
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of oxygen-releasing dextran microgels by droplet-based microfluidic method†
Daisuke Tomioka‡
a,
Shannon Anna Jung‡bc,
Andrij Pich *bc and
Michiya Matsusaki*a
*bc and
Michiya Matsusaki*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka, 565-0871, Japan. E-mail: m-matsus@chem.eng.osaka-u.ac.jp
bDWI-Leibniz Institute for Interactive Materials, RWTH Aachen University, Forckenbeckstraße 50, 52074 Aachen, Germany
cInstitute for Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 22nd August 2024
Abstract
In the tissue engineering field, the supply of oxygen to three-dimensional (3D) tissues is an important aspect to avoid necrosis due to hypoxia. Although oxygen-releasing bulk materials containing calcium peroxide (CaO2, CP) have attracted much attention, micrometer-sized oxygen-releasing soft materials would be advantageous because of their highly controllable structures, which can be applied for cell scaffolds, injectable materials, and bioink components in 3D bioprinting. In this study, oxygen-releasing microgels were fabricated via a droplet-based microfluidic system. Homogeneous, monodisperse and stable oxygen-releasing microgels were obtained by photo-crosslinking of droplets composed of biocompatible dextran modified with methacrylate groups and CP nanoparticles as an oxygen source. We also used our microfluidic system for the in situ amorphous calcium carbonate (CaCO3, ACC) formation on the surface of CP nanoparticles to achieve the controlled release of oxygen from the microgel. Oxygen release from an ACC–CP microgel in a neutral cell culture medium was suppressed because incorporation of CP in the ACC suppressed the reaction with water. Strikingly, stimuli to dissolve ACC such as a weak acidic conditions triggered the oxygen release from microgels loaded with ACC–CP, as the dissolution of CaCO3 allows CP to react. Taken together, applications of this new class of biomaterials for tissue engineering are greatly anticipated. In addition, the developed microfluidic system can be used for a variety of oxygen-releasing microgels by changing the substrates of the hydrogel network.
1. Introduction
In tissue engineering and regenerative medicine, construction and transplantation of thick 3D tissues are still major challenges because limited diffusion of oxygen inside these tissues can cause cell death.1–4 To address this problem, oxygen-releasing materials have attracted much attention in recent years.5–7 Although perfluorocarbons8,9 and heme proteins such as hemoglobin10,11 are some of the most common oxygen sources for these types of materials, the low payload of oxygen is an issue.7 To overcome this limitation, calcium peroxide (CaO2, CP) is often used for oxygen-releasing materials because it can carry a higher oxygen payload than perfluorocarbons and heme proteins.7 Since CP is an inorganic solid which generates oxygen, hydrogen peroxide and calcium hydroxide (Ca(OH)2) in its reaction with water, many researchers have reported the production of oxygen-releasing hydrogels by mixing CP into the hydrogels.4,12–16 In addition, some researchers have incorporated CP in microparticles composed of hydrophobic synthetic polymers such as polycaprolactone17,18 for the sustained oxygen release from CP by suppressing the reaction with water. However, a downside of using these synthetic polymers is their hydrophobic properties that are not suitable for direct contact with cells compared to water rich hydrogels.19 Therefore, these hydrophobic oxygen-releasing microparticles were finally incorporated in bulk hydrogels for tissue engineering applications.17,18Recently, micrometer-sized hydrogels, microgels, have also attracted much attention in the tissue engineering field because microgel assemblies can be used for cell scaffolds, injectable materials, and 3D bioprinting.20–22 Scaffold properties such as stiffness, microporosity and degradability are controllable at the micro-scale by modulating individual microgels. Unlike the 3D cell culture in bulk hydrogels, 3D cell culture in microgel assembly improves cell infiltration and nutrient diffusion via its tunable microporous structure, which improves long term cell survival, tissue repair and regeneration.21 Furthermore, microgels are preferable to hydrophobic polymer microparticles because their hydrophilicity is suitable for cells. By taking advantage of oxygen-releasing materials and microgels, oxygen-releasing microgels are expected to be very useful biomaterials for tissue engineering applications. However, to the best of our knowledge, no one has yet reported oxygen-releasing microgels using CP as an oxygen source.
In this study, we report the fabrication of oxygen-releasing microgels using a droplet-based microfluidic system. A microfluidic system is suitable for the reproducible and controlled fabrication of uniform microgels based on its highly tuned channel in the micrometer range.21–23 Herein, a water-in-oil emulsion with two aqueous phases and one oil phase was used for microgel fabrication (Fig. 1). In one of the aqueous phases, dextran methacrylate (Dex-MA) was used for the polymer network of the microgel.24 Since dextran is a biocompatible and biodegradable natural polysaccharide, dextran-based materials are suitable for tissue engineering applications.25,26 In addition, synthesized CP nanoparticles were dispersed in the Dex-MA aqueous phase as an oxygen source. The Dex-MA aqueous phase containing CP nanoparticles was mixed with another aqueous phase containing a photoinitiator at the junction point and then droplets were formed by the continuous phase. After the photo-crosslinking of droplets, homogeneous, monodisperse and stable oxygen-releasing microgels were successfully obtained. Furthermore, we also applied our microfluidic system for the controlled release of oxygen from the microgel. Herein, in situ amorphous calcium carbonate (ACC) formation was also performed to incorporate CP nanoparticles in ACC in the microgel (Fig. 1). Recently, we have reported controlled release of oxygen from CP microparticles in a weak acidic condition by stabilized ACC coating for biomedical applications.27 It is now commonly considered that calcium carbonate (CaCO3) crystals are produced via dissolution–reprecipitation process of a kinetically stabilized ACC.28–34 On the other hand, stabilization of ACC by additives such as Mg34,35 and phosphates36–38 has also been reported in recent years, which suppresses the dissolution–reprecipitation process of ACC. Therefore, stabilized ACC was produced on CP microparticle by the reaction between calcium and carbonate ions in the presence of phosphate ions.27 Based on the chemistry of CaCO3, sodium hydrogen carbonate (NaHCO3) and sodium dihydrogen phosphate (NaH2PO4) were used for the aqueous phase including a photoinitiator in this microfluidic system. Since Ca(OH)2 is already released from CP nanoparticles in the Dex-MA aqueous phase, stabilized ACC formation by mixing these two aqueous phases is expected as shown in Fig. 1. Although the microgel containing CP released oxygen quickly in a neutral cell culture medium, oxygen release from the ACC–CP microgel was suppressed because incorporation of CP in ACC suppressed the reaction between CP and water. On the other hand, oxygen release from the ACC–CP microgel was observed in the weak acidic acetate buffer because the dissolution of ACC due to lower pH and the lack of calcium and carbonate ions triggered the CP reaction.39 It is well known that the environment in ischemic tissue is weakly acidic.40 Thus, this controlled oxygen release property of an ACC–CP microgel based on CaCO3 chemistry is expected to provide selective oxygen supply in hypoxic environments. The developed oxygen releasing microgels by microfluidic system therefore show great potential for solving the oxygen shortage problem in tissue engineering field.
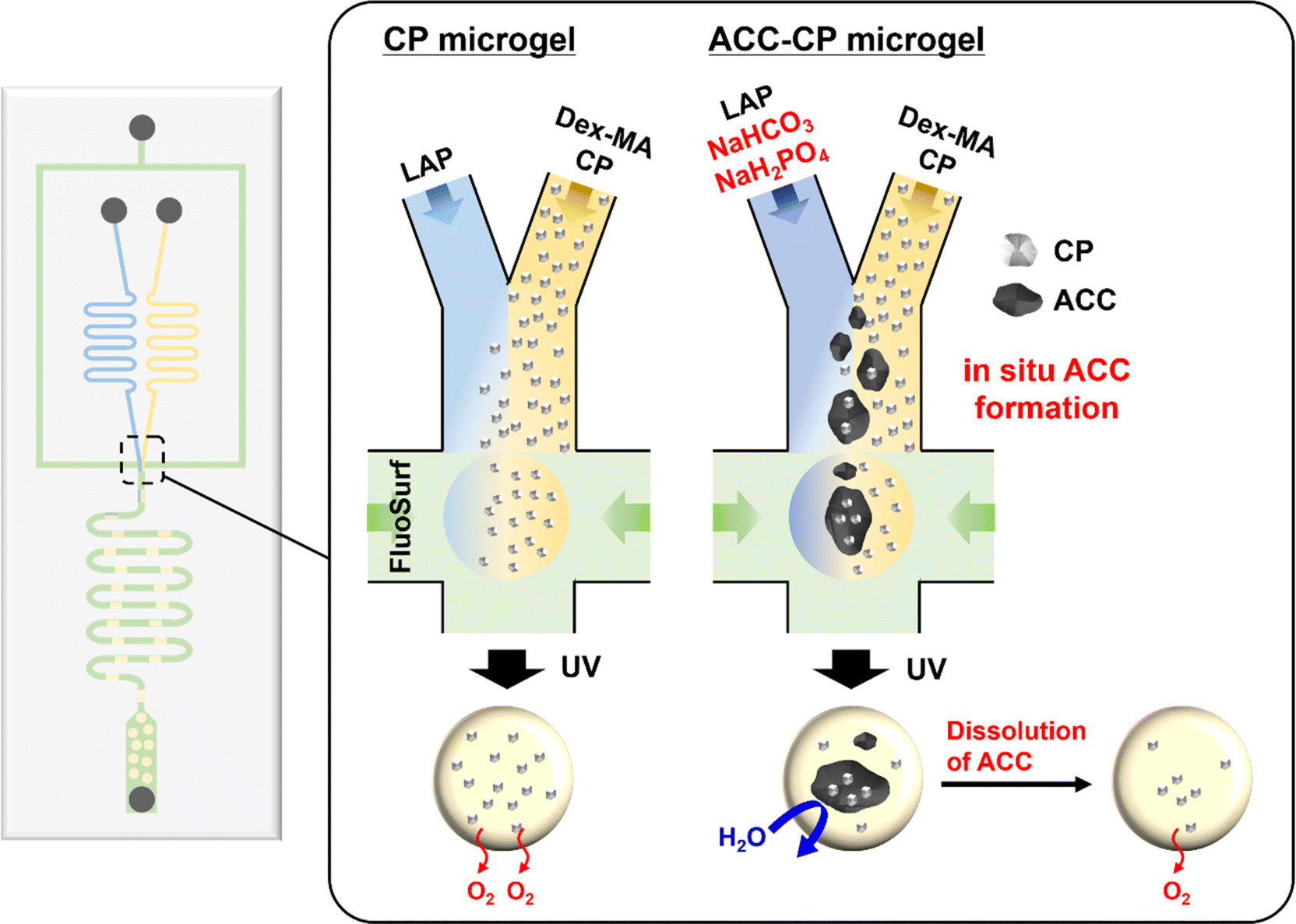 | ||
| Fig. 1 Schematic illustration of oxygen-releasing microgel synthesis and in situ ACC formation via a droplet-based microfluidic system. | ||
2. Experimental section
2.1. Materials
Glycidyl methacrylate, dimethyl sulfoxide, 4-dimethylaminopyridine, lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), catalase from bovine liver and calcium peroxide (CP) (particle size < 200 mesh (74 μm)) were purchased from Sigma-Aldrich. Dextran 150 kDa and pure HPLC water were purchased from VWR International. Sodium hydrogen carbonate (NaHCO3) and Dulbecco's modified Eagle's medium (DMEM) (transparent; 08489-45, cell culture; 08458-16) were purchased from Nacalai Tesque. Sodium dihydrogen phosphate (NaH2PO4) and sodium acetate were purchased from FUJIFILM Wako Pure Chemical Corporation. Sodium hydroxide (NaOH) and aluminum oxide were purchased from Honeywell International Inc. Aquapel was purchased from PWG Auto Glass, LLC. FluoSurf was purchased from Emulseo. Novec oil (Novec 7500) was purchased from 3M. Hexane was purchased from Fisher Scientific International, Inc. Acetic acid was purchased from Kishida Chemical Co., Ltd. Calcium chloride (CaCl2) was purchased from Sigma-Aldrich and Nacalai Tesque. Ammonium hydroxide solution (28–30%) was purchased from Sigma-Aldrich and Kishida Chemical Co., Ltd. Poly(ethylene glycol)-average mol wt 200 (PEG200) was purchased from Sigma-Aldrich and FUJIFILM Wako Pure Chemical Corporation. Hydrogen peroxide was purchased from Carl Roth and Kishida Chemical Co., Ltd. Normal human dermal fibroblasts (NHDF, CC-2509) was purchased from LONZA (Basel, Switzerland). Fetal bovine serum (FBS) and antibiotics were purchased from Thermo Fisher Scientific (MA, USA).2.2. Synthesis of CP nanoparticles
CP nanoparticles were synthesized using a previously reported method with slight modifications.41 3 mL of 0.9 M CaCl2 solution, 12 mL of PEG200 and 100 μL of ammonium hydroxide solution were mixed at room temperature. Then, 1.5 mL of hydrogen peroxide (30%) was added at a speed of 1 drop per 5 s while stirring the solution. After 2 h stirring, 0.1 M NaOH was added until the pH value of the solution reached 11.5. The obtained white suspension was centrifuged for 5 min at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, and sequentially washed with NaOH (0.1 M), water, and ethanol. Then, obtained CP nanoparticles were dispersed in ethanol. After 3 h incubation, CP nanoparticles dispersed in supernatants were collected. Collected CP nanoparticles were kept in ethanol until use. CP nanoparticle concentration in ethanol was calculated by measuring the dry mass of CP nanoparticle suspension. Scanning electron microscope (SEM) images of dried CP nanoparticles were taken using an Ultra-high Resolution Scanning Electron Microscope SU9000 (Hitachi-High Technologies). For the SEM, samples were sputter coated with carbon (Leica EM ACE600 sputter coater). The crystal structure of dried CP nanoparticles was measured by X-ray powder diffraction using a Nano-inXider SW/L Xenocs diffractometer equipped with two PILATUS3 hybrid detector arrays.
000 rpm, and sequentially washed with NaOH (0.1 M), water, and ethanol. Then, obtained CP nanoparticles were dispersed in ethanol. After 3 h incubation, CP nanoparticles dispersed in supernatants were collected. Collected CP nanoparticles were kept in ethanol until use. CP nanoparticle concentration in ethanol was calculated by measuring the dry mass of CP nanoparticle suspension. Scanning electron microscope (SEM) images of dried CP nanoparticles were taken using an Ultra-high Resolution Scanning Electron Microscope SU9000 (Hitachi-High Technologies). For the SEM, samples were sputter coated with carbon (Leica EM ACE600 sputter coater). The crystal structure of dried CP nanoparticles was measured by X-ray powder diffraction using a Nano-inXider SW/L Xenocs diffractometer equipped with two PILATUS3 hybrid detector arrays.
2.3. Synthesis of Dex-MA
The Dex-MA was synthesized according to a previously reported method.26,42,43 In brief, glycidyl methacrylate was passed through glass wool (Carl Roth) containing aluminum oxide for the activation. Under an inert atmosphere, dextran (10.01 g, 0.062 mol glucose unit, 1 eq.) was dissolved in dimethyl sulfoxide (70 mL) and then 4-dimethylaminopyridine (2.00 g, 0.016 mol, 0.3 eq.) was added. The reaction was initiated by adding glycidyl methacrylate (10 mL, 10.7 g, 0.075 mol, 1.2 eq.). The solution was stirred at room temperature for 48 h. The reaction solution was dialyzed for 5 days at 4 °C. The product was obtained through lyophilization and yielded a white powder. 1H-NMR spectra of dextran and the modified dextran were recorded with a Bruker DPX-400 FT-NMR spectrometer at a frequency of 400 MHz. All measurements were conducted in D2O as a solvent. The data analysis was carried out by MestReNova software. The compositions of dextran and modified dextran were characterized by an FTIR machine Spectrum 3 from PerkinElmer with a GladiATR accessory.2.4. Fabrication of the microfluidic device
A master mold for the microfluidic device was fabricated via photolithography as previously reported.44–46 The master mold was then used to fabricate microfluidic devices as previously reported.24 In brief, a two-component system of siloxane and a cross-linker/curing agent Sylgard 184 Elastomer Kit was mixed. The master molds filled with the polydimethylsiloxane (PDMS)/cross-linker mixture was then placed in an oven at 60 °C overnight. The hardened PDMS device was removed from the master mold and inlets and outlets for the connection of tubes were generated with a biopsy puncher (0.75 mm). The PDMS device and a glass slide were then washed three times with isopropyl alcohol and water and then placed into an oven at 60 °C for drying. After treating the PDMS chip and glass slide with oxygen plasma (PVA TePla 100 plasma system), they were adhered by pressing together. Finally, Aquapel solution was filled into the channels and flushed out with air.2.5. Microfluidic synthesis of oxygen-releasing microgels
Microfluidic experiments were conducted on a microfluidic station consisting of three syringe pumps (Harvard Apparatus, Holliston, MA, USA), to control the flow rates of the different phases. The microfluidic synthesis was observed using a microscope (Motic AE2000, TED PELLA, INC., Redding, CA) equipped with a camera (Flea3, Point Grey, Richmond, CA). The PDMS microfluidic device was connected to the syringe containing aqueous phases and a continuous phase (fluorinated oil with 2% surfactant) using fine bore polyethylene tubing. The microchannels had a rectangular cross-junction with a uniform height of 80 μm. The CP nanoparticle suspension of ethanol was centrifuged at 14000 rpm for 5 min and then dispersed in 15 wt% Dex-MA in deionized water so that the final concentrations of CP nanoparticles were 0, 10 and 20 mg mL−1. For monodisperse droplet formation, one aqueous phase containing Dex-MA and CP nanoparticles was injected into the inlet of the PDMS device at a flow rate of 150 μL h−1. Another aqueous phase containing 20 mg mL−1 LAP in deionized water was also injected at a same flow rate. For in situ ACC formation, one aqueous phase containing 15 wt% Dex-MA and 20 mg mL−1 CP nanoparticles in deionized water and another aqueous phase containing 20 mg mL−1 LAP in 88 mM NaHCO3 solution with 0.2 mg mL−1 NaH2PO4 were injected in the same way. The continuous phase containing a fluorinated oil with surfactant (FluoSurf) was injected into the third inlet at a flow rate of 600 μL h−1. Two aqueous phases were mixed at the joint point initially, then an immiscible continuous phase provided water-in-oil droplets (Fig. S1†). Droplets were collected from the outlet connected to the sample tube using fine bore polyethylene tubing. Collected droplets were exposed to UV light (365 nm) for 1 min to initiate cross-linking of the precursor solution and yielded monodisperse microgels. The obtained microgels were washed by Novec oil and hexane and their sizes were determined with ImageJ (average diameter of 50 microgels). Purified microgels were freeze-dried for storage. Morphology of purified and freeze-dried microgels were observed in deionized water by microscopy. Surface morphology of freeze-dried microgels was investigated by SEM (Hitachi-High Technologies) after sputter coated with carbon. Crystal structures of microgels were evaluated by XRD (AERIS, Malvern Panalytical, Malvern, U.K.). To clarify the structure of CP nanoparticles and ACC–CP in the microgels, microgels were crashed by a spatula. Microgels were sputter-coated with osmium using an HPC-30 Plasma Coater (Vacuum Device, Ibaraki, Japan) and then SEM and energy dispersive X-ray spectrometry (EDX) measurements were performed by Phenom ProX Desktop SEM (Thermo Fisher Scientific, MA) for the morphology and elemental analysis. CP nanoparticles leakage from microgels was also evaluated by SEM-EDX after sputter-coating with osmium. 5 mg of microgel containing 10 mg mL−1 CP nanoparticles was dispersed in 500 μL of ethanol 3 times. After freeze drying samples, elemental analysis of microgels before and after dispersing in ethanol was performed.
2.6. Oxygen release behaviors of microgels
Oxygen release behaviors of microgels were evaluated by measuring the dissolved oxygen amount in a solution containing microgels under a hypoxic condition as previously reported.4 An SDRSensor Dish Reader (SDRSensor Dish Reader, PreSens, Regensburg, Germany) was placed in a hypoxia cell culture airtight bag (6-8669-03, As one, Osaka, Japan). 15 mg mL−1 microgels were dispersed in DMEM containing 100 U mL−1 catalase and then 500 μL of microgel suspension was added into a 24-well sensor dish. As a control, a DMEM solution containing 100 U mL−1 catalase was also added into a 24-well sensor dish. Soon after the sample preparation, the 24-well sensor dish was placed on the Sensor Dish Reader. To create a hypoxic condition, three AnaeroPack O2 absorbers (MITSUBISHI GAS CHEMICAL COMPANY INC., Tokyo, Japan) were placed in the bag and it was then completely sealed using parafilm. In this system, the oxygen concentration in the air went down below 1% at around 2 h. The dissolved oxygen amount in DMEM was then monitored at 37 °C. For the ACC–CP microgel, 15 mg mL−1 ACC–CP microgel was dispersed in DMEM and 100 mM acetate buffer (pH 5.0) containing 100 U mL−1 catalase. Then, the dissolved oxygen amount in the solution including microgels was measured under a hypoxic condition as mentioned above. For the morphology changes of the ACC–CP microgel, 1 mg mL−1 ACC–CP microgel was dispersed in DMEM and 100 mM acetate buffer (pH 5.0). After 1 h incubation at 37 °C, phase contrast images of microgels were taken with a microscope. 100 mM acetate buffer (pH 5.0) was prepared by mixing 100 mM acetic acid solution and 100 mM sodium acetate solution so that the final pH was 5.0. For the evaluation of stability of ACC–CP microgel in DMEM model solution, ACC–CP microgels were dispersed in 44 mM NaHCO3 solution containing 0.1 mg mL−1 NaH2PO4, which is the buffer component of DMEM. Morphology of microgels were observed by microscopy.Oxygen release behaviors of CP nanoparticles and ACC–CP were evaluated in the same way. Briefly, ACC–CP was fabricated by immersing 0.5 mg CP nanoparticles in 50 μL of 44 mM NaHCO3 including 0.1 mg mL−1 NaH2PO4 for 1 hour. After collecting ACC–CP by centrifugation, obtained ACC–CP and 0.5 mg CP nanoparticles were dispersed in 500 μL of DMEM containing 100 U mL−1 catalase. Dissolved oxygen concentration in DMEM was measured under hypoxic condition as mentioned above. SEM images of prepared ACC–CP and CP nanoparticles immersed in DMEM for 1 hour at a concentration of 1 mg mL−1 were observed by Phenom ProX Desktop SEM after sputter-coated with osmium.
2.7. Hydrogen peroxide release from microgels
Hydrogen peroxide release from microgels was evaluated in a DMEM model solution (44 mM NaHCO3 solution containing 0.1 mg mL−1 NaH2PO4) and 100 mM acetate buffer (pH 5.0). 1.5 mg mL−1 microgels were dispersed in solution. After 60 min incubation at room temperature, the hydrogen peroxide concentration in each solution was measured using an Oxiselect Hydrogen Peroxide/Peroxidase Assay Kit (Fluorometric) (STA-344, Cell Biolabs Inc., San Diego, USA) as previously reported.4 Due to the limitation of the detection range for this assay kit, sample solutions of microgels containing 5 and 10 mg mL−1 CP nanoparticles were diluted 10 times.2.8. Cytocompatibility test of oxygen releasing microgels and pH changes of cell culture medium containing microgel
Cell proliferation in the presence of oxygen releasing microgel containing 10 mg mL−1 CP nanoparticles was evaluated by WST-8 (CCK-8) kit assay. Briefly, 1 × 104 cells of NHDF were seeded on a 96-well plate (3860-096, IWAKI, Shizuoka, Japan) using 100 μL of DMEM containing 10% FBS and 1% antibiotics. After 1 day incubation at 37 °C in a 5% CO2 incubator, 2 mg of microgels containing 10 mg mL−1 CP nanoparticles was dispersed in 50 μL of DMEM containing 1000 U mL−1 catalase, 10% FBS and 1% antibiotics. The 96-well plate inserts (E-plate Insert 16, Agilent, CA, USA) containing microgel suspension was placed in the 96-well plate with cells and then 150 μL of DMEM containing 1000 U mL−1 catalase, 10% FBS and 1% antibiotics was added. The 96-well plate was incubated in a gas barrier box (AnaeroPack Rectangular Jar, MITSUBISHI GAS CHEMICAL COMPANY INC., Tokyo, Japan) with one AnaeroPack O2 absorbers at 37 °C to create a hypoxic condition. As a control, a cell culture without microgels was also prepared and cultured under hypoxic and normoxic conditions (5% CO2 incubator). After 1 day incubation, pH in DMEM containing microgel was measured using LAQUAtwin-pH-22B (HORIBA, Kyoto, Japan). WST assay reagent was prepared by mixing DMEM and cell count reagent SF (Nacalai Tesque, Kyoto, Japan) at a ratio of 9:1. Cells were washed by 200 μL of PBS and then 100 μL of WST assay reagent was added. The cells were incubated in a 5% CO2 incubator for 30 min at 37 °C. Then, 80 μL of WST solutions from each well were collected in a 96-well plate and absorbance at 450 nm was measured using a microplate reader (SYNERGY/HTX multi-mode reader, BioTek Instruments, Winooski, USA). The mitochondrial activity of each sample was standardized from the relative values of absorbance at 450 nm as compared to 100% for that of a cell culture without microgel at normoxic condition.
2.9. Weight loss and pH change of CP after the dispersion in water
10 mg of commercially available CP microparticles were dispersed in 1 mL of ultrapure water (Milli-Q, Merck, Darmstadt, Germany). After 1 hour incubation, the pH value of solution was measured using pH meter (F-72, HORIBA, Kyoto, Japan) and then CP was collected by centrifugation at 14000 rpm for 5 min. Obtained CP was dried under reduced pressure and then weight loss of CP was measured.
3. Results and discussion
3.1. Synthesis of CP nanoparticles and Dex-MA
As commercially available CP microparticles (size < 74 μm) are too large for the microfluidic system, leading to clogging of the channels, CP nanoparticles with smaller sizes were synthesized using a previously reported method with slight modifications.41 Synthesized CP nanoparticles were analyzed by X-ray diffraction (XRD) for crystal structure and SEM for the morphology. Fig. 2a shows the XRD spectrum of synthesized CP nanoparticles. The peaks at 30, 36, 48, and 53° were observed, which were assigned to the (002), (110), (112) and (103) reflections of CP.47,48 SEM images showed aggregates composed of nanoparticles with a size around tens of nm (Fig. 2b). These aggregates were formed probably during drying of aqueous dispersion on the microscopy grid.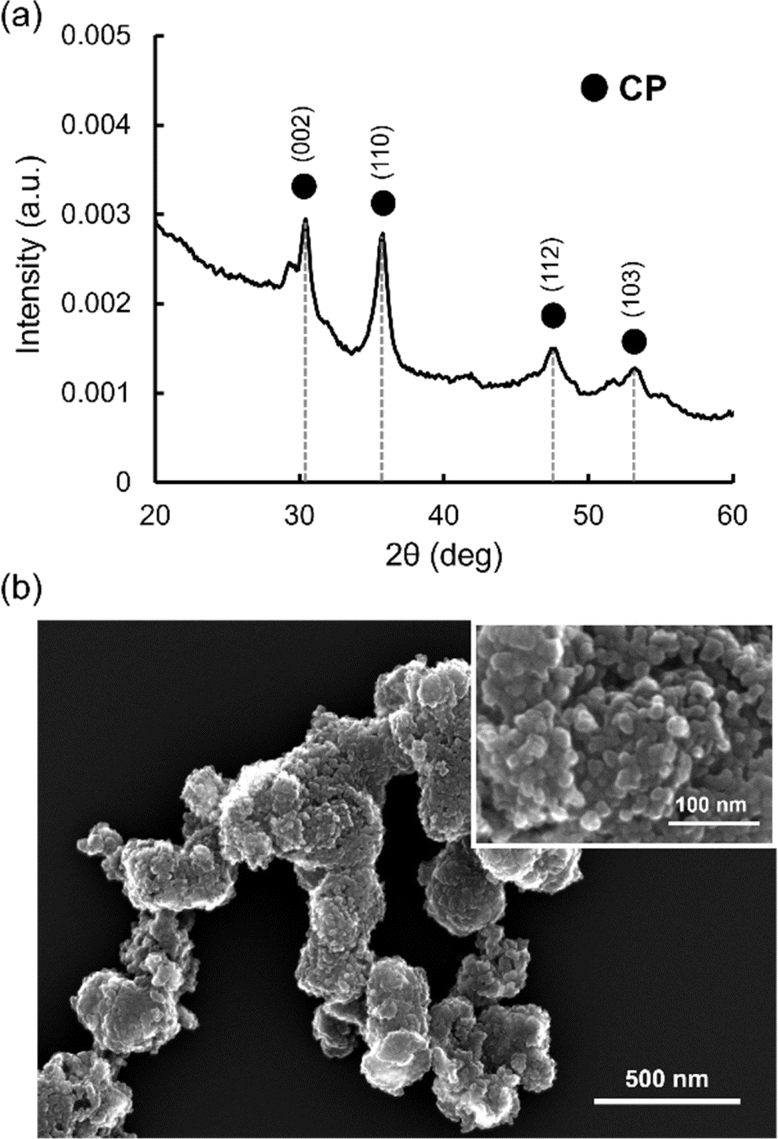 | ||
| Fig. 2 (a) XRD spectrum and (b) SEM image of synthesized CP nanoparticles. | ||
Dex-MA was synthesized by coupling glycidyl methacrylate to dextran (Mw = 150 kDa) in dimethylsulfoxide.26,42,43 The grafting degree of the methacrylate group was 48 mol% according to the integration of relevant protons in the 1H-NMR analysis (Fig. S2†). The chemical composition of Dex-MA was also evaluated by Fourier transform infrared spectroscopy (FT-IR) measurement (Fig. S3†). Dex-MA showed bands derived from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration (blue line) at around 1630 cm−1 and CO stretching vibration (red line) at around 1710 cm−1, suggesting the successful coupling of glycidyl methacrylate to dextran.
C stretching vibration (blue line) at around 1630 cm−1 and CO stretching vibration (red line) at around 1710 cm−1, suggesting the successful coupling of glycidyl methacrylate to dextran.
3.2. Microfluidic synthesis of Dex-MA microgel containing CP nanoparticles
Microgels were fabricated by free radical cross-linking reaction of droplets containing Dex-MA, CP nanoparticles and a photoinitiator (LAP) initiated through UV-light exposure after the microfluidic production of uniform aqueous droplets. LAP is known as a biocompatible photoinitiator and is often used for the fabrication of hydrogels in the tissue engineering field.25,49 Fig. 3a shows microfluidic synthesis of Dex-MA microgels containing 0, 5 and 10 mg mL−1 CP nanoparticles at the junction point. An aqueous phase containing 15 wt% Dex-MA and 0, 10 and 20 mg mL−1 CP nanoparticles was mixed with an aqueous phase containing 20 mg mL−1 LAP. Subsequently, monodisperse droplets were fabricated in a continuous phase consisting of the inert fluorinated oil “FluoSurf”. A flow rate of 150 μL h−1 was used for both of the aqueous phases and 600 μL h−1 was used for the continuous phase to generate monodisperse droplets. As shown in Fig. 3b, homogeneous droplets of black suspension were obtained using CP nanoparticles while transparent droplets were obtained without CP. This result suggests the encapsulation of CP nanoparticles in the droplets in this system. After the droplet formation, collected droplets were exposed to UV light for 1 min and subsequently purified and redispersed in water (Fig. 3c). In all conditions, homogeneous and monodisperse microgels with a size of 114.2 ± 4.1 μm for 0 mg mL−1 CP, 111.4 ± 4.1 μm for 5 mg mL−1 CP and 110.5 ± 5.3 μm for 10 mg mL−1 CP were obtained. The amount of CP nanoparticles in the microgel derived from 5 mg mL−1 CP was much smaller than that before UV crosslinking. This is probably because many CP nanoparticles were decomposed by UV irradiation.50 In the case of 10 mg mL−1 CP, CP remained in the microgel even after UV crosslinking. According to these results, fabrication of the Dex-MA microgel containing CP nanoparticles was confirmed especially for the higher concentration of CP nanoparticles.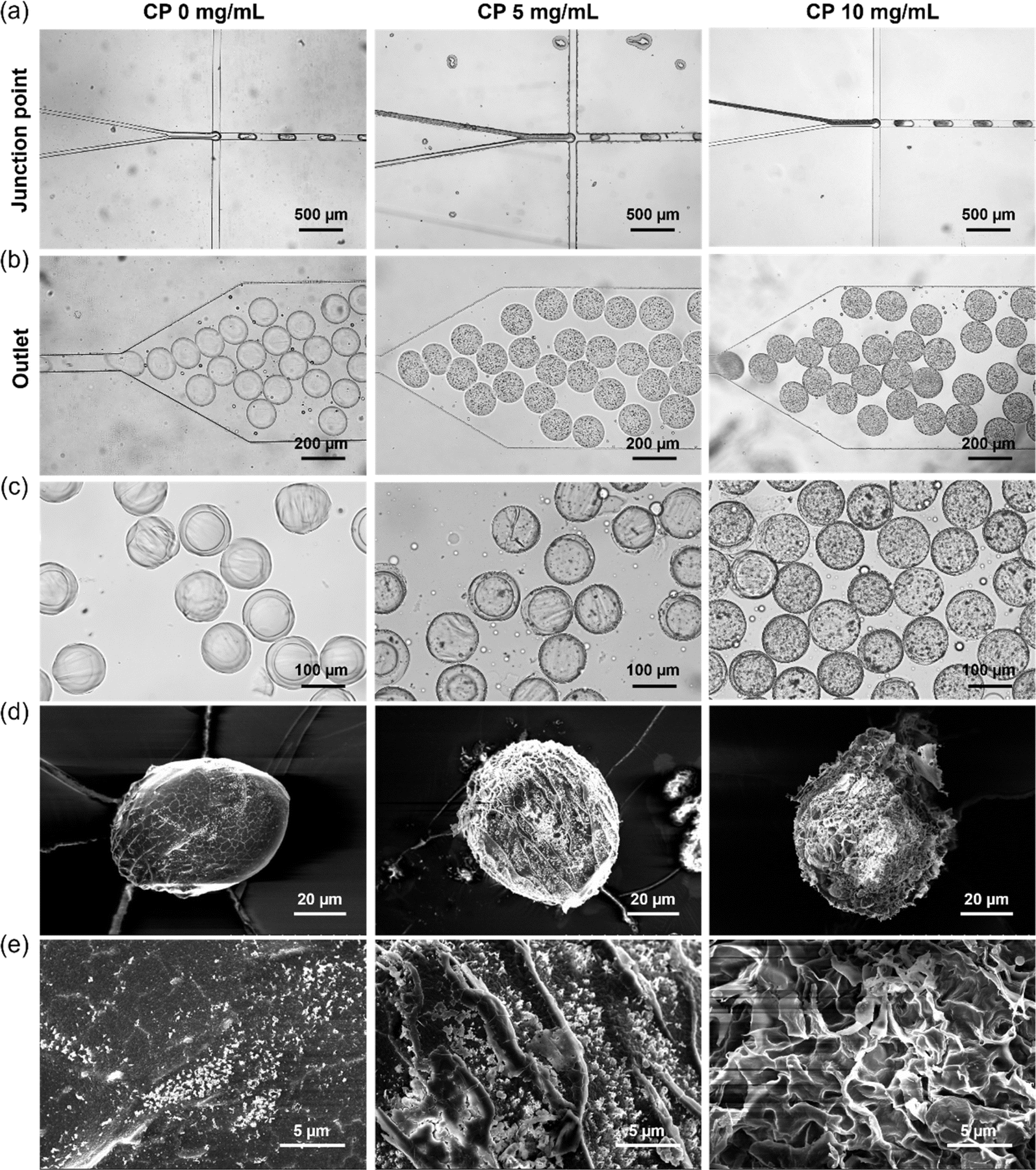 | ||
| Fig. 3 Bright field images of microfluidic synthesis of Dex-MA microgels containing 0, 5 and 10 mg mL−1 CP nanoparticles at (a) junction point and (b) outlet. (c) Bright field images of a purified Dex-MA microgel containing 0, 5 and 10 mg mL−1 CP nanoparticles. SEM images of a freeze-dried Dex-MA microgel containing 0, 5 and 10 mg mL−1 CP nanoparticles at (d) lower and (e) higher magnification. | ||
For the storage of oxygen-releasing microgels, the obtained microgels were freeze-dried. Fig. 3d and e shows SEM images of a freeze-dried Dex-MA microgel containing 0, 5 and 10 mg mL−1 CP nanoparticles taken at lower and higher magnifications. Freeze-dried microgels without CP showed a smooth surface structure, suggesting that the crosslinking degree of the polymer network is high. On the other hand, the freeze-dried microgel containing 5 mg mL−1 CP nanoparticles showed a porous structure compared to the case without CP. This is probably because oxygen and hydrogen peroxide released from CP interfered with the free radical cross-linking reaction in the droplet, which provided the microgel with a lower crosslinking degree. Furthermore, the freeze-dried microgel containing 10 mg mL−1 CP nanoparticles showed a larger porous structure compared to the case of 5 mg mL−1 CP nanoparticles. This is probably because higher concentrations of CP released a greater amount of oxygen and hydrogen peroxide, which interfered more greatly with the radical cross-linking reaction.
To investigate the leakage of CP nanoparticle from microgels, elemental analysis of microgels was performed after dispersing them in ethanol 3 times. The ethanol was selected as a solvent since CP nanoparticles in microgels react with water and then dissolve over time. The EDX spectra of microgels before and after dispersion in ethanol showed the peak derived from calcium, suggesting that CP nanoparticles didn't diffuse out after dispersing the microgels in solution (Fig. S4†).
3.3. Stability of freeze-dried oxygen-releasing microgels
To evaluate the stability of the freeze-dried microgels, they were dispersed in water. As shown in Fig. 4a, all freeze-dried microgels retained their spherical morphology in water. Furthermore, after 7 days incubation in water, none of the freeze-dried microgels changed their spherical morphologies compared to soon after the dispersion in water, suggesting that microgels were not decomposed by hydrogen peroxide and oxygen released from CP (Fig. 4b). These results suggested that oxygen-releasing microgels were stable even after freeze-drying. Therefore, these freeze-dried microgels are storable in a dry state until their use.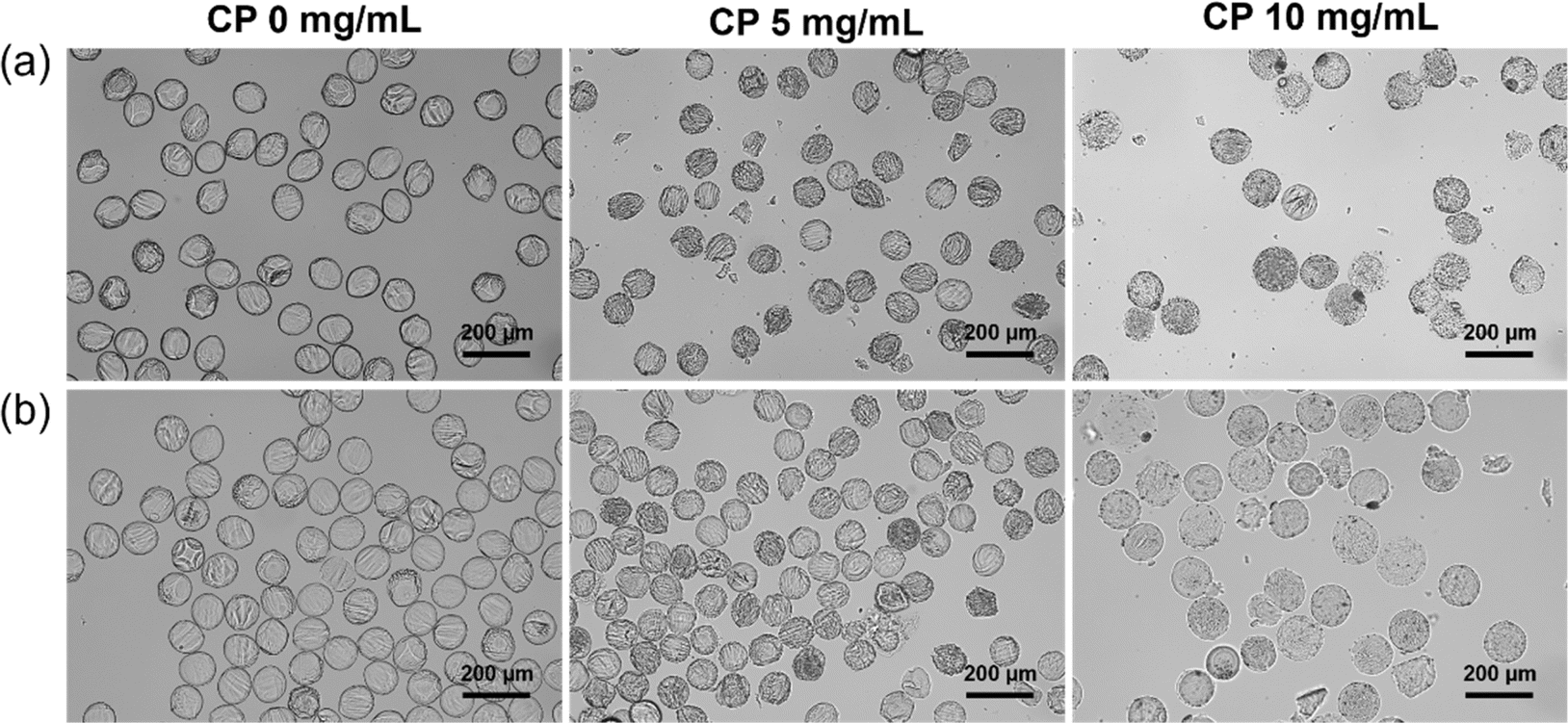 | ||
| Fig. 4 Bright field images of freeze-dried Dex-MA microgels containing 0, 5 and 10 mg mL−1 CP nanoparticles after (a) the dispersion in water and (b) 7 days incubation in water. | ||
3.4. Oxygen release behaviors of microgels
Oxygen release behaviors of freeze-dried microgels in a cell culture medium (DMEM) containing 100 U mL−1 catalase were evaluated by measuring dissolved oxygen concentration in the solution after the dispersion of microgels under a hypoxic condition as previously reported (Fig. 5a).4 Catalase is an enzyme which decomposes hydrogen peroxide into oxygen.51 Thus, larger amounts of oxygen release from microgels containing CP nanoparticles can be observed since CP releases not only oxygen but also hydrogen peroxide.52 Herein, 100% indicates equilibrium with oxygen in the atmosphere. Without microgels, dissolved oxygen concentration decreased rapidly because of the hypoxic condition (black line). On the other hand, dissolved oxygen concentration in the solution containing a microgel with 5 mg mL−1 CP nanoparticles increased to 108% and showed a higher value until 200 min compared to without a microgel (yellow line). Furthermore, dissolved oxygen concentration in the solution containing a microgel with 10 mg mL−1 CP nanoparticles increased over 250% (detection limit of the oxygen measuring machine) and showed a higher value until 300 min compared to without a microgel (red line). These results confirm oxygen release from microgels containing CP nanoparticles. We also evaluated hydrogen peroxide release from microgels. Since the components of DMEM such as methionine decompose hydrogen peroxide,4,53 44 mM NaHCO3 solution including 0.1 mg mL−1 NaH2PO4 was used as a model solution for DMEM to evaluate the detailed hydrogen peroxide release behavior. As expected, the microgel without CP nanoparticles did not release hydrogen peroxide (below 1 μM), as shown in Fig. 5b. The microgel containing 5 mg mL−1 CP nanoparticles released 33.9 ± 1.2 μM hydrogen peroxide while the microgel containing 10 mg mL−1 CP nanoparticles released 213.2 ± 3.3 μM hydrogen peroxide in a DMEM model solution for 60 min. The value of oxygen and hydrogen peroxide release from the microgel containing 5 mg mL−1 CP nanoparticles was much smaller than that of 10 mg mL−1 CP nanoparticles. As explained earlier, this is probably because many CP nanoparticles decomposed in the microgel containing 5 mg mL−1 CP nanoparticles during the radical cross-linking reaction. Thus, when enough CP nanoparticles were contained in the microgel, larger amounts of oxygen and hydrogen peroxide release can be seen. Since CP release basic calcium hydroxide as a byproduct, pH changes of DMEM containing microgels were evaluated. The pH value in DMEM didn't change after dispersing 10 mg mL−1 microgel containing 10 mg mL−1 CP nanoparticles (pH 7.4), suggesting that 44 mM NaHCO3 in DMEM suppressed pH changes in this condition. Cytocompatibility of oxygen releasing microgel was also evaluated since microgel release not only oxygen but also toxic hydrogen peroxide (Fig. S5†). Mitochondria activities of cells didn't change in the presence of 10 mg mL−1 microgel containing 10 mg mL−1 CP nanoparticles using catalase under hypoxic condition compared to the cells without microgels under normoxic and hypoxic conditions. These results suggested that oxygen releasing microgels are biocompatible in the presence of catalase.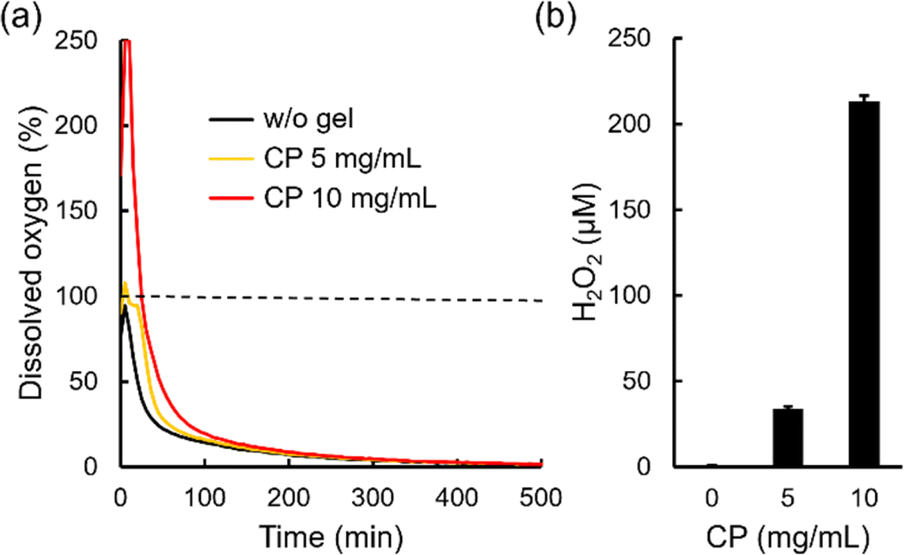 | ||
| Fig. 5 (a) Dissolved oxygen concentration changes in 500 μL DMEM containing 100 U mL−1 catalase and 15 mg mL−1 Dex-MA microgel encapsulating 5 and 10 mg mL−1 CP nanoparticles under a hypoxic condition. (b) Hydrogen peroxide concentration in a DMEM model solution (44 mM NaHCO3 solution containing 0.1 mg mL−1 NaH2PO4) after the dispersion of 1.5 mg mL−1 microgels containing 0, 5 and 10 mg mL−1 CP nanoparticles for 60 min. Data presented as mean ± SD, n = 3. | ||
3.5. Microfluidic synthesis of a Dex-MA microgel containing ACC–CP by in situ ACC formation
For the in situ ACC formation in the microfluidic system, an aqueous phase containing 15 wt% Dex-MA and 20 mg mL−1 CP nanoparticles was mixed with another aqueous phase containing 20 mg mL−1 LAP, 88 mM NaHCO3 and 0.2 mg mL−1 NaH2PO4. The concentrations of NaHCO3 and NaH2PO4 were derived from our previous report about ACC coating on CP microparticles.27 As shown in Fig. 6a, aggregates were formed at the interfaces of two aqueous phases probably because of ACC formation by the reaction between Ca(OH)2 derived from CP nanoparticles in the Dex-MA aqueous phase and NaHCO3 containing NaH2PO4 in the other aqueous phase. After the droplet formation (Fig. 6b), collected droplets were exposed to UV light for 1 min and subsequently purified and redispersed in water (Fig. 6c). Interestingly, purified microgels showed aggregates inside, suggesting the formation of ACC in the microgel. As with the microgel containing CP nanoparticles, SEM images of the freeze-dried ACC–CP microgel showed a porous structure (Fig. 6d). In addition, the freeze-dried ACC–CP microgel was stable even after dispersion in water and 7 days incubation in water (Fig. 6e and f). | ||
| Fig. 6 Bright field images of microfluidic synthesis of an ACC–CP microgel at (a) junction point and (b) outlet. (c) Bright field image of a purified ACC–CP microgel. (d) SEM images of a freeze-dried ACC–CP microgel. Bright field images of a freeze-dried ACC–CP microgel after (e) the dispersion in water and (f) 7 days incubation in water. | ||
For the elemental analysis on the microgels, SEM-EDX measurements were performed. To clarify the structure of CP nanoparticles and ACC–CP in the microgels, microgels were crashed by a spatula before sputter-coating with osmium. The elemental mapping of microgels containing 10 mg mL−1 CP nanoparticles showed calcium throughout the cross section of microgels, suggesting that CP nanoparticles were distributed homogeneously in the microgel (Fig. S6a and b†). From a high magnification SEM image, CP nanoparticles with a size below 1 μm were observed (Fig. S6c†). On the other hand, the elemental mapping of microgels containing ACC–CP showed calcium locally, suggesting the formation of aggregates composed of CP and ACC (Fig. S6d and e†). This result is consistent with the observation of aggregates in Fig. 6. High magnification SEM image of these aggregates showed small particles of around 1 μm in size (Fig. S6f†). To clarify the crystal structure of inorganic compound in microgels, XRD measurements was performed. Microgel containing 10 mg mL−1 CP nanoparticles showed small peaks at 30 and 36°, which were assigned to the (002) and (110) reflections of CP (Fig. S7a†). On the other hand, ACC–CP microgel showed peaks at 30, 36, 40, 43, 48 and 49°, which were assigned to the (104), (110), (113), (202), (024), and (116) reflections of calcite, one of the crystal structures of calcium carbonate (Fig. S7b†). Due to the overlap of peaks, the peaks derived from CP could not be identified at ACC–CP microgel. XRD results suggested the existence of calcite at the aggregates in ACC–CP microgel. It is well known that typical calcite shows cubic structure.27,28,31,34,37 However, cubic structure was not clearly observed in ACC–CP microgel in the SEM image (Fig. S6f†). Therefore, it was suggested that most of the aggregates in microgels were ACC.
3.6. Controlled release of oxygen and hydrogen peroxide from an ACC–CP microgel
In our previous study, CP microparticles coated with ACC did not release oxygen in a neutral cell culture medium because ACC suppressed the reaction between CP and water. On the other hand, ACC–CP released oxygen in a weak acidic condition because dissolution of ACC started the CP reaction.27 To evaluate such a controlled release of oxygen derived from the dissolution of ACC, the oxygen release behavior of the ACC–CP microgel was measured in neutral DMEM and acetate buffer (pH 5.0) containing 100 U mL−1 catalase under a hypoxic condition. Fig. 7a and S8† show dissolved oxygen concentration in DMEM after the addition of the ACC–CP microgel. Unlike the microgels containing CP nanoparticles (Fig. S9†), the time dependent changes of dissolved oxygen concentration in DMEM containing the ACC–CP microgel showed a similar trend to the case without a microgel, suggesting that oxygen release from the ACC–CP microgel was suppressed. On the other hand, dissolved oxygen concentration in a weakly acidic acetate buffer (pH 5.0) containing the ACC–CP microgel increased until 150 min compared to the case without a microgel, suggesting oxygen release from the ACC–CP microgel (Fig. 7b). Compared to the microgel containing 10 mg mL−1 CP nanoparticles (Fig. 5a), the increase value of dissolved oxygen concentration was much smaller for the ACC–CP microgel. It has been reported that CP is stable in a basic condition while it dissolves quickly in acidic and neutral conditions.54 For the fabrication process of a microgel containing CP nanoparticles, the pH in water used for aqueous phases increased because basic Ca(OH)2 was released from CP. Thus, it is considered that further CP reaction was suppressed in this basic aqueous phase. For 10 mg mL−1 CP microparticles in water, the pH value in water reached 12.0 after 1 hour incubation. As a result, the weight loss of CP was below 1% as expected. On the other hand, in the fabrication process of the ACC–CP microgel, NaHCO3 solution was used for one aqueous phase. Since NaHCO3 has a buffering effect, the pH change in the aqueous phase was suppressed compared to water. Therefore, it is considered that many CP nanoparticles were consumed during the in situ ACC formation process. We also evaluated hydrogen peroxide release from the ACC–CP microgel in a DMEM model solution (44 mM NaHCO3 and 0.1 mg mL−1 NaH2PO4) and acetate buffer (pH 5.0). In our previous study, we reported that the buffer component of DMEM (44 mM NaHCO3 and 0.1 mg mL−1 NaH2PO4) played an important role in the stabilization of ACC.27 As shown in Fig. 7c, the ACC–CP microgel released around a 7-times higher concentration of hydrogen peroxide in a weakly acidic acetate buffer (pH 5.0) compared to the DMEM model solution. To understand these oxygen and hydrogen peroxide release behaviors of the ACC–CP microgel, the microgel morphologies were observed after immersing in DMEM and a weakly acidic acetate buffer (pH 5.0). In DMEM, the aggregates derived from ACC were observed inside the microgels (Fig. 7d), suggesting that incorporation of CP nanoparticles in ACC suppressed the reaction with water. On the other hand, in the weakly acidic acetate buffer (pH 5.0), aggregates inside the microgel had disappeared (Fig. 7e). These results suggest that the dissolution of ACC triggered the CP reaction with water, which started oxygen and hydrogen peroxide release from the ACC–CP microgel. In addition, aggregates inside the ACC–CP microgels were observed in DMEM model solution for 7 days, suggesting the long-term stability of aggregates composed of CP nanoparticles and ACC (Fig. S10†).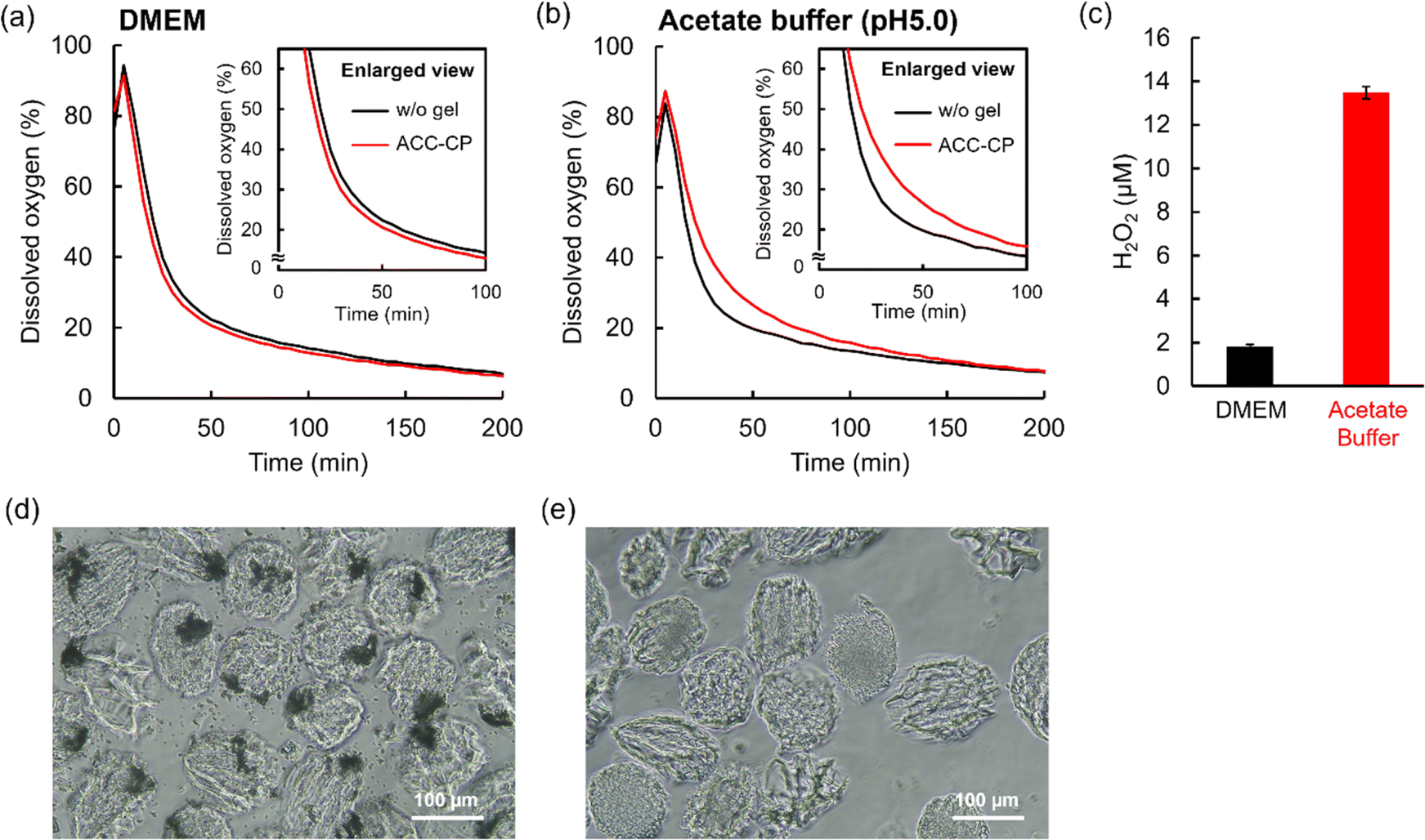 | ||
| Fig. 7 Variation of the dissolved oxygen concentration with time in 500 μL (a) DMEM and (b) 100 mM acetate buffer (pH 5.0) containing 100 U mL−1 catalase and 15 mg mL−1 ACC–CP microgel under a hypoxic condition. (c) Hydrogen peroxide concentration in a DMEM model solution (44 mM NaHCO3 solution containing 0.1 mg mL−1 NaH2PO4) and 100 mM acetate buffer (pH 5.0) after the dispersion of 1.5 mg mL−1 ACC–CP microgel for 60 min. Data presented as mean ± SD, n = 3. Phase contrast images of a freeze-dried ACC–CP microgel dispersed in (d) DMEM and (e) 100 mM acetate buffer (pH 5.0) for 60 min. | ||
To investigate the effect of encapsulating oxygen sources in microgels, oxygen release behaviors of ACC–CP and CP nanoparticles were evaluated in DMEM (Fig. S11a†). Unlike ACC–CP microgel (Fig. 7a), ACC–CP released oxygen in DMEM. To understand these differences, SEM image of ACC–CP was observed (Fig. S11b†). ACC–CP showed some cubic calcite, suggesting that crystallization of ACC to calcite triggered oxygen release from ACC–CP. On the other hand, cubic calcite was not observed in ACC–CP microgel as shown in Fig. S6.† Thus, it is suggested that polymer network of microgel suppressed the crystallization of ACC, which prevented oxygen release from ACC–CP microgel in DMEM. ACC–CP released higher amount of oxygen than CP nanoparticle in DMEM while some CP was already consumed during the preparation process of ACC–CP. Unlike ACC–CP, SEM image of CP nanoparticles immersed in DMEM didn't show cubic calcite (Fig. S11c†). In the measurement of oxygen release behavior, 1 mg mL−1 CP nanoparticles were dispersed in DMEM. Since DMEM includes 44 mM NaHCO3 and 0.1 mg mL−1 NaH2PO4, ACC formation on CP nanoparticle is expected based on our previous study,27 which suppressed the CP reaction. On the other hand, 10 mg mL−1 CP nanoparticles were dispersed in 44 mM NaHCO3 solution including 0.1 mg mL−1 NaH2PO4 for the preparation of ACC–CP in this study. Thus, it is suggested that the difference of CP concentration in carbonate solution affected ACC formation. Since some crystallization was observed for ACC–CP, ACC–CP released higher amount of oxygen than CP nanoparticle in DMEM. Compared to CP nanoparticles, microgel containing CP nanoparticles showed burst release of oxygen in DMEM (Fig. 5a). This is probably because incorporation of CP in polymer network prevented the ACC formation on CP.
4. Conclusions
In this study, we reported the fabrication of oxygen-releasing microgels using a droplet-based microfluidic system. Homogeneous and monodisperse microgels containing CP nanoparticles as an oxygen source were obtained by photo-crosslinking of Dex-MA in aqueous droplets. Under optimized synthesis conditions, we were able to vary the loading of CP nanoparticles into microgels. Obtained oxygen-releasing microgels were stable even after freeze drying, suggesting that they are storable in a dry condition. Since oxygen release from the microgel containing CP nanoparticles was observed, application of this new class of biomaterials for tissue engineering is highly expected. Furthermore, in situ ACC formation in the microfluidic system was also performed for the controlled release of oxygen from the microgel. The obtained ACC–CP microgel did not release oxygen in neutral DMEM because incorporation of CP in ACC suppressed the reaction with water. However, the ACC–CP microgel released oxygen in a weakly acidic acetate buffer because the dissolution of ACC started the reaction between CP and water. Since the environment in ischemic tissue is weakly acidic40 and the solubility of ACC increases in an acidic condition,39 this controlled oxygen release property of the ACC–CP microgel based on CaCO3 chemistry would be an attractive function. We believe that application of this new class of biomaterials for tissue engineering are greatly anticipated. In addition, oxygen releasing materials using CP are used not only for tissue engineering but also for cancer therapies55 and water and soil treatments.56 Thus, application of oxygen-releasing microgels to these research fields are also expected.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
D. T. and S. A. J. contributed equally to this work. D. T. and S. A. J. carried out the experiments. D. T. wrote the manuscript. All authors edited the manuscript. All authors contributed to the design and implementation of the research and the analysis of results. All authors have seen and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge XRD and SEM analyses for Sven Buschmann and Stefan Hauk in DWI-Leibniz Institute for Interactive Materials and Prof. T. Kida in Osaka University, respectively. This study was financially supported by COI-NEXT (JPMJPF2009) from JST, Grant-in-Aid for Scientific Research (A) (20H00665), Grant-in-Aid for Challenging Exploratory Research (22K19918), Grant-in-Aid for JSPS Fellows (22J20533), Grants-in-Aid for Scientific Research “KAKENHI” The Promotion of Joint International Research, “International Leading Research” (22K21348) from JSPS and Deutsche Forschungsgemeinschaft (DFG) in the framework of the joint project “Towards a model-based control of biohybrid implant maturation” (PAK961) by the grant NE1650/4-2 and PI614/13-2 and the Excellence Strategy of the Federal Government and the Länder by the grant G:(DE-82)EXS-SF-OPSF761.Notes and references
- T. Shimizu, H. Sekine, J. Yang, Y. Isoi, M. Yamato, A. Kikuchi, E. Kobayashi and T. Okano, FASEB J., 2006, 6, 708 CrossRef PubMed.
- M. Radisic, J. Malda, E. Epping, W. Geng, R. Langer and G. V. Novakovic, Biotechnol. Bioeng., 2006, 93, 332 CrossRef CAS.
- Y. Mizukami, Y. Takahashi, K. Shimizu, S. Konishi, Y. Takakura and M. Nishikawa, Biol. Pharm. Bull., 2021, 44, 1458 CrossRef CAS PubMed.
- D. Tomioka, S. Fujita, J. Groll and M. Matsusaki, Chem. Mater., 2023, 35, 5378 CrossRef CAS.
- A. L. Farris, A. N. Rindone and W. L. Grayson, J. Mater. Chem. B, 2016, 4, 3422 RSC.
- T. Agarwal, S. Kazemi, M. Costantini, F. Perfeito, C. R. Correia, V. Gaspar, L. Montazeri, C. D. Maria, J. F. Mano, M. Vosough, P. Makvandi and T. K. Maiti, Mater. Sci. Eng., C, 2021, 121, 111896 CrossRef PubMed.
- N. G. A. Willemen, S. Hassan, M. Gurian, J. Li, I. E. Allijn, S. R. Shin and J. Leijten, Trends Biotechnol., 2021, 39, 1144 CrossRef CAS PubMed.
- H. Y. Lee, H. W. Kim, J. H. Lee and S. H. Oh, Biomaterials, 2015, 53, 583 CrossRef CAS.
- X. Fu, S. Ohta, M. Kamihira, Y. Sakai and T. Ito, Langmuir, 2019, 35, 4094 CrossRef CAS.
- A. Paciello, G. Amalfitano, A. Garziano, F. Urciuolo and P. A. Netti, Adv. Healthcare Mater., 2016, 5, 2655 CrossRef CAS.
- S. Ohta, K. Hashimoto, X. Fu, M. Kamihira, Y. Sakai and T. Ito, J. Biosci. Bioeng., 2018, 126, 533 CrossRef CAS PubMed.
- S. Park and K. Min Park, Biomaterials, 2018, 182, 234 CrossRef CAS PubMed.
- N. Alemdar, J. Leijten, G. C. Unal, J. Hjortnaes, J. Ribas, A. Paul, P. Mostafalu, A. K. Gaharwar, Y. Qiu, S. Sonkusale, R. Liao and A. Khademhosseini, ACS Biomater. Sci. Eng., 2017, 3, 1964 CrossRef CAS.
- B. Newland, M. Baeger, D. Eigel, H. Newland and C. Werner, ACS Biomater. Sci. Eng., 2017, 3, 787 CrossRef CAS PubMed.
- Z. Lu, X. Jiang, M. Chen, L. Feng and Y. J. Kang, Biofabrication, 2019, 11, 045012 CrossRef CAS PubMed.
- M. Razavi, R. Primavera, B. D. Kevadiya, J. Wang, P. Buchwald and A. S. Thakor, Adv. Funct. Mater., 2020, 1902463 CrossRef CAS.
- A. Farzin, S. Hassan, L. S. M. Teixeira, M. Gurian, J. F. Crispim, V. Manhas, A. Carlier, H. Bae, L. Geris, I. Noshadi, S. R. Shin and J. Leijten, Adv. Funct. Mater., 2021, 2100850 CrossRef CAS PubMed.
- N. G. A. Willemen, S. Hassan, M. Gurian, M. F. J. Salazar, K. Fan, H. Wang, M. Becker, I. E. Allijn, A. B. Öztürk, J. Leijten and S. R. Shin, Adv. Healthcare Mater., 2022, 11, 2102697 CrossRef CAS PubMed.
- D. Mondal, M. Griffith and S. S. Venkatraman, Int. J. Polym. Mater. Polym. Biomater., 2016, 65, 255 CrossRef CAS.
- M. Xie, J. Wang, S. Wu, S. Yan and Y. He, Biomater. Sci., 2024, 12, 1950 RSC.
- Q. Feng, D. Li, Q. Li, X. Cao and H. Dong, Bioact. Mater., 2022, 9, 105 CAS.
- Z. Wei, S. Wang, J. Hirvonen, H. A. Santos and W. Li, Adv. Healthcare Mater., 2022, 11, 2200846 CrossRef CAS.
- K. O. Rojek, M. Ćwiklińska, J. Kuczak and J. Guzowski, Chem. Rev., 2022, 122, 16839 CrossRef CAS PubMed.
- S. Bulut, S. H. Jung, T. Bissing, F. Schmitt, M. Bund, S. Braun and A. Pich, Small, 2023, 2303783 CrossRef CAS PubMed.
- S. Bulut, D. Günther, M. Bund, C. Haats, T. Bissing, C. Bastard, M. Wessling, L. De Laporte and A. Pich, Adv. Healthcare Mater., 2023, 2302957 CrossRef PubMed.
- S. A. Jung, H. Malyaran, D. E. Demco, A. Manukanc, L. S. Häser, V. Kučikas, M. V. Zandvoort, S. Neuss and A. Pich, Biomacromolecules, 2023, 24, 3972 CrossRef CAS.
- D. Tomioka, S. Fujita and M. Matsusaki, ACS Omega, 2024, 9, 5903 CrossRef CAS PubMed.
- Y. Q. Niu, J. H. Liu, C. Aymonier, S. Fermani, D. Kralj, G. Falini and C. H. Zhou, Chem. Soc. Rev., 2022, 51, 7883 RSC.
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819 CrossRef CAS PubMed.
- J. J. D. Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- M. H. Nielsen, S. Aloni and J. J. D. Yoreo, Science, 2014, 345, 1158 CrossRef CAS PubMed.
- J. Ihli, W. C. Wong, E. H. Noel, Y. Y. Kim, A. N. Kulak, H. K. Christenson, M. J. Duer and F. C. Meldrum, Nat. Commun., 2014, 5, 3169 CrossRef PubMed.
- P. Bots, L. G. Benning, J. D. R. Blanco, T. R. Herrero and S. Shaw, Cryst. Growth Des., 2012, 12, 3806 CrossRef CAS.
- Z. Liu, Z. Zhang, Z. Wang, B. Jin, D. Li, J. Tao, R. Tang and J. J. D. Yoreo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 3397 CrossRef CAS.
- M. Albéric, L. Bertinetti, Z. Zou, P. Fratzl, W. Habraken and Y. Politi, Adv. Sci., 2018, 5, 1701000 CrossRef.
- Z. Zou, X. Yang, M. Albéric, T. Heil, Q. Wang, B. Pokroy, Y. Politi and L. Bertinetti, Adv. Funct. Mater., 2020, 30, 2000003 CrossRef CAS.
- Z. Zou, J. Xie, E. M. Sanchez and Z. Fu, Cryst. Growth Des., 2021, 21, 414 CrossRef CAS.
- S. Kababya, A. Gal, K. Kahil, S. Weiner, L. Addadi and A. Schmidt, J. Am. Chem. Soc., 2015, 137, 990 CrossRef CAS PubMed.
- M. Wang, B. Zhou, L. Wang, F. Zhou, N. Smith, D. Saunders, R. A. Towner, J. Song, J. Qu and W. R. Chen, J. Mater. Chem. B, 2020, 8, 8261 RSC.
- G. X. Yan and A. G. Kléber, Circ. Res., 1992, 71, 460 CrossRef CAS PubMed.
- Y. Hu, X. Wang, P. Zhao, H. Wang, W. Gu and L. Ye, Biomater. Sci., 2020, 8, 2931 RSC.
- W. N. E. van Dijk-Wolthuis, O. Franssen, H. Talsma, M. J. van Steenbergen, J. J. Kettenes-van den Bosch and W. E. Hennink, Macromolecules, 1995, 28, 6317 CrossRef CAS.
- W. N. E. van Dijk-Wolthuis, J. J. Kettenes-van den Bosch, A. van der Kern Hoof and W. E. Hennink, Macromolecules, 1997, 30, 3411 CrossRef CAS.
- S. H. Jung, F. Meyer, S. Hörnig, M. Bund, B. Häßel, L. P. B. Guerzoni, L. D. Laporte, G. B. Messaoud, S. P. Centeno and A. Pich, Small, 2024, 20, 2303444 CrossRef CAS.
- S. H. Jung, S. Bulut, L. P. B. B. Guerzoni, D. Günther, S. Braun, L. D. Laporte and A. Pich, J. Colloid Interface Sci., 2022, 617, 409 CrossRef CAS PubMed.
- L. P. B. Guerzoni, J. C. Rose, D. B. Gehlen, A. Jans, T. Haraszti, M. Wessling, A. J. C. Kuehne and L. D. Laporte, Small, 2019, 15, 1900692 CrossRef.
- D. M. D. Santos, L. M. Dias, A. K. Surur, D. A. D. Moraes, A. C. Pavarina, C. R. Fontana and D. S. Correa, ACS Appl. Nano Mater., 2022, 5, 14425 CrossRef.
- X. Zhao, M. C. Nguyen, C. Z. Wang and K. M. Ho, RSC Adv., 2013, 3, 22135 RSC.
- N. Monteiro, G. Thrivikraman, A. Athirasala, A. Tahayeri, C. M. França, J. L. Ferracane and L. E. Bertassoni, Dent. Mater., 2018, 34, 389 CrossRef CAS PubMed.
- S. Zhang, B. Song, R. Wang, Z. Zhan, M. Saakes, R. D. van der Weijden, C. J. N. Buisman and Y. Lei, ACS ES&T Water, 2023, 3, 866 Search PubMed.
- M. A. Prieto, X. Biarnes, P. Vidossich and C. Rovira, J. Am. Chem. Soc., 2009, 131, 11751 CrossRef PubMed.
- H. Wang, Y. Zhao, T. Li, Z. Chen, Y. Wang and C. Qin, Chem. Eng. J., 2016, 303, 450 CrossRef CAS.
- S. Boonvisut, A. Aksnes and L. R. Njaa, Food Chem., 1982, 9, 183 CrossRef CAS.
- A. Northup and D. Cassidy, J. Hazard. Mater., 2008, 152, 1164 CrossRef CAS PubMed.
- Y. Xiao, Z. Li, A. Bianco and B. Ma, Adv. Funct. Mater., 2023, 33, 2209291 CrossRef CAS.
- S. Lu, X. Zhang and Y. Xue, J. Hazard. Mater., 2017, 337, 163 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04356a |
| ‡ These authors have contributed equally to the study. |
| This journal is © The Royal Society of Chemistry 2024 |