
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew multifunctional hybrids as modulators of apoptosis markers and topoisomerase II in breast cancer therapy: synthesis, characterization, and in vitro and in silico studies†
Heba W. Alhamdia,
Mohammad Y. Alfaifibc,
Ali A. Shatibc,
Serag Eldin I. Elbehairibcd,
Mohammed Er-rajye,
Reda F. M. Elshaarawy *f,
Yasser A. Hassang and
Rozan Zakryah
*f,
Yasser A. Hassang and
Rozan Zakryah
aCollege of Sciences, Biology Department, King Khalid University, Abha 61413, Saudi Arabia
bKing Khalid University, Faculty of Science, Biology Department, Abha 9004, Saudi Arabia
cTissue Culture and Cancer Biology Research Laboratory, King Khalid University, Abha, 9004, Saudi Arabia
dCell Culture Lab, Egyptian Organization for Biological Products and Vaccines (VACSERA Holding Company), 51 Wezaret El-Zeraa St., Agouza, Giza, Egypt
eLIMAS Laboratory, Faculty of Sciences Dhar El Mahraz, Sidi Mohamed Ben Abdellah University, Fez, Morocco
fDepartment of Chemistry, Faculty of Science, Suez University, 43533 Suez, Egypt. E-mail: reda.elshaarawy@suezuniv.edu.eg
gDepartment of Pharmaceutics, Faculty of Pharmacy, Delta University for Science and Technology, Gamasa, Egypt
hChemistry Department, Faculty of Science, Port-Said University, Port-Said, Egypt. E-mail: rzakrya@yahoo.com
First published on 6th September 2024
Abstract
Recently, molecular hybrids of two or more active pharmacophores have shown promise for designing and synthesizing anticancer drugs. Herein, a new multifunctional hybrid (PAHMQ), combining azobenzene and quinoline pharmacophores, and its M(II) complexes (MPAHMQ) have been successfully developed and structurally characterized. The MTT assay revealed CuBHTP as the most efficient and safe breast cancer treatment, with an IC50 of 11.18 ± 0.39 μg mL−1 and a high selectivity index (SI) of 5.63 for cancer MCF-7 cells over healthy MCF10A cells. Moreover, the CuPAHMQ-treated MCF-7 cells experience a dramatic impact with regard to key apoptotic markers, including an increase in P53 and Bax expression, with a decrease in Bcl-2 expression levels compared to the untreated MCF-7 cells. Additionally, CuPAHMQ effectively halted the growth and division of MCF-7 cells by inducing cell cycle arrest in the crucial G1 and S phases, ultimately inhibiting both Topo II activity and cell proliferation. Molecular docking investigations validated the CuPAHMQ complex's groove binding and topoisomerase II binding, establishing it as a potent anticancer drug.
1. Introduction
Breast cancer is a devastating disease that affects millions of women worldwide. According to the WHO, breast cancer is the most common cancer among women, with an estimated 2.3 million new cases diagnosed in 2020 alone. This alarming statistic highlights the urgent need for effective prevention and treatment strategies.1 Surgery, radiation, hormone therapy, and chemotherapy are some of the therapeutic options accessible to people with breast cancer.2 Although chemotherapy has shown promise in treating breast cancer, it is not without its challenges and limitations.3 One of the main challenges of breast cancer chemotherapy is its side effects on healthy cells. Another challenge is drug resistance. Still, there is reason to be optimistic about enhancing chemotherapy's efficacy while decreasing its adverse effects. One approach is the research and development of selective and targeted therapies, which aim to eradicate cancer cells by destroying their particular molecular targets while sparing healthy cells from harm.Molecular hybridization (MH) could revolutionize the pharmaceutical industry with its application to multitarget medication development. This method can potentially improve the efficiency and effectiveness of therapies for complicated diseases by combining several pharmacophores in a single molecule, which can help overcome medication resistance and optimize pharmacological characteristics.4–6 A significant benefit of MH is its capacity to concurrently target multiple disease pathways, such as diabetes, cancer, and Alzheimer's, in addition to its capability to overcome pharmaceutical resistance. Interestingly, the MH approach has emerged as a promising strategy for developing cancer-targeted and selective therapy. Its ability to specifically target cancer cells, increase efficacy, and potentially personalize treatment make it a highly attractive option for cancer treatment.6–8
The quinoline ring is a key pharmacophoric molecule that has been used extensively in the development of various clinical drugs (see Fig. 1).9 One of the main reasons for the widespread use of the quinoline ring in drug development is its ability to interact with a wide range of biological targets. This is due to the presence of a nitrogen atom in the ring, which can form hydrogen bonds with various functional groups in proteins and enzymes. As a result, quinoline-based drugs have been shown to exhibit diverse pharmacological activities, including antibacterial, antimalarial, antiviral, anticancer, and anti-inflammatory properties.10 Furthermore, the quinoline ring is highly versatile and can be easily modified to fine-tune its pharmacological properties. For instance, by introducing different substituents at specific positions on the ring, researchers can control the lipophilicity, polarity, and overall shape of the molecule, which can greatly influence its interaction with biological targets. This flexibility has allowed pharmaceutical researchers to develop a wide range of quinoline-based drugs with varying potencies, selectivity, and mechanisms of action. Interestingly, the quinoline derivatives have shown great potential in cancer therapy through their effects on topoisomerase II (an essential enzyme involved in DNA replication and repair in cancer cells),11 AKT1 (essential for cell survival and growth),12 P53 (tumour suppressor protein),13 and Bcl-2 (anti-apoptotic protein).12,14 Its ability to inhibit the activity of topoisomerase II, AKT1, and p53 proteins, while overexpressing Bcl-2 and inducing cell death makes them promising candidates for the development of new anticancer drugs.
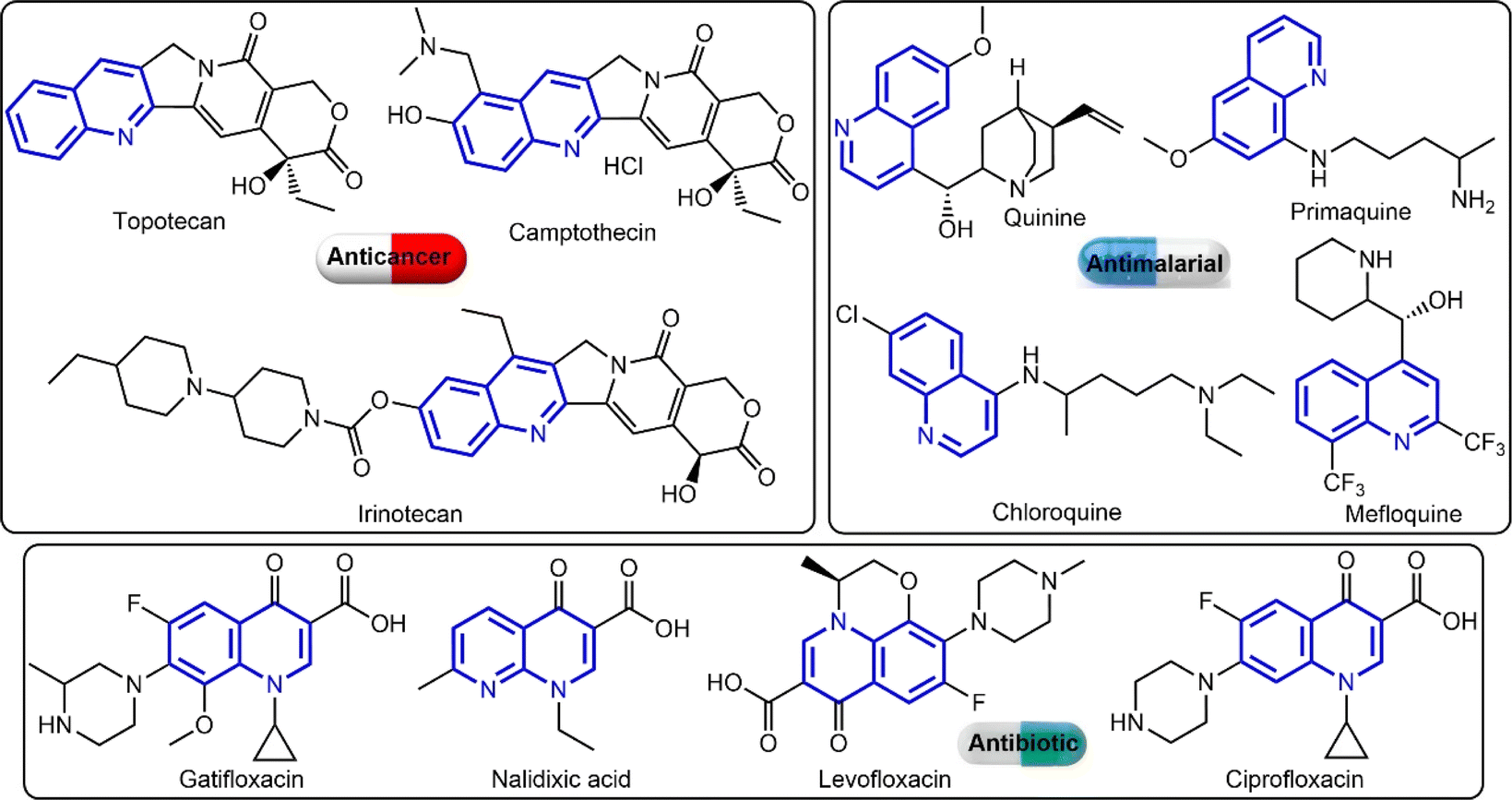 | ||
| Fig. 1 Examples for quinoline-based clinical drugs. | ||
On the other hand, azobenzene is an important pharmacophore and could serve as a versatile building block for a variety of pharmaceuticals. This could be due to its inherent photoisomerization (cis ↔ trans transformation) capability, which makes it an ideal building block for photopharmaceuticals, or drugs that are sensitive to light.15,16 Furthermore, the azo group in azobenzene can act as a redox center, making it a valuable component in developing prodrugs.17,18 Another significant application of azobenzene in drug development is its role as a molecular switch. When incorporated into a drug molecule, the azo group can be used to control the release of the active drug. By switching between its cis and trans forms, the azobenzene moiety can alter the drug's solubility, stability, and binding affinity, thereby regulating its release and duration of action.19 This property is particularly useful for developing sustained-release formulations, minimizing the frequency of drug administration, and improving patient compliance. Moreover, azobenzene derivatives have shown promising results in the treatment of various diseases. For instance, studies have demonstrated the potential of azobenzene-based compounds in treating cardiovascular diseases, inflammatory disorders, and even neurodegenerative diseases. These compounds can act as potent antioxidants and anti-inflammatory agents, making them valuable for therapeutic applications.20
These astounding results have sparked a new phase in our continuing endeavor to develop hybrid bioactive compounds with various pharmacological uses. To combat cancer, this research aims to create new multifunctional hybrid molecules (PAHMQ) by fusing two powerful pharmacophores: azobenzene and quinoline. In addition, M-PAHMQ complexes will be prepared to explore the capacity of PAHMQ and its metal complexes to selectively target human breast carcinoma cells (MCF-7) over healthy human breast cells (MCF-10A). An insight into the mode of anticancer action of these new compounds was investigated by studying their impacts on topoisomerase II, P53, Bax, and Bcl-2.
2. Materials and methods
Information regarding the solvents and important starting chemicals that were utilized in the current study, as well as the providers of those substances, was supplied in the ESI.† The preparation procedures for benzenediazonium chloride and 7-hydroxy-4-methylquinolin-2(1H)-one (HMQ) were also described in ESI.† Additionally, the ESI† included a description of the instrumental methods that were utilized to thoroughly characterize the produced compounds.2.1. Synthesis of PAHMQ
The bis-azo derivative of HMQ was synthesized by dissolving 0.175 g (1 mmol) of HMQ in 1.5 mL of 10% aqueous NaOH. The solution obtained was cooled to a temperature range of 0–5 °C. Next, a solution containing 0.28 grams (2 mmoles) of benzenediazonium chloride was gradually introduced into the reaction mixture over a period of 30 min, while ensuring that the temperature remained below 5 °C and the pH value in the range 8–9. Following the addition of the diazonium salt, the reaction mixture was agitated for 2 h until the diazonium salt was fully depleted. Following the completion of the azo coupling reaction, the reaction mixture was treated with 10% aqueous HCl until achieving an acidic pH. The resulting solid was separated by filtration, rinsed with deionized water, and then subjected to a 24 h drying process at 25 °C in a vacuum oven. The crude product was purified by re-crystallization from ethanol. PAHMQ obtained as a reddish-brown powder in 83% yield. FTIR (KBr; ν, cm−1): 3433 (s, br), 3174 (m, sh), 3059 (m, sh), 2979 (m, sh), 1718 (s, sh, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1595 (m, sh), 1487 (s, sh), 1272 (s, sh), 1186 (s, sh), 1008 (m, sh), 839 (m, sh), 758 (m, sh). 1H NMR (200 MHz, DMSO-d6) δ 15.11 (s, 1H), 7.80–7.09 (m, 11H), 6.02 (s, 1H), 3.74 (s, 1H), 2.48 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 177.52, 166.83, 153.94, 153.74, 149.39, 142.16, 141.81, 134.98, 132.26, 131.70, 131.20, 130.29, 129.76, 128.48, 127.15, 126.72, 125.75, 125.06, 123.16, 122.56, 121.40, 120.10, 25.64. EI-MS: m/z at 383.40 corresponding to C22H17N5O2 [M]+. Anal. calcd for C22H17N5O2 (M = 383.41 g mol−1): C, 68.92; H, 4.47; N, 18.27%. Found: C, 68.89; H, 4.51; N, 18.18%.
O), 1595 (m, sh), 1487 (s, sh), 1272 (s, sh), 1186 (s, sh), 1008 (m, sh), 839 (m, sh), 758 (m, sh). 1H NMR (200 MHz, DMSO-d6) δ 15.11 (s, 1H), 7.80–7.09 (m, 11H), 6.02 (s, 1H), 3.74 (s, 1H), 2.48 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 177.52, 166.83, 153.94, 153.74, 149.39, 142.16, 141.81, 134.98, 132.26, 131.70, 131.20, 130.29, 129.76, 128.48, 127.15, 126.72, 125.75, 125.06, 123.16, 122.56, 121.40, 120.10, 25.64. EI-MS: m/z at 383.40 corresponding to C22H17N5O2 [M]+. Anal. calcd for C22H17N5O2 (M = 383.41 g mol−1): C, 68.92; H, 4.47; N, 18.27%. Found: C, 68.89; H, 4.51; N, 18.18%.
2.2. Synthesis of M-PAHMQ complexes
A solution containing the respective metal(II) chlorides (CoCl2·6H2O, CuCl2·2H2O, and ZnCl2) (2 mmol, in 15 mL ethanol) was gradually added to a hot solution containing 1 mmol of PAHMQ ligand in (25 mL) ethanol. Following the addition of ammonia solution to bring the pH of the resultant combinations to 10, they were refluxed under stirring at 80 °C for 2 h. They then vaporized to half their original volume. The mixtures were cooled for the entire night, at which point the solid complexes precipitated, filtered, and were then rinsed with ethanol. The produced complexes were re-crystallized from ethanol and then vacuum-dried over anhydrous CaCl2. Thin-layer chromatography was used to verify the purity.O), 1583 (s, sh), 1516 (m, sh), 1450 (m, sh), 1372 (m, sh), 1256 (s, sh, Ar–O) (Δν = −16 cm−1), 1172 (m, sh, NN) (Δν = −15 cm−1), 1026 (m, sh), 839 (m, sh), 754 (m, sh, NN) (Δν = −4 cm−1), 693 (m, sh), 587 (m, sh, Co–N), 506 (m, sh, Co–N), 428 (m, sh, Co–O). Anal. calcd for C22H30Cl3Co2N7O6 (M = 712.74 g mol−1): C, 37.07; H, 4.24; N, 13.76%. Found: C, 36.98; H, 4.27; N, 13.69%.O), 1589 (s, sh), 1516 (m, sh), 1451 (m, sh), 1354 (m, sh), 1257 (s, sh, Ar–O) (Δν = −15 cm−1), 1180 (m, sh, NN) (Δν = −7 cm−1), 1021 (m, sh), 851 (m, sh), 750 (m, sh, NN) (Δν = −8 cm−1), 694 (m, sh), 597 (m, sh, Cu–N), 501 (m, sh, Cu–N), 446 (m, sh, Cu–O). Anal. calcd for C22H22Cl3Cu2N5O5 (M = 669.89 g mol−1): C, 39.45; H, 3.31; N, 10.45%. Found: C, 39.37; H, 3.34; N, 10.33%.O), 1590 (s, sh), 1516 (m, sh), 1486 (m, sh), 1322 (m, sh), 1249 (s, sh, Ar–O) (Δν = −23 cm−1), 1208 (m, sh, NN), 1179 (m, sh, NN) (Δν = −8 cm−1), 1008 (m, sh), 899 (m, sh), 752 (m, sh, NN) (Δν = −6 cm−1), 689 (m, sh), 506 (m, sh, Zn–N), 463 (m, sh, Zn–N), 428 (m, sh, Zn–O). Anal. calcd for C22H20Cl3N5O4Zn2 (M = 655.54 g mol−1): C, 40.31; H, 3.08; N, 10.68%. Found: C, 40.28; H, 3.12; N, 10.65%.2.3. In vitro cytotoxicity assay
The cytotoxic effects of the PAHMQ ligand and its metal complexes were examined on two human cell lines, specifically the breast cancer (MCF-7) and breast epithelial (MCF10A) cell lines, which obtained from the VACSERA–Cell Culture Unit (Dokky, Giza, Egypt). Mycoplasma-free cells were used for all experiments. The cytotoxicity was tested in vitro using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to determine the compounds' capacity to induce endothelial cell death.21,22 A positive control was established using the therapeutic anticancer drug (5-fluorouracil, 5-Fu). To conduct the experiment, MCF-7 and MCF10A cells were separately seeded in 96-well Corning plates in triplicate. Each cell was then treated with 50 μM of the designated compound. After 72 hours, the MTT reagent was added and the cells were incubated for 4 hours. The resulting formazan crystals were dissolved in 100 μL of DMSO and the absorbance was measured at both 570 nm and 630 nm using a ROBONIK P2000 ELIZA multi-detection microplate reader located in MIDC Industrial Area, Mahape, Navi Mumbai – 400710, INDIA. The data was then calculated using a predetermined equation to determine the effect of the compound on cell viability. The cytotoxic effect of each sample on cell viability was calculated using the following equation:A, A0, and ANC are the mean absorbances of the tested compound, blank, and growth control (only cells), respectively. Cell viability variations and IC50 values were monitored as indicators of cytotoxicity.
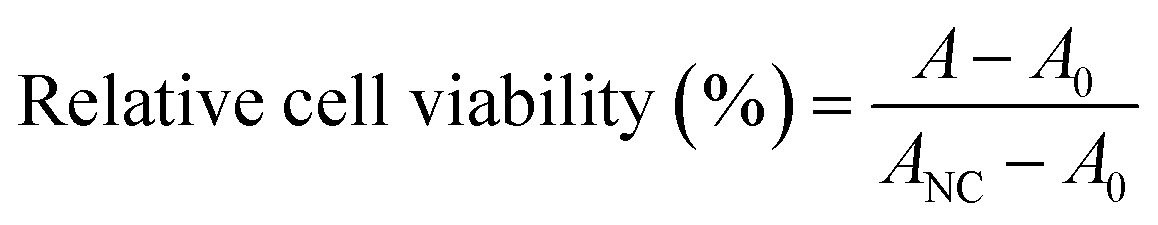
2.4. Effect of CuPAHMQ on gene expressions
The real-time PCR (RT-PCR) method was used to examine the levels of the P53 BAX, and Bcl-2 gene expression in the CuPAHMQ-treated and untreated MCF-7 cells. The experiment involved cultivating MCF-7 cells in tissue culture dishes and subsequently subjecting them to CuPAHMQ at a level of IC50 dose (11.18 μg mL−1) for a duration of 24 h. RNA was extracted using a Qiqamp mini kit (Qiagen; USA) according to the manufacturer's instructions. The entire cellular RNA was subsequently analyzed using one-step QuantiTecht SYBR green (Qiagen; USA) RT-PCR to measure the expression of P53 and Bcl-2 genes before and after CuPAHMQ treatment, following a prior study.23 Table S1 (ESI†) contains the primer sequences and experimental conditions utilized in this investigation. The 2−ΔΔCT technique was utilized to ascertain the relative expression of each gene.232.5. Topoisomerase inhibitory
The efficacy of the most powerful anti-cancer compound (CuPAHMQ) was evaluated for its ability to hinder human DNA topoisomerase II (Topo II) by the utilization of an enzyme-linked immunosorbent assay (ELISA) kit, in accordance with the instructions provided by the kit. The setup of all the samples and reagents followed the established work processes.24,252.6. DNA flow cytometry assay
The MCF-7 cells were grown in a multiwell plate containing DMEM with 10% FBS (2 × 105 cells per mL) and then kept in a humidified 5% CO2 environment at 37 °C for 24 h. Following this, the growth medium was replaced with DMSO solution (1% v/v) containing the most effective anticancer agent (CuPAHMQ) at its IC50 dose, and the cells were re-incubated for an additional 24 h. Next, the cells were fixed with cold ethanol (75%) after being rinsed twice with cold phosphate-buffered saline (PBS). Following a 30 min wash with PBS at 37 °C, the treated cells were collected by centrifugation at 2000 rpm for 5 min. Finally, A FACS Caliber flow cytometer (Becton Dickinson, Heidelberg, Germany) was used for analysis after the plate had been incubated for 20 minutes in a dark environment at room temperature.2.7. Apoptosis detection by annexin-V assay
After incubation of MCF-7 cells with an IC50 dosage of most effective anticancer agent (CuPAHMQ) in a multiwell plate (2 × 105 cells per well) for 48 h, the cells were collected and washed two times with PBS (100) at 4 °C for 10 min. Next, 5 μL of annexin V-FITC and 100 μL of PBS were added to every well. The apoptosis of MCF-7 cells was detected using the annexin V-FITC Apoptosis Detection Kit, manufactured by BioVisionResearch Products in the USA. After 5 μL of annexin V-FITC and PI solution was added to the cells, they were incubated at room temperature for 15 minutes in a dark environment. The cells were examined using the FACS Calibur Flow cytometer, and the data were analyzed using Cell-Quest software (Becton and Dickinson, Heidelberg, Germany).2.8. Molecular docking studies
Before commencing the docking study, the synthesized compounds underwent visualization with ChemDraw 16.0,26 followed by geometry optimization employing the MM2 method. Molecular Operating Environment (MOE) software was used to examine the ligand–protein receptor interaction, remove water molecules, rectify missing side-chain residues, consolidate non-polar hydrogens, perform molecular anchoring, and visualize the results obtained.27 We utilized the RSCBPD (https://www.rcsb.org/) to procure the targeted proteins.28 This database encompasses tens of thousands of genetic targets.To initiate the molecular docking procedure, the synthesized compounds need to undergo initial configuration, involving the addition of hydrogen atoms and removal of water molecules surrounding their structures. Subsequently, the potential energy must be established, incorporating atomic charges and other pertinent parameters adjusted according to the MMFF94x force field. Following this, the oriented compounds should be saved in MDB format, thus preparing them for the docking process.29
For the co-crystallized proteins, the PDB files are configured by adding hydrogen atoms, selecting the receptor, and generating α-site spheres using the site finder. The docking inhibitors will target the interior grooves of the protein surfaces through a series of 30 trials until achieving the most stable docking complexes. Scoring energies, calculated as the mean values of trials utilizing the London dG scoring function, were further enhanced by two independent refinements employing the triangular Matcher methods. Interaction complexes and electrostatic maps of interacting surfaces were extracted, along with essential interaction parameters. The degree of inhibition was assessed on the basis of the extracted parameters, including ligand, receptor backbones (amino acids), interaction type, and internal and evaluation energies.
In this work, we chose the ligand (CuPAHMQ) and its complex (CuPAHMQ) of the highest anticancer activity with the aim of identifying their anticancer activity against inhibitors of AKT1 kinase and topoisomerase II.29,30
2.9. Statistical analysis
The measurements were taken three times in every experiment. We provide the data as the mean plus or minus the standard deviation (SD). The significance of the differences between the experimental groups was determined using Student's t-test, which has a significance level of P < 0.05. Values were considered statistically significant when p < 0.05, highly significant when p < 0.01 and extremely highly significant when p < 0.001.3. Results and discussion
3.1. Synthesis protocol
A sequential procedure that combines azobenzene and quinoline to create the target hybrid molecule (PAHMQ) is shown in Scheme 1. Initially, the reaction between m-aminophenol and ethyl acetoacetate (EAA) proceeds via a cyclocondensation reaction to produce 7-hydroxy-4-methylquinolin-2(1H)-one (HMQ). Concurrently, the process of converting aniline into benzendiazonium chloride was performed through a diazotization reaction using a combination of NaNO2 and HCl. Finally, coupling of HMQ with benzendiazonium chloride leads to the creation of the desired bis-phenyazo-HMQ (PAHMQ). On the other hand, the synthesis of metal complexes using the new ligand (PAHMQ) involved refluxing a mixture of the ammonia-mediated pre-deprotonated ligand with metal chlorides in ethanol. The new compounds were obtained in high yields and pure states. The elemental analyses and spectral methods were used to elucidate the structures of these compounds.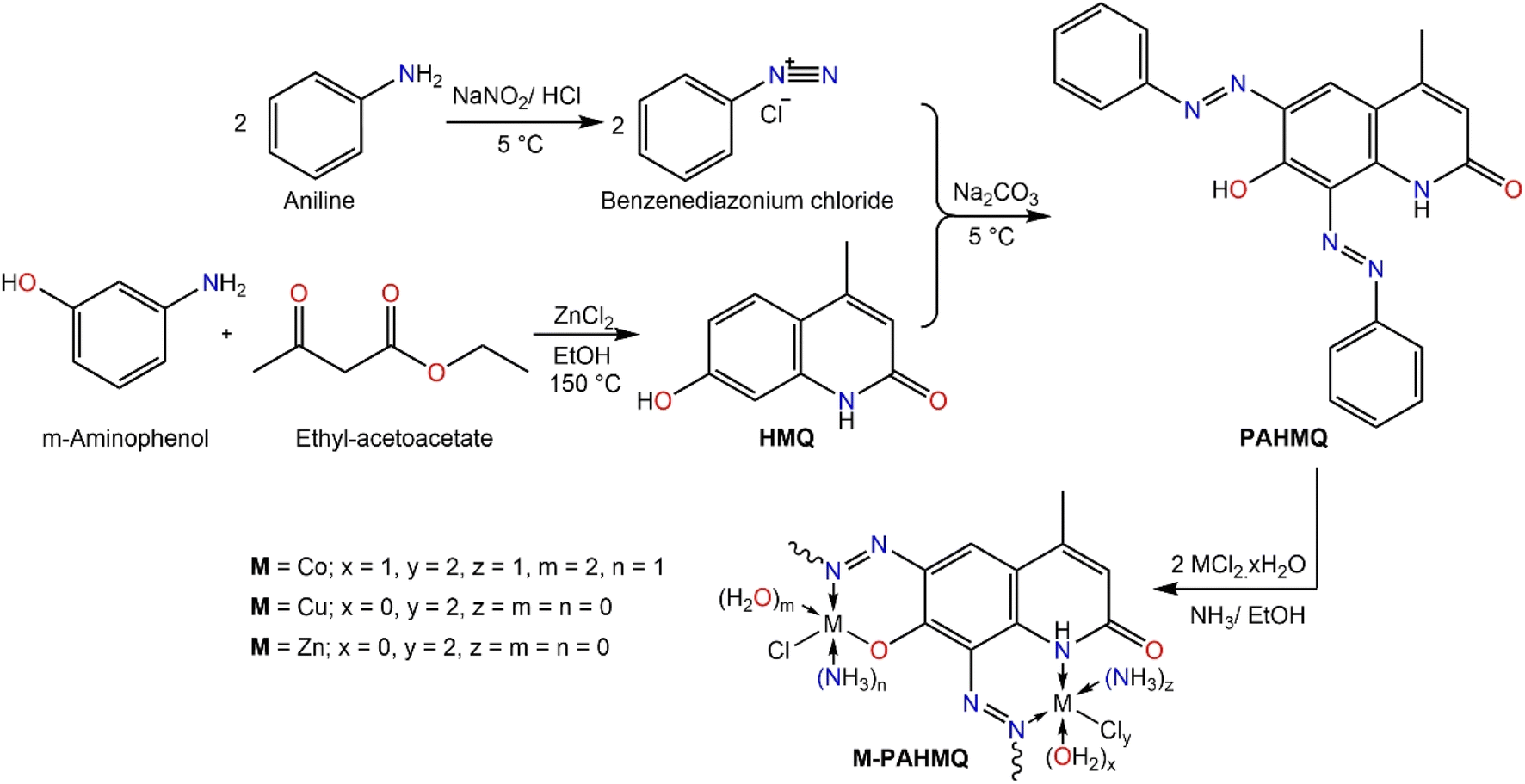 | ||
| Scheme 1 A stepwise synthesis protocol of PAHMQ and its metal complexes. | ||
3.2. Structural characterization
As demonstrated in the experimental section, the microanalytical data obtained from the elemental analyses (CHN) of PAHMQ and its metal complexes are in perfect harmony with their proposed structural formula. Furthermore, the elemental composition of the new CuPAHMQ complex was comprehensively analyzed using Energy Dispersive X-ray (EDX) spectroscopy (Fig. S1, ESI†), which revealed distinct peaks corresponding to carbon (C), nitrogen (N), oxygen (O), copper (Cu), and chlorine (Cl). The presence of these specific elements, particularly copper and chlorine, confirms their integration into the molecular framework, corroborating the proposed chemical structure of the PAHMQ ligand coordinated to a central Cu ion. Furthermore, this data validates the purity of the synthesized compound by confirming that no extraneous elemental contaminants were detected in significant quantities.The FTIR spectroscopy of new compounds is a powerful tool in proofing their successful formation as well as determining their molecular structures and functional groups present.6,31 In this scenario, the FTIR spectrum of PAHMQ (Fig. 2A) displays several characteristic peaks that are indicative of its functional groups.32 For example, a broad peak centred at 3433 cm−1, which corresponds to the phenolic OH group. This broadness could be attributed to the involvement of OH group in intermolecular H-bonding. Furthermore, a sharp peak observed at 3174 cm−1 could be assignable to the NH stretching vibration. Notably, a peak near 1718 cm−1 is observed, which is attributed to the CO stretching vibration from the quinolinone moiety. The sharp peaks observed at approximately 1492 and 1186 cm−1 are due to NN vibrations, highlighting the phenyldiazenyl substituent. Furthermore, C–H stretching and bending vibrations associated with methyl groups contribute peaks around 2979 and 1385 cm−1, respectively. As compared to the FTIR spectrum of the free ligand, the spectra of its metal complexes show characteristic peaks that indicate successful coordination with metal ions. Upon complexation with metals (Co(II), Cu(II), or Zn(II)) the main characteristic peaks of the ligand (OH, NH, and –NN–) undergo remarkable changes in their positions and intensities due to changes in electron density distribution around the coordinating sites. For instance, the phenol O–H stretching peak typically shifts to a lower wavenumber (around 3325–3346 cm−1), indicating deprotonation and participation of the hydroxyl group in metal binding. In contrast, the NH stretching band was shifted upwards by approximately 7–18 cm−1 upon complex formation due to coordination through the nitrogen atom. In addition, the peaks distinctive of the azo (–NN–) group was shifted downwards by approximately 7–15 cm−1 upon complexation, indicating the involvement of this group in the coordination sphere of metal ion. Similarly, Ar–O peak was shifted downwards by approximately 15–23 cm−1 upon complex formation due to coordination through oxygen atoms. Additionally, new peaks emerge in the spectra within the range of 400–600 cm−1 attributable to M–N and M–O bonds, confirming metal–ligand interaction. The presence of these characteristic shifts and new peaks collectively supports the formation of stable metal–ligand complexes.
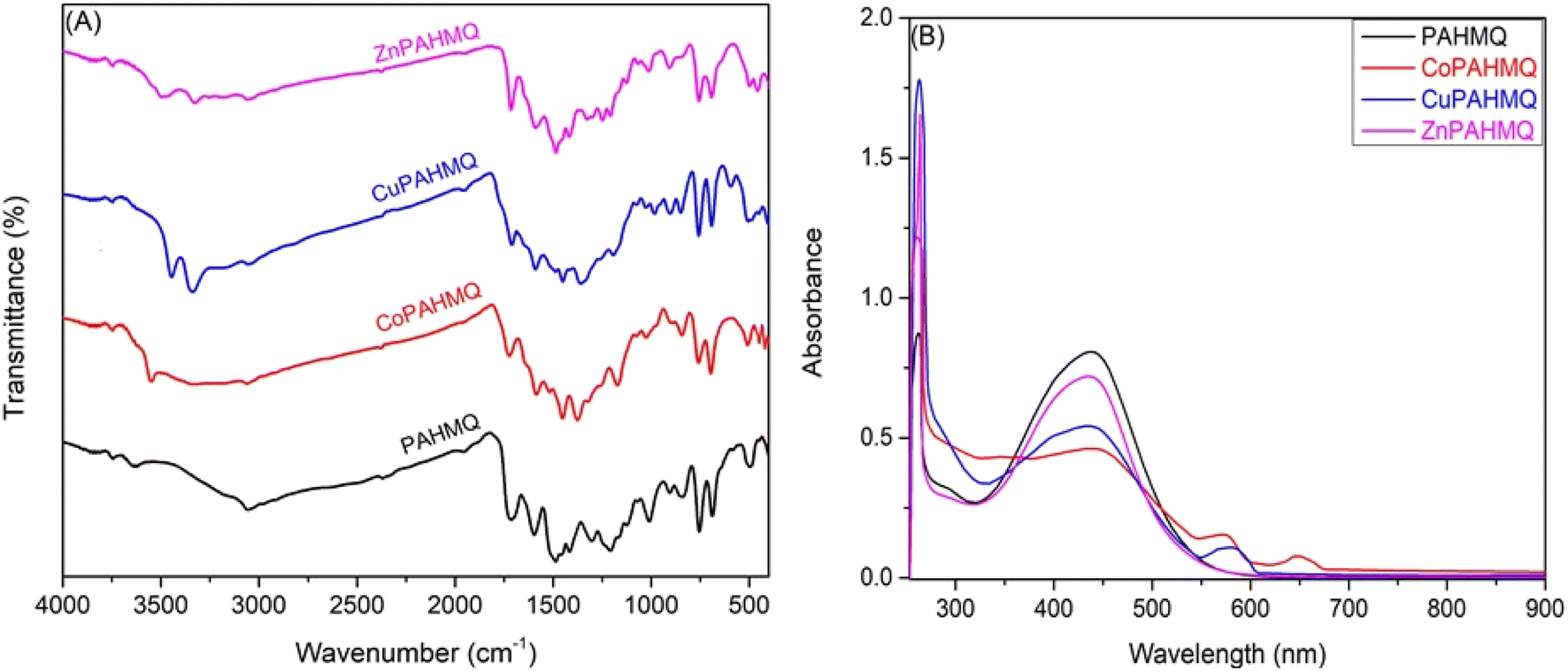 | ||
| Fig. 2 (A) FTIR & (B) UV-vis spectra of the new ligand (PAHMQ) and its metal complexes. | ||
The UV-vis spectrum of the free ligand (PAHMQ) exhibits two characteristic absorption peaks corresponding to π → π* and n → π* transitions (Fig. 2B). The first peak observed at 261 nm can be attributed to π → π* transitions within the aromatic quinoline ring system and the azo groups (NN). Furthermore, a significant peak observed at 436 nm could be ascribed to n → π* transitions associated with lone pairs on NN and CO groups. These spectral characteristics not only offer insight into the electronic structure of the free ligand but also serve as diagnostic tools for studying its binding behavior and complex formation properties. As compared to the UV-vis spectrum of the free ligand, the spectra of its metal complexes display notable shifts in peak positions and intensities as well as the emergence of new peaks due to coordination with metal ions. These changes are frequently the outcome of interactions between the ligand's donor atoms (such N and O atoms) and the metal center's unoccupied d-orbitals, which alter the electron density distribution. For instance, notable blue shifts coupled with hypochromic effects in the n → π* absorption peaks are discernible in the complex's spectra. These alterations may be attributed to the coordination of the NN group with metal ions. Furthermore, additional bands at 575 and 650 nm have been observed in the CoPAHMQ complex spectra, which may be attributed to d–d transitions, namely 4T1g(F) → 4A2g(F), 4T1g(F) → 4T2g(F), which are characteristic of the Co(II) ion in an octahedral setting. In a line with this hypothesis, the effective room temperature magnetic moment of CoPAHMQ was found to be μeff = 5.11 B.M that is typical of high spin octahedral Co(II) complexes.33,34 On the other hand, the spectrum of the CuPAHMQ complex revealed a new d–d transition peak at 580 nm, which corresponds to 2E → 2T2, and a magnetic moment of 1.74 B.M. These findings support the tetrahedral geometry of CuPAHMQ.5,35
The 1H NMR spectrum of the newly synthesized ligand (PAHMQ) exhibits characteristic peaks indicative of its distinct chemical structure (Fig. S1, ESI†). The presence of the NH group is confirmed by a broad singlet around δ 15.11 ppm, which is typical for an amide NH group engaged in strong intermolecular hydrogen bonding. Notably, the aromatic protons in the phenyl and quinoline rings contribute to multiple signals within the δ 7.80–7.09 ppm range, demonstrating complex splitting patterns due to their interactions with neighboring protons. Furthermore, the OH group is responsible for the downfield peak that appears as a singlet between δ 3.74 ppm. Additionally, the methyl group on the quinoline ring generates a sharp singlet at δ 2.48 ppm. These spectral features collectively confirm this new ligand's successful synthesis and structural integrity. Noteworthy, to confirm the position of H in –OH and –NH groups, a minimal amount of D2O is added to the sample, and the 1H NMR was then re-measured (Fig. S2, ESI†). The deuterium atoms of D2O will be rapidly exchanged for the labile protons previously mentioned, as shown below:
| –OH + D2O ↔ –OD + HOD |
| –NH– + D2O ↔ –ND– + HOD |
As can be seen in Fig. S2 (ESI†), the characteristic peaks of NH (15.11 ppm) and OH (3.74 ppm) were disappeared. On the other hand, the 13C NMR spectrum of PAHMQ shows several characteristic peaks corresponding to its carbon skeleton (Fig. S3, ESI†). For example, A distinct peak near 177.52 ppm could be assigned to the carbonyl carbon (CO). Furthermore, significant deshielding effects are observed around 142–154 ppm where peaks are associated with the carbons in the quinoline and phenyl rings which possibly attached directly to nitrogen atoms in diazenyl groups. Multiple peaks within the region of 120.10–141.81 ppm can be attributed to the aromatic carbons of both the quinoline and phenyl rings. Finally, a peak at 25.64 ppm corresponds to the methyl carbon (–CH3) attached to the quinoline core.
The thermogravimetric analysis (TGA) and differential thermogravimetry (DTG) techniques offer invaluable insights into the ligand and its metal complexes' decomposition patterns, thermal stability, and compositional nuances. This analysis was conducted in an inert environment at temperatures between 40 and 1000 °C (see Fig. S4–S7, ESI†). Results from the TG curves show that the novel ligand PAHMQ is thermally stable up to 198.7 °C, with crystallization water and volatile molecule losses occurring at approximately 100 °C. After that, it underwent a single thermal deterioration phase between 200 and 400 °C, resulting in its complete decomposition and leaving 20% residual carbon. In contrast, the M(II)-PAHMQ complexes demonstrate altered thermal behaviors indicating enhanced stability due to metal–ligand coordination. For instance, the CoPAHMQ exhibits two separate thermal decomposition phases in the temperature ranges of 401.32–553.41 and 587.72–856.78 °C and is thermally stable at about 390 °C. After the experiment, the residual mass was 18.78%. Similarly, the CuPAHMQ is thermally stable up to a temperature around 420 °C and then underwent three separate thermal degradation phases (421.82–504.75, 513.39–649.65, and 653.49–839.23 °C, respectively), leaving a residual mass of 32.78% when the experiment is over. CuCO3 could be responsible for this residue. Similar thermal behavior was observed for the ZnPAHMQ complex.
3.3. Cytotoxicity
By utilizing the MTT assay, the cytotoxic effects of the novel hybrid Ligand (PAHMQ) and its metal complexes on the cancerous MCF-7 and healthy MCF10A cell lines were assessed in vitro, compared to a therapeutic medication (5-fluorouracil, 5-Fu). According to the dose–cytotoxicity correlation curve (Fig. 3), all new chemotherapeutic agents exhibited toxic impacts on the MCF-7 cells, however, with performances depending on the structural features of the tested sample and its dose. Overall, the metal complexes (M(II)-PAHMQ) showed elevated levels of cytotoxicity as compared to the parent ligand (PAHMQ). For instance, as shown in Fig. 3A and B, the survival of MCF-7 cells has significantly declined from 100% to around 4% (96% cytotoxicity) after treatment with a 62.5 μg mL−1 dose of the most potent anti-breast cancer agent (CuPAHMQ). In contrast, the treatment with the same dosage of parent ligand has reduced the survival of MCF-7 cells to around 16% (84% cytotoxicity). Interestingly, the IC50 value of the CuPAHMQ complex against MCF-7 cells was determined to be 11.18 ± 0.39 μg mL−1, which is significantly less than the IC50 values of other complexes and the clinical anticancer drug 5-FU (28.34 ± 1.89 μg mL−1) as well as the parent ligand (19.79 ± 0.31 μg mL−1).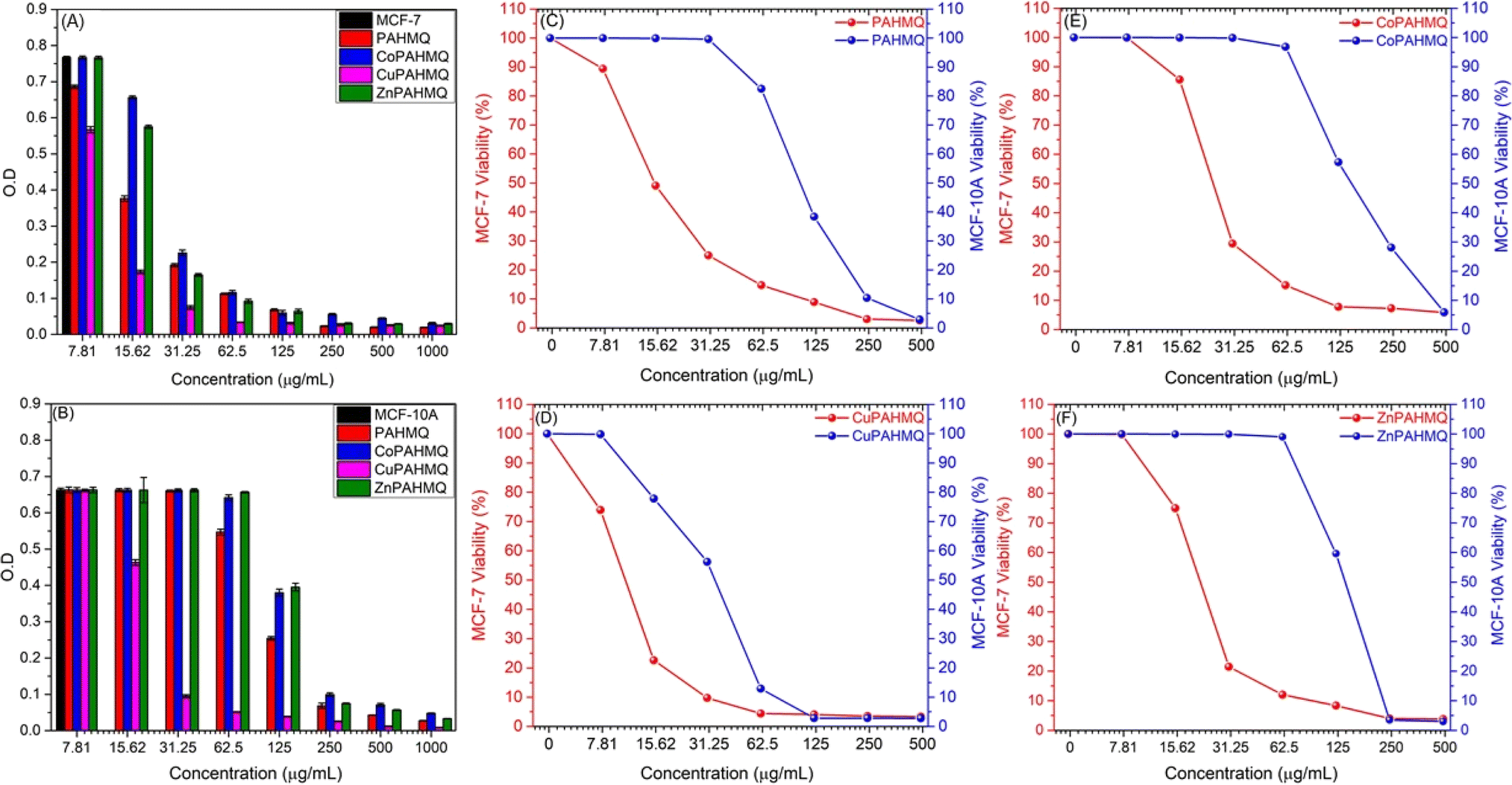 | ||
| Fig. 3 The samples' dosages–cytotoxicity relationships in terms of (A and B) changes in the optical density (O.D) of the MCF-7 and MCF-10A cultures, respectively; and (C–F) cell viability after 24 h treatment with (C) PAHMQ, (D) CuPAHMQ, (E) CoPAHMQ, and (F) ZnPAHMQ. | ||
The higher anti-breast cancer activity of the new Cu(II) complex (CuPAHMQ) as compared to the parent ligand (PAHMQ) as well as other complexes could be attributed to several crucial factors. Firstly, the thermal measurements have shown that the novel Cu(II) complex outperforms both the parent ligand and previous complexes in terms of stability. This stability allows for a longer duration of action, ensuring that the complex remains active in the body for longer. This is of the utmost importance when dealing with cancer, as the disease demands continuous and sustained action to eradicate cancer cells.8,36 Moreover, Cu(II) complexes have a high binding affinity towards cancer cells due to their ability to mimic natural enzymes. These complexes can act as enzyme inhibitors, disrupting vital cellular processes and leading to cell death. This is particularly effective against cancer cells, which have increased metabolic activity and rely on enzymes for their survival and proliferation.5,37 Furthermore, copper's redox activity is essential in producing reactive oxygen species (ROS) within the cell. Elevated ROS levels cause oxidative stress in cancer cells, which have a greater metabolic rate and impaired redox balance than normal cells, increasing Cu's lethal efficiency(II).38 Additionally, the preferential accumulation of copper in malignant cells through overexpressed transporters, such as Ctr1 (copper transporter 1), further amplifies this selective cytotoxicity towards cancer cells, minimizing its effects on healthy cells.39 These combined factors underscore why Cu(II) complexes exhibit significantly higher binding affinities toward cancer cells than their parent ligands or other non-copper metal complexes.
When developing new anticancer compounds, it is crucial to ensure that they specifically target cancer cells and do not harm healthy cells. To validate the specificity of these compounds, it is important to conduct experiments that examine their effects on both cancer cells and healthy cells. To that end, the viability of healthy breast (MCF-10A) cells was monitored after treatment with increasing doses of the new compounds for 24 h. The outcomes (Fig. 3B–F) demonstrated that, in contrast to their effects on cancer cells, the novel chemicals had minimal, dose-dependent effects on MCF-10A cell viability. The compounds had a negligible impact on healthy cells, so they preferentially targeted MCF-7 tumor cells. Finding that the compounds have a high degree of selectivity and are less likely to damage healthy cells is encouraging. Specifically, the Selectivity Index (SI) provides a quantitative measure to determine whether these agents preferentially inhibit cancerous cells while sparing normal cells, thus offering an insight into their therapeutic window and potential safety profile. The equation utilized for calculating the SI:40
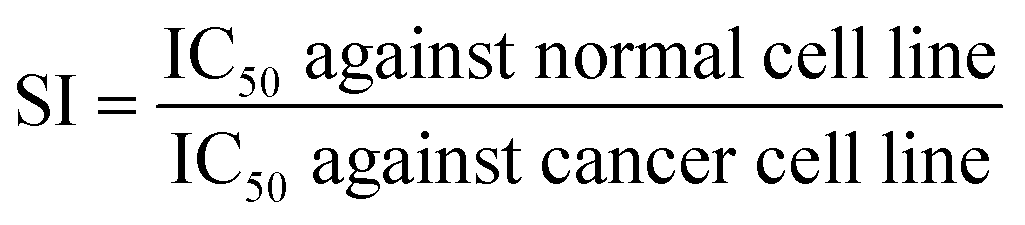
| Cells | IC50 (μg mL−1) ± SD | ||||
|---|---|---|---|---|---|
| PAHMQ | CoPAHMQ | CuPAHMQ | ZnPAHMQ | 5-FU | |
| MCF-7 | 19.79 ± 0.31 | 25.79 ± 0.32 | 11.18 ± 0.39 | 23.83 ± 0.18 | 28.34 ± 1.89 |
| MCF-10A | 101.29 ± 0.58 | 149.37 ± 2.1 | 59.97 ± 0.06 | 128.84 ± 2.32 | 41.03 ± 1.89 |
| SI | 5.11 | 5.79 | 5.63 | 5.40 | 1.45 |
3.4. Morphological changes in treated MCF-7 and MCF-10A cells
Studying morphological changes in cells is essential to understanding the effects of various treatments on cellular structures. Phase contrast microscopy (PCM), a noninvasive imaging technique, has been a valuable tool in observing changes in cell morphology.41 The use of PCM in this study allows for a detailed analysis of the morphological changes in MCF-7 cells after treatment with the new ligand and its complexes to get insightful data on the compounds' potential cytotoxic and therapeutic effects. As shown in Fig. 3 and S8–S12 (ESI†), all treatments typically induce notable changes in the cell membrane, cytoskeleton, and organelles, which indicate cellular stress, apoptosis, or necrosis. For instance, treated MCF-7 cells may exhibit significant shrinkage due to loss of cellular components, membrane blebbing, and changes in the distribution and density of cellular components (see Fig. 4). Other morphological alterations like chromatin condensation and nuclear fragmentation were also observed under these conditions. These changes indicate apoptosis, a programmed cell death mechanism crucial in cancer treatment strategies. Furthermore, the loss of adherence to culture plates suggested detachment-mediated apoptosis (anoikis), bolstering evidence for the complex's efficacy in destabilizing cancer cell architecture and function. Noteworthy, some cells underwent nuclear swelling, chromatin flocculation, and loss of nuclear basophilia as indicative of necrosis. Compared to untreated control cells which maintained a typical epithelial morphology with intact cellular boundaries and dense cytoplasm, the treated cells exhibited significant disintegration of structural integrity. | ||
| Fig. 4 Morphological alterations of untreated MCF-7 cells (control) (A), as well as after 24 h treatment with serial concentrations PAHMQ (B–D) and CuPAHMQ (E–G). PCM was used to track modifications in the cell morphologies (magnification 200 μm). | ||
Contrastingly, when subjected to the same treatment conditions, the non-tumorigenic mammary epithelial cell line MCF-10A showed only slight morphological changes (see Fig. 5 and S13–S17 (ESI†)). This minimal impact suggests that the new anticancer agents possess a degree of selectivity for malignant cells over healthy ones, thereby reducing collateral damage to normal tissues. The reduced sensitivity in MCF-10A cells may be attributable to their intact regulatory mechanisms and lower proliferative rates compared to MCF-7 cells, which often exhibit dysregulated pathways driving unchecked growth (source not provided). Such findings underscore the importance of continued research into targeted therapies that maximize efficacy against cancerous cells while preserving healthy tissue integrity.
 | ||
| Fig. 5 Morphological alterations of untreated MCF-10A cells (control) (A), as well as after 24 h treatment with 31.25 μg mL−1 concentrations (>IC50) PAHMQ (B) and CuPAHMQ (C). | ||
Interestingly, the novel Cu complex (CuPAHMQ) demonstrates superior anti-breast cancer activity compared to traditional platinum-based anticancer drugs such as cisplatin, carboplatin, and oxaliplatin, as well as the most potent Cu(II) complexes documented in recent studies. This is evident from a comparative analysis of their IC50 values presented in Table S2 (ESI†). The new Cu complex exhibits significantly lower IC50 value than those of cisplatin (IC50 = 12.25 μM), carboplatin (IC50 = 73.48 μM), and oxaliplatin (IC50 = 21.92 μM), highlighting its enhanced cytotoxic efficiency against breast cancer cells. Additionally, it surpasses even the most effective Cu(II) complexes identified so far, underscoring its potential as a formidable candidate for anti-breast cancer therapy. These findings suggest that the structural modifications implemented in this new Cu complex potentially confer heightened bioactivity and selectivity toward malignant cells while possibly reducing systemic toxicity.
3.5. Effect of CuPAHMQ on DNA content in cancer cells and cell cycle phases
The efficacy of new compounds in suppressing MCF-7 cell proliferation largely hinges on their capacity to diminish DNA content and modulate the cell cycle. This modulation manifests through two key mechanisms: cell cycle arrest and inhibition of cell growth in MCF-7 cells. The cell cycle regulates the delicate balance between cellular growth and apoptosis, crucial to carcinogenesis. When this equilibrium is broken, cell division and tumor growth often begin.42 This prompted us to look into the possibility that the suppression of cell growth in MCF-7 cells could be associated with cell cycle arrest, focusing specifically on the 24 hour post-treatment period using the most effective anticancer drug (CuPAHMQ). MCF-7 cells were treated with CuPAHMQ at a concentration equal to its IC50 dosage for 24 h and subsequently analyzed by flow cytometry. The data depicted in Fig. 6A–C convincingly illustrates that the administration of the Cu(II) complex substantially affects the distribution of the cell cycle and triggers apoptosis in MCF-7 cells. More precisely, the proportion of cells undergoing programmed cell death in the sub-G1 phase significantly rose from 1.73% in untreated cells to 33.89% after treatment, indicating a significant increase in cell death caused by the Cu(II) complex. In addition, this treatment caused changes in the progression of different cell cycle phases. Specifically, there was a slight increase in the percentage of cells in the G0/G1 phase from 52.96% to 56.01%, which suggests a possible halt or delay at this checkpoint. At the same time, there was a significant decrease in the percentage of cells transitioning from the G2/M phase, dropping from 8.39% to 1.58%. This indicates a suppression of mitotic entry or progression. The percentage of cells in the S phase increased from 38.56% to 42.41%, indicating a buildup of DNA caused by the halt of the cell cycle in this phase.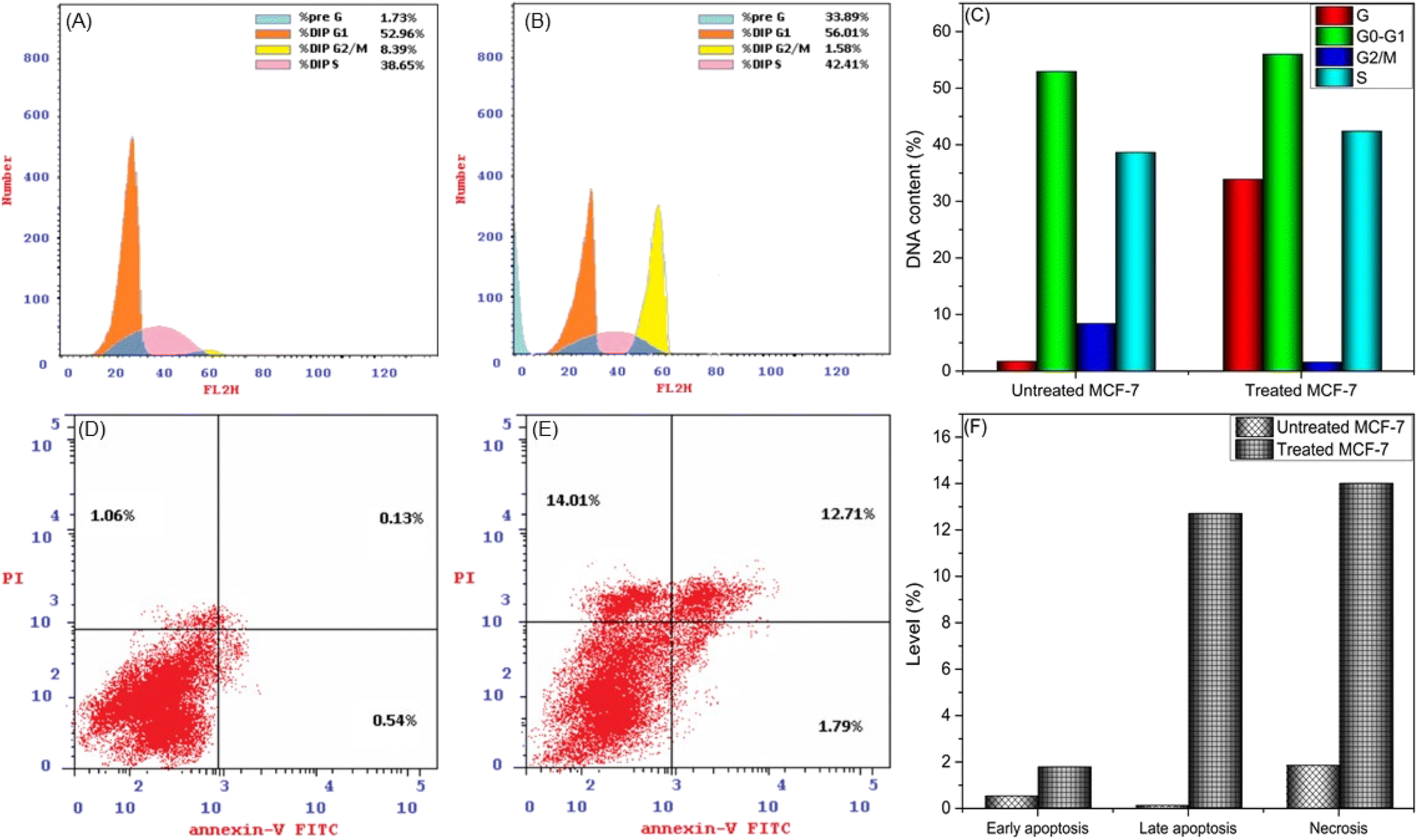 | ||
| Fig. 6 Impacts of CuPAHMQ on the MCF-7 cell cycle and apoptosis using flow cytometry analysis. (A and B) Cell cycle phases for (A) untreated MCF-7 cells, (B) treated MCF-7 cells, and (C) the DNA content. (D–F) Impacts of a 24 hour treatment with an IC50 dose of the CuPAHMQ complex on the distribution of MCF-7 cells across four distinct quadrants, early apoptosis (Q4; Ann V+/PI), viable cells (Q3; Ann V, PI), late apoptosis (Q2; Ann V+, PI+), and necrosis (Q1; Ann V, PI+) using annexin V-FITC assay. | ||
These findings suggest that the Cu(II) complex can induce apoptosis and interfere with the normal cell cycle regulation in MCF-7 breast cancer cells. It does this by affecting critical checkpoints and progression points, providing insight into how it may work as an anti-cancer agent.
3.6. CuPAHMQ-induced apoptosis performance
Flow cytometry was employed to investigate the impact of a 24 hour treatment with an IC50 dose of the CuPAHMQ complex on the distribution of MCF-7 cells across four distinct quadrants, early apoptosis (Q4; Ann V+/PI), viable cells (Q3; Ann V, PI), late apoptosis (Q2; Ann V+, PI+), and necrosis (Q1; Ann V, PI+), in comparison to untreated MCF-7 cells, after staining with annexin V-FITC/PI. The analysis revealed significant changes in cell distribution post-treatment, as illustrated in Fig. 5. The viability of cells (Q3) significantly decreased, suggesting that the complex exhibits a heightened cytotoxic effect, resulting in cell death. Concurrently, there was a marked increase in both early apoptotic (Q4, from 0.54% for untreated MCF-7 to 1.79% for treated MCF-7) and late apoptotic (Q2, from 0.13% for untreated MCF-7 to 12.71% for treated MCF-7) populations, signifying that the treatment induced substantial programmed cell death among MCF-7 cells. In addition, there was a notable increase in necrotic cells (Q1, from 1.06% for untreated MCF-7 to 14.01% for treated MCF-7), indicating that although apoptosis was the main outcome of this therapy, there was also a considerable level of cell membrane damage resulting in necrosis. These findings underscore the CuPAHMQ complex's potential utility as an anti-cancer agent that effectively disrupts cellular homeostasis and promotes apoptosis in breast cancer cells.3.7. Impacts of CuPAHMQ on the P53, Bax, and Bcl-2 gene expressions
P53 (tumor suppressor), Bax (pro-apoptotic), and Bcl2 (anti-apoptotic) genes are pivotal in regulating apoptosis in MCF-7 cells, effectively orchestrating the balance between cell survival and programmed cell death.43 Consequently, any dysregulation among these molecules can lead to enhanced apoptotic responses and contribute to cancer therapy. Hence, it was necessary to examine the impact of IC50 dosage CuPAHMQ on P53, Bax, and Bcl2 levels in the treated MCF-7 cells (see Fig. 7A). The increase in the apoptotic marker P53 in MCF-7 cancer cells treated with the CuPAHMQ complex indicates a notable activation of apoptosis, which could be a significant advancement in targeted cancer treatments. The data shows that the levels of P53 increased by 5.5-fold in response to the treatment, indicating a strong and significant response. This significant rise not only emphasizes the strength of the complex but also emphasizes its focus on malignant cells without harming non-cancerous ones, reducing the usual harm caused by traditional chemotherapy. The overexpression of the P53 protein was responsible for a notable decrease in the G2/M phase of the MCF7 cell cycle, elucidating its pivotal role in cell cycle regulation and tumor suppression.44 When P53 is overexpressed, it makes it very difficult to go from the G2 to the mitotic transition. Various biochemical routes hinder G2/M transition, but one of the most important ones is the transcriptional activation of p21 (Cip1/Waf1) by P53. Thus, p21 builds up and stops cells from entering mitosis, giving them time to fix damaged DNA or, in the worst-case scenario, to initiate programmed cell death.45,46 Similarly, Bax level in complex-treated MCF7 cells increased 4.3-fold, suggesting that apoptotic signaling pathways were greatly enhanced in these cancer cells. The significant increase in Bax levels highlights its critical function in triggering apoptosis by activating the mitochondrial pathway.47 Given the prevalence of anti-apoptotic signals in malignant contexts, the observed increase implies that the treatment successfully tips the scales in favor of cell death. When overexpressed in MCF7 cells, the elevated levels of Bax cause an increase in the population of cells in the G1 phase and induce a G1 cell cycle arrest, thereby inhibiting DNA synthesis and cell proliferation.48 In contrast, the observation of a significant decrease in Bcl2 expression in CuPAHMQ-treated MCF7 cells, which showed a 5.4-fold reduction compared to untreated cells, underscores the potential efficacy of the treatment in modulating apoptotic pathways. The downregulation of Bcl2 expression suggests that the treatment induced apoptotic mechanisms, making cancer cells more susceptible to programmed cell death.49 By reducing Bcl2 levels by a factor of four, the CuPAHMQ may be able to inhibit cancer cells' ability to avoid programmed cell death by interfering with anti-apoptotic pathways.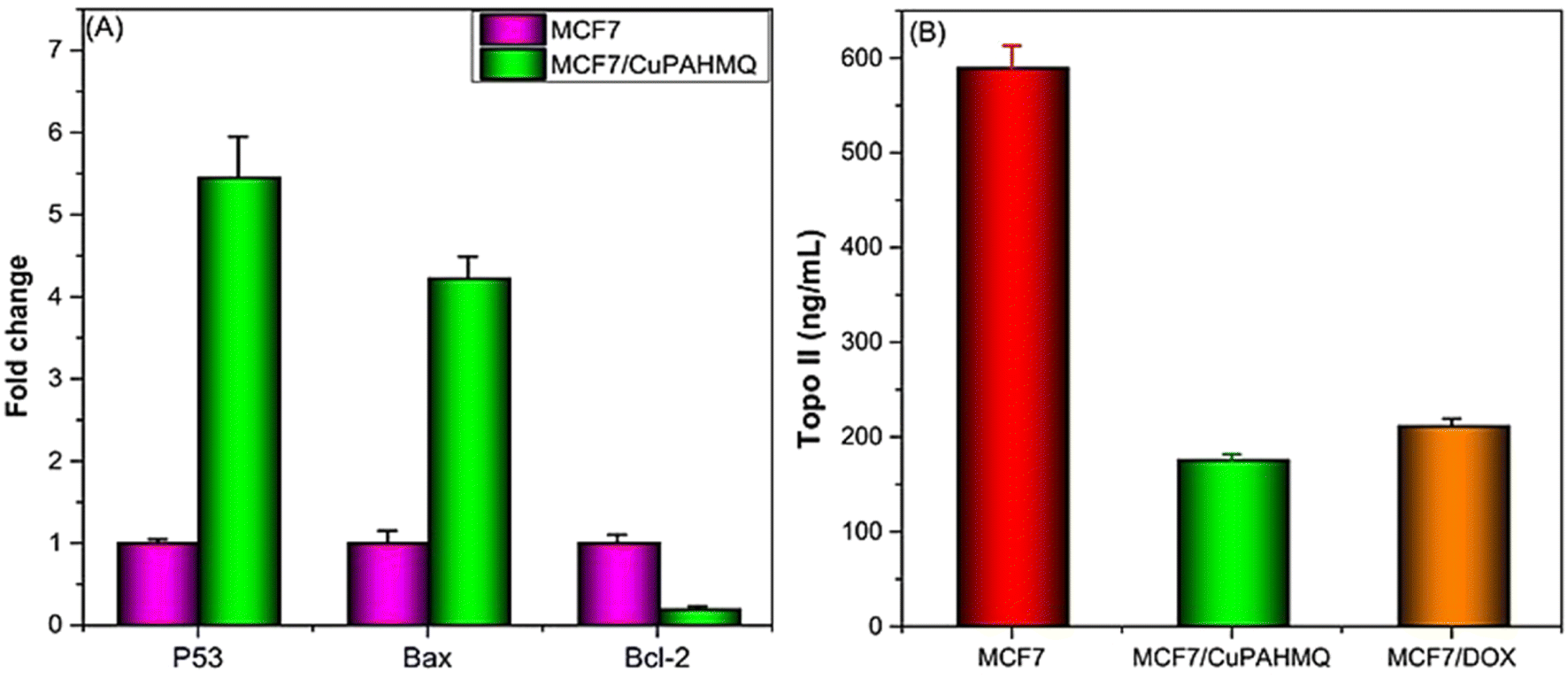 | ||
| Fig. 7 Impacts of a 24 hours treatment with an IC50 dose of the CuPAHMQ complex on the (A) p53, Bax, and Bcl-2 gene expression, and (B) Topo II activity in MCF-7 cells. | ||
3.8. Topoisomerase II inhibition by CuPAHMQ
The enzyme topoisomerase II (Topo II) plays a crucial role in DNA replication and transcription, making it a potential target for anticancer treatment. Blocking this enzyme can hinder the unwinding of DNA required for cell division, thereby inducing cytotoxicity in rapidly proliferating cancer cells.50 Thus, the potential of the new copper(II) complex (CuPAHMQ) as a potent Topo II inhibitor should be investigated. The significant decrease in Topo II activity in MCF7 cells treated with the CuPAHMQ complex compared to control cells elucidates the profound impact of the complex on cellular function. Specifically, Topo II activity in untreated MCF7 cells was measured at 591.18 ± 24.15 ng mL−1, whereas treatment with CuPAHMQ reduced this activity dramatically to 175.99 ± 5.97 ng mL−1 (see Fig. 7B), signifying that CuPAHMQ effectively inhibits Topo II. These findings strongly suggest that CuPAHMQ could serve as a potent chemotherapeutic agent by specifically targeting and inhibiting Topo II activity within breast cancer cells, underscoring its potential for therapeutic application in oncology to inhibit tumor growth through mechanistic disruption of DNA topology management.513.9. Molecular docking
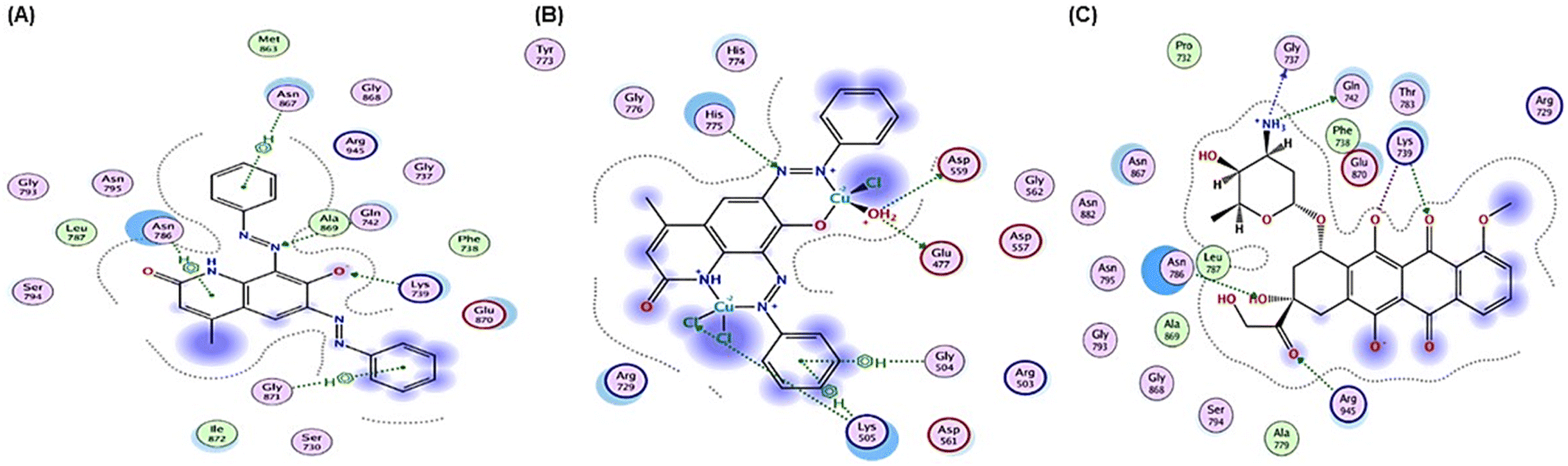 | ||
| Fig. 8 2D positioning and interactions of (A) PAHMQ, (B) CuPAHMQ, (C) and reference drug (Doxorubicin, DOX) with Topoisomerase II (3QX3). | ||
These findings suggest that CuPAHMQ could potentially be a more effective inhibitor of Topo II than the parent ligand and the reference drug. Thus, the Cu(II) complex may effectively inhibit the enzymatic function by stabilizing DNA-enzyme complexes or interfering with ATP hydrolysis, mechanisms often targeted in chemotherapeutic strategies.
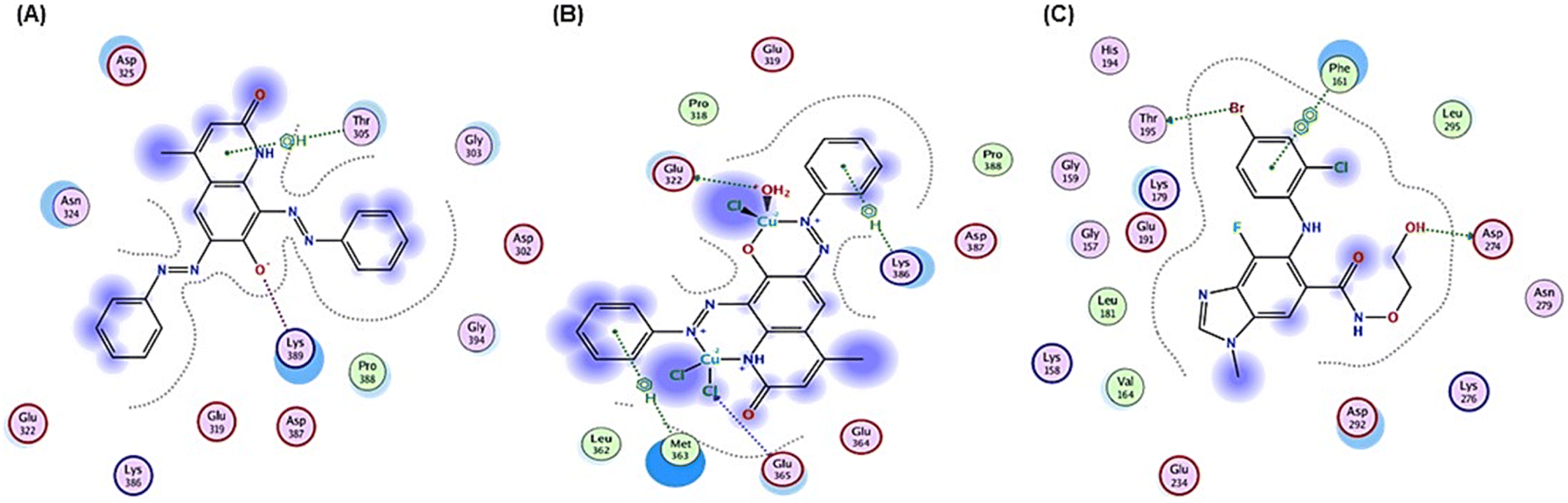 | ||
| Fig. 9 2D positioning and interactions of (A) PAHMQ, (B) CuPAHMQ, (C) and reference drug (Selumetinib, SEL) with AKT1. | ||
4 Conclusion
To develop novel non-platinum anticancer agents with lower toxicity, molecular hybridization has gained great interest. Intriguingly, copper complexes because of their endogenous nature and potential to induce DNA damage. A novel multifunctional hybrid, named PAHMQ, which combines azobenzene and quinoline pharmacophores, has been successfully developed and structurally characterized. Additionally, its M(II) complexes, referred to as MPAHMQ, have also been synthesized and characterized. The MTT assay demonstrated that CuBHTP is a highly effective and safe treatment for breast cancer, with an IC50 of 11.18 ± 0.39 μg mL−1 and a significant selectivity index (SI) of 5.63 for cancer MCF-7 cells compared to healthy MCF10A cells. In addition, the MCF-7 cells treated with CuPAHMQ show significant changes in apoptotic markers. This includes an upregulation of P53 and Bax expression and a downregulation of Bcl-2 expression when compared to the untreated MCF-7 cells. The CuPAHMQ compound effectively arrested the growth and division of MCF-7 cells by inducing cell cycle arrest in the G1 and S phases. This inhibition of cell cycle progression resulted in suppressing both Topo II activity and cell proliferation. The binding of the CuPAHMQ complex to the groove and topoisomerase II has been confirmed through molecular docking studies, indicating its potential as an effective anticancer medication.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Deanship of Scientific Research at King Khalid University for funding this work through large Groups (Project under grant numbers R.G.P. 2/217/44).References
- M. Arnold, E. Morgan, H. Rumgay, A. Mafra, D. Singh, M. Laversanne, J. Vignat, J. R. Gralow, F. Cardoso and S. Siesling, Breast, 2022, 66, 15–23 CrossRef PubMed.
- C. W. S. Tong, M. Wu, W. C. S. Cho and K. K. W. To, Front. Oncol., 2018, 8, 227 CrossRef PubMed.
- Y. Chen, Y. Qi and K. Wang, Front. Oncol., 2023, 13, 1169010 CrossRef PubMed.
- P. de Sena Murteira Pinheiro, L. S. Franco, T. L. Montagnoli and C. A. M. Fraga, Expert Opin. Drug Discovery, 2024, 19, 451–470 CrossRef CAS PubMed.
- R. F. M. Elshaarawy and C. Janiak, Arabian J. Chem., 2016, 9, 825–834 CrossRef CAS.
- R. F. M. Elshaarawy, T. B. Mostafa, A. A. Refaee and E. A. El-Sawi, RSC Adv., 2015, 5, 68260–68269 RSC.
- X. Xie, N. Zhang, X. Li, H. Huang, C. Peng, W. Huang, L. J. Foster, G. He and B. Han, Bioorg. Chem., 2023, 106721 CrossRef CAS PubMed.
- Y. A. Hassan, M. Y. Alfaifi, A. A. Shati, S. E. I. Elbehairi, R. F. M. Elshaarawy and I. Kamal, J. Drug Delivery Sci. Technol., 2022, 69, 103151 CrossRef CAS.
- O. O. Ajani, K. T. Iyaye and O. T. Ademosun, RSC Adv., 2022, 12, 18594–18614 RSC.
- B. S. Matada, R. Pattanashettar and N. G. Yernale, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef CAS PubMed.
- M. M. Hammoud, A. S. Nageeb, M. A. Morsi, E. A. Gomaa, A. A. Elmaaty and A. A. Al-Karmalawy, New J. Chem., 2022, 46, 11422–11436 RSC.
- R. Z. Batran, S. M. El-Daly, W. A. El-Kashak and E. Y. Ahmed, Chem. Biol. Drug Des., 2022, 99, 470–482 CrossRef CAS PubMed.
- A. Mrozek-Wilczkiewicz, M. Kuczak, K. Malarz, W. Cieślik, E. Spaczyńska and R. Musiol, Eur. J. Med. Chem., 2019, 177, 338–349 CrossRef CAS PubMed.
- R. Hamdy, S. A. Elseginy, N. I. Ziedan, A. T. Jones and A. D. Westwell, Molecules, 2019, 24, 1274 CrossRef PubMed.
- A. Paramanya, S. Sharma, R. B. Bagdat and A. Ali, in Nanomaterials for Agriculture and Forestry Applications, Elsevier, 2020, pp. 435–467 Search PubMed.
- D. Mulatihan, T. Guo and Y. Zhao, Photochem. Photobiol., 2020, 96, 1163–1168 CrossRef CAS PubMed.
- J. Zhu, T. Guo, Z. Wang and Y. Zhao, J. Controlled Release, 2022, 345, 475–493 CrossRef CAS PubMed.
- D. Xiao, L. Liu, F. Xie, J. Dong, Y. Wang, X. Xu, W. Zhong, H. Deng, X. Zhou and S. Li, Angew. Chem., 2024, e202310318 CAS.
- J. S. Boruah and D. Chowdhury, Appl. Nanosci., 2022, 12, 4005–4017 CrossRef CAS.
- H. B. Cheng, S. Zhang, J. Qi, X. J. Liang and J. Yoon, Adv. Mater., 2021, 33, 2007290 CrossRef CAS PubMed.
- O. A. A. Ali, W. Abd El-Fattah, M. Y. Alfaifi, A. A. Shati, S. E. I. Elbehairi, A. H. A. Almaaty, R. F. M. Elshaarawy and E. Fayad, Inorg. Chim. Acta, 2023, 551, 121460 CrossRef.
- W. N. El-Sayed, J. Alkabli, R. F. M. Elshaarawy and Y. A. Hassan, Arabian J. Chem., 2024, 17, 105655 CrossRef CAS.
- O. A. Abu Ali, W. Abd El-Fattah, M. Y. Alfaifi, A. A. Shati, S. E. I. Elbehairi, A. H. Abu Almaaty, R. F. M. Elshaarawy and E. Fayad, Inorg. Chim. Acta, 2023, 551, 121460 CrossRef CAS.
- B. H. Asghar, R. K. A. Hassan, L. A. A. Barakat, A. Alharbi, M. El Behery, R. F. M. Elshaarawy and Y. A. Hassan, J. Drug Delivery Sci. Technol., 2023, 83, 104388 CrossRef CAS.
- Z. Topcu, J. Clin. Pharm. Ther., 2001, 26, 405–416 CrossRef CAS PubMed.
- N. Mills, Comput. Software Rev., 2006, 128, 13649 CAS.
- S. Vilar, G. Cozza and S. Moro, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
- T. Noguchi and Y. Akiyama, Nucleic Acids Res., 2003, 31, 492–493 CrossRef CAS PubMed.
- I. Althagafi, N. El-Metwaly and T. A. Farghaly, Molecules, 2019, 24, 1741 CrossRef PubMed.
- C. Yuan, M.-H. Wang, F. Wang, P.-Y. Chen, X.-G. Ke, B. Yu, Y.-F. Yang, P.-T. You and H.-Z. Wu, Life Sci., 2021, 270, 119105 CrossRef CAS PubMed.
- H. K. Ibrahim, S. H. El-Tamany, R. F. El-Shaarawy and I. M. El-Deen, Maced. J. Chem. Chem. Eng., 2008, 27, 65–79 CrossRef CAS.
- J. Michalski, I. Bryndal, J. Lorenc, K. Hermanowicz, J. Janczak and J. Hanuza, Spectrochim. Acta, Part A, 2018, 191, 521–531 CrossRef CAS PubMed.
- N. Mondal, D. K. Dey, S. Mitra and K. M. A. Malik, Polyhedron, 2000, 19, 2707–2711 CrossRef CAS.
- A. R. E. Mahdy, O. A. A. Ali, W. M. Serag, E. Fayad, R. F. M. Elshaarawy and E. M. Gad, J. Mol. Struct., 2022, 1259, 132726 CrossRef CAS.
- I. Warad, S. Musameh, I. Badran, N. N. Nassar, P. Brandao, C. J. Tavares and A. Barakat, J. Mol. Struct., 2017, 1148, 328–338 CrossRef CAS.
- K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422–487 CrossRef CAS PubMed.
- T. J. P. McGivern, S. Afsharpour and C. J. Marmion, Inorg. Chim. Acta, 2018, 472, 12–39 CrossRef CAS.
- S. Sarfraz, A. Javed, S. S. Mughal, M. Bashir, A. Rehman, S. Parveen, A. Khushi and M. K. Khan, Int. J. Comput. Theor. Chem., 2020, 8, 40–46 CrossRef CAS.
- R. H. G. Teles, A. E. Graminha, C. M. Rivera-Cruz, D. H. Nakahata, A. L. B. Formiga, P. P. Corbi, M. L. Figueiredo and M. R. Cominetti, Toxicol. in Vitro, 2020, 67, 104922 CrossRef CAS PubMed.
- A. A. Awaji, M. A. Rizk, R. A. Alsaiari, N. F. Alqahtani, F. A. Al-Qadri, A. S. Alkorbi, H. S. Hafez and R. F. M. Elshaarawy, Pharmaceuticals, 2023, 16, 1711 CrossRef CAS PubMed.
- D. Roitshtain, L. Wolbromsky, E. Bal, H. Greenspan, L. L. Satterwhite and N. T. Shaked, Cytometry, Part A, 2017, 91, 482–493 CrossRef CAS PubMed.
- H. K. Matthews, C. Bertoli and R. A. M. de Bruin, Nat. Rev. Mol. Cell Biol., 2022, 23, 74–88 CrossRef CAS PubMed.
- M. A. O'Brien and R. Kirby, J. Vet. Emerg. Crit. Care, 2008, 18, 572–585 CrossRef.
- L.-F. Shyur, S.-H. Lee, S.-T. Chang, C.-P. Lo, Y.-H. Kuo and S.-Y. Wang, Phytomedicine, 2010, 18, 16–24 CrossRef CAS PubMed.
- P. H. Shaw, Pathol., Res. Pract., 1996, 192, 669–675 CrossRef CAS PubMed.
- A. K. Shendge, D. Chaudhuri, T. Basu and N. Mandal, Clin. Transl. Oncol., 2021, 23, 718–730 CrossRef CAS PubMed.
- A. Peña-Blanco and A. J. García-Sáez, FEBS J., 2018, 285, 416–431 CrossRef PubMed.
- S. Shirali, M. Aghaei, M. Shabani, M. Fathi, M. Sohrabi and M. Moeinifard, Tumor Biol., 2013, 34, 1085–1095 CrossRef CAS PubMed.
- J. C. Reed, Hematol./Oncol. Clin. North Am., 1995, 9, 451–474 CrossRef CAS PubMed.
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 338–350 CrossRef CAS PubMed.
- V. M. Matias-Barrios and X. Dong, Pharmaceuticals, 2023, 16, 94 CrossRef CAS PubMed.
- C. Bailly, Chem. Rev., 2012, 112, 3611–3640 CrossRef CAS PubMed.
- D. E. Arthur, Radiol. Infect. Dis., 2019, 6, 68–79 CrossRef.
- N. Hinz and M. Jücker, Cell Commun. Signaling, 2019, 17, 1–29 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04219k |
| This journal is © The Royal Society of Chemistry 2024 |