
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, biological evaluation, and docking studies of novel triazolo[4,3-b]pyridazine derivatives as dual c-Met/Pim-1 potential inhibitors with antitumor activity†
Mohamed E. Mahmouda,
Eman M. Ahmedb,
Hamdy M. Ragabb,
Rania Farag A. Eltelbanyc and
Rasha A. Hassan *b
*b
aDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Modern University for Technology and Information (MTI), Cairo, Egypt
bDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Cairo University, Cairo, Egypt. E-mail: rasha.hasan@pharma.cu.edu.eg
cDepartment of Biochemistry, Faculty of Pharmacy, Modern University for Technology and Information (MTI), Cairo, Egypt
First published on 24th September 2024
Abstract
Interest has been piqued in c-Met and Pim-1, potential new cancer treatment targets. A variety of triazolo[4,3-b]pyridazine derivatives were synthesized to create powerful dual c-Met/Pim-1 inhibitors having the pharmacophoric elements of both enzyme inhibitors. All derivatives were screened for their cytotoxic effects on 60 cancer cell lines. Compounds 4g and 4a, had strong antiproliferative cytotoxic impacts on tumor cells, with mean GI% values of 55.84 and 29.08%, respectively. Research revealed that 4g has more powerful inhibitory activity against c-Met and Pim-1, with IC50 of 0.163 ± 0.01 and 0.283 ± 0.01 μM, respectively than the reference and derivative 4a. Moreover, compound 4g was the subject of an additional investigation into biological processes. The findings showed that compound 4g caused MCF-7 cells to arrest in the S stage of the cell cycle. Also, it accelerated the progress of apoptosis 29.61-fold more than the control. Compound 4g demonstrated a significantly higher level of caspase-9 and a decreased level of p-PI3K, p-AKT, and p-mTOR compared to staurosporine. Later, analysis of 4g showed good drug-ability and pharmacokinetic properties. A similar mode of interaction at the ATP-binding site of c-Met and Pim-1 compared to the docked ligands was suggested by additional docking studies of compound 4g.
1. Introduction
Breast cancer is the most prevalent cancer in women diagnosed globally.1 Most of them can be identified early enough to be successfully treated; nevertheless, many patients may not benefit from the medication, and metastasis ultimately contributes to death.2 The life expectancy of patients with breast cancer has not been substantially raised by the now-available therapeutics, which include surgery, radiotherapy, and chemotherapy, and they frequently have detrimental side effects. Most pharmaceutical cancer therapies are cytotoxic. Additionally, multiple complications, such as neuropathy, axillary vein thrombosis, and cardiovascular disease, may develop after radiation or surgical treatments for breast cancer.3Hepatocyte growth factor (HGF) binding stimulates the tyrosine kinase receptor c-Met kinase, which leads to receptor dimerization and subsequent signaling.4 This pathway is crucial for healthy adult wound healing and normal embryonic development in normal cells.5 Based on reports, the overexpression of the c-Met kinase receptor deregulates the c-Met kinase pathway in several human cancers. Poor prognosis, tumor proliferation, angiogenesis, and metastasis are all attributed to this deregulation.6 The analogous proto-oncogenic serine/threonine protein kinases known as Pim kinases 1, 2, and 3 are positive controllers of cell cycle progression and constitutively active enzymes.7 As oncogenic survival factors, they can inhibit apoptosis. They are crucial in controlling cell proliferation, differentiation, migration, and survival.8 Pim kinases are abnormally overexpressed in a wide range of solid (including prostate and breast tumors) and hematologic (including multiple myeloma) tumors.9 Ultimately, this leads to cancer progression, metastasis, drug resistance, and, frequently, a poor prognosis. Notably, drug efflux transporter expression, activation, and stabilization have all been linked to Pim kinases, which contribute to multidrug resistance. Moreover, Pim-1 phosphorylates the breast cancer-resistant protein, which promotes its multimerization and stable membrane expression.10 Furthermore, due to the duplicated functions of the three Pim isoforms, plenty of interest is shown in developing pan-Pim inhibitors.11
Cellular signaling pathways are closely correlated with one another to enable multicellular functions like cell division, cell fate determination, and cell migration. Pathologies, including cancer, may result from erroneous signal transfer within the cell, and this dysregulation results in the incorrect signaling of many other protein degradation pathways, such as PI3K/AKT/mTOR (phosphoinositide 3-kinase/AKT/mechanistic target of rapamycin) signaling.12
One of the most frequent genomic mutations in breast cancer across different subtypes is imperfections in the PI3K/AKT/mTOR pathway, which ultimately fosters tumor-cell growth.13 The PI3K/AKT/mTOR signaling pathway, which is frequently dysregulated in cancer, is essential for regulating cell metabolism, proliferation, and survival.13,14 PI3Ks are available in 4 isoforms (α, β, δ, and γ) and are heterodimers made up of regulatory (p85) and catalytic (p110) subunits.13 The agonist effect of receptor tyrosine kinases initiates PI3K activation, which in turn causes phosphorylation of AKT and mTOR complex 1 (mTORC1) to trigger the process. mTOR is an abnormal serine/threonine protein kinase made up of mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2).15
Although several genes participate in apoptosis, caspases are the principal mediators of the process. The carboxyl side of the aspartate residue is where caspases, aspartate-specific cysteine proteases, fragment their substrates. At present, it is known that there are at least 14 different types of caspases, of which 2/3 are involved in apoptosis. The two primary groups of caspases involved in apoptosis are the upstream initiator caspases (such as caspases 2, 3, and 6) and the downstream effector caspases (such as caspases 8, 9, and 10). The representatives of the latter class are responsible for the morphological changes that occur during apoptosis and destroy numerous cellular proteins.3
1,2,4-Triazolo[4,3-b]pyridazine derivatives exhibit substantial therapeutic properties like anxiolytic,16,17 anticonvulsant,18 antimicrobial,19,20 and antiviral properties.21,22 Additionally, they are used as various enzyme inhibitors, such as leucine-rich repeat kinase 2 (LRRK2),23,24 phosphodiesterase (PDE4),25 and TRAF6 E3 ligase in autoimmune diseases.26 During the past twenty years, more attention has focused on the use of triazolopyridazine to design ligands that could be potential anticancer drugs by targeting enzymatic pathways involved in the different pathogenesis stages adopted in the progression of cancer, including metastasis, angiogenesis, and others. The c-Met and Pim-1 enzymes are the most promising novel targeted enzymes for designing new antitumor compounds. The therapeutic potential for c-Met and Pim-1 inhibitors is significant due to their critical roles in cancer progression and therapy resistance. Dual inhibition of c-Met and Pim-1 produces a synergistic effect, where a dual inhibitor can target multiple pathways simultaneously. This can result in greater therapeutic efficacy than targeting either kinase alone. Also, a dual c-Met/Pim-1 inhibitor can minimize the chances of resistance development and offer a promising strategy to enhance anticancer efficacy.27
A promising c-Met and Pim-1 inhibitory activity was reported for the previously synthesized compounds I–III (ref. 28) and IV–VI (ref. 29) (Fig. 1).
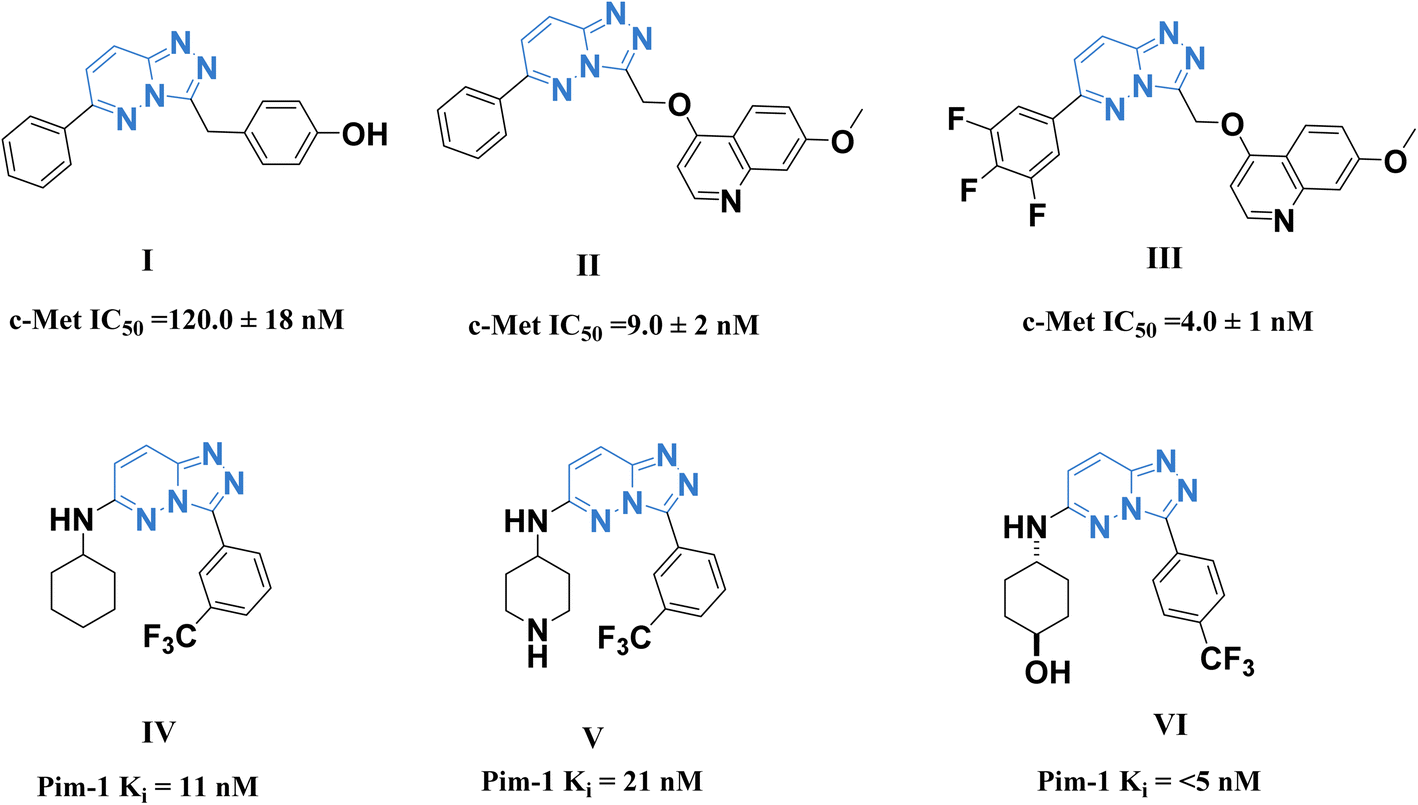 | ||
| Fig. 1 Some reported 1,2,4-triazolo[4,3-b]pyridazine derivatives as c-Met (I–III) and Pim-1 (IV–VI) inhibitors. | ||
Following the above data, it was interesting to continue our research to create potent new anticancer drugs with high therapeutic equivalence.30–37 The main goal of the current study was to generate new 1,2,4-triazolo[4,3-b]pyridazine derivatives that are dual inhibitors of the c-Met and Pim-1 kinases with anticancer activity. The NCI (USA) 60-panel cell line was used as a model to assess the anticancer activities of the developed derivatives, and in vitro inhibitory studies using c-Met and Pim-1 enzymes were conducted on the most promising derivatives. We investigated the total concentration and phosphorylated concentration of the PI3K-Akt-mTOR pathway, assessed through the derivatives produced, to delve deeper into the mechanistic pathways of antiproliferative activity. In addition, the effects of these substances, including their impact on the typical cell cycle profile, caspase-9 activity, and apoptosis induction, have been studied in the breast cancer cell line MCF7. In a concluding study, molecular docking confirmed these molecules' binding modes and action targets.
The primary objective of the molecular design was to customize the lead compounds I and IV to generate new antiproliferative drugs that target c-Met and Pim-1 and have greater dual inhibitory activity. The triazolopyridazine core, which formed crucial interactions with important amino acids in the c-Met and Pim-1 active pockets, was preserved as part of the modification strategy (Fig. 2). The replacement of the 3-hydroxybenzyl group in compound I and the 3-trifluoromethylphenyl group in compound IV was accomplished using a phenyl group (4a–c, 5, and 6a) or divergent para-substituted phenyl groups, ranging from hydrophilic groups like OCH3 (4e–g and 6b) to hydrophobic substituents like CH3 in compound 4d. On the other hand, the phenyl ring at C6 in compound I and the cyclohexyl ring in compound IV were replaced by different para-substituted phenyl groups in derivatives 4, the dimethyl pyrazole group in derivative 5, or the acetyl group in derivatives 6. Finally, the amino linker in compound IV was replaced by an amino azomethine group in derivatives 4, a hydrazine group in compounds 6a,b, or removed as in compound 5. Such replacements were adopted so that the triazolopyridazine core was retained in the proper position inside the active pockets of the two enzymes. The enzymatic interactions were enhanced by the different groups located in position 6, either the donating groups (OH and N(CH3)2) or the withdrawing groups (NO2). Finally, hydrogen bond donors and acceptors were incorporated through the OH and NH in various positions or the carbonyl group in acetohydrazide derivatives 6a,b.
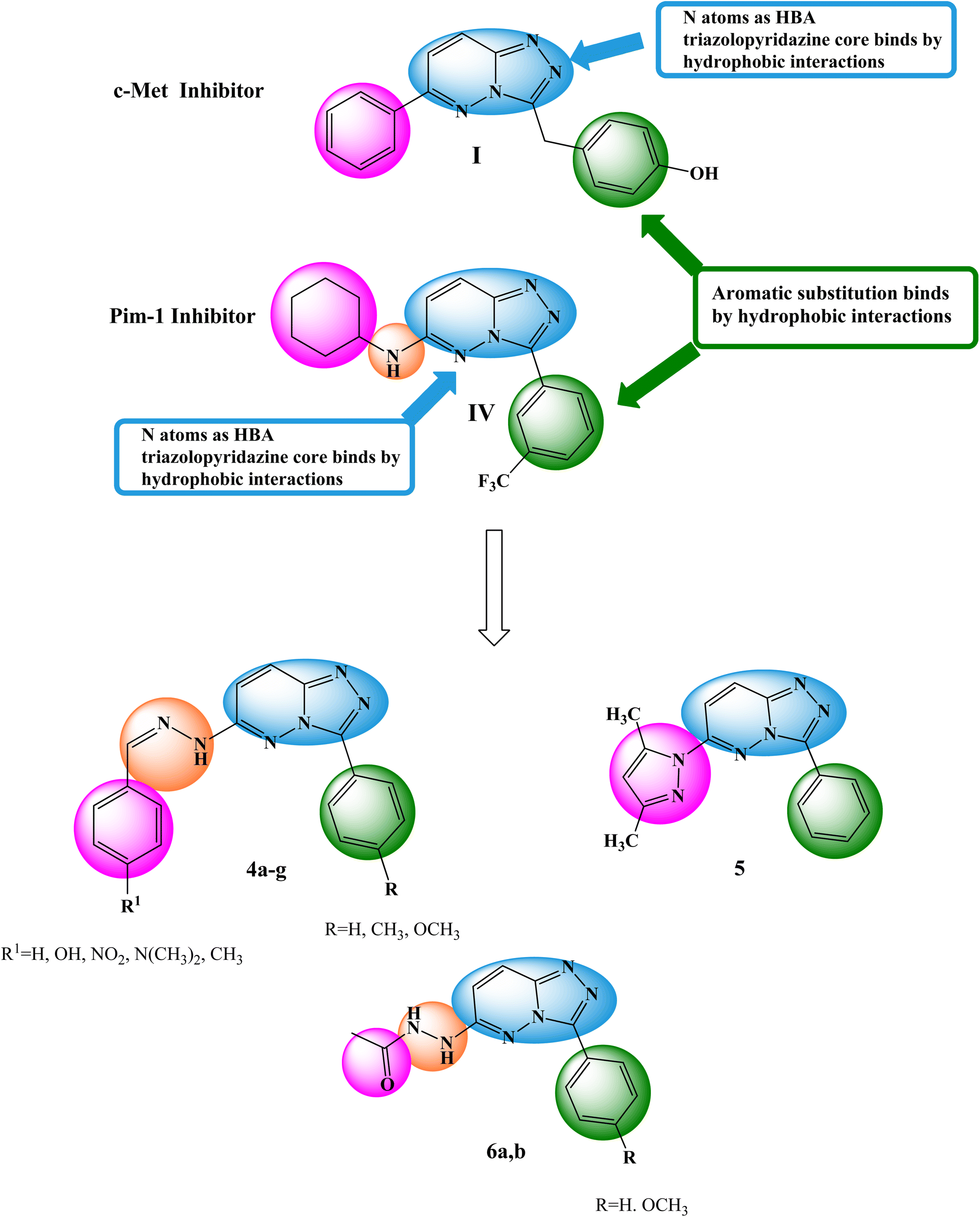 | ||
| Fig. 2 Basic structural elements of compounds I and IV and modifications to the newly synthesized derivatives. | ||
2. Experimental
2.1 Chemistry
2.1.1.1 General procedure for the preparation of 6-arylidenehydrazino-3-aryl-[1,2,4]triazolo[4,3-b]pyridazine (4a–g). 6-Hydrazineyl-3-(4-aryl)-[1,2,4]triazolo[4,3-b]pyridazine derivatives 3a–c (0.68 g (3a), 0.72 g (3b), or 0.76 g (3c), 0.003 mol) was heated in 15 mL of absolute ethanol for 10 min. Following complete dissolution, the appropriate aromatic aldehyde such as benzaldehyde, 4-hydroxybenzaldehyde, 4-nitrobenzaldehyde, 4-methylbenzaldehyde or N,N-dimethylbenzaldehyde (0.003 mol) was added and the reaction mixture was refluxed at 80 °C for 4 h. The reaction was cooled, the crude product was collected through filtration and purified via recrystallization from hot ethanol.
2.1.1.2 3-((2-(3-Phenyl-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)hydrazono)methyl)phenol (4a). Dark yellow solid: 72% yield; m.p. 229–230 °C; IR (KBr, cm−1) 3425 (OH), 3298 (NH), 3051 (CH aromatic), 1631 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1604 (CC); 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H, NH, D2O exchangeable), 9.97 (s, 1H, OH, D2O exchangeable), 8.47 (s, 2H, Ar–H), 8.09 (s, 2H, Ar–H), 7.56–7.49 (m, 6H, Ar–H), 6.86 (s, 2H, Ar–H); 13C NMR (100 MHz, DMSO-d6) δ 159.4, 153.8, 143.6, 130.6, 130.2, 129.1 (2C), 128.7 (2C), 127.8, 127.5 (2C), 126.0, 125.3, 116.2 (2C), 114.2, 109.8. Anal. calcd for C18H14N6O (330.35): C, 65.44; H, 4.27; N, 25.44; found, C, 65.31; H, 4.52; N, 25.70%.
N), 1604 (CC); 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H, NH, D2O exchangeable), 9.97 (s, 1H, OH, D2O exchangeable), 8.47 (s, 2H, Ar–H), 8.09 (s, 2H, Ar–H), 7.56–7.49 (m, 6H, Ar–H), 6.86 (s, 2H, Ar–H); 13C NMR (100 MHz, DMSO-d6) δ 159.4, 153.8, 143.6, 130.6, 130.2, 129.1 (2C), 128.7 (2C), 127.8, 127.5 (2C), 126.0, 125.3, 116.2 (2C), 114.2, 109.8. Anal. calcd for C18H14N6O (330.35): C, 65.44; H, 4.27; N, 25.44; found, C, 65.31; H, 4.52; N, 25.70%.
2.1.1.3 6-(2-(4-Nitrobenzylidene)hydrazinyl)-3-phenyl-[1,2,4]triazolo[4,3-b]pyridazine (4b). Canary yellow solid: 70%; m.p. 226–227 °C; IR (KBr, cm−1) 3390 (NH), 3051 (CH aromatic), 1631 (C
N), 1554 (CC), 1530, 1338 (NO2); 1H NMR (400 MHz, DMSO-d6) δ 12.15 (s, 1H, NH, D2O exchangeable), 8.45 (d, J = 7.6 Hz, 2H, Ar–H), 8.31 (d, J = 8.0 Hz, 1H, Ar–H), 8.24 (s, 1H, CH = N), 8.22 (d, J = 6.4 Hz, 2H, Ar–H), 7.95 (d, J = 8.0 Hz, 2H, Ar–H), 7.67 (d, J = 8.0 Hz, 1H, Ar–H), 7.63–7.59 (m, 2H, Ar–H), 7.56 (d, J = 7.6 Hz, 1H, Ar–H); 13C NMR (100 MHz, DMSO-d6) δ 160.7, 153.8, 147.6, 146.4, 141.4, 130.3, 130.0, 129.1 (2C), 127.6 (2C), 127.4 (2C), 126.9, 126.0, 124.4 (2C), 113.9. Anal. calcd for C18H13N7O2 (359.35): C, 60.16; H, 3.65; N, 27.29; found, C, 60.37; H, 3.84; N, 27.11%.
2.1.1.4 N,N-Dimethyl-4-((2-(3-phenyl-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)hydrazono)methyl)aniline (4c). Reddish brown solid: 72%; m.p. 271–272 °C; IR (KBr, cm−1) 3425 (NH), 3061 (CH aromatic), 2966, 2893 (CH aliphatic), 1612 (C
N), 1566 (CC); 1H NMR (400 MHz, DMSO-d6) δ 11.36 (s, 1H, NH, D2O exchangeable), 8.50 (d, J = 6.8 Hz, 2H, Ar–H), 8.20 (d, J = 9.2 Hz, 1H, Ar–H), 8.05 (s, 1H, CH = N), 7.61–7.55 (m, 6H, Ar–H), 6.76 (d, J = 8.0 Hz, 2H, Ar–H), 2.98 (s, 6H, N(CH3)2); 13C NMR (100 MHz, DMSO-d6) δ 151.6, 146.4, 144.1, 130.1, 129.1 (2C), 128.3 (2C), 127.3 (2C), 125.6, 122.5, 121.6, 114.1, 113.0, 112.4 (2C), 106.1, 40.4 (2C). Anal. calcd for C20H19N7 (357.42): C, 67.21; H, 5.36; N, 27.43; found, C, 67.43; H, 5.50; N, 27.69%.
2.1.1.5 6-(2-Benzylidenehydrazinyl)-3-(p-tolyl)-[1,2,4]triazolo[4,3-b]pyridazine (4d). Yellow solid: 74% yield; m.p. 269–270 °C; IR (KBr, cm−1) 3429 (NH), 3151, 3028 (CH aromatic), 2920, 2893 (CH aliphatic), 1631 (C
N), 1562 (CC); 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H, NH, D2O exchangeable), 8.39 (d, J = 6.8 Hz, 2H, Ar–H), 8.25 (d, J = 9.5 Hz, 1H, Ar–H), 8.17 (s, 1H, CH = N), 7.76 (d, J = 6.4 Hz, 2H, Ar–H), 7.60 (d, J = 8.0 Hz, 1H, Ar–H), 7.47–7.41 (m, 5H, Ar–H), 2.42 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 153.9, 146.5, 144.5, 142.9, 139.8, 135.1, 129.8, 129.7 (2C), 129.3 (2C), 127.2 (2C), 127.0 (2C), 126.0, 124.5, 113.6, 21.5. Anal. calcd for C19H16N6 (328.38): C, 69.50; H, 4.91; N, 25.59; found, C, 69.67; H, 5.05; N, 25.78%.
2.1.1.6 6-(2-Benzylidenehydrazinyl)-3-(4-methoxyphenyl)-[1,2,4]triazolo[4,3-b]pyridazine (4e). Off-white solid: 75% yield; m.p. 267–268 °C; IR (KBr, cm−1) 3425 (NH), 3151, 3039 (CH aromatic), 2997, 2920 (CH aliphatic), 1631 (C
N), 1556 (CC); 1H NMR (400 MHz, DMSO-d6) δ 11.67 (s, 1H, NH, D2O exchangeable), 8.42 (d, J = 8.4 Hz, 2H, Ar–H), 8.22 (d, J = 9.8 Hz, 1H, Ar–H), 8.16 (s, 1H, CH = N), 7.75 (d, J = 6.8 Hz, 2H, Ar–H), 7.58 (d, J = 9.8 Hz, 1H, Ar–H), 7.47–7.39 (m, 3H, Ar–H), 7.14 (d, J = 8.8 Hz, 2H, Ar–H), 3.86 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ 160.7, 153.9, 146.4, 144.4, 142.8, 135.1, 129.8, 129.3 (2C), 128.9 (2C), 127.0 (2C), 125.9, 119.7, 114.5 (2C), 113.4, 55.8. Anal. calcd for C19H16N6O (344.38): C, 66.27; H, 4.68; N, 24.40; found, C, 66.09; H, 4.79; N, 24.63%.
2.1.1.7 3-(4-Methoxyphenyl)-6-(2-(4-methylbenzylidene)hydrazinyl)-[1,2,4]triazolo[4,3-b]pyridazine (4f). Dark brown solid: 72% yield; m.p. 230–231 °C; IR (KBr, cm−1) 3425 (NH), 3151, 3036 (CH aromatic), 2916, 2893 (CH aliphatic), 1616 (C
N), 1562 (CC); 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H, NH, D2O exchangeable), 8.43 (d, J = 4.8 Hz, 2H, Ar–H), 8.20 (d, J = 6.4 Hz, 1H, Ar–H), 8.13 (s, 1H, CH = N), 7.63–7.53 (m, 3H, Ar–H), 7.26 (d, J = 4.8 Hz, 2H, Ar–H), 7.15 (d, J = 5.6 Hz, 2H, Ar–H), 3.87 (s, 3H, OCH3), 2.34 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 160.7, 153.9, 152.4, 146.5, 142.9, 139.5, 132.4 (2C), 129.9 (2C), 128.9 (2C), 126.9, 125.9, 119.7, 114.5 (2C), 113.5, 55.8, 21.5. Anal. calcd for C20H18N6O (358.41): C, 67.02; H, 5.06; N, 23.45; found, C, 67.23; H, 5.29; N, 23.73%.
2.1.1.8 4-((2-(3-(4-Methoxyphenyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)hydrazono)methyl)phenol (4g). Yellow solid: 70% yield; m.p. 224–225 °C; IR (KBr, cm−1) 3491 (OH), 3332 (NH), 3055 (CH aromatic), 2966, 2931 (CH aliphatic), 1631 (C
N), 1562 (CC); 1H NMR (400 MHz, DMSO-d6) δ 11.42 (s, 1H, NH, D2O exchangeable), 9.96 (s, 1H, OH, D2O exchangeable), 8.42 (d, J = 8.8 Hz, 2H, Ar–H), 8.17 (d, J = 9.6 Hz, 1H, Ar–H), 8.07 (s, 1H, CH = N), 7.58 (d, J = 8.4 Hz, 2H, Ar–H), 7.52 (d, J = 9.6 Hz, 1H, Ar–H), 7.14 (d, J = 8.4 Hz, 2H, Ar–H), 6.85 (d, J = 8.4 Hz, 2H, Ar–H), 3.86 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.3, 153.9, 146.4, 144.3, 143.5, 129.0 (2C), 128.7 (2C), 126.1, 125.6, 119.6, 116.2 (2C), 114.5 (2C), 113.8, 55.8. Anal. calcd for C19H16N6O2 (360.38): C, 63.32; H, 4.48; N, 23.32; found, C, 63.57; H, 4.41; N, 23.60%.
2.1.1.9 Preparation of 6-(3,5-dimethyl-1H-pyrazol-1-yl)-3-phenyl-[1,2,4]triazolo[4,3-b]pyridazine (5). Acetyl acetone (0.7 g, 0.007 mol) was added to a solution of 6-hydrazineyl-3-(4-aryl)-[1,2,4]triazolo[4,3-b]pyridazine derivatives 3a–c (0.007 mol) in absolute ethanol (10 mL), and the reaction mixture was refluxed at 80 °C for 6 h. The mixture was allowed to cool at room temperature, and the product was filtered, dried, and recrystallized from ethanol to give a buff solid: 80% yield; m.p. 139–140 °C; IR (KBr, cm−1) 3047 (CH aromatic), 2962, 2927 (CH aliphatic), 1624 (C
N), 1543 (CC); 1H NMR (400 MHz, DMSO-d6) δ 8.48 (d, J = 9.2 Hz, 1H, Ar–H), 8.29 (s, 2H, Ar–H), 7.96 (d, J = 8.8 Hz, 1H, Ar–H), 7.62 (d, J = 3.6 Hz, 3H, Ar–H), 6.26 (s, 1H, CH pyrazole), 2.63 (s, 3H, CH3), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 151.5, 150.4, 147.8, 144.3, 142.1, 130.7, 129.2 (2C), 127.8 (2C), 127.1, 126.3, 117.3, 111.3, 14.8, 13.8. Anal. calcd for C16H14N6 (290.33): C, 66.19; H, 4.86; N, 28.95; found, C, 65.98; H, 5.02; N, 29.17%.
2.1.1.10 General procedure for the preparation of [1,2,4]triazolo[4,3-b]pyridazine acetohydrazide (6a,b). Acetic anhydride (0.714 g, 0.007 mol) was added to a solution of 6-hydrazineyl-3-(4-aryl)-[1,2,4]triazolo[4,3-b]pyridazine derivatives 3a–c (0.007 mol) in glacial acetic acid (10 mL), and the reaction mixture was heated under reflux at 120 °C for 6 h. The mixture was poured into 20 mL of ice-cold water. After filtration, drying, and recrystallization from ethanol, the final crude product was produced.
2.1.1.11 N′-(3-Phenyl-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)acetohydrazide (6a). Buff solid: 75% yield, m.p. 159–160 °C; IR (KBr, cm−1) 3383, 3232 (2 NH), 3097 (CH aromatic), 2993 (CH aliphatic), 1685 (C
O), 1631 (CN), 1589 (CC); 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H, NH, D2O exchangeable), 8.44 (d, J = 7.6 Hz, 3H, Ar–H), 8.11 (d, J = 9.8 Hz, 1H, Ar–H), 7.59–7.53 (m, 3H, Ar–H + NH, D2O exchangeable), 6.97 (d, J = 9.8 Hz, 1H, Ar–H), 2.01 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 174.3, 169.4, 154.8, 146.3, 144.4, 130.1, 129.1 (2C), 127.2 (2C), 125.2, 115.0, 21.0. Anal. calcd for C13H12N6O (268.28): C, 58.20; H, 4.51; N, 31.33; found, C, 58.47; H, 4.63; N, 31.50%.
2.1.1.12 N′-(3-(4-methoxyphenyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)acetohydrazide (6b). Brown solid: 75% yield m.p. 164–165 °C; IR (KBr, cm−1) 3483, 3240 (2 NH), 3008 (CH aromatic), 2947, 2908 (CH aliphatic), 1685 (C
O), 1620 (CN), 1577 (CC); 1H NMR (400 MHz, DMSO-d6) δ 9.83 (s, 1H, NH, D2O exchangeable), 8.39 (d, J = 8.4 Hz, 3H, Ar–H + NH, D2O exchangeable), 8.10 (d, J = 9.8 Hz, 1H, Ar–H), 7.12 (d, J = 8.4 Hz, 2H, Ar–H), 6.96 (d, J = 9.8 Hz, 1H, Ar–H), 3.85 (s, 3H, OCH3), 2.02 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 174.1, 169.5, 160.7, 154.7, 146.3, 144.1, 128.8 (2C), 125.3, 119.7, 114.5 (2C), 55.8, 21.1. Anal. calcd for C14H14N6O2 (298.31): C, 56.37; H, 4.73; N, 28.17; found, C, 56.70; H, 4.95; N, 28.41%.
2.2 Biological testing
The biological assays were completed using the previously disclosed methods, and they are included in the ESI;† antiproliferative activity screening by NCI,41–45 MTT assay,46 PI3K-Akt-mTOR pathway evaluation,31 caspase-9 activity assay,47 cell cycle analysis,48 and apoptosis assay.492.3 In silico studies
3. Results and discussion
3.1 Chemistry
The new series of triazolo[4,3-b]pyridazine compounds 4a–g, 5, and 6a,b is depicted in Scheme 1. First, 3-(2-arylidenehydrazinyl)-6-chloropyridazine derivatives 1a–c were generated by refluxing 3-chloro-6-hydrazinopyridazine with the relevant aromatic aldehydes in absolute ethanol and glacial acetic acid.38 Moreover, 6-chloro-3-arylidene-[1,2,4]triazolo[4,3-b]pyridazine derivatives 2a–c were obtained by heating compounds 1a–c under reflux at 80 °C in a mixture of ferric chloride and ethanol.39 Compounds 2a–c were heated under reflux at 80 °C with hydrazine hydrate in absolute ethanol to yield 6-hydrazinyl-3-arylidene-[1,2,4]triazolo[4,3-b]pyridazine derivatives 3a–c.40 New derivatives of 6-(2-arylidenehydrazinyl)-3-aryl-[1,2,4]triazolo[4,3-b]pyridazine (4a–g) were delivered by heating compounds 3a–c under reflux at 80 °C with the appropriate aromatic aldehydes in absolute ethanol. Besides, 6-(3,5-dimethyl-1H-pyrazol-1-yl)-3-phenyl-[1,2,4]triazolo[4,3-b]pyridazine (5) was created by reacting compounds 3a with acetylacetone in absolute ethanol.50 Finally, [1,2,4]triazolo[4,3-b]pyridazine acetohydrazide compounds (6a,b) were obtained by refluxing acetic anhydride with the key intermediates 3a and 3c at 120 °C, respectively, in glacial acetic acid.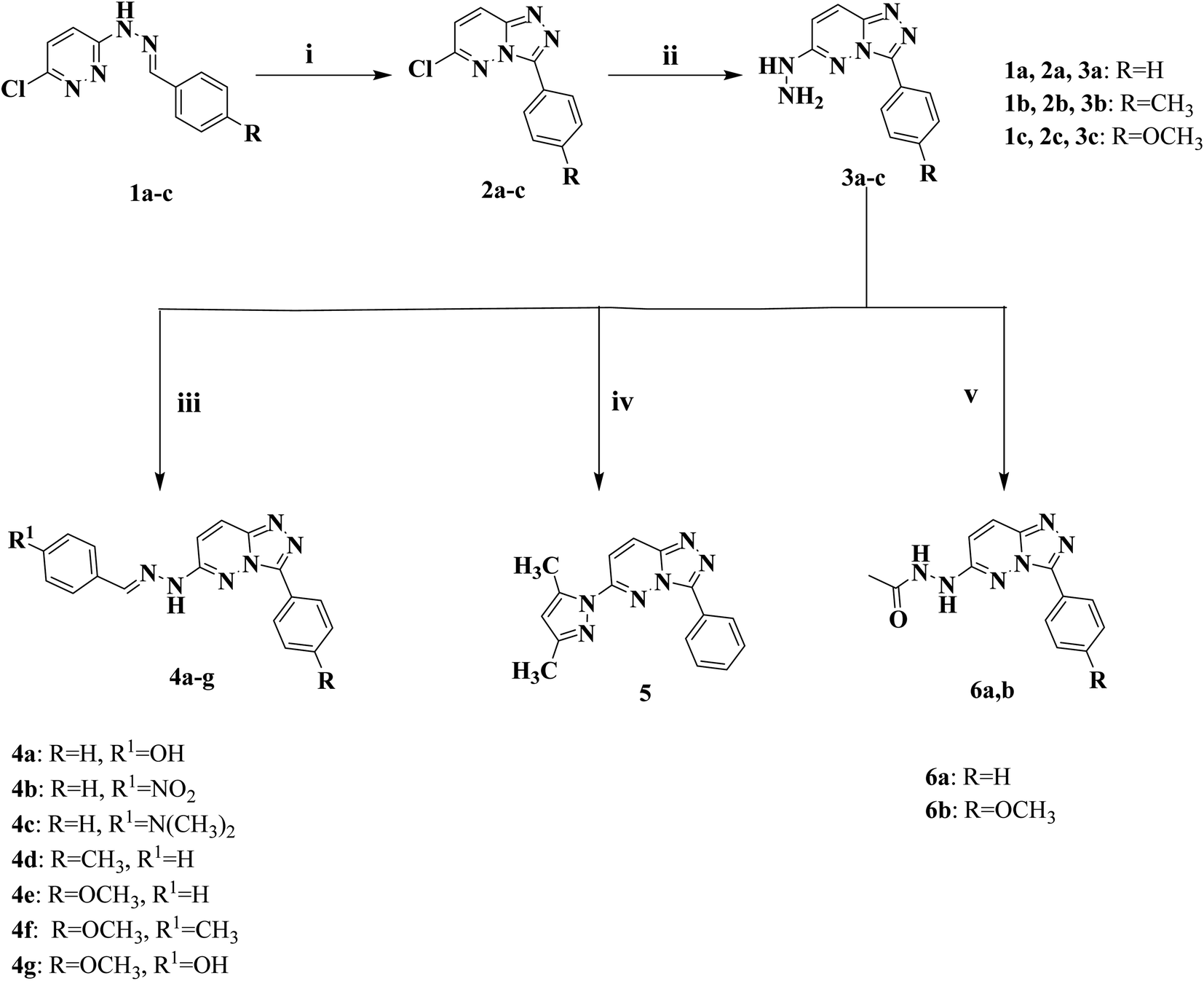 | ||
| Scheme 1 Synthetic pathway of novel triazolo[4,3-b]pyridazine 4a–g, 5, and 6a,b. | ||
The retrieved derivatives' structures were supported by spectral and elemental data. The 1H NMR spectra of compounds 4a–g showed distinctive downfield singlet signals corresponding to the NH group of the hydrazone moiety, with δ values ranging from δ 11.36 to 12.15 ppm. As a stark example, the stretching bands in the IR spectrum for compound 4a were located at 3425 cm−1 for the OH group and 3298 cm−1 for the NH group. Two downfield singlet signals, corresponding to the NH and OH groups, were noticed in the 1H NMR spectrum of compound 4a at δ = 11.65 and 9.97 ppm, respectively. On the other hand, the 1H NMR spectrum of compound 5 demonstrated a sharp singlet peak at δ = 6.26 ppm representing the CH of the pyrazole ring, in addition to two strong up-field peaks at δ = 2.63 and 2.23 ppm corresponding to the two CH3 groups attached to the pyrazole ring, which were confirmed by the 13C NMR spectrum showing two peaks at δ = 14.8 and 13.8 ppm. Moreover, the 1H NMR spectra of derivatives 6a,b exhibited one singlet peak at δ = 9.72 ppm, corresponding to one of the NH of the acetohydrazide group in addition to a strong singlet peak at δ = 2.01 ppm in 6a and 2.02 ppm in 6b representing the CH3 group of the acetohydrazide part. The previously mentioned data were confirmed by the IR spectra, which showed two bands at the range of 3483–3232 cm−1 for the two NH groups and 1685 cm−1 for the amidic CO group in 6a,b, in addition to the 13C NMR spectra, which displayed characteristic peaks at δ = 174.3 ppm in 6a and 174.1 ppm in 6b corresponding to the amidic CO and a peak at δ = 21.0 ppm corresponding to the CH3 groups in 6a,b.
3.2 Biological evaluation
| Panel/cell lines | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | 4a | 4b | 4c | 4d | 4e | 4f | 4g | 5 | 6a | 6b |
| a — GI% is below 15%.b Mean inhibition: GI% values over all NCI-60 cell lines.c Bold: GI% from 70 to 100%. | ||||||||||
| Leukemia | ||||||||||
| CCRF-CEM | 82.45 | — | — | — | — | — | 85.88 | — | — | — |
| RPMI-8226 | 72.66 | — | — | 19.37 | — | — | 86.88 | — | — | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||||
| NSC lung cancer | ||||||||||
| A549/ATCC | 24.99 | — | — | — | 21.90 | 66.62 | 41.49 | — | — | — |
| EKVX | 37.00 | — | 21.18 | 23.07 | — | 31.76 | 62.20 | — | — | — |
| HOP-62 | — | — | — | — | — | 29.57 | — | — | — | — |
| HOP-92 | 31.71 | — | 29.78 | 29.46 | 29.15 | 81.11 | 68.67 | — | — | — |
| NCI-H226 | — | — | — | — | 42.42 | 45.38 | 22.48 | — | — | — |
| NCI-H23 | 27.96 | — | 38.62 | 39.55 | — | 49.25 | 67.47 | — | — | — |
| NCI-H322M | 17.16 | — | — | — | — | — | 38.06 | — | — | — |
| NCI-H460 | 25.61 | — | 18.89 | 31.51 | — | 48.83 | 55.73 | — | — | — |
| NCI-H522 | 31.41 | — | 40.80 | — | — | — | 53.35 | 25.34 | — | — |
|
||||||||||
| Colon cancer | ||||||||||
| COLO-205 | — | — | — | — | — | 20.07 | 43.51 | — | — | — |
| HCC-2998 | — | — | — | — | — | 20.85 | 85.07 | — | — | — |
| HCT-116 | 37.12 | — | — | — | — | 21.17 | 72.71 | — | — | — |
| HCT-15 | 34.22 | — | 19.56 | 30.81 | — | 50.02 | 73.30 | — | — | — |
| HT29 | — | — | — | — | — | 17.40 | — | — | — | — |
| KM12 | 46.62 | — | 28.28 | 42.77 | — | 31.89 | 71.51 | — | — | — |
| SW-620 | — | — | — | — | — | — | 32.85 | — | — | — |
|
||||||||||
| CNS cancer | ||||||||||
| SF-268 | 34.95 | — | 57.96 | 42.23 | — | 40.91 | 64.19 | — | — | — |
| SF-295 | 16.99 | — | — | 47.66 | — | 43.09 | 56.41 | — | — | — |
| SF-539 | 43.13 | — | — | 19.19 | 17.17 | 38.74 | 63.61 | — | — | — |
| SNB-19 | 21.93 | — | 30.50 | — | 17.10 | — | 32.46 | — | — | — |
| SNB-75 | 25.31 | — | 65.40 | >100 | 62.97 | >100 | 46.38 | — | — | — |
| U251 | — | — | 24.58 | 24.9 | 22.16 | 55.13 | 32.41 | — | — | — |
|
||||||||||
| Melanoma | ||||||||||
| LOX IMIV | 51.37 | — | 62.55 | 52.47 | 29.87 | 76.27 | >100 | 22.70 | — | — |
| MALME-3M | — | — | — | — | — | — | 35.83 | — | — | — |
| M14 | — | — | — | 32.66 | — | 16.52 | 43.64 | — | — | — |
| MDA-MB-435 | 37.50 | — | — | 23.87 | — | 35.32 | 54.20 | — | — | — |
| SK-MEL-2 | — | — | — | — | — | — | 26.79 | — | — | — |
| SK-MEL-28 | — | — | — | — | — | — | 30.86 | — | — | — |
| UACC-257 | — | — | — | — | — | — | 17.70 | — | — | — |
| UACC-62 | 16.98 | — | — | — | — | — | 41.19 | — | — | — |
|
||||||||||
| Ovarian cancer | ||||||||||
| IGROV1 | 21.21 | — | — | — | — | — | 60.66 | — | — | — |
| OVCAR-3 | 35.66 | — | 34.65 | 69.65 | — | 84.42 | 62.40 | — | — | — |
| OVCAR-4 | 26.85 | — | 41.36 | 30.86 | — | 63.09 | 52.11 | — | — | — |
| OVCAR-5 | — | — | — | — | — | — | — | — | — | — |
| OVCAR-8 | 25.02 | — | 57.57 | — | 34.25 | 58.61 | 54.60 | — | — | — |
| NCI/ADR-RES | 41.13 | — | 55.19 | 15.93 | 30.10 | 74.21 | 73.98 | 23.93 | — | — |
| SK-OV-3 | — | — | — | — | — | 20.16 | — | — | — | — |
|
||||||||||
| Renal cancer | ||||||||||
| 786-0 | 46.01 | — | 18.27 | — | — | 43.09 | 77.10 | — | — | — |
| A498 | — | — | — | — | — | — | — | — | — | — |
| ACHN | 25.31 | — | 21.61 | — | — | 41.02 | 56.52 | — | — | — |
| CAKI-1 | 57.00 | — | 32.25 | 16.69 | — | 45.37 | 78.54 | 25.28 | — | — |
| RXF 393 | 22.59 | — | 15.29 | — | — | 37.84 | 57.34 | — | — | — |
| SN12C | 39.51 | — | — | — | — | 23.07 | 57.71 | — | — | — |
| TK-10 | 30.62 | — | — | — | — | 17.28 | 61.05 | — | — | — |
| UO-31 | 49.79 | — | 18.89 | 15.52 | — | 66.50 | 69.74 | >100 | — | — |
|
||||||||||
| Prostate cancer | ||||||||||
| PC-3 | 27.08 | — | 18.94 | — | — | 27.83 | 59.52 | — | — | — |
| DU-145 | — | — | 26.64 | 24.06 | — | 60.93 | 48.11 | — | — | — |
|
||||||||||
| Breast cancer | ||||||||||
| MCF7 | 71.51 | — | 34.47 | 52.06 | 28.54 | 42.84 | 86.77 | — | — | — |
| MDA-MB-231/ATCC | — | — | — | — | 34.41 | 34.65 | 39.44 | — | — | — |
| HS 578T | — | — | 33.19 | 53.45 | 17.35 | 70.93 | 53.18 | — | — | — |
| T-47D | 16.17 | — | 22.66 | — | — | 24.01 | 54.64 | — | — | — |
| MDA-MB-468 | 30.50 | — | — | — | — | — | 51.76 | — | — | — |
| Mean inhibitionb | 29.08 | <0 | 17.68 | 18.27 | 7.15 | 34.83 | 55.84 | <0 | — | <0 |
In addition, it displayed moderate activity against 35 cell lines with a GI% range between 30.86 and 69.74 and lethal activity against melanoma LOX IMIV. Substituting the p-hydroxy group in 4g with the p-methyl group and maintaining the p-methoxy group yielded the derivative 4f with a mean growth inhibition percent of 34.83. Compound 4f demonstrated strong antiproliferative activity against NSC lung cancer (HOP-92 with GI% 81.11), ovarian cancer (OVCAR-3 with GI% 84.42 and NCI/ADR-RES with GI% 74.21), and breast cancer (HS 578 T with GI% 70.93).
Moreover, it produced moderate activity against 22 cell lines with GI% ranging from 31.76 to 66.62 and lethal activity against the CNS cancer SNB-75. On the other hand, compound 4a, which resulted from the removal of the p-methoxy group of 4g, displayed a mean growth inhibition percent of 29.08 and displayed strong anticancer activity against leukemia (CCRF-CEM with GI% 82.45 and RPMI-8226 with GI% 72.66) and breast cancer (MCF7 with GI% 71.51) as well as moderate activity against 18 cancer cell lines with GI% ranging from 30.62 to 57.00. The absence of both p-methoxy and p-hydroxy substituents resulted in derivatives with low to moderate cytotoxic activity (compound 4b). Replacement of the hydrazone moiety with either a pyrazole ring (compound 5) or acetohydrazide group (derivatives 6a,b) resulted in compounds with demolished cytotoxicity, which confirmed the importance of the hydrazone part in the anticancer activity of the synthesized compounds.
Structure–activity relationship studies (SAR) were studied for the synthesized triazolo[4,3-b]pyridazine derivatives 4a–g, 5, and 6a,b. The percentage inhibition pattern for the compounds showed that the benzylidene group is essential for showing any degree of antiproliferative activity, and methoxy substitution on the phenyl ring shows the best cytotoxicity except for the unsubstituted benzylidene derivative. Moreover, hydroxy substitution on the benzylidene ring displayed optimal antiproliferative activity. Electron-donating groups on the benzylidene moiety were required to show cytotoxic activity, while substitution with electron-withdrawing groups resulted in demolished cytotoxicity, as shown in Fig. 3.
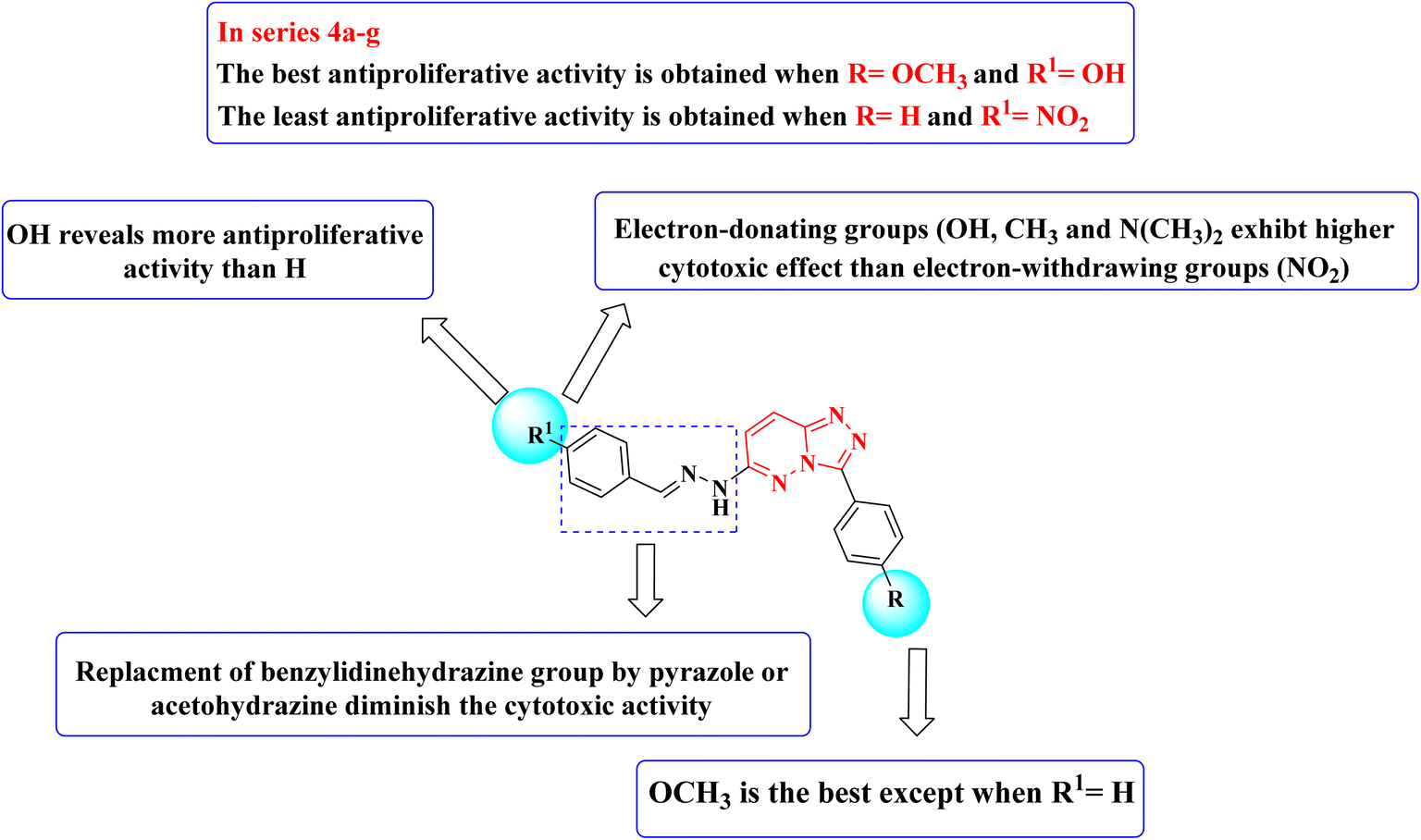 | ||
| Fig. 3 Structure–activity relationship (SAR) of the synthesized triazolo[4,3-b]pyridazine derivatives. | ||
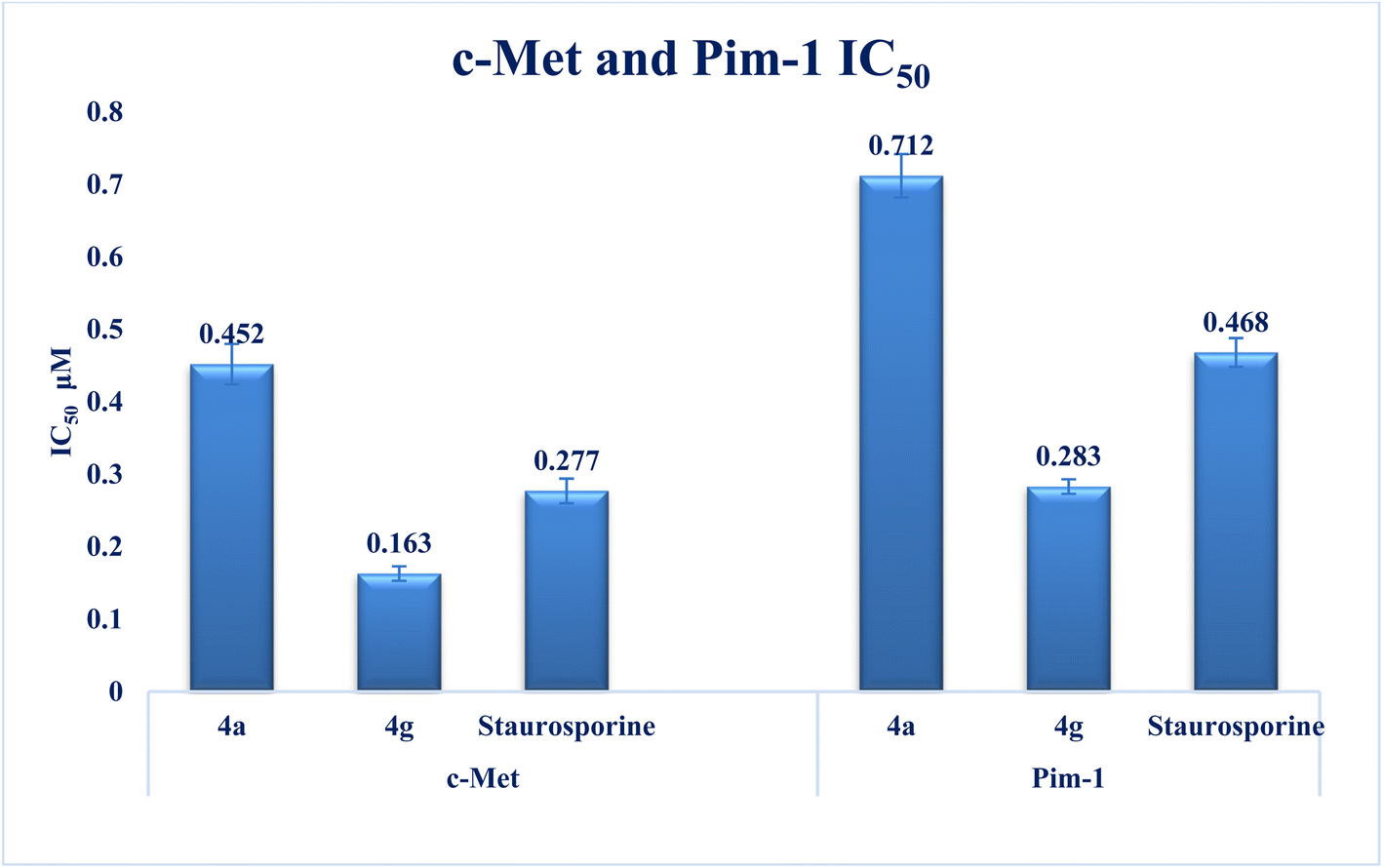 | ||
| Fig. 4 Graph showing IC50 inhibition of both c-Met and Pim-1 kinases of compounds 4a and 4g in comparison to staurosporine. | ||
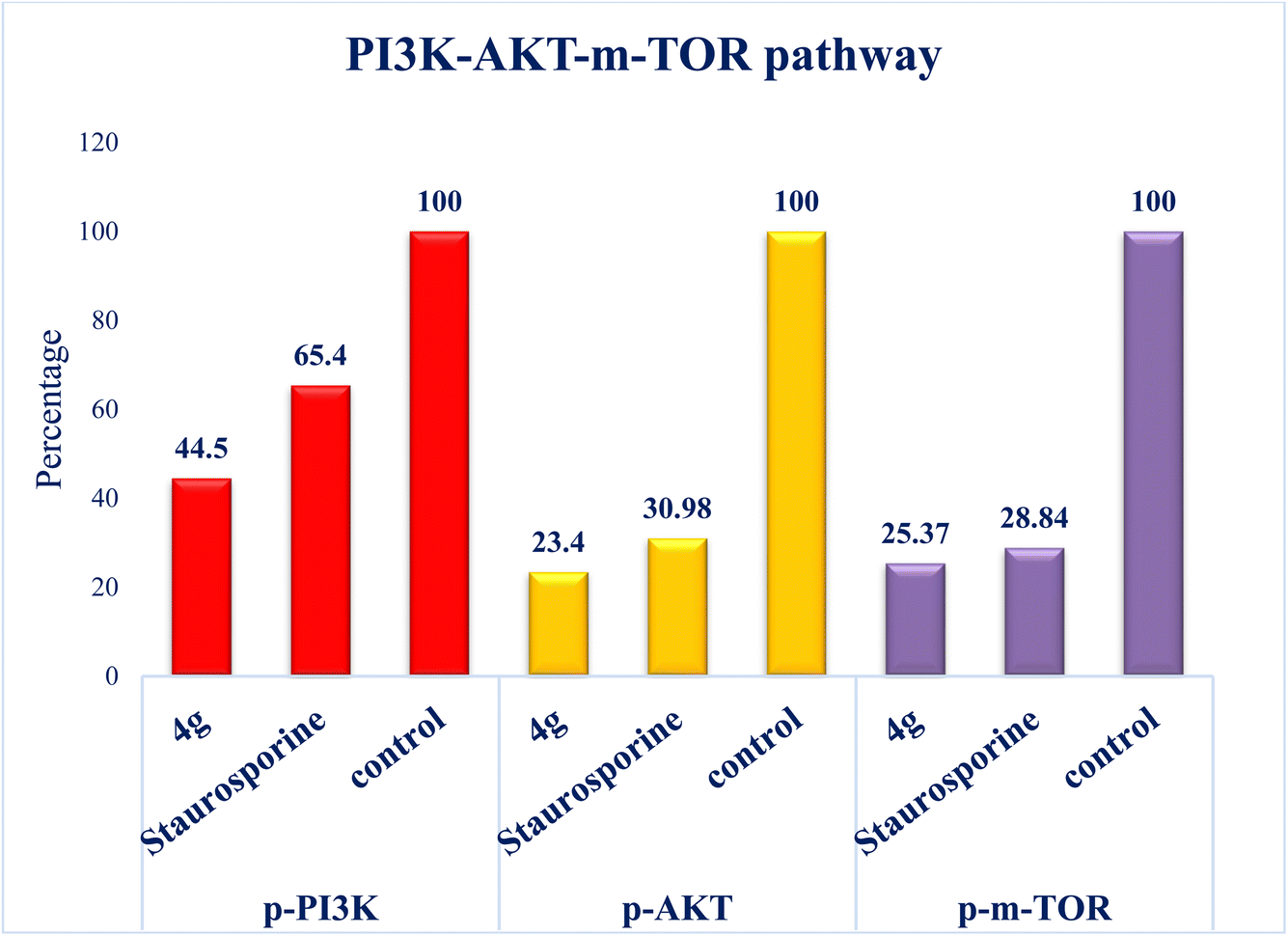 | ||
| Fig. 5 The plot shows the down-regulation of phosphorylation of PI3K-Akt-mTOR pathway in breast cell line MCF7 cells following the application of compound 4g, versus staurosporine and untreated control cells. | ||
 | ||
| Fig. 6 Graphical representation for active caspase-9 assays of compound 4g compared to staurosporine as a control. | ||
 | ||
| Fig. 7 Cell cycle analysis in the breast MCF7cell line. (A) Graphical diagram for the distribution of cell cycle analysis in treated and control cells. (B) Control MCF7. (C) Treatment with compound 4g. | ||
| Compound | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 4g/MCF7 | 48.56 | 25.16 | 18.13 | 5.27 |
| Cont. MCF7 | 1.64 | 0.36 | 0.18 | 1.1 |
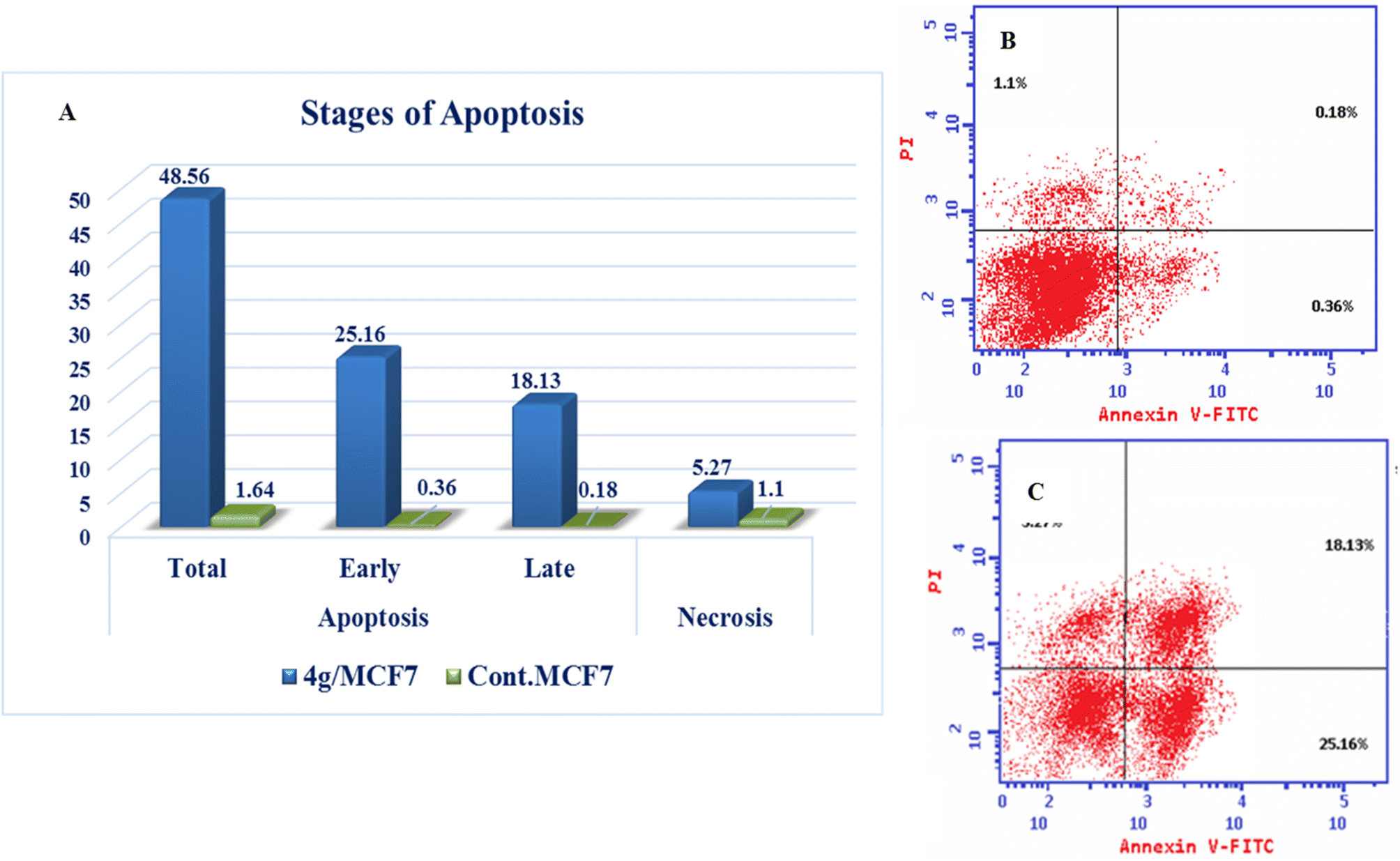 | ||
| Fig. 8 Annexin V positive staining in MCF7 breast cells after compound 4g treatment. (A) Diagram showing the apoptosis and necrosis in treated and control cells. (B) Control MCF7. (C) Treatment with compound 4g. | ||
3.3 In silico studies
| Compound/enzyme | S (kcal mol−1) | Amino acids | Interaction types | Distance (Å) |
|---|---|---|---|---|
| 4a/c-Met | −7.49 | Ile1084 | π–H | 4.49 |
| Tyr1230 | π–π | 3.59 | ||
| Met1160 | HB acceptor | 2.76 | ||
| Pro1158 | HB donor | 2.70 | ||
| 4g/c-Met | −8.55 | Ile1084 | π–H | 4.24 |
| Ile1084 | π–H | 3.93 | ||
| Met1160 | HB acceptor | 3.11 | ||
| Met1160 | HB acceptor | 3.20 | ||
| I/c-Met | −9.45 | Tyr1230 | π–H | 4.08 |
| Val1092 | π–H | 4.01 | ||
| Tyr1230 | π–π | 3.95 | ||
| Tyr1230 | π–π | 3.73 | ||
| Met1160 | HB acceptor | 3.06 | ||
| Asp1222 | HB acceptor | 3.40 | ||
| Val1092 | π–H | 4.15 | ||
| Met1211 | π–H | 3.68 | ||
| 4a/Pim-1 | −7.97 | Lys67 | HB acceptor | 3.42 |
| Lys67 | HB acceptor | 3.44 | ||
| Val52 | π–H | 3.91 | ||
| Val52 | π–H | 4.27 | ||
| Val52 | π–H | 3.99 | ||
| Ile185 | π–H | 4.08 | ||
| Ile185 | π–H | 4.09 | ||
| Phe49 | π–π | 3.73 | ||
| Asp128 | HB donor | 3.09 | ||
| 4g/Pim-1 | −8.24 | Lys67 | HB acceptor | 3.27 |
| Val52 | π–H | 4.23 | ||
| Val52 | π–H | 4.01 | ||
| Asp128 | HB donor | 3.17 | ||
| Asp131 | HB donor | 3.25 | ||
| IV/Pim-1 | −9.05 | Lys67 | HB acceptor | 3.02 |
| Lys67 | HB acceptor | 3.11 | ||
| Val52 | π–H | 4.56 | ||
| Ile185 | π–H | 4.04 | ||
| Ile185 | π–H | 4.26 | ||
| Ile185 | π–H | 4.46 | ||
| Phe49 | π–π | 3.95 |
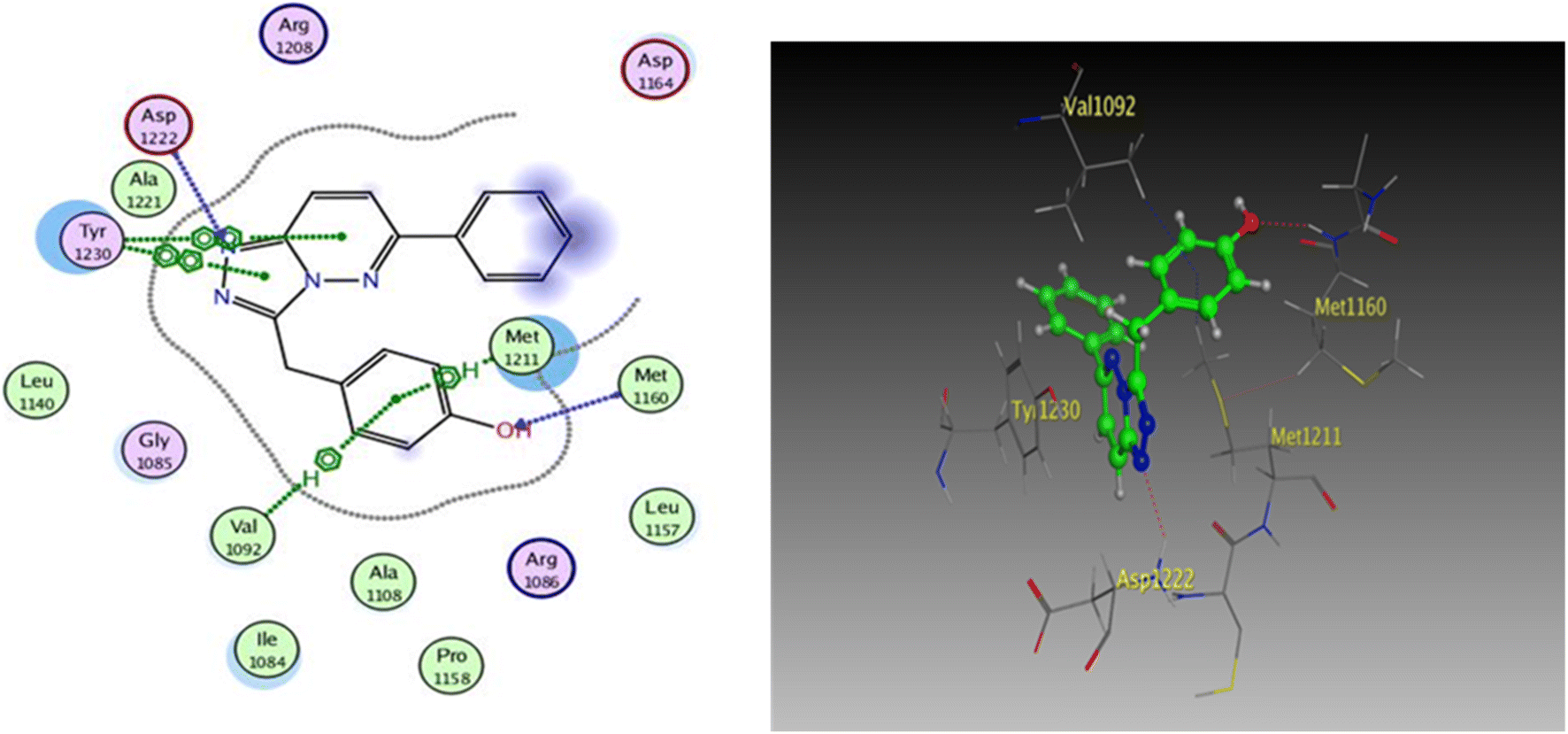 | ||
| Fig. 9 2D and 3D diagrams of compound I interactions with c-Met (PDB ID: 3CCN) (in 3D figure hydrogen bonds are represented in red and hydrophobic interactions in blue). | ||
One hydrogen bond was created between the amino acid residue Asp1222 and the hydrogen bond acceptor (N1 of the triazole ring). Due to the hydrophobic interaction, two hydrophobic bonds were formed between Tyr1230 and the triazolopyridazine core group. Additionally, the phenol moiety formed two hydrophobic interactions with Met1211 and Val1092 through the phenyl ring, forming a hydrogen bond via its O with Met1160. These results closely match the described data.28 Similar to the vital interactions of compound I, the target molecules interacted with the active site of c-Met in a manner that demonstrated binding mode. The binding affinity of compound 4a was −7.49 kcal mol−1. The critical amino acid residue Tyr1230 formed a π–π stack interaction with the triazolopyridazine core. The terminal phenol group formed a bifurcated hydrogen bond interaction with Met1160 and Pro1158 through the HBA/HBD (OH) substituent and was incorporated in a hydrophobic interaction with Ile1084 through its phenyl group. Compound 4g exhibited binding energy up to −8.55 kcal mol−1. First, the N1 and N2 of the triazolo ring functioned as hydrogen bond acceptors and formed two hydrogen bonds with Met1160 which may contribute to its potency. Second, the phenoxy side chain's triazolopyridazine core and phenyl moiety formed three hydrophobic bonds with Ile1084 and Val1092. Third, the phenyl group of the phenol moiety formed a π–π stack interaction with the crucial amino acid residue Tyr1230 (Fig. 10).
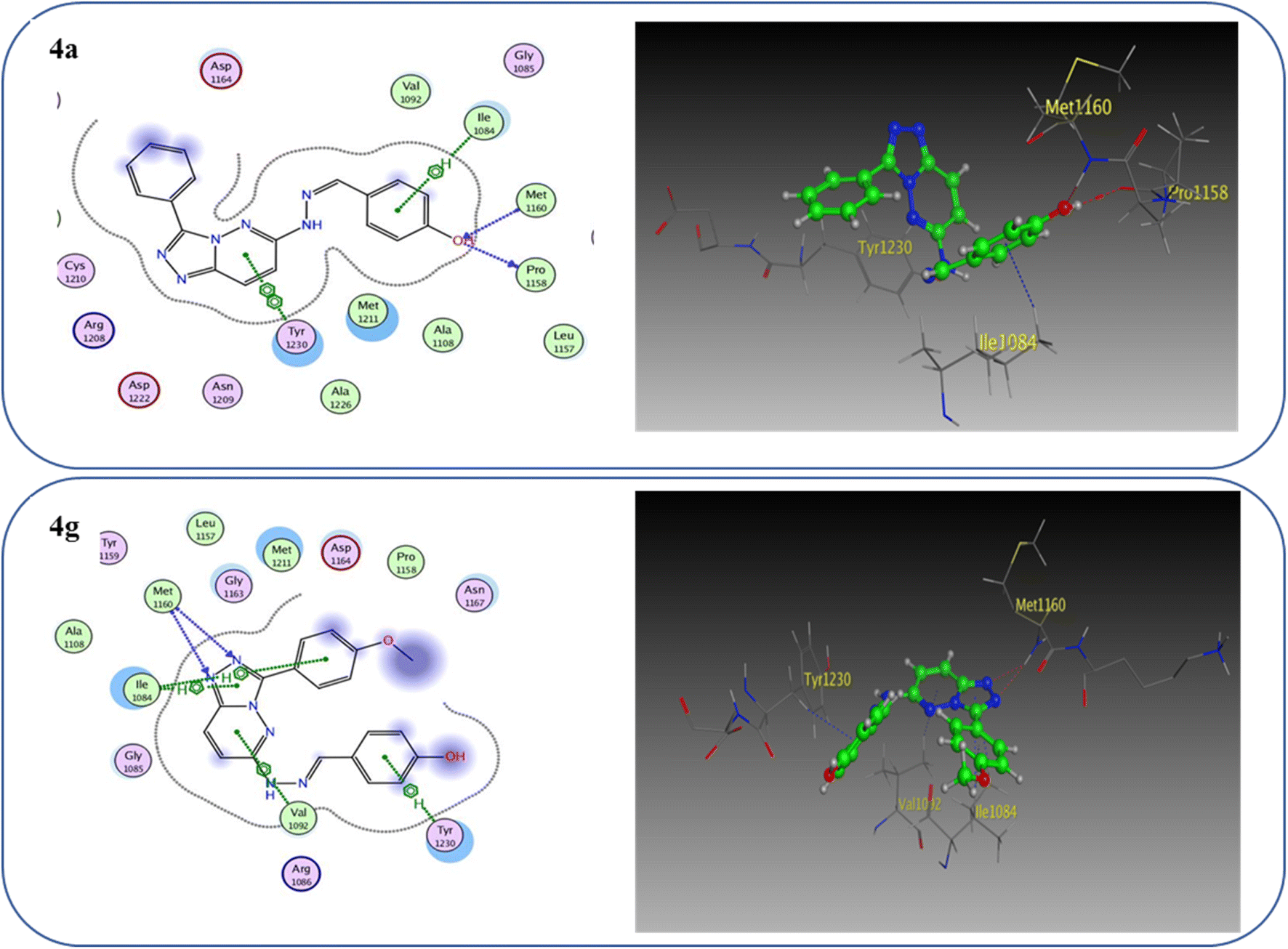 | ||
| Fig. 10 2D and 3D interaction diagrams of derivative 4a and 4g interactions with c-Met in 2D and 3D diagrams. | ||
To prove the dual action of our derivatives, a docking study was carried out on Pim-1 (PDB ID 3BGQ) using compound IV as a reference ligand.29 The docking process was validated, and the ligand achieved −9.05 kcal mol−1 binding free energy with an RMSD value of 1.22. There were two crucial interactions, a hydrogen bond with Lys67 and a hydrophobic interaction with Val52 amino acid residues, besides other hydrophobic interactions (Fig. 11). It was found that compound 4a bound to the active pocket with a binding energy of −7.97 kcal mol−1 through three hydrogen bond interactions, two with Lys67 via N1 of the triazole moiety as HBA, and the third one between the Asp128 residue and the OH of the phenol ring as HBD. In addition to the hydrogen bonds, the derivative 4a formed many hydrophobic interactions with Val52, Phe49, and Ile185 amino acids. In the same context, compound 4g was effectively linked to the active pocket through different types of hydrogen bonds with Lys67, Asp128, and Asp131, which could be the reason for its potency, besides two hydrophobic interactions with Val52 (Fig. 12). The binding energy with the receptor was −8.24 kcal mol−1.
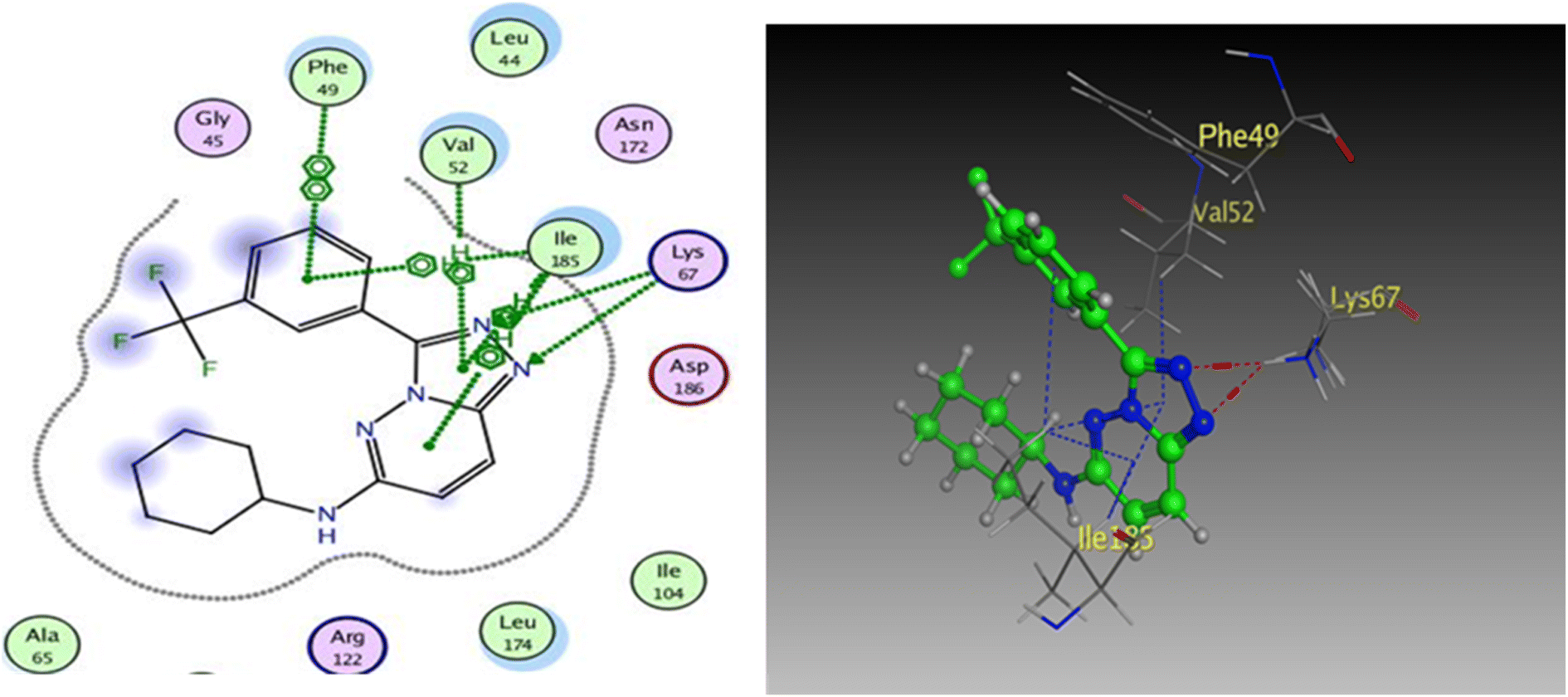 | ||
| Fig. 11 2D and 3D diagrams of compound IV interactions with Pim-1 (PDB ID: 3BGQ). | ||
 | ||
| Fig. 12 2D and 3D diagrams of compound 4a and 4g interactions with Pim-1. | ||
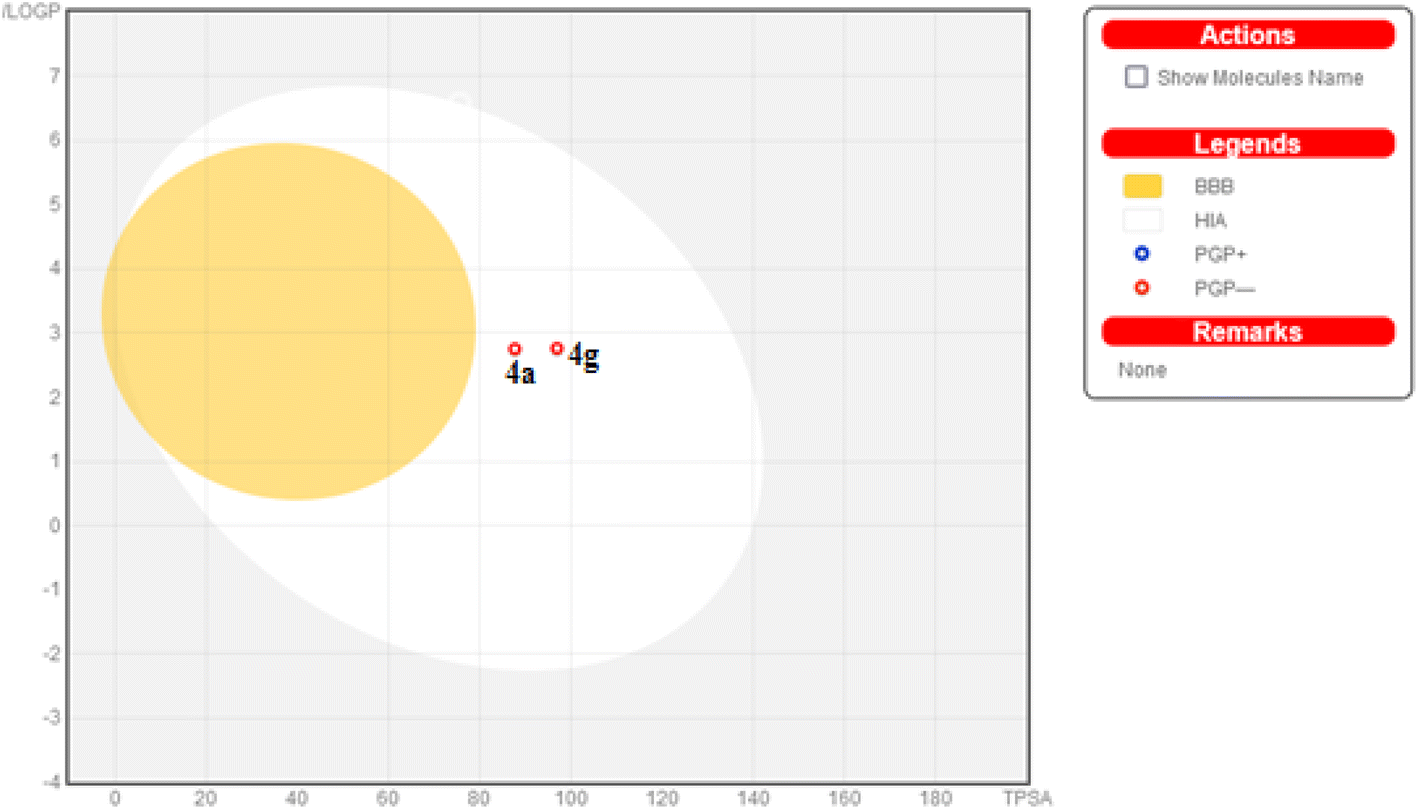 | ||
| Fig. 13 Predicted boiled-egg plot from SwissADME online web tool for compounds 4a and 4g. | ||
Additionally, this graph demonstrates that compounds 4a and 4g are not P-glycoprotein substrates (PGP), which means they are immune to the transporter's efflux system, which many tumor cell lines use as a mechanism for drug resistance. Compounds 4a and 4g, according to the SwissADME online web tool, fulfill the criteria for drug-likeness established by the major pharmaceutical companies due to their high molecular weight and logP. According to the computational analysis of the physicochemical and pharmacokinetic properties, compounds 4a and 4g have promising pharmacokinetic and biological efficacy characteristics.
4. Conclusion
New series of triazolo[4,3-b]pyridazine derivatives 4a–g, 5, and 6a,b were successfully created. The NCI-60 human cancer cell line panel was used to screen the synthesized compounds' anticancer activity. Compounds 4a and 4g demonstrated extensive antiproliferative activity on various cell lines. The most effective derivatives against the breast cancer cell line MCF7 were compound 4a and compound 4g, with IC50 values of 2.97 ± 0.18 and 0.56 ± 0.03 μM, respectively. While compound 4g displayed higher inhibition on c-Met with an IC50 of 0.163 ± 0.01 and Pim-1 with an IC50 of 0.283 ± 0.01 μM, compound 4a displayed lower dual inhibition of c-Met and Pim-1 with IC50 values of 0.452 ± 0.028 and 0.712 ± 0.03 μM, respectively. According to an annexin V-FITC/PI assay, the most potent compound, 4g, significantly increased the early and late apoptosis phases in MCF-7 cells, respectively, from 0.36 to 25.16% and from 0.18 to 18.13%. An increase of 11.6-fold in the amount of apoptotic caspase-9 served as additional support for this evidence. In addition, cell cycle analysis results revealed that derivative 4g ceased the proliferation of MCF-7 cancer cells in the S phase. These findings confirm 4g's cytotoxic potential and could make it a candidate for additional biological testing. According to the findings of a molecular docking study, the binding pattern of compound 4g is consistent with its dual c-Met and Pim-1 inhibitory activity. Based on the promising antiproliferative activities demonstrated by the synthesized benzylidene derivatives, future work will involve synthesizing new derivatives by incorporating heteroaryl aldehydes such as pyridine, pyrimidine, pyrazole, and imidazole. Additionally, various aryl groups with electron-withdrawing and electron-donating properties will be introduced to further explore and test their biological activities.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have no conflicts of interest. This manuscript's content and writing are the sole responsibility of its authors.Acknowledgements
The authors are grateful to the National Institute of Health, for their support of antiproliferative screening.References
- A. K. Kwakye, S. Kampo, J. Lv, M. N. Ramzan, S. A. Richard, A. A. Falagán, J. Agudogo, E. Atito-Narh, Q. Yan and Q. P. Wen, BMC Res. Notes, 2020, 13, 386 CrossRef CAS.
- E. Y. Ahmed, O. M. Abdelhafez, D. Zaafar, A. M. Serry, Y. H. Ahmed, R. F. A. El-Telbany, Z. Y. Abd Elmageed and H. I. Ali, Arch. Pharm., 2022, 355, 2100327 CrossRef CAS PubMed.
- R. Wang, J. Wang, T. Dong, J. Shen, X. Gao and J. Zhou, Oncol. Lett., 2019, 17, 1217–1222 Search PubMed.
- X. Liu, W. Yao, R. C. Newton and P. A. Scherle, Expert Opin. Investig. Drugs, 2008, 17, 997–1011 Search PubMed.
- C. Schmidt, F. Bladt, S. Goedecke, V. Brlnkmann, W. Zschiesche, M. Sharpet, E. Gherardlt and C. Birchmelert, Nature, 1995, 373, 699–702 CrossRef PubMed.
- E. M. Ahmed, N. A. Khalil, A. T. Taher, R. H. Refaey and Y. M. Nissan, Bioorg. Chem., 2019, 92, 103272 CrossRef.
- Y. Tursynbay, J. Zhang, Z. Li, T. Tokay, Z. Zhumadilov, D. Wu and Y. Xie, Biomed. Rep., 2016, 4, 140–146 CrossRef PubMed.
- C. O. ning Leung, C. C. lui Wong, D. N. yin Fan, A. K. lun Kai, E. K. kwan Tung, I. M. jing Xu, I. O. lin Ng and R. C. lam Lo, Oncotarget, 2015, 6, 10880–10892 CrossRef.
- N. A. Keane, M. Reidy, A. Natoni, M. S. Raab and M. O'Dwyer, Blood Cancer J., 2015, 5, e325 CrossRef CAS.
- R. A. Darby, A. Unsworth, S. Knapp, I. D. Kerr and R. Callaghan, Cancer Chemother. Pharmacol., 2015, 76, 853–864 CrossRef CAS PubMed.
- B. Oyallon, M. Brachet-Botineau, C. Logé, T. Robert, S. Bach, S. Ibrahim, W. Raoul, C. Croix, P. Berthelot, J. Guillon, N. Pinaud, F. Gouilleux, M.-C. Viaud-Massuard and C. Denevault-Sabourin, Molecules, 2021, 26, 867 CrossRef CAS PubMed.
- D. Reddy, R. Kumavath, P. Ghosh and D. Barh, Biomolecules, 2019, 9, 792 CrossRef CAS PubMed.
- R. L. B. Costa, H. S. Han and W. J. Gradishar, Breast Cancer Res. Treat., 2018, 169, 397–406 CrossRef CAS PubMed.
- F. André, E. Ciruelos, G. Rubovszky, M. Campone, S. Loibl, H. S. Rugo, H. Iwata, P. Conte, I. A. Mayer, B. Kaufman, T. Yamashita, Y.-S. Lu, K. Inoue, M. Takahashi, Z. Pápai, A.-S. Longin, D. Mills, C. Wilke, S. Hirawat and D. Juric, N. Engl. J. Med., 2019, 380, 1929–1940 CrossRef.
- N. C. Turner, E. Alarcón, A. C. Armstrong, M. Philco, Y. A. López Chuken, M. P. Sablin, K. Tamura, A. Gómez Villanueva, J. A. Pérez-Fidalgo, S. Y. A. Cheung, C. Corcoran, M. Cullberg, B. R. Davies, E. C. De Bruin, A. Foxley, J. P. O. Lindemann, R. Maudsley, M. Moschetta, E. Outhwaite, M. Pass, P. Rugman, G. Schiavon and M. Oliveira, Ann. Oncol., 2019, 30, 774–780 CrossRef PubMed.
- R. W. Carling, A. Madin, A. Guiblin, M. G. N. Russell, K. W. Moore, A. Mitchinson, B. Sohal, A. Pike, S. M. Cook, I. C. Ragan, R. M. McKernan, K. Quirk, P. Ferris, G. Marshall, S. A. Thompson, K. A. Wafford, G. R. Dawson, J. R. Atack, T. Harrison, J. L. Castro and L. J. Street, J. Med. Chem., 2005, 48, 7089–7092 CrossRef PubMed.
- R. W. Carling, K. W. Moore, L. J. Street, D. Wild, C. Isted, P. D. Leeson, S. Thomas, D. O'Connor, R. M. McKernan, K. Quirk, S. M. Cook, J. R. Atack, K. A. Wafford, S. A. Thompson, G. R. Dawson, P. Ferris and J. L. Castro, J. Med. Chem., 2004, 47, 1807–1822 CrossRef.
- L. P. Guan, X. Sui, X. Q. Deng, Y. C. Quan and Z. S. Quan, Eur. J. Med. Chem., 2010, 45, 1746–1752 CrossRef.
- J. S. Ruso, N. Rajendiran, C. Srinivas, K. N. Murthy and K. Soumya, J. Korean Chem. Soc., 2014, 58, 377–380 CrossRef.
- M. Islam and A. A. Siddiqui, Acta Pol. Pharm. – Drug Res., 2010, 67, 555–562 Search PubMed.
- A. E. Rashad, A. H. Shamroukh, M. A. Ali and F. M. Abdel-Motti, Heteroat. Chem., 2007, 18, 274–282 CrossRef.
- A. H. Shamroukh and M. A. Ali, Arch. Pharm., 2008, 341, 223–230 CrossRef.
- P. Galatsis, J. L. Henderson, B. L. Kormos, S. Han, R. G. Kurumbail, T. T. Wager, P. R. Verhoest, G. S. Noell, Y. Chen, E. Needle, Z. Berger, S. J. Steyn, C. Houle and W. D. Hirst, Bioorg. Med. Chem. Lett., 2014, 24, 4132–4140 CrossRef PubMed.
- M. Franzini, X. M. Ye, M. Adler, D. L. Aubele, A. W. Garofalo, S. Gauby, E. Goldbach, G. D. Probst, K. P. Quinn, P. Santiago, H. L. Sham, D. Tam, A. Truong and Z. Ren, Bioorg. Med. Chem. Lett., 2013, 23, 1967–1973 CrossRef PubMed.
- A. P. Skoumbourdis, C. A. LeClair, E. Stefan, A. G. Turjanski, W. Maguire, S. A. Titus, R. Huang, D. S. Auld, J. Inglese, C. P. Austin, S. W. Michnick, M. Xia and C. J. Thomas, Bioorg. Med. Chem. Lett., 2009, 19, 3686–3692 CrossRef.
- J. K. Brenke, G. M. Popowicz, K. Schorpp, I. Rothenaigner, M. Roesner, I. Meininger, C. Kalinski, L. Ringelstetter, O. R’kyek, M. Vincendeau, O. Plettenburg, M. Sattler, D. Krappmann, K. Hadian and G. Jürjens, J. Biol. Chem., 2018, 293, 13191–13203 CrossRef.
- F. Jin, Y. Lin, W. Yuan, S. Wu, M. Yang, S. Ding, J. Liu and Y. Chen, Eur. J. Med. Chem., 2024, 272, 116477 CrossRef.
- B. K. Albrecht, J. C. Harmange, D. Bauer, L. Berry, C. Bode, A. A. Boezio, A. Chen, D. Choquette, I. Dussault, C. Fridrich, S. Hirai, D. Hoffman, J. F. Larrow, P. Kaplan-Lefko, J. Lin, J. Lohman, A. M. Long, J. Moriguchi, A. O'Connor, M. H. Potashman, M. Reese, K. Rex, A. Siegmund, K. Shah, R. Shimanovich, S. K. Springer, Y. Teffera, Y. Yang, Y. Zhang and S. F. Bellon, J. Med. Chem., 2008, 51, 2879–2882 CrossRef PubMed.
- R. Grey, A. C. Pierce, G. W. Bemis, M. D. Jacobs, C. S. Moody, R. Jajoo, N. Mohal and J. Green, Bioorg. Med. Chem. Lett., 2009, 19, 3019–3022 CrossRef.
- S. E. Seif, Z. Mahmoud, W. W. Wardakhan, A. M. Abdou and R. A. Hassan, Drug Dev. Res., 2023, 84, 839–860 CrossRef.
- M. T. M. Sayed, P. A. Halim, A. K. El-Ansary and R. A. Hassan, Drug Dev. Res., 2023, 84, 1299–1319 CrossRef PubMed.
- W. E. Elgammal, S. S. Shaban, E. M. Eliwa, A. H. Halawa, S. M. A. El-gilil, R. A. Hassan, A. M. Abdou, G. A. M. Elhagali and M. Abdel Reheim, Future Med. Chem., 2024, 1–19 Search PubMed.
- M. M. Zeid, O. M. El-Badry, S. Elmeligie and R. A. Hassan, Chem. Biodivers., 2024, 21, e202400077 CrossRef.
- S. H. Emam, R. A. Hassan, E. O. Osman, M. I. A. Hamed, A. M. Abdou, M. M. Kandil, E. M. Elbaz and D. S. Mikhail, Drug Dev. Res., 2023, 84, 475–499 CrossRef.
- E. O. Osman, S. H. Emam, A. Sonousi, M. M. Kandil, A. M. Abdou and R. A. Hassan, Drug Dev. Res., 2023, 84, 888–906 CrossRef PubMed.
- A. Sonousi, R. A. Hassan, E. O. Osman, A. M. Abdou and S. H. Emam, J. Enzyme Inhib. Med. Chem., 2022, 37, 2644–2659 CrossRef.
- S. Y. Ewieda, R. A. Hassan, E. M. Ahmed, A. M. Abdou and M. S. A. Hassan, Bioorg. Chem., 2024, 150, 107623–107635 CrossRef.
- Z. Arghiani, S. M. Seyedi, M. Bakavoli and H. Eshghi, J. Heterocycl. Chem., 2015, 52, 1099–1107 CrossRef.
- W. R. Carling, J. L. C. Pineiro, I. J. Collins, A. R. Guiblin, T. Harrison, A. Madin, K. W. Moore, M. G. Russell, G. Scott and L. J. Street, US Pat., 6828322, 2004 Search PubMed.
- R. Aggarwal, Mamta, G. Sumran and M. C. Torralba, J. Mol. Struct., 2019, 1185, 379–391 CrossRef.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef PubMed.
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef PubMed.
- M. R. Boyd and K. D. Paull, Drug Dev. Res., 1995, 34, 91–109 Search PubMed.
- M. C. Alley, D. A. Scudiere, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601 Search PubMed.
- M. R. Grever, S. A. Schepartz and B. A. Chabner, Semin. Oncol., 1992, 19, 622–638 Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef PubMed.
- L. Gong, Y. Tang, R. An, M. Lin, L. Chen and J. Du, Cell Death Dis., 2017, 8, e3080 CrossRef PubMed.
- M. F. Tolba, A. Esmat, A. M. Al-Abd, S. S. Azab, A. E. Khalifa, H. A. Mosli, S. Z. Abdel-Rahman and A. B. Abdel-Naim, IUBMB Life, 2013, 65, 716–729 CrossRef CAS PubMed.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutellingsperger, J. Immunol. Methods, 1995, 184, 39–51 CrossRef CAS PubMed.
- T. H. Ibrahim, Y. M. Loksha, H. A. Elshihawy, D. M. Khodeer and M. M. Said, Arch. Pharm., 2017, 350, e1700093 CrossRef.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef.
- A. Daina and V. Zoete, ChemMedChem Commun., 2016, 11, 1117–1121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04036h |
| This journal is © The Royal Society of Chemistry 2024 |