
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking the potential of water-soluble gold(I)–NHC complexes: unveiling the role of carboxylic acid in cycloisomerization of alkynyl amino acid derivatives†
Liddier O. Péreza,
Tomás A. Dóminaa,
Gabriela A. Fernándezb,
Gustavo F. Silbestri *b and
Sebastián A. Testero*a
*b and
Sebastián A. Testero*a
aInstituto de Química Rosario-IQUIR (CONICET), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, Rosario, S2002LRK, Argentina. E-mail: testero@iquir-conicet.gov.ar
bDepartamento de Química, INQUISUR, Universidad Nacional del Sur (UNS)-CONICET, B8000CPB Bahía Blanca, Argentina. E-mail: gsilbestri@uns.edu.ar
First published on 6th August 2024
Abstract
Hydrocarboxylation of alkyne-containing amino acid derivatives using water-soluble gold(I)–NHC complexes in an aqueous biphasic system at room temperature is described. Addition of silver salts is not required as the carboxylic acid group of the substrate is responsible for the activation of the gold catalyst at room temperature. Our results confirm that the steric bulk of the N-heterocyclic carbene ligands is an important factor in both the stability and the catalytic activity of gold(I) complexes in aqueous medium, and consequently in the recycling (at least 15 times without any loss of activity). The catalytic activity of our most active water-soluble gold(I)–NHC complex has been demonstrated in the cycloisomerization of amino acids derivatives with terminal and internal alkynes in aqueous media at room temperature.
Introduction
The use of water as a solvent in organic reactions has spread in recent years showing its versatility in a wide range of chemical transformations.1 Water is considered a green solvent, the main advantages are its low cost, safety, non-flammability, renewability and high heat capacity that make it a very attractive solvent for chemical processes. To carry out catalytic reactions in aqueous medium, the availability of water-soluble catalysts must be considered. Nowadays, various transition metal complexed with N-heterocyclic carbenes (NHCs) bearing hydrophilic groups are known.2,3 NHC ligands provide higher stability and reactivity to the transition metal catalyst compared with phosphine-based ligands since the metal–NHC bonds exhibit a strong σ donor contribution and π back-donation ability.4 In particular, a number of water-soluble NHC-based gold complexes were reported by us5–7 and other authors.8–12 These complexes proved to be efficient and reusable catalysts in aqueous media.Until the beginning of the 21st century, gold was considered to be less reactive than its neighbors in the periodic table, and there was a perception that it was unreactive in catalytic applications.13 However, the discovery of gold's ability to activate C–C unsaturated groups to promote the addition of a variety of nucleophiles to these multiple bonds,14,15 together with gold catalysts tolerance to a wide range of functional groups and mild reaction conditions,16–18 has made gold-based complexes highly sought after.19 These characteristics have led to an exponential growth of homogeneous gold catalysis in the last twenty years and paved the way for the development of numerous of new catalytic transformations, including tandem reactions and asymmetric catalysis using gold,20–22 turning it into a powerful tool in mainstream organic chemistry.
In this context and as part of our research program concerning the search for new water-soluble gold complexes, we have synthesized, with excellent yields (90–95%), a series of symmetrical and asymmetrical Au(I)–NHC (C1–5, Fig. 1) complexes containing at least one sulfonate group in its structure.5 These water-soluble gold(I)–NHC complexes proved to be efficient and highly recyclable precatalysts in the alkyne hydration in water/methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 100 °C, even in the absence of silver salts. Our results indicate that the bulkier complex (C1) is the most effective and that the addition of methanol as co-solvent not only shortens reaction times but also stabilizes the less bulky complexes.6
:1) at 100 °C, even in the absence of silver salts. Our results indicate that the bulkier complex (C1) is the most effective and that the addition of methanol as co-solvent not only shortens reaction times but also stabilizes the less bulky complexes.6
 | ||
| Fig. 1 Water-soluble Au(I)–NHC complexes. Water solubility at 25 °C (g L−1): C1 111, C2 80, C3 180, C4 645, C5 (stable in a methanol solution). | ||
On the other hand, γ-alkylidene lactones are found in an extensive array of biologically active natural compounds.23–25 They are usually obtained through an atom-economical approach such as the cycloisomerization of γ-acetylenic acids.26–28 In recent years, the intramolecular addition of acids to alkynes have been performed utilizing a variety of transition metals, among which are Hg,29,30 Ag,31,32 Cu,33,34 and Pd.35,36 Unfortunately, most of these methods have certain drawbacks, including: (i) the use of high catalyst loadings, (ii) the use of organic solvents, (iii) extended reaction times, (iv) the use of additives, and/or (v) the need for refluxing solvents. Moreover, a significant limitation of the majority of the reported approaches is their inability to recycle the catalyst.
Environmentally friendly alternatives for the cycloisomerization of alkynoic acids have been reported. These include the use of a heterogeneous Pd(II) catalyst,37 an iminophosphorane–Pd(II) complex,38 an NCN–pincer Pd-complex-bound norvaline-based supramolecular gel.39 Additionally, alternatives involving heterogeneous gold catalysis through gold nanoparticles stabilized by PEG-tagged imidazolium salts,40 or thiolstabilized in the pores of siliceous mesocellular foam,41 a sol–gel immobilized NHC gold complex in a biphasic system42 or in combination with deep eutectic solvents43 and gold catalysts with a solvent free process with a digold complex44 are also reported.
Herein, we present five water-soluble gold catalysts (C1–5) that efficiently cycloisomerize acetylenic acids at 1 mol% at room temperature in a biphasic system, without requiring silver salts. This research compares these catalysts with previous data, identifying a reactivity trend influenced by the steric volume of substituents attached to the NHC backbone. Additionally, it underscores the important role of the carboxylic acid group in activating the gold catalyst, aiding in the dissociation of the Au–Cl bond at room temperature.
Results and discussion
We wanted to explore and expand the scope of our water-soluble precatalysts (C1–5) and apply them in a more complex reaction than an alkyne hydration. In 2012, Cadierno et al. published the cycloisomerization (hydrocarboxylation of alkynes) of γ-alkynoic acids into γ-alkylidenelactones using a pyridine-substituted water-soluble Au(III)–NHC complex in a biphasic media such as toluene/water (Scheme 1).10,11 The biphasic media has the advantage that the product is soluble in the organic layer and can be isolated with a simple phase separation. On the contrary, the hydrophilic catalyst remains in the aqueous phase facilitating the recycling and reuse of the catalytic active gold species. In this way, a new cycle can be carried out simply adding a fresh load of substrate and toluene to the aqueous phase containing the Au–NHC catalyst (Fig. 2). | ||
| Scheme 1 Water-soluble gold–NHC complexes in hydration and hydrocarboxylation of alkynes. | ||
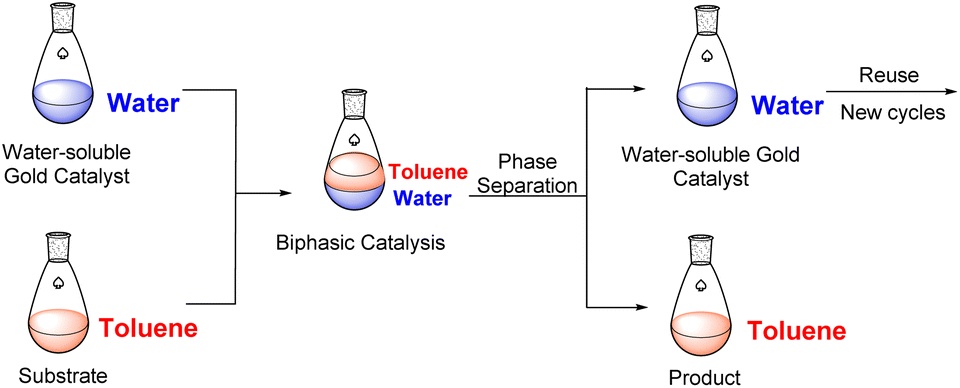 | ||
| Fig. 2 Biphasic catalysis using water-soluble gold complexes. | ||
To prove the value of the water-soluble gold(I)–NHC complexes (C1–5) in the hydrocarboxylation of alkynes we chose as substrates alkyne-containing amino acid derivatives (Scheme 1). They can be easily prepared by a number of methods using few transformations from economical starting materials.28,45
The optimization of the reaction conditions began with a screening of the hydrophilic gold-catalysts C1–5 (1 mol%) using the N-Boc-propargylglycine 1a in a biphasic system or on neat water at room temperature (Table 1).
|
|||
|---|---|---|---|
| Entry | Catalyst (1 mol%) | Conditions | Yieldb (%) |
| a Reactions were carried out starting from 0.18 mmol of 1a in 1 mL of toluene and 2 mL of distilled water.b Determined by 1H NMR. In brackets are shown the isolated yields after phase separation. | |||
| 1 | C1 | Toluene/water | 95 (81) |
| 2 | Water | 99 (72) | |
| 3 | Water/K2CO3 | 99 (89) | |
| 4 | C2 | Toluene/water | 87 (89) |
| 5 | C3 | Toluene/water | 94 (82) |
| 6 | C4 | Toluene/water | 95 (85) |
| 7 | C5 | Toluene/water | 98 (86) |
| 8 | AuCl | Toluene/water | 99 (86) |
| 9 | — | Toluene/water | 0 |

In general, the reactions were completed in 1 h (with exception of the reaction catalyzed by C2, entry 4) and showed a high 5-exo-dig regioselectivity. Under these conditions at room temperature, the product of alkyne hydration is not detected and once the phases are separated, the product does not require further purification.46 As can be seen from Table 1, all complexes are active and effective catalysts in toluene/water mixture showing conversions around 95% (entries 1, 5, 6 and 7). The reaction can be performed on water without any organic solvent but the isolated yields of the enol-lactone 2a are lower than the use of a biphasic system (entry 2). Furthermore, it has to be mentioned that the use of only water as reaction solvent narrows the scope of the reaction since many substrates are insoluble. The addition of catalytic amounts of K2CO3 is necessary to obtain good yields of the enol-lactones in organic solvents.27,28,47 In our case when we incorporated 0.1 equivalent of K2CO3 using water as solvent, this led to similar yields of the isolated product 2a (entry 3). Therefore, the presence of this additive is not essential. Curiously, taking into account that C5 is unstable in water, a lower performance was expected under the conditions used; however, its catalytic performance was remarkably good (entry 7). The utilization of a simpler source of gold as AuCl converts efficiently the N-Boc-propargylglycine into the enol lactone 2a (entry 8) but there are no possibilities of recycling or reusing the gold catalyst, as AuCl would be taken up by the organic phase. As expected, in absence of catalyst (entry 9), the cycloisomerization does not occur confirming that the activation of the alkyne by gold is necessary to the hydrocarboxylation of alkynes. Next, an evaluation of the catalyst concentration was investigated using 1 mol% of C1 as a model.
As can be seen in Table 2, performing the reaction at different concentrations (from 0.30 to 0.84 mM) of C1 does not affect the yields of the product (entries 1–3). The short reaction time led us to evaluate the loading of C1 in the gold catalyzed cycloisomerization of 1a. The cyclization proceeds with catalyst loadings as low as 0.01 mol% without loss of yield albeit requiring 2 hours for full completion (entries 4–7).
|
|||||
|---|---|---|---|---|---|
| Entry | C1 (mol%) | Toluene/water ratio (mL) | C1 (mM) | TOF (h−1) | Yieldb (%) |
| a Reactions were carried out starting from 0.14 mmol of 1a.b Determined by 1H NMR. In brackets are shown the isolated yields after phase separation.c The reaction was run until disappearing of starting material (2 h). | |||||
| 1 | 1 | 1:1 |
0.84 | 95 | 94 (82) |
| 2 | 1 | 1:2 |
0.62 | 96 | 95 (81) |
| 3 | 1 | 1:3 |
0.30 | 96 | 95 (81) |
| 4 | 0.5 | 1:2 |
0.24 | 187 | 93 (75) |
| 5 | 0.25 | 1:2 |
0.13 | 376 | 94 (77) |
| 6 | 0.1 | 1:2 |
0.06 | 938 | 99 (88) |
| 7c | 0.01 | 1:2 |
0.006 | 4829 | 99 (90) |
Next, in order to compare the efficiency of our water-soluble catalysts C1–C5 in the hydrocarboxylation of 1a in aqueous media, we decreased the catalyst loading of C2–C5 as shown in Table 3.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | (mol%) | TOF (h−1) | Yieldb (%) |
| a Reactions were carried out starting from 0.14 mmol of 1a in 1 mL of toluene and 2 mL of distilled water.b Determined by 1H NMR. In brackets are shown the mass recovery after phase separation, which is composed of the starting material and the product.c The reaction was run for 2 h. | ||||
| 1 | C2 | 0.1 | 182 | 30 (61) |
| 2 | C3 | 0.1 | 597 | 95 (73) |
| 3c | C3 | 0.01 | 2005 | 53 (85) |
| 4 | C4 | 0.1 | 367 | 67 (59) |
| 5c | C4 | 0.01 | 1051 | 31 (73) |
| 6 | C5 | 0.1 | 345 | 50 (85) |

The results obtained suggest that an increase in steric hindrance on the substituent directly attached to the NHC ring caused an increase in the catalytic activity of gold(I) complexes with the bulkier NHC complex (C1) being the most effective catalyst in the studied reaction (Fig. 3). This trend is in accordance with our previous results in the alkyne hydration with these precatalyst. To best of our knowledge, C1 is the most active gold catalyst in aqueous medium to the date (0.14 mmol, TOF = 4829 h−1).
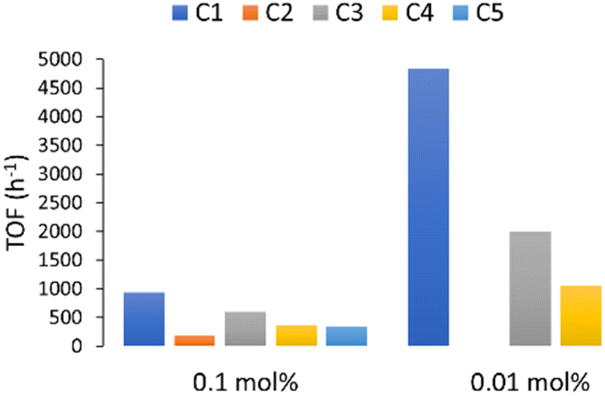 | ||
| Fig. 3 Relative effectiveness of our water-soluble catalysts C1–C5 in the hydrocarboxylation of 1a in aqueous media. | ||
Thereafter, in order to obtain some kinetic information about the cycloisomerization catalyzed by C1, we monitored the reaction over time using a GC-FID. The kinetic plot in Fig. 4 shows a rapid activation with no induction period detected, reaching maximum conversion around 10 minutes (0.14 mmol, TOF = 575 h−1). To confirm this, we carried out a reaction at room temperature for 10 min. After that time, we separated the organic phase and a conversion of 95% by NMR and 83% isolated yield was observed, values almost identical for 1 h reaction (Table 1, entry 1).
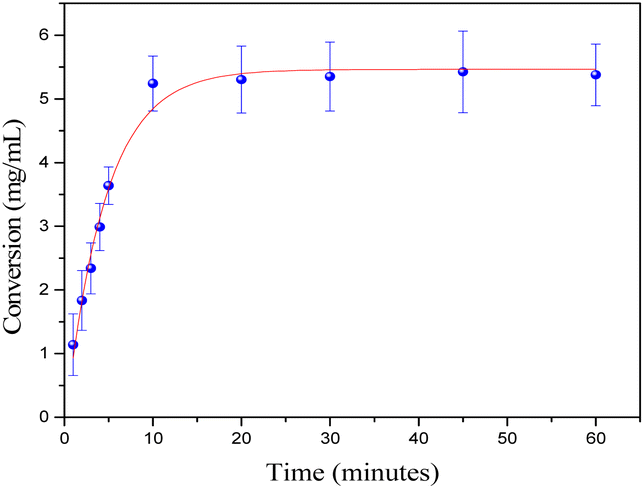 | ||
| Fig. 4 Time-course of the conversion of 1a (0.14 mmol) into 2a in toluene/water (2 mL:2 mL) at room temperature with 1 mol% of C1. | ||
Usually, the gold complexes precatalyst are activated by heating the reaction or with the addition of silver salts which remove the chlorine from the gold center to generate in situ the active gold species. Nevertheless, it is proved that the silver salts can influence the reaction selectivity, rate and yield in gold catalysis.48 In our case, the reaction conditions indicate that adding silver salts or heating the reaction was not required to generate the active gold species [Au(I)–NHC]+ responsible for the catalytic activity in the aqueous media. This means that the Au–Cl bond in the precatalysts C1–5 is quickly dissociated in the aqueous media at room temperature without the need of any additive.49 In order to shed light on the role of the carboxylic acid group in the mechanism of reaction and to understand the absence of product of alkyne hydration, we performed a reaction with the propargyl ester 3 at room temperature and at 100 °C (Scheme 2). After stirring for 4 h at room temperature compound 3 in a mixture toluene/water in the presence of 1 mol% of precatalyst C1, we recovered the totality of propargyl ester 3, which means that hydration of alkyne does not occur at this temperature. When this reaction was carried out at 100 °C, in four h we obtained the methyl ketone 4 (product of alkyne hydration) with 78% yield. This might imply that the carboxylic group in 1a is involved in the activation of the gold catalyst at room temperature and hydration of alkyne (intermolecular reaction) takes place only at 100 °C (activation by heating) in absence of a good intramolecular nucleophile.
 | ||
| Scheme 2 Hydration of the propargyl ester 3 at room temperature and at 100 °C. | ||
Taking into account the experimental results, we envisaged the reaction mechanism consisting in three steps as shown in Scheme 3. The first step is the pre-equilibrium, the second step is the nucleophilic attack of the carboxylic O–H to the gold–alkyne complex and the third step is the protodeauration.50
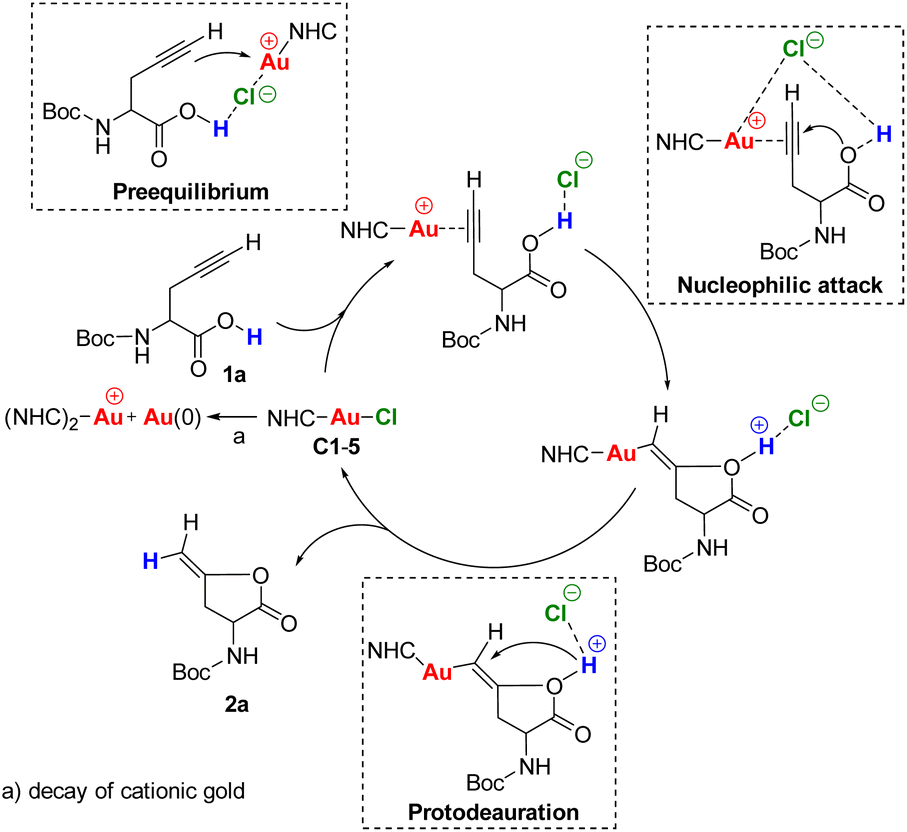 | ||
| Scheme 3 Proposed reaction mechanism for hydrocarboxylation of alkyne-containing amino acids derivatives. | ||
In the NHC–Au–Cl precatalysts, the counterion chlorine has a strong affinity for gold, therefore it coordinates strongly to it. Although, usually the addition of silver salts is used to remove the chlorine from the gold complexes, there are other ways to activate the precatalyst. For example, it has been reported that an H-bond donor in the substrate is able to assist the Au–Cl bond activation.51 In our substrate we have a carboxylic O–H that can interact with the chlorine to facilitate the Au–Cl dissociation allowing the alkyne to coordinate with the gold center (pre-equilibrium step). In the nucleophilic attack, the chlorine may hold the alkyne in the right position and boost the nucleophilicity of the carboxylic O–H through a long-range proton–counterion interaction. Finally, the protodeauration may be assisted by the chlorine to cleavage the O–H bond.
Furthermore, we decided to study the recycling and reuse of the catalysts under the optimized conditions (Fig. 5). Regarding the recyclability and reuse of the water-soluble Au(I)–NHC, Cadierno' group was able to recycle and reuse his gold(I) catalysts (5b and 5c, Fig. 6) only 4 times11 while we were able to recycle and reuse our gold(I) catalyst C1 at least 15 times without any loss of activity. Conversely, catalysts C2, C3 and C4 began to lose efficiency from the 6th or 7th recycling cycle resulting in the formation of inactive purple colloidal gold. Surprisingly, given its low stability, C5 could be reused up to 9 times. Based on these results and to the best of our knowledge, C1 is the water-soluble gold(I)–NHC catalyst that can be recycled the largest number of times.
 | ||
| Fig. 5 Recycling experiment. Number of cycles vs. conversion percentage of different [Au] complexes. | ||
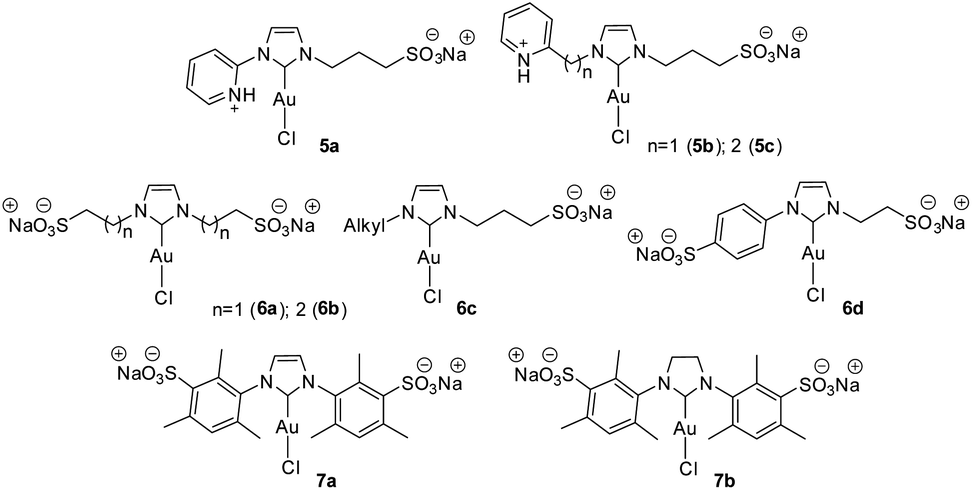
In the gold–NHC complexes and of course in the water-soluble analogs, the substituents directly attached on the NHC moiety play an important role in the stability and activity of the catalyst. Cadierno claimed that his pyridinium-substituted Au(I)–NHC complexes with alkyl chains directly connected to the imidazole-2-ylidene unit were more stable in water in comparison with those complexes containing aryl-substituent (Figure 6, 5a–c).11 In our opinion, this is not a general trend since Cadierno's10,11 alkyl-pyridinium catalysts are able to form a chelate between the pyridyl unit (uncommon NHC ligand) and the gold atom. This is not the case for the catalyst that has the pyridinium directly attached to the NHC ring. On the other hand, Joó et al.8 synthesized (Fig. 6) a series of sulfonated bis N-alkyl (6a–c) and N-alkyl-N-aryl-substituted (6d) gold(I)–NHC complexes and observed that the bis N-alkyl derivate (6a–c) complexes discompose in aqueous solution more slowly than their N-alkyl-N-aryl-substituted NHC analogs (6d). It is important to note that the N-aryl groups employed by Joó are benzenesulfonic acid and therefore they are not hindered. In a later report, Joó described the synthesis of sulfonated IMes (7a) and SIMes (7b) gold(I)–NHC complexes.9 These more hindered precatalysts proved to be more active than the N-alkyl substituted analogs (6a–c) reported by them earlier. According to Joó,9 7a and 7b are stable in solid state but there is no mention of their stability in aqueous solution nor is there any comparison of their stability in water solution with that of the sulfoalkyl-substituted imidazolylidene complexes 6a–d. Of note, Nolan found that among the analogues of gold complexes without the sulfonate group, the bulky complex [(IPr)AuCl] is much more efficient than the less bulky [(IMes)AuCl] in the hydration of alkynes.52 We have previously reported that, the induction is more favored with increasing size of the N-substituents.6 As it is shown by our catalysts C1–5, the stability of the catalyst depends on the steric volume of the substituents bound to the NHC backbone. The higher the steric hindrance, the more stable the complex. In our case, the most stable and efficient catalyst (C1) was the one with two hindered aromatic rings bound to the NHC core. Catalysts with at least one alkyl chain directly bound to the NHC core showed to be less stable (C3–5) or less efficient (C2) in the cycloisomerization and consequently in the recycling.
Nolan et al.53 discuss the use of two different methods to quantify and analyze the steric impact of NHC ligands, along with some examples of their use in organometallic chemistry and catalysis. The percent buried volume (% Vbur) provides a single number to measure the overall steric impact of a ligand and the steric maps provide a graphical representation of the steric profile of a ligand and also provide per quadrant information of the steric impact of them. These two molecular descriptors have been used to understand and correlate with reactivity trends. In the article, it is observed that, during the transition from simple and flexible N-alkyl substituents to IMes to IPr (analogue to the N-substituent in C1) on the corresponding [AuCl(NHC)] species, there is a notable increase in the buried volume. It rises from around 28% to 36.5% to 45.4% respectively. Taking into account the catalytic activity and stability of our gold complexes C1–5, those of Cadierno10,11 (5a–c) and those of Joó8,9 (6a–d and 7a–b), we can postulate that higher % Vbur correlates with higher catalytic activity in the gold complexes (IPr ligands > IMes ligands > N-alkyl NHCs). This confirms that the steric bulk is an important factor in both the stability and the catalytic activity of gold(I) complexes in aqueous medium, with the bulkier NHC complex (C1) being the most effective catalyst in the studied reaction.
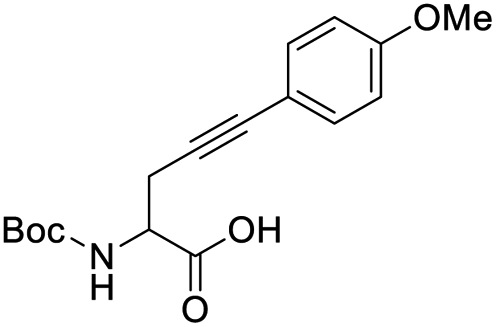
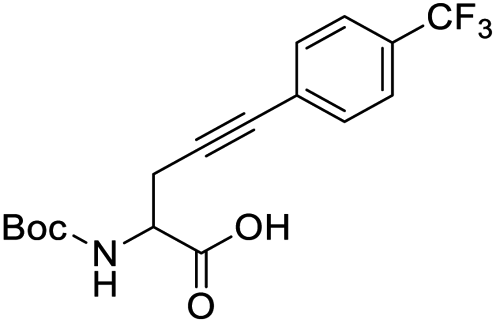
Finally, to evaluate the scope of our most promising catalyst C1, amino acids derivatives with internal alkynes were subjected to the cycloisomerization conditions in aqueous media. It is known that the internal alkynes are less reactive than terminal alkynes,8,10,11 this is reflected in longer reaction times and/or more vigorous conditions to obtain the enol lactones. The cycloisomerizations with internal alkynes are also a good measure of the catalyst's activity. For instance, Joó's8,9 catalyst 7a and 7b with hindered aryl substituents bound to the NHC backbone are more active than his N-alkyl substituted analogs 6a–d since catalysts 7 react with internal alkynes while catalysts 6 are not reactive. In the case of an alkane substituent such as a methyl (Table 4, entry 1) directly bound to sp-carbon of the triple bond, the cycloisomerization at room temperature using 1 mol% of catalyst C1 required only 1 h to complete (99% yield, 0.1329 mmol, TOF = 68 h−1 and TON = 68). 1H NMR spectra of the crude showed a mixture of the exo and endo cyclization products in a ratio 5:1 respectively (see ESI†). Internal alkynes substituted with aromatic rings are less reactive than the ones with alkyl chains. In the case of a phenyl substituent directly bound to the triple bond (Table 4, entry 2), complete conversion with 1 mol% of C1 required 5 h at room temperature. In this case, the 1H NMR spectra of the crude revealed only one product coming from the 5-exo-dig cycloisomerization in a 79% yield (0.1033 mmol, TOF = 9.6 h−1 and TON = 48).

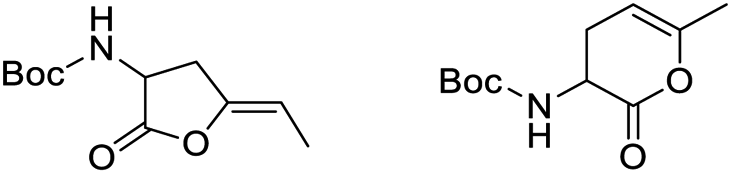
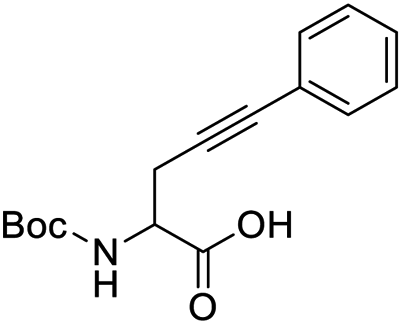

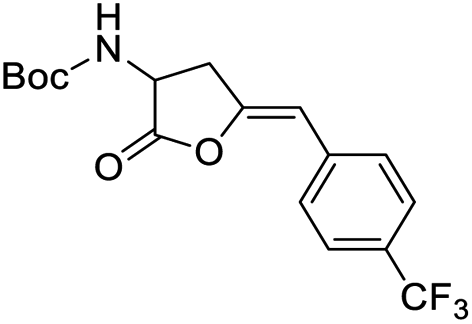
It is worth noting that this is the first example of a phenyl-substituted internal alkyne successfully cycled at room temperature with a water-soluble gold complex. In a similar example, a phenyl-substituted internal alkyne required heating for 2 h at 80 °C with 2.5 mol% of a water-soluble Au(III)–NHC catalyst to obtain a 28% yield of the enol lactone,10 while Joó's most active catalysts 7a–b required 5 mol% of catalyst loading and an acid co-catalyst to transform diphenylacetylene in 2-phenylacetophenone in 38%.9
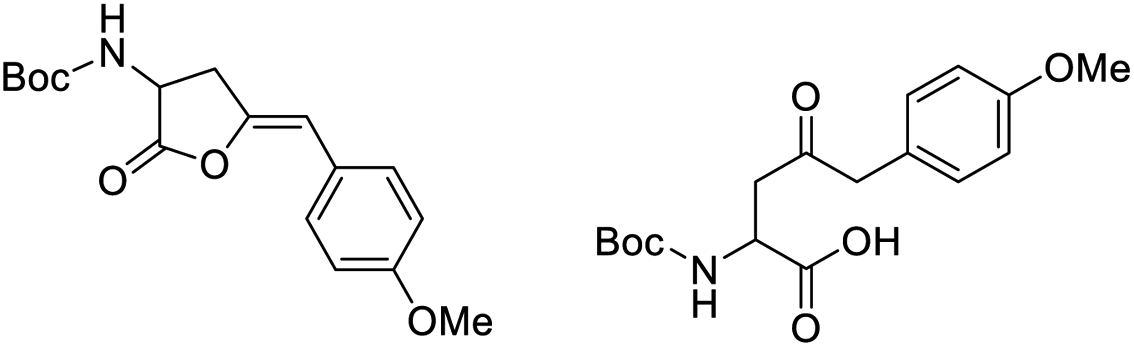
When an alkyne substituted with p-methoxybenzene was treated with our cycloisomerization conditions, we found out a mixture of the corresponding enol lactone 2d and ketone in a ratio of 1:2.6 respectively (Table 4, entry 3). Interestingly, when an internal alkyne derivative with an electron-withdrawing group such as a trifluoromethyl attached to the aromatic ring was tested (Table 4, entry 4), the reaction resulted only in the enol lactone 2e that come from gold-catalyzed cycloisomerization without observing any trace of ketone. This might be attributed to the electronic effect of the substituent in para position in the aromatic ring, electron-withdrawing groups make the gold–alkyne complex more electron deficient and thus favouring the enol lactone formation. On the other hand, electron-donating substituents deactivate gold–alkyne complex benefiting the intermolecular alkyne hydrofunctionalization.
Conclusions
In summary, we have proved that the water-soluble gold(I)–NHC complexes C1–5 are active in the hydrocarboxylation of alkyne-containing amino acid derivatives in a biphasic media toluene/water. The catalyst C1 that has the bulkiest ligand (1,3-bis-(2,6-diisopropylphenyl) imidazole-2-ylidene (IPr)) is the most active of them. C1 is able to catalyze cycloisomerization of terminal alkynes with loadings as low as 0.01 mol% (0.14 mmol, TOF = 4829 h−1). The carboxylic acid group of the substrate is responsible for the gold catalyst's activation at room temperature in this kind of cycloisomerization; a mechanism for this activation (dissociation Au–Cl bond) of the gold catalyst at room temperature has been postulated. As a result, the addition of silver salts is not necessary. On the other hand, C1 can be reuse at least 15 times without any loss of activity which convert it in the water-soluble gold(I)–NHC catalyst that can be recycled the largest number of times. In the case of less reactive internal alkynes, only 1 mol% of C1 was able to successfully cycloisomerize methyl and phenyl alkynes until completion without need of heating the reaction. Interestingly, the electronic effect of the substituent attached to the aromatic ring plays an important role in the reaction product. Electron-withdrawing groups favor the formation of enol lactones, while electron-donating substituents benefit the ketone formation. There is no other report of phenyl-substituted internal alkynes reacting with water-soluble gold catalysts at room temperature which certainly makes C1 the most active water-soluble gold catalyst.Experimental
General
Chemical reagents were purchased from commercial sources and were used without further purification unless noted otherwise. Solvents were of analytical grade or were purified by standard procedures prior to use. The chemical identity of the compounds was confirmed by recording 1H and 13C NMR spectra on a Bruker Avance III HD at 400 MHz spectrometer in CDCl3. Chemical shifts (δ) are reported in ppm using tetramethylsilane (δ = 0.00 ppm) as an internal standard. 13C NMR assignments were made on the basis of chemical shifts and proton multiplicities (from inverse HSQC spectra). The kinetic studies were performed is a Gas Chromatograph Shimadzu GC-2010 Plus with a FID detector. Synthesis reactions were monitored by analytical thin-layer chromatography (TLC) on 0.20 mm E. Merck silica gel 60 F254 pre-coated aluminum sheets with visualization of product bands by UV fluorescence at 254 nm and/or staining with aqueous p-anisaldehyde followed by heating. Flash column chromatography was performed using silica gel 60 (230–400 mesh) employing gradient of solvent (hexanes/EtOAc) polarity techniques, under positive pressure.Representative experimental procedure for the gold catalyzed cycloisomerization of alkyne-containing amino acids derivatives
To a mixture of 0.14 mmol of alkyne-containing amino acids derivative (1a–e) in 3 mL of toluene/distilled water (1:2) was added 0.0014 mmol of the water-soluble Au(I)–NHC complex (C1–5). The biphasic system was stirred at room temperature for 1 h or until disappearance of the starting material was observed by TLC. The organic phase was separated using a Pasteur pipette and freezing the aqueous phase to facilitate the phase separation. Toluene was concentrated under reduced pressure to give the corresponding enol-lactone in high purity. In the case of internal alkynes (1b–e), the aqueous layer was extracted with diethyl ether (2 × 10 mL). The combined organic phases were dried on Na2SO4, filtered and concentrated under reduced pressure. As described in the literature,10,11,26 due to the instability of these enol-lactones, when required the compounds were purified by passage through a short pad of silica (eluent: EtOAc).
For the reactions using 0.5, 0.25, 0.1 and 0.01 mol% of water-soluble Au(I)–NHC complex C1, a stock solution of 0.0014 mmol of catalyst in 1 mL of distilled water was prepared. Then, using a Hamilton syringe 500 μL, 250 μL, 100 μL and 10 μL were respectively taken from the stock solution and added to the biphasic system containing 0.14 mmol of N-Boc-propargylglycine (1a).
Hydration of the propargyl ester 3 at room temperature
To a mixture of 20.2 mg (0.119 mmol) of the propargyl ester 3 in 3 mL of toluene/distilled water (1:2) was added 1 mg (0.0012 mmol) of catalyst C1. The biphasic system was stirred at room temperature for 4 h. The toluene was separated using a Pasteur pipette and the aqueous phase was extracted with diethyl ether (2 × 10 mL). The combined organic phases were dried on Na2SO4, filtered and concentrated under reduced pressure to afford 19.8 mg (0.117 mmol) of unaltered propargyl ester 3 (98% recovery).
Hydration of the propargyl ester 3 at 100 °C
To a mixture of 20.2 mg (0.119 mmol) of the propargyl ester 3 in 3 mL of toluene/distilled water (1:2) was added 1 mg (0.0012 mmol) of catalyst C1. The biphasic system was stirred at 100 °C for 4 h. Then, the toluene was separated using a Pasteur pipette and the aqueous phase was extracted with diethyl ether (2 × 10 mL). The combined organic phases were dried on Na2SO4, filtered and concentrated under reduced pressure to afford 15.8 mg (0.093 mmol, 78%) of methyl ketone 4.
Reuse process of catalysts C1–5
The study of catalysts C1–5 reusability was conducted using the catalytic cycloisomerization of N-Boc-propargylglycine (1a) with 1 mol% of catalyst as model reaction.Representative procedure for catalyst C1
To 40 mg (0.186 mmol) of substrate (1a) in 3 mL of a biphasic solvent system made up of toluene/distilled water (1:2) was added 0.0018 mmol of complex C1. The biphasic system was stirred at room temperature for 1 h and the organic phase was separated using a Pasteur pipette and freezing the aqueous phase to facilitate the phase separation. Toluene was concentrated under reduced pressure to give the corresponding enol-lactone 2a which was analyzed by NMR to confirm complete transformation. To the aqueous phase containing hydrophilic catalyst, 1 mL of toluene and 40 mg (0.186 mmol) of N-Boc-propargylglycine (1a) was added. The mixture was stirred at room temperature for 1 h. This procedure was repeated number of times until the transformation was not efficient. In the case of C1 the procedure was repeated 15 times without a significant loss of yield (see ESI, Table S1, page S30†).
For the reusability study of C2, 31.9 mg (0.150 mmol) of substrate (1a) and 0.0019 mmol of C2 were employed. For C3, 50 mg (0.23 mmol) of 1a and 0.0023 mmol of C3 were employed. For C4, 30 mg (0.14 mmol) of 1a and 0.0014 mmol of C4 were employed. For C5, 30.1 mg (0.141 mmol) of 1a and 0.0018 mmol of C5 were used in the reusability assessment. This procedure was repeated until the transformation was not efficient, with C2 being recycled 6 times (see ESI, Table S2, page S39†), with C3 6 times (see ESI, Table S3, page S44†), C4 7 times (see ESI, Table S4, page S49†) and C5 9 times (see ESI, Table S5, page S54†).
Time-course of the conversion of 1a into 2a monitored by GC-FID
The gold-catalyzed transformation of N-Boc-propargylglycine (1a) into the enol-lactone 2a was monitored over time using GC-FID. The reaction was performed using a 29.9 mg (0.140 mmol) of N-Boc-propargylglycine (1a) and 1.6 mg (0.00194 mmol) of catalyst C1 in 4 mL of toluene/distilled water (2:2). The biphasic system was stirred at room temperature for 1 h. 2,3-Dimethoxytoluene (10 μL, 0.0657 mmol) was employed as internal standard. Throughout the experiment, 10 μL samples of the organic phase were collected at one-minute intervals during the initial 5 minutes, and subsequently at 10, 20, 30, 45, and 60 minutes. These samples were put in a vial and 990 μL of toluene was added to reach a final volume of 1 mL. The experiments were run by triplicate (see ESI, Table S6, page S60†).
Abbreviations
| IPr | 1,3-bis(2,6-diisopropylphenyl) imidazole-2-ylidene |
| IMes | 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene |
| SIMes | 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
LPH performed the screenings, the recycling experiments, the kinetic experiments and synthesized compounds. TAD performed the screenings and synthesized compounds. GAF synthesized the catalysts. SAT and GFS designed the experiments and wrote the manuscript, with contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We would like to thank Fernanda Villalonga, Sergio Tindiglia and Pablo Duche from IQUIR for the 400 Mhz NMR analyses, and Leticia I. Llarrull for helpful discussion about kinetics. The authors would also like to thank ANPCyT (PICT 2019-2522 and PICT 2018-03362), ASaCTei (PEICID-2021-078) and the Universidad Nacional del Sur (PGI 24/Q108), Bahía Blanca, Argentina. L. O. P. and T. A. D. gratefully acknowledge CONICET for their doctoral fellowship.Notes and references
- C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68–82 RSC.
- H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC.
- J. C. Flores, G. F. Silbestri and E. de Jesús, in Adv. Organomet. Chem., ed. P. J. Pérez, Academic Press, 2022, vol. 77, pp. 169–242 Search PubMed.
- L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407–5413 CrossRef CAS.
- G. A. Fernández, A. n. S. Picco, M. R. Ceolín, A. B. Chopa and G. F. Silbestri, Organometallics, 2013, 32, 6315–6323 CrossRef.
- G. A. Fernández, A. B. Chopa and G. F. Silbestri, Catal. Sci. Technol., 2016, 6, 1921–1929 RSC.
- G. A. Fernández, M. A. Schiel and G. F. Silbestri, J. Organomet. Chem., 2020, 923, 121452 CrossRef.
- A. Almássy, C. E. Nagy, A. C. Bényei and F. Joó, Organometallics, 2010, 29, 2484–2490 CrossRef.
- C. E. Czégéni, G. Papp, Á. Kathó and F. Joó, J. Mol. Catal. A: Chem., 2011, 340, 1–8 CrossRef.
- E. Tomas-Mendivil, P. Y. Toullec, J. Diez, S. Conejero, V. Michelet and V. Cadierno, Org. Lett., 2012, 14, 2520–2523 CrossRef CAS PubMed.
- E. Tomas-Mendivil, P. Y. Toullec, J. Borge, S. Conejero, V. Michelet and V. Cadierno, ACS Catal., 2013, 3, 3086–3098 CrossRef CAS.
- P. Crochet and V. Cadierno, Catalysts, 2023, 13, 436 CrossRef CAS.
- S. P. Nolan, Nature, 2007, 445, 496–497 CrossRef CAS PubMed.
- A. S. K. Hashmi, Gold Bull., 2003, 36, 3–9 CrossRef CAS.
- C. Praveen, Chem. Rec., 2021, 21, 1697–1737 CrossRef CAS PubMed.
- H. C. Shen, Tetrahedron, 2008, 64, 3885–3903 CrossRef CAS.
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232–5241 CrossRef CAS PubMed.
- A. La-Venia, N. S. Medran, V. Krchňák and S. A. Testero, ACS Comb. Sci., 2016, 18, 482–489 CrossRef CAS PubMed.
- A. Collado, D. J. Nelson and S. P. Nolan, Chem. Rev., 2021, 121, 8559–8612 CrossRef CAS PubMed.
- A. B. Gade, Urvashi and N. T. Patil, Org. Chem. Front., 2024, 11, 1858–1895 RSC.
- Y.-L. Du, Y. Hu, Y.-F. Zhu, X.-F. Tu, Z.-Y. Han and L.-Z. Gong, J. Org. Chem., 2015, 80, 4754–4759 CrossRef CAS PubMed.
- F. Zhao, N. Li, Y.-F. Zhu and Z.-Y. Han, Org. Lett., 2016, 18, 1506–1509 CrossRef CAS PubMed.
- X. Yang, Y. Shimizu, J. R. Steiner and J. Clardy, Tetrahedron Lett., 1993, 34, 761–764 CrossRef CAS.
- D. Kuhnt, T. Anke, H. Besl, M. Bross, R. Herrmann, U. Mocek, B. Steffan and W. Steglich, J. Antibiot., 1990, 43, 1413–1420 CrossRef CAS PubMed.
- X.-p. Fang, J. E. Anderson, C.-j. Chang and J. L. McLaughlin, Tetrahedron, 1991, 47, 9751–9758 CrossRef CAS.
- E. Genin, P. Y. Toullec, S. Antoniotti, C. Brancour, J. P. Genet and V. Michelet, J. Am. Chem. Soc., 2006, 128, 3112–3113 CrossRef CAS PubMed.
- H. Harkat, J. M. Weibel and P. Pale, Tetrahedron Lett., 2006, 47, 6273–6276 CrossRef CAS.
- N. S. Medran, M. Villalba, E. G. Mata and S. A. Testero, Eur. J. Org Chem., 2016, 2016, 3757–3764 CrossRef CAS.
- R. A. Amos and J. A. Katzenellenbogen, J. Org. Chem., 1978, 43, 560–564 CrossRef CAS.
- M. Yamamoto, J. Chem. Soc., Chem. Commun., 1978, 649–650 RSC.
- M. M. Rammah, M. Othman, K. Ciamala, C. Strohmann and M. B. Rammah, Tetrahedron, 2008, 64, 3505–3516 CrossRef CAS.
- T. Yoshikawa and M. Shindo, Org. Lett., 2009, 11, 5378–5381 CrossRef CAS PubMed.
- T. L. Mindt and R. Schibli, J. Org. Chem., 2007, 72, 10247–10250 CrossRef CAS PubMed.
- C. H. Sun, Y. W. Fang, S. Li, Y. Zhang, Q. W. Zhao, S. N. Zhu and C. Z. Li, Org. Lett., 2009, 11, 4084–4087 CrossRef CAS PubMed.
- D. Bouyssi, J. Gore and G. Balme, Tetrahedron Lett., 1992, 33, 2811–2814 CrossRef CAS.
- R. Rossi, F. Bellina and L. Mannina, Tetrahedron Lett., 1998, 39, 3017–3020 CrossRef CAS.
- A. Nagendiran, O. Verho, C. Haller, E. V. Johnston and J.-E. Bäckvall, J. Org. Chem., 2014, 79, 1399–1405 CrossRef CAS PubMed.
- J. García-Álvarez, J. Díez and C. Vidal, Green Chem., 2012, 14, 3190–3196 RSC.
- K. Ogata, D. Sasano, T. Yokoi, K. Isozaki, H. Seike, H. Takaya and M. Nakamura, Chem. Lett., 2012, 41, 498–500 CrossRef CAS.
- G. Fernández, L. Bernardo, A. Villanueva and R. Pleixats, New J. Chem., 2020, 44, 6130–6141 RSC.
- K. Eriksson, O. Verho, L. Nyholm, S. Oscarsson and J. E. Backvall, Eur. J. Org Chem., 2015, 2250–2255 CrossRef CAS.
- M. Ferré, X. Cattoën, M. Wong Chi Man and R. Pleixats, ChemCatChem, 2016, 8, 2824–2831 CrossRef.
- M. J. Rodriguez-Alvarez, C. Vidal, J. Diez and J. Garcia-Alvarez, Chem. Commun., 2014, 50, 12927–12929 RSC.
- D. Gasperini, L. Maggi, S. Dupuy, R. M. P. Veenboer, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Adv. Synth. Catal., 2016, 358, 3857–3862 CrossRef CAS.
- R. J. Brea, M. P. Lopez-Deber, L. Castedo and J. R. Granja, J. Org. Chem., 2006, 71, 7870–7873 CrossRef CAS PubMed.
- The enol-lactones were obtained in high purity, and due to their instability, they were purified by passing through a short silica pad (eluent: EtOAc) when required..
- E. Marchal, P. Uriac, B. Legouin, L. Toupet and P. van de Weghe, Tetrahedron, 2007, 63, 9979–9990 CrossRef CAS.
- B. Ranieri, I. Escofet and A. M. Echavarren, Org. Biomol. Chem., 2015, 13, 7103–7118 RSC.
- F. Li, N. Wang, L. Lu and G. Zhu, J. Org. Chem., 2015, 80, 3538–3546 CrossRef CAS PubMed.
- Z. Lu, T. Li, S. R. Mudshinge, B. Xu and G. B. Hammond, Chem. Rev., 2021, 121, 8452–8477 CrossRef CAS PubMed.
- O. Seppänen, S. Aikonen, M. Muuronen, C. Alamillo-Ferrer, J. Burés and J. Helaja, Chem. Commun., 2020, 56, 14697–14700 RSC.
- N. Marion, R. S. Ramón and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448–449 CrossRef CAS PubMed.
- A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, analytical data of individual compounds, 1H, 13C NMR spectra of the synthesized compounds, kinetic study data and reuse of catalyst information. See DOI: https://doi.org/10.1039/d4ra04876h |
| This journal is © The Royal Society of Chemistry 2024 |