
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of sulfonamide antibiotics in forage grasses by an improved QuEChERS and multi-plug filtration cleanup method combined with UHPLC-MS/MS†
Min Xie a,
Jun Xu*ab,
Dan Feia,
Ye-Lan Guanga,
Yao-Min Zhoua,
Fan Lib and
Li-Zhen Hub
a,
Jun Xu*ab,
Dan Feia,
Ye-Lan Guanga,
Yao-Min Zhoua,
Fan Lib and
Li-Zhen Hub
aInstitute for Quality & Safety and Standards of Agricultural Products Research, Jiangxi Academy of Agricultural Sciences, Key Laboratory of Agro-product Quality and Safety of Jiangxi Province, Nanchang, Jiangxi, China. E-mail: xujun0125@163.com; Fax: +86-791-87090291; Tel: +86 18172871823
bAnimal Husbandry and Veterinary Science of Jiangxi Academy of Agricultural Sciences, Nanchang, Jiangxi, China
First published on 29th October 2024
Abstract
An improved QuEChERS method combined with Multi-Plug Filtration Cleanup (m-PFC) clean-up procedure was developed for the simultaneous determination of 16 different sulfonamides in forage grasses using ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS). The forage grass sample was extracted with 0.1 mol L−1 Na2EDTA-Mcllvaine buffer and acetonitrile solution, purified by the Navo U-QuE column tube, separated by Agilent ZORBAX Eclipse Plus C18 column, and analyzed by MRM (multiple reaction monitoring) mode and the internal standard method was utilized for quantification. The 16 different sulfonamides display good linearity within the range of 0.5–50 μg L−1 (R2 > 0.9967), the limit of detection was 0.02–0.5 μg kg−1, and the limit of quantification was 0.05–1.0 μg kg−1. Under the additive amounts of 1, 2, and 10 μg kg−1, the recoveries were in the range of 72.3–116.9%, and the relative standard deviation was between 1.4% and 10.3%. The presented method showed the advantages of simple operation, excellent selectivity and high sensitivity. It was well-suited for both qualitative and quantitative analyses of sulfonamides in forage grass. Overall, this method provided a scientific basis for risk assessment, enforcement of quality and safety standards, and detection of sulfonamide residues in forage grass for import and export.
1 Introduction
Sulfonamides are a class of antibiotics with a p-aminobenzene sulfonamide moiety and were the earliest synthetic bacteriostatic antibiotics.1 Commonly, the different sulfonamide derivatives were formed by replacing the R group on the p-aminobenzene sulfonamide with various heterocycle groups (Fig. 1), such as sulfacetamide (SA), sulfamethizol (SMTZ) and sulfisoxazole (SIZ), which exerted more broad-spectrum antibacterial activities and higher potency.2 Sulfonamides were widely used in livestock and poultry farming to prevent and treat bacterial infections due to their efficient broad-spectrum antibacterial and low-cost advantages.3 However, the farmers often used sulfonamides beyond the recommended scope and dosage to reduce animal mortality and improve breeding benefits. Sulfonamides could accumulate in the human body when not correctly used, leading to antibiotic resistance, toxic reactions and the occurrence of many diseases, such as gut disorders, allergies and cancer.4 | ||
| Fig. 1 Chemical structures of the sulfonamides. | ||
At the same time, sulfonamides are not fully metabolised by livestock and poultry; around 50–70% of these antibiotics will be excreted into environments in the form of parent compounds or derivatives along with urine and feces of animals, directly causing contamination to soil and water.5,6 Studies have shown that the concentration of antibiotics in soil has reached from μg kg−1 level to mg kg−1 level.7,8 Moreover, during the cultivation of forage grass, the application of organic fertilizers could easily lead to the accumulation of antibiotics in forage grass, severely affecting the safety of roughage for the ruminant. The collection of antibiotic residues in the forage grass would migrate to animal bodies through the food chain, resulting in antibiotic or metabolite residues in animal products such as meat, eggs and raw milk, which poses a potential threat to the quality and safety of livestock products and human health.9,10 Meanwhile, the antibiotic residue in the forage grass is also considered a critical indicator to assess the forage quality in the import and export trade. However, the method for determining antibiotic residues in forage grass has been reported infrequently. Therefore, it is necessary to establish an accurate and rapid detection method for sulfonamides residue in forage grass.
Currently, the analytical methods of sulfonamides have included enzyme-linked immunosorbent assays, microbiological methods, capillary electrophoresis, electrochemical methods and high-performance liquid chromatography-mass spectrometry.11–13 Studies on sulfonamides residues have focused on vegetables and animal products such as meat, fish and egg.14–16 However, to the best of our knowledge, the detection method of sulfonamides in the forage grass has not yet been reported in the literature. Thus, there is a need for further studies on multi-residue detection methods of sulfonamides in the forage grass. Unfortunately, the complex matrix of the forage grass, such as cellulose, pigment and organic acids, will co-extract with sulfonamides during the solvent extraction step. Therefore, sample purification is a crucial step for the quantitative and qualitative detection of sulfonamides in the forage grass. Many effective purification techniques for sulfonamides residue analysis have been reported, such as solid phase extraction (SPE),17 solid-phase microextraction (SPME),18 matrix dispersive solid phase extraction (d-SPE)19 and QuEChERS (quick, easy, cheap, effective, rugged, and safe).11,15,16 The QuEChERS method has been extensively applied for detecting antibiotic residues in animal-derived foods due to its simplicity, higher recovery and accuracy.9 Subsequently, a more practical way for sample cleanup, called the multi-plug filtration cleanup (m-PFC) method, was developed based on the QuEChERS approach.20,21 In the m-PFC procedure set by the group of Professor Canping Pan, the solid-phase sorbents included multi-walled carbon nanotubes (MWCNTs) and other sorbents such as primary secondary amine-modified silica (PSA), graphitic carbon black (GCB) and C18 were packed in a short syringe cartridge. Then, the interfering substances and water could be adsorbed by pushing and pulling the syringe.20,21 The m-PFC method could shorten the purification completion time and increase sample preparation efficiency for pesticide detection.21–23 However, the m-PFC method for detecting antibiotics has only been reported infrequently.
Overall, the aim of this study is to establish innovative strategies for simultaneously determining 16 sulfonamides in forage grass using the m-PFC cleanup method combined with ultra-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS). The UHPLC-MS/MS detection conditions, extraction solvent and purification method were optimized to improve analytical accuracy and sensitivity. The proposed method was effectively employed for the analysis of fundamental samples.
2 Materials and methods
2.1 Chemicals and reagents
Sulfacetamide (SA), sulfamethizol (SMTZ), sulfisoxazole (SIZ), sulfachloropyridazine (SCP), sulfadiazine (SD), sulfamethoxazole (SMZ), sulfathiazole (ST), sulfamonomethoxine (SMM), sulfamerazine (SM1), sulfadoxine (SDX), sulfapyridine (SPD), sulfameter (SMT), sulfamethoxypyridazine (SMP), sulfamethazine (SM2), sulfaphenazole (SPZ), sulfadimethoxine (SDM) and internal standard sulfadoxine-D3 (SDX-D3), sulfadimethoxine-D6 (SDM-D6) were purchased from Sigma-Aldrich (St. Louis, MO, USA). HPLC grade formic acid was obtained by Thermo Fisher Scientific (USA). HPLC-grade methanol, acetonitrile and ammonium acetate were supplied by Aladdin, Inc. (Shanghai, China). Primary secondary amine-modified silica (PSA) (size of 40–60 μm) and C18 sorbent (size of 50 μm 60 Å) were obtained from Agela Technologies (Delaware, USA). Graphitic carbon black (GCB) was obtained from Macklin (Shanghai, China). Anhydrous magnesium sulfate (MgSO4), anhydrous sodium sulfate (Na2SO4) and sodium chloride (NaCl), all analytical grade, were purchased from Aladdin, Inc. (Shanghai, China). Ultrapure water was obtained from a Milli-Q purification system (Millipore, Molsheim, France). Navo U-QuE column tube and Anavo-U Syringe Filter were purchased from Beijing Zhenxiang Technology Co. (Beijing, China). Navo U-QuE column tubes were packed with multi-walled carbon nanotubes (MWCNTs), PSA, C18, GCB and Na2SO4. All other reagents and solvents were of analytical reagent (AR) or ultragradient HPLC grade.2.2 Preparation of stock and working solutions
Individual stock standards were prepared by dissolving 1 mg (±0.01 mg) of each compound in methanol, resulting in a final concentration of 1.0 mg mL−1. The mixtures of all the standards were prepared at 10 mg L−1 in methanol. The standard working solutions at 1.0 mg L−1 were prepared in methanol by diluting the mixture of standard solutions. The internal standards (SDM-D6 and SDM-D3) were prepared at 1.0 mg L−1 in methanol. They were later diluted with methanol to achieve a series of standard solutions with varying concentrations of 0.5, 1, 2, 5, 10, 20, 50 μg L−1, each contained 30 μg L−1 of the internal standards. All stock solutions were stored in the dark at −20 °C, while the working solutions were stored at 0 °C.0.1 M Na2EDTA-Mcllvaine (pH 4.0 ± 0.05) was prepared by dissolving 10.92 g disodium hydrogen phosphate, 12.93 g citric acid monohydrate and 37.23 g ethylenediamine tetraacetic acid disodium salt (Na2EDTA) in 1 L of Milli-Q water. Then, the pH was adjusted to 4.0 ± 0.05 with 0.1 mol L−1 hydrochloric acid solution or 0.1 mol L−1 NaOH solution.
2.3 UHPLC-MS/MS analysis
The target sulfonamides were analyzed by a UHPLC-MS/MS system, which consisted of Agilent UHPLC (1290 Infinity) coupled with a quadrupole tandem mass spectrometer (QQQ, 6465) (California, USA). All the target compounds were separated by a ZORBAX RRHD Eclipse Plus-C18 column (2.1 × 100 mm, 1.8 μm, Agilent, California, USA). Mobile phase A was an aqueous solution containing 0.1% (v/v) formic acid, and mobile phase B was acetonitrile. The injection volume was 10 μL, and the column temperature was maintained at 35 °C. The flow rate was 0.40 mL min−1. The gradient elution was programmed as follows: 5–15% B for 0–4 min, 15–25% B for 4–8 min, 25–32% B for 8–11 min, 32–95% B for 11–12 min, 95–5% B for 12–13 min, 5% B for 13–14 min.The tandem mass spectrometer was operated in electrospray ionization (ESI) and positive ion multiple reaction monitoring (MRM) modes, with a capillary voltage of 3 kV, an ion source temperature of 150 °C and a gas flow rate of 15 L min−1. The other operating conditions, including sheath gas temperature, sheath gas flow rate, nebulizer pressure and nozzle voltage, were set to 300 °C, 11 L min−1, 30 psi and 0 V, respectively.
2.4 Sample treatment and clean-up method
The forage grass samples were freeze-dried and cut to <1 cm in length. Subsequently, the dried samples were powdered in liquid nitrogen and stored in a desiccator until use.A 1.0 g of homogenized forage grass samples were transferred into a 50 mL polypropylene centrifuge tube and spiked with 300 μL of the internal standard solution (1.0 mg L−1). 5 mL of 0.1 M Na2EDTA-Mcllvaine buffer was added to the samples, and the mixture was vortexed for 1 min. Then 10 mL of acetonitrile was added, and the combination was vortexed for 1 min and sonicated for 10 min. Afterwards, 4 g of anhydrous Na2SO4 and 1 g of NaCl were added, the mixture was vortexed for 1 min and centrifuged at 5000 rpm for 4 min. The 2 mL supernatant was used for further sample purification.
The sample purification was carried out by the Navo U-QuE column tube according to the m-PFC method.20,21 The m-PFC setups were performed as reported by Liu et al.21 2 mL of the supernatant was transferred to the Navo U-QuE column tube to purify according to the m-PFC procedure. The syringe piston was pulled and pushed to allow the extracts to pass through the sorbents for cleaning. Then, 1 mL of the supernatant was placed in a 15 mL centrifuge tube and evaporated to near dryness under a gentle stream of nitrogen. Finally, the residue was re-dissolved in 1 mL 0.1% of formic acid aqueous solution/acetone (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and filtered through a 0.22 μm Anavo-U Syringe Filter into an autosampler vial for UPLC-MS/MS analysis.
:1, v/v) and filtered through a 0.22 μm Anavo-U Syringe Filter into an autosampler vial for UPLC-MS/MS analysis.
2.5 Method validation
The analytical method was validated for linearity, recovery, precision, matrix effect (ME), limit of detection (LOD), and limit of quantification (LOQ). The linearity test was checked using matrix-matched calibration curves by spiking blank forage grass matrix at seven concentration levels (0.5, 1, 2, 5, 10, 20, 50 μg L−1, each contained 30 μg L−1 of the internal standards) ranging from 0.5 μg L−1 to 50 μg L−1. The calibration curves were obtained by plotting the peak areas of the standards against their concentration. For spiking blank samples, recovery and precision were carried out in six replicates at three concentration levels (1, 2, 10 μg kg−1). The recovery of each compound was calculated by comparing the measured concentration with the spiked concentration of each compound.24,25 The precision was expressed as the relative standard deviation (RSD). According to the SANTE/12682/2019.26 The limit of detection (LOD) and quantification (LOQ) were calculated as the concentrations with 3-fold and 10-fold of the signal-to-noise (S/N) rations, respectively. The limit of detection (LOD, mg kg−1) is determined by multiplying the concentration at a 3-fold signal-to-noise ratio injected into high-performance liquid chromatography (μg L−1) with the dilution volume (L), and then dividing it by the sample mass (g). The limit of quantification (LOQ, mg kg−1) is determined by multiplying the concentration measured at a 10-fold signal-to-noise ratio injected into high-performance liquid chromatography (μg L−1) with the dilution volume (L), and then dividing it by the sample mass (g).The matrix effect (ME) of each compound was evaluated using the extracted blank matrix spiked with standard solutions and unextracted standard solutions from 0.5 ng mL−1 to 50 ng mL−1. The matrix effect was calculated by following Chawla et al.,27 and the equation was as follows:
| ME% = (Sm − Ss)/Ss × 100 |
3 Results and discussion
3.1 Optimization of UHPLC-MS/MS
To optimize the chromatographic conditions for the target compounds, a comparative analysis was conducted on the separation efficiency of three different chromatographic columns (ZORBAX RRHD Eclipse Plus-C18, ZORBAX SB-Aq, Poroshell 120 EC-C18) for these compounds. The ZORBAX SB-Aq and Poroshell 120 EC-C18 columns exhibited broader and trailing peaks for the target compounds. There was insufficient separation between the SMP and SMT compounds on the Poroshell 120 EC-C18 column. Compared to the other two columns, the ZORBAX RRHD Eclipse Plus-C18 column demonstrated significantly superior separation with sharp and symmetrical peak shapes, accompanied by higher abundance. Simultaneously, the different mobile phase compositions (acetonitrile/water, methanol/water, acetonitrile/water with 0.1% formic acid, acetonitrile/water with 0.1% formic acid and 5 mM ammonium acetate) were investigated and compared. The poorer separation efficiency and lower response abundance of the target compound was observed when using a mobile phase composition of methanol/water. The introduction of 0.1% formic acid into the mobile phase significantly enhanced the response abundance of the target compound. However, the responses abundance for all analytes diminished upon incorporating 5 mM ammonium acetate in the mobile phase. Considering the responses of all target analytes, 0.1% formic acid in acetonitrile/water was selected as the optimal mobile phase for sulfonamides analysis.The MS parameters were optimized by investing a standard solution of 1 mg L−1 of each compound in positive and negative ionization modes to obtain a method with both high sensitivity and separation efficiency. The base peak selected for quantification of the target compounds was the protonated molecule [M + H]+ and deprotonated molecule [M − H]− for positive and negative ionization modes, respectively.25 Results showed that all the target compounds reveal high responses and lower noise in positive ionization mode. The MS2 Scan mode was performed to obtain the precursor ion of the target compound. Then, fragment ions were selected in the product scan mode. The most intense fragment ions were used as the quantitative ion, and the second was used as the qualitative ion. Finally, Multiple Reaction Monitoring (MRM) was selected to optimize the mass spectrometry (MS) parameters, including collision energy, fragmentor, etc. The optimized MS parameters of the target analytes are detailed in Table 1. The total ion current chromatogram of a blank forage grasses matrix spiked at 20 μg L−1, as shown in Fig. 2, demonstrates that the selected UHPLC-MS/MS conditions exhibit exceptional peak separation and satisfactory chromatographic resolution within a reduced timeframe.
| Compounds | Retention time (min) | Polarity | Parent ion (m/z) | Production (m/z) | Collision energy | Fragment |
|---|---|---|---|---|---|---|
| SA | 2.36 | Positive | 215.1 | 156.1/92.0 | 5/20 | 380 |
| SCP | 6.36 | Positive | 285.0 | 156.0/108.0 | 10/25 | 380 |
| SD | 2.81 | Positive | 251.1 | 156.0/108.0 | 10/22 | 380 |
| SDM | 9.35 | Positive | 311.0 | 155.9/108.0 | 20/32 | 380 |
| SDX | 7.06 | Positive | 311.1 | 92.0/155.9 | 30/20 | 380 |
| SIZ | 7.81 | Positive | 268.1 | 156.0/113.0 | 10/10 | 380 |
| SM1 | 3.84 | Positive | 265.1 | 172.0/156.0 | 12/15 | 380 |
| SM2 | 4.82 | Positive | 279.1 | 186.1/156.1 | 15/16 | 380 |
| SMM | 6.13 | Positive | 281.1 | 156.1/108.1 | 15/26 | 380 |
| SMP | 5.22 | Positive | 281.1 | 156.0/108.0 | 15/25 | 380 |
| SMT | 5.09 | Positive | 281.0 | 156.0/108.0 | 15/25 | 380 |
| SMTZ | 5.09 | Positive | 271.0 | 156.0/108.0 | 10/22 | 380 |
| SMZ | 7.06 | Positive | 254.1 | 156.0/108.0 | 14/24 | 380 |
| SPD | 3.52 | Positive | 250.1 | 184.0/156.0 | 15/10 | 380 |
| SPZ | 9.60 | Positive | 315.0 | 222.0/158.0 | 15/30 | 380 |
| ST | 3.40 | Positive | 256.0 | 156.0/108.0 | 10/21 | 380 |
| SDM-D6 | 9.24 | Positive | 317.0 | 155.9 | 20 | 380 |
| SDX-D3 | 7.00 | Positive | 314.0 | 156.1 | 15 | 380 |
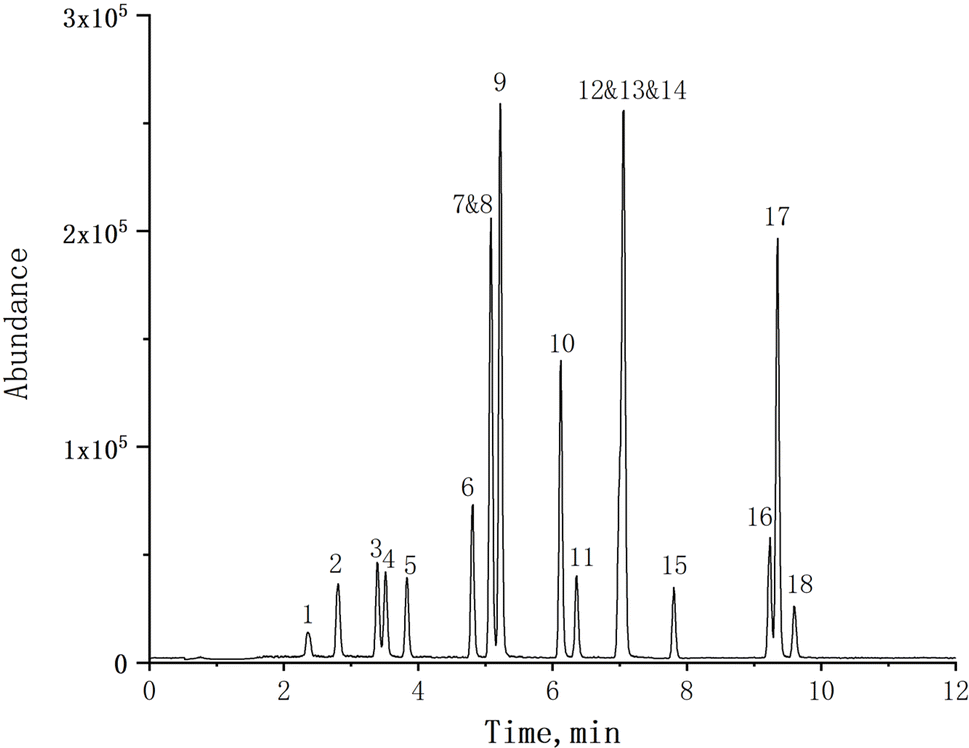 | ||
| Fig. 2 The total ion flow chromatogram of blank forage grasses matrix spiked at 20 μg L−1. (1) SA, (2) SD, (3) ST, (4) SPD, (5) SM1, (6) SM2, (7) SMTZ, (8) SMT, (9) SMP, (10) SMM, (11) SCP, (12) SDX-D3, (13) SDX, (14) SMZ, (15) SIZ, (16) SDM-D6, (17) SDM, (18) SPZ. | ||
3.2 Optimization of the extraction process
The sulfonamides are weak polarity compounds and are easily extracted by organic solvents, such as acidified acetonitrile, acetonitrile, methanol, and ethyl acetate. Based on previous studies,14,30 we selected five extraction solvents including 1% acetic acid in acetonitrile, methanol, acetonitrile–methanol (50:50, v/v) and acetonitrile–methanol (15:85, v/v), to compared the extraction efficiency of the sulfonamides. The blank forage samples were fortified with 100 μL of a 1.0 μg mL−1 mixed standard solution, followed by extraction and purification procedures as described above. As shown in Fig. 3, acetonitrile has better extraction recovery, the recoveries of 16 sulfonamides ranged from 62.7% to 98.9%. When methanol, acetonitrile–methanol (50:50, v/v) and acetonitrile–methanol (15:85, v/v) were used as the extraction solvent, the recoveries of most of the sulfonamides were below 60%. The recoveries of SDM and SCP were 42.5% and 57.5% when acetonitrile containing 1% acetic acid was used as the extraction solvent. Therefore, acetonitrile was selected as the optimal extraction solvent.
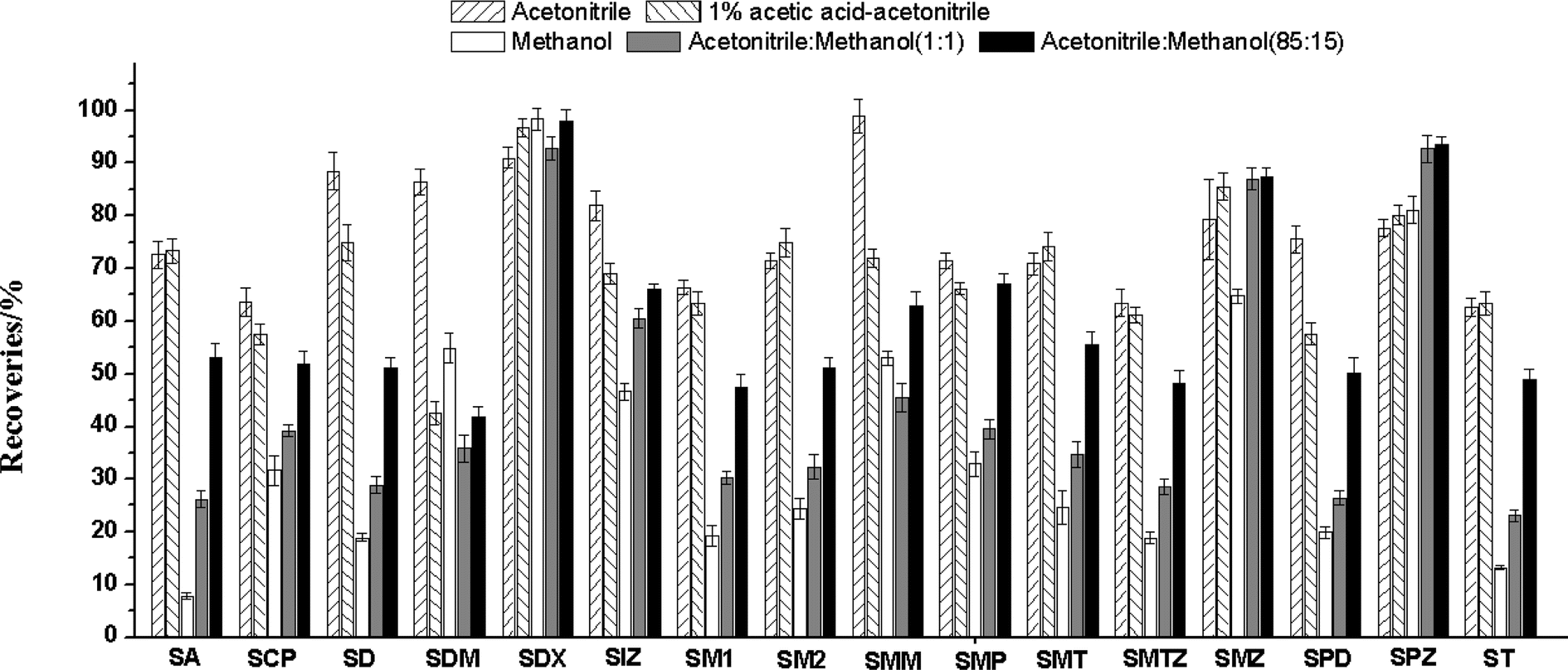 | ||
| Fig. 3 Recoveries of the sulfonamides with different extractants. | ||
3.3 Optimization of the salt
Anhydrous MgSO4 and Na2SO4 were usually used for salting-out in the QuEChERS method aimed to decrease analytes' solubility in the aqueous phase and enhance their partitioning into the organic phase.31,32 This study investigated the impact of extraction recovery of the analytes when anhydrous MgSO4 and Na2SO4 were added. As shown in Fig. 4, Na2SO4 has better extraction recovery. The extraction recovery of the sulfonamides ranged from 62.7% to 98.9% when Na2SO4 was added. The recoveries of most of the sulfonamides were below 60% when MgSO4 was added. The sulfonamides could quickly form a chelation structure with Mg2+ due to the carboxyl and carbonyls in the sulfonamides. Therefore, Na2SO4 was added in all the subsequent experiments.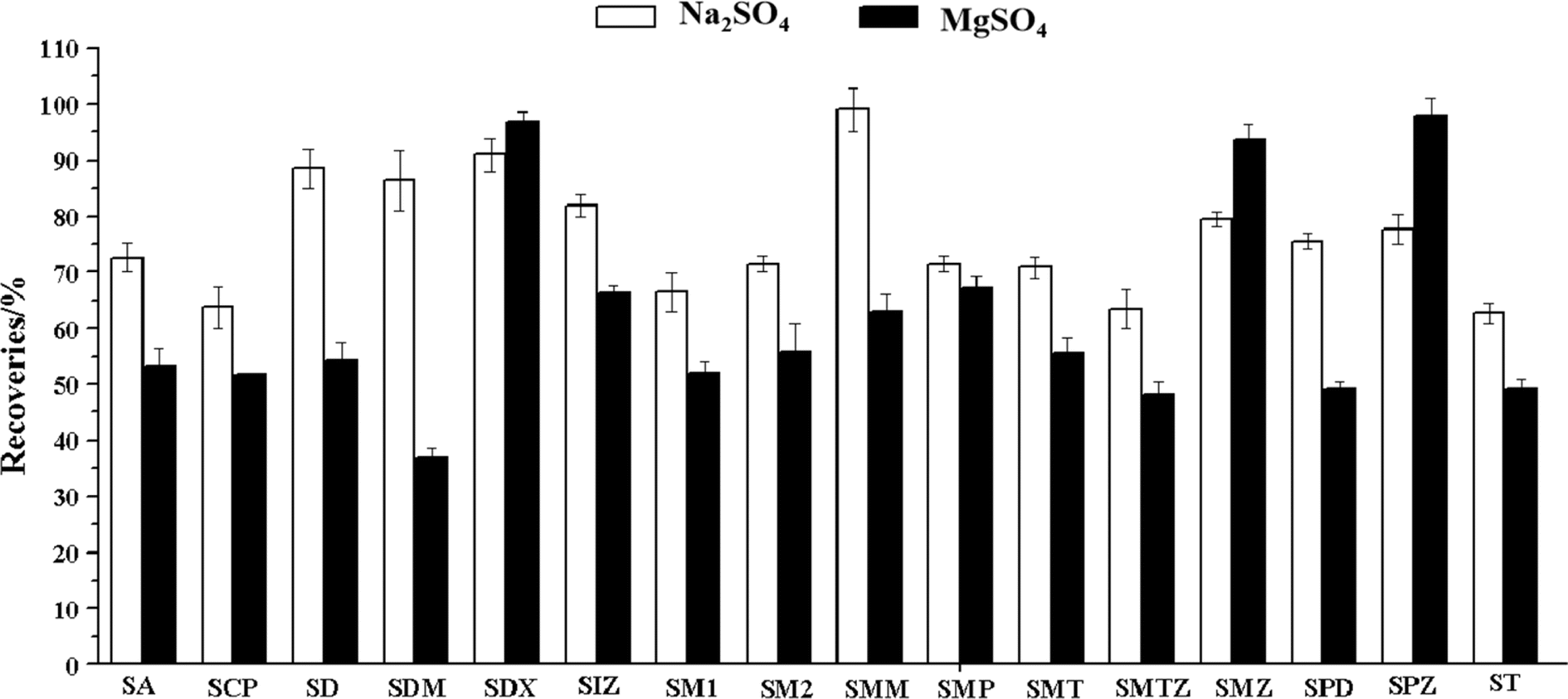 | ||
| Fig. 4 Recoveries of the sulfonamides with different salt. | ||
3.4 Optimization of the purification procedure
The forage grass contains many complex matrices, such as pigments, organic acids, proteins and crude fat. The substances in the matrix will affect the contaminated target compounds' ionisation and response values, and may contaminate the ion source. Therefore, it is necessary further to purify the extraction residues before UHPLC-MS/MS detection. The dispersive solid-phase extraction (d-SPE) was used to purify the samples using the original QuEChERS method. The purification adsorbents, including PSA, C18, GCB and multi-walled carbon nanotubes (MWCNTs), usually are used in the d-SPE procedure.33 PSA can absorb polar substances, such as organic acids, carbohydrates, sugars and fatty acids, whereas C18 can effectively remove non-polar substances, such as crude fat and steroids.11,34 GCB has strong adsorption capacity for pigments.35 MWCNTs have a strong cleanup capacity to remove stains and lipids.21,34 In recent years, the m-PFC methods have been developed for detecting residues of veterinary drugs and pesticides.21,22 Previous studies have shown that the m-PFC method was very rapid, taking about ten seconds to perform without solvent evaporation.21–23In this study, the purifying effects of the d-SPE and the m-PFC methods was compared. PSA, C18 and GCB were selected for the purification of extracting solution in d-SPE methods. The Navo U-QuE column tube, including MWCNTs, C18 and PSA, was set for the purification procedure in m-PFC methods. As shown in Fig. 5, the better extraction recovery of the sulfonamides was observed when the Navo U-QuE column tube was used, which ranged from 74.3% to 112.7%. The PSA sorbent showed a strong adsorption capacity to SMT, SDM, SMP, SPD and ST with low recovery (0–51.7%) and signal enhancement (>180%) for SA and SPZ. The GCB sorbent showed a strong adsorption capacity to SMT, SMTZ, SMZ and SMP with low recoveries (32.4–57.7%). At the same time, the recovery of SMZ and SMT was below 62% when the C18 sorbent was used. Therefore, the Navo U-QuE column tube in the m-PFC methods was selected for further purification.
 | ||
| Fig. 5 Recoveries of the sulfonamides with different purification sorbents. | ||
3.5 Method validation
| Compounds | Regression equation | R2 value | Matrix effects | LOD (μg kg−1) | LOQ (μg kg−1) |
|---|---|---|---|---|---|
| SA | Y = 0.035935X − 0.003737 | 0.9999 | −70 | 0.05 | 0.10 |
| SCP | Y = 0.007381X − 0.001339 | 0.9967 | −87 | 0.50 | 1.00 |
| SD | Y = 0.068011X + 0.002958 | 0.9999 | −46 | 0.02 | 0.05 |
| SDM | Y = 0.082643X − 0.003478 | 0.9960 | −87 | 0.10 | 0.30 |
| SDX | Y = 0.039106X + 0.000987936 | 0.9995 | −84 | 0.10 | 0.20 |
| SIZ | Y = 0.025699X + 0.003080 | 0.9994 | −88 | 0.30 | 0.50 |
| SM1 | Y = 0.90099X − 0.002357 | 0.9991 | −39 | 0.10 | 0.30 |
| SM2 | Y = 0.080536X +0.0025086 | 0.9999 | −63 | 0.10 | 0.50 |
| SMM | Y = 0.017509X + 0.003316 | 0.9966 | −78 | 0.05 | 0.10 |
| SMP | Y = 0.067212X + 0.004520 | 0.9983 | −69 | 0.05 | 0.10 |
| SMT | Y = 0.034280X + 0.001002 | 0.9981 | −62 | 0.05 | 0.10 |
| SMTZ | Y = 0.028957X − 0.000564767 | 0.9997 | −56 | 0.20 | 0.50 |
| SMZ | Y = 0.010607X − 0.000195817 | 0.9987 | −84 | 0.20 | 0.50 |
| SPD | Y = 0.043999X + 0.003179 | 0.9990 | −61 | 0.02 | 0.05 |
| SPZ | Y = 0.017106X − 0.000015879 | 0.9986 | −95 | 0.10 | 0.50 |
| ST | Y = 0.255073X − 0.041229 | 0.9988 | −55 | 0.05 | 0.10 |
| Compounds | Recovery/% | RSD/% | ||||
|---|---|---|---|---|---|---|
| 1 μg kg−1 | 2 μg kg−1 | 10 μg kg−1 | 1 μg kg−1 | 2 μg kg−1 | 10 μg kg−1 | |
| SA | 114.2 | 113.0 | 90.4 | 3.7 | 3.1 | 9.1 |
| SCP | 105.4 | 107.7 | 72.3 | 5.2 | 2.8 | 7.4 |
| SD | 99.8 | 110.7 | 86.5 | 6.2 | 6.4 | 7.0 |
| SDM | 112.0 | 111.0 | 83.4 | 5.9 | 2.2 | 3.2 |
| SDX | 103.4 | 113.2 | 95.9 | 8.1 | 6.1 | 7.1 |
| SIZ | 92.6 | 107.9 | 87.5 | 1.4 | 2.7 | 4.5 |
| SM1 | 103.7 | 110.0 | 77.8 | 4.5 | 3.0 | 9.3 |
| SM2 | 83.7 | 104.6 | 84.2 | 8.7 | 4.8 | 4.1 |
| SMM | 98.5 | 106.0 | 83.8 | 5.4 | 4.0 | 6.0 |
| SMP | 92.6 | 104.0 | 73.0 | 3.3 | 3.7 | 4.4 |
| SMT | 92.4 | 104.7 | 79.5 | 1.4 | 6.3 | 4.2 |
| SMTZ | 95.0 | 103.1 | 70.1 | 3.0 | 6.0 | 8.3 |
| SMZ | 116.9 | 116.0 | 91.8 | 1.3 | 3.8 | 7.7 |
| SPD | 92.2 | 101.9 | 76.2 | 8.9 | 6.6 | 10.3 |
| SPZ | 92.9 | 105.0 | 78.5 | 1.7 | 5.3 | 3.5 |
| ST | 104.8 | 103.8 | 73.0 | 3.7 | 7.9 | 7.5 |
3.6 Method performance comparison
The comparative data between this QuEChERS-UHPLC-MS/MS method and other reported analytical methods from the viewpoint of analytical method, pretreatment method, analytes, sample, linearity, LOD, RSD, and extraction time were shown in Table 4. Among them, QuEChERS and SPE were the most used pretreatment methods for the detection of sulfonamides. However, QuEChERS method could not only reduce testing procedure and reagent consumption but also reduce the extraction time compared with SPE and MSPE.17,37,38 In these comparative methods, the samples are mostly pork, fish and other animal-derived food.37–39 The relevant detection of sulfonamides in forage grass has rarely been reported. As shown in Table 4, the improved QuEChERS method combined with m-PFC clean-up procedure was developed for the determination of 16 sulfonamides in forage grass with comparable linearity, LODs, and RSDs and shorter extraction time, which showed satisfactory sensitivity and accuracy with simple pretreatment procedure.| Analytical method | Pretreatment method | Analytes | Sample | Linearity (μg L−1 or μg kg−1) | LOD (μg L−1 or μg kg−1) | RSD (%) | Pretreatment time (min) | Ref. |
|---|---|---|---|---|---|---|---|---|
| LC-MS/MS | On-line SPE | 15 sulfonamides | Pork and fish | 0.1–100 | 0.125–2.00 | <10 | 15 min | 39 |
| LC-MS | SPE | 5 sulfonamides | Meat | 0.5–200 | 0.10–0.23 | 2.3–12.4 | 21 min | 37 |
| LC-MS/MS | SPE | 16 sulfonamides | Beeswax | 2–50 | 1–2 | <24.2 | >30 min | 17 |
| HPLC-MS/MS | MSPE | 8 sulfonamides | Water and animal-derived food | 2–1000 | 0.20–1.50 | 3.5–8.7 | >40 min | 38 |
| UPLC-MS/MS | QuEChERS | 12 sulfonamides | Fish | 0.5–200 | 0.79–2.71 | 3.0–9.2 | 15 min | 15 |
| UHPLC-MS/MS | QuEChERS | 16 sulfonamides | Forage grass | 0.5–50 | 0.02–0.50 | 1.4–10.3 | 17 min | This work |
3.7 Application to actual sample analysis
The method established in this study was applied to analyze forage grasses samples collected from a different farm in Jiangxi, China. A total of 24 actual samples were analyzed following preparation as the procedure mentioned above. Among these forage grass samples, SCP was detected in 8 out of 24 samples with concentrations ranging from 32.8 μg kg−1 to 77.2 μg kg−1. SM2 was observed in 6 out of 24 samples, exhibiting concentrations ranging from 10.6 μg kg−1 to 49.4 μg kg−1. SMM was detected in 7 out of 24 samples, with concentrations varying between 25.4 μg kg−1 and 66.7 μg kg−1. SMZ was detected in 4 out of 24 samples with concentrations ranging from 13.4 μg kg−1 to 40.7 μg kg−1. However, the maximum residue limits (MRLs) of sulfonamides in forage grass have not been established widely. The proposed method is also applicable for detecting the trace amounts of sulfonamides in forage grass samples.4 Conclusion
In this study, the 16 different sulfonamides in forage grass were extracted and purified by an improved QuEChERS method combined with efficient m-PFC multi-residue clean-up procedures and quantified by UHPLC-MS/MS. This method has proved to be highly sensitive and has provided quantitative results for all the analytes, with appropriate validation parameters such as linearity, LODs, LOQs and precision. This is advantageous in terms of simplicity, rapidity, efficiency, environmental friendliness, and low reagent usage. Simultaneously, compared with the d-SPE clean-up method, the m-PFC method was a simple, rapid clean-up without any solvent evaporation, vortexing or centrifugation procedure. This method was successfully applied in detecting sulfonamides in forage grass. It provided a scientific basis for sulfonamides residues risk assessment, quality and safety supervision, and detection for import and export of forage grass.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
All authors declare they have no conflicts of interest concerning this work.Acknowledgements
This work was supported by the Jiangxi Key Research and Development Program (20201BBF61008), the Science and Technology Cooperation Program of Jiangxi (2021BDH80013) and the Key Laboratory of Agro-product Quality and Safety of Jiangxi Province (2024SSY04191).References
- R. Hoff, Anal. Biochem., 2020, 611, 114011 CrossRef.
- I. N. Kolesnikova, A. N. Rykov, V. V. Kuznetsov and I. F. Shishkov, Struct. Chem., 2020, 31, 1353–1362 CrossRef.
- R. Xu, C. Yang, L. Huang, W. Lv, W. Yang, Y. Wu and F. Fu, J. Agric. Food Chem., 2022, 70, 11804–11812 CrossRef.
- F. Mor, F. S. Kocasari, G. Ozdemir and B. Oz, Food Chem., 2012, 134, 1645–1649 CrossRef.
- J. Q. Xiong, M. B. Kurade and B. H. Jeon, Trends Biotechnol., 2018, 36, 30–44 CrossRef PubMed.
- F. Hernandez, N. Calısto-Ulloa, C. Gómez-Fuentes, M. Gómez, J. Ferrer, G. González-Rocha, H. Bello-Toledo, A. M. Botero-Coy, C. Boıx and M. Ibáñez, J. Hazard. Mater., 2019, 363, 447–456 CrossRef.
- Y. W. Li, X. L. Wu, C. H. Mo, Y. P. Tai, X. P. Huang and L. Xiang, J. Agric. Food Chem., 2011, 59, 7268–7276 CrossRef PubMed.
- X. Hu, Q. Zhou and Y. Luo, Environ. Pollut., 2010, 158, 2992–2998 CrossRef CAS.
- L. Aljerf, N. AlMasri and U. Prince, Int. J. Case Rep. Stud, 2018, 2(2), 88–91 Search PubMed.
- L. Aljerf and N. Aljerf, Prog. Nutr., 2023, 25, e2023024 CAS.
- S. Li, C. Zhang, H. X. Tang, Y. Gu, A. J. Guo, K. Wang and K. Q. Lian, J. Food Drug Anal., 2023, 31, 73 CrossRef PubMed.
- R. Galarini, F. Diana, S. Moretti, B. Puppini, G. Saluti and L. Persic, Food Control, 2014, 35, 300–310 CrossRef.
- N. F. Semail, A. S. A. Keyon, B. Saad, S. Kamaruzaman and D. D. Y. Chen, Talanta, 2022, 236, 122833 CrossRef PubMed.
- O. Tadi, V. Matamoros and J. M. Bayona, Anal. Bioanal. Chem., 2019, 411, 5209–5222 CrossRef PubMed.
- Y. Lu, Z. Cheng, C. Liu and X. Cao, Food Anal. Methods, 2016, 9, 1857–1866 CrossRef.
- C. H. Wen, S. L. Lin and M. R. Fuh, Talanta, 2017, 164, 85–91 CrossRef PubMed.
- K. Mitrowska and M. Antczak, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2015, 1006, 179–186 CrossRef.
- R. Duan, L. Sun, H. Y. Yang, Y. R. Ma, X. Y. Deng, C. Peng, C. Zheng, L. Y. Dong and X. H. Wang, J. Chromatogr., A, 2020, 1609, 460510 CrossRef CAS PubMed.
- D. Lu, C. Liu, M. Qin, J. Deng, G. Shi and T. Zhou, Anal. Chim. Acta, 2020, 1133, 88–98 CrossRef CAS.
- X. Dong, T. Lan, X. Tian, Y. Li, Y. Zhao, Q. Zong, S. Liu and C. Pan, J. Environ. Sci. Health B, 2021, 56, 771–781 CrossRef CAS.
- S. Liu, A. Bai, L. Song, N. Zou, Y. Han, L. Zhou, C. Yu, C. Li and C. Pan, Foods, 2021, 10, 2731 CrossRef CAS.
- L. Ma, L. Cao, Y. Feng, L. Jia, C. Liu, Q. Ding, J. Liu, P. Shao and C. Pan, J. Chromatogr. Sci., 2023, 61, 559–568 CAS.
- Y. H. Qin, P. Y. Zhao, S. F. Fan, Y. T. Han and Y. J. Li, J. Chromatogr. A, 2015, 1385, 1–11 CrossRef CAS.
- L. Aljerf and A. Mashlah, Microchem. J., 2017, 132, 411–421 CrossRef CAS.
- L. Aljerf and I. Alhaffar, Biochem. Res. Int., 2017, 2017(1), 9596202 Search PubMed.
- T. Pihlström and M. Gamón, Sante 11813, 2017, pp. 21–22 Search PubMed.
- C. Suchi, H. K. Patel, H. N. Gor, K. M. Vaghela, P. P. Solanki and P. G. Shah, J. AOAC Int., 2017, 100, 616–623 CrossRef.
- D. C. M. Helena, C. H. Collins and S. F. J. Cristina, Food Chem., 2018, 249, 77–83 CrossRef.
- A. Economou, H. Botitsi, S. Antoniou and D. Tsipi, J. Chromatogr. A, 2009, 1216, 5856–5867 CrossRef CAS PubMed.
- Y. H. Chuang, Y. Zhang, W. Zhang, S. A. Boyd and H. Li, J. Chromatogr. A, 2015, 1404, 1–9 CrossRef CAS.
- M. Anastassiades, S. J. Lehotay, D. Štajnbaher and F. J. Schenck, J. Chromatogr. A, 2003, 86, 412–431 CAS.
- A. Wilkowska and M. Biziuk, Food Chem., 2011, 125, 803–812 CrossRef.
- C. Zhang, Y. Deng, J. Zheng, Y. Zhang and A. Luo, Trac. Trends Anal. Chem., 2019, 118, 517–537 CrossRef.
- K. Pyrzynska, Chemosphere, 2011, 83, 1407–1413 CrossRef PubMed.
- Z. Y. He, Y. H. Wang, Y. P. Xu and X. W. Liu, Food Anal. Methods, 2018, 11, 2857–2864 CrossRef.
- S. Kittlaus, J. Schimanke, G. Kempe and K. Speer, J. Chromatogr. A, 2011, 1218, 8399–8410 CrossRef PubMed.
- R. Shen, L. Huang, R. Liu and Q. Shuai, J. Chromatogr. A, 2021, 1655, 462518 CrossRef.
- Y. Zhao, R. Wu, H. Yu, J. Li, L. Liu, S. Wang, X. Chen and T. W. D. Chan, J. Chromatogr. A, 2020, 1610, 460543 CrossRef PubMed.
- F. S. Ma J, L. Sun, L. He, Y. Zhang and Q. Li, Food Sci. Hum. Wellness, 2020, 9, 363–369 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04330h |
| This journal is © The Royal Society of Chemistry 2024 |