
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoumarin–amino acid hybrids as promising anticancer agents: design, synthesis, docking studies and CK2 inhibition†
Abd-Allah S. El-Etrawyab,
Ahmad Ramadanc,
Farag F. Sherbiny *d,
I. F. Zeidc,
A. A.-H. Abdel-Rahmanc and
Mohamed A. Hawatac
*d,
I. F. Zeidc,
A. A.-H. Abdel-Rahmanc and
Mohamed A. Hawatac
aDepartment of Chemistry, Basic Science Center, Misr University for Science and Technology (MUST), Al-Motamayez District, 6th of the October City 77, Egypt
bDepartment of Pharmaceutical Organic Chemistry College of Pharmaceutical Science & Drug Manufacturing, Misr University for Science and Technology (MUST), Al-Motamayez District, 6th of the October City 77, Egypt
cDepartment of Chemistry, Faculty of Science, Menoufia University, Shebin El-Koam, Egypt
dPharmaceutical Organic Chemistry Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt. E-mail: dr-farag-sherbiny@azhar.edu.eg
First published on 6th August 2024
Abstract
A series of mono-peptide, di-peptide and tri-peptide derivatives linked to a coumarin scaffold (5a–c, 7a–c, and 9a–c) were synthesized via the azide-coupling method from corresponding hydrazides 4, 6, and 8. These compounds were tested for anticancer activity against HepG-2, PC-3, and Hct-116 cell lines. Compounds, 7c, and 5b showed significant cytotoxicity, outperforming doxorubicin, with IC50 values of 34.07, 16.06, and 16.02 μM for 7c and 42.16, 59.74, and 35.05 μM for 5b. Compound 7b also displayed promising results with IC50 values of 72.13, 70.82, and 61.01 μM. Moreover, the key structural features of amino acids indicated that mono-peptide and di-peptide derivatives play a key role in increasing their anticancer activities compared with tri-peptides. In addition, the most potent compound 5b also exhibited strong CK2 kinase inhibition with an IC50 value of 0.117 ± 0.005 μM compared with roscovetine as a control drug with an IC50 value of 0.251 ± 0.011 μM. Finally, the binding mode of the chemical inhibitors at the active site of CK2 receptor was also investigated using a docking study which confirmed that the presence of the amino acid functionality is an important feature for anticancer activity and the synthesized compounds showed favorable ADME properties. Besides that, SAR analysis was implemented for the target compounds.
1 Introduction
For the time being, cancer is considered one of the leading causes of mortality worldwide and therefore, the evolution of potent and robustly effective anticancer agents is one of the most important challenges in advanced therapeutics due to the unique pathophysiology of tumors, systemic toxicity, and the prospective emergence of resistance to chemical therapy.1 Traditional methods for cancer medication encompass radiotherapy, immunotherapy, chemotherapy, and cancer starvation therapy each with their own limitations.2Heterocyclic compounds are known as interesting scaffolds to incorporate bioactive small molecules, due to their crucial roles in many physiological processes.3 In this context, coumarins have several attractive chemical features, such as low molecular weight, high solubility, high bioavailability, and low toxicity. Therefore, prosperous coumarin structures have been widely used as an effective template for drug discovery due also to their abilities to interact with many diverse enzymes and receptors and their anticancer properties on diverse cancerous cells.4 In addition, the versatility of coumarin scaffold has been of great interest because of their potential roles in preventing and treating various diseases.5 Many products contain coumarin derivatives exhibit a plethora of pharmacological effects including antioxidant,6,7 carbonic anhydrase inhibitor,8,9 antibacterial activity,10 antifungal activity,11,12 antiviral activity,13,14 anti-inflammatory activity,15,16 neuro-protection,17 anti-convulsant activity,18,19 anti-coagulant activity,20,21 as well as anti-diabetic activity,22,23 and anti-cancer activity.24,25
On the other hand, amino acid conjugations are pharmacological agents strategically used to improve water solubility and enhance the anti-cancer activity.26 Thus, coumarin derivatives containing amino acid are worthy target as the coumarin moiety allows them to be used as fluorescent probes.27,28 In addition, several amino acids bearing heterocyclic moieties are reported as highly potential antitumor agents.29–31
Molecular hybridization of two or more bioactive pharmacophores is a tremendous strategy for drug discovery to give new chemical entities with various and new biological activities.32,33 In the present study, we assumed that the merging of coumarin nucleus and amino acid moieties in a single chemical structure was able to drive new potential anticancer drug candidate.
Therefore, this study has been focused on the development of coumarin derivatives with particular amino acid residues which may led to novel hybrid compounds with potential anticancer agents. Thence, the synthesized compounds have been examined for their cytotoxic effects against a panel of human cancer cell lines using MTT assay technique and the most potent compounds were further selected and evaluated for their activities against CK2 kinase inhibitors. In addition, the molecular docking was applied to evaluate the binding modes of examined synthesized compounds.
1.1. Rational drug design
Coumarin is an aromatic compound that has a bicyclic chemical structure with lactone moiety. The presence of an electronegative atom is substantially effective for hydrogen bonding interactions and enhancing solubility. Besides, aromatic ring is responsible for having hydrophobic and aromatic stacking interactions as well as the lactone form is responsible for its anticancer activity.34 The substitution of coumarin nucleus makes its more significant and effective for bioactivity. Abundant types of coumarins have been synthesized and also are present in nature. Different chemical structures of coumarins have been produced due to the various types of substitution patterns or pharmacophoric features in their basic bicyclic structures which are significant in showing effective and diverse classes of biological activities (Fig. 1). The anticancer properties of coumarin derivatives have also been extensively investigated in several studies which are closely related to its anticancer activity.35–40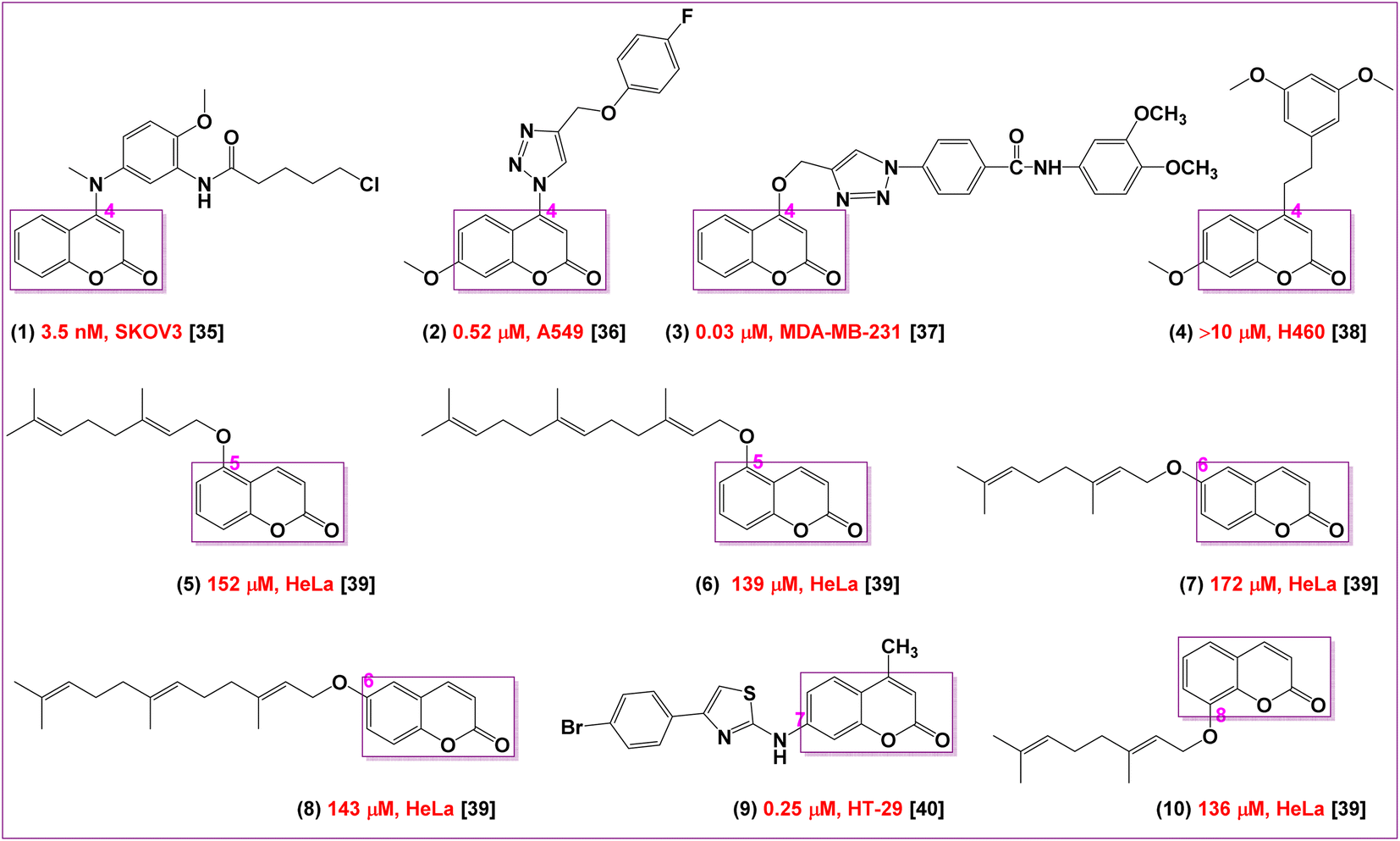 | ||
| Fig. 1 The basic structural substitution requirements for coumarin moiety as reported. | ||
Many SAR studies proposed that the substitution at the 7-position of coumarin moiety are likely promising structural features to obtain excellent antitumor activities38–42 including many clinical trials or drugs have already been used in the treatment of various cancers.43,44 In addition, it was found that the cytotoxic properties of substituted coumarins depend on the length of the chain, which increases the lipophilicty and thus facilitating their penetrations into the cells. SAR studies also demonstrated that the linker between coumarin nucleus and the substituent at the 7-position influences the anticancer activity.44 Moreover, the presence of hydrogen bonding acceptors and donors are essential for their anticancer activities.45
On the other hand, the incorporation of coumarin moiety into the side chain of amino acid is expected to improve the achievement of anticancer activity and reduce their adverse side effects. Furthermore, amino acid ester pro-drugs significantly improve the cellular uptake of the parent drugs via peptide transport mechanism and some epithelial cancer cells are wealthy in these transporters permits their use for the delivery of peptidomimetic anticancer agents.46,47 Also, amino acid ester decreases extensive degradation of anti-proliferative chemical drugs which increase their affinity to oligopeptide transports that are overexpressed in the tumor cells.48 Therefore, L-amino acids are used to design the amino acid pro-drugs therapy.
On the other hand, modern researches have been demonstrated that 7-hydroxycoumarin derivatives inhibit the secretion of cyclin D1, which is overexpressed in abundant types of cancer cells49–51 and thus could be successfully utilized in cancer therapy. Therefore, the essential core of rational design was prepared by chemical pharmacophoric modifications including the substitution at the 7-position of coumarin moiety with amino acid residues namely, glycine, phenyl glycine and leucine having hydrophobic side chains. Furthermore, the derivatization procedure including formation of mono-peptide, di-peptide and tri-peptide derivatives as chemical pharmacophoric features that has crucial role to form hydrogen bonding acceptors and donors as a naturally present in the human body. These hybrid chemical modifications are hopefully expected to improve their cytotoxic activities and therefore reduce their side effects.
Therefore, the main core of rational design encompasses the following bioisosteric chemical features which include; coumarin moiety, amide linker at the 7-position of coumarin nucleus, connected with phenyl ring, aliphatic moiety, or hydrophilic moiety (Fig. 2). Thus, the rational drug design generates new target hybrid structures with different biological isosteres with the hope that these new hybrid compounds might lead to a potential safe, effective, and potent as anticancer activity.
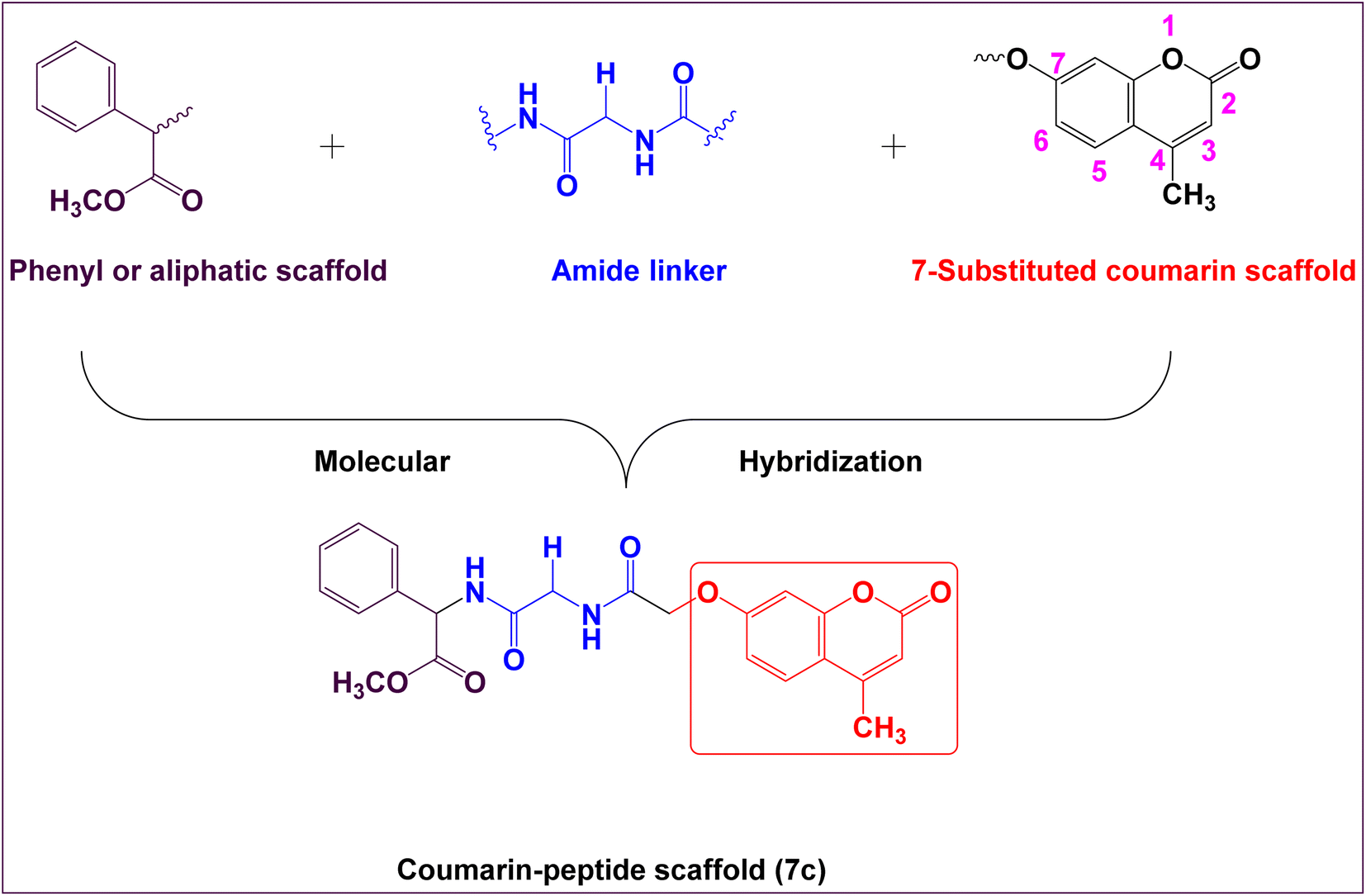 | ||
| Fig. 2 Planned design of novel coumarin derivatives for antitumor activity. | ||
2 Results and discussion
2.1. Chemistry
The synthesis of target compounds 5a–c, 7a–c and 9a–c were presented in Scheme 1 and 2. Firstly, the key starting intermediate compound 2 was synthesized according to the most extensively applied method, Pechmann condensation reaction of coumarin synthesis,52,53 by stirring a mixture of activated resorcinol 1 and ethyl acetoacetate in the presence of Conc. H2SO4 as a catalyst under solvent-free conditions for 18 h to obtain corresponding 7-hydroxy-4-methyl-2H-chromen-2-one (2). Then compound 2 was allowed to react with ethyl chloroacetate (β-ketoester) in the presence of dry acetone and anhydrous potassium carbonate to give ester derivative, compound 3. The corresponding acid hydrazide derivative 4 can be synthesized by the reaction of compound 3 with hydrazine hydrate in the presence of ethanol. The target peptide derivatives 5a–c was prepared by a coupling reaction of compound 4 with appropriate amino acids, via azide-coupling method54 in good yields (Scheme 1). The hydrazide is converted to acyl azide in the presence of HNO2. Then amino acid esters were introduced which reacts with the acyl azide to form an amide bond, which typically takes several hours. Low temperature is required to prevent decomposition of azide which releases nitrogen gas. Furthermore, the coupling step is achieved under basic a condition which prevents the formation of hydrazoic acid (HN3). The structures of the newly synthesized compounds 5a–c were established on the basis of elemental analysis and spectral data.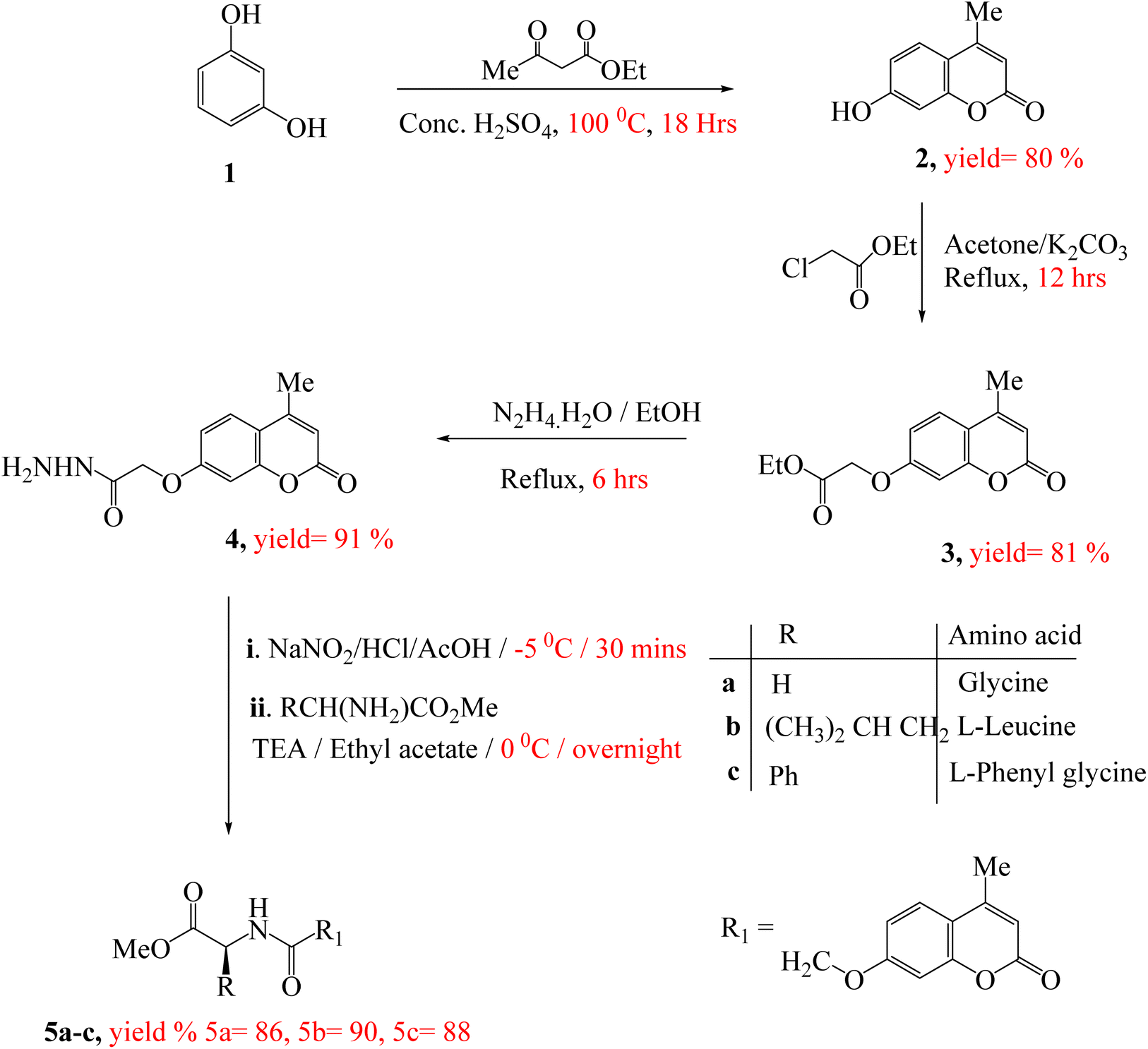 | ||
| Scheme 1 Synthetic route of target compounds 5a–c. | ||
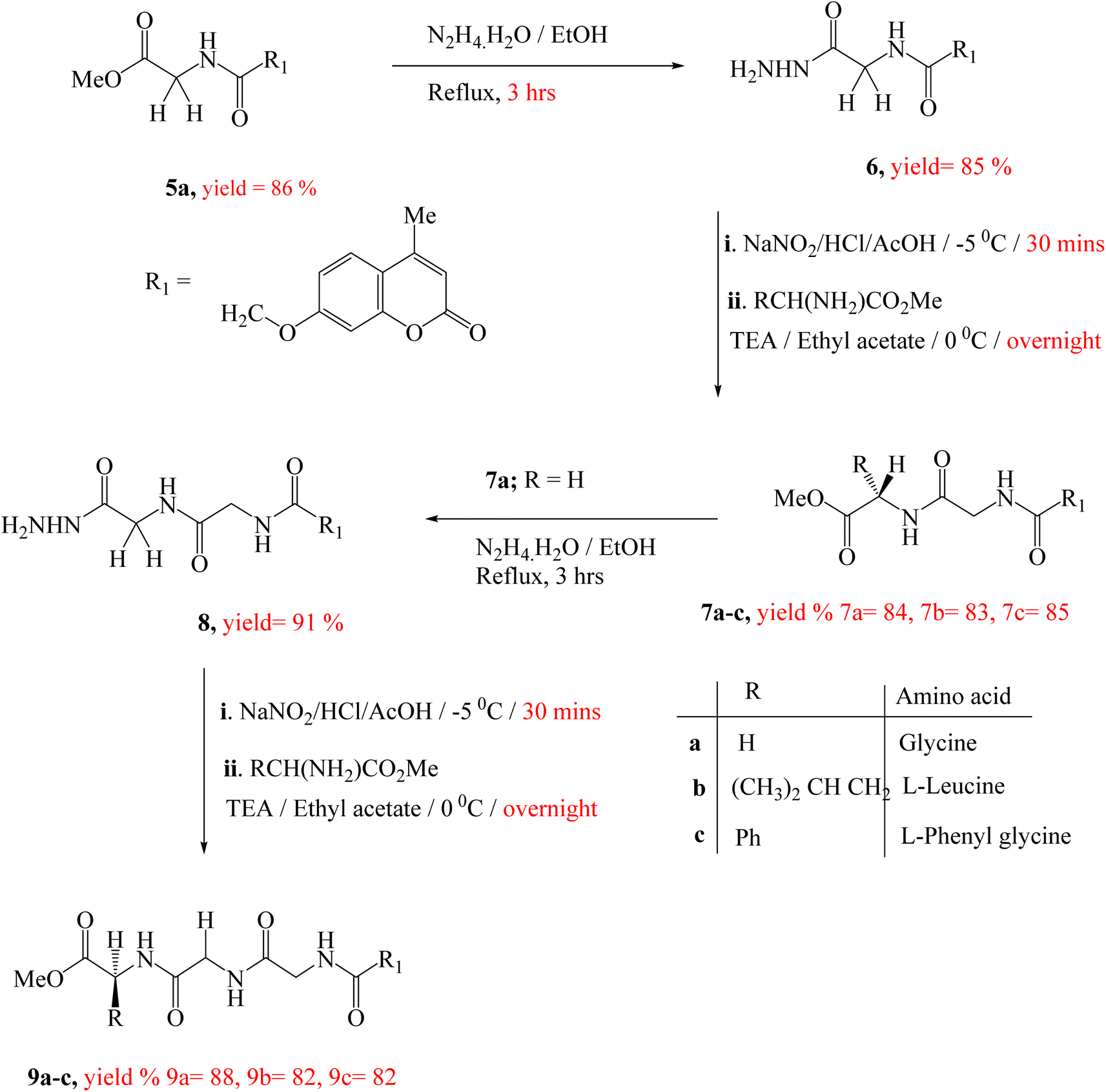 | ||
| Scheme 2 Synthetic route of target compounds 7a–c & 9a–c. | ||
Treatment of the peptide derivative, compounds 5a and 7a with excess hydrazine hydrate in ethanol afforded the corresponding hydrazide 6 and 8 in a good yield, respectively. The di-peptides 7a–c and tri-peptides 9a–c were successfully obtained in very good yields from compounds 6 and 8, respectively by similar general methodology mentioned in the synthesis of mono-peptides 5a–c under the same condition (Scheme 2). The chemical structures of these set of novel compounds 7a–c and 9a–c were established by their spectral and elemental analyses.
2.2. Biological evaluation
The obtained results of anticancer activities indicated that the screened chemical compounds showed obviously different levels of cytotoxic activity ranging from potent, moderate, and weak cytotoxicity in the treatment of all tested tumor cell lines. Therefore, data represented in (Table 1) revealed that, compound 7c distinctly appeared to be the most potent cytotoxic candidate, and efficient than doxorubicin with IC50 values of 34.07, 16.06, and 16.02 μM against three human cancer cell lines (HepG-2, PC-3, and HCT-116) indicating broad-spectrum anticancer activity. These results displayed that the hybrid structure possesses di-peptide linker with coumarin nucleus and substituted at the 7-position with mainly aromatic ring was more potent than those possessing mono-peptide and tri-peptide linkers substituted with aliphatic group 5b, 7b, 5a leading to a significant decrease in cytotoxic activity.
| Compound no. | In vitro cytotoxicitya IC50 (μM) | ||
|---|---|---|---|
| HePG2 | PC-3 | HCT-116 | |
| a IC50 values are the mean ± standard deviation (SD) of three separate experiments. | |||
| Doxorubicin | 108 ± 0.6 | 57.34 ± 0.6 | 61.01 ± 0.3 |
| 5a | 101.18 ± 0.3 | 105.74 ± 5.3 | 63.81 ± 3.6 |
| 5b | 42.16 ± 0.3 | 59.74 ± 4.2 | 35.05 ± 2.8 |
| 5c | 153.79 ± 2.1 | 173.89 ± 1.8 | 124.06 ± 4.4 |
| 6 | 168.32 ± 4.4 | 168.35 ± 4.3 | 171.34 ± 4.2 |
| 7a | 173.82 ± 3.4 | 172.27 ± 1.4 | 137.45 ± 2.4 |
| 7b | 72.13 ± 3.7 | 70.82 ± 2.1 | 61.01 ± 2.7 |
| 7c | 34.07 ± 4.2 | 16.06 ± 4.6 | 16.02 ± 4.1 |
| 8 | 172.24 ± 4.3 | 63.74 ± 4.3 | 94.74 ± 4.0 |
| 9a | 377.48 ± 2.5 | 115.01 ± 1.6 | 219.12 ± 2.8 |
| 9b | 298.76 ± 0.3 | 85.5 ± 0.4 | 169.13 ± 0.3 |
| 9c | 221.0 ± 0.9 | 147.29 ± 0.4 | 139.94 ± 0.7 |
Moreover, compound 5b, substituted with aliphatic moiety appeared to be the most potent derivative against three cell lines with IC50 values of 42.16, 59.74, and 35.05 μM in comparison to an anticancer drug, doxorubicin as standard drug. Besides, compounds 7b, and 5a possessed excellent anti-proliferative activities against three tested cell lines with IC50 values ranging from 61.01 μM to 105.74 μM indicating that the number of peptide bond can affect the anticancer property and gave the highest activity with the most potent compound with di-peptide bond, 7c.
Furthermore, several analogs such as 8, 5c, 6, and 7a demonstrated moderate anti-proliferative activities with IC50 values ranging from 63.74 μM to 173.89 μM, which probably due to the fact that incorporation of a hydrophilic group leading to unfavorable interaction with target receptor.
Finally, various compounds 9a, 9b, and 9c, exhibited relatively weak cytotoxicity towards three cell lines, indicating that the tri-peptide moiety did not play a key role in increasing their anti-cancer activities.
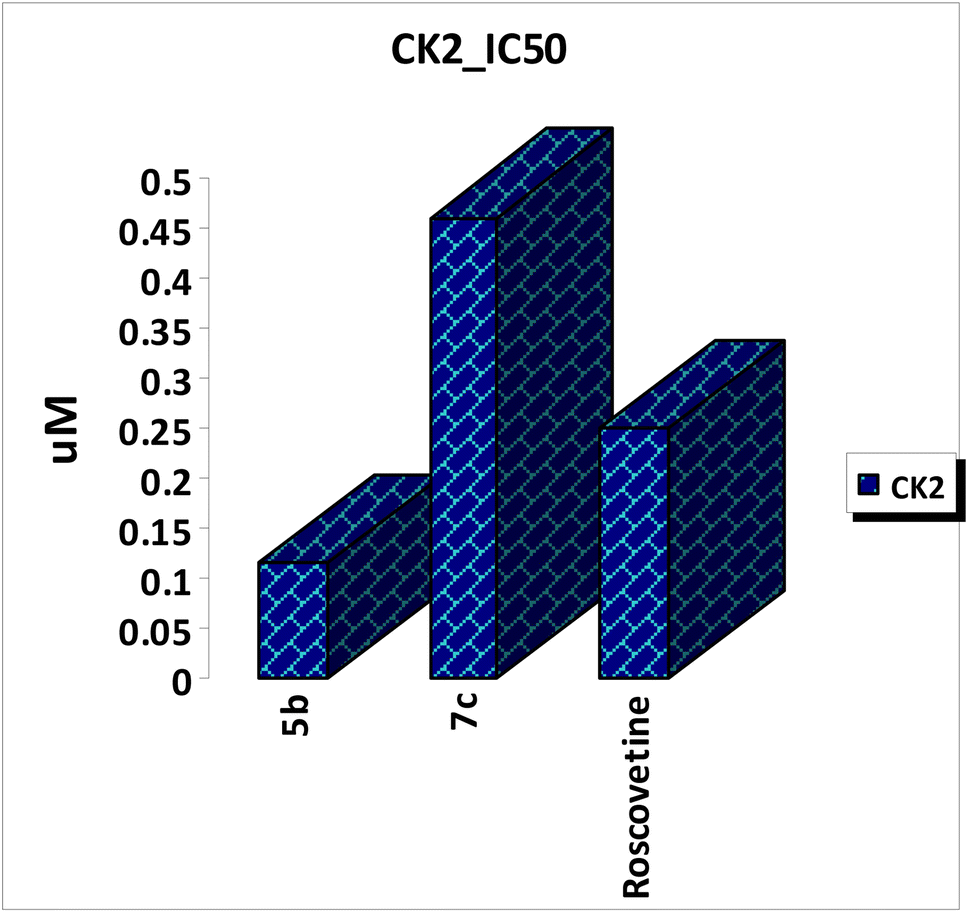 | ||
| Fig. 3 In vitro enzymatic inhibitory activities of the most potent amino acid derivatives against CK2 kinase. | ||
The obtained binding mode of co-crystalized ligand, G12 revealed that the hydroxyl group at the 7-position was involved in a hydrogen bonding interaction with Lys68 and formed water-mediated interactions with Glu81 and backbone amino group of Asp175. The chromene core of co-crystalized ligand was also stabilized by an aromatic stacking interaction with Phe113 and located in the hydrophobic pocket formed by primarily with Val45, Val53, Ile66, Val95, Val116, Met163, and Ile174.
Compound 7c as a representative example demonstrated a binding mode like that of co-crystalized ligand, G12, with multiple hydrophilic and hydrophobic interactions was found to facilitate the binding interactions of this compound to CK2 binding site. The carbonyl group at the 2-position was involved in the hydrogen bonding interaction with Lys68. And the oxygen atom at the 7-position of chromene nucleus was formed a water-mediated interaction with Asn118, with a water molecule that is conserved in all deposited CK2 structure. The methyl group at the 4-position was stabilized by hydrophobic interactions with Ile66, Val95 and Phe113. Furthermore, the two amino groups of acetamido moieties were located in hydrophilic interactions with the carbonyl backbone of His60 and formed a water-mediated interaction with Val45. Chromene moiety was also stabilized by an aromatic stacking interaction with Phe113 and located in the hydrophobic pocket formed by Val45, Val116, Met163, and Ile174. The carbonyl group of acetate moiety was located in a hydrogen bonding interaction with backbone amino group of Arg47 as well as the phenyl ring was stabilized by cationic π interactions with Arg47 and Lys158, which enhancing the affinity of compound 7c towards CK2 binding site (Fig. 4).
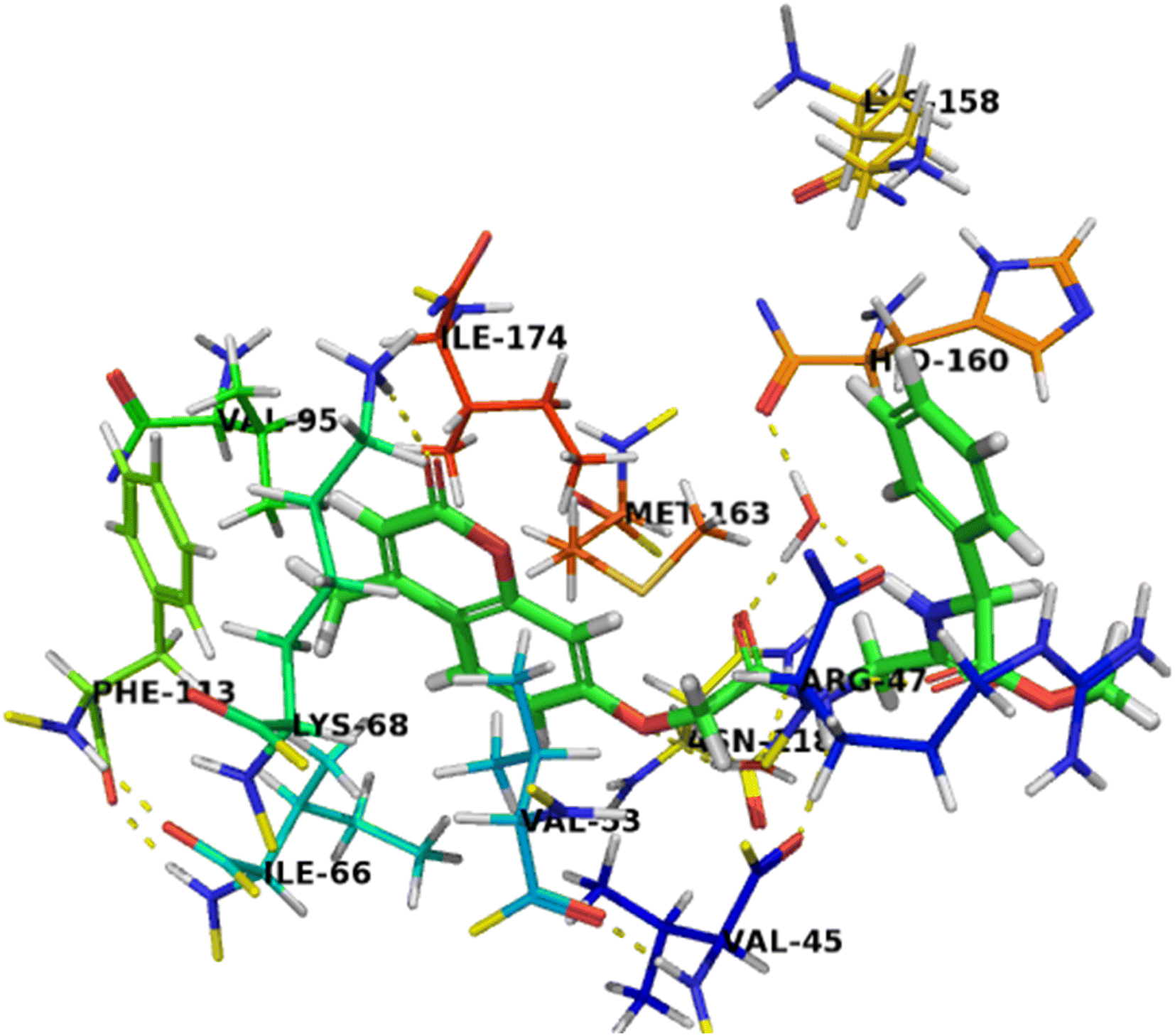 | ||
| Fig. 4 Crystal structure of CK2 receptor (PDB ID: 2QC6) with compound 7c. The hydrogen bonding interactions are presented by dashed lines. Residues contacting ligand are demonstrated as lines. | ||
The binding mode of compound 5b was like that of co-crystalized ligand and compound 7c. The amino group of acetamide moiety was involved in a hydrogen bonding interaction with backbone carbonyl group of Val45 and the other carbonyl group of acetamide moiety was formed a water-mediated interaction with His160 and these network interactions with water molecule seems to have a crucial role in the recognition process. The hydrophobic leucinate group was located in an unfavorable hydrophilic interaction with Arg47, which may explain its lower affinity of compound 5b compared with compound 7c (Fig. 5).
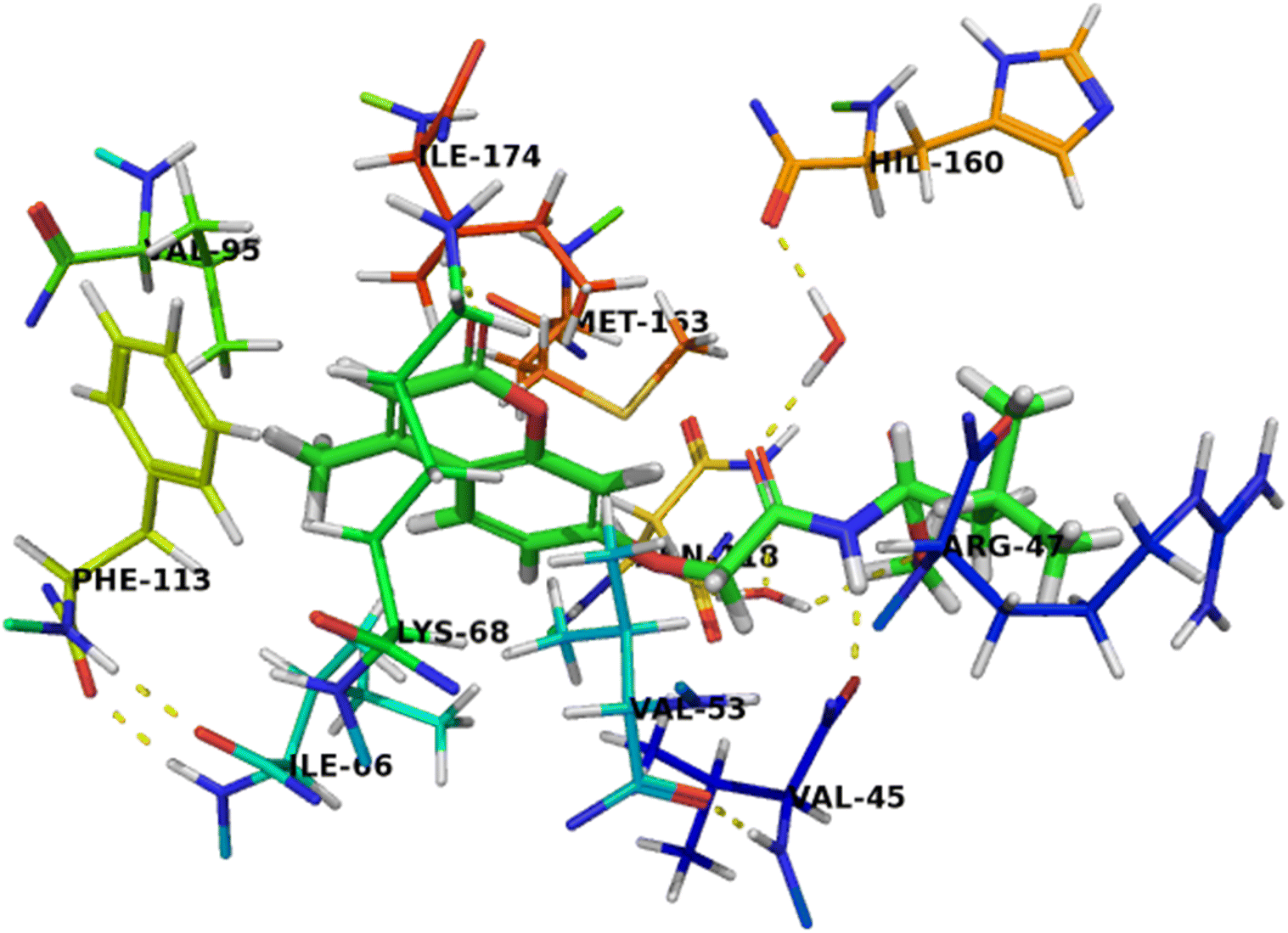 | ||
| Fig. 5 Crystal structure of CK2 receptor (PDB ID: 2QC6) with compound 5b. The hydrogen bonding interactions are presented by dashed lines. Residues contacting ligand are demonstrated as lines. | ||
The binding mode of compound 7b (Fig. 6) was similar to compound 7c that can completely accommodate the binding site. In addition, the hydrophobic glycylleucinate was located in an unfavorable hydrophilic interaction with Arg47. The molecular docking study results with protein kinase 2 receptor indicated that co-crystalized ligand, reference drug and synthesized compounds have similar binding mode patterns (Fig. 7) and our study has confirmed that most of the synthesized compounds have good binding affinities towards the receptor target ranging from −7.61 to −10.15 kcal mol−1. Furthermore, the computed values of free energy of binding of the synthesized compounds reflect the overall trend (Table 3).
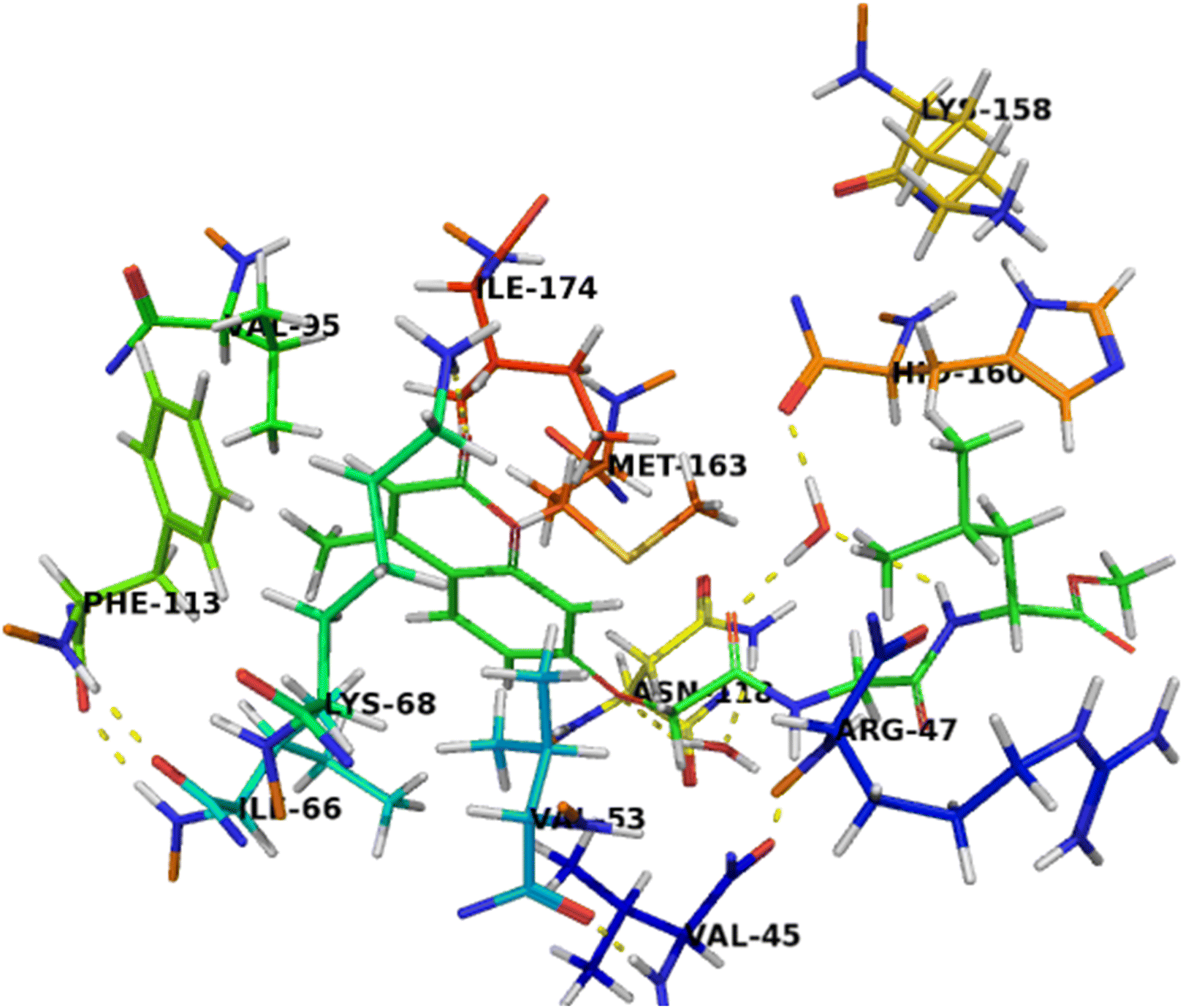 | ||
| Fig. 6 Crystal structure of CK2 receptor (PDB ID: 2QC6) with compound 7b. The hydrogen bonding interactions are presented by dashed lines. Residues contacting ligand are demonstrated as lines. | ||
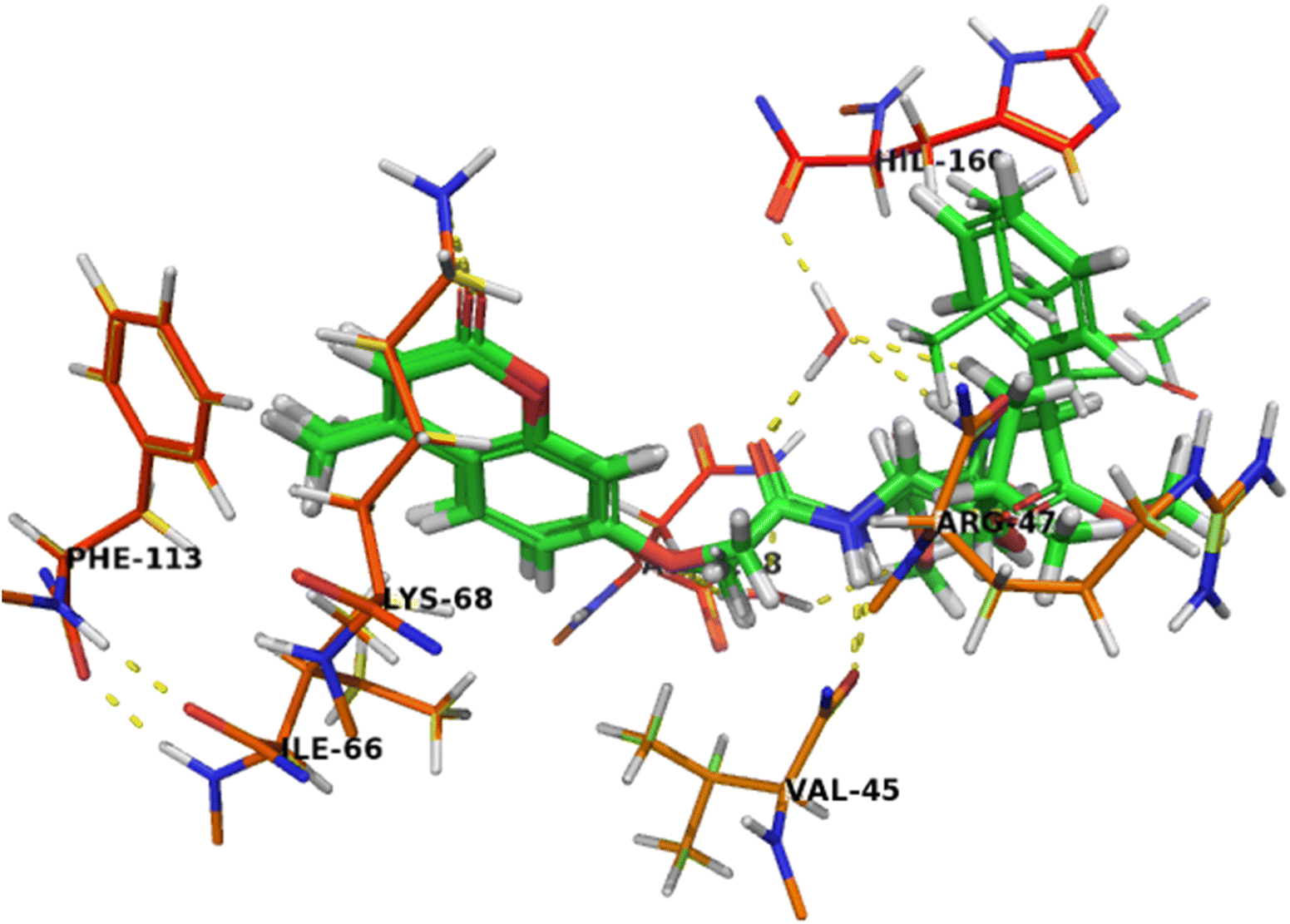 | ||
| Fig. 7 Crystal structure of CK2 receptor (PDB ID: 2QC6) with aligned compounds, 7c, 5b, 7b and 5a. The hydrogen bonding interactions are presented by dashed lines. Residues contacting ligand are demonstrated as lines. | ||
| Comp. no. | Total energy (kcal mol−1) | Hydrophilic and hydrophobic interactions | Hydrogen bonding interactions |
|---|---|---|---|
| 5a | −8.20 | Val45, Val53, Ile66, Val95, Phe113, Val116, Met163, and Ile174 | Arg47, Lys68, Asn118, and His160 |
| 5b | −8.28 | Val45, Val53, Ile66, Val95, Phe113, Val116, Met163, and Ile174 | Val45, Arg47, Lys68, Asn118 and His160 |
| 5c | −7.68 | Val45, Arg47, Val53, Ile66, Val95, Phe113, Val116, Phe121, Met163, and Ile174 | Val45, Lys68, Asn118, His160, and Asp175 |
| 6 | −8.43 | Val45, Val53, Ile66, Val95, Phe113, Val116, Met163, Phe121, and Ile174 | Val45, Lys68, Asn118, Asp120, Asp175, and His160 |
| 7a | −7.94 | Val45, Val53, Ile66, Val95, Phe113, Val116, Met163, Phe121, and Ile174 | Val45, Lys68, Asn118 and His160 |
| 7b | −8.24 | Val45, Val53, Ile66, Val95, Phe113, Val116, Phe121, Met163, and Ile174 | Val45, Arg47, Lys68, and Asn118 |
| 7c | −10.15 | Val45, Val53, Ile66, Val95, Phe113, Val116, Met163, and Ile174 | Val45, Arg47, Lys68, Asn118 and His160 |
| 8 | −7.92 | Val45, Val53, Ile66, Val95, Phe113, Phe121, Met163, and Ile174 | Arg47, Lys68, Asn118, Asp120, and His160 |
| 9a | −7.61 | Val45, Val53, Ile66, Val95, Phe113, Val116, Phe121, Met163, and Ile174 | Val45, Arg47, Lys68, Asn118, and His160 |
| 9b | −7.79 | Val45, Val53, Ile66, Val95, Phe113, Val116, Phe121, Met163, and Ile174 | Val45, Lys68, Asn118, Asp120, and His160 |
| 9c | −7.82 | Val45, Val53, Ile66, Val95, Phe113, Val116, Phe121, Met163, and Ile174 | Val45, Lys68, Asn118 Asp120, and His160 |
Analyzing the favorable binding pose, five different hydrogen bonding interactions were found, including, side-chains and backbones, Val45, Arg47, Lys68, Asn118 and His160 residues. In addition, higher hydrophobic contributions were found, due to a more deeply located binding pose inside the binding site of protein kinase 2 receptor. The mainly hydrophobic amino acid residues seem to be responsible for the affinity and selectivity of protein kinase 2 receptor, in particular, the interactions with Val45, Val53, Ile66, Val95, Phe113, Val116, Met163, and Ile174 could be responsible for the selectivity of protein kinase 2 receptor ligands and play an important role in protein kinase 2 inhibition which indicates the hydrophobic CK2 ATP-binding pocket. These fingerprints of interaction features can be used to improve and design new chemical compounds to yield better amino acid derivatives attached to coumarin nucleus in the search for the anticancer agents.
Among which were molecular weight, hydrogen bond-donors, hydrogen bond-acceptors, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, logp MDCK according to Lipinski's rule of five65 that characterizes molecular properties which are climacteric for drug's pharmacokinetics in the human body including its ADME parameter.
P, logp MDCK according to Lipinski's rule of five65 that characterizes molecular properties which are climacteric for drug's pharmacokinetics in the human body including its ADME parameter.
The partition coefficient (QPlogPo/w) and aqueous solubility (QPlogS), are critical for estimating the absorption, transport and distribution in the body. The absorption and distribution levels (QPlog(o/w)) of all synthesized compounds were ranged between 1.005 to 2.685 and −0.504 to −1.257 respectively. As a result, all synthesized compounds appeared to be good range.
Furthermore, the levels of cell permeability (QPPCaCO-2) and QPPMDCK, key factors governing drug metabolism and its access to biological membranes were ranged from 25.835 to 213.401 and 35.052 to 187.87 respectively. Thus, all the synthesized compounds have good cell permeability except compounds 7a, 9a, 9b, and 9c were predicted to get poor cell permeability.
In addition, the levels of (QPlogBB) penetration of the most compounds were expected to be safe to CNS, while compounds 9a, 9b, and 9c were expected to have moderate (QPlogBB) penetration. Furthermore, the new synthesized compounds have favorable ADME properties and the percentage human oral absorption was predicted to be more than 81.47% and 62.284 for all biologically potent compounds (5b and 7b). Therefore, all these pharmacokinetic parameters were within the acceptable range that defined for human use especially for active compounds, thereby indicating their potentials as drug-like molecules (Table 1, and ESI data†).
Therefore, structure activity relationship containing pharmacophoric features that are essential for explanation of their cytotoxic effects can be explained by a compound having a substitution at the 7-position of coumarin scaffold substituted with lengthy linker containing an aromatic amino acid (7c) was more potent than those possessing short linker (5b), or lengthy linker containing an aliphatic amino acid (7b), leading to a significant decrease in cytotoxic activity and indicated that the substitution with lengthy aromatic amino acid linker is more preferred biologically. These results are in accordance with coumarin–amino acid generated by Naik et al. and Sousa et al. which indicated that conjugation with aromatic amino acids showed potent inhibition whereas aliphatic amino acids showed compelling inhibition.66,67
Moreover, the key structural features of amino acid indicated that di-peptide (7c) was more potent than those possessing mono-peptide and tri-peptide linkers leading to a significant decrease in cytotoxic activities (5b, 9a–c) indicating that the number of peptide bond can affect the anticancer property and gave the highest activity with the most potent compound with di-peptide bond, (7c).68 On the other hand,69,70 the incorporation of a hydrophilic group seems to be a contributing factor for decreasing antitumor activity (6, 8) (Fig. 8).
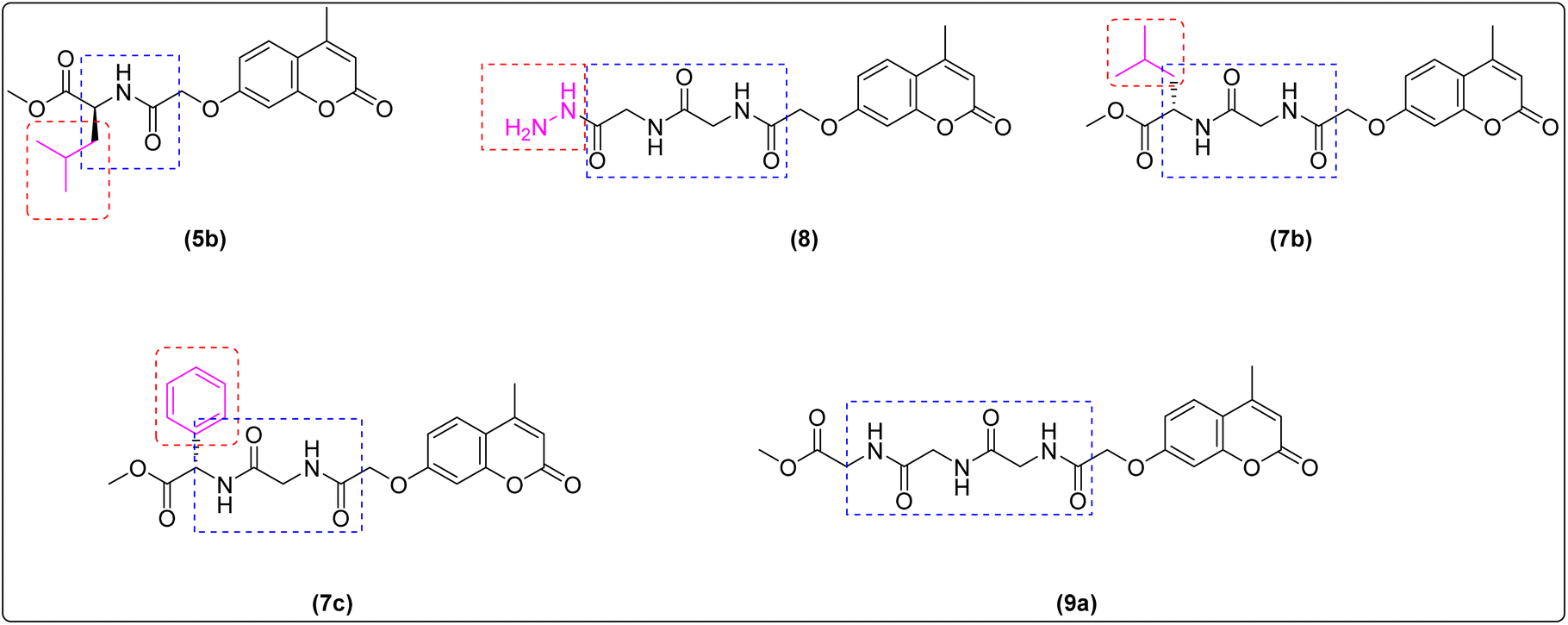 | ||
| Fig. 8 SAR of newly synthesized compounds. | ||
3 Experimental
3.1. Chemistry
All the chemical compounds which employed in this study were commercially available with analytical grade and used without any further purification. Solvents were purified and freshly distilled before using according to the standard procedures. The progress of the reaction mixtures was monitored by thin layer chromatography (TLC). The spots on the TLC plates were visualized with a UV lamp (254 nm). Melting points were measured using Thermo Fisher Scientific. IR spectra were recorded Bruker tensor 27, FT-IR Spectrophotometer. All 1H NMR and 13C NMR spectra were recorded on a Bruker 400 and 100 MHz Spectrophotometer. Chemical shifts (δ) are reported in parts per million (ppm) using tetramethylsilane (TMS) as an internal standard. Ultraviolet-visible (UV-vis) absorption spectra were recorded on PerkinElmer spectrophotometer at the wavelength of maximum absorption (kmax) in a range of DMSO at same concentrations (1 × 10−6 M). The mass spectra were run on a Shimadzu QP 5050 Ex Spectrometer. The microanalyses for C, H and N were performed on PerkinElmer elemental analyzer. All compounds were within ±0.4 of the theoretical values.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester), 1612 (CC).266 (NH), 3079 (CH aromatic), 2978 (CH aliphatic), 1675 (CO, amide), 1610 (CC), 1509 (C–N).O, lactone), 1660 (CO, amide), 1614 (CC), 1548 (C–N).O, amide), 1611 (CC), 1550 (C–N).:1) to afford the corresponding product 5a–c, 7a–c and 9a–c.O ester), 1673 (CO amide), 1610 (CC), 1507 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.41 (s, 3H, H4–CH3); 3.65 (s, 3H, O–CH3); 3.92–3.94 (d, 2H, J = 8 Hz, (CO)CH2NH); 4.70 (s, 2H, O–CH2(CO); 6.24 (s, 1H, H-3 coumarin)); 6.98 (d, 1H, J = 2.8 Hz, H-8 coumarin); 7.01–7.04 (dd, 1H, J = 8.8 Hz 2.8 Hz, H-6 coumarin), 7.71–7.73 (d, 1H, J = 8.8 Hz, H-5 coumarin) 8.64 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 45.8, 52.2, 67.4, 102.2, 111.9, 113.0, 114.1, 126.9, 153.8, 154.9, 160.5, 161.0, 168.1, 170.4; MS (EI, 70 eV) m/z (%): 307 (M+ + 2, 2.74), 306 (17.33), 305 (100), 297 (10.51), 246 (9.43), 217 (4.38), 189 (35.47), 178 (13.76), for C15H15NO6 (305.28): calcd% C 59.01, H 4.95, N 4.59, O 31.45; found: % C 59.12, H 4.85, N 4.3.O, ester), 1665 (CO, amide), 1613 (CC), 1507 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 1.08–1.12 (dd, 6H, J = 8, 16 Hz, (CH3)2 CH), 1.49 (m, 1H, (CH3)2 CH), 1.89 (m, 2H, CHCH2CH), 2.35 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 4.41 (s, 2H, O–CH2(CO); 6.95 (s, 1H, H-3 coumarin)); 7.00 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.4 Hz, 2.4 Hz, H-6 coumarin), 7.70–7.73 (d, 1H, J = 12 Hz, H-5 coumarin) 8.30–8.32 (d, 1H, J = 8 Hz, (CO)CHNH); 8.35 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 22.1, 45.5, 67.78, 101.6, 111.1, 113.0, 114.1, 126.9, 153.8, 154.9, 160.5, 161.0, 168.1, 170.4; MS (EI, 70 eV) m/z (%): 363 (M+ + 2, 0.14), 362 (0.88), 361 (100), 305 (13.43), 234 (69.60), 205 (41.75), 189 (9.41), 176 (10.24), for C19H23NO6 (361.39): calcd% C 63.15, H 6.41, N 3.88; found: % C 63.22, H 6.38, N 3.93.O, ester), 1684 (CO, amide), 1613 (CC), 1531 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.40 (s, 3H, H4–CH3); 3.65 (s, 3H, O–CH3); 4.77 (s, 2H, O–CH2(CO)); 5.50–5.52 (d, 1H, J = 8 Hz, (CO)CHNH); 6.23 (s, 1H, H-3 coumarin); 6.94 (d, 1H, J = 2.8 Hz, H-8 coumarin); 6.97–7.00 (dd, 1H, J = 8.8 Hz 2.8 Hz, H-6 coumarin), 7.36–7.44 (m, 5H, Ar–H), 7.68–7.70 (d, 1H, J = 8.8 Hz, H-5 coumarin) 8.64 (d, 1H, J = 7.2 Hz, (CO)CHNH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 52.8, 56.5, 67.0, 101.9, 111.8, 112.9, 114.0, 126.9, 128.2, 128.8, 129.1, 136.4, 153.8, 154.9, 160.5, 161.3, 167.5, 171.1; MS (EI, 70 eV) m/z (%): 382 (M+ + 1, 5.30), 381 (20.23), 323 (22.45), 322 (100), 189 (17.58), 176 (18.58), for C21H19NO6 (381.38): calcd% C 66.13, H 5.04, N 3.67; found: % C 66.16, H 5.09, N 3.59.O, ester), 1664 (CO, amide), 1622 (CC), 1546 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.41 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 3.82–3.83 (d, 2H, J = 4 Hz, (CO)CH2NH); 3.86–3.88 (d, 2H, J = 4 Hz, (CO)CH2NH); 4.68 (s, 2H, O–CH2(CO); 6.24 (s, 1H, H-3 coumarin)); 7.01 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.4 Hz 2.4 Hz, H-6 coumarin), 7.71–7.74 (d, 1H, J = 12 Hz, H-5 coumarin) 8.37 (t, 1H, J = 8 Hz, (CO)CH2NH); 8.45 (t, 1H, J = 8 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 41.0, 41.9, 52.1, 67.5, 102.1, 111.9, 112.9, 114.1, 127.0, 153.8, 155.0, 160.5, 161.0, 167.8, 169.5, 170.6; MS (EI, 70 eV) m/z (%): 364 (M+ + 2, 1.84), 362 (44.69), 330 (18.70), 274 (31.03), 246 (18.63), 187 (100), 177 (30.98), 148.05 (20.22), for C17H18N2O6 (362.33): calcd% C 56.35, H 5.01, N 7.73; found: % C 56.45, H 5.08, N 7.78.O, ester), 1672 (CO, amide), 1625 (CC), 1536 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 0.83–0.89 (dd, 6H, J = 6.4, 12 Hz, (CH3)2 CH), 1.49 (m, 1H, (CH3)2 CH), 1.63 (m, 2H, CHCH2CH), 2.41 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 3.82–3.83 (d, 2H, J = 3.6 Hz, (CO)CH2NH); 3.86–3.88 (m, 2H, 2CHNH); 4.65 (s, 2H, O–CH2(CO); 6.95 (s, 1H, H-3 coumarin)); 7.00 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.4 Hz, 2.4 Hz, H-6 coumarin), 7.70–7.73 (d, 1H, J = 12 Hz, H-5 coumarin) 8.30–8.32 (d, 1H, J = 8 Hz, (CO)CHNH); 8.35 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 21.7, 23.1, 24.6, 41.7, 46.0, 50.6, 52.3, 67.5, 102.1, 111.9, 112.9, 114.1, 127.0, 153.8, 155.0, 160.5, 161.1, 167.7, 169.0, 173.3; MS (EI, 70 eV) m/z (%): 419 (M+ + 1, 1.60), 418 (6.26), 330 (15.15), 274 (7.64), 246 (12.97), 233 (39.07), 189 (32.41), 176 (33.91), 148 (57.14), 86 (100) for C21H26N2O6 (418.44): calcd% C 60.28, H 6.26, N 6.69; found: % C 60.34, H 6.29, N 6.75.O, ester), 1671 (CO, amide), 1627 (CC), 1541 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.41 (s, 3H, H4–CH3); 3.60 (s, 3H, O–CH3); 3.98–3.91 (d, 2H, J = 5.6 Hz, (CO)CH2NH); 4.68 (s, 2H, O–CH2(CO); 5.45–5.46 (d, 1H J = 702 Hz, PhCHNH), 6.24 (s, 1H, H-3 coumarin)); 6.99 (s, 1H, H-8 coumarin); 7.01–7.04 (dd, 1H, J = 8.8 Hz 2.4 Hz, H-6 coumarin), 7.29–7.40 (m, 5H, Ar–H), 7.71–7.73 (d, 1H, J = 8.8 Hz, H-5 coumarin) 8.38 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 8.83–8.85 (d, 1H, J = 702 Hz, (CO)CHNH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 41.7, 52.7, 56.6, 67.5, 102.2, 111.9, 112.9, 114.1, 127.0, 128.1, 128.7, 129.1, 136.6, 153.8, 155.0, 160.5, 161.1, 167.8, 168.9, 171.3; MS (EI, 70 eV) m/z (%): 440 (M+ + 2, 0.06), 438 (0.65), 378 (14.06), 274 (1.43), 246 (4.88), 189 (7.53), 176 (4.91), 148 (6.42), 106 (100), for C23H27N2O7 (438.43): calcd% C 63.01, H 5.06, N 6.39; found: % C 63.07, H 5.09, N 6.35.O, ester), 1647 (CO, amide), 1607 (CC), 1557 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.40 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 3.76–3.77 (d, 2H, J = 6 Hz, (CO)CH2NH); 3.83–3.85 (m, 4H, 2(CO)CH2NH); 4.68 (s, 2H, O–CH2(CO); 6.23 (s, 1H, H-3 coumarin)); 7.00 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.4 Hz 2.4 Hz, H-6 coumarin), 7.71–7.73 (d, 1H, J = 8.8 Hz, H-5 coumarin) 8.37 (m, 2H, 2 NH (CO)CH2); 8.45 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 40.9, 42.1, 42.2, 52.1, 67.5, 102.2, 111.9, 112.9, 114.1, 127.0, 153.8, 155.0, 160.5, 161.0, 167.9, 169.3, 169.8, 170.6; MS (EI, 70 eV) m/z (%): 421 (M+ + 2, 1.95), 419 (29.07), 387 (65.69), 330 (59.51), 274 (100), 246 (98.35), 189 (78.22), 177 (95.15), 148 (44.98), for C19H21N3O8 (319.39): calcd% C 54.41, H 5.05, N 10.02; found: % C 54.46, H 5.09, N 9.92.O, ester), 1660 (CO, amide), 1622 (CC), 1544 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 0.83–0.89 (dd, 6H, J = 6.4, 12 Hz, (CH3)2 CH), 1.13 (t, 2H, J = 12 Hz, CHCH2CH), 1.25 (m, 1H, (CH3)2 CH), 2.41 (s, 3H, H4–CH3); 3.61 (s, 3H, O–CH3), 3.61–3.76 (m, 4H, 2CH2NH), 4.42 (m, 1H, CHNH), 4.67 (s, 2H, O–CH2(CO), 6.24 (s, 1H, H-3 coumarin)), 7.00 (s, 1H, H-8 coumarin), 7.01–7.04 (dd, 1H, J = 8.4 Hz 2.4 Hz, H-6 coumarin), 7.71–7.73 (d, 1H, J = 12 Hz, H-5 coumarin), 8.19 (t, 1H, J = 8 Hz, (CO)CH2NH), 8.37 (d, 1H, J = 6 Hz, (CO)CHNH); 8.44 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 21.7, 22.8, 23.1, 24.5, 42.0, 46.0, 50.6, 52.1, 67.5, 102.2, 111.9, 112.9, 114.1, 127.0, 153.8, 155.0, 160.5, 161.0, 167.8, 169.3, 173.3; MS (EI, 70 eV) m/z (%): 477 (M+ + 2, 0.08), 475 (0.81), 443 (7.22), 416 (9.31), 330 (5.36), 274 (23.46), 246 (20.81), 189 (26.17), 177 (26.36), 148.05 (32.69), 86 (100), for C17H18N2O6 (475.49): calcd% C 58.10, H 6.15, N 8.84; found: % C 58.15, H 6.12, N 8.81.O, ester), 1653 (CO, amide), 1618 (CC), 1552 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.41 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 3.81–3.85 (m, 4H, 2CH2NH); 4.67 (s, 2H, O–CH2(CO); 5.43 (d, 1H, J = 7.2 Hz, PhCHNH), 6.24 (s, 1H, H-3 coumarin)); 7.00 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.8 Hz 2.4 Hz, H-6 coumarin), 7.29–7.40 (m, 5H, Ar–H), 7.71–7.73 (d, 1H, J = 8.8 Hz, H-5 coumarin), 8.19 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 8.43 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 8.75–8.77 (d, 1H, J = 7.2 Hz, (CO)CHNH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 41.9, 42.1, 52.7, 56.6, 67.5, 102.2, 111.9, 112.9, 114.1, 127.0, 128.1, 128.7, 129.1, 136.6, 153.8, 155.0, 160.5, 161.0, 167.9, 169.1, 169.2, 171.3; MS (EI, 70 eV) m/z (%): 497 (M+ + 2, 0.07), 495 (0.16), 463 (13.19), 362 (5.70), 290 (13.27), 274 (15.07), 246 (11.77), 189 (26.38), 176 (14.57), 148 (21.40), 106 (100), for C25H25N3O8 (495.38): calcd% C 60.60, H 5.09, N 8.48; found: % C 60.64, H 5.06, N 8.42.
O ester), 1612 (CC).266 (NH), 3079 (CH aromatic), 2978 (CH aliphatic), 1675 (CO, amide), 1610 (CC), 1509 (C–N).O, lactone), 1660 (CO, amide), 1614 (CC), 1548 (C–N).O, amide), 1611 (CC), 1550 (C–N).:1) to afford the corresponding product 5a–c, 7a–c and 9a–c.O ester), 1673 (CO amide), 1610 (CC), 1507 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.41 (s, 3H, H4–CH3); 3.65 (s, 3H, O–CH3); 3.92–3.94 (d, 2H, J = 8 Hz, (CO)CH2NH); 4.70 (s, 2H, O–CH2(CO); 6.24 (s, 1H, H-3 coumarin)); 6.98 (d, 1H, J = 2.8 Hz, H-8 coumarin); 7.01–7.04 (dd, 1H, J = 8.8 Hz 2.8 Hz, H-6 coumarin), 7.71–7.73 (d, 1H, J = 8.8 Hz, H-5 coumarin) 8.64 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 45.8, 52.2, 67.4, 102.2, 111.9, 113.0, 114.1, 126.9, 153.8, 154.9, 160.5, 161.0, 168.1, 170.4; MS (EI, 70 eV) m/z (%): 307 (M+ + 2, 2.74), 306 (17.33), 305 (100), 297 (10.51), 246 (9.43), 217 (4.38), 189 (35.47), 178 (13.76), for C15H15NO6 (305.28): calcd% C 59.01, H 4.95, N 4.59, O 31.45; found: % C 59.12, H 4.85, N 4.3.O, ester), 1665 (CO, amide), 1613 (CC), 1507 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 1.08–1.12 (dd, 6H, J = 8, 16 Hz, (CH3)2 CH), 1.49 (m, 1H, (CH3)2 CH), 1.89 (m, 2H, CHCH2CH), 2.35 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 4.41 (s, 2H, O–CH2(CO); 6.95 (s, 1H, H-3 coumarin)); 7.00 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.4 Hz, 2.4 Hz, H-6 coumarin), 7.70–7.73 (d, 1H, J = 12 Hz, H-5 coumarin) 8.30–8.32 (d, 1H, J = 8 Hz, (CO)CHNH); 8.35 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 22.1, 45.5, 67.78, 101.6, 111.1, 113.0, 114.1, 126.9, 153.8, 154.9, 160.5, 161.0, 168.1, 170.4; MS (EI, 70 eV) m/z (%): 363 (M+ + 2, 0.14), 362 (0.88), 361 (100), 305 (13.43), 234 (69.60), 205 (41.75), 189 (9.41), 176 (10.24), for C19H23NO6 (361.39): calcd% C 63.15, H 6.41, N 3.88; found: % C 63.22, H 6.38, N 3.93.O, ester), 1684 (CO, amide), 1613 (CC), 1531 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.40 (s, 3H, H4–CH3); 3.65 (s, 3H, O–CH3); 4.77 (s, 2H, O–CH2(CO)); 5.50–5.52 (d, 1H, J = 8 Hz, (CO)CHNH); 6.23 (s, 1H, H-3 coumarin); 6.94 (d, 1H, J = 2.8 Hz, H-8 coumarin); 6.97–7.00 (dd, 1H, J = 8.8 Hz 2.8 Hz, H-6 coumarin), 7.36–7.44 (m, 5H, Ar–H), 7.68–7.70 (d, 1H, J = 8.8 Hz, H-5 coumarin) 8.64 (d, 1H, J = 7.2 Hz, (CO)CHNH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 52.8, 56.5, 67.0, 101.9, 111.8, 112.9, 114.0, 126.9, 128.2, 128.8, 129.1, 136.4, 153.8, 154.9, 160.5, 161.3, 167.5, 171.1; MS (EI, 70 eV) m/z (%): 382 (M+ + 1, 5.30), 381 (20.23), 323 (22.45), 322 (100), 189 (17.58), 176 (18.58), for C21H19NO6 (381.38): calcd% C 66.13, H 5.04, N 3.67; found: % C 66.16, H 5.09, N 3.59.O, ester), 1664 (CO, amide), 1622 (CC), 1546 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.41 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 3.82–3.83 (d, 2H, J = 4 Hz, (CO)CH2NH); 3.86–3.88 (d, 2H, J = 4 Hz, (CO)CH2NH); 4.68 (s, 2H, O–CH2(CO); 6.24 (s, 1H, H-3 coumarin)); 7.01 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.4 Hz 2.4 Hz, H-6 coumarin), 7.71–7.74 (d, 1H, J = 12 Hz, H-5 coumarin) 8.37 (t, 1H, J = 8 Hz, (CO)CH2NH); 8.45 (t, 1H, J = 8 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 41.0, 41.9, 52.1, 67.5, 102.1, 111.9, 112.9, 114.1, 127.0, 153.8, 155.0, 160.5, 161.0, 167.8, 169.5, 170.6; MS (EI, 70 eV) m/z (%): 364 (M+ + 2, 1.84), 362 (44.69), 330 (18.70), 274 (31.03), 246 (18.63), 187 (100), 177 (30.98), 148.05 (20.22), for C17H18N2O6 (362.33): calcd% C 56.35, H 5.01, N 7.73; found: % C 56.45, H 5.08, N 7.78.O, ester), 1672 (CO, amide), 1625 (CC), 1536 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 0.83–0.89 (dd, 6H, J = 6.4, 12 Hz, (CH3)2 CH), 1.49 (m, 1H, (CH3)2 CH), 1.63 (m, 2H, CHCH2CH), 2.41 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 3.82–3.83 (d, 2H, J = 3.6 Hz, (CO)CH2NH); 3.86–3.88 (m, 2H, 2CHNH); 4.65 (s, 2H, O–CH2(CO); 6.95 (s, 1H, H-3 coumarin)); 7.00 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.4 Hz, 2.4 Hz, H-6 coumarin), 7.70–7.73 (d, 1H, J = 12 Hz, H-5 coumarin) 8.30–8.32 (d, 1H, J = 8 Hz, (CO)CHNH); 8.35 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 21.7, 23.1, 24.6, 41.7, 46.0, 50.6, 52.3, 67.5, 102.1, 111.9, 112.9, 114.1, 127.0, 153.8, 155.0, 160.5, 161.1, 167.7, 169.0, 173.3; MS (EI, 70 eV) m/z (%): 419 (M+ + 1, 1.60), 418 (6.26), 330 (15.15), 274 (7.64), 246 (12.97), 233 (39.07), 189 (32.41), 176 (33.91), 148 (57.14), 86 (100) for C21H26N2O6 (418.44): calcd% C 60.28, H 6.26, N 6.69; found: % C 60.34, H 6.29, N 6.75.O, ester), 1671 (CO, amide), 1627 (CC), 1541 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.41 (s, 3H, H4–CH3); 3.60 (s, 3H, O–CH3); 3.98–3.91 (d, 2H, J = 5.6 Hz, (CO)CH2NH); 4.68 (s, 2H, O–CH2(CO); 5.45–5.46 (d, 1H J = 702 Hz, PhCHNH), 6.24 (s, 1H, H-3 coumarin)); 6.99 (s, 1H, H-8 coumarin); 7.01–7.04 (dd, 1H, J = 8.8 Hz 2.4 Hz, H-6 coumarin), 7.29–7.40 (m, 5H, Ar–H), 7.71–7.73 (d, 1H, J = 8.8 Hz, H-5 coumarin) 8.38 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 8.83–8.85 (d, 1H, J = 702 Hz, (CO)CHNH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 41.7, 52.7, 56.6, 67.5, 102.2, 111.9, 112.9, 114.1, 127.0, 128.1, 128.7, 129.1, 136.6, 153.8, 155.0, 160.5, 161.1, 167.8, 168.9, 171.3; MS (EI, 70 eV) m/z (%): 440 (M+ + 2, 0.06), 438 (0.65), 378 (14.06), 274 (1.43), 246 (4.88), 189 (7.53), 176 (4.91), 148 (6.42), 106 (100), for C23H27N2O7 (438.43): calcd% C 63.01, H 5.06, N 6.39; found: % C 63.07, H 5.09, N 6.35.O, ester), 1647 (CO, amide), 1607 (CC), 1557 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.40 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 3.76–3.77 (d, 2H, J = 6 Hz, (CO)CH2NH); 3.83–3.85 (m, 4H, 2(CO)CH2NH); 4.68 (s, 2H, O–CH2(CO); 6.23 (s, 1H, H-3 coumarin)); 7.00 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.4 Hz 2.4 Hz, H-6 coumarin), 7.71–7.73 (d, 1H, J = 8.8 Hz, H-5 coumarin) 8.37 (m, 2H, 2 NH (CO)CH2); 8.45 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 40.9, 42.1, 42.2, 52.1, 67.5, 102.2, 111.9, 112.9, 114.1, 127.0, 153.8, 155.0, 160.5, 161.0, 167.9, 169.3, 169.8, 170.6; MS (EI, 70 eV) m/z (%): 421 (M+ + 2, 1.95), 419 (29.07), 387 (65.69), 330 (59.51), 274 (100), 246 (98.35), 189 (78.22), 177 (95.15), 148 (44.98), for C19H21N3O8 (319.39): calcd% C 54.41, H 5.05, N 10.02; found: % C 54.46, H 5.09, N 9.92.O, ester), 1660 (CO, amide), 1622 (CC), 1544 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 0.83–0.89 (dd, 6H, J = 6.4, 12 Hz, (CH3)2 CH), 1.13 (t, 2H, J = 12 Hz, CHCH2CH), 1.25 (m, 1H, (CH3)2 CH), 2.41 (s, 3H, H4–CH3); 3.61 (s, 3H, O–CH3), 3.61–3.76 (m, 4H, 2CH2NH), 4.42 (m, 1H, CHNH), 4.67 (s, 2H, O–CH2(CO), 6.24 (s, 1H, H-3 coumarin)), 7.00 (s, 1H, H-8 coumarin), 7.01–7.04 (dd, 1H, J = 8.4 Hz 2.4 Hz, H-6 coumarin), 7.71–7.73 (d, 1H, J = 12 Hz, H-5 coumarin), 8.19 (t, 1H, J = 8 Hz, (CO)CH2NH), 8.37 (d, 1H, J = 6 Hz, (CO)CHNH); 8.44 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 21.7, 22.8, 23.1, 24.5, 42.0, 46.0, 50.6, 52.1, 67.5, 102.2, 111.9, 112.9, 114.1, 127.0, 153.8, 155.0, 160.5, 161.0, 167.8, 169.3, 173.3; MS (EI, 70 eV) m/z (%): 477 (M+ + 2, 0.08), 475 (0.81), 443 (7.22), 416 (9.31), 330 (5.36), 274 (23.46), 246 (20.81), 189 (26.17), 177 (26.36), 148.05 (32.69), 86 (100), for C17H18N2O6 (475.49): calcd% C 58.10, H 6.15, N 8.84; found: % C 58.15, H 6.12, N 8.81.O, ester), 1653 (CO, amide), 1618 (CC), 1552 (C–N); 1H NMR (400 MHz; DMSO-d6) δ (ppm): 2.41 (s, 3H, H4–CH3); 3.63 (s, 3H, O–CH3); 3.81–3.85 (m, 4H, 2CH2NH); 4.67 (s, 2H, O–CH2(CO); 5.43 (d, 1H, J = 7.2 Hz, PhCHNH), 6.24 (s, 1H, H-3 coumarin)); 7.00 (s, 1H, H-8 coumarin); 7.02–7.05 (dd, 1H, J = 8.8 Hz 2.4 Hz, H-6 coumarin), 7.29–7.40 (m, 5H, Ar–H), 7.71–7.73 (d, 1H, J = 8.8 Hz, H-5 coumarin), 8.19 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 8.43 (t, 1H, J = 5.6 Hz, (CO)CH2NH); 8.75–8.77 (d, 1H, J = 7.2 Hz, (CO)CHNH); 13C NMR (100 MHz; DMSO-d6) δ (ppm): 18.6, 41.9, 42.1, 52.7, 56.6, 67.5, 102.2, 111.9, 112.9, 114.1, 127.0, 128.1, 128.7, 129.1, 136.6, 153.8, 155.0, 160.5, 161.0, 167.9, 169.1, 169.2, 171.3; MS (EI, 70 eV) m/z (%): 497 (M+ + 2, 0.07), 495 (0.16), 463 (13.19), 362 (5.70), 290 (13.27), 274 (15.07), 246 (11.77), 189 (26.38), 176 (14.57), 148 (21.40), 106 (100), for C25H25N3O8 (495.38): calcd% C 60.60, H 5.09, N 8.48; found: % C 60.64, H 5.06, N 8.42.3.2. Biological evaluation
3.3. Molecular docking
The new synthesized compounds were subjected to molecular docking in order to recognize their binding modes and free energies of binding towards CK2 receptor. Therefore, crystallographic structure of CK2 receptor in complex with co-crystalized ligand, G12 was retrieved from Protein Data Bank (PDB ID: 2QC6). The docking analysis was performed using AutoDock program. The 2D chemical structures of the synthesized compounds and co-crystalized ligand were sketched using ChemBioDraw Ultra 14.0. In order to assess the efficacy of the method used for docking, we performed molecular redocking of co-crystalized ligand, G12 which validated by getting low RMSD values between the docked and X-ray structures. Then the docking of co-crystalized ligand and the synthesized compounds were performed using default protocol parameters. The docking results from AutoDock program was further analyzed and visualized using Pymol software to investigate the putative interaction mechanism with CK2 target. The complexes of synthesized compounds were subjected to energy minimization to optimize the complexes and subsequent MD simulations to predict the energetically most favorable binding mode for the respective compounds and to decrease in the number of false positive and negative docking results. Therefore, the complexes were subjected to MD simulation at 300 K during 400 ps. The time step of the simulation was 2.0 fs with a cutoff 10 Å for the non-bonded interactions. The MD simulation is performed at constant temperature and pressure. During the MD simulation all backbone atoms of the complexes were restrained to their starting positions with harmonic force constant 2.0 kcal (mol−1 Å−2).4 Conclusion
Coumarin scaffold have been widely used as an effective template for drug discovery. Thus, a series of novel hybrid coumarin derivatives were designed and chemically synthesized in synthetically useful yields. All the novel synthesized compounds were biologically evaluated in vitro for their cytotoxic activities against a panel of three human cancer cell lines, namely HepG-2, PC-3, and HCT-116. The results of cytotoxic estimation indicated that the anticancer activity of the synthesized compounds showed excellent to weak anticancer activity. In particular, compound 7c appeared to be significantly more potent than doxorubicin with IC50 values of 34.07, 16.06, and 16.02 μM against tested cell lines. Furthermore, compound 5b displayed the highest cytotoxic effect against all tested cell lines with IC50 values of 42.16, 59.74, and 35.05 μM compared with an anticancer drug, doxorubicin as a control drug. Besides, compounds 7b, and 5a possessed good anti-proliferative activities against the three cell lines with IC50 values ranging from 61 μM to 105 μM. Pharmacophoric features indicated that coumarin scaffold having a dipeptide linker substituted with phenyl moiety was more potent than other analogs leading to significant decrease in cytotoxic activity. In addition, the most potent compounds were further evaluated for their activities against CK2 kinase inhibitors. The most potent compound 5b displayed the most promising inhibitory activity against CK2 kinase with IC50 value of 0.117 ± 0.005 μM compared with roscovetine as control drug with IC50 value of 0.251 ± 0.011 μM. Finally, molecular docking studies were conducted to explain the anticancer results with the prospective target and the ADME properties were calculated to predict pharmacokinetic and toxic properties of the synthesized compounds.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- M. J. Thun, J. O. DeLancey, M. M. Center, A. Jemal and E. M. Ward, The global burden of cancer: priorities for prevention, Carcinogenesis, 2009, 31, 100–110 CrossRef PubMed.
- S. Yu, Z. Chen, X. Zeng, X. Chen and Z. Gu, Advances in nanomedicine for cancer starvation therapy, Theranostics, 2019, 26, 8026–8047 CrossRef PubMed.
- J. Jampilek, Heterocycles in medicinal chemistry, Molecules, 2019, 21, 3839 CrossRef PubMed.
- A. Stefanachi, F. Leonetti, L. Pisani, M. Catto and A. Carotti, Coumarin: A natural, privileged and versatile scaffold for bioactive compounds, Molecules, 2018, 2, 250 CrossRef PubMed.
- F. Annunziata, C. Pinna, S. Dallavalle, L. Tamborini and A. Pinto, An overview of coumarin as a versatile and readily accessible scaffold with broad-ranging biological activities, Int. J. Mol. Sci., 2020, 13, 7370201 Search PubMed.
- A.-K. Singh, P. K. Patel, K. Choudhary, J. Joshi, D. Yaday and J. O. Jin, Quercetin and coumarin inhibit dipeptidyl peptidase-IV and exhibits antioxidant properties: in silico, in vitro, ex vivo, Biomolecules, 2020, 2, 207 CrossRef PubMed.
- G. Wang, Y. Liu, L. Zhang, L. An, R. Chen, Y. Liu, Q. Luo, Y. Li, H. Wang and Y. Xue, Computational study on the antioxidant property of coumarin-fused coumarins, Food Chem., 2020, 340, 125446 CrossRef PubMed.
- C. T. Supuran, Coumarin carbonic anhydrase inhibitors from natural sources, J. Enzyme Inhib. Med. Chem., 2020, 1, 1462–1470 CrossRef PubMed.
- D. Cuffaro, E. Nuti and A. Rossello, An overview of carbohydrate-based carbonic anhydrase inhibitors, J. Enzyme Inhib. Med. Chem., 2020, 1, 1906–1922 CrossRef PubMed.
- R. Nagamallu, B. Srinivasan, M. B. Ningappa and A. K. Kariyappa, Synthesis of novel coumarin appended bis(formylpyrazole) derivatives: studies on their antimicrobial and antioxidant activities, Bioorg. Med. Chem. Lett., 2016, 2, 690–694 CrossRef PubMed.
- C. Jia, J. Zhang, L. Yu, C. Wang, Y. Yang, X. Rong, K. Xu and M. Chu, Antifungal activity of coumarin against candida albicans is related to apoptosis, Front. Cell. Infect. Microbiol., 2018, 8, 445 CrossRef CAS PubMed.
- R. Elias, R. I. Benhamou, Q. Z. Jaber, O. Dorot, S. L. Zada, K. Oved, E. Pichinuk and F. Micha, Antifungal activity, mode of action variability, and subcellular distribution of coumarin-based antifungal azoles, Eur. J. Med. Chem., 2019, 179, 779–790 CrossRef CAS PubMed.
- S. K. Chidambaram, D. Saud-Alarifi, S. Radhakrishnan and I. Akbar, In silico molecular docking: evaluation of coumarin based derivatives against SARS-CoV-2, J. Infect. Public Health, 2020, 11, 1–1677 Search PubMed.
- S. Mishra, A. Pandey and S. Manvanti, Coumarin: an emerging antiviral agent, Heliyon, 2000, 1, e03217 Search PubMed.
- M. Bian, Q. Q. Ma, Y. Wu, H. H. Du and G. Guo-hua, Small molecule compounds with good anti-inflammatory activity reported in the literature from 01/2009 to 05/2021: a review, J. Enzyme Inhib. Med. Chem., 2021, 1, 2139–2159 CrossRef PubMed.
- H. Yu, Z. Hou, X. Yang, Y. Mou and C. Guo, Design, synthesis, and mechanism of dihydroartemisinin-coumarin hybrids as potential anti-neuroinflammatory agents, Molecules, 2019, 9, 1672 CrossRef PubMed.
- H. W. Querfurth and F. M. Laferla, Alzheimer's disease, N. Engl. J. Med., 2010, 4, 329–344 CrossRef PubMed.
- M. X. Song and X. Q. Deng, Recent development on triazole nucleus in anticonvulsant compounds: a review, J. Enzyme Inhib. Med. Chem., 2018, 1, 453–478 CrossRef PubMed.
- C. X. Wei, M. Bian and G. H. Gong, Current research on antiepileptic compounds, Molecules, 2015, 11, 20471–20776 Search PubMed.
- M. Greaves, Pharmacogenetics in the management of coumarin anticoagulant therapy: the way forward or an expensive diversion?, PLoS Med., 2005, 10, e342 CrossRef PubMed.
- G. Lippi, R. Gosselin and E. J. Favaloro, Current and emerging direct oral anticoagulants state-of-the-art, Semin. Thromb. Hemostasis, 2019, 5, 490–501 Search PubMed.
- A. M. Abu-Odeh and W. H. Talib, Middle east medicinal plants in the treatment of diabetes: a review, Molecules, 2021, 3, 742 CrossRef PubMed.
- Y. Hu, B. Wang, J. Yang, T. Liu, J. Sun and X. Wang, Synthesis and biological evaluation of 3-arylcoumarin derivatives as potential anti-diabetic agents, J. Enzyme Inhib. Med. Chem., 2019, 1, 15–30 CrossRef PubMed.
- D. Hanahan and R. A. Weinberg, The hallmarks of cancer, Cell, 2000, 1, 57–70 CrossRef PubMed.
- A. Herrera-R, W. Castrillon, E. Otero, E. Ruiz, M. Carda, R. Agut, T. Naranjo, G. Moreno, M. E. Maldonado and W. Cardona-G, Synthesis and antiproliferative activity of 3- and 7-styrylcoumarins, Med. Chem. Res., 2018, 27, 1893–1905 CrossRef CAS.
- D. Gupta, S. V. Gupta, K. D. Lee and G. L. Amidon, Chemical and enzymatic stability of amino acid produrgs containing methoxy, ethoxy and propylene glycol linkers, Mol. Pharmaceutics, 2009, 5, 1604–1611 CrossRef PubMed.
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Coumarin-based small-molecule fluorescent, Chem. Rev., 2019, 18, 10403–10519 CrossRef PubMed.
- J. Ge, L. Li and S. Q. Yao, Amino acid that mimics phosphotyrosine, Chem. Commun., 2011, 39, 10939–10941 RSC.
- Y. L. Lo, C. T. Ho and F. L. Tsai, Inhibit multidrug resistance and induce apoptosis by using glycocholic acid and epirubicin, Eur. J. Pharm. Sci., 2008, 35, 52–67 CrossRef CAS PubMed.
- F. Chu, X. Xu, G. Li, S. Gu, K. Xu, Y. Gong, B. Xu, M. Wang, H. Zhang, Y. Zhang, P. Wang and H. Lei, Amino acid derivatives of ligustrazine-oleanolic acid as new cytotoxic agents, Molecules, 2014, 19, 18215–18231 CrossRef PubMed.
- C. S. Francisco, L. R. Rodrigues, M. F. S. A. Cerqueira Nuno, M. F. Oliverira-Campo Ana and L. M. Rodrigues, Synthesis of novel benzofurocoumarin analogues and their anti-proliferative effect on human cancer cell lines, Eur. J. Med. Chem., 2012, 47, 370–376 CrossRef CAS PubMed.
- I. Fotopoulos and D. Hadjipavlou-Litina, Hybrids of coumarin derivatives as potent and multifunctional bioactive agents: a review, Med. Chem., 2020, 3, 272–306 CrossRef PubMed.
- R. Bhatia and R. K. Rawal, Coumarin hybrids: promising scaffolds in the treatment of breast cancer, Mini-Rev. Med. Chem., 2019, 17, 1443–1458 CrossRef PubMed.
- A. V. Yurkovetskiy and R. J. Fram, XMT-1001, a novel polymeric camptothecin pro-drug in clinical development for patients with advanced cancer, Adv. Drug Delivery Rev., 2009, 61, 1193–1202 CrossRef CAS PubMed.
- D. Cao, Y. Liu, W. Yan, C. Wang, P. Bai, T. Wang, M. Tang, X. Wang, Z. Yang, B. Ma, L. Ma, L. Lei, F. Wang, B. Xu, Y. Zhou, T. Yang and L. Chen, Design, synthesis, and evaluation of in vitro and in vivo anticancer activity of 4-substituted coumarins: a novel class of potent tubulin polymerization, J. Med. Chem., 2016, 12, 5721–5739 CrossRef PubMed.
- W. Zhang, Z. Li, M. Zhou, F. Wu, X. Hou, H. Luo, H. Liu, X. Han, G. Yan, Z. Ding and R. Li, Synthesis and biological evaluation of 4-(1,2,3-triazol-1-1yl)coumarin derivatives as potential antitumor agents, Bioorg. Med. Chem. Lett., 2014, 3, 799–807 CrossRef PubMed.
- R. An, Z. Hou, J. T. Li, H. N. Yu, Y. H. Mou and C. Guo, Design, synthesis and biological evaluation of novel 4-substituted coumarin derivatives as antitumor agents, Molecules, 2018, 9, 2281 CrossRef PubMed.
- F. Belluti, G. Fontana, L. D. Bo, N. Carenini, C. Giommarelli and F. Zunino, Design, synthesis and anticancer activities of stilbene-coumarin hybrid compounds: identification of novel proapoptoic agents, Bioorg. Med. Chem., 2010, 10, 3543–3550 CrossRef PubMed.
- E. H. Maleki, A. Z. Bahrami, H. Sadeghian and M. M. Matin, Discovering the structure activity relationships of different o-prenylated coumarin derivatives as effective anticancer agents in human cervical cancer cells, Toxicol. In Vitro, 2020, 63, 104745 CrossRef CAS PubMed.
- A. Rawat and B. V. A. Reddy, Recent advances on anticancer activity of coumarin derivatives, Eur. J. Med. Chem., 2022, 5, 100038 CAS.
- A. Herrera-R, W. Castrillón, E. Otero, E. Ruiz, M. Carda, R. Agut, T. Naranjo, G. Moreno and M. E. Maldonado, Cardona-G W. Synthesis and antiproliferative activity of 3- and 7-styrylcoumarins, Med. Chem. Res., 2018, 27, 1893–1905 CrossRef CAS.
- Q. P. Diao, H. Guo and G. Q. Wang, Design, Synthesis and In Vitro Anticancer Activities of Diethylene Glycol Tethered Isatin-1,2,3-triazole-coumarin Hybrids, J. Heterocycl. Chem., 2019, 56, 1667–1671 CrossRef CAS.
- Z. H. Li, X. Q. Liu, P. F. Geng, F. Z. Suo, J. L. Ma, B. Yu, T. Q. Zhao, Z. Q. Zhou, C. X. Huang and Y. C. Zheng, Discovery of [1,2,3]Triazolo[4,5-d]pyrimidine Derivatives as Novel LSD1 Inhibitors, ACS Med. Chem. Lett., 2017, 8, 384–389 CrossRef CAS PubMed.
- H. Yu, Z. Hou, Y. Tian, Y. Mou and C. Guo, Design, synthesis, cytotoxicity and mechanism of novel dihydroartemisinin-coumarin hybrids as potential anti-cancer agents, Eur. J. Med. Chem., 2018, 151, 434–449 CrossRef CAS PubMed.
- Z. Xu, X. F. Song, Y. Q. Hu, M. Qiang and Z. S. Lv, Azide-alkyne cycloaddition towards 1H-1,2,3-triazole-tethered gatifloxacin and isatin conjugates: Design, synthesis and in vitro anti-mycobacterial evaluation, Eur. J. Med. Chem., 2017, 138, 66–71 CrossRef CAS PubMed.
- B. S. Vig, K. M. Huttunen, K. Laine and J. Rautio, Amino acids as promoities in prodrug design and development, Adv. Drug Delivery Rev., 2013, 65, 1370–1385 CrossRef CAS PubMed.
- T. Nakanishi, I. Tamai, A. Takaki and A. Tsuji, Cancer cell-targeted drug delivery utilizing oligopeptide transport activity, Int. J. Cancer, 2000, 88, 274–280 CrossRef CAS PubMed.
- X. Song, P. L. Lorenzi, C. P. Landowski, B. S. Vig, J. M. Hilfinger and G. L. Amidon, Amino acid ester prodrug of the anticancer agent gemcitabine: Synthesis, bioconversion, metabolic bioevasion and hPEPT1-mediated transport, Mol. Pharm., 2005, 2, 157–167 CrossRef CAS PubMed.
- A. Lacy and R. O. Kennedy, Studies on coumarins and coumarin-related compounds to determine their therapeutic role in the treatment of cancer, Curr. Pharm. Des., 2004, 30, 3797–37811 CrossRef PubMed.
- A. Chilin, R. Battistutta, A. Bortolato, G. Cozza, S. Zanatta, G. Poletto, M. Mazzorana, G. Zagotto, E. Uriarte, A. Guiotto, L. A. Pinna, F. Meggio and S. Moro, Coumarin as attractive casein kinase 2 (CK2) inhibitors scaffold: an integrate approach to elucidate the putative binding motif and explain structure activity relationships, J. Med. Chem., 2008, 4, 752–759 CrossRef PubMed.
- N. Zhang and R. Zhong, Docking and 3D-QSAR studies of 7-hydroxycoumarin derivatives as CK2 inhibitors, Eur. J. Med. Chem., 2010, 1, 292–297 CrossRef PubMed.
- V. S. V. Satyanarayana, P. Sreevani, A. Sivakumar and V. Vijayakumar, Synthesis and antimicrobial activity of new Schiff bases containing coumarin moiety and their spectral characterization, ARKIVOC, 2008, 17, 221–233 Search PubMed.
- K. A. Alheety, N. M. Jamel and B. J. Ahmed, Synthesis of coumarin by Pechman reaction a review, J. Pharm. Sci. Res., 2019, 9, 3344–3347 Search PubMed.
- A. H. Abdel-Rahman, W. A. El-Sayed, H. M. Abdel-Bary, A. S. Abdel-Megied and M. I. Morcy, Amino acid derivatives, VIII [1]: synthesis and antimicrobial evaluation of alpha-amino acid esters bearing an indole side chain, Monatsh. Chem., 2008, 139, 1095–1101 CrossRef CAS.
- J. Van Meerloo, G. J. Kaspers and J. Cloos, Cell sensitivity assays: the MTT assay, Methods Mol. Biol., 2011, 731, 237–245 CrossRef CAS PubMed.
- C. Borgo and M. Ruzzene, Role of protein kinase CK2 in antitumor drug resistance, J. Exp. Clin. Cancer Res., 2019, 38, 287 CrossRef PubMed.
- N. Zhang, W. J. Chen, Y. Zhou, H. Zhao and R. G. Zhong, Rational design of coumarin derivatives as CK2 inhibitors by improving the interaction with the hinge region, Mol. Inf., 2016, 35, 15–18 CrossRef CAS PubMed.
- N. Zhang and R. Zhong, Docking and 3D-QSAR studies of 7-hydroxycoumarin derivatives as CK2 inhibitors, Eur. J. Med. Chem., 2010, 45, 292–297 CrossRef CAS PubMed.
- J. E. Chojnowski, E. A. McMillan and T. Strohlic, Identification of novel CK2 kinase substrates using a versatile biochemical approach, J. Visualized Exp., 2019, 21, 59037 Search PubMed.
- L. Meijer and E. Raymond, Roscovitine and other purines as kinase inhibitors from starfish oocytes to clinical trials, Acc. Chem. Res., 2003, 36, 417–425 CrossRef CAS PubMed.
- C. Borgo and M. Ruzzene, Role of protin kinase CK2 in antitumor drug resistanc, J. Exp. Clin. Cancer Res., 2019, 38, 287 CrossRef PubMed.
- B. Guerra and O.-G. Issinger, Role of protein kinase CK2 in aberrant lipid metabolism in cancer, Pharmaceuticals, 2020, 13(10), 292 CrossRef CAS PubMed.
- G. Loele, M. Chieffallo, M. A. Occhiuzzi, M. D. Luca, A. Garofalo, G. Ragno and F. Grande, Anticancer drugs: recent strategies to improve stability profile, pharmacokinetic and pharmacodynamics properties, Molecules, 2022, 27, 5436 CrossRef PubMed.
- Small-Molecule Drug Discovery Suite, QikProp, Version 4.2, New York, Schrodinger, LLC, 2014 Search PubMed.
- C. M. Chagas, S. Moss and L. Alisaraie, Drug metabolites and their effects on the development of adverse reactions: revisiting Lipinski's Rule of five, Int. J. Pharm., 2018, 549, 133–149 CrossRef CAS PubMed.
- M. D. Naik, Y. D. Bodke, K. M. Vijay and B. C. Revanasiddappa, An efficient one-pot synthesis of coumarin-amino acid derivatives as potential anti-inflammatory and anti-oxidant agents. Syn, Commun, 2020, 1735442 Search PubMed.
- B. L. Sousa, P. V. Leite, A. O. Mendes, V. V. Varejao, C. S. Chaves, J. G. Silva, A. P. Agrizzi, P. G. Ferreira, E. J. Pilau, E. Silva and M. H. Santos, Inhibition of acetylcholinesterase by cumarin-linked amino acids synthetized via triazole associated with molecule partition coefficient, J. Braz. Chem. Soc., 2021, 32, 652–664 Search PubMed.
- M. S. Gomaa, I. A. I. Ali, G. El Enany, E. H. El Ashry, S. M. El Rayes, W. Fathalla, A. H. A. Ahmed, S. A. Abushait, H. A. Abushait and M. S. Nafie, Facile synthesis of some coumarin derivatives and their cytotoxicity through VEGFR2 and topoisomerase II inhibition, Molecules, 2022, 27, 8279 CrossRef CAS PubMed.
- V. F. Morales, A. P. Villasana-Ruiz, I. G. Veloz, S. G. Delgado and M. L. Martinez-Fierro, Theraspeutic effects of coumarins with different substitution patterns, Molecules, 2023, 28, 2413 CrossRef PubMed.
- M. Koley, J. Han, V. A. Soloshonok, S. Mojumdr, R. Javahershenas and A. Makarem, Latest developments in coumarin-based anticancer agents: mechanism of action and structure-activity relationship studies, RSC Med. Chem., 2024, 15, 10–54 RSC.
- V. S. V. Satyanarayana, P. Sreevani, A. Sivakumar and V. Vijayakumar, Synthesis and antimicrobial activity of new Schiff bases containing coumarin moiety and their spectral characterization, Arkivoc, 2008, 17, 221–233 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04226c |
| This journal is © The Royal Society of Chemistry 2024 |