
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homotrinuclear ruthenium(II) and rhodium(I) complexes of redox-active tris(ferrocenyl)arene-based tris-phosphanes†
Axel Straube‡
 ,
Peter Coburger§
and
Evamarie Hey-Hawkins*
,
Peter Coburger§
and
Evamarie Hey-Hawkins*
Institute of Inorganic Chemistry, Leipzig University, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
First published on 6th August 2024
Abstract
Homotrinuclear complexes of the C3-symmetric tris(ferrocenyl)arene-based tris-phosphanes 1a–d with ruthenium(II) ([1a–d(Ru)3]) and rhodium(I) ([1a–d(Rh)3]) were prepared and fully characterised. Complexes [1a–d(Ru)3] and [1a–d(Rh)3] are electrochemically active. The nature of the arene core in 1a–d ranging from benzene, 1,3,5-trifluorobenzene and mesitylene to s-triazine allows to fine-tune the exact oxidation potentials for tailoring the electrochemical response. With a BArF4−-based supporting electrolyte, a distinct separation of the three iron-centred oxidations of the ligand backbone is observable. Under these conditions, these oxidations are mostly reversible but, especially for the third oxidation, already show signs of irreversibility. In general, while the coordinated metal complex fragment does not strongly alter the electrochemical response of the arene-trisferrocenyl core 1a–d, there are observable differences. Rhodium(I) complexes are oxidised at slightly higher potentials than ruthenium(II) complexes. In both cases, individual oxidation states for the C6H3(CH2)3-based ligand (1d) are difficult to address and the C3N3-based ligand (1c) shows the most complicated and least reversible electrochemistry with severely broadened third oxidations and reduced reversibility in cyclic voltammetry. The most well-suited system for potential applications in redox-switchable catalysis, in all cases, is the C6H3-based ligand (1a), showing entirely reversible and well-separated redox events.
Introduction
The last decades have seen C3-symmetry increasingly being adopted into ligand design due to enhanced stability and outstanding performances in asymmetric catalysis due to a reduced possible number of transition states.1,2 Next to countless nitrogen-containing compounds,3–6 several tris-phosphanes have been reported,7–11 often based on the archetypical triphos ligand by Hewertson and Watson.12 The corresponding C3-symmetric ligands featuring three ferrocenylene groups were reported by Butler and co-workers in 2003.13 Ferrocene is highly suitable for ligand design, owing to its amenability to synthetic modification and favourable, while modifiable, redox properties.14,15 Constructing multi-ferrocene systems is usually motivated by the redox properties of the individual ferrocene moieties which, when assembled, can add up to more than the mere sum of its parts.16,17 Motivated by the prominence of C3-symmetry in modern-day ligand design and the potential to exploit the use of three ferrocenyl groups for redox-switchable catalysis (RSC), we have built upon our previous works on ferrocenylphosphanes18–26 and their applications in RSC27–29 and reported a new family of tris-phosphanes (1a–d, Scheme 1) based on a redox-active, C3-symmetric tris(ferrocenyl)arene backbone. By incorporating an electron-withdrawing (b and c) or a tris-benzylic arene core (d), the electrochemical response as well as more subtle influences on the coordination behaviour of the corresponding phosphanes become adjustable. Thus, tris-phosphanes 1a–d were employed in the formation of mono- and trinuclear gold(I) complexes, and the latter were shown to act as four-state redox-switchable catalysts.30–32 We have now extended this synthetic concept to homotrinuclear rhodium(I) and ruthenium(II) complexes.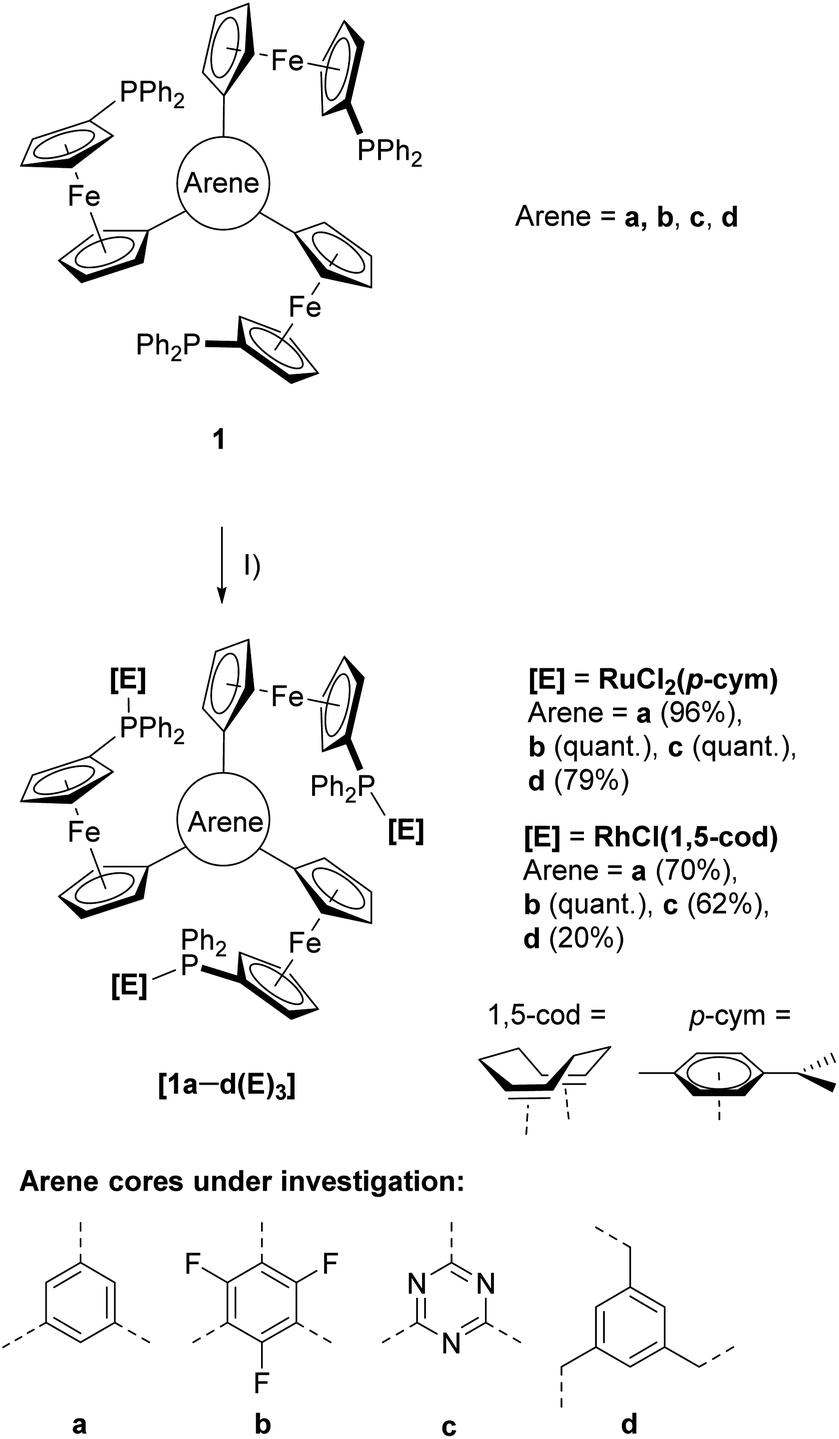 | ||
| Scheme 1 Preparation of ruthenium(II) ([1a–d(Ru)3]) and rhodium(I) ([1a–d(Rh)3]) complexes from tris-phosphanes 1a–d and (I) [{RuCl2(p-cym)}2] or [{RhCl(1,5-cod)}2] in CH2Cl2 at r.t. | ||
Experimental general procedures
Syntheses
All reactions and manipulations were carried out under an atmosphere of either nitrogen or argon using standard Schlenk line techniques unless stated otherwise. Thin-layer chromatography (TLC) with silica gel 60 F254 on glass or aluminium sheets available from Merck KGaA was used for monitoring the ligand synthesis. Column chromatography of the ruthenium complexes was performed using silica gel (Macherey-Nagel 60, 0.04–0.063 mm) and dried solvents purged with nitrogen prior to use. Molecular sieves (4 Å) were activated at 300 °C in vacuo for a minimum of 3 h. Dry, oxygen-free solvents (THF, CH2Cl2, Et2O, hexanes, and toluene) were obtained from an MBraun Solvent Purification System MB SPS-800 and directly stored over 4 Å molecular sieves, except for THF, which was further distilled from potassium/benzophenone and stored over 4 Å molecular sieves. CD2Cl2 was dried by stirring over P2O5 at room temperature for several days, followed by vacuum transfer into a storage flask, degassing by the freeze–pump–thaw method, and storage over 4 Å molecular sieves. THF-d8 was distilled from potassium/benzophenone and stored over 4 Å molecular sieves after degassing by the freeze–pump–thaw method. Tris-phosphanes 1a,30 1b,30 1c,31 and 1d,30 (nBu4N)[B{3,5-C6H3(CF3)2}4] (=(nBu4N)BArF4),33 and the transition metal precursors [{Rh(μ-Cl)(1,5-cod)}2]34 (1,5-cod = η4-cycloocta-1,5-diene) and [{RuCl(μ-Cl)(p-cym)}2]35 (p-cym = η6-p-cymene) were prepared according to previously published procedures. All other chemicals were used as purchased.NMR spectra were recorded with a BRUKER Avance III HD 400 MHz NMR spectrometer at 25 °C (frequencies of 1H: 400.13 MHz; 13C 100.63 MHz; 19F: 376.53 MHz; 31P: 161.99 MHz). Pseudo-triplets and -quadruplets (due to additional coupling to heteronuclei like 19F and 31P) of ferrocenyl protons are abbreviated as pt/pq and their observable coupling constants J are given. Quintuplets are abbreviated as “quint”. Assignment of 1H and 13C signals to the respective chemical entities are based on 1H,1H COSY, phase-sensitive 1H,13C HSQC and 1H,13C HMBC NMR experiments. TMS was used as the internal standard in the 1H and 13C{1H}/13C{31P,1H} NMR spectra, and spectra of all other nuclei were referenced to TMS using the Ξ scale.36 The numbering schemes for the assignment of specific nuclei is given in the ESI.†
Electrospray ionisation (ESI) mass spectrometry was performed with an ESI ESQUIRE 3000 PLUS spectrometer with an IonTrap analyser from Bruker Daltonics, or a MicroTOF spectrometer from Bruker Daltonics with a ToF analyser in positive mode. As solvents for the measurements, pure degassed CH2Cl2 or mixtures of degassed CH2Cl2 and CH3CN were used. Elemental analyses were performed with a VARIO EL elemental analyser from Heraeus. Melting points were determined with a Gallenkamp MPD350 BM2.5 melting point device and are reported uncorrected. FTIR spectra were obtained with a PerkinElmer FT-IR spectrometer Spectrum 2000 as KBr pellets and with a Thermo Scientific Nicolet iS5 with an ATR unit in the range from 4000 to 400 cm−1.
Crystallography
The data were collected on a Gemini-CCD diffractometer (RIGAKU INC.) using Mo-Kα radiation (λ = 0.71073 Å), ω-scan rotation. Data reduction was performed with CrysAlis Pro37 including the program SCALE3 ABSPACK38 for empirical absorption correction. The structure solution for [1a(Rh)3] was performed with SHELXS-97 (direct methods).39 The anisotropic full-matrix least-squares refinement on F2 of all non-hydrogen atoms was performed with SHELXL-97.40 All non-hydrogen atoms were refined with anisotropic thermal parameters. The structure figures were generated with Mercury (versions 3.8 and 3.10)40 and POV-Ray (Version 3.7).41 CCDC 1990280 contains the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.Electrochemistry
Cyclic voltammetry (CV) measurements on 1.0 mmol per L analyte solutions in dry, oxygen-free dichloromethane containing 0.1 mol per L (nBu4N)BF4 or (nBu4N)BArF4 as supporting electrolyte were conducted in a three-electrode setup (GAMRY Instruments, SP-50 potentiostat by BioLogic Science Instruments) under a blanket of nitrogen at room temperature. The glassy-carbon working electrode (ALS; surface area 0.07 cm2) and the counter electrode (neoLab; platinum wire, 99.9%) were immersed in the analyte solution, while the reference electrode (ALS; Ag/AgNO3 (0.01 mol L−1) in 0.1 mol per L tetrabutylammonium hexafluorophosphate in dry, oxygen-free CH3CN) was connected to the cell via a bridge tube (filled with the supporting electrolyte) through Vycor tips. The reference electrode was calibrated against decamethylferrocene as an internal standard at the end of the CV experiment,42 and the results were converted to the FcH/[FcH]+ scale in accordance with the IUPAC requirements.43Synthesis of μ3-[1,3,5-tris(1-diphenylphosphanyl-1′-ferrocenylene)benzene-1κ1P,2κ1P,3κ1P]-tris[dichlorido-η6-(p-cymene)ruthenium(II)] ([1a(Ru)3])
In a Schlenk flask, 1a (100 mg, 84.7 μmol, 1.00 eq.) and [{RuCl(μ-Cl)(p-cym)}2] (77.7 mg, 127 μmol, 1.50 eq.) were dissolved in CH2Cl2 (10 mL) and stirred overnight at room temperature. The reaction mixture was concentrated to half the original volume and filtered over degassed silica, using THF to elute the product. Complex [1a(Ru)3] was obtained as a red microcrystalline solid (170 mg, 96%) after removal of the volatiles in vacuo.M.p.: 210 °C (decomposition; from THF); 1H NMR (CD2Cl2): δ [ppm] = 7.91–7.84 (m, 12H, H10), 7.51–7.37 (m, 18H, H11 + 12), 7.17 (s, 3H, H2), 5.08 (m, 12H, H14 + 15), 4.43 (pq, J = 1.7 Hz, 6H, H8), 4.38 (pt, JH,H = 1.9 Hz, 6H, H4/5), 4.14 (pq, J = 1.7 Hz, 6H, H7), 3.83 (pt, JH,H = 1.9 Hz, 6H, H5/4), 2.46 (sept,3JH,H = 6.9 Hz, 3H, H18), 1.76 (s, 9H, H17), 0.94 (d, 3JH,H = 6.9 Hz, 18H, H19); 13C{1H} NMR (CD2Cl2): δ [ppm] = 138.2 (s, C1), 136.2 (d, 1JC,P = 47.3 Hz, C9), 134.2 (d, 2JC,P = 9.3 Hz, C10), 130.1 (d, 4JC,P = 2.4 Hz, C12), 127.5 (d, 3JC,P = 9.8 Hz, C11), 122.2 (s, C2), 108.8 (d, 2JC,P = 1.0 Hz, C13), 95.2 (s, C16), 90.1 (d, 2JC,P = 4.4 Hz, C15), 86.2 (s, C3), 86.1 (d, 2JC,P = 6.0 Hz, C14), 78.1 (d, 1JC,P = 47.5 Hz, C6), 75.6 (d, 3JC,P = 10.5 Hz, C8), 73.4 (d, 2JC,P = 7.9 Hz, C7), 72.4 (s, C4/5), 67.9 (s, C5/4), 30.1 (s, C18), 21.6 (s, C19), 17.1 (s, C17); 31P{1H} NMR (CD2Cl2): δ [ppm] = 18.9 (s); IR (neat): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1] = 3077 (w), 3052 (w), 2957 (m), 2923 (m), 2867 (m, all ν(C–H)), 1594 (w), 1559 (w), 1540 (w), 1507 (w), 1498 (w), 1481 (w), 1472 (w), 1457 (w), 1432 (m, ν(C–P)), 1381 (w), 1361 (w), 1318 (w), 1306 (w), 1198 (w), 1188 (w), 1157 (m), 1094 (m), 1058 (m), 1027 (m), 999 (w), 921 (w), 888 (w), 827 (m), 799 (m), 744 (m), 695 (s), 669 (m), 623 (m), 562 (w), 540 (m), 519 (m), 509 (m), 492 (s), 471 (s), 414 (m); HRMS (ESI): m/z calcd for C102H99Cl4Fe3P3Ru3 1015.5458 [M − 2Cl]2+; found 1015.5474; elemental analysis calcd [%] for C102H99Cl6Fe3P3Ru3: C 58.30, H 4.75, found: C 55.95, H 4.72.
[cm−1] = 3077 (w), 3052 (w), 2957 (m), 2923 (m), 2867 (m, all ν(C–H)), 1594 (w), 1559 (w), 1540 (w), 1507 (w), 1498 (w), 1481 (w), 1472 (w), 1457 (w), 1432 (m, ν(C–P)), 1381 (w), 1361 (w), 1318 (w), 1306 (w), 1198 (w), 1188 (w), 1157 (m), 1094 (m), 1058 (m), 1027 (m), 999 (w), 921 (w), 888 (w), 827 (m), 799 (m), 744 (m), 695 (s), 669 (m), 623 (m), 562 (w), 540 (m), 519 (m), 509 (m), 492 (s), 471 (s), 414 (m); HRMS (ESI): m/z calcd for C102H99Cl4Fe3P3Ru3 1015.5458 [M − 2Cl]2+; found 1015.5474; elemental analysis calcd [%] for C102H99Cl6Fe3P3Ru3: C 58.30, H 4.75, found: C 55.95, H 4.72.
Synthesis of μ3-[2,4,6-tris(1-diphenylphosphanyl-1′-ferrocenylene)-1,3,5-trifluorobenzene-1κ1P,2κ1P,3κ1P]-tris[dichlorido-η6-(p-cymene)ruthenium(II)] ([1b(Ru)3])
[1b(Ru)3] was prepared analogously to [1a(Ru)3] using 1b (50.0 mg, 40.4 μmol, 1.00 eq.) and obtained as a red amorphous solid in quantitative yield (87.0 mg).M.p.: 188 °C (decomposition; from THF); 1H NMR (CD2Cl2): δ [ppm] = 7.99–7.71 (m, 12H, H10), 7.54–7.28 (m, 18H, H11 + 12), 5.09 (pq, J = 6.2 Hz, 12H, H14 + 15), 4.44 (m, 6H, H8), 4.37 (m, 6H, H4), 4.31 (m, 6H, H7), 3.96 (pt, JH,H = 1.9 Hz, 6H, H5), 2.45 (hept, 3JH,H = 7.0 Hz, 3H, H18), 1.76 (s, 9H, H17), 0.94 (d, 3JH,H = 7.0 Hz, 18H, H19); 13C{1H} NMR (CD2Cl2):44 δ [ppm] = 155.2 (s, C1), 135.8 (d, 1JC,P = 47.3 Hz, C9), 134.1 (d, 2JC,P = 9.4 Hz, C10), 130.1 (d, 4JC,P = 1.9 Hz, C12), 127.4 (d, 3JC,P = 9.7 Hz, C11), 111.5 (s, C2), 108.6 (d, 2JC,P = 0.7 Hz, C13), 95.0 (s, C16), 90.2 (d, 2JC,P = 4.4 Hz, C15), 85.9 (d, 2JC,P = 5.8 Hz, C14), 78.1 (d, 1JC,P = 47.7 Hz, C6), 75.7 (d, 3JC,P = 10.2 Hz, C8), 74.6 (s, C3), 72.9 (d, 2JC,P = 7.9 Hz, C7), 71.7 (s, C5), 71.2 (m, C4), 30.0 (s, C18), 21.5 (s, C19), 17.0 (s, C17); 19F{1H} (CD2Cl2): δ [ppm] = −107.5 (s); 31P{1H} (CD2Cl2): δ [ppm] = 19.4 (s); IR (neat): [cm−1] = 3077 (w), 3052 (w), 2957 (m), 2923 (m), 2867 (m, all ν(C–H)), 1594 (w), 1559 (w), 1540 (w), 1507 (w), 1498 (w), 1483 (m), 1472 (w), 1457 (w), 1432 (m, ν(C–P)), 1421 (m, ν(C–F)), 1387 (m), 1361 (w), 1318 (w), 1308 (m), 1229 (m), 1198 (w), 1188 (w), 1157 (m), 1094 (m), 1058 (m), 1027 (m), 999 (w), 921 (w), 888 (w), 827 (m), 799 (m), 744 (m), 695 (s), 669 (m), 623 (m), 562 (w), 540 (m), 520 (m), 509 (m), 492 (s), 469 (vs.), 454 (s), 414 (m), 405 (s); HRMS (ESI): m/z calcd for C102H96Cl6F3Fe3P3Ru3 [M]+ 2156.0003, calcd for C102H96Cl5F3Fe3P3Ru3 [M − Cl]+ 2120.0323; found 2156.0060, 2120.0362; elemental analysis calcd [%] for C102H96Cl6F3Fe3P3Ru3: C 56.84, H 4.49, found: C 56.43, H 4.11.
Synthesis of μ3-[2,4,6-tris(1-diphenylphosphanyl-1′-ferrocenylene)-1,3,5-triazine-1κ1P,2κ1P,3κ1P]-tris[dichlorido-η6-(p-cymene)-ruthenium(II)] ([1c(Ru)3])
[1c(Ru)3] was prepared analogously to [1a(Ru)3] using 1c (50.0 mg, 42.2 μmol, 1.00 eq.) and obtained as a dark-red amorphous solid in quantitative yield (84.9 mg).M.p.: 175 °C (decomposition; from THF); 1H NMR (CD2Cl2): δ [ppm] = 7.94–7.84 (m, 12H, H9), 7.58–7.34 (m, 18H, H10 + 11), 5.10 (pq, J = 6.0 Hz, 12H, H13 + 14), 4.84 (m, 6H, H3), 4.51 (m, 6H, H7), 4.07 (m, 6H, H6), 3.99 (m, 6H, H4), 2.46 (hept, 3JH,H = 6.9 Hz, 3H, H17), 1.78 (s, 9H, H16), 0.93 (d, 3JH,H = 6.9 Hz, 18H, H18); 13C{1H} NMR (CD2Cl2): δ [ppm] = 175.0 (s, C1), 136.1 (d, 1JC,P = 47.3 Hz, C8), 134.1 (d, 2JC,P = 9.4 Hz, C9), 130.2 (d, 4JC,P = 2.4 Hz, C11), 127.5 (d, 3JC,P = 9.7 Hz, C10), 108.7 (s, C12), 95.1 (s, C15), 90.2 (d, 2JC,P = 4.4 Hz, C14), 86.0 (d, 2JC,P = 6.0 Hz, C13), 80.4 (s, C2), 78.3 (d, 1JC,P = 47.0 Hz, C5), 76.0 (d, 3JC,P = 10.2 Hz, C7), 75.2 (s, C4), 73.3 (d, 2JC,P = 7.7 Hz, C6), 70.7 (s, C3), 30.0 (s, C17), 21.5 (s, C18), 17.0 (s, C16); 31P{1H} (CD2Cl2): δ [ppm] = 18.8 (s); IR (neat): [cm−1] = 3077 (w), 3052 (w), 2957 (m), 2923 (m), 2867 (m, all ν(C–H)), 1506 (s), 1498 (w), 1483 (m), 1472 (w), 1457 (w), 1432 (m, ν(C–P)), 1380 (m), 1357 (m), 1319 (m), 1308 (m), 1229 (m), 1198 (w), 1188 (w), 1157 (m), 1094 (m), 1058 (m), 1028 (m), 999 (w), 925 (w), 888 (w), 827 (m), 799 (m), 744 (m), 695 (s), 669 (m), 623 (m), 562 (w), 540 (m), 509 (m), 492 (s), 469 (s), 454 (s), 424 (m), 409 (m); HRMS (ESI): m/z calcd for C99H96Cl5Fe3N3P3Ru3 [M − Cl]+ 2069.0462, calcd for C101H99Cl5Fe3N4P3Ru3 [M − Cl + CH3CN]+ 2110.0728; found 2069.0479, 2110.0723; elemental analysis calcd [%] for C99H96Cl6Fe3N3P3Ru3: C 56.51, H 4.60, N 2.00, found: C 55.77, H 4.41, N 1.95.
Synthesis of μ3-[1,3,5-tris{(1-diphenylphosphanyl-1′-ferrocenylene)methyl}benzene -1κ1P,2κ1P,3κ1P]-tris[dichlorido-η6-(p-cymene)-ruthenium(II)] ([1d(Ru)3])
[1d(Ru)3] was prepared analogously to [1a(Ru)3] using 1d (50.0 mg, 40.8 μmol, 1.00 eq.) and obtained as a dark-red amorphous solid (69.0 mg, 79%).M.p.: >160 °C decomp. (from THF/diethyl ether); 1H NMR (CD2Cl2): δ [ppm] = 7.92–7.81 (m, 12H, H11), 7.47–7.34 (m, 18H, H12 + H13), 6.48 (s, 3H, H1), 5.14–5.06 (m, 12H, H15 + H16), 4.37 (pq, JH,H = 1.7 Hz, 6H, H9), 4.27 (pq, JH,H = 1.7 Hz, 6H, H8), 3.69 (pt, JH,H = 1.8 Hz, 6H, H5), 3.57 (pt, JH,H = 1.8 Hz, 6H, H6), 3.23 (s, 6H, H3), 2.49 (hept, 3JH,H = 7.0 Hz, 3H, H18), 1.79 (s, 9H, H20), 0.97 (d, 3JH,H = 7.0 Hz, 18H, H19); 13C{1H} NMR (CD2Cl2): δ [ppm] = 141.3 (s, C2), 136.2 (d, 1JC,P = 47.3 Hz, C10), 134.1 (d, 2JC,P = 9.3 Hz, C11), 130.0 (d, 4JC,P = 2.3 Hz, C13), 127.3 (d, 3JC,P = 9.7 Hz, C12), 125.7 (s, C1), 108.6 (s, C14), 95.0 (s, C17), 90.1 (d, 2JC,P = 4.4 Hz, C16), 89.3 (s, C4), 85.9 (d, 2JC,P = 5.9 Hz, C15), 77.4 (d, 1JC,P = 48.4 Hz, C7), 75.1 (d, 3JC,P = 10.5 Hz, C9), 71.2 (d, 2JC,P = 8.0 Hz, C8), 70.29 (s, C5/C6), 70.27 (s, C6/C5), 35.0 (s, C3), 30.0 (s, C18), 21.5 (s, C19), 17.0 (s, C20); 31P{1H} NMR (CD2Cl2): δ [ppm] = 18.8 (s); IR (neat, ATR): [cm−1] = 3077 (m), 3053 (m), 2959 (s), 2926 (s), 2867 (m, all ν(C–H)), 1599 (m), 1574 (w), 1537 (w), 1481 (m), 1468 (m), 1433 (s, ν(C–P)), 1385 (m), 1305 (m), 1236 (m), 1190 (m), 1157 (s), 1095 (m), 1058 (m), 1026 (s), 1000 (w), 981 (w), 925 (w), 890 (w), 827 (m), 800 (m), 745 (m), 697 (s), 670 (m), 541 (m), 520 (m), 491 (s), 471 (s), 457 (m), 437 (w), 423 (w); HRMS (ESI): m/z calcd for C105H105Cl6Fe3P3Ru3 [M]+ 2143.0760, for C105H105Cl5Fe3P3Ru3 [M − Cl]+ 2108.1077; found 2143.0737, 2108.1052; elemental analysis calcd [%] for C105H105Cl6Fe3P3Ru3: C 58.84, H 4.94, found: C 58.17, H 4.97.
Synthesis of μ3-[1,3,5-tris(1-diphenylphosphanyl-1′-ferrocenylene)benzene-1κ1P,2κ1P,3κ1P]-tris[chlorido-1,2,5,6-η4-(cycloocta-1,5-diene)rhodium(I)] ([1a(Rh)3])
Under stirring, a solution of 1a (120 mg, 101 μmol, 1.00 eq.) in THF (10 mL) was added to [{Rh(μ-Cl)(1,5-cod)}2] (76.0 mg, 154 μmol, 1.52 eq.) in THF (5 mL) and kept stirring at room temperature overnight. Complex [1a(Rh)3] was precipitated from the clear orange solution using diethyl ether (45 mL), yielding a fine orange powder which was dried in vacuo at 40 °C (136 mg, 70%). Crystals suitable for XRD were obtained from vapour diffusion of diethyl ether into 0.7 mL of the reaction mixture in an NMR tube at room temperature.M.p.: >185 °C decomp. (from THF/diethyl ether); 1H NMR (THF-d8): δ [ppm] = 7.65 (s, 3H, H2), 7.62–7.48 (m, 12H, H11), 7.39–7.14 (m, 18H, H10 + 12), 5.56 (m, 6H, H13/14/17/18), 5.10 (pt, JH,H = 1.9 Hz, 6H, H4/5), 4.78 (m, 6H, H8), 4.55 (pt, JH,H = 1.9 Hz, 6H, H5/4), 4.25 (m, 6H, H7), 3.03 (m, 6H, H14/13/18/17), 2.49–2.34 (m, 12H, H15 + 16,19 + 20), 2.07 (m, 6H, H15 + 16,19 + 20), 1.92 (m, 6H, H15 + 16,19 + 20); 13C{1H} NMR (THF-d8): δ [ppm] = 138.7 (s, C1), 133.7 (d, 3JC,P = 11.0 Hz, C11), 133.6 (d, 1JC,P = 42.5 Hz, C9), 129.2 (d, 4JC,P = 1.9 Hz, C12), 127.1 (d, 2JC,P = 9.5 Hz, C10), 122.1 (s, C2), 103.8 (dd, 2JC,P = 13.5 Hz, 1JC,Rh = 7.5 Hz, C13/14/17/18), 86.4 (s, C3), 75.7 (d, 3JC,P = 10.5 Hz, C8), 73.5 (d, 2JC,P = 6.9 Hz, C7), 73.4 (d, 1JC,P = 47.5 Hz, C6), 72.0 (s, C4/5), 69.5 (d, 1JC,Rh = 13.3 Hz, C14/13/18/17), 68.3 (s, C5/4), 32.60 (s, C15/16/19/20), 32.58 (s, C15/16/19/20), 28.3 (s, C15/16/19/20), 25.0 (s, C15/16/19/20); 31P{1H} NMR (THF-d8): δ [ppm] = 22.9 (d, 1JP,Rh = 152.4 Hz); IR (KBr): [cm−1] = 3105 (w), 3092 (w), 3074 (w), 3054 (w), 2968 (m), 2934 (m), 2915 (m), 2831 (m, all ν(C–H)), 1970 (w), 1899 (w), 1769 (w, all aromatic overtones), 1597 (m), 1479 (m), 1434 (s, ν(C–P)), 1384 (m), 1333 (m), 1304 (m), 1261 (w), 1164 (s), 1111 (s), 1095 (s), 1061 (s), 1035 (s), 1029 (s), 997 (m), 961 (w), 921 (w), 899 (w), 871 (w), 832 (m), 813 (m), 746 (m), 702 (s), 694 (s), 626 (m), 541 (m), 522 (m), 498 (s), 470 (m), 445 (w), 430 (m); HRMS (ESI): m/z calcd for C96H93Cl3Fe3P3Rh3 [M]+ 1922.0776; found 1922.0763; elemental analysis calcd [%] for C96H93Cl3Fe3P3Rh3: C 59.98, H 4.83, found: C 60.16, H 4.97.
Synthesis of μ3-[2,4,6-tris(1-diphenylphosphanyl-1′-ferrocenylene)-1,3,5-trifluorobenzene-1κ1P,2κ1P,3κ1P]-tris[chlorido-1,2,5,6-η4-(cycloocta-1,5-diene)rhodium(I)] ([1b(Rh)3])
[1b(Rh)3] was prepared analogously to [1a(Rh)3] using 1b (50.0 mg, 40.4 μmol, 1.00 eq.) and obtained as a fine orange powder in quantitative yield (80.0 mg) after precipitation by adding diethyl ether (THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O = 1:8 v/v).
:Et2O = 1:8 v/v).
M.p.: 188–191 °C (from THF/diethyl ether); 1H NMR (CD2Cl2): δ [ppm] = 7.77–7.49 (m, 12H, H11), 7.40–7.34 (m, 6H, H12), 7.33–7.26 (m, 12H, H10), 5.50 (s (br), ω1/2 = 11.3 Hz, 6H, H13/14/17/18), 5.01 (pt, JH,H = 1.9 Hz, 6H, H4), 4.75 (pt, JH,H = 1.9 Hz, 6H, H5), 4.70 (pq, J = 2.0 Hz, 6H, H8), 4.40 (pt, J = 2.0 Hz, 6H, H7), 3.13 (s (br), ω1/2 = 10.5 Hz, 6H, H14/13/18/17), 2.53–2.36 (m, 12H, H15/16/19/20), 2.15–2.03 (m, 6H, H15/16/19/20), 2.00–1.87 (m, 6H, H15/16/19/20); 13C{1H} NMR (CD2Cl2):44 δ [ppm] = 155.7 (s, C1), 133.8 (d, 3JC,P = 11.2 Hz, C11), 133.3 (d, 1JC,P = 42.6 Hz, C9), 130.0 (d, 4JC,P = 2.2 Hz, C12), 127.7 (d, 2JC,P = 9.6 Hz, C10), 112.3 (s, C2), 104.9 (dd, 2JC,P = 12.9 Hz, 1JC,Rh = 6.9 Hz, C13/14/17/18), 75.9 (d, 3JC,P = 9.9 Hz, C8), 74.7 (s, C3) 73.7 (d, 2JC,P = 6.7 Hz, C7), 73.3 (d, 1JC,P = 47.1 Hz, C6), 71.9 (s, C5), 71.7 (m, C4), 70.3 (d, 1JC,Rh = 13.7 Hz, C14/13/18/17), 33.04 (s, C15/16/19/20), 33.01 (s, C15/16/19/20), 28.8 (s, C15/16/19/20); 19F{1H} NMR (CD2Cl2): δ [ppm] = −107.4 (s); 31P{1H} NMR (CD2Cl2): δ [ppm] = 22.7 (d, 1JP,Rh = 151.8 Hz); IR (neat, ATR): [cm−1] = 3098 (w), 3068 (w), 3049 (w), 2959 (w), 2935 (m), 2914 (m), 2874 (m), 2828 (m, all ν(C–H)), 1479 (m), 1433 (s, ν(C–P)), 1422 (m, ν(C–F)), 1388 (m), 1332 (m), 1304 (m), 1273 (w), 1230 (w), 1218 (w), 1204 (w), 1177 (m, sh), 1163 (m), 1096 (m), 1066 (m), 1024 (s), 997 (m), 960 (w), 912 (w), 888 (w), 859 (w), 832 (m), 815 (m), 763 (m, sh), 745 (s), 692 (vs.), 627 (m), 540 (m), 521 (s), 497 (vs.), 488 (vs.), 468 (vs.), 428 (s); HRMS (ESI): m/z calcd for C96H90Cl3F3Fe3P3Rh3 [M − Cl]+ 1941.0813; found 1941.0819; elemental analysis calcd [%] for C96H90Cl3F3Fe3P3Rh3: C 58.34, H 4.59, found: C 58.02, H 4.69.
Synthesis of μ3-[2,4,6-tris(1-diphenylphosphanyl-1′-ferrocenylene)-1,3,5-triazine-1κ1P,2κ1P,3κ1P]-tris[chlorido-1,2,5,6-η4-(cycloocta-1,5-diene)rhodium(I)] ([1c(Rh)3])
[1c(Rh)3] was prepared analogously to [1a(Rh)3] using 1c (50.0 mg, 42.2 μmol, 1.00 eq.) and obtained as a red powder (50.0 mg, 62%) which already partly precipitated from the reaction mixture.M.p.: 246–248 °C (from THF/diethyl ether); 1H NMR (CD2Cl2): δ [ppm] = 7.63–7.50 (m, 12H, H10), 7.42–7.26 (m, 18H, H9 + H11), 5.55 (s (br), ω1/2 = 11.0 Hz, 6H, H12/13/16/17), 5.53 (pt, JH,H = 2.0 Hz, 6H, H3), 4.93 (pt, JH,H = 2.0 Hz, 6H, H4), 4.71 (pq, J = 2.0 Hz, 6H, H7), 4.26 (pt, J = 2.0 Hz, 6H, H6), 3.19 (s (br), ω1/2 = 10.6 Hz, 6H, H13/12/17/16), 2.56–2.42 (m, 12H, H14/15/18/19), 2.20–2.06 (m, 6H, H14/15/18/19), 2.05–1.94 (m, 6H, H14/15/18/19); 13C{1H} NMR (CD2Cl2): δ [ppm] = 175.5 (s, C1), 133.9 (d, 3JC,P = 11.1 Hz, C10), 133.1 (d, 1JC,P = 42.4 Hz, C8), 130.0 (d, 4JC,P = 2.2 Hz, C11), 127.8 (d, 2JC,P = 9.7 Hz, C9), 105.0 (dd, 2JC,P = 12.7 Hz, 1JC,Rh = 6.9 Hz, C12/13/16/17), 81.2 (s, C2), 76.0 (d, 3JC,P = 9.7 Hz, C7), 74.9 (s, C4), 74.2 (d, 1JC,P = 46.4 Hz, C6), 73.9 (d, 2JC,P = 6.6 Hz, C8), 71.4 (s, C3), 70.3 (d, 1JC,Rh = 13.7 Hz, C13/12/17/16), 33.12 (s, C14/15/18/19), 33.09 (s, C14/15/18/19), 28.8 (s, C14/15/18/19); 31P{1H} NMR (CD2Cl2): δ [ppm] = 22.4 (d, 1JP,Rh = 152.0 Hz); IR (neat, ATR): [cm−1] = 3092 (w), 3071 (w), 3050 (w), 2992 (w), 2936 (w), 2914 (w), 2875 (m), 2828 (m, all ν(C–H)), 1506 (vs.), 1481 (s), 1434 (m, ν(C–P)), 1394 (w), 1379 (m), 1355 (m), 1332 (w), 1319 (m), 1305 (m), 1218 (w), 1161 (m), 1096 (m), 1072 (w), 1054 (w), 1027 (m), 997 (m), 960 (w), 926 (w), 889 (w), 859 (w), 832 (m), 760 (m), 743 (s), 691 (s), 626 (m), 502 (vs.), 471 (s), 425 (s); HRMS (ESI): m/z calcd for C93H90Cl2Fe3N3P3Rh3 [M − Cl]+ 1890.0952; found 1890.0957; elemental analysis calcd [%] for C93H90Cl3Fe3N3P3Rh3: C 58.02, H 4.71, N 2.18, found: C 57.25, H 4.66, N 2.16.
Synthesis of μ3-[1,3,5-tris{(1-diphenylphosphanyl-1′-ferrocenylene)methyl}benzene-1κ1P,2κ1P,3κ1P]-tris[chlorido-1,2,5,6-η4-(cycloocta-1,5-diene)rhodium(I)] ([1d(Rh)3])
[1d(Rh)3] was prepared analogously to [1a(Rh)3] using 1d (50.0 mg, 40.8 μmol, 1.00 eq.). For precipitation, significantly larger amounts of diethyl ether, hexanes (THF:Et2O:hexanes = 1:10:4 v/v/v) and low temperatures (−20 °C) had to be used. The very fine, light yellow precipitate was filtered over degassed Celite and recovered by dissolution in THF. Removal of the volatiles gave [1d(Rh)3] in 20% yield (16 mg).
M.p.: >160 °C decomp. (from THF); 1H NMR (THF-d8): δ [ppm] = 7.65–7.55 (m, 12H, H12), 7.40–7.27 (m, 18H, H11 + H13), 6.86 (s, 3H, H1), 5.49 (s (br), ω1/2 = 11.2 Hz, 6H, H14/15/18/19), 4.71 (pq, J = 2.0 Hz, 6H, H9), 4.36–4.29 (m, 6H, H8), 4.28 (s, 12H, H5 + H6), 3.72 (s, 6H, H3), 3.06 (s, 6H, H14/15/18/19), 2.43–2.27 (m, 12H, H16/17/20/21), 2.07–1.97 (m, 6H, H16/17/20/21), 1.92–1.83 (m, 6H, H16/17/20/21); 13C{1H} NMR (THF-d8): δ [ppm] = 142.4 (s, C2), 135.1 (d, 1JC,P = 42.5 Hz, C10), 135.0 (d, 3JC,P = 10.9 Hz, C12), 130.5 (d, 4JC,P = 2.1 Hz, C13), 128.4 (d, 2JC,P = 9.5 Hz, C11), 126.9 (s, C1), 105.0 (dd, 2JC,P = 13.2 Hz, 1JC,Rh = 7.1 Hz, C14/15/18/19), 90.8 (s, C4), 76.4 (d, 3JC,P = 10.5 Hz, C9), 74.1 (d, 1JC,P = 47.9 Hz, C7), 72.6 (d, 2JC,P = 6.9 Hz, C8), 71.5 (s, C5/C6), 71.2 (s, C6/C5), 70.7 (d, 1JC,Rh = 13.6 Hz, C15/14/19/18), 36.4 (s, C3), 33.88 (s, C16/17/20/21), 33.86 (s, C16/17/20/21), 29.6 (s, C16/17/20/21); 31P{1H} NMR (THF-d8): δ [ppm] = 22.9 (d, 1JP,Rh = 152.2 Hz); IR (neat, ATR): [cm−1] = 3073 (m), 3051 (m), 2996 (m), 2934 (m), 2912 (s), 2876 (s), 2828 (s, all ν(C–H)), 1979 (w), 1600 (m), 1572 (w), 1526 (w), 1480 (m), 1433 (vs., ν(C–P)), 1387 (w), 1333 (m), 1305 (m), 1229 (m), 1160 (s), 1096 (s), 1059 (m), 1027 (s), 997 (m), 961 (m), 925 (w), 899 (w), 857 (m), 829 (m), 815 (m), 744 (s), 702 (s), 693 (s), 625 (m), 540 (m), 522 (m), 496 (s), 467 (s), 445 (m), 426 (m), 406 (m); HRMS (ESI): m/z calcd for C96H99Cl2Fe3P3Rh3 [M − Cl]+ 1929.1567; found 1929.1592; elemental analysis calcd [%] for C99H99Cl3Fe3P3Rh3: C 60.53, H 5.08, found: C 59.16, H 5.16.45
Results and discussion
Synthesis and characterisation
Reacting the tris-phosphanes 1a–d, prepared according to the published procedure,30,31 with the suitable precursor compounds [{RuCl2(p-cym)}2] (p-cym = η6-p-cymene) or [{RhCl(1,5-cod)}2] (1,5-cod = η4-cycloocta-1,5-diene) in CH2Cl2 at r.t. in slight stoichiometric excess (1:3) afforded, after simple work-up procedures, the homotrinuclear metal complexes [1a–d(Ru)3] and [1a–d(Rh)3] in good to excellent yields (Scheme 1).
NMR spectroscopy (31P{1H} NMR chemical shifts for all complexes are presented in Table 1) does not suggest hindered rotation about the arene–ferrocenylene bonds, and all complexes remain homotrinuclear in the gas phase as assessed by high-resolution electrospray-ionisation mass spectrometry (HR-ESI MS, see ESI†).
Single crystals of [1a(Rh)3] suitable for XRD were obtained by slow diffusion of diethyl ether into a THF solution, confirming the trinuclear nature of the complex in the solid state (Fig. 1). As most of the few reported crystal structures of Rh3P3 complexes contain triangular or linear Rh3-derived cores,46–48 [1a(Rh)3] is, to the best of our knowledge, only the second tris-phosphane-based example. The other entry features rhodium(III) atoms coordinated by tris(2-diphenylphosphanylethyl)amine and includes one Rh–N bond.49 Balakrishna and co-workers have prepared C3-symmetric rhodium(I) tris–phosphane complexes, yet have not been able to determine their solid-state molecular structures.50 Tris(N-heterocyclic carbene)51 and tris(pyridyl) ligands52,53 have also been used for the preparation of C3-symmetric trinuclear rhodium complexes. Surprisingly, even though many ferrocenylphosphane rhodium complexes are listed in the CSD, the simple diphenylferrocenylphosphane cyclooctadiene (cod) rhodium(I) chloride moiety has not yet been crystallographically described; cationic complexes like the planar chiral diphenylphosphinoferrocenylthioether-derived rhodium(cod) complexes by Manoury and co-workers are not well comparable to [1a(Rh)3].54 The closest analogue, a [1]phosphaferrocenophane-derived complex by Breher and co-workers,55 compares favourably with [1a(Rh)3] (Table 2; more information in the ESI†) regarding the Rh–P bond lengths of 2.304–2.314 Å (their work: 2.296 Å).
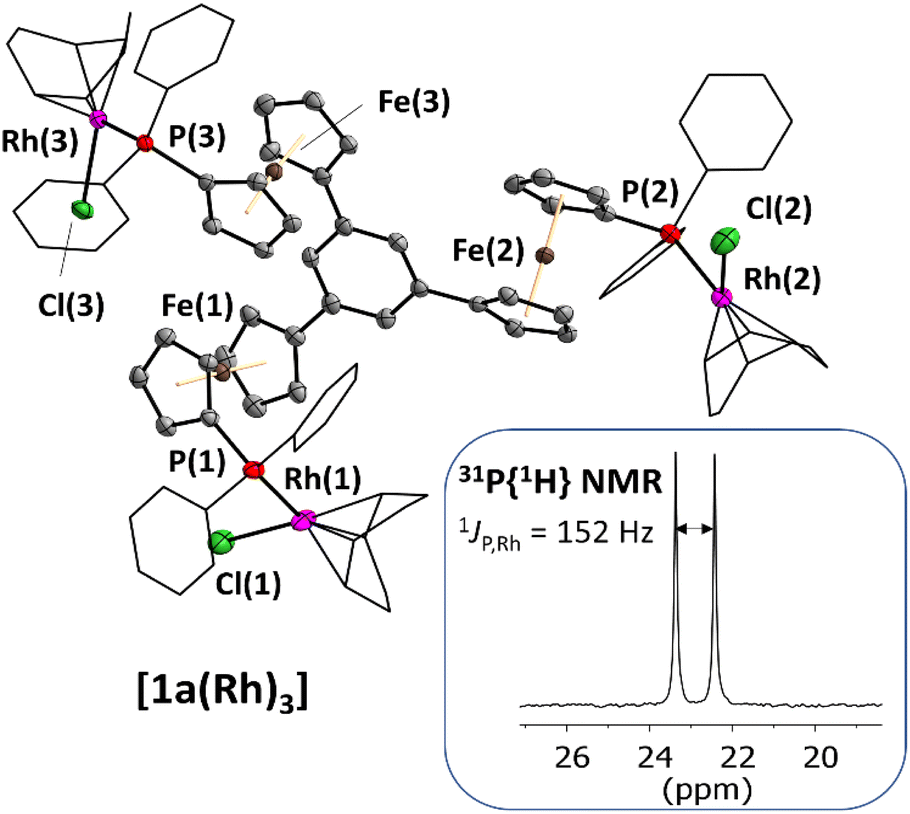 | ||
| Fig. 1 Molecular structure of the homotrinuclear rhodium(I) complex [1a(Rh)3] with part of the atom-numbering scheme and the 31P{1H} NMR signal in THF-d8 solution, featuring the characteristic 1J coupling between 31P and 103Rh. Thermal ellipsoids are set at the 50% probability level. For clarity, the phenyl rings and 1,5-cyclooctadiene ligands are depicted in wireframe style, and co-crystallised solvent and hydrogen atoms have been omitted. | ||
| [1a(Rh)3] | |
|---|---|
| a Intramolecular distances; the shortest intermolecular distance is given italicised and in brackets. | |
| Rh(1,2,3)–P(1,2,3) | 2.314(1)/2.382(1)/2.307(1) |
| Rh(1,2,3)–Cl(1,2,3) | 2.355(1)/2.304(1)/2.370(1) |
| P(1,2,3)–Rh(1,2,3)–Cl(1,2,3) | 91.28(4)/88.98(4)/91.38(4) |
| Rh(1,2,3)⋯Rh(2,3,1)a | 11.5266(7)/15.9121(7)/11.5422(5)/(6.1288(6)) |
| Rh(1,2,3)⋯Fe(1,2,3) | 4.289(1)/4.428(1)/4.460(1) |
No single crystals suitable for XRD analysis could be obtained for the ruthenium(II) complexes [1a–d(Ru)3] which were, however, fully characterised spectroscopically and by HR-ESI MS. In CD2Cl2 or THF-d8 they undergo a slow chemical transformation, liberating p-cymene, exemplarily shown by 1H and 31P{1H} NMR spectroscopy of [1a(Ru)3] (cf. ESI). This process is also solvent-dependent. We speculate that the loss of p-cymene is induced by either intra- or intermolecular η6-coordination of one of the phenyl rings in the PPh2 moiety. The addition of three equivalents of p-cymene slowed down this degradation, which is thus most likely connected to the intra- or intermolecular substitution by a P-bound phenyl ring,56,57 a process we have recently employed to prepare tethered P-chiral ruthenium(II) complexes.23 In the present case, the resulting products are hardly soluble and likely oligomeric or polymeric. These findings notwithstanding, compounds [1a–d(Ru)3] expand the scope of trinuclear ruthenium complexes and are the first non-cluster examples to incorporate more than one ferrocenyl moiety in the complex.58
Cyclic voltammetry
Complexes [1a–d(Ru)3] and [1a–d(Rh)3] are electrochemically active (Fig. 2). As a general observation, the electrochemistry in a BF4−-based supporting electrolyte (SE) system is more complicated, less reversible, and hence more difficult to interpret. Accordingly, the BArF4−-based SE was used which enables a distinct separation of the three iron-centred oxidations of the ligand backbone. Under these conditions, these oxidations are mostly reversible but, especially for the third oxidation, already show signs of irreversibility. The system with no deviations from (quasi-)reversibility, in all cases, is the C6H3-based ligand (1a); all other systems show reduced reversibility, most likely arising from follow–up reactions (for a detailed discussion of the complexes' electrochemistry as probed by cyclic voltammetry and, in selected cases, differential pulse voltammetry, see the ESI, Section 5†), which makes them less well-suited for potential applications in redox-switchable catalysis which relies on fully reversible redox events of the catalyst to ensure full control.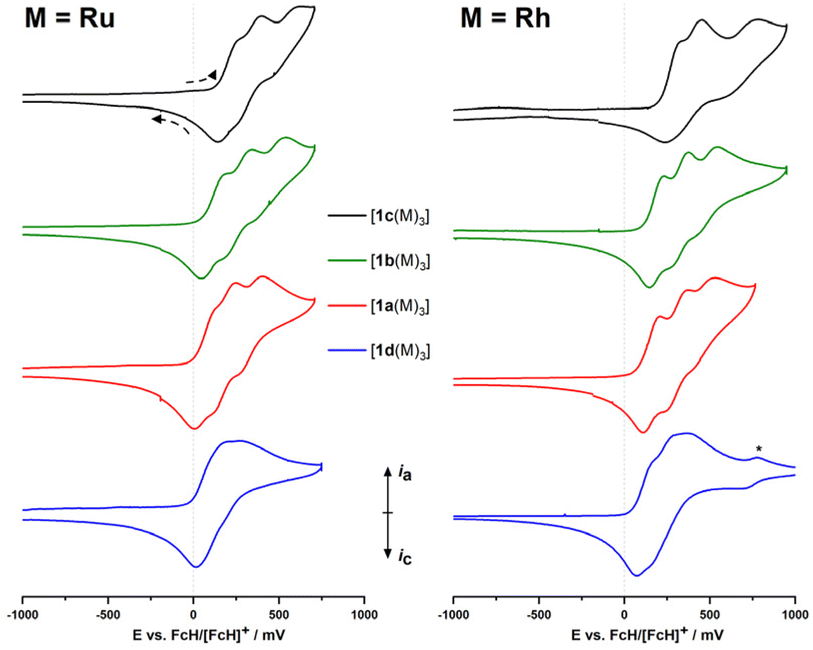 | ||
| Fig. 2 Partial cyclic voltammograms (iron-centred oxidations only) of ruthenium (left) and rhodium (right) complexes [1a–d(M)3] at ca. 1 mmol L−1 in 0.1 mol per L (nBu4N)BArF4/CH2Cl2 (scan rate: 100 mV s−1, working electrode: glassy carbon, counter electrode: platinum wire). The 2nd of three cycles is shown for all compounds, recorded currents are shown normalised for easier comparison. Scanning direction as indicated. The rhodium-centred oxidation event for [1d(Rh3)] is marked with an asterisk (*). For full voltammograms, see ESI (Section 5).† | ||
In general, while the coordinated metal complex fragment does not strongly alter the electrochemical response of the arene-trisferrocenyl core, there are observable differences. Rhodium(I) complexes are oxidised at slightly higher potentials than ruthenium(II) complexes (Table 3). For both metal fragments, individual oxidation states for the C6H3(CH2)3-based complexes are difficult to address and the C3N3-based complexes show severely broadened third, Fe-centred oxidations and reduced reversibility, meaning that the initial system cannot be fully restored under these conditions.
| E01 (ΔEp)a [mV] | |||||
|---|---|---|---|---|---|
| a Potentials vs. the FcH/[FcH]+ couple at a glassy carbon working electrode (scan rate 100 mV s−1). Determined on 1 mmol per L samples in anhydrous 0.1 mol per L (nBu4N)BArF4/CH2Cl2 as SE (working electrode: glassy carbon). The difference between oxidation and reduction potential, ΔEp, is given in brackets.b Determined from square-wave voltammetry due to close peak-to-peak separation, leaving ΔEp inaccessible. | |||||
| 1a30 | 138 (98) | [1a(Ru)3] | 64 (118) | [1a(Rh)3] | 158 (99) |
| 1b30 | 206 (116) | [1b(Ru)3] | 116 (140) | [1b(Rh)3] | 190 (86) |
| 1c31 | 275 (160) | [1c(Ru)3] | 204 (122) | [1c(Rh)3] | 283 (86) |
| 1d30 | 113b | [1d(Ru)3] | 91 (148) | [1d(Rh)3] | 116 (93) |
Oxidation events that are likely associated with the coordinated metal complex fragments can be well separated from the ligand-centred oxidations. Thus, a fourth, likely Rh-centred oxidation can be observed for the less electron-withdrawing ligands, but for ligands with a C6F3 or C3N3 core, the fourth oxidation event is outside the window of the electrochemical stability of the supporting electrolyte (see ESI,† Section 5). [1a–d(Rh)3] show metal-centred oxidations owing to the redox-active rhodium(I) centres. The peak potentials for the RhI/RhII couple in [1a(Rh)3] (751 mV with BF4−, 854 mV with BArF4−) are significantly higher and the oxidations less reversible than for a related P-ferrocenophane-derived chlorido(cyclooctadiene)rhodium(I) complex (E0 = 390 mV vs. FcH/[FcH]+) reported by Breher and co-workers.55 This reduced reversibility might be tied to the fact that the oxidation of the rhodium centre will most likely generate a quadruply charged cation.
In general, all ruthenium(II) complexes show a very similar electrochemical fingerprint under the given conditions, but more oxidation events associated with the coordinated metal are observed within the available electrochemical window. When measured in the BArF4−-based SE, the ruthenium-centred oxidations split into two distinct yet irreversible oxidation events, apparently consisting of one 1e−- and one 2e−-transfer steps. As an electrochemical comparison for [1a(Ru)3], a tethered (1′-methoxy-1-ferrocenylene)-based diarylphosphane ruthenium(II) complex, reported previously by us, is well suited and shows similar redox properties with E0(FeII/FeIII) = 110 mV and Eox(RuII/RuIII) = 700 mV vs. FcH/[FcH]+ in (nBu4N)PF6/CH2Cl2.59
For both metal complex fragments, the onset of ligand oxidation is determined by the arene substitution pattern and follows the expected order with 1d being the easiest, 1c the hardest to oxidise. None of the Ru/Rh-centred oxidations appear to be electrochemically reversible (see ESI,† Section 5). Consequently, redox state control of the metal centre, another variation of redox-switchable catalysis, is not possible with these complexes. In conclusion, this means that, among the systems investigated in this study, fully reversible ligand-centred redox control is most promising for [1a(M)3] (M = Ru, Rh).
Conclusions
We have demonstrated the synthesis of rhodium(I) and ruthenium(II) complexes with the tris(ferrocenyl)arene-based tris-phosphanes 1a–d, which form well-defined, C3-symmetric homotrinuclear transition metal complexes with four accessible oxidation states relating to the tris(ferrocenyl)arene backbone. With a BArF4−-based supporting electrolyte, a distinct separation of the three iron-centred oxidations of the ligand backbone was observed, making these complexes potentially suitable for redox-switchable catalysis or other applications in which control of the charge state of the system could be of interest, such as electrochromic films or materials with switchable surface characteristics.59,60Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. S. has carried out the syntheses and characterisation of the compounds, including the electrochemical experiments. P. C. has acquired and solved the solid-state structure. E. H.-H. has supervised and administered the project, helped in acquiring funding for A. S. and P. C.; E. H.-H. and A. S. wrote the first draft and revised the manuscript according to the referee comments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Studienstiftung des deutschen Volkes (doctoral fellowships to P. C. and A. S.), the Deutsche Forschungsgemeinschaft (DFG, He 1376/51-1), and the Graduate School BuildMoNa is gratefully acknowledged.Notes and references
- S. E. Gibson and M. P. Castaldi, Angew. Chem., Int. Ed., 2006, 45, 4718–4720 CrossRef CAS PubMed.
- C. Moberg, Angew. Chem., Int. Ed., 1998, 37, 248–268 CrossRef CAS PubMed.
- L. R. Gahan, Coord. Chem. Rev., 2016, 311, 168–223 CrossRef CAS.
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- A. G. Blackman, Polyhedron, 2005, 24, 1–39 CrossRef CAS.
- A. G. Blackman, Eur. J. Inorg. Chem., 2008, 2633–2647 CrossRef CAS.
- J. Wassenaar, M. A. Siegler, A. L. Spek, B. de Bruin, J. N. H. Reek and J. I. van der Vlugt, Inorg. Chem., 2010, 49, 6495–6508 CrossRef CAS PubMed.
- S. Bontemps, G. Bouhadir, W. Gu, M. Mercy, C.-H. Chen, B. M. Foxman, L. Maron, O. V. Ozerov and D. Bourissou, Angew. Chem., Int. Ed., 2008, 47, 1481–1484 CrossRef CAS PubMed.
- M. J. Burk and R. L. Harlow, Angew. Chem., Int. Ed. Engl., 1990, 29, 1462–1464 CrossRef.
- Z. Xu, M. F. Cain, A. V. Rupert, D. S. Glueck, J. A. Golen and A. L. Rheingold, Tetrahedron: Asymmetry, 2015, 26, 1459–1468 CrossRef CAS.
- P. Scherl, A. Kruckenberg, S. Mader, H. Wadepohl and L. H. Gade, Organometallics, 2012, 31, 7024–7027 CrossRef CAS.
- W. Hewertson and H. R. Watson, J. Chem. Soc., 1962, 1490–1494 RSC.
- I. R. Butler, M. G. Drew, A. G. Caballero, P. Gerner and C. H. Greenwell, J. Organomet. Chem., 2003, 679, 59–64 CrossRef CAS.
- D. Astruc, Eur. J. Inorg. Chem., 2017, 2017, 6–29 CrossRef CAS.
- Ferrocenes. Ligands, Materials and Biomolecules, ed. P. Štěpnička, J. Wiley, Chichester, England, Hoboken, NJ, 2008 Search PubMed.
- S. Santi, A. Bisello, R. Cardena and A. Donoli, Dalton Trans., 2015, 44, 5234–5257 RSC.
- P. Debroy and S. Roy, Coord. Chem. Rev., 2007, 251, 203–221 CrossRef CAS.
- R. Kalio, P. Lönnecke and E. Hey-Hawkins, J. Organomet. Chem., 2008, 693, 590–600 CrossRef CAS.
- J. R. F. Pritzwald-Stegmann, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2016, 45, 2208–2217 RSC.
- J. Popp, S. Hanf and E. Hey-Hawkins, Chem.–Eur. J., 2020, 26, 5765–5769 CrossRef CAS PubMed.
- M. Madalska, P. Lönnecke and E. Hey-Hawkins, J. Mol. Catal. A: Chem., 2014, 383–384, 137–142 CrossRef CAS.
- M. Madalska, P. Lönnecke, V. Ivanovski and E. Hey-Hawkins, Organometallics, 2013, 32, 5852–5861 CrossRef CAS.
- J. Popp, S. Hanf and E. Hey-Hawkins, ACS Omega, 2019, 4, 22540–22548 CrossRef CAS PubMed.
- A. Schmied, A. Straube, T. Grell, S. Jähnigen and E. Hey-Hawkins, Dalton Trans., 2015, 44, 18760–18768 RSC.
- C. Limburg, P. Lönnecke, S. Gómez-Ruiz and E. Hey-Hawkins, Organometallics, 2010, 29, 5427–5434 CrossRef CAS.
- S. Tschirschwitz, P. Lönnecke and E. Hey-Hawkins, Organometallics, 2007, 26, 4715–4724 CrossRef CAS.
- P. Neumann, H. Dib, A. Sournia-Saquet, T. Grell, M. Handke, A.-M. Caminade and E. Hey-Hawkins, Chem.–Eur. J., 2015, 21, 6590–6604 CrossRef CAS PubMed.
- J. Popp, A.-M. Caminade and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2020, 17, 1654–1669 CrossRef.
- P. Neumann, H. Dib, A.-M. Caminade and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2015, 54, 311–314 CrossRef CAS PubMed.
- A. Straube, P. Coburger, L. Dütsch and E. Hey-Hawkins, Chem. Sci., 2020, 11, 10657–10668 RSC.
- A. Straube, P. Coburger, M. R. Ringenberg and E. Hey-Hawkins, Chem.–Eur. J., 2020, 26, 5758–5764 CrossRef CAS PubMed.
- A. Straube, P. Coburger, M. Michak, M. R. Ringenberg and E. Hey-Hawkins, Dalton Trans., 2020, 49, 16667–16682 RSC.
- R. Taube and S. Wache, J. Organomet. Chem., 1992, 428, 431–442 CrossRef CAS.
- G. Giordano, R. H. Crabtree, R. M. Heintz, D. Forster and D. E. Morris in Inorganic Syntheses, ed. R. J. Angelici, John Wiley & Sons, Inc, Hoboken, NJ, USA, 1990, vol. 28, pp. 88–90 Search PubMed.
- M. A. Bennett, T.-N. Huang, T. W. Matheson, A. K. Smith, S. Ittel and W. Nickerson in Inorganic Syntheses, ed. J. P. Fackler, vol. 21, John Wiley & Sons, Inc, Hoboken, NJ, USA, 1982, pp. 74–78 Search PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- Rigaku Corporation, CrysAlisPro Software System, Rigaku Oxford Diffraction, Wroclaw, Poland, 1995–2023 Search PubMed.
- SCALE3 ABSPACK: Empirical Absorption Correction Using Sperical Harmonics, included in the CrysAlisPro Software System, Rigaku Oxford Diffraction, Wroclaw, Poland, 1995–2023 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- C. Cason, T. Fröhlich and C. Lipka, POV-Ray – The Persistence of Vision Raytracer, Persistence of Vision Pty. Ltd, 2013 Search PubMed.
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, J. Phys. Chem. B, 1999, 103, 6713–6722 CrossRef CAS.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461–466 CrossRef.
- The 13C resonances of the C6F3 core have only been identified through 19F-decoupling and are not visible in the standard 13C{1H} NMR spectrum, even using a high number of scans. Their multiplicities are indicated with “s” as they are singlets in the 13C{19F} NMR spectrum.
- An editorial about elemental analysis: F. P. Gabbaï, P. J. Chirik, D. E. Fogg, K. Meyer, D. J. Mindiola, L. Schafer and S.-L. You, Organometallics, 2016, 35(19), 3255–3256 CrossRef . https://www.perkinelmer.com/lab-solutions/resources/docs/APP_011267_01_TheElementalAnalysisofVariousClassesofChemicalCompoundsUsingCHN.pdf.
- Y. Matsusaka, S. Shitaya, K. Nomura and A. Inagaki, Inorg. Chem., 2017, 56, 1027–1030 CrossRef CAS PubMed.
- A. Preetz, W. Baumann, H.-J. Drexler, C. Fischer, J. Sun, A. Spannenberg, O. Zimmer, W. Hell and D. Heller, Chem.–Asian J., 2008, 3, 1979–1982 CrossRef CAS PubMed.
- C. Fischer, C. Kohrt, H.-J. Drexler, W. Baumann and D. Heller, Dalton Trans., 2011, 40, 4162–4166 RSC.
- P. Dapporto, P. Stoppioni and P. M. Maitlis, J. Organomet. Chem., 1982, 236, 273–280 CrossRef CAS.
- S. Naik, M. Kumaravel, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2014, 53, 1370–1381 CrossRef CAS PubMed.
- E. Mas-Marzá, E. Peris, I. Castro-Rodríguez and K. Meyer, Organometallics, 2005, 24, 3158–3162 CrossRef.
- P. Chellan, K. M. Land, A. Shokar, A. Au, S. H. An, D. Taylor, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 4793–4804 CrossRef CAS.
- P. Chellan, K. M. Land, A. Shokar, A. Au, S. H. An, D. Taylor, P. J. Smith, T. Riedel, P. J. Dyson, K. Chibale and G. S. Smith, Dalton Trans., 2014, 43, 513–526 RSC.
- E. M. Kozinets, O. Koniev, O. A. Filippov, J.-C. Daran, R. Poli, E. S. Shubina, N. V. Belkova and E. Manoury, Dalton Trans., 2012, 41, 11849–11859 RSC.
- A. Feyrer, M. K. Armbruster, K. Fink and F. Breher, Chem.–Eur. J., 2017, 23, 7402–7408 CrossRef CAS PubMed.
- J. C. McConway, A. C. Skapski, L. Phillips, R. J. Young and G. Wilkinson, J. Chem. Soc., Chem. Commun., 1974, 327–328 RSC.
- T. Wilczewski, J. Organomet. Chem., 1985, 297, 331–340 CrossRef CAS.
- B. Milde, T. Rüffer and H. Lang, Inorg. Chim. Acta, 2012, 387, 338–345 CrossRef CAS.
- M. Gallei and C. Rüttiger, Chem.–Eur. J., 2018, 24, 10006–10021 CrossRef CAS PubMed.
- Y. Kim and K. Kubo, Pure Appl. Chem., 2023, 95, 707–731 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Assignment of specific nuclei in NMR spectra, NMR spectra, X-ray crystallographic data, CV data. CCDC 1990280. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra03822c |
| ‡ New address: Dr A. Straube, Wiley-VCH, Boschstr. 12, D-69469 Weinheim, Germany. |
| § New address: Dr P. Coburger, Department of Inorganic Chemistry, TU München, Lichtenbergstraße 4, 85747 Garching, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |