical measurements were performed using an Autolab potentio-galvanostat (Metrohm Autolab, PGSTAT302N) in a flow injection system coupled with a three-electrode setup. The system shown in Fig. 1 consists of a peristaltic pump to drive the carrier buffer solution (0.04 M Britton–Robinson (BR) buffer, pH 2), and a six-port injection valve to inject the working solution while controlling the sample volume. A laboratory-built flow cell was constructed from acrylic plastic containing the MIL-100(Fe)/GO/PGE working electrode, Ag/AgCl in a 3 M KCl reference electrode, and a stainless-steel tube counter electrode. The sample flow rate and volume, detection potential and ratio between the MIL-100(Fe) and GO were optimized to obtain the highest response, good repeatability, and reproducibility. All the electrochemical procedures were performed at room temperature. The output data were recorded and analyzed using the NOVA 1.11 software.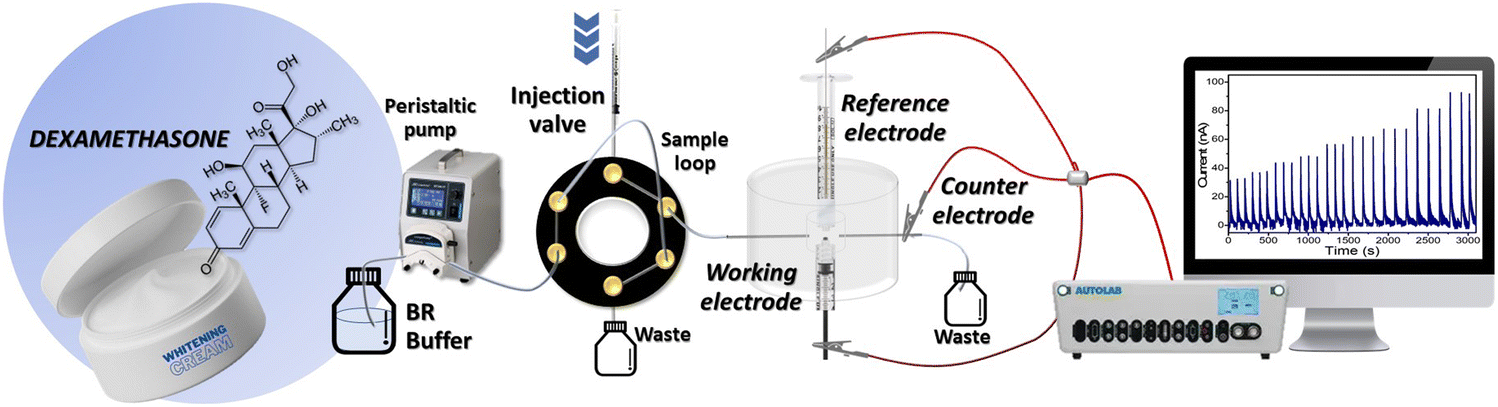 | ||
| Fig. 1 Flow injection amperometric detection system. | ||
Real sample analysis
Three dexamethasone pharmaceutical products and five whitening cream samples were obtained from local drugstores and cosmetics shops. All samples were prepared by the standard addition method using 2.00 g of samples dissolved in 0.04 M BR buffer pH 2, and the volume was adjusted to 25.0 mL.Standard DEX concentrations of 10, 20, 50, 100, and 200 μM were spiked into 1.0 mL of prepared solutions. The flow injection amperometric analysis was performed under optimal conditions, and the standard addition calibration curve was plotted. The calculated % relative error and % recovery were used to estimate the accuracy of the method. The real samples were confirmed by high-performance liquid chromatography coupled with diode array detection (HPLC-DAD) at a detection wavelength of 236 nm. The analytical column is a Vertical C18 column (5 μm particle size, 250 mm × 4.6 mm I.D.). Water and acetonitrile solution at a ratio of 50:50 was used as the mobile phase at a flow rate of 0.8 mL min−1 using isocratic elution.
Results and discussion
Materials characterization
The morphologies of prepared materials were characterized using SEM and TEM. Fig. 2A shows the SEM image of GO, revealing thin and wrinkled sheets of GO. The SEM image of MIL-100(Fe) nanocrystals (Fig. 2B) shows an octahedral morphology belonging to cubic (isometric) crystals with a particle size range of 70–200 nm.18 In addition, from the SEM images, some small irregularly shaped particles can be observed, which are probably due to the structural collapse and agglomeration of MIL-100(Fe). Owing to the agglomeration of MIL-100(Fe) particles, an aggregated spread of MIL-100(Fe) occurred on the GO sheet when a composite MIL-100(Fe)/GO material was generated (Fig. 2C). The anti-aggregation effect of GO emphasizes the more exposed active edges of MIL-100(Fe), allowing a high specific surface area to be obtained. The specific surface area of GO, MIL-100(Fe), and MIL-100(Fe)/GO samples was studied using the nitrogen adsorption–desorption isotherm, as depicted in Fig. S1.† The nitrogen adsorption–desorption experiment revealed a Type IV isotherm for all samples, suggesting a mesoporous structure.19 MIL-100(Fe) exhibited a BET surface area of 303.09 m2 g−1, while GO displayed a surface area of 297.12 m2 g−1. When MIL-100(Fe) was combined with GO, the surface area increased to 669.18 m2 g−1. The TEM images of the MIL-100(Fe)/GO composite (Fig. 2D and E) show the sheet-like graphene oxide structure and anchoring of MIL-100(Fe) resulted in a rough surface in a random manner. When zooming into the surface, an octahedral morphology with a smooth surface is observed. Additionally, EDS elemental analysis further confirmed the presence of Fe in the MIL-100(Fe) sample (Fig. S2†). | ||
| Fig. 2 SEM images of (A) GO, (B) MIL-100(Fe), and (C) MIL-100(Fe)/GO, (D) and (E) TEM images of MIL-100(Fe)/GO. | ||
The PXRD crystallinity pattern of the synthesized MIL-100(Fe) was compared to the simulated MIL-100(Fe) (Fig. 3A).18 The main characteristic diffraction peaks of synthesized MIL-100(Fe) were 2θ equal to 10.3, 11.0, and 20.1 which matched well with the simulated MIL-100(Fe),18 confirming the high purity and good crystallinity of MIL-100(Fe). The patterns of the MIL-100(Fe)/GO composites are similar to those observed for MIL-100(Fe), indicating that the structure of MIL-100(Fe) is well preserved in the MIL-100(Fe)/GO composites. The GO peak in the composite materials is barely observed. This could be due to the overlapping of the signals, as the XRD peak of GO appeared close to the peak of MIL-100(Fe). The MIL-100(Fe) and GO phases existing in the MIL-100(Fe)/GO composite is further confirmed by comparing the FTIR spectra of the MIL-100(Fe), GO, and MIL-100(Fe)/GO composite (Fig. 3B). The peaks at approximately 3450 cm−1 in all three spectra were assigned to the stretching vibrations of the surface-adsorbed H2O. The FTIR spectrum of the synthesized MIL-100(Fe) showed a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration peak at 1710–1720 cm−1 from the residual trimesic acid ligand,8 indicating deprotonation of the ligand and coordination with Fe3+ ions, which is also supported by the presence of the absorption band of Fe–O stretching vibrations at 615 cm−1.20,21 Additionally, in the low wavenumber (ν < 1000 cm−1) region, the characteristic vibrations appearing at 755 and 709 cm−1 for MIL-100(Fe) can be ascribed to the characteristic C–H bond of the benzene ring. The characteristic peaks appeared in both the MIL-100(Fe) and MIL-100(Fe)/GO composites, demonstrating the preservation of the MIL-100(Fe) structure.
O stretching vibration peak at 1710–1720 cm−1 from the residual trimesic acid ligand,8 indicating deprotonation of the ligand and coordination with Fe3+ ions, which is also supported by the presence of the absorption band of Fe–O stretching vibrations at 615 cm−1.20,21 Additionally, in the low wavenumber (ν < 1000 cm−1) region, the characteristic vibrations appearing at 755 and 709 cm−1 for MIL-100(Fe) can be ascribed to the characteristic C–H bond of the benzene ring. The characteristic peaks appeared in both the MIL-100(Fe) and MIL-100(Fe)/GO composites, demonstrating the preservation of the MIL-100(Fe) structure.
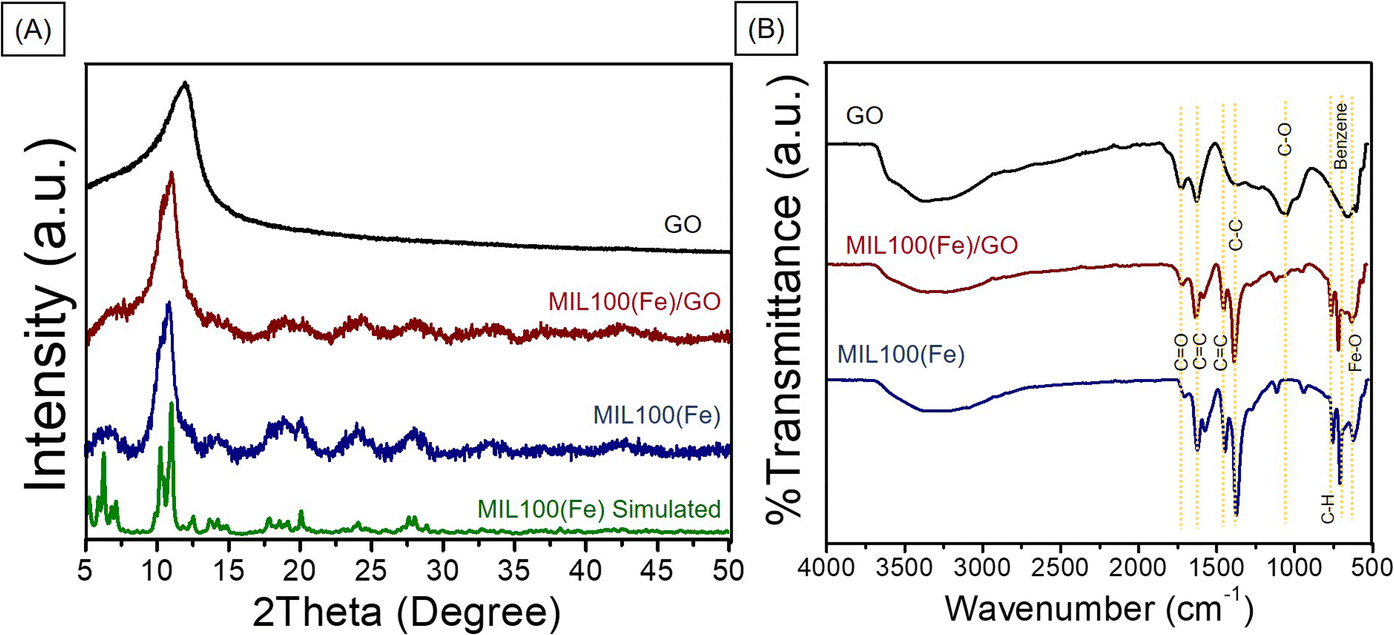 | ||
| Fig. 3 (A) PXRD patterns and (B) FTIR spectra of GO, MIL-100(Fe), and MIL-100(Fe)/GO composites. | ||
Cyclic voltammetry (CV) of ferric/ferrocyanide was performed to evaluate the electroactivity of the modified surfaces i.e., GO, MIL-100(Fe), and MIL-100(Fe)/GO composites. The CV parameters were set at a 50 mV s−1 scan rate and applied potentials from −0.5 to +1.0 V in 10 mM ferric/ferro-cyanide redox couples in 0.1 M KCl. The voltammograms in Fig. S3† show that GO/PGE has the highest faradaic and background currents among the other electrodes because of its effective surface area and high electrical conductivity.22 In comparison, MIL-100(Fe)/PGE exhibited the lowest current because of the limited performance of MIL-100(Fe), which has low electrical conductivity. The peak current of MIL-100(Fe)/GO/PGE was higher than that of MIL-100(Fe)/PGE, emphasizing the increasing conductivity of the electrode materials in the presence of GO. However, the current is still lower than pure GO electrode, this could be due to the agglomeration of MIL-100(Fe) with GO on the working electrode surface, which can limit the electrochemical transfer.23
The electrochemical impedance spectroscopy (EIS) analysis was further conducted to investigate the electron transfer resistance between the solution and electrode surface (Ret). EIS was studied by setting a frequency range from 100 kHz to 0.1 Hz using an alternative voltage with an amplitude of 10 mV, superimposed on a DC potential of 0.20 V (vs. Ag/AgCl). The Nyquist plots were shown in Fig. S4.† The resistance of the electron transfer between the solution and electrode surface (Ret), resistance of the solution (Rs), constant phase element (CPE), and Warburg element (ZW) of the Randles equivalent circuit model (inset) were obtained from the fitted data of the Nyquist plots. GO/PGE had a Ret value of 1.15 kΩ. This was lower than 5.15 kΩ and 17.50 kΩ for the MIL-100(Fe)/GO/PGE and MIL-100(Fe)/PGE, respectively. GO possesses an obviously low resistance. Its high electrical conductivity significantly decreased the resistance of MIL-100(Fe)/GO, thereby enhancing the electrical conductivity of the material used to construct the electrode surface.
Electrocatalytic behavior of DEX at MIL-100(Fe)/GO modified PGE
The electrocatalytic activity of the modified electrodes toward DEX was evaluated using CV with 1.0 mM DEX in 0.04 M BR buffer at pH 2 at a scan rate of 50 mV s−1, as shown in Fig. S5† using bare PGE, GO/PGE, MIL-100(Fe)/PGE, and MIL-100(Fe)/GO/PGE. Considering the anodic part of the bare PGE (Fig. S5A†) and GO/PGE (Fig. S5B†), the oxidation peak of DEX occurred at +0.62 V. However, the anodic current from the GO/PGE was higher than the bare PGE. This is because the superior electrical conductivity and strong adsorption ability of GO are responsible for the excellent electrocatalytic activity.23 For MIL-100(Fe) modified PGE, shown in Fig. S5C,† and MIL-100(Fe)/GO/PGE (Fig. S5D†), two oxidation peaks were observed around +0.25 V and +0.60 V when measured 1.0 mM DEX in 0.04 M BR buffer pH 2. Similar to bare and GO/PGE, at around +0.60 V due to the direct oxidation of DEX on the surface of electrode. A redox couple with formal potential of +0.15 V (vs. Ag/AgCl) were observed in the CV of blank BR buffer, indicating the presence of Fe(II)-MOF/Fe(III)-MOF redox couple.24 After the addition of DEX, the significantly increased anodic and cathodic currents of Fe(II)-MOF/Fe(III)-MOF implied that DEX diffused from the solution to the electrode surface and reduced Fe(III)-MOF to Fe(II)-MOF, whereas DEX was oxidized to 21-dehydro-dexamethasone.25 To verify the influence of DEX on the detection system, the concentration of DEX was decreased from 1.0 mM to 0.5 mM. As illustrated in Fig. S6,† reducing the concentration of DEX resulted in a decrease in peak current, which affected both the direct oxidation potential of DEX at +0.6 V and the Fe(III)-MOF/Fe(II)-MOF peak at +0.15 V vs. Ag/AgCl. Therefore, the amount of DEX can be determined by the increase in the anodic current of the MIL-100(Fe)-MOF. The presence of the Fe(II)-MOF/Fe(III)-MOF redox couple in the detection system was validated by analyzing the chemical state of iron using X-ray photoelectron spectroscopy (XPS). Fig. 4 illustrates the high-resolution scanning XPS spectra of Fe2p of the as-prepared MIL-100(Fe) and used to modified electrode. In the freshly prepared MIL-100(Fe), the binding energy positions of Fe2p1/2 and Fe2p3/2 peaks were centered at 725.8 eV and 711.8 eV, respectively and their separation was 14 eV, which were attributed to the +3 oxidation chemical state of Fe.24,26 After using MIL-100(Fe) as the electrode material and electrochemically cleaned by CV, the binding energies of Fe2p1/2 and Fe2p3/2 of Fe(III) species remained unchanged. Nevertheless, new peaks at 723.5 and 709.5 eV were detected, corresponding to the Fe2p1/2 and Fe2p3/2 of Fe(II), respectively.24,26 The results suggested the existence of mixed-valence iron in the MIL-100(Fe) under detection conditions. Based on this observation, the following detection mechanism is proposed (Fig. 5). At the applied potential, Fe(II)-MOF on the PGE surface was reduced to Fe(III)-MOF. In the presence of DEX, Fe(III)-MOF is oxidized by DEX, which causes a shift in the reaction equilibrium and accelerates electron transfer at the electrode interface. This is advantageous because DEX can be detected at low applied potential. Furthermore, when PGE was modified with MIL-100(Fe)/GO, the peak current increased significantly, implying that the outstanding sensing performance could be attributed to the large specific surface area of MIL-100(Fe) MOF-based with highly conductive GO materials due to the synergistic effect.27,28 It can be concluded that the MIL-100(Fe)/GO composite material is an excellent alternative for amperometric detection of DEX coupled with a flow injection system for convenient and fast measurement. In addition, the electrochemical reaction of DEX based on different pH solutions were studied in the pH range from 2.0 to 8.0 using MIL-100(Fe)/GO/PGE. As shown in Fig. S7,† the anodic peak of MIL-100 (Fe) at +0.25 V and DEX at +0.6 V decreased and disappeared as the pH of the BR buffer increased. It is evident from our findings that the BR buffer at pH 2 provides the optimal condition for detecting DEX.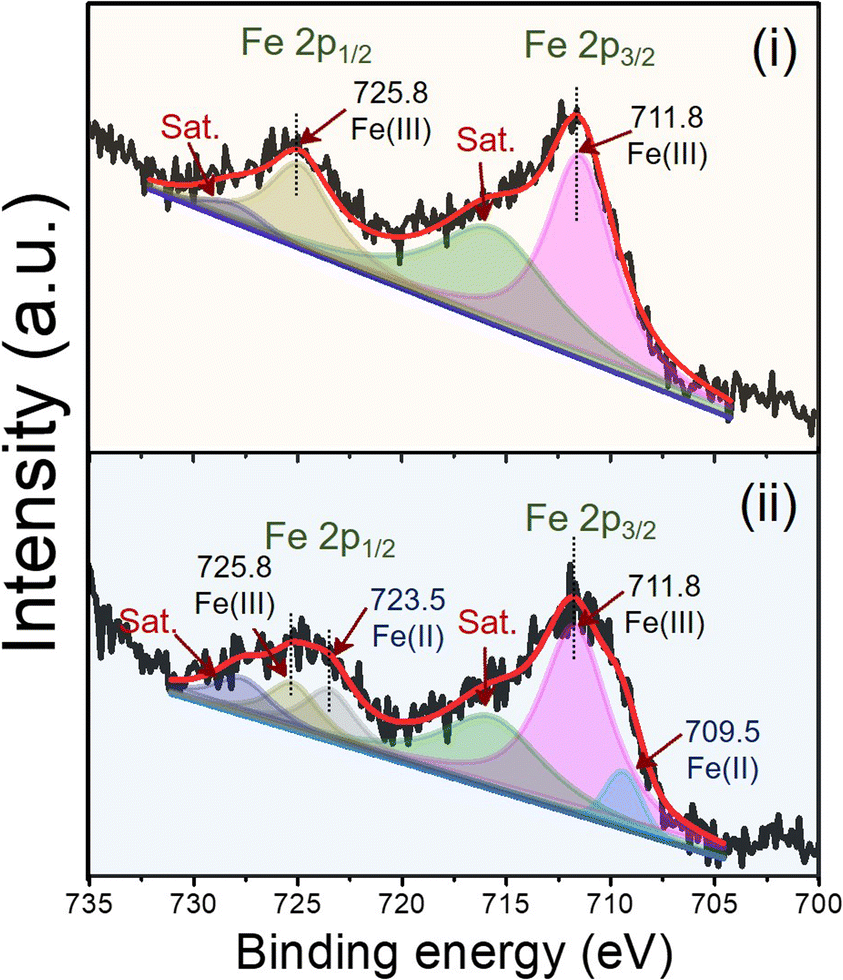 | ||
| Fig. 4 Deconvoluted XPS spectra of Fe2p of (i) as-prepared MIL-100(Fe) and (ii) MIL-100(Fe) as the electrode material and electrochemically cleaned by CV. | ||
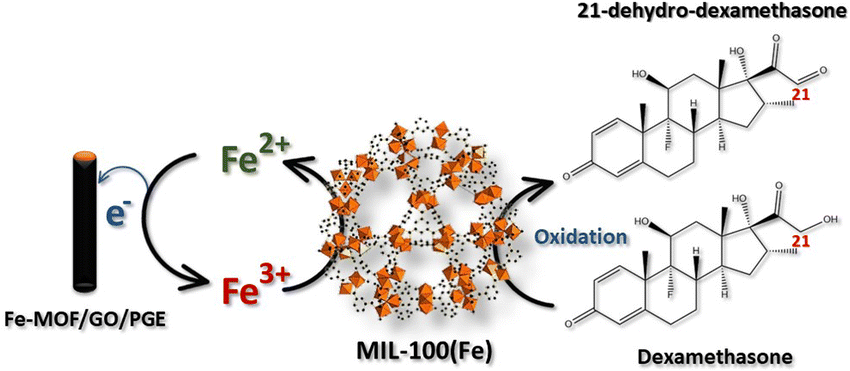 | ||
| Fig. 5 Proposed mechanism of the electrochemical behavior of DEX on the MIL-100(Fe)/GO/PGE. | ||
Amperometric flow injection analysis optimizations
Flow injection amperometric sensors offer several benefits over traditional electrochemical techniques, such as high sensitivity and selectivity. This enables the detection of low concentrations of analytes in complex matrices. The continuous flow of the sample enhances mass transport to the working electrode, resulting in rapid response times and improved signal-to-noise ratios.29,30 To obtain the conditions for the best performance of the amperometric flow injection analysis for DEX, the ratio of MIL-100(Fe) to GO, detection potential and sample volume and flowrate were optimized by injecting 1.0 mM DEX using 0.04 M BR (pH 2) as a carrier buffer. The optimizations were performed by varied single parameter while kept other parameters constant. First, the sample volume was studied with respect to flow rate. As shown in Fig. 6A, the signal increased when the sample volume increased to 300 μL and then decreased. These higher sample loop volumes most likely intensified electrode deactivation via adsorption of DEX or its electro-oxidation products to the point where it was insufficient to support surface electrochemical cleaning.31 The same trend was observed for the sample flow rate, the response increased up to 1250 μL min−1 then decreased. This is due to at high flow rate, mass transport increase by time unit on the electrode surface making the analytical signal (current) increased and the Nernst diffusion layer decreased. However, beyond a certain flow rate, the thickness of the layer stabilizes, causing the current to plateau despite further increases in flow rate.32 However, our research, the current decreased after a flow rate of 1250 μL min−1. This may be this may be attributed to the insufficient contact time between DEX and MIL-100(Fe)/GO at higher flow rates or the detachment of some MIL-100(Fe)/GO from the surface.33,34 Although we did not directly observe the MIL-100(Fe)/GO detaching from the PGE surface. Therefore, a flow rate of 1250 μL min−1 and a sample volume of 300 μL were chosen as the optimum conditions.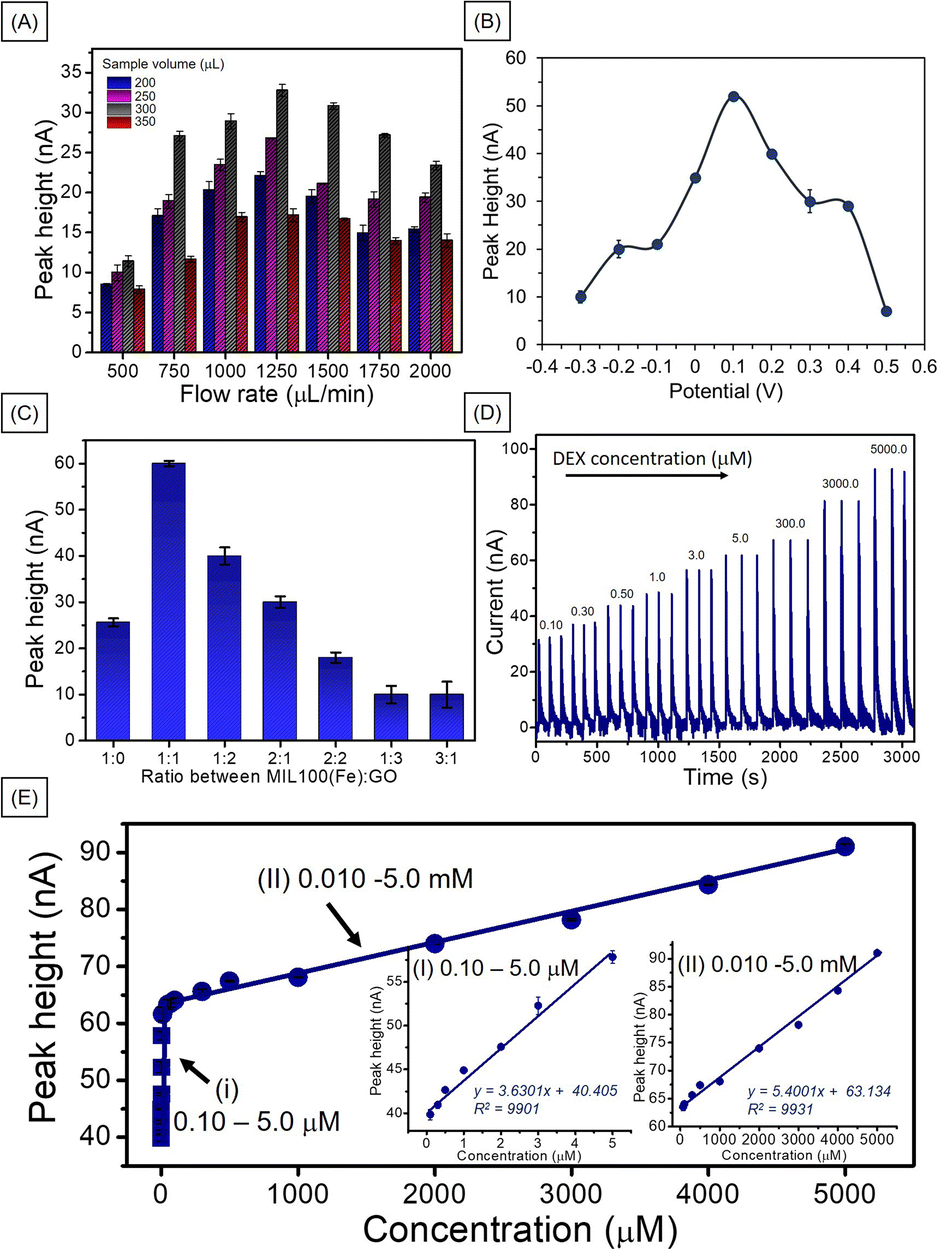 | ||
| Fig. 6 (A) Histograms represent effect of flow rate and sample volume on current response of 1.0 mM DEX in a carrier stream of 0.04 M BR buffer (pH 2), (B) peak height of MIL-100(Fe)/GO/PGE at different working potentials, (C) peak heights of different material ratios of MIL-100(Fe) and GO, (D) examples of flow injection amperometric responses of different concentrations of DEX measured at MIL-100(Fe)/GO/PGE under optimal conditions, and (E) corresponding calibration plot of amperometric signal versus DEX concentration. Inset (I) in the range of 0.1–5.0 μM and (II) 0.01–5 mM of DEX in BR (pH 2). | ||
The detection potential was the next parameter to be studied. The amperometric detection of the MIL-100(Fe)/GO-modified PGE working electrode at a constant potential was tested between −0.3 and +0.5 V of the MIL-100(Fe)/GO modified PGE (Fig. 6B). The response current increased as the applied potential increase from −0.3 to +0.1 V. Any further increase in the applied potential caused the response current to decrease, indicating that the best performance occurred at +0.1 V. The system requires a low applied potential to detect DEX, which is advantageous for real-sample applications. Due to in cosmetic sample the electroactive interferences such as glucose, methyl paraben (MP), ethyl paraben (EP), propyl paraben (PP), butyl paraben (BP), butylated hydroxyanisole (BHA), and butylated hydroxytoluene (BHT) may presented and normally they have oxidation potential around 0.2 to 0.8 V.35,36
The ratio of MIL-100(Fe) to GO was optimized. As shown in Fig. 6C, the best performance was obtained at MIL-100(Fe):GO ratio of 1:1. When the material ratio increased, a noticeable decrease in the signal was observed, which may be attributed to the increased presence of MIL-100(Fe), causing a decrease in the electrical conductivity of the electrode. In addition, an excessive increase in the amount of GO could result in the overlapping of the GO sheet layers, obstructing electron flow.18 When comparing the response of MIL-100(Fe)/GO/PGE and MIL-100(Fe)/PGE at applied potential +0.1 V with bare PGE, and GO/PGE at applied potential +0.25 V, the result shows that the MIL-100(Fe)/GO/PGE demonstrated the highest response to dexamethasone detection (Fig. S8†). The MIL-100(Fe)/GO/PGE exhibited a 7.9 times improvement over the bare PGE and approximately 2.9 and 2.3 times better performance than GO and MIL-100(Fe), respectively.
To verify the working range of MIL-100(Fe)/GO/PGE with flow injection analysis, DEX detection was performed under optimum conditions. Each DEX concentration was analyzed in 3 replicates. Example responses of the MIL-100(Fe)/GO-modified electrode to various DEX concentrations are shown in Fig. 6D. A linear relationship between the peak height (nA) and the DEX concentrations between 0.10–5.0 μM and 0.01–5.0 mM (Fig. 6E). The limit of detection (LOD) was 0.030 μM based on 3(sd/slope). This LOD value was lower than the recommended safe value (<1.27 mM) for low-potency steroids as the safest agents for long-term use on large surface areas, on the face or areas of the body with thinner skin, and in children.37 Therefore, this method could certainly be used to determine whether DEX in cosmetics is above this safety value.
Method validation
Compared to the previous research in Table 1, the developed DEX sensor has the lowest detection potential and the limit of detection in this work (0.03 μM) is in the same range as some other works. This is a result of utilizing MIL-100(Fe) as a sensing material for amperometric analysis coupled with a flow injection system, as well as a high sensitivity with a broad detection range. The outstanding sensing performance can be attributed to the large specific surface area of MOF-based nanomaterials resulting from the combination of MIL-100(Fe) with highly conductive materials, which is GO enhancing sensor performance via synergistic effects.27,28| Electrodes | Ep (V) | LOD | Linear range | Applications | Ref. |
|---|---|---|---|---|---|
| a Pencil graphite electrode (PGE), carbon ionic liquid electrode (CILE), electrochemical paper-based analytical devices (ePADs), amperometry (AM), glassy carbon electrode (GCE), cyclic voltammetry (CV), multiwalled carbon nanotubes (MWCNTs), square wave voltammetry (SWV), amalgam film silver-based electrode (Hg(Ag)FE), hanging mercury electrode (HMDE). | |||||
| MIL-100(Fe)/GO/PGE | +0.1 | 0.03 μM | 0.1–5.0 μM and 0.01–5 mM | Applied to pharmaceutical and cosmetic samples compared with HPLC, recovery = 93–111% | This work |
| MWCNTs/PGE | +0.8 | 0.09 μM | 0.15–100 μM | Applied to pharmaceutical and human urine samples, recovery = 99–103% | 11 |
| Graphene/GCE | −1.3 | 0.015 μM | 0.1–50 μM and 0.05–5 mM | Applied to human blood serum | 12 |
| Fe3O4/PANI–Cu(II)/CILE | +0.72 | 0.003 μM | 0.05–30 μM | Applied to pharmaceutical samples, recovery 97.0–102.0% | 38 |
| Fe3O4/PANI–Cu(II)/α-Fe2O3/CILE | +0.6 | 0.015 μM | 0.05–100 μM | Applied to human serum and urine samples, recovery 97.5–103.5% | 39 |
| ePADs | −0.95 | 3.59 μM | 10–500 μM | Applied to herbal medicine samples compared with HPLC, recovery = 99–122%. | 40 |
| Hg drop electrode | −0.6 | 0.002 μM | 49.8 nM to 0.61 μM | Applied to pharmaceutical samples, recovery = 94.14–112.41% | 41 |
| Hg(Ag)FE | −1.0 | 0.002 μM | 2.50 nM to 0.22 μM | Applied to pharmaceutical samples, recovery = 98–101% | 42 |
| HMDE | −1.1 | 7.6 μM | 0.85–1.4 μM | Applied to pharmaceutical samples, recovery = 99.8% | 43 |
For repeatability of the developed method, five different electrodes were prepared at the same time under the same conditions. The electrochemical performance for 2.0 mM DEX using MIL-100(Fe)/GO/PGE of each electrode were illustrated in Fig. 7A. The calculated percentage of relative standard deviation (% RSD) was 0.6–5.1 which is lower than acceptable value of 7.3 by AOAC Official Methods of Analysis.44 To ensure reproducibility of the developed method, five different electrodes were prepared under the same conditions at different times. The electrochemical performance of the 2.0 mM DEX is shown in Fig. 7B. The calculated percentage of relative standard deviation (% RSD) was 0.7–5.3 which is also lower than AOAC acceptable value. Therefore, the developed method provided acceptable repeatability and reproducibility in response to high precision.
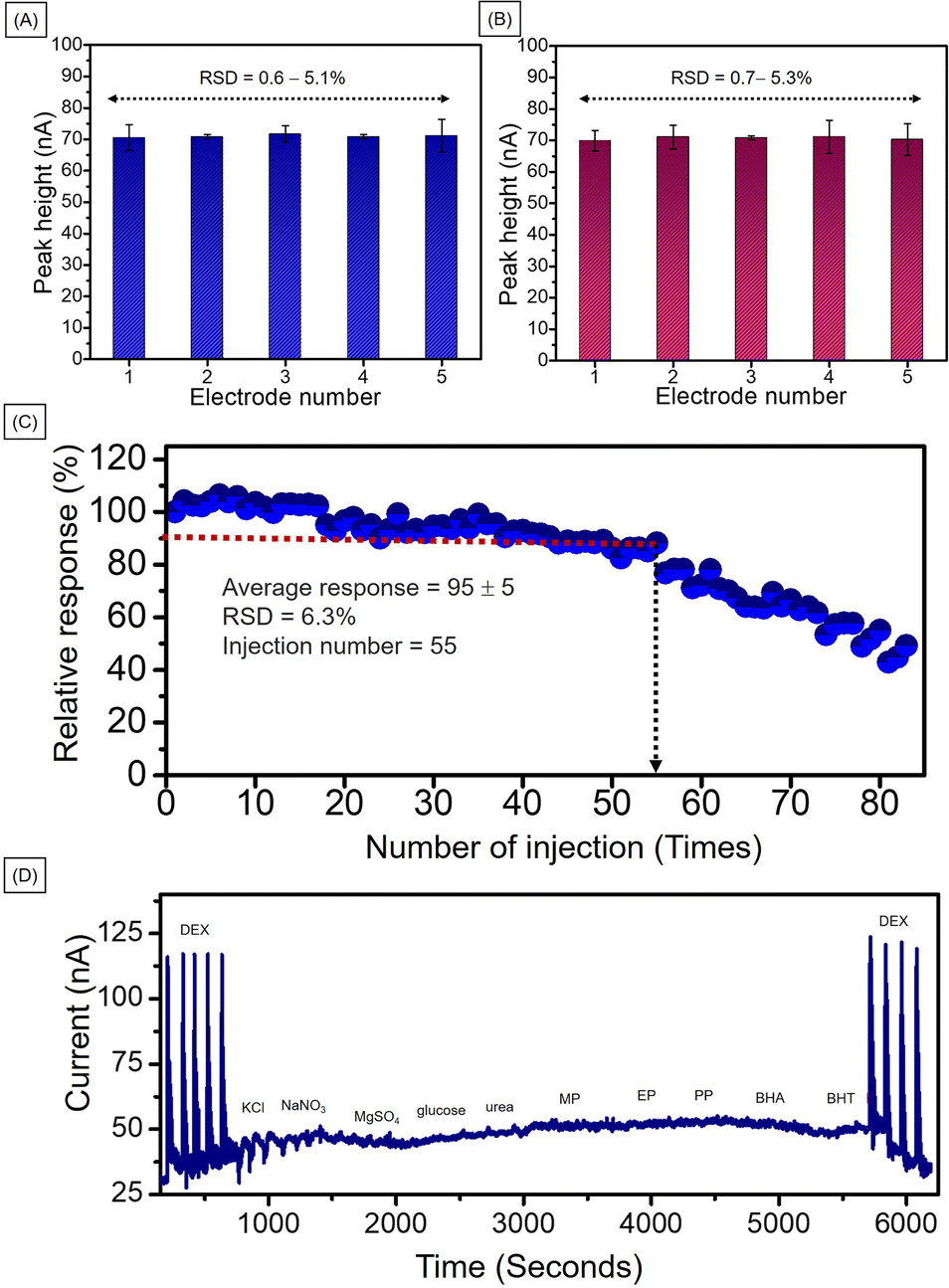 | ||
| Fig. 7 Histograms represent (A) the measured peak height using 5 different MIL-100(Fe)/GO/PGE electrodes prepared at the same time, (B) the measured peak height using 5 different MIL-100(Fe)/GO/PGE electrodes prepared in different batches, and different time, (C) the scatter graph represents the remaining measured height of anodic peak current for 85 cycles in the presence of 2.0 mM DEX in 0.04 M BR buffer (pH 2), (D) amperogram of 2.0 mM DEX and 10 mM of each interference including KCl, NaNO3, MgSO4, glucose, urea, MP, EP, PP, BP, BHA, and BHT. | ||
The operational stability of the MIL-100(Fe)/GO modified PGE was studied by repeatedly injecting 2.0 mM DEX in 0.04 M BR buffer (pH 2) under optimal conditions. The peak height obtained from each injection was converted into a percentage response based on the 100% response of the first injection (Fig. 7C). The plot of the percentage response against the number of injection cycles showed an average response of 90 ± 15% for first 55 injection cycles. For the 55th cycle, the response decreased to 80% and then continuously decreased. In order to confirm that each modified electrode afforded good activity, in the subsequent experiments each modified electrode was only used for 40 injections. The number of injections is sufficient for each analysis set.
For the interference study, the developed method was used to detect ions, compounds and other preservatives used in cosmetics including KCl, NaNO3, MgSO4, glucose, urea, MP, EP, PP, BP, BHA, and BHT. As shown in Fig. 7D, when a the 5-fold concentration of interfering species (10.0 mM) was injected into the system, no significant changes were observed in the amperogram. Thus, it can be concluded that the developed technique has a high selectivity for DEX. The results of the experiment reinforce the advantages of the system that require low detection potential.
Application in real sample
In order to evaluate the efficiency of flow injection using MIL-100(Fe)/GO/PGE electrochemical sensors for real sample analysis, the system was applied to detect DEX in three pharmaceutical and five whitening cream samples. All samples were prepared using a standard addition method. A standard addition calibration curve was plotted. The calculated % relative error and % recovery was utilized to determine the accuracy of the method for pharmaceutical and cosmetic samples, respectively as shown in Table S1.† The real samples were also confirmed by HPLC-DAD (Table S2†). The results obtained for the pharmaceutical samples were compared with the labelled concentrations. The results showed that, for both methods, the percentage relative error for the pharmaceutical samples was <8%. For whitening cream samples, the results from the developed method and HPLC-DAD showed no detectable DEX in any of the cosmetic samples. This may be due to the absence of DEX or because the concentration of DEX is lower than the limit of detection (0.030 μM). The calculated percentage recovery was 93–111% (with RSDs ranging from 0.30–0.87%) which is within the acceptable range of 75–120% by AOAC Official Methods of Analysis.44 These values were similar to the HPLC-DAD recoveries of 84–107% with RSD = 0.01–0.23%. The HPLC results are in good agreement with those obtained using the developed method. Consequently, it is reasonable to conclude that the developed method is highly accurate and can be used to detect DEX in real-world samples, without complicated sample preparation steps. Furthermore, using a flow-based amperometric sensor with a MIL-100(Fe)/GO modified PGE has a significantly shorter analysis time (100 seconds for 1 injection as opposed to 10 min for the HPLC-DAD method) and a much lower analytical cost than HPLC-DAD because of the use of a pencil lead. In addition, the developed sensor can be reused up to 55 times and is suitable for practical applications.Conclusions
We demonstrated the first DEX detection using a PGE modified with a MIL-100(Fe)/GO composite material. The detection principle relies on an increase in the anodic current of MIL-100(Fe) when DEX is present in the solution. The peak current of MIL-100(Fe)/GO/PGE exhibited excellent sensing performance, which can be ascribed to large specific surface area of MIL-100(Fe) sensing element and highly conductive GO material, which can be attributed to a synergistic effect. When coupled with a flow-based amperometric system, it requires a low applied potential to detect DEX, and high accuracy, precision, and time-efficient results were obtained, which is beneficial for real-world applications. Under optimal conditions, the sensitivity of DEX detection and operational stability were excellent. Finally, the successful determination of DEX in real samples revealed the efficacy of the electrochemical sensor, and the results agreed well with those obtained from HPLC analysis.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†Author contributions
CJ: conceptualization; data curation; formal analysis; methodology; visualization; roles/writing – original draft; and writing – review & editing. FM: data curation; formal analysis; methodology; and writing – review & editing. PN: conceptualization; data curation; formal analysis; methodology. MS: conceptualization; validation; and writing – review & editing. SP: conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; supervision; validation; and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely thank the Walailak University Graduate Research Fund (CGS-RF-2022/12) for financial support. The scholarship from the Development and Promotion of Science and Technology Talents Project (DPST) is acknowledged through Chanida Jakkrawhad. We also acknowledge Functional Materials and Nanotechnology Center of Excellence, Walailak University and NanoQAM, Le Centre de Caractérisation Microscopique des Matériaux.References
- Y. S. Nam, I. K. Kwon, Y. Lee and K.-B. Lee, Forensic Sci. Int., 2012, 220, e23–e28 CrossRef CAS PubMed.
- V. Giaccone, G. Polizzotto, A. Macaluso, G. Cammilleri and V. Ferrantelli, Int. J. Anal. Chem., 2017, 2017, 3531649 Search PubMed.
- M. Hichens and A. F. Hogans, Clin. Chem., 1974, 20, 266–271 CrossRef CAS.
- J. M. Hawley, L. J. Owen, M. Debono, J. Newell-Price and B. G. Keevil, Ann. Clin. Biochem., 2018, 55, 665–672 CrossRef CAS PubMed.
- M. Urban, R. Mainardes and M. Gremiao, Braz. J. Pharm. Sci., 2009, 45, 87–92 CrossRef CAS.
- A. Khataee, A. Hasanzadeh, R. Lotfi, R. Pourata and S. W. Joo, Spectrochim. Acta, Part A, 2015, 150, 63–71 CrossRef CAS PubMed.
- B. Das, K. Datta and S. K. Das, J. Liq. Chromatogr., 1985, 8, 3009–3016 CrossRef CAS.
- R. N. Goyal, V. K. Gupta and S. Chatterjee, Biosens. Bioelectron., 2009, 24, 3562–3568 CrossRef CAS PubMed.
- L. Zhang, S. Li, J. Xin, H. Ma, H. Pang, L. Tan and X. Wang, Microchim. Acta, 2018, 186, 9 CrossRef PubMed.
- S. Fu, Y. Zhu, Y. Zhang, M. Zhang, Y. Zhang, L. Qiao, N. Yin, K. Song, M. Liu and D. Wang, Microchem. J., 2021, 171, 106776 CrossRef CAS.
- B. Rezaei, s. Z. Mirahmadi-Zare and A. A. Ensafi, J. Braz. Chem. Soc., 2011, 1–8 Search PubMed.
- S. Alimohammadi, M. A. Kiani, M. Imani, H. Rafii-Tabar and P. Sasanpour, Sci. Rep., 2019, 9, 11775 CrossRef PubMed.
- T. Ma, H. Li, J.-G. Ma and P. Cheng, Dalton Trans., 2020, 49, 17121–17129 RSC.
- Y. Chang, J. Lou, L. Yang, M. Liu, N. Xia and L. Liu, Nanomaterials, 2022, 12, 3248 CrossRef CAS PubMed.
- S. Patra, T. Hidalgo Crespo, A. Permyakova, C. Sicard, C. Serre, A. Chaussé, N. Steunou and L. Legrand, J. Mater. Chem. B, 2015, 3, 8983–8992 RSC.
- G. Vinodha, L. Cindrella and P. D. Shima, Mater. Res. Express, 2019, 6, 085548 CrossRef CAS.
- J. N'Diaye, S. Poorahong, O. Hmam, G. C. Jiménez, R. Izquierdo and M. Siaj, Membranes, 2020, 10, 340 CrossRef PubMed.
- G. Chen, X. Leng, J. Luo, L. You, C. Qu, X. Dong, H. Huang, X. Yin and J. Ni, Molecules, 2019, 24, 1211 CrossRef PubMed.
- S. S. Chang, B. Clair, J. Ruelle, J. Beauchêne, F. Di Renzo, F. Quignard, G. J. Zhao, H. Yamamoto and J. Gril, J. Exp. Bot., 2009, 60, 3023–3030 CrossRef CAS PubMed.
- B. T. Le, D. D. La and P. T. H. Nguyen, ACS Omega, 2023, 8, 1262–1270 CrossRef CAS PubMed.
- J. Shi, S. Hei, H. Liu, Y. Fu, F. Zhang, Y. Zhong and W. Zhu, J. Chem., 2013, 2013, 792827 Search PubMed.
- V. Ganeshan, L. Cindrella and S. P Damodaran, Mater. Res. Express, 2019, 6, 085548 CrossRef.
- D. Antuña-Jiménez, M. B. González-García, D. Hernández-Santos and P. Fanjul-Bolado, Biosensors, 2020, 10, 9 CrossRef PubMed.
- M. Kurd, A. Salimi and R. Hallaj, Mater. Sci. Eng., C, 2013, 33, 1720–1726 CrossRef CAS PubMed.
- M. Matura, A. Goossens and M. Matura, Allergy, 2000, 55, 698–704 CrossRef CAS PubMed.
- J. Tang and J. Wang, RSC Adv., 2017, 7, 50829–50837 RSC.
- J. Liao, D. Wang, A. Liu, Y. Hu and G. Li, Analyst, 2015, 140, 8165–8171 RSC.
- M. Nemiwal and D. Kumar, Inorg. Chem. Commun., 2020, 122, 108279 CrossRef CAS.
- M. Trojanowicz and K. Kołacińska, Analyst, 2016, 141, 2085–2139 RSC.
- M. Trojanowicz and M. Pyszynska, Molecules, 2022, 27, 1410 CrossRef CAS PubMed.
- M. A. Franco, D. A. G. Araújo, L. H. Oliveira, M. A. G. Trindade, R. M. Takeuchi and A. L. Santos, Anal. Methods, 2016, 8, 8420–8426 RSC.
- A. M. Santos, F. C. Vicentini, L. C. S. Figueiredo-Filho, P. B. Deroco and O. Fatibello-Filho, Diamond Relat. Mater., 2015, 60, 1–8 CrossRef CAS.
- P. Reanpang, P. Mool-am-kha, J. Upan and J. Jakmunee, Talanta, 2021, 232, 122493 CrossRef CAS PubMed.
- K. Torrarit, S. Kongkaew, K. Samoson, P. Kanatharana, P. Thavarungkul, K. H. Chang, A. F. L. Abdullah and W. Limbut, ACS Omega, 2022, 7, 17679–17691 CrossRef CAS PubMed.
- L. Goodnight, D. Butler, T. Xia and A. Ebrahimi, Biosensors, 2021, 11, 409 CrossRef CAS PubMed.
- N. Malarat, W. Oin, K. Kanjana, F. Makkliang, M. Siaj and S. Poorahong, Microchem. J., 2023, 188, 108473 CrossRef CAS.
- J. D. Ference and A. R. Last, Am. Fam. Physician, 2009, 79, 135–140 Search PubMed.
- A. Fatahi, R. Malakooti and M. Shahlaei, RSC Adv., 2017, 7, 11322–11330 RSC.
- B. Li and P. Wang, Int. J. Electrochem. Sci., 2024, 19, 100622 CrossRef CAS.
- V. Primpray, O. Chailapakul, M. Tokeshi, T. Rojanarata and W. Laiwattanapaisal, Anal. Chim. Acta, 2019, 1078, 16–23 CrossRef CAS PubMed.
- T. M. B. F. Oliveira, F. W. P. Ribeiro, J. E. S. Soares, P. de Lima-Neto and A. N. Correia, Anal. Biochem., 2011, 413, 148–156 CrossRef CAS PubMed.
- J. Smajdor, R. Piech and B. Paczosa-Bator, J. Electroanal. Chem., 2018, 809, 147–152 CrossRef CAS.
- C. Jeyaseelan and A. Joshi, Anal. Bioanal. Chem., 2002, 373, 772–776 CrossRef CAS PubMed.
- G. W. Latimer, in Official Methods of Analysis of AOAC International, Oxford University Press, 2023, DOI:10.1093/9780197610145.005.006.
Footnote |
| † Electronic supplementary information (ESI) available: Iron-based metal–organic frameworks/graphene oxide composite electrodes for efficient flow-injection amperometric detection of dexamethasone. See DOI: https://doi.org/10.1039/d4ra03815k |
| This journal is © The Royal Society of Chemistry 2024 |