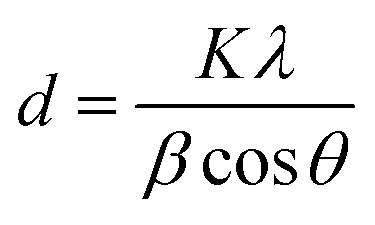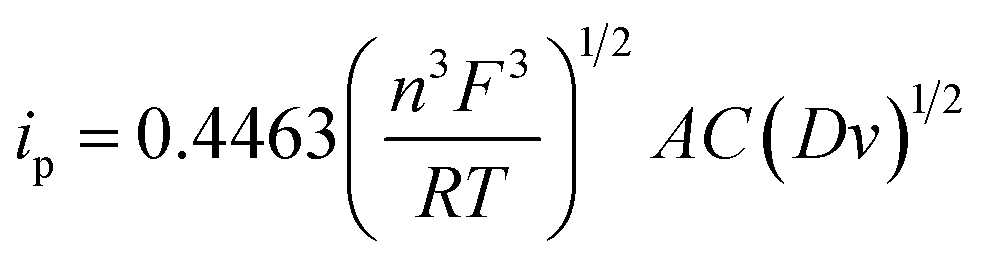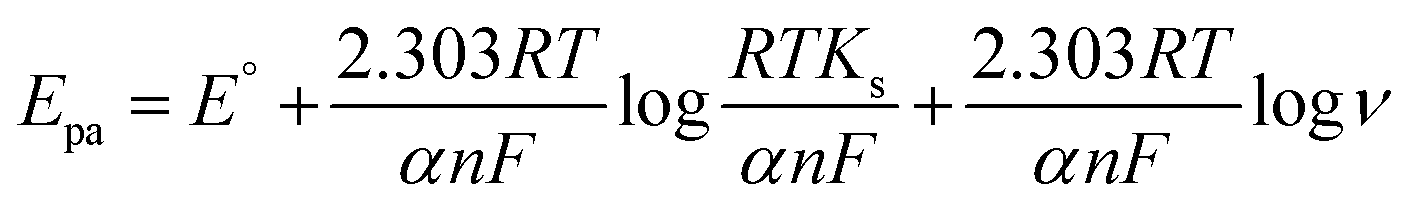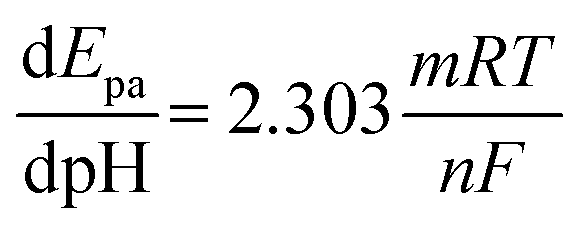DOI:
10.1039/D4RA04046E
(Paper)
RSC Adv., 2024,
14, 36073-36083
From e-waste to eco-sensors: synthesis of reduced graphene oxide/ZnO from discarded batteries for a rapid electrochemical bisphenol A sensor
Received
2nd June 2024
, Accepted 25th October 2024
First published on 11th November 2024
Abstract
Improper disposal of used dry cell batteries and the leaching of bisphenol A (BPA), a prevalent endocrine disruptor present in food packaging, into surface water pose a significant threat to both the environment and drinking water, threatening the sustainability of the ecosystem. Thus, it is imperative to manage detrimental e-waste and regularly monitor BPA using a sensitive and reliable technique. This study proposes a cost-effective reduced graphene oxide/zinc oxide (rGO/ZnO) nanohybrid, entirely synthesized from electronic waste, for electrochemically detecting BPA in an aqueous medium. Graphite and metallic Zn precursors obtained from discarded batteries were employed to synthesize rGO/ZnO. The successful characterization of the prepared rGO and rGO/ZnO nanohybrid was conducted through different state-of-the-art techniques. An rGO/ZnO-modified glassy carbon electrode (GCE) exhibited superior conductivity and a larger surface area. Voltammetric study at the rGO/ZnO-modified GCE successfully detected BPA in an aqueous medium, demonstrating a one-electron and proton pathway for BPA oxidation. The sensor demonstrated a linear response within the concentration range of 1–30 μM, with a limit of detection of 0.98 nM and sensitivity of 0.055 μA μM−1. The developed electrode could also detect BPA in real water samples with reasonable recovery. These findings imply that the developed sensor has the potential to be a sensitive, practical, and economical monitoring system for BPA in water.
1 Introduction
Bisphenol A (BPA), also known as 2,2′-bis(4-hydroxyphenyl)propane, functions as a monomer in the synthesis of diverse plastic materials utilized across various industries, notably in the production of food and water packaging, from which it leaches into food stuffs.1–3 The widespread application of BPA in these industries raises concerns regarding the regular ingestion of minute quantities of BPA by people, which could lead to higher vulnerability to cardiovascular diseases, obesity, diabetes, and adverse effects on sexual differentiation, immunological function, and cardiovascular health.1,4 Therefore, it is crucial to establish an accurate and highly sensitive analytical system for the detection of minute quantities of BPA.
Several methods can be employed for detecting BPA, such as gas chromatography-mass spectrometry, high-performance liquid chromatography, chemiluminescence, and capillary electrophoresis.5,6 The utilization of these techniques is highly limited by the requirement for complex instrumentation and substantial time spent, despite their accuracy and sensitivity.3,6,7 Electrochemical technique, on the other hand, has become increasingly popular in the field of sensing applications owing to its many advantages, which include ease of operation, real-time detection, high sensitivity, and selectivity.8–10 However, achieving these advantages is dependent upon the choice of appropriate materials for modification of substrate electrodes.
Numerous attempts have been made to develop effective materials for the fabrication of electrochemical sensors used for the detection of BPA.7–9,11–17 To improve the current performance of BPA sensing electrodes, several endeavors have been undertaken, such as employing a range of carbon-based materials, including biochar (BC),13 multi-walled carbon nanotubes (MWCNTs),9,18 single-walled carbon nanotubes (SWCNTs),12 graphene oxide (GO),4,19–22 and reduced graphene oxide (rGO).7,8,11,15–17,23 Additionally, metal oxides such as zinc oxide (ZnO),8,9,11,13,18,19,24 nickel oxide (NiO),11,25 iron oxide (Fe3O4),10,26 copper oxide (CuO),14 and molybdenum oxide (MoO3)27,28 have garnered considerable attention in these efforts. ZnO has garnered significant interest in electrocatalysis due to its notable catalytic activity,29,30 stability, and non-toxic nature.31 Despite these advantages, ZnO faces significant drawbacks, including inadequate electrical conductivity and agglomeration stemming from its inherent polarity,31 making it a suboptimal choice for electroanalysis. Additionally, the limited adsorption capacity of organic compounds on the ZnO surface32,33 imposes limitations on its utility in electro-sensors. Consequently, the integration of a good supporting material becomes essential to overcome these challenges. Among carbon nanomaterials, graphene-based materials such as GO and rGO have demonstrated promising potential as supports for metal oxides.34–40 This is attributed to their favorable surface-area ratio, high organic compound adsorption capacity (e.g., BPA), and excellent electrical conductivity and electrocatalytic properties.2,34,36,41 Therefore, the incorporation of ZnO onto the surface of rGO holds promise for enhancing the electrochemical performance of BPA sensing.
To our knowledge, this study represents the first use of rGO/ZnO derived from discarded batteries for electrochemical detection of BPA in aqueous media. We successfully synthesized a rGO/ZnO nanohybrid using precursor materials obtained from graphite and zinc extracted from discarded dry cells, enabling a cost-effective and innovative source for the synthesis of this nanohybrid material. The utilization of electronic waste (e-waste) as the exclusive source of precursors not only enhances cost-effectiveness but also addresses environmental concerns by repurposing and management of detrimental e-waste. Analytical measurements were performed using cyclic and linear sweep voltammetry techniques. The prepared rGO/ZnO nanohybrid offers advantages such as environmental friendliness, low cost, scalability, and high sensitivity.
2 Experimental
2.1 Reagents and chemicals
H2SO4 (CAS No. 7664-93-9) and KMnO4 (CAS No. 7722-64-7) were purchased from Scharlau, Spain. H3PO4 (CAS No. 7664-38-2) was purchased from Janssen Chemica, Belgium. NaOH (CAS No. 1310-73-2) and HCl (CAS No. 7647-01-0) were purchased from Sigma-Aldrich, USA. N2H4·H2O (CAS No. 7803-578), KHPO4, K2HPO4, and KOH were purchased from AppliChem, Germany. BPA was collected from Sigma Aldrich, Switzerland. All reagents were of analytical grade and used without further purification. All solutions were prepared using ultrapure deionized water (18 MΩ cm).
2.2 Instruments
The morphology of the prepared samples was studied by scanning electron microscopy (SEM) (JEOL, JSM 6490LA, USA), and photographs were taken by changing different magnifications by stereo SEM at 30 tilt and 10 kV. Transmission electron micrographs were obtained with transmission electron microscopy (TEM) (JEOL, JEM 2100F, Japan) operated at 200 kV FE (field emission) accelerating voltage. An energy-dispersive X-ray (EDX) spectrometer (JEOL, JED 2200, Japan) was utilized for the elemental composition determination of the prepared samples. Fourier transform infrared (FT-IR) spectra were acquired in the transmittance mode using a Fourier transform spectrophotometer (Frontier FT-NIR/MIR, PerkinElmer, USA). The X-ray diffraction (XRD) patterns of the materials were acquired with ARL, Equinox (Thermo Scientific) diffractometer running at 40 kV and 30 mA using a Cu Kα1 (λ = 1.5406 Å) source. The CHI660 (USA) instrument was used to perform all electrochemical analysis, and glassy carbon (CHI104, Φ = 3 mm), Ag/AgCl/KCl (sat.) (CHI111) and spiral Pt wire (BAS Inc, 23 cm) were used as the working electrode, reference and counter electrodes, respectively.
2.3 Recovery of graphite and Zn-metal from waste dry cell
Graphite rods and zinc metal were obtained from abandoned dry cells obtained from residential houses. Scheme 1 shows the schematic diagram of the recovery of graphite and Zn from the wasted dry cell and the synthesis of rGO/ZnO nanohybrid. To maintain the integrity of the graphite rods and Zn shell, the dry cells were carefully disassembled. Then, in order to remove the adherent paste made of MnO2, NH4Cl, carbon powder, etc., the collected graphite rods and Zn shells were cleaned with DI water. The extraction of graphite powder from the collected graphite rods was described in detail in our previous publications.42–44 For the Zn metal, sandpaper etching and thorough washing with Triton-X-100 surfactant were performed to remove any adherent impurities, resulting in a shining Zn metal, as shown in Scheme 1.
 |
| | Scheme 1 Synthesis of rGO/ZnO utilizing graphite and Zn collected from wasted dry cells. | |
2.4 Synthesis of GO
GO was prepared from the obtained graphite using the modified Hummers' method. Briefly, a solution of H2SO4 and H3PO4 was mixed with KMnO4 and graphite powder, heated to 50 °C for six hours, cooled, and then gradually DI water and H2O2 were added while stirring continuously at almost 4 °C. After the reaction was halted, the product was separated using centrifugation and then underwent a washing process. The GO powder was obtained after drying the freshly prepared GO.
2.5 Synthesis of rGO/ZnO nanohybrid
100 mg of GO was suspended thoroughly in 100 mL of DI water using an ultrasonic bath for about 30 minutes at room temperature. In a separate beaker, 25 mg of Zn metal was completely dissolved in a sufficient amount of concentrated HCl (eqn (1)), and then 1 M NaOH was added dropwise until Zn(OH)2 was completely precipitated (eqn (2)). The contents from the two beakers were mixed thoroughly.| | |
Zn (s) + 2HCl (aq) = ZnCl2 (aq) + H2 (g)
| (1) |
| | |
ZnCl2 (aq) + 2NaOH (aq) = Zn(OH)2 (s)↓ + 2NaCl (aq)
| (2) |
To reduce GO, 10 mL of 1 M N2H4·H2O was slowly added. The entire mixture was stirred at 80 °C for 2 hours in an oil bath at 400 rpm. The resulting nanohybrid was separated through centrifugation and washed with DI water several times until a pH of 7 was reached. Finally, the mixture was washed with ethanol to remove any organic residue present. The prepared nanohybrid was dried at 80 °C for 24 hours in a vacuum drying oven.
2.6 Fabrication of the rGO/ZnO modified GCE
The glassy carbon electrode (GCE) (CHI104, Φ = 3 mm) underwent meticulous polishing using a polishing microcloth and an aqueous slurry containing fine alumina particles. After 5 minutes of ultrasonication in DI water, any remaining alumina particle was extracted from the GCE. Sonication was employed to dissolve 1 mg mL−1 of active materials in DI water for a duration of 30 minutes in order to modify the GCE. Drop-casting the homogeneous suspension onto the GCE and allowing it to dry at room temperature resulted in the formation of a film composed of uniform active materials (1.13 mg cm−2). The modified electrode was utilized in subsequent electrochemical analyses. Before each electrochemical experiment, the electrolyte was saturated with Ar gas to remove dissolved oxygen, ensuring the highest standard of experimental consistency. Although oxygen does not interfere within the specific potential window studied, this step eliminates any possibility of unforeseen interactions between oxygen and BPA.
3 Results and discussion
We synthesized rGO/ZnO nanohybrid from the wasted batteries and used it in the electrochemical BPA sensors. The thorough characterization of the prepared materials, as well as their electrochemical performance, are discussed.
3.1 Characterization
The prepared rGO/ZnO nanohybrid was characterized using various techniques, including SEM, EDX, CHNS, EDX, FTIR, and XRD, as described in the following subsections.
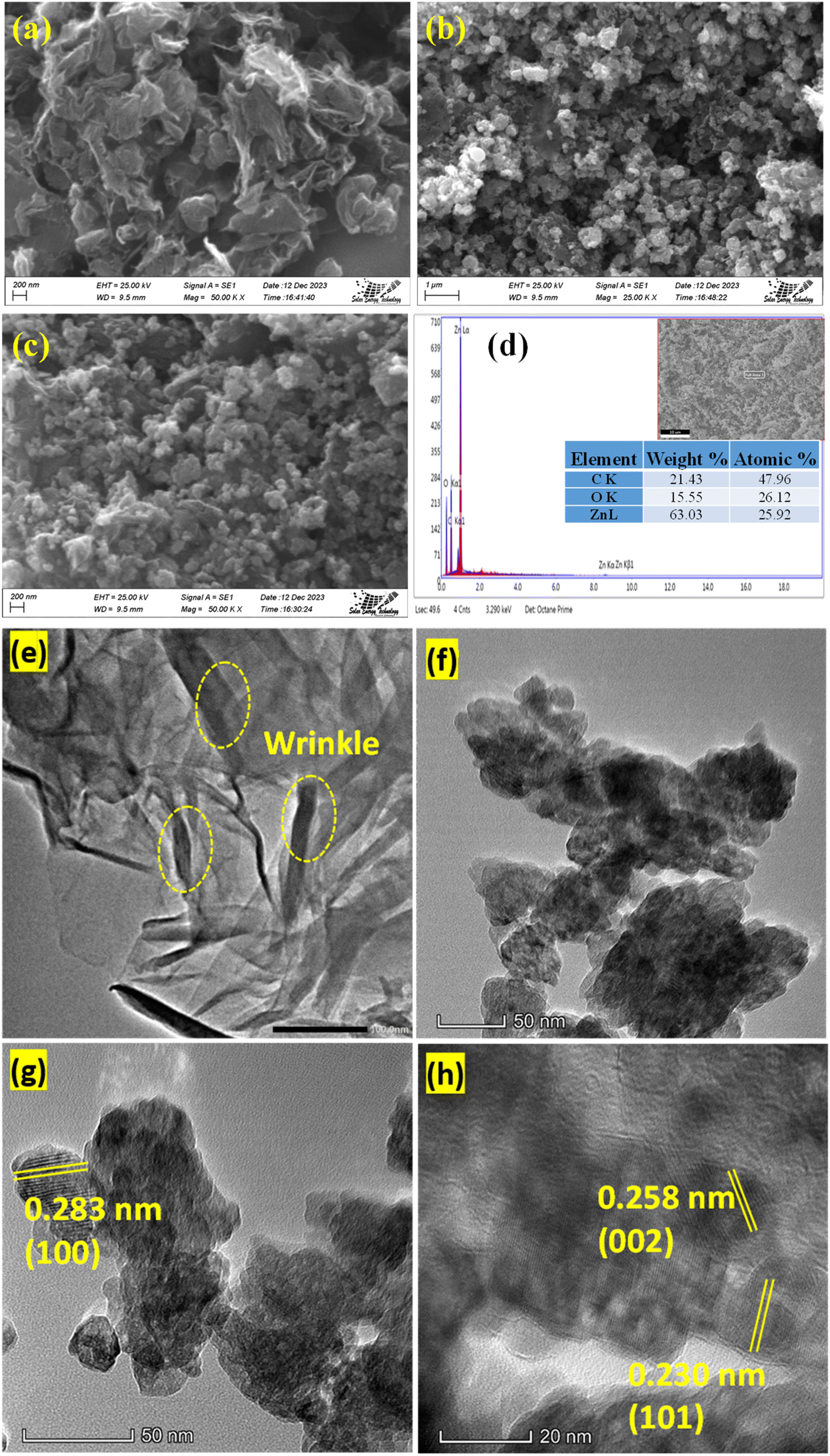
3.1.1 Physical morphology and elemental analysis. The morphology and microstructure of the prepared rGO and rGO/ZnO nanohybrid were determined by analyzing the SEM images. The SEM images of rGO, shown in Fig. 1a, elucidate the morphology of rGO, revealing a network of thin, sheet-like structures with distinct wrinkles and folds.40 These characteristics signify a substantial surface area, a pivotal attribute for electrochemical applications due to the abundance of surface available for interaction.45 Fig. 1b and c show the SEM images of the rGO/ZnO nanocomposite at two different magnifications. These images unveil ZnO nanoparticles intricately attached to the surface of the rGO sheets. The morphology displays ZnO nanoparticles in spherical form, uniformly dispersed across the rGO surface. This distribution effectively mitigates ZnO nanoparticle agglomeration while concurrently reducing their individual particle sizes. Such a morphology optimizes the utilization of numerous active sites present on both ZnO and rGO components, facilitating enhanced electrochemical sensing capabilities in ZnO/rGO nanocomposite-based sensors. Fig. 1d shows the EDX spectrum of the rGO/ZnO nanohybrid indicating the elemental composition within the material. Analysis of weight and atomic percent data reveals substantial amounts of carbon (C) at 21.43% (with an atomic percent of 47.96%), oxygen (O) at 15.55% (with an atomic percent of 26.12%), and zinc (Zn) at 63.03% (with an atomic percent of 25.92%). These results indicate the successful integration of ZnO into the rGO matrix, with graphene sheet structures abundant in carbon, likely accompanied by oxygen-containing functional groups on the surface, and zinc predominating the weight of the nanohybrid. Furthermore, CHNS elemental analysis shows that GO and rGO exhibit C-to-H ratios (C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H) of 27.3:1 and 49.5:1, respectively. The higher C:H in rGO suggests an effective reduction process that eliminates certain oxygen-containing functional groups inherent in GO, which was also evidenced in the FTIR spectrum (Fig. 2a) and XRD pattern (Fig. 2b).
:H) of 27.3:1 and 49.5:1, respectively. The higher C:H in rGO suggests an effective reduction process that eliminates certain oxygen-containing functional groups inherent in GO, which was also evidenced in the FTIR spectrum (Fig. 2a) and XRD pattern (Fig. 2b).
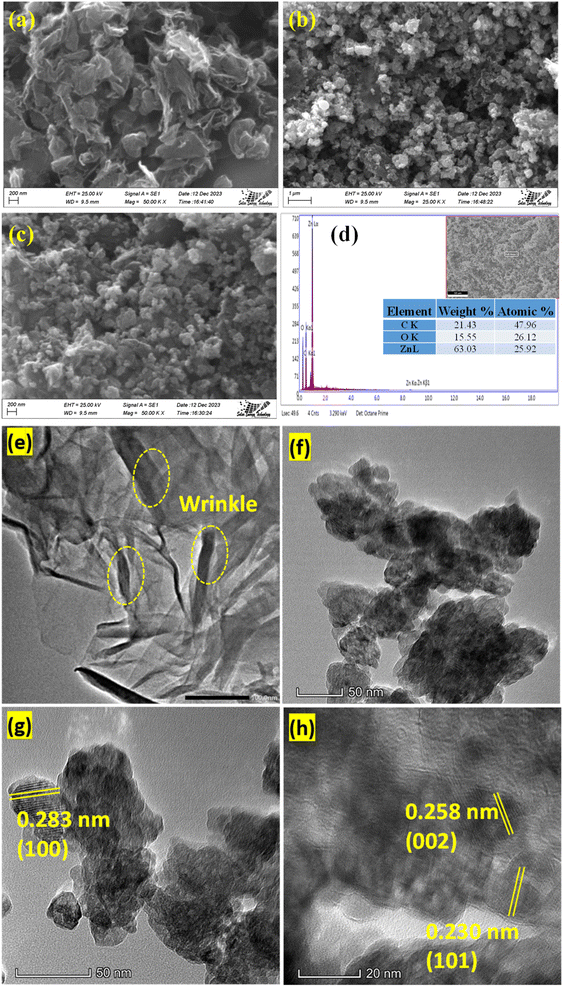 |
| | Fig. 1 SEM images of (a) rGO and (b and c) rGO/ZnO. (d) EDX spectrum of rGO/ZnO; TEM images of (e) rGO and (f–h) rGO/ZnO at different magnifications. | |
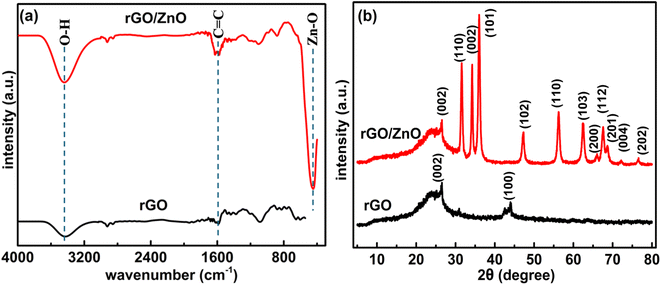 |
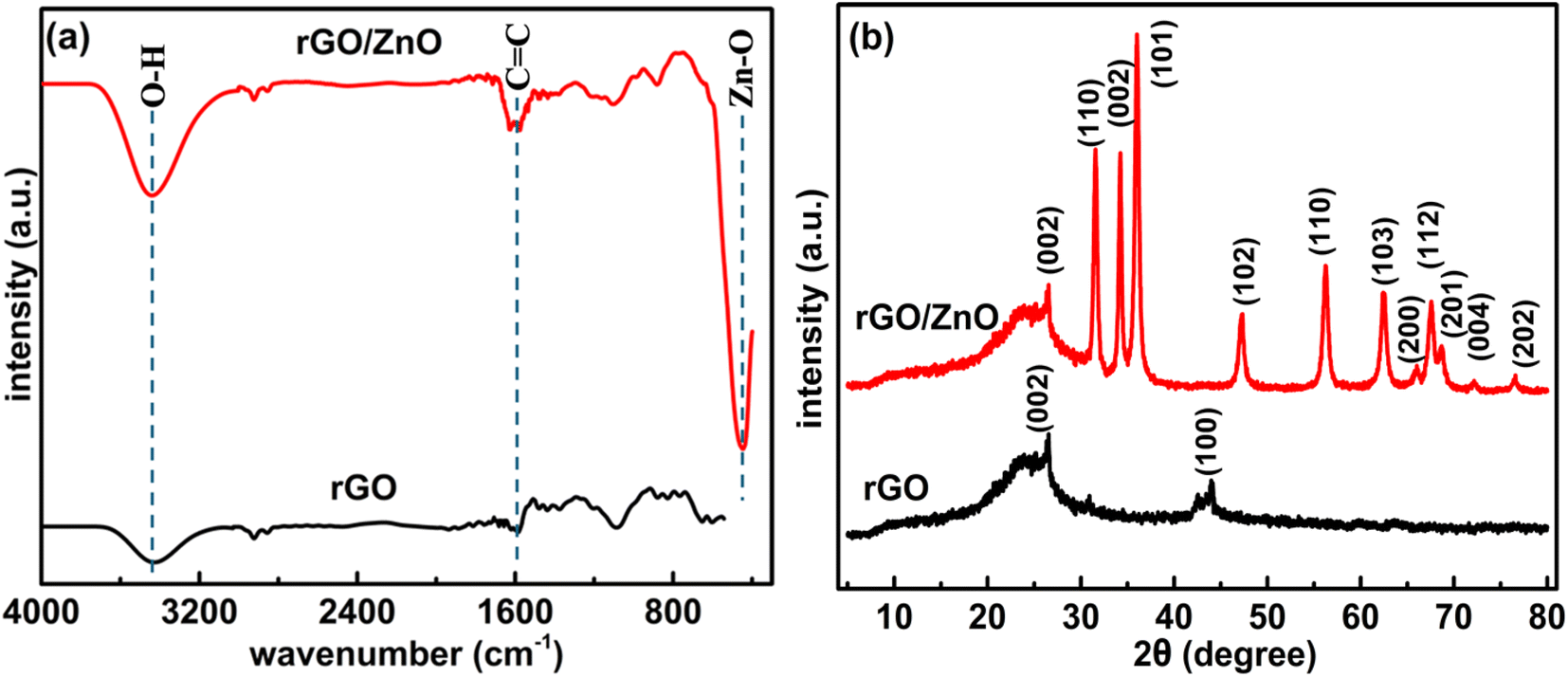
| | Fig. 2 (a) FTIR spectra and (b) XRD patterns of rGO and rGO/ZnO nanohybrids. | |
To obtain further insights into the nanostructure morphology, ZnO particle size, dispersion, and lattice structure, and TEM images were analyzed. TEM images in Fig. 1e–h depict the unique surface characteristics of rGO and rGO/ZnO.40,45 Fig. 1e illustrates the layered planar structure with a wrinkled surface of rGO as indicated by a yellow-colored circle, consistent with SEM images and previous study.43 Fig. 1f–h shows the TEM images of rGO/ZnO at different magnifications. The TEM images revealed that the ZnO nanoparticles were well integrated into the rGO nanosheet with minimal agglomeration. The average diameter of the ZnO nanoparticle was estimated to be ca. 35 nm analyzed by ImageJ software (64 bit). Furthermore, the lattice fringes of rGO and ZnO with the spacings of 0.283, 0.258 and 0.230 nm correspond to the (100), (002) and (101) crystal planes, respectively, of ZnO, which were visible in the TEM images (Fig. 1g and h).43,46–50 The presence of these planes was also supported by the XRD pattern (Fig. 2b).
3.1.2 Functional group analysis. Surface functional groups of the synthesized rGO and rGO/ZnO nanohybrid were characterized using FTIR spectra, as depicted in Fig. 2a. The FTIR analysis of rGO revealed low-intensity peaks within the 2000–1000 cm−1 range (Fig. 2a), indicating the reduction of most oxygen-containing functional groups present in GO.42,43,51,52 The peak at 2937 cm−1 suggests the reduction of carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) to methylene (–CH2–) groups. Specifically, this transition reflects the change from the stretching frequency of the carbonyl group, typically observed around 1700–1750 cm−1, to the stretching frequency of the methylene group, which appears around 2850–2950 cm−1. The appearance of the peak at 2937 cm−1 indicates the successful reduction of CO groups, characteristic of the carbonyl compounds, to –CH2– groups, a key step in the reduction process. Peaks observed around 3405 and 1620 cm−1 in rGO correspond to –OH groups on the basal plane of graphene sheets and CC aromatic bonds, respectively.43 The FTIR spectrum of rGO/ZnO as shown in Fig. 2a displays distinct peaks associated with rGO as mentioned and the peaks corresponding to ZnO, where the peaks at 875 and 435 cm−1 are attributed to the bending and stretching of the Zn–O bond, respectively, within ZnONPs.46,47,51–54 This observation provides compelling evidence for the successful formation of ZnONPs. Furthermore, peaks at 3405 and 1615 cm−1 confirm the presence of –OH groups on the surface of ZnONPs.52 The confirmation of ZnONP formation was also substantiated through XRD analysis (Section 3.1.3) and morphological and elemental analyses (Section 3.1.1). The presence of peaks associated with both rGO and ZnO in the FTIR spectrum of rGO/ZnO further supports the successful formation of the rGO/ZnO nanohybrid.
O) to methylene (–CH2–) groups. Specifically, this transition reflects the change from the stretching frequency of the carbonyl group, typically observed around 1700–1750 cm−1, to the stretching frequency of the methylene group, which appears around 2850–2950 cm−1. The appearance of the peak at 2937 cm−1 indicates the successful reduction of CO groups, characteristic of the carbonyl compounds, to –CH2– groups, a key step in the reduction process. Peaks observed around 3405 and 1620 cm−1 in rGO correspond to –OH groups on the basal plane of graphene sheets and CC aromatic bonds, respectively.43 The FTIR spectrum of rGO/ZnO as shown in Fig. 2a displays distinct peaks associated with rGO as mentioned and the peaks corresponding to ZnO, where the peaks at 875 and 435 cm−1 are attributed to the bending and stretching of the Zn–O bond, respectively, within ZnONPs.46,47,51–54 This observation provides compelling evidence for the successful formation of ZnONPs. Furthermore, peaks at 3405 and 1615 cm−1 confirm the presence of –OH groups on the surface of ZnONPs.52 The confirmation of ZnONP formation was also substantiated through XRD analysis (Section 3.1.3) and morphological and elemental analyses (Section 3.1.1). The presence of peaks associated with both rGO and ZnO in the FTIR spectrum of rGO/ZnO further supports the successful formation of the rGO/ZnO nanohybrid.
3.1.3 X-ray diffraction. Fig. 2b depicts the XRD patterns of both the synthesized rGO and rGO/ZnO nanohybrid. In the XRD pattern of rGO, illustrated in Fig. 2b, a prominent peak at 2θ = 26.2° and a smaller peak at 2θ = 44.3° were observed, corresponding to the (002) and (100) planes present in rGO, respectively.55 The (002) plane is indicative of graphitic carbon, while the (100) plane corresponds to the in-plane ordering of carbon atoms within the graphene layers of rGO. Notably, the absence of a peak around 2θ = 10–12°, which would correspond to oxygen-containing functional groups such as hydroxyls, epoxides, and carboxyl groups between the graphene layers in GO, indicates the successful reduction of GO to rGO.43 This inference is also supported by the analysis of functional groups (Fig. 2a) as well as morphological and elemental analysis (Section 3.1.1). The XRD pattern of the rGO/ZnO nanohybrid, as depicted in Fig. 2b, reveals distinct diffraction peaks of ZnO at 2θ values of 31.90°, 34.57°, 36.41°, 47.70°, 56.76°, 62.97°, 66.58°, 68.07°, 69.21°, 72.75°, 76.98°, and 81.51°, corresponding to the lattice planes of (100), (002), (101), (102), (110), (103), (200), (112), (201), (004), (202), and (104), respectively.48–54 The narrow and strong diffraction peaks, particularly for (100), (002), and (101), signify the high crystallinity of ZnO, consistent with SEM and TEM analyses. Notably, no additional diffraction peaks were observed, indicating the purity of the sample. The average crystallized size (d) of the ZnONPs was calculated using Debye–Scherrer's eqn (3).| |
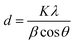 | (3) |
where, K = 0.9, λ = 0.15406 nm for CuKα, β is the FWHM (the line width at half-maximum height) and θ is the diffraction angle. The average d of the ZnONPs, calculated from the width of (101) peak using the eqn (3), was found to be ca. 34 nm. This finding is aligned with the TEM analysis. Furthermore, the presence of a peak at 26.2°, associated with the (002) planes of rGO, confirms the successful integration of rGO with ZnO, forming the rGO/ZnO nanohybrid. However, in the XRD pattern of the rGO/ZnO nanohybrid, the peak at 2θ = 44.3°, present in the XRD pattern of rGO, is absent. This absence could be attributed to an interaction between ZnO and rGO, possibly resulting in deformation or dislocation of the (100) planes of rGO. This interaction is also evident in the FTIR spectrum (Fig. 2a).
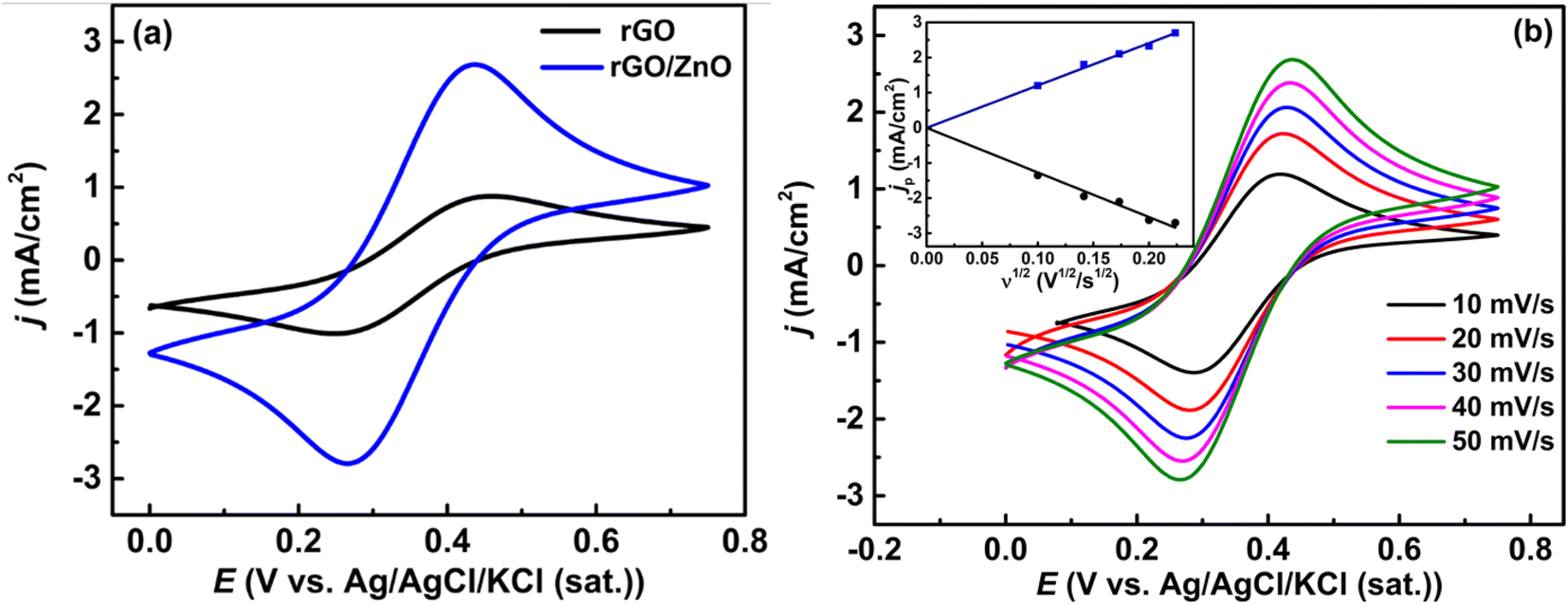
3.1.4 Electrochemical analysis. The redox activity and electrochemical surface area of the prepared materials were investigated by cyclic voltammetric studies. Fig. 3a shows the cyclic voltammograms (CVs) at rGO and rGO/ZnO-modified GCE in a 5 mM [Fe(CN)6]3−/[Fe(CN)6]4− solution containing 0.1 M KCl in the potential range of 0 to + 0.5 V with a scan rate (ν) of 50 mV s−1. The increased current density (j) at rGO/ZnO-modified GCE compared to rGO-modified GCE indicated enhanced catalytic activity of rGO/ZnO-modified GCE. Additionally, the ratio of the cathodic to anodic peak current density (jpc/jpa) for rGO, and rGO/ZnO was 1.03 and 1.07, respectively, showing that the Fe2+/Fe3+ couples were undergoing reversible electron transfer.56 It is seen clearly from the ν dependent CVs of the rGO/ZnO modified electrode (Fig. 3b) that both peak current densities (jp) increase as the ν increases. The plot of jp vs. v1/2 as presented in the inset of Fig. 3b suggests that the redox kinetics of the Fe2+/Fe3+ pair are diffusion controlled.43,56
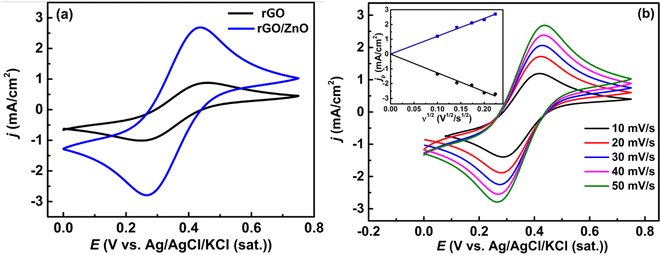 |
| | Fig. 3 (a) CVs obtained at the rGO and rGO/ZnO-modified GCE at a ν of 50 mV s−1 and (b) CVs obtained at rGO/Ag at different ν values (inset: plot of the variation in anodic and cathodic jp as a function of ν1/2) in a 5 mM [Fe(CN)6]3−/[Fe(CN)6]4− solution containing 0.1 M KCl. | |
The electrochemically active surface area of the rGO/ZnO nanohybrid modified GCE was determined by calculating the slope of the ip vs. v1/2 plot, depicted in the inset of Fig. 3b, and employing the Randles–Sevcik eqn (4).
| |
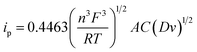 | (4) |
where,
n = number of electrons transferred (1),
F = Faraday's constant (96
485.339 C mol
−1),
R = universal gas constant (8.31447 J K
−1 mol
−1),
T = absolute temperature (298 K),
A = surface area of electrode (cm
2),
C = molar concentration of redox-active species (5 mmol L
−1), and
D = diffusion coefficient (0.06 cm
2 s
−1). The surface area of rGO/ZnO nanohybrid modified GCE was measured at
ca. 0.103 cm
2, representing a 45% increase compared to that of the bare GCE (0.071 cm
2). This finding implies a notable augmentation in the surface area, attributable to the presence of the rGO/ZnO nanohybrid. The improved electrocatalytic activity and high surface area will greatly benefit the electrochemical sensing of BPA in aqueous media.
38,40,43
3.2 BPA sensing application
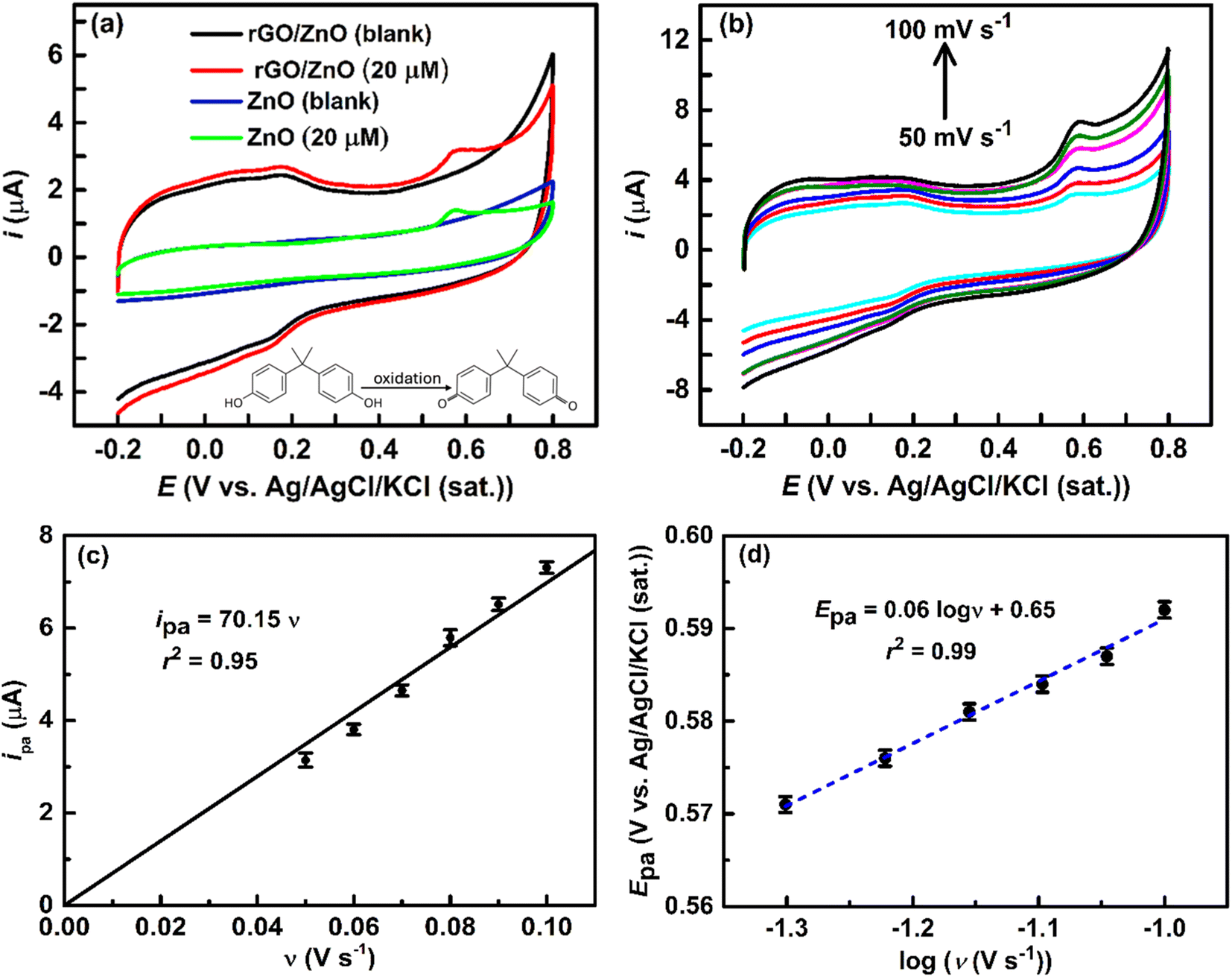
3.2.1 Electrochemical behavior of rGO/ZnO modified GCE towards BPA oxidation. The sensing performance of an electrode is determined by its electrochemical redox capability with the target analyte (i.e., BPA) at a desired rate. The high surface area of rGO, together with its capacity to absorb BPA through a variety of interactions, including hydrogen bonding and π–π interactions,2,57 and the existence of active sites for the electrochemical oxidation of BPA on the surface of ZnO,58 can be utilized to improve the sensing performance of rGO/ZnO-modified GCE. Fig. 4a shows the comparison of CVs of 20 μM BPA in an Ar-saturated 0.1 M PBS solution (pH = 7) at ZnO and rGO/ZnO-modified GCE with a ν of 50 mV s−1. In the absence of BPA, the ZnO and rGO/ZnO-modified GCE did not show a redox response in the PBS solution. On the other hand, in the presence of 20 μM BPA, the ZnO and rGO/ZnO-modified GCE showed anodic peak currents (ipa) (oxidation) of 1.4 and 3.2 μA at an anodic peak potential (Epa) of 0.58 V, respectively. The ipa at rGO/ZnO-modified GCE was nearly 2.3 times higher than at ZnO-modified GCE, on the other hand, no such ipa was observed at rGO-modified GCE (data not given). The substantial increase in the ipa at the rGO/ZnO-modified GCE could be attributed to its large surface area (45% increased), excellent oxidation capability as shown in the catalytic activity of iron couples, and improved interaction between the electrode surface and BPA molecules, resulting in higher BPA accumulation on the electrode surface. In contrast, pure metal oxide has a low surface adsorption capacity for BPA, which slows down its total reaction rate.59 After removing background currents from the CVs, the peak currents for BPA oxidation at the ZnO and rGO/ZnO-modified electrodes were found to be 0.35 and 0.61 μA cm−2, respectively. This result further confirms the enhanced performance of the rGO/ZnO electrode compared to ZnO. However, in the case of the rGO/ZnO-modified electrode, distinct oxidation–reduction peaks at lower potential were observed both in the absence and presence of BPA (Fig. 4a). This distinctive CV pattern has been previously reported by Shi et al. for Cu2O/rGO-modified electrodes in a PBS solution.60 The separation of these peak potentials is ∼0.03 V, suggesting a highly reversible redox process occurring on the electrode surface—recognized as surface redox reactions—a phenomenon commonly associated with electrodes modified with graphene-based materials.61 The presence of oxygen-containing functional groups on the rGO/ZnO modified GCE surface, as evidenced by FTIR spectra (Fig. 2a) and XRD patterns (Fig. 2b), facilitates the participation of these groups in the redox process, thereby contributing to the observed characteristic CVs. However, no reduction peak was observed associated with BPA or its oxidized form on these modified electrodes, as shown in Fig. 4a, indicating an irreversible electrochemical redox reaction at these modified electrodes. Moreover, irreversible reactions or electrode surface fouling frequently cause cyclic voltammetry loops to stay open, impeding the reverse process. This observation aligns with prior reports,3,59,60 emphasizing the propensity of the modified electrodes to favor an irreversible electrochemical pathway for the electrochemical process of BPA. Moreover, we conducted 10 sequential cyclic voltammetric experiments at the same conditions mentioned in Fig. 4a. The results showed that the CV shape remained consistent across all measurements, with only a 3.7% decrease in peak current after the 10th cycle. This indicates that the electrode is robust and reliable for BPA sensing.
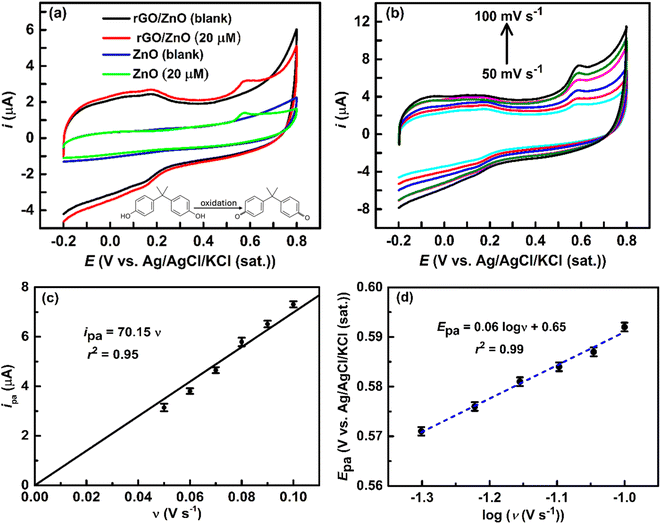 |
| | Fig. 4 (a) CVs at the ZnO and rGO/ZnO-modified GCE in the absence and presence of 20 μM BPA in an Ar-saturated 0.1 M PBS solution at a ν of 50 mV s−1. (b) CVs at the rGO/ZnO-modified GCE in the presence of 20 μM BPA in an Ar-saturated 0.1 M PBS solution at different ν. A plot of ipa vs. ν (c) and Epa vs. logν (d) was obtained from (b) (error bar indicates the standard deviations of the three replicate experiments). | |
3.2.2 Kinetics of BPA oxidation at the rGO/ZnO electrode. The charge transfer characteristics provide vital information about the electrochemical sensing mechanism, and the number of electron transfers at the rate-determining step (RDS) is a key performance indicator for the electrochemical oxidization of BPA. An understanding of this aspect can be achieved through the analysis of ν dependent ipa and Epa of BPA for the rGO/ZnO-modified GCE electrode. Fig. 4b shows CVs at different ν (i.e., 50, 60, 70, 80 and 100 mV s−1) for 20 μM BPA in Ar-saturated 0.1 M PBS solution at rGO/ZnO-modified GCE. The CVs indicate an increase in ipa and a shift of Epa towards more positive values with higher ν, consistent with the findings of other researchers.3,56,59,60 Generally, increasing the ν typically leads to higher currents. As the ν increases, the diffusion layer thickness reduces, resulting in increased peak and background currents.62 Fig. 4c shows a linear relationship of ipa and ν (ipa = 70.15 ν, coefficient of determination, r2 = 0.95). The linear relationships of ipa and ν indicate that the electrochemical process involved with BPA and the surface of rGO/ZnO is an adsorption-controlled process as aligned with the previous report.56 Moreover, there is a linear relation between the Epa and log of ν (logν) as shown in Fig. 4d, and expressed by the eqn (5).| | |
Epa = 0.06logν + 0.65
| (5) |
For an irreversible faradaic process, the Epa is related to the logν following eqn (6).63
| |
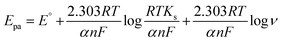 | (6) |
where,
E°,
α (0.5 for an irreversible electrode process),
n and
Ks are formal redox potential, charge transfer coefficient, number of electrons involved in the rate-determining step (RDS) and standard rate constant of the reaction, respectively. The other quantities are the same as mentioned above. Comparing
eqn (5) and
(6), the value of
n was calculated to be 1.17, which is close to 1 as reported in a previous study.
60 This indicates that the RDS of the oxidation process of BPA involved one electron at rGO/ZnO-modified GCE in an Ar-saturated 0.1 M PBS solution.
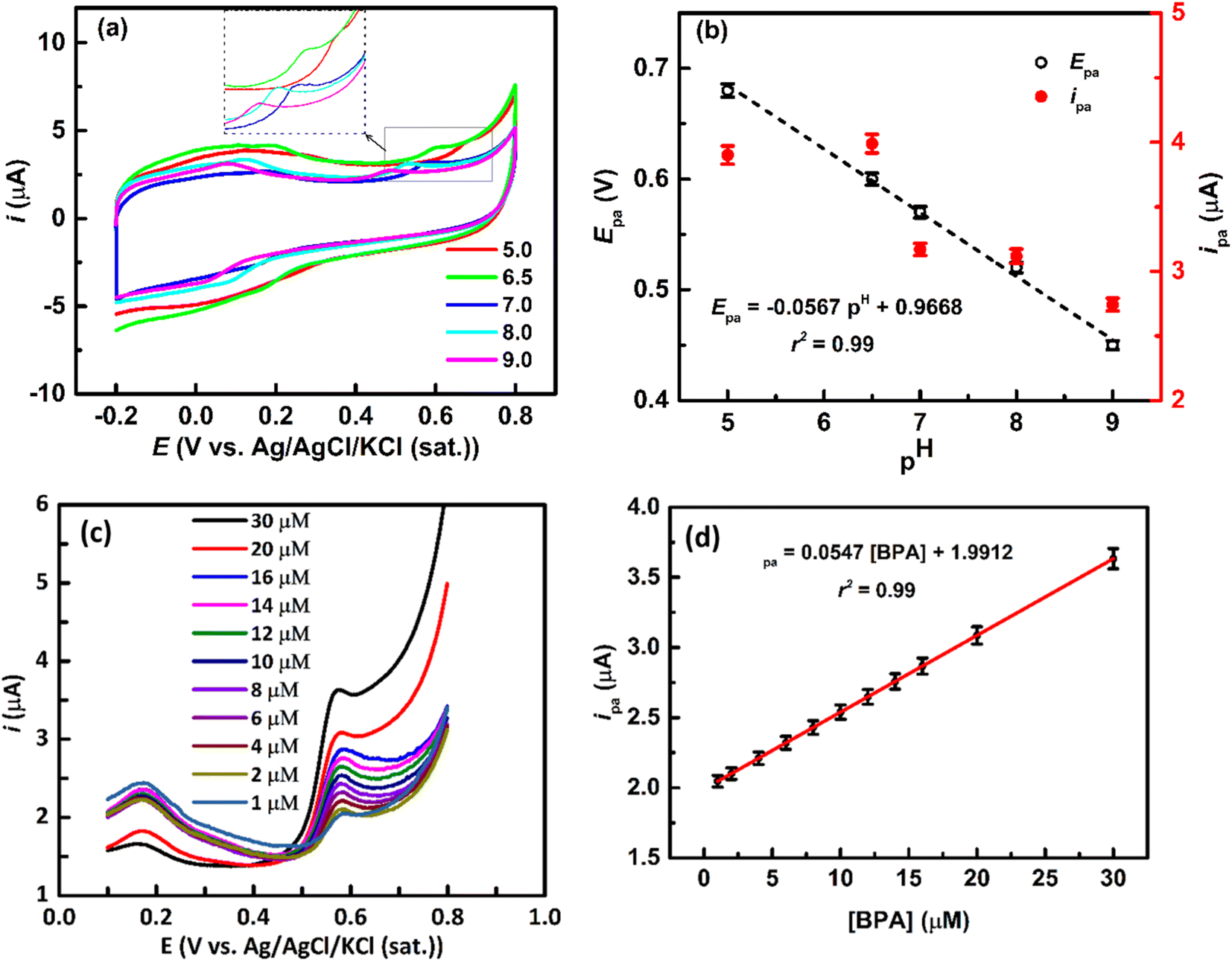
The pH of the solution is a critical factor influencing the electrochemical oxidation of BPA, impacting both Epa and ipa. Generally, the Epa shifts towards more positive and negative directions with lower and higher pH, respectively.3,56,59
Fig. 5a shows that CVs at the rGO/ZnO-modified GCE in a 20 μM BPA solution of various pH, revealing a negative shift in Epa with increasing pH, coupled with an increase in ipa at lower pH (Fig. 5b). These observations align with previous reports3,56 and can be rationalized by considering the pH-dependent ionization state of BPA and its adsorption interactions with the surface of rGO/ZnO. The phenolic –OH groups of BPA undergo pH-dependent ionization; in acidic conditions, they become fully protonated (BPA + 2H+ → BPAH2+), while in basic conditions, they become fully deprotonated (BPA → BPA2− + 2H+). These ionization states such as BPAH2+, BPA0 and BPAH2−, influence the redox properties of BPA, affecting its electrochemical Epa. Additionally, the pH of the solution affects the ionization of functional groups on the rGO/ZnO surface, altering the nature of the surface charge. Changes in pH can thus modify electrostatic interactions between charged surface groups of rGO and BPA.2,57 Even, at higher pH, metal ions on metal oxide may form hydroxide complexes, potentially influencing BPA adsorption.59 Therefore, the decrease in ipa at higher pH values may be attributed to electrostatic repulsion between negatively charged BPA2− and the rGO/ZnO surface. However, a linear relationship (r2 = 0.99) is observed between Epa and the pH as shown in Fig. 5b following the eqn (7).
| | |
Epa = −0.0567pH + 0.9919
| (7) |
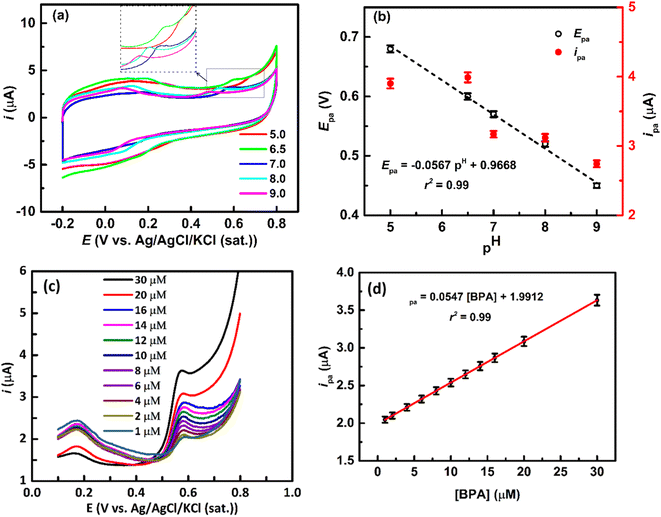 |
| | Fig. 5 (a) CVs at the rGO/ZnO-modified GCE in the presence of 20 μM BPA in an Ar-saturated 0.1 M PBS solution at different pH values at a ν of 50 mV s−1. (b) Plot of Epa vs. pH and ipa vs. pH was obtained from (a). (c) LSVs at the rGO/ZnO-modified GCE at different concentrations of BPA solution in an Ar-saturated 0.1 M PBS solution at a ν of 50 mV s−1. (d) A plot of ipa vs. concentrations of BPA was obtained from (c) (the error bar indicates the standard deviations of the three replicate experiments). | |
The ratio of proton to electron transfer in the oxidation of BPA can be calculated by comparing eqn (7) and (8).
| |
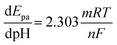 | (8) |
where,
m and
n are the number of protons and electrons, respectively. The other quantities are the same as mentioned above. The
m/
n calculated to be 0.96 for the oxidation of BPA, which is close to 1, indicating that the number of protons and electrons involved in the oxidation of BPA were equal, signifying a one-electron-one-proton electrochemical process.
60
It has been observed that there is no significant increase in the ipa, even after a 30 minute of the accumulation time, during the oxidation of BPA (data not provided) and BPA is a non-polar organic compound, therefore, its accumulation would not be affected by applying a potential (known as accumulation potential).3 The lack of a need for accumulation time can be ascribed to the ultrafast adsorption64 capability of rGO/ZnO towards BPA.65 This indicates a rapid sensing process for BPA in aqueous media employing rGO/ZnO-modified electrodes, which is crucial for practical applications.
3.2.3 Quantitative determination of BPA. Fig. 5c depicts the linear sweep voltammograms (LSVs) for BPA concentrations ranging from 1 to 30 μM at the rGO/Zn-modified GCE in an Ar-saturated 0.1 M PBS solution of pH = 7 at a ν of 50 mV s−1. The peaks at 0.58 V in the LSVs are responsible for the oxidation of BPA, as seen in the CVs (Fig. 4a), which is further confirmed by these LSVs as the ipa increases with increasing concentrations of BPA ([BPA]), following a linear relationship (calibration line) as depicted in Fig. 5d. The linear equation of the calibration line is ipa = 0.0547 [BPA] + 1.9912 with an r2 = 0.99. The limit of detection (LOD), defined as LOD = 3S/m (where, S is the standard deviation of the blank solution and m is the slope of the calibration curve), is estimated to be 0.98 nM and the sensitivity is calculated to be 0.055 μA μM−1. The LOD is below the predicted-no-effect-concentration concentration of BPA in water for aquatic organisms.3 It is observed that the rGO/ZnO nanohybrid-modified electrode has achieved a low LOD and excellent sensitivity for the detection of BPA. The improved electrochemical detection performance of BPA at the rGO/ZnO nanohybrid fabricated electrode can be attributed to the highly effective surface area of the modified electrode and enhanced adsorption interactions, i.e., hydrogen bonding, π–π interactions, Lewis acid–base interactions, between BPA and the surface of the modified electrode. Xu et al. reported that graphene shows the highest BPA adsorption capacity (182 mg g−1) among various carbonaceous materials.2 Additionally, Wang et al. found that BPA can form hydrogen bonding and π–π interactions with GO, along with a Lewis acid–base interaction between anionic BPA and Fe2O3.57 Table 1 presents a comparison of ZnO and/or graphene-based electrodes for detecting BPA in water, revealing that our approach of synthesizing rGO/ZnO from e-waste leads to a lower LOD for electrochemical BPA detection. Although certain alternative electrodes have demonstrated comparable or superior detection performance of BPA, the rGO/ZnO prepared in our study offers notable advantages, including its affordability, non-toxic nature, and straightforward preparation process. Additionally, the use of e-waste as the only source of precursors for the synthesis of rGO/ZnO nanohybrid not only contributes to cost-effectiveness but also addresses environmental concerns by managing and repurposing detrimental e-waste.
Table 1 Comparison of the ZnO and graphene-based electrode for the detection of BPA in watera
| Electrode |
Method |
Sensitivity μA μM−1 |
LOD μM |
Reference |
| BMO: barium oxide/molybdenum oxide; CTAB: cetyltrimethylammonium bromide; DPV: differential pulse voltammetry; ErGO: electrochemically reduced graphene oxide; MCPE: modified carbon paste electrode; MOF: metal organic framework; MXene: Ti3C2; NDC: 1,4-naphthalenedicarboxylate; SWV: square-wave voltammetry; UiO-66: Zr(IV) dicarboxylate; *: μA (log nM)−1 cm−2; **: μA (log μM)−1 cm−2. |
| CTAB/Au/ZnO/rGO |
DPV |
— |
0.00495 |
8 |
| NiO/ZnO/rGO |
DPV |
— |
0.004 |
11 |
| ZnO/GO/MCPE |
Voltammetric |
— |
2.760 |
19 |
| Gd@ZnO-MWCNTs |
DPV |
0.02633 |
0.2 |
9 |
| UiO-66-NDC/GO |
DPV |
— |
0.025 |
20 |
| Pt@SWCNTs-Mxene-rGO |
|
|
0.0028 |
12 |
| Ag@Fe3O4-rGO |
DPV |
— |
0.028 |
10 |
| BMO/rGO |
DPV |
— |
0.004 |
7 |
| ZnO/ZnCo2O4 |
DPV |
— |
0.01 |
24 |
| MWCNTs/Ag–ZnO |
Voltammetric |
— |
0.006 |
18 |
| ZnO/BC |
DPV |
0.092 |
0.1 |
13 |
| CuO/GO |
DPV |
|
0.093 |
14 |
| rGO/MoO3NPs |
DPV |
13.96* |
0.00012 |
27 |
| Ce-MOF-ErGO |
|
|
0.0019 |
15 |
| TiN-rGO |
|
|
0.00019 |
23 |
| AgNPs/rGO |
|
2.56** |
0.14 |
16 |
| I-rGO |
DPV |
— |
0.02 |
17 |
| rGO/ZnO |
Voltammetric |
0.055 |
0.00098 |
Present study |
3.2.4 Real water sample analysis. The practical utility of the rGO/ZnO electrode for the detection of BPA was evaluated by measuring the concentration of the BPA in real tap water samples. Tap water was utilized without further treatment to maintain the presence of ionic species that could interfere with BPA detection. The recovery studies were carried out utilizing the cyclic voltammetric technique and the standard addition method. In this study, tap water samples were spiked with a 2 μM BPA in 0.1 M PBS solution (pH = 7) to prepare different concentrations of BPA, as shown in Table 2. The background current, representing the current without any BPA addition, was corrected to accurately calculate the percentage recovery of BPA, and three consecutive experiments were conducted and recorded. Table 2 shows that the developed sensor recovered a reasonable amount of BPA (95–107%) using the simple cyclic voltammetric technique. These findings suggest that the proposed sensor is successful in detecting BPA in real water samples.
Table 2 Recovery of BPA from tap water using the rGO/ZnO-based electrochemical sensor
| Sample no. |
Concentration of BPA (μM) |
Recovery (%) |
| Added |
Found |
| 1 |
0.50 |
0.53 ± 0.02 |
106 |
| 2 |
1.00 |
1.03 ± 0.02 |
103 |
| 3 |
1.50 |
1.42 ± 0.01 |
95 |
| 4 |
2.50 |
2.68 ± 0.03 |
107 |
| 5 |
5.00 |
4.90 ± 0.02 |
98 |
4 Conclusions
A cheaper method is unveiled for the synthesis of ZnO nanoparticles using metallic Zn shells collected from the waste dry cells via the precipitation method. A simple, low-cost and sensitive electrode material for the electrochemical detection of BPA based on rGO/ZnO nanohybrid was fabricated. The fabricated electrode demonstrated excellent electrochemical sensing performance towards the detection of BPA with a limit of detection of 0.98 nM and sensitivity of 0.055 μA μM−1 in water, which is below the predicted-no-effect-concentration in water for aquatic organisms. Furthermore, the suggested sensor has the ability to detect BPA in real water samples. The oxidation of BPA at the electrode surface followed one electron reaction with enhanced sensitivity due to the highly effective surface area and enhanced interaction of the BPA molecules with the rGO/ZnO electrode. The low cost of the precursors for the synthesized rGO/ZnO nanohybrid, derived from discarded dry cell batteries, makes it a feasible solution for detecting BPA in economically challenged and resource-limited regions, while also contributing to the environmentally sustainable management of e-waste.
Data availability
Data will be made available on request.
Conflicts of interest
The authors declare no competing interest.
Acknowledgements
The authors are grateful to the Bangladesh Council of Scientific and Industrial Research for financial support (R&D ref. no. 39.02.0000.011.14.169.2023/877, date: 17.09.2023) and facilities. The assistance from IERD, CARF, and BAEC F&PRD to perform SEM, XRD, TEM, and FTIR is appreciated.
References
- F. Liguori, C. Moreno-Marrodan and P. Barbaro, Chem. Soc. Rev., 2020, 49, 6329–6363 RSC.
- J. Xu, L. Wang and Y. Zhu, Langmuir, 2012, 28, 8418–8425 CrossRef CAS PubMed.
- A. U. Alam and M. J. Deen, Anal. Chem., 2020, 92, 5532–5539 CrossRef CAS.
- S. Almeida, A. Raposo, M. Almeida-González and C. Carrascosa, Compr. Rev. Food Sci. Food Saf., 2018, 17, 1503–1517 CrossRef PubMed.
- K. V. Ragavan, N. K. Rastogi and M. S. Thakur, TrAC, Trends Anal. Chem., 2013, 52, 248–260 CrossRef CAS.
- F. Sun, L. Kang, X. Xiang, H. Li, X. Luo, R. Luo, C. Lu and X. Peng, Anal. Bioanal. Chem., 2016, 408, 6913–6927 CrossRef CAS PubMed.
- R. Sundaresan, V. Mariyappan, T.-W. Chen, S.-M. Chen, M. Akilarasan, M. A. Alsaigh, M. A. Ali, M. S. Elshikh and J. Yu, Mater. Today Chem., 2023, 30, 101602 CrossRef CAS.
- A.-Y. Zha, Q.-B. Zha, Z. Li, H.-M. Zhang, X.-F. Ma, W. Xie and M.-S. Zhu, Rare Met., 2023, 42, 1274–1282 CrossRef CAS.
- S. Agrahari, A. K. Singh, R. K. Gautam and I. Tiwari, J. Appl. Electrochem., 2023, 53, 345–358 CrossRef CAS.
- M. Shen, W. Li, F. Chen, L. Chen, Y. Chen, S. Chen, S. Ren and D. Han, Microchem. J., 2023, 186, 108315 CrossRef CAS.
- J. A. Buledi, H. Shaikh, A. R. Solangi, A. Mallah, Z.-H. Shah, M. M. Khan, A. L. Sanati, H. Karimi-Maleh, C. Karaman, M. B. Camarada and D. E. Niculina, Ind. Eng. Chem. Res., 2023, 62, 4754–4764 CrossRef CAS.
- G. Qu, Y. Zhang, J. Zhou, H. Tang, W. Ji, Z. Yan, K. Pan and P. Ning, Chemosphere, 2023, 337, 139315 CrossRef CAS PubMed.
- J. Hu, D. Mao, P. Duan, K. Li, Y. Lin, X. Wang and Y. Piao, Chemosensors, 2022, 10, 163 CrossRef CAS.
- F. Ebrahimi-Tazangi, H. Beitollahi, H. Hekmatara and J. Seyed-Yazdi, J. Iran. Chem. Soc., 2021, 18, 191–199 CrossRef CAS.
- X. Wang, Y. Shi, J. Shan, H. Zhou and M. Li, Ionics, 2020, 26, 3135–3146 CrossRef CAS.
- D. Verma, T. K. Dhiman, M. D. Mukherjee and P. R. Solanki, J. Electrochem. Soc., 2021, 168, 097504 CrossRef CAS.
- K.-P. Wang, J.-M. Hu and X. Zhang, Microchem. J., 2022, 173, 107047 CrossRef CAS.
- K. Kunene, M. Sabela, S. Kanchi and K. Bisetty, Waste Biomass Valorization, 2020, 11, 1085–1096 CrossRef CAS.
- L. S. Manjunatha, B. E. K. Swamy, S. C. Sharma and C. Krithika, Mater. Today Commun., 2024, 38, 108012 CrossRef.
- T. S. Sunil Kumar Naik, S. Singh, P. N, R. Varshney, B. Uppara, J. Singh, N. A. Khan, L. Singh, M. Zulqarnain Arshad and P. C. Ramamurthy, Chemosphere, 2023, 311, 137104 CrossRef CAS PubMed.
- S. Yasmin, M. H. Kabir, N. Roy and S. Jeon, ECS Adv., 2023, 2, 024504 CrossRef CAS.
- S. Yasmin, N. Roy, M. H. Kabir and S. Jeon, Appl. Surf. Sci. Adv., 2022, 9, 100235 CrossRef.
- W. Xu, Y. Zhang, X. Yin, L. Zhang, Y. Cao, X. Ni and W. Huang, Anal. Bioanal. Chem., 2021, 413, 1081–1090 CrossRef CAS PubMed.
- M. Amiri and H. Mahmoudi-Moghaddam, Microchem. J., 2021, 160, 105663 CrossRef CAS.
- A. K. Mohiuddin, S. Yasmin and S. Jeon, Sens. Actuators, A, 2023, 355, 114314 CrossRef CAS.
- S. Yasmin, M. S. Ahmed and S. Jeon, J. Electrochem. Soc., 2015, 162, B363 CrossRef CAS.
- D. Verma, A. K. Yadav, M. D. Mukherjee and P. R. Solanki, J. Environ. Chem. Eng., 2021, 9, 105504 CrossRef CAS.
- N. Roy, S. Yasmin and S. Jeon, Microchem. J., 2020, 153, 104501 CrossRef CAS.
- Md. F. Ehsan, H. R. Barai, Md. M. Islam, Md. A. Bin Hasan Susan, S. W. Joo and M. S. Miran, Mater. Today Commun., 2023, 36, 106563 CrossRef CAS.
- M. Y. Pabel, M. F. Ehsan, M. S. Miran and M. M. Islam, Emerging Applications of Nanomaterials, 2023, vol. 141, pp. 101–123 Search PubMed.
- D. K. Sharma, S. Shukla, K. K. Sharma and V. Kumar, Mater. Today: Proc., 2022, 49, 3028–3035 CAS.
- Y.-H. Chiu, K.-D. Chang and Y.-J. Hsu, J. Mater. Chem. A, 2018, 6, 4286–4296 RSC.
- K. O. Sodeinde, S. O. Olusanya, O. S. Lawal, M. Sriariyanun and A. A. Adediran, Sci. Rep., 2022, 12, 17054 CrossRef CAS.
- S. Z. Hussain, M. Ihrar, S. B. Hussain, W. C. Oh and K. Ullah, SN Appl. Sci., 2020, 2, 764 CrossRef CAS.
- M. M. Islam, M. Y. A. Mollah, M. A. B. H. Susan and M. M. Islam, RSC adv., 2020, 10, 44884–44891 RSC.
- M. Galib, M. M. Hosen, J. K. Saha, Md. M. Islam, S. H. Firoz and Md. A. Rahman, J. Mol. Model., 2020, 26, 251 CrossRef CAS PubMed.
- A. Musa, M. M. Islam, M. A. B. H. Susan and M. M. Islam, ECS J. Solid State Sci. Technol., 2023, 12, 061005 CrossRef CAS.
- S. Yasmin, M. S. Ahmed, D. Park and S. Jeon, J. Electrochem. Soc., 2016, 163, B491 CrossRef CAS.
- S. Yasmin, M. S. Ahmed and S. Jeon, J. Nanosci. Nanotechnol., 2017, 17, 3959–3966 CrossRef CAS.
- S. Yasmin, Y. Joo and S. Jeon, Appl. Surf. Sci., 2017, 406, 226–234 CrossRef CAS.
- N. Roy, S. Yasmin, A. Ejaz, H. Soon Han and S. Jeon, Appl. Surf. Sci., 2020, 533, 147500 CrossRef CAS.
- Md. G. Azam, M. H. Kabir, Md. A. A. Shaikh, S. Ahmed, M. Mahmud and S. Yasmin, J. Water Process Eng., 2022, 46, 102597 CrossRef.
- Md. Yeasin Pabel, S. Yasmin, M. A. A. Shaikh and M. H. Kabir, Sens. Actuators, A, 2024, 366, 115028 CrossRef CAS.
- B. P. Upoma, S. Yasmin, Md. A. Ali Shaikh, T. Jahan, Md. A. Haque, M. Moniruzzaman and M. H. Kabir, ACS Omega, 2022, 7, 29655–29665 CrossRef CAS.
- S. Yasmin, S. Cho and S. Jeon, Appl. Surf. Sci., 2018, 434, 905–912 CrossRef CAS.
- S. Faisal, H. Jan, S. A. Shah, S. Shah, A. Khan, M. T. Akbar, M. Rizwan, F. Jan, Wajidullah, N. Akhtar, A. Khattak and S. Syed, ACS Omega, 2021, 6, 9709–9722 CrossRef CAS PubMed.
- Z. Wang, M. R. Bockstaller and K. Matyjaszewski, ACS mater. lett., 2021, 3, 599–621 CrossRef CAS.
- M. Zare, K. Namratha, S. Ilyas, A. Sultana, A. Hezam, S. L, M. A. Surmeneva, R. A. Surmenev, M. B. Nayan, S. Ramakrishna, S. Mathur and K. Byrappa, ACS Food Sci. Technol., 2022, 2, 763–781 CrossRef CAS.
- C. R. Mendes, G. Dilarri, C. F. Forsan, V. de, M. R. Sapata, P. R. M. Lopes, P. B. de Moraes, R. N. Montagnolli, H. Ferreira and E. D. Bidoia, Sci. Rep., 2022, 12, 2658 CrossRef CAS.
- Y. Khan, B. Singh, R. Ullah, M. Shoeb, A. Naqvi and S. Abidi, PLoS One, 2015, 10(7), e0133086 CrossRef.
- F. Yousefimehr, S. Jafarirad, R. Salehi and M. S. Zakerhamidi, Sci. Rep., 2021, 11, 11900 CrossRef CAS PubMed.
- F. A. Alharthi, A. A. Alsyahi, S. G. Alshammari, H. A. AL-Abdulkarim, A. AlFawaz and A. Alsalme, ACS Omega, 2022, 7, 2786–2797 CrossRef CAS PubMed.
- G. M. Abdelghani, A. B. Ahmed and A. B. Al-Zubaidi, Sci. Rep., 2022, 12, 20016 CrossRef CAS.
- S. Al-Ariki, N. A. A. Yahya, S. A. Al-A’nsi, M. H. H. Jumali, A. N. Jannah and R. Abd-Shukor, Sci. Rep., 2021, 11, 11948 CrossRef CAS.
- S. Yasmin, M. Shamsuddin Ahmed and S. Jeon, Int. J. Hydrogen Energy, 2017, 42, 1075–1084 CrossRef CAS.
- J. Huang, T. Zhang, G. Dong, S. Zhu, F. Yan and J. Liu, Front. Chem., 2022, 10, 900282 CrossRef CAS PubMed.
- Y. Wang, X. Wei, Y. Qi and H. Huang, Chemosphere, 2021, 263, 127563 CrossRef CAS PubMed.
- A. Qurashi, J. A. Rather, K. D. Wael, B. Merzougui, N. Tabet and M. Faiz, Analyst, 2013, 138, 4764–4768 RSC.
- D.-N. Pei, A.-Y. Zhang, X.-Q. Pan, Y. Si and H.-Q. Yu, Anal. Chem., 2018, 90, 3165–3173 CrossRef CAS PubMed.
- R. Shi, J. Liang, Z. Zhao, A. Liu and Y. Tian, Talanta, 2017, 169, 37–43 CrossRef CAS PubMed.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, 3rd edn, https://books.wiley.com/titles/9781119334064/, accessed January 25, 2024 Search PubMed.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 CrossRef CAS.
- M. Sohag Hossain, M. Humayun Kabir, M. A. A. Shaikh, M. Anamul Haque and S. Yasmin, RSC Adv., 2024, 14, 1431–1444 RSC.
- T. Dong, C. Ling, L. Fu, Y. Xue, Y. Pan, Y. Zhang and C. Zhu, J. Hazard. Mater., 2023, 445, 130562 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Md Yeasin Pabela,
Nishat Tasnim Bristyab,
Md. Abdus Salamb,
Muhammad Shahriar Basharc and
Sabina Yasmin
*a,
Md Yeasin Pabela,
Nishat Tasnim Bristyab,
Md. Abdus Salamb,
Muhammad Shahriar Basharc and
Sabina Yasmin