
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaction contest: hydrolysis versus intramolecular cyclisation reaction in alkyl squaramate esters†
Marta Ximenis ,
Santiago Cañellas,
Rosa M. Gomila,
Bartomeu Galmés,
Antonio Frontera,
Antonio Costa and
Carmen Rotger*
,
Santiago Cañellas,
Rosa M. Gomila,
Bartomeu Galmés,
Antonio Frontera,
Antonio Costa and
Carmen Rotger*
Universitat de les Illes Balears, Cra. Valldemossa Km 7.5, Palma de Mallorca, 07122, Spain. E-mail: carmen.rotger@uib.es
First published on 11th October 2024
Abstract
The stability and hydrolytic behavior of squaramate esters in aqueous solutions have been investigated. The structure of squaramates and the nature of adjacent groups significantly influence their aqueous stability and reactivity towards nucleophiles. Squaramate esters, lacking or containing weakly basic neighboring group participation (NGP) substitutions, remain stable up to pH 9. Their hydrolysis rate (kOH ≈ 10−1 M−1 s−1) is 1000 times faster than that of squaramides, following a second-order rate law. Squaramate esters functionalized with basic NGP groups, such as amines, display a pH-dependent hydrolysis rate due to anchimeric assistance of the terminal amino group, reducing stability to pH 5. However, when the squaramate ester has a terminal nucleophilic group in the γ position of the alkyl chain, it undergoes rapid intramolecular cyclization, forming cyclic squaramides.
Introduction
Alkyl squaramates, the 3-amino, 4-alkoxy derivatives of squaric acid, are directly synthesised from squaramate diesters by condensation with amines.1 This reaction, typically conducted at room temperature, without catalyst or coupling reagents has drawn much attention due to its straightforward protocols and high yields.2Squaric acid esters have emerged as valuable tools for synthesising compounds with diverse applications, including dyes,3–5 catalysts,6,7 sensors,8–12 anion transporters,13–17 and bioactive compounds.18–21 In general, their reactivity with primary or secondary amines and a certain resistance to hydrolysis makes them suitable for preparing amino-saccharide, protein, and nucleic acid conjugates with significant biomedical applications,22 such as neoglycoproteins for cell targeting,23 synthetic vaccines,24 and lectin recognition2,25 among others. Furthermore, squaric acid esters modify proteins through highly chemoselective reactions with the amino acid cysteine.26,27
However, despite the widespread use of squaric esters, understanding their fate and degradation pathways in biological-like environments remains limited. Unlike squaramides, which exhibit kinetic stability for more than 100 days in the pH range of 3–10 at 37 °C, squaramate esters suffer hydrolysis under milder conditions.28 Studies have shown that the hydrolysis of squaramate alkyl esters occurs over days at pH 9, with greater stability observed for longer alkyl chains.29
Additionally, the influence of Neighbouring Group Participation (NGP) on the kinetics of various reactions has been well-documented.30,31 Specifically, the hydrolysis of carboxylic esters is enhanced by the NGP through the formation of a cyclic intermediate.32,33 Similar behaviour is anticipated for squaramate esters. However, an intramolecular cyclisation reaction, driven by a nucleophilic attack from a group of the squaramate substituent, might compete with the hydrolysis, Chart 1.
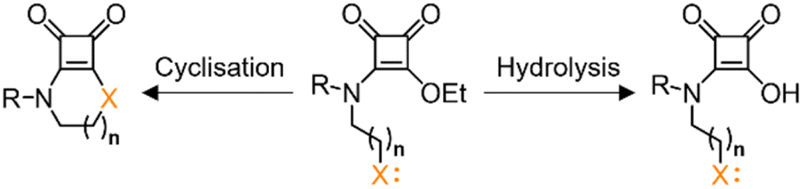 | ||
| Chart 1 Hydrolysis vs. intramolecular cyclisation pathways for squaramate esters. | ||
The distinctive conformational properties of squaric acid derivatives could play a key role in this context. Previous studies have demonstrated that these conformational properties of secondary and tertiary squaramate esters facilitate a rapid intramolecular cyclisation reaction through the nucleophilic attack of a group located at the γ position of the squaramide substituent.34,35
This study investigates the kinetic competition between hydrolysis and intramolecular cyclisation reactions in squaramate esters. We examine the effect of NGP on the hydrolysis rate and the role of different nucleophiles (N, S) in the cyclisation reaction, using a primary amine and a thiol group to mimic the behaviour of the amino acids lysine and cysteine, respectively, in peptide or protein bioconjugation.
First, we investigated the hydrolysis reaction in squaramate ethyl esters. Similar to carboxylic esters, we assumed that the hydrolysis of squaramate esters would proceed under both acid and alkaline catalysis (see Scheme S1†).
Therefore, we synthesized squaramate esters 1a–7a and 8b to assess the hydrolytic stability within the pH range of 3 to 9, Fig. 1. The structure of the studied esters differs from the nature of the N-alkyl chain. Compounds 2a–7a and 8b were functionalized with chemical groups that potentially assist the hydrolytic reaction (amine, carboxyl, alcohol, and pyridine groups). Compound 1a featuring an n-butyl chain, served as a negative control for NGP.
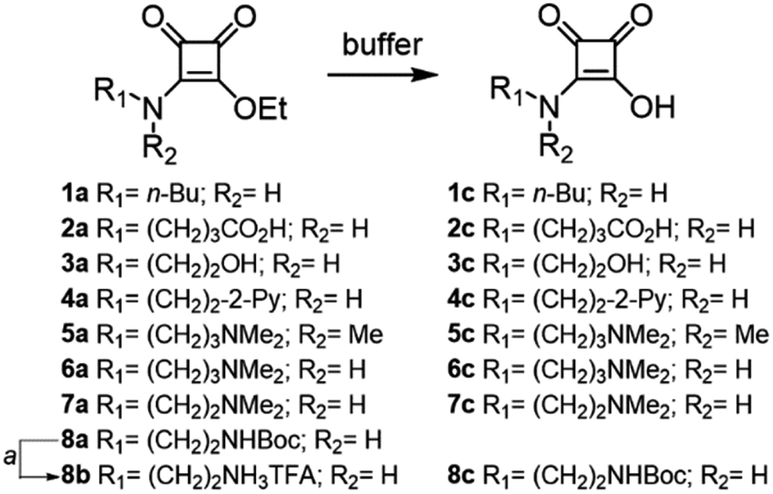 | ||
| Fig. 1 Structure of the squaramate esters 1a–8b and the corresponding squaramic acids 1c–8c obtained from their buffered hydrolysis at 37 °C. aTFA in CH2Cl2. | ||
Squaramate esters 1a–8a were synthesised by condensing diethyl squarate with one equivalent of the corresponding amine in acetonitrile (see ESI†). The amino-protecting group of compound 8a was cleaved with trifluoroacetic acid (TFA) in dichloromethane to yield the corresponding ammonium salt 8b.
The kinetics of the hydrolysis of compounds 1a–7a and 8b to give the corresponding squaramic acids 1c–8c were studied by UV-spectroscopy. In aqueous solution, squaramic esters exhibit a strong absorption UV band (ε > 6 × 104 M−1 cm−1) with a maximum at 272 nm, independent of the pH, while the squaramate acid derivatives display a maximum absorption band at 283 nm. Capitalizing on this, we monitored the time-dependent changes in the UV-vis spectra of esters 1a–7a and 8b at 37 °C to mimic physiological conditions, Fig. 2a. The experiments were conducted under pseudo-first-order conditions, and the apparent rate constants (kobs) were determined through global absorption spectra analysis‡ (see ESI eqn (2′)†).
 | ||
| Fig. 2 (a) Changes observed in the UV spectra of 1a (blue trace) due to hydrolysis (30 μM in 10 mM carbonate buffer, pH 10, 37 °C) as a representative example of the studied squaramate esters. Red trace represents the corresponding hydrolysed squaric acid. (b) Calculated disappearance of 1a (30 μM, 37 °C) from the UV spectrum at different pH (10 mM carbonate buffer). From pH 3 to 8, no hydrolysis was observed after one week. | ||
The calculated kobs values are summarized in Table 1. Based on these data, the squaramate esters studied can be classified into two groups.
| pH | |||||
|---|---|---|---|---|---|
| 3 | 5 | 7 | 8 | 9 | |
| 1a | — | — | — | — | 1.10 ± 0.04 |
| 2a | — | — | — | — | 0.34 ± 0.03 |
| 3a | — | — | — | — | 0.86 ± 0.01 |
| 4a | — | — | — | — | 0.73 ± 0.04 |
| 5a | — | — | 0.02 ± 0.01 | 0.43 ± 0.03 | 3.62 ± 0.02 |
| 6a | — | — | 0.31 ± 0.01 | 1.03 ± 0.01 | 4.85 ± 0.02 |
| 7a | — | — | 0.46 ± 0.01 | 2.38 ± 0.03 | 8.56 ± 0.14 |
| 8b | — | — | 1.01 ± 0.01 | 2.91 ± 0.01 | 16.1 ± 0.2 |
The first group comprises esters 1a–4a, which exhibit kinetic stability under pH 9, and the second group includes esters 5a–7a and 8b, undergoing hydrolysis from pH 7 onwards. Noticeably, at pH 9, the hydrolysis for esters 5a–7a and 8b occurs significantly faster compared to esters 1a–4a, highlighting the role of the basic NGPs on the reaction kinetics.
Ester 1a hydrolysis was further examined under pseudo-first-order conditions (3 × 10−5 M and 37 °C) at the pH range of 8.8–10, as illustrated in Fig. 2b. In contrast to observations in acidic or neutral media, the hydrolysis of 1a occurs within hours under alkaline conditions, yielding acid 1c. The obtained kobs values show a linear dependence within the pH range studied. This linear relationship discards the ionization of the squaramidic NH within this pH interval, as such ionization would lead to a non-linear dependence of the obtained kobs. From the data fitting, we could calculate a second-order rate constant kOH of 0.47 ± 0.02 M−1 s−1 for the hydrolysis reaction.36
The same linear dependency was observed when conducting the aforementioned experiments in a D2O solution of 1a (3 × 10−5 M, pD 9.1–10.2, 37 °C), obtaining a value of kOD of 0.21 ± 0.02 M−1 s−1. The solvent Kinetic Isotope Effect (KIE) calculated as kOH/kOD was found to be 2.24 ± 0.02 falling within the typical range of 1.5–3 that corresponds to solvent KIEs resulting from the transfer of a proton from nitrogen or oxygen atoms (see Fig. S3†).37 Therefore, the obtained KIE value supports the involvement of a water molecule in the rate-determining step (RDS) of the reaction. These results align with the proposed general base-catalysed mechanism for the hydrolysis of carboxylic esters, suggesting that an analogous mechanism operates for the hydrolysis of the squaramate ester 1a (see ESI Scheme S1†).
In analogy to ester 1a and assuming the same reactivity, we conducted the kinetic studies for 2a–4a at the same pH interval as 1a and fitted the data to obtain the corresponding constants k(OH). The fitted values of 0.34 ± 0.03 M−1 s−1 for 2a, 0.86 ± 0.01 M−1 s−1 for 3a, and 0.73 ± 0.04 M−1 s−1 for 4a all fall within the same order of magnitude than the k(OH) obtained for 1a. This indicates that the presence of poor nucleophilic/basic groups does not enhance the reaction rate when compared to 1a. Thus, in this scenario, hydrolysis is likely governed solely by the electrophilic character of the squaryl ester carbon.
Similar to esters 1a–4a, we observed that compounds 5a–7a and 8a withstand hydrolysis in moderate acidic media (pH 3–5, 37 °C) for over three weeks. However, their notably accelerated hydrolysis in neutral or alkaline media (7–10.5) at 37 °C suggests that the amine group on the N-alkyl chain could act as anchimeric assistance (NGP), thereby enhancing the hydrolysis reaction rate. We assumed that the terminal alkyl amine group would participate in an N-protonation equilibrium in water, where only the free amino group would be available to catalyse the reaction. Therefore, the hydrolysis would be sensitive to the pH.
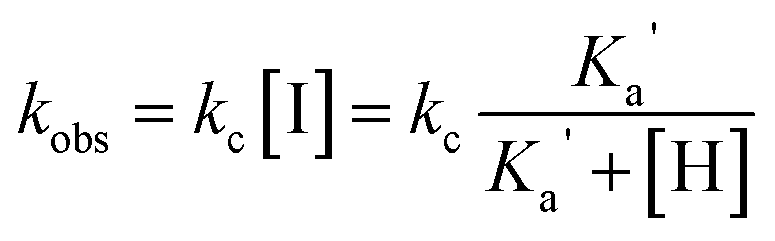 | (1) |
To confirm our hypothesis, we ran the kinetic experiments on ester 5a within the pH range of 7 to 10.5 at 37 °C, Fig. 3. Data fitting provided the corresponding kobs for each pH studied. The rate constant k1 found for the assisted hydrolysis is 5.2 ± 0.9 × 10−5 M−1 s−1, and the rate constant for the direct hydrolysis kOH is 1.03 ± 0.04 M−1 s−1. Despite the calculated k1 value being five orders of magnitude smaller than kOH, it significantly contributes to the hydrolysis rate, particularly with increasing pH, where both proposed mechanisms operate simultaneously.
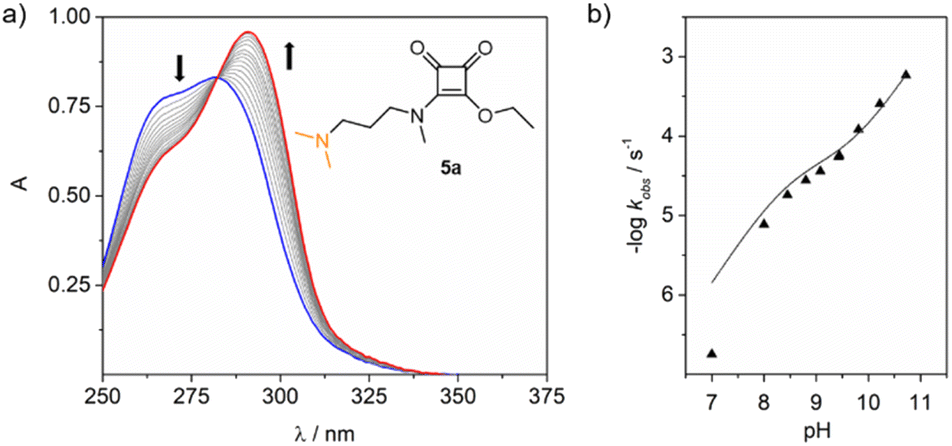 | ||
| Fig. 3 (a) UV changes observed for the hydrolysis of ester 5a (30 μM) at pH 9 (10 mM borate buffer) and 37 °C. (b) Data fitting to obtain k1 and kOH. | ||
Consequently, we hypothesised that the rate law for the reaction would depend on the amine pKa (eqn (1)), and the direct hydrolysis would compete with the NGP process.§
Similarly, we calculated the kinetic constant k1 for squaramate esters 6a–7a and 8b using the kOH value obtained for 5a in the fitting. The calculated k1 values are listed in Table 2 and are within the same order of magnitude as that determined for 5a, indicating the involvement of both primary and secondary amines as an NGP and thereby increasing the overall hydrolysis reaction rate.
| pKaa | kOH, M−1 s−1 | k1, 10−5 M−1 s−1 | |
|---|---|---|---|
| a Obtained by potentiometric titration and fitted with Hyperquad2013 (Protonic Software. http://www.hyperquad.co.uk). To calculate k1 we assumed the same kOH found for 5a.b To calculate k1, we assumed the same kOH as for 5a. | |||
| 5a | 8.82 | 1.03 ± 0.04 | 5.2 ± 0.9 |
| 6a | 8.61 | 1.03b | 12 ± 2 |
| 7a | 7.95 | 1.03b | 3.9 ± 0.3 |
| 8b | 8.50 | 1.03b | 30 ± 6 |
In line with these results, a similar behaviour has been reported for carboxylic ester analogues. Mautner and Bruice studied the hydrolytic stability of the benzoyl choline ester at 25 °C, showing a kinetic profile akin to that found for compound 1a (kOH 0.56 M−1 s−1), discarding the assistance of the tetraalkyl ammonium group to the hydrolysis reaction.38 However, the hydrolytic constants obtained for the 2-(dimethylamino)ethyl benzoate (kOH = 0.06 M−1 s−1 and k1 = 4.8 10−5 M−1 s−1) show faster hydrolysis like in compounds 5a–7a and 8b, due to the intramolecular assistance of the amino-terminal group. Our results suggest that squaramate esters exhibit moderate resistance to hydrolysis compared to their carboxylic analogs. In comparison with carbamates, squaramate esters like 1a hydrolyse more rapidly under neutral and basic conditions. However, under acidic conditions, squaramate esters exhibit high stability, with no observable reaction over several days, while carbamate ester analogs hydrolyze within a few hours. Conversely, squaramate esters undergo hydrolysis around 1000 times faster than squaramides, mirroring the behavior observed in carboxylic acids and their corresponding amides.
After studying the hydrolysis of squaramate esters 1a–8b, we delved into the competition of the intramolecular cyclisation reaction and the hydrolysis reaction to ascertain whether the intramolecular cyclization occurs more rapidly than the hydrolysis. To explore this further, we analysed the kinetic behaviour of squaramate esters 9a–10b, whose structure could potentially yield cyclic squaramide compounds, Fig. 4a.34 Esters 9b and 10b were synthesised using a methodology similar to that for 8b (see ESI†). The cyclisation reaction is expected to be pH sensitive due to the effect of the acid–base equilibrium on the nucleophile. Accordingly, we observed an acceleration of the reaction when the pH increases.35 Notably, at 37 °C the conversion of 9b into 12a occurs rapidly, even under moderate acidic (pH 5, kobs = 0.65 ± 0.01 10−5 s−1) and neutral conditions (pH 7, kobs = 3.85 ± 0.05 10−5 s−1). At pH > 8 the reaction proceeds too rapidly to be accurately measured, Fig. 4b. Furthermore, no hydrolytic degradation was observed for the cyclic products 12a and 12b.
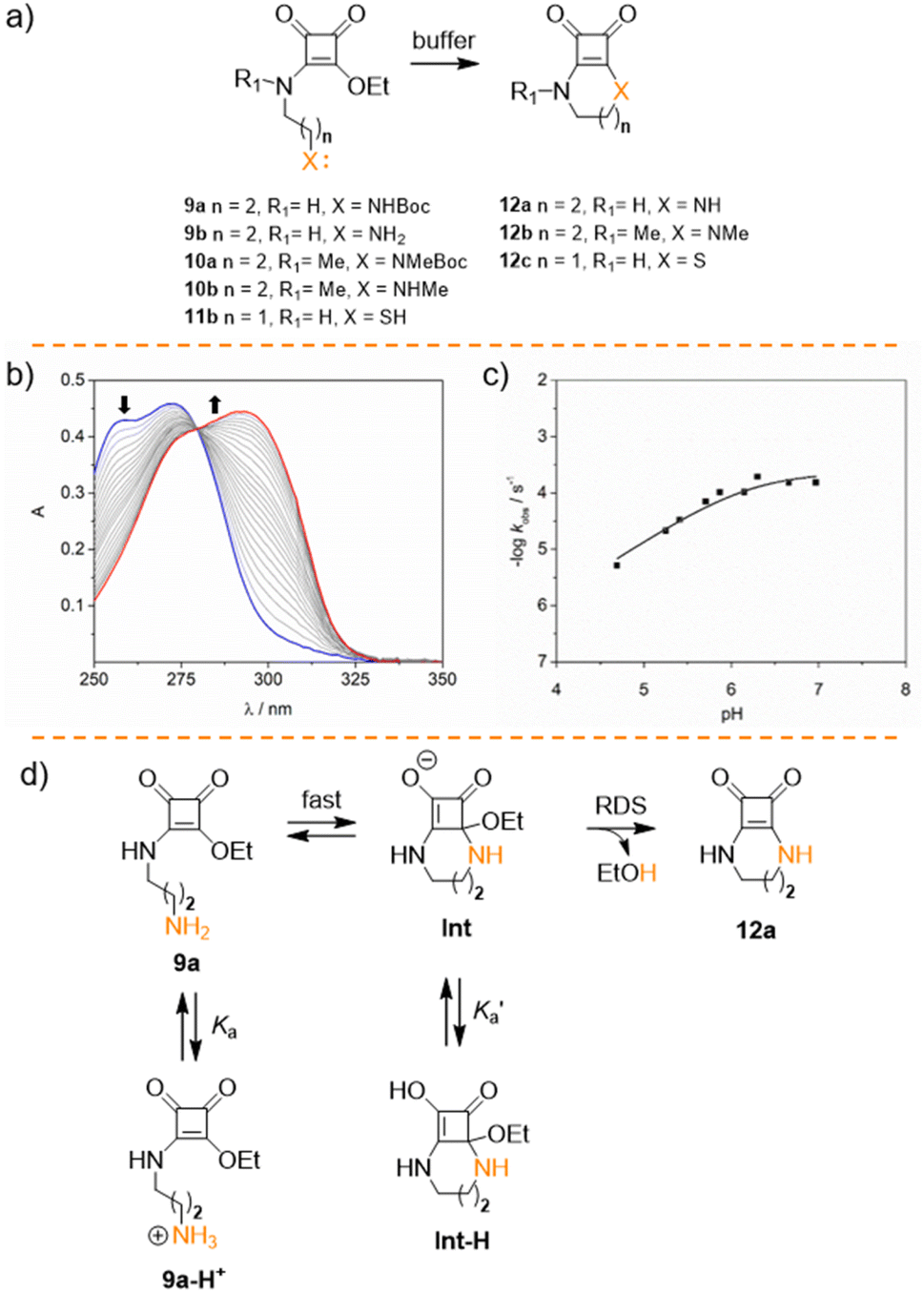 | ||
| Fig. 4 (a) Structure of the squaramate esters 9a–10c and cyclic compounds 12a–12c. (b) Representative example of the UV changes observed for the cyclisation of 9a (30 μM at 10 mM cacodylate buffer pH 5.7 at 37 °C). (c) Plot of the kobs obtained at pH range 4.5–7. The curve shows the fitting of the experimental data. (d) Proposed intramolecular addition–elimination mechanism for ester 9a to give cyclic squaramide 11a under mildly acidic conditions. | ||
Promoting the squaramate ester E,Z conformation significantly affects the cyclization reaction rate. Consequently, the cyclisation reaction accelerates for the N-methylated analogue 10b, becoming too rapid to be accurately measured even at pH 7 by the UV experiments.
To complement the kinetics data for the cyclisation reaction, we studied 9b (30 μM) cyclisation in the pH range of 4.5–7 at 37 °C. As anticipated, plotting the observed rate constants against pH resulted in an asymptotic profile, indicating a strong pH dependence due to the nucleophile sensitivity to pH, Fig. 4c.
The rapid cyclisation of compound 9b hindered the determination of the amine group's pKa. Homologous compounds of 9b featuring the amino group on shorter alkyl chains have a pKa around 8.5, as evaluated by potentiometric titration and fitted with Hyperquad (see Fig. S1†). However, within the pH range studied, the amine in compound 9b should be fully protonated, thus behaving as a poor nucleophile and potentially slowing down the formation of the cyclic compound 12a. To address this, we fitted the kinetic data to obtain the apparent cyclisation kinetic constant kc and the Ka values using eqn (1). Good fitting was obtained for both constants with Ka = 6.3 × 10−7 (corresponding to a pKa of 6.2) and kc = 2.3 × 10−4 s−1. The calculated pKa is surprisingly low for an alkylamine. Therefore, we assumed that the obtained Ka represents an apparent acidic constant 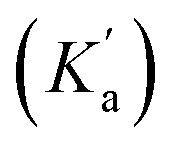 associated with the equilibrium between other species, that determines the reaction rate-determining step (RDS) rather than the nucleophilic amine attack step. Consequently, we propose that the elimination of the leaving group from any tautomeric species with a pKa of 6.2 becomes the limiting step for this reaction, Fig. 4d. The intermediate species resulting from the rapid intramolecular nucleophilic addition reaction (Int) undergoes a prototropic equilibrium and eliminates an ethanol molecule to facilitate the gain of aromaticity of the tetra-membered ring, to give the cyclic compound. This could be the driving force behind the acidity enhancement of the protonated species up to a pKa of 6.
associated with the equilibrium between other species, that determines the reaction rate-determining step (RDS) rather than the nucleophilic amine attack step. Consequently, we propose that the elimination of the leaving group from any tautomeric species with a pKa of 6.2 becomes the limiting step for this reaction, Fig. 4d. The intermediate species resulting from the rapid intramolecular nucleophilic addition reaction (Int) undergoes a prototropic equilibrium and eliminates an ethanol molecule to facilitate the gain of aromaticity of the tetra-membered ring, to give the cyclic compound. This could be the driving force behind the acidity enhancement of the protonated species up to a pKa of 6.
To further investigate the effect of the nucleophile nature in the cyclisation reaction kinetics, we synthesised squaramate ester 11b, incorporating a 2-amino ethane thiol as a substituent, Fig. 4a. We hypothesized that the larger size of the sulfur atom would compensate for the shorter length of the alkyl chain and facilitate the formation of the cyclic compound 12c.
Compound 11b was synthetized first by reacting cystamine dihydrochloride with two equivalents of diethyl squarate in acetonitrile in the presence of DiPEA to yield the bis escuaramate ester 11a. Then, before starting the kinetic studies, ester 11b was prepared in situ by reducing 11a with tris(2-carboxyethyl)phosphine (TCEP) (see ESI†).
As anticipated, the UV studies performed with 11b in acidic and neutral buffered solutions (pH 5–7) showed a band at 309 nm corresponding to the tethered cyclothiosquaramide 12c. However, in mild alkaline solutions (pH 8), the 309 nm band diminished over time, and a new band appeared at 332 nm, suggesting the formation of a different product. To elucidate the structure of this unexpected compound, we synthesized the cyclothiosquaramide 12c by reaction of diethyl squarate with cysteamine in a water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol mixture (v/v, 2:3). After isolating and characterizing 12c, we let to stand in water at pH 8 (NaOH 1 N) at 50 °C for 5 h to complete its conversion. This yielded a pale yellow solid compound, which was characterised by mass spectrometry and 1D-and 2D-NMR experiments, allowing us to propose the structure of the α-ketocarboxylate 13 shown in Fig. 5a. The transformation of the cyclic compound 12c into the α-ketocarboxylate 13 in alkaline media occurs due to the ring-opening of the cyclobutenedione, provoked by the nucleophilic attack of the hydroxide anion on the C4 carbonyl. The regioselectivity of the ring-opening reaction to yield 13 was confirmed by 2D NMR experiments (see ESI Fig. S12–S17†).
:ethanol mixture (v/v, 2:3). After isolating and characterizing 12c, we let to stand in water at pH 8 (NaOH 1 N) at 50 °C for 5 h to complete its conversion. This yielded a pale yellow solid compound, which was characterised by mass spectrometry and 1D-and 2D-NMR experiments, allowing us to propose the structure of the α-ketocarboxylate 13 shown in Fig. 5a. The transformation of the cyclic compound 12c into the α-ketocarboxylate 13 in alkaline media occurs due to the ring-opening of the cyclobutenedione, provoked by the nucleophilic attack of the hydroxide anion on the C4 carbonyl. The regioselectivity of the ring-opening reaction to yield 13 was confirmed by 2D NMR experiments (see ESI Fig. S12–S17†).
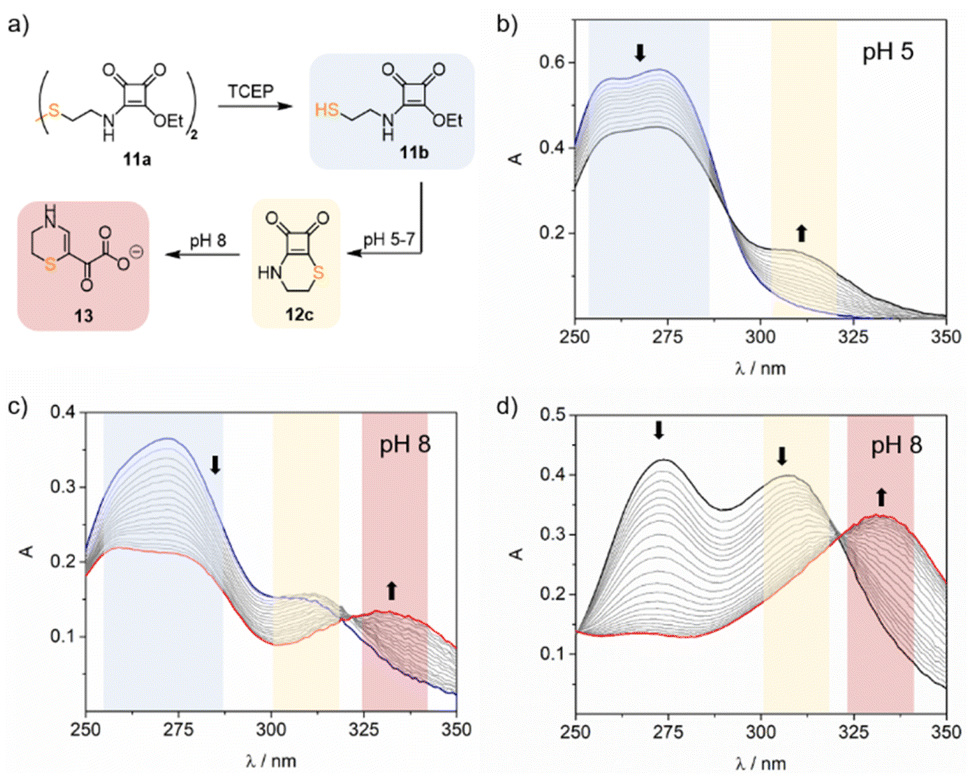 | ||
| Fig. 5 (a) Synthetic scheme for synthesizing squaramate ester 11a, cyclothiosquaramide 11c, and the α-keto carboxylate 13. UV changes observed for the hydrolysis of 11b after in situ reduction of 11a at (b) 30 μM, 10 mM acetate buffer, pH 5, 37 °C, and (c) 30 μM, 10 mM PBS buffer, pH 8, 37 °C. (d) UV changes of the degradation of the isolated 12c at 30 μM, 10 mM PBS buffer, pH 8, 37 °C. | ||
A similar reaction was described by Horri and collaborators for 3,4-diphenylcyclobut-3-ene-1,2-diones, resulting in the formation of the corresponding α-ketoacid.39
We used DFT calculations to elucidate why the opening of the four-membered cyclobutadiene-dione ring occurs in compound 12c but not in compounds 12a and 12b at pH ≥ 8. In our computational study, we also included compound 12d to investigate whether the observed experimental differences are attributed to the fused ring size or the presence of the sulfur atom, Fig. 6a.
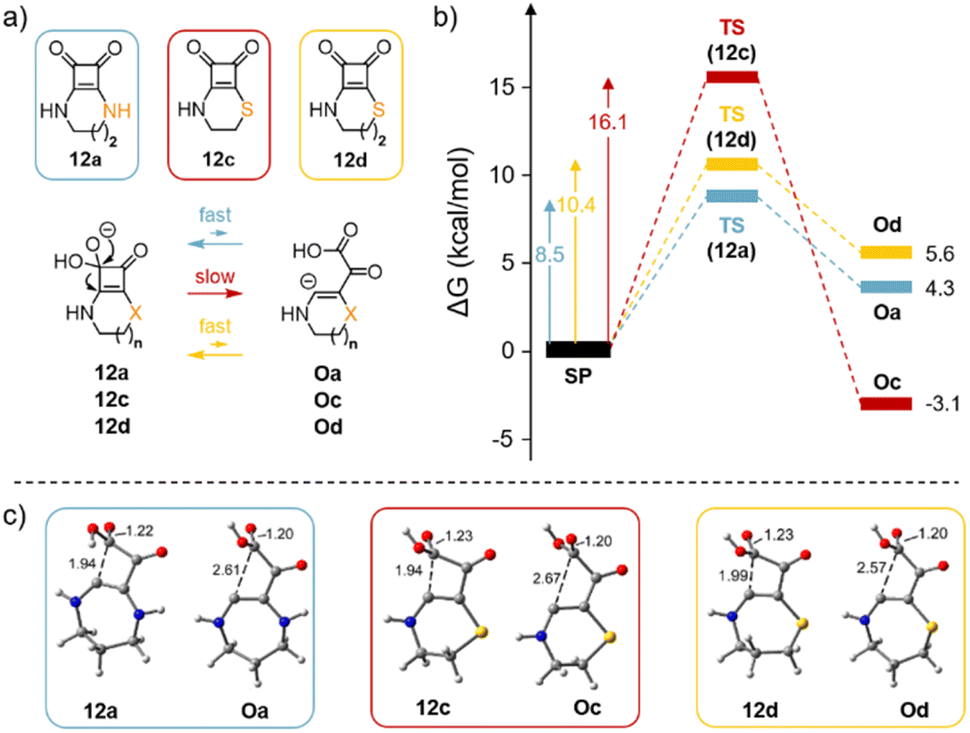 | ||
| Fig. 6 (a) Cyclobutenediones 12a, 12c and 12d, the reactions studied (1) to (3) and (b) their energetic profiles. (c) The optimised geometries of the transition states and final products are also represented. Distances in Å. | ||
Remarkably, the energetic profile obtained for the ring opening of compound 12c upon the addition of the hydroxide anion to the carbonyl differs significantly from that of 12a or 12d. That is, for 12c, the ring opening step has a larger barrier (16.1 kcal mol−1, red profile) than either 12a (8.5 kcal mol−1, blue profile) or 12d (10.4 kcal mol−1, yellow profile) Fig. 6b. Additionally, the reaction is exergonic for compound 12c. Interestingly, for compound 12a, although the barrier is lower, the resulting opened intermediate is energetically higher (4.3 kcal mol−1) than the starting product (SP) (blue profile in Fig. 6), thus indicating that any potential fast equilibrium (low barrier) is predominantly shifted towards the starting material. Furthermore, compound 12d (yellow profile in Fig. 6b) behaves similarly to compound 12a, suggesting that the ring opening in compound 12c is likely attributed to the size of the fused ring rather than the presence of the sulphur atom. The geometries of the final products resulting from the opening of the four-membered ring are also represented in Fig. 6c (Oa, Oc and Od). Notably, the carbanion is stabilised by a C(−)⋯COOH π-hole interaction.40 The results align closely with the experimental results as they indicate that only in the case of compound 12c, the reaction is both exergonic and irreversible.
As observed, the evolution of the squaramate ester 11b in alkaline media follows a two-step reaction. Firstly, an intramolecular cyclisation yields squaramide thioester 12c, governed by the cyclisation reaction constant kc. Secondly, the degradation of 12c occurs with a pH-dependent rate constant kOH. To investigate this, compound 11b was studied in the pH range of 3 to 9 at 37 °C, monitoring the spectral UV changes registered upon time to define kc and kOH.
The formation of the α-ketocarboxylate 13 was not detected at pH 3 and 5. However, in neutral and alkaline media, the reaction rate increased from pH 7 (kobs 0.74 ± 0.05 × 10−5 s−1) to pH 9 (kobs 33.4 ± 0.7 × 10−5 s−1) as expected. The obtained kobs values exhibited a linear dependence on the hydroxide ion concentration. Using this correlation, we determined the corresponding pseudo-first-order rate constant for the reaction as kOH = 33.5 ± 0.7 M−1 s−1 for the reaction (see ESI eqn (2′)†).
In addition, we calculated the apparent cyclisation rate constant kc for each studied pH, incorporating the observed kOH, to fit the experimental data. Results indicate that the cyclisation rate increases with the pH, ranging from 0.72 ± 0.02 × 10−5 s−1 at pH 3 to 16.1 ± 0.3 × 10−5 s−1 at pH 8. However, the lower value of the kc (1.23 ± 0.02 × 10−5 s−1) at pH 9 suggests that the cyclisation slows down at this pH compared with pH 8, consistent with significant competition from squaramate hydrolysis. By calculating the apparent rate constants kc we were able to simulate the distribution and evolution of the three species: the squaramide thioester 11b, the cyclic compound 12c, and the α-ketocarboxylate 13 over time. For example, at pH 8, ester 11b rapidly transforms to form the cyclic intermediate 12c reaching the maximum concentration after 3 h, corresponding to a molar fraction of 0.6. Within approximately 20 hours, the cyclic compound is fully converted into α-ketocarboxylate 13.
Conclusions
In summary, the squaramate esters investigated in this study exhibit a strong relationship between the alkyl chain nature and their inherent stability in water at 37 °C. Squaramate esters 1a–4a laking o featuring weak basic NGP groups substitutions remain stable up to pH 9, beyond which hydrolysis becomes significant. The hydrolytic degradation of these compounds (kOH ≈ 10−1 M−1 s−1) is 1000-fold faster than the corresponding rate for squaramides, and follows a conventional second-order rate law, with a linear dependency on the hydroxide species concentration. Conversely, squaramate esters 5a–7a and 8b, functionalised with basic NGP groups such as amines, showed a pH-dependent hydrolysis rate, influenced by the anchimeric assistance of the terminal amino group on the alkyl chain. This mechanism coexists with direct hydroxide hydrolysis reducing their hydrolytic stability to pH 5. However, in squaramate esters 9b–11b having a terminal nucleophilic group on a sufficiently long alkyl chain, the undergo hydrolysis is overridden by a rapid intramolecular cyclization reaction, resulting in the formation of the corresponding cyclic squaramides 12a–12b and squaramide-thioester 12c. Hence, the structure of squaramate and the nature of nearby groups significantly influence their stability in water, thereby affecting their reactivity towards nucleophiles.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
Marta Ximenis (conceptualization, investigation); Santiago Canellas (investigation); Rosa M. Gomila (computational studies), Bartomeu Galmés (investigation), Antonio Frontera (supervision), Antonio Costa (supervision), Carmen Rotger (supervision, writing and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the MICIU/AEI (project PID2020-115637GB-I00 FEDER funds) for financial support. M. X. acknowledges the support from Govern de les Illes Balears; FSE funds for a predoctoral fellowship, and B. G. acknowledges the support from Ministerio de Universidades for a FPU19/05598 fellowship.Notes and references
- L. F. Tietze, M. Arlt, M. Beller, K. -H. Gl üsenkamp, E. Jähde and M. F. Rajewsky, Anticancer agents, 15. Squaric acid diethyl ester: a new coupling reagent for the formation of drug biopolymer conjugates. Synthesis of squaric acid ester amides and diamides, Chem. Ber., 1991, 124, 1215–1221 CrossRef.
- L. F. Tietze, C. Schroter, S. Gabius, U. Brinck, A. Goerlach-Grawj and H.-J. Gabius, Conjugation of p-Aminophenyl glycosides with squaric acid diester to a carrierprotein and use of neoglycoprotein in the histochemical detection of lectins, Bioconjugate Chem., 1991, 2, 148–153 CrossRef PubMed.
- M. Ávila-Costa, C. L. Donnici, J. D. dos Santos, R. Diniz, A. Barros-Barbosa, A. Cuin and L. F. C. de Oliveira, Synthesis, vibrational spectroscopy and X-ray structural characterization of novel NIR emitter squaramides, Spectrochim. Acta, Part A, 2019, 223, 117354–117366 CrossRef PubMed.
- T. S. Jarvis, C. G. Collins, J. M. Dempsey, A. G. Oliver and B. D. Smith, Synthesis and structure of 3,3-dimethylindoline squaraine rotaxanes, J. Org. Chem., 2017, 82, 5819–5825 CrossRef PubMed.
- Y. Chen, Y. Li, X. Gao and M. Cui, Squaraine dye based prostate-specific membrane antigen probes for near-infrared fluorescence imaging of prostate cancer, Dyes Pigm., 2022, 208, 110822–110830 CrossRef.
- J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Squaramides: bridging from molecular recognition to bifunctional organocatalysis, Chem.–Eur. J., 2011, 17, 6890–6899 CrossRef PubMed.
- F. R. Wurm and H. A. Klok, Be squared: expanding the horizon of squaric acid-mediated conjugations, Chem. Soc. Rev., 2013, 42, 8220–8236 RSC.
- M. N. Piña, B. Soberats, C. Rotger, P. Ballester, P. M. Deyà and A. Costa, Selective sensing of competitive anions by non-selective hosts: The case of sulfate and phosphate in water, New J. Chem., 2008, 32, 1919–1923 RSC.
- C. López, E. Sanna, L. Carreras, M. Vega, C. Rotger and A. Costa, Molecular recognition of zwitterions: enhanced binding and selective recognition of miltefosine by a squaramide-based host, Chem.–Asian J., 2013, 8, 84–87 CrossRef PubMed.
- R. Prohens, G. Martorell, P. Ballester and A. Costa, A squaramide fluorescent ensemble for monitoring sulfate in water, Chem. Commun., 2001, 1456–1457 RSC.
- D. Jagleniec, Ł. Dobrzycki, M. Karbarz and J. Romański, Ion-pair induced supramolecular assembly formation for selective extraction and sensing of potassium sulfate, Chem. Sci., 2019, 10, 9542–9547 RSC.
- J. Zhou, H. Lin, X. F. Cheng, J. Shu, J. H. He, H. Li, Q. F. Xu, N. J. Li, D. Y. Chen and J. M. Lu, Ultrasensitive and robust organic gas sensors through dual hydrogen bonding, Mater. Horiz., 2019, 6, 554–562 RSC.
- L. A. Marchetti, T. Krämer and R. B. P. Elmes, Amidosquaramides - a new anion binding motif with pH sensitive anion transport properties, Org. Biomol. Chem., 2022, 20, 7056–7066 RSC.
- G. Picci, M. Kubicki, A. Garau, V. Lippolis, R. Mocci, A. Porcheddu, R. Quesada, P. C. Ricci, M. A. Scorciapino and C. Caltagirone, Simple squaramide receptors for highly efficient anion binding in aqueous media and transmembrane transport, Chem. Commun., 2020, 56, 11066–11069 RSC.
- I. Marques, P. M. R. Costa, M. Q. Miranda, N. Busschaert, E. N. W. Howe, H. J. Clarke, C. J. E. Haynes, I. L. Kirby, A. M. Rodilla, R. Pérez-Tomás, P. A. Gale and V. Félix, Full elucidation of the transmembrane anion transport mechanism of squaramides using: In silico investigations, Phys. Chem. Chem. Phys., 2018, 20, 20796–20811 RSC.
- L. Q. Deng, Y. M. Lu, C. Q. Zhou, J. X. Chen, B. Wang and W. H. Chen, Synthesis and potent ionophoric activity of a squaramide-linked bis(choloyl) conjugate, Bioorg. Med. Chem. Lett., 2014, 24, 2859–2862 CrossRef PubMed.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Squaramides as potent transmembrane anion transporters, Angew Chem. Int. Ed. Engl., 2012, 51, 4426–4430 CrossRef.
- P. Villalonga, S. Fernández de Mattos, G. Ramis, A. Obrador-Hevia, A. Sampedro and C. R. A. Costa, Cyclosquaramides as kinase inhibitors with anticancer activity, ChemMedChem, 2012, 7, 1472–1480 CrossRef PubMed.
- F. Olmo, C. Rotger, I. Ramírez-Macías, L. Martínez, C. Marín, L. Carreras, K. Urbanová, M. Vega, G. Chaves-Lemaur, A. Sampedro, M. J. Rosales, M. Sánchez-Moreno and A. Costa, Synthesis and biological evaluation of N,N'-squaramides with high in vivo efficacy and low toxicity: toward a low-cost drug against Chagas disease, J. Med. Chem., 2014, 57, 987–999 CrossRef CAS PubMed.
- A. Sampedro, R. Villalonga-Planells, M. Vega, G. Ramis, S. Fernández de Mattos, P. Villalonga, A. Costa and C. Rotger, Cell Uptake and Localization Studies of Squaramide Based Fluorescent Probes, Bioconjugate Chem., 2014, 25, 1537–1546 CrossRef CAS.
- V. Molodtsov, P. R. Fleming, C. J. Eyermann, A. D. Ferguson, M. A. Foulk, D. C. McKinney, C. E. Masse, E. T. Buurman and K. S. Murakami, X-ray Crystal Structures of Escherichia coli RNA Polymerase with Switch Region Binding Inhibitors Enable Rational Design of Squaramides with an Improved Fraction Unbound to Human Plasma Protein, J. Med. Chem., 2015, 58, 3156–3171 CrossRef.
- L. A. Marchetti, L. K. Kumawat, N. Mao, J. C. Stephens and R. B. P. Elmes, The versatility of squaramides: from supramolecular chemistry to chemical biology, Chem, 2019, 5, 1398–1485 Search PubMed.
- S. Böcker, D. Laaf and L. Elling, Galectin binding to neo-glycoproteins: LacDiNAc conjugated BSA as ligand for human galectin-3, Biomolecules, 2015, 5, 1671–1696 CrossRef PubMed.
- H. Kunz, S. Dziadek, S. Wittrock and T. Becker, Synthetic glycopeptides for the construction of anticancer vaccines, in Carbohydrate-based Vaccines, ed. R. Roy, ACS Symposium Series, 2008, vol. 989, pp. 293–310 Search PubMed.
- A. Bergh, B. G. Magnusson, J. Ohlsson, U. Wellmar and U. J. Nilsson, Didecyl squarate - A practical amino-reactive cross-linking reagent for neoglycoconjugate synthesis, Glycoconjugate J., 2002, 18, 615–621 CrossRef PubMed.
- P. Sejwal, Y. Han, A. Shah and Y. Y. Luk, Water-driven chemoselective reaction of squarate derivatives with amino acids and peptides, Org. Lett., 2007, 9, 4897–4900 CrossRef PubMed.
- D. Cui, D. Prashar, P. Sejwal and Y.-Y. Luk, Water-driven ligations using cyclic amino squarates: a class of useful SN1-like reactions, Chem. Commun., 2011, 47, 1348–1350 RSC.
- M. Ximenis, E. Bustelo, A. G. Algarra, M. Vega, C. Rotger, M. G. Basallote and A. Costa, Kinetic analysis and mechanism of the hydrolytic degradation of squaramides and squaramic acids, J. Org. Chem., 2017, 82, 2160–2170 CrossRef CAS PubMed.
- A. Chernyak, A. Karavanov, Y. Ogawa and P. Kováč, Conjugating oligosaccharides to proteins by squaric acid diester chemistry: Rapid monitoring of the progress of conjugation, and recovery of the unused ligand, Carbohydr. Res., 2001, 330, 479–486 CrossRef CAS PubMed.
- Y. Shalitin and S. A. Bernhard, Neighboring group effects on ester hydrolysis. I. Neighboring hydroxyl groups, J. Am. Chem. Soc., 1964, 86, 2291–2292 CrossRef CAS.
- Y. Shaltin and S. A. Berhard, Neighboring group effects on ester hydrolysis. II. Neighboring carbonyl groups, J. Am. Chem. Soc., 1964, 86, 2292 CrossRef.
- J. R. Yerabolu, C. L. Liotta and R. Krishnamurthy, Anchimeric-assisted spontaneous hydrolysisof cyanohydrins under ambient conditions: implicationsfor cyanide-initiated selective transformations, Chem.–Eur. J., 2017, 23, 8756–8765 CrossRef.
- J. L. García Ruano, A. M. Martín Castro and J. H. Rodriguez Ramos, Anchimeric assistance of the sulfinyl group in the hydrolysis of cyano groups: a new mild method for the reduction of sulfoxides, Tetrahedron Lett., 1996, 37, 4569–4572 CrossRef.
- M. Ximenis, J. Piterch-Jarque, S. Blasco, C. Rotger, E. García-España and A. Costa, Water-soluble squaramide dihydrates: N-methylation modulates the occurrence of one- and two-dimensional water clusters through hydrogen bonding and dipolar interactions, Cryst. Growth Des., 2018, 18, 4420–4427 CrossRef.
- M. Ximenis, A. Sampedro, L. Martínez-Crespo, G. Ramis, F. Orvay, A. Costa and C. Rotger, Introducing a squaramide-based self-immolative spacer for controlled drug release, Chem. Commun., 2021, 57, 2736–2739 RSC.
- K. Covington, M. I. A. Ferra and R. A. Robinson, Ionic product and enthalpy of ionization of water from electromotive force measurements, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1721–1730 RSC.
- P. F. Fitzpatrick, Combining solvent isotope effects with substrate isotope effects in mechanistic studies of alcohol and amine oxidation by enzymes, Biochim. Biophys. Acta, 2015, 1854, 1746–1755 CrossRef PubMed.
- P. Yurkanis Bruice and H. G. Mautner, Intramolecular catalysis and the involvement of tetrahedral intermediate partitioning in the hydrolysis of benzoylcholine, benzoylthionocholine, and their dimethylamino analogs, J. Am. Chem. Soc., 1973, 95, 1582–1585 CrossRef.
- A. Al-Najjar, K. Bowden and M. V. Horri, Reactions of carbonyl compounds in basic solutions. Part 25.1 The mechanism of the base-catalysed ring fission of 3,4-diphenylcyclobut-3-ene-1,2-diones, J. Chem. Soc., Perkin Trans., 1997, 2, 993–996 RSC.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, σ-Holes, π-holes and electrostatically-driven interactions, J. Mol. Model., 2012, 18, 541–548 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General information, experimental details, characterization data for products, NMR spectra of products, and cartesian coordinates of computed structures. See DOI: https://doi.org/10.1039/d4ra04362f |
| ‡ Data vas fitted using ReactLabTM Kinetics (Jplus Consulting Ltd) software package. |
| § Amine pKa values for compounds 5a–7a and 8b were obtained by potentiometric titration and data fitting with the Hyperquad2013 program. |
| This journal is © The Royal Society of Chemistry 2024 |