
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfonamide based pyrimidine derivatives combating Plasmodium parasite by inhibiting falcipains-2 and falcipains-3 as antimalarial agents†
Abdur Rahman‡
a,
Shazia Anjum‡a,
Jaimin D. Bhattb,
Bharat C. Dixitb,
Anju Singha,
Sabiha khana,
Sadaf Fatimaa,
Tarosh S. Patel *b and
Nasimul Hoda*a
*b and
Nasimul Hoda*a
aDrug Design and Synthesis Lab., Department of Chemistry, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India. E-mail: nhoda@jmi.ac.in; Fax: +0091-11-26985507; Tel: +0091-9910200655
bChemistry Department, V. P. & R. P. T. P Science College, Affiliated to Sardar Patel University, Vallabh Vidyanagar 388120, Gujarat, India. E-mail: tarosh_patel@yahoo.com; Tel: +91-2692-230011#31
First published on 7th August 2024
Abstract
In this report, we present the design and synthesis of a novel series of pyrimidine-tethered spirochromane-based sulfonamide derivatives aimed at combating drug resistance in malaria. The antimalarial effectiveness of these compounds was assessed in vitro. Structural validation of the synthesized compounds was conducted using mass spectrometry and NMR spectroscopy. Strong antimalarial activity against CQ-sensitive (3D7) and CQ-resistant (W2) strains of Plasmodium falciparum was demonstrated by the majority of the compounds. Notably, compounds SZ14 and SZ9 demonstrated particularly potent effects, with compound SZ14 showing IC50 values of 2.84 μM and SZ9 3.22 μM, indicating single-digit micromolar activity. The compounds exhibiting strong antimalarial activity were assessed through enzymatic tests against the cysteine protease enzymes of P. falciparum, falcipain-2 and falcipain-3. The results indicated that SZ14 and SZ9 inhibited PfFP-2 (IC50 values: 4.1 and 5.4 μM, respectively), and PfFP-3 (IC50 values: 4.9 and 6.3 μM, respectively). To confirm the compounds' specificity towards the parasite, we investigated their cytotoxicity against Vero cell lines, revealing strong selectivity indices and no significant cytotoxic effects. Additionally, in vitro hemolysis testing showed these compounds to be non-toxic to normal human blood cells. Moreover, predicted in silico ADME parameters and physiochemical characteristics demonstrated the drug-likeness of the synthetic compounds. These collective findings suggest that sulfonamide derivatives based on pyrimidine-tethered oxospirochromane could serve as templates for the future development of potential antimalarial drugs.
1. Introduction
Malaria is a severe deadly infective disease that causes illness and deaths across many tropical and subtropical countries.1,2 Plasmodium parasites are transmitted to humans by the bites of infected female Anopheles mosquitoes.3 The World Health Organization (WHO) estimated 249 million cases and 608![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths from malaria in 2022 with an increase of 5 million new cases and a slight decline in malarial deaths from 2021, indicating that malaria is still difficult to control.4 Artemisinin-based combination therapies (ACTs) are an increasingly common treatment for preventing malaria. However, the effectiveness of ACTs is reduced gradually on the grounds of the emergence of resistance against partner medications, especially in Southeast Asia5,6 and presently in Sub-Saharan Africa.7 Consequently, these situations have prompted medicinal chemists to design and develop novel inhibitors with new modes of action targeting several crucial enzymes that exhibit a vital role in the parasitic life cycle to tackle the emergence of drug resistance to some extent.
000 deaths from malaria in 2022 with an increase of 5 million new cases and a slight decline in malarial deaths from 2021, indicating that malaria is still difficult to control.4 Artemisinin-based combination therapies (ACTs) are an increasingly common treatment for preventing malaria. However, the effectiveness of ACTs is reduced gradually on the grounds of the emergence of resistance against partner medications, especially in Southeast Asia5,6 and presently in Sub-Saharan Africa.7 Consequently, these situations have prompted medicinal chemists to design and develop novel inhibitors with new modes of action targeting several crucial enzymes that exhibit a vital role in the parasitic life cycle to tackle the emergence of drug resistance to some extent.
Cysteine protease is one of the most promising chemotherapeutic targets for various infectious diseases caused by parasites which include Leishmania, Entamoeba histolytica, Toxoplasma gondii, Trypanosoma brucei, Cryptosporidium and T. cruzi.8–11 Cysteine protease enzymes in the Plasmodium genus play an important role in parasite organisms to catalyze the host hemoglobin deterioration in the acidic food vacuole, thereby offering essential amino acids for the survivability and development of malaria parasites.12,13 Four subtypes of falcipains in plasmodium are identified: falcipains-1, falcipains-2, falcipains-2′, and falcipains-3. Gene-knockout investigations and the FPs expression by erythrocytic parasites have shown that FP-2 and FP-3 found a 68% homology, which is likely important in regulating the parasite's erythrocytic stage.14,15 However, FP-1 develops during the parasite's schizont/merozoite stage and does not seem necessary for the malaria parasite during its erythrocytic stage.15 The study by the Sijwali group demonstrated that disruption in the FP-2 gene has a potential impact on the degradation of hemoglobin. However, the disruption of the FP-2′ has no impact on the degradation of hemoglobin, and the FP-3 was established to be crucial for the development of the erythrocytic parasite.16 Therefore, efforts to synthesize drugs targeting FP-2 were made because it seems to serve as the primary hemoglobinase, breaking down hemoglobin at several sites and contributing to the degradation of the proteins that make up the membrane of an erythrocyte skeletal system. For FP-2 and FP-3, several peptides and non-peptide inhibitors, including leupeptin and E64, have been identified; however, neither of these individuals has reached clinical trials because of poor bioavailability and toxicological profile.14,17–21 Therefore, inhibition against cysteine protease particularly for falcipains could lead to the arrest of parasitic growth, highlighting an attractive target for antimalarial drug development.22–24
Thus, two distinct biologically active pharmacophores were hybridized to develop single and potent hybrid antimalarial agents. This developed hybrid molecule could have the potential of low risk of drug resistance, enhanced solubility, and lower toxicity than the parent pharmacophore.25,26 In addition, the two partner medications may interact more effectively with the target protein and have greater activity when an appropriate linker links their active moieties together than when they are administered individually.27 The pharmacokinetic characteristics of a hybrid medication are therefore simpler to forecast and control when compared to two separate medications.28 Therefore, expanding our fascinating results with molecular hybrids for developing new malarial medications, herein we introduced a rational design of a hybrid set of pyrimidine-tethered spirocyclic chromane as antimalarial agents according to various pharmacophore combination approaches.
1.1. Design strategy
Pyrimidines, a heterocyclic nitrogen-containing scaffolds that are commonly found in several natural bioactive products and have diverse biological activities such as antitumor, antitubercular, anti-allergic, antipyretic, antibacterial, anti-inflammatory, and antimalarial activities.29,30 Pyrimethamine, a derivative of pyrimidine, was used to treat malaria, targeting the enzyme DHFR.31 Kareem et al.32 have explored the optimization of pyrimidine-based compounds that demonstrated in vitro anti-plasmodial activity and act as dual inhibitors of plasmodial PfGSK3/PfPK6. Similarly, Coteron et al.33 have developed 5-substituted-2-cyanopyrimidine-based derivatives (Fig. 1) as cysteine protease inhibitors particularly for falcipains (FP-2 and FP-3) of the Plasmodium parasite, showing significant activity. Recent studies34–36 established that pyrimidine is an attractive bioactive scaffold in the development of new antimalarial agents. | ||
| Fig. 1 Rationale for the design of sulfonamide derivatives based on pyrimidine-tethered spirocyclic chromane. | ||
The incorporation of spirocyclic scaffolds has been found in recently approved drugs which has led to the escape from flatland to provide less planar molecule with improved potency.37 In addition, the spirocyclic compound with a well-defined three-dimensional arrangement has a greater possibility of being the drug candidate appearing in clinical trials.38 A recent investigation by Brindisi et al.39 presented that spirocyclic compounds possessed improved antimalarial activity. Spirocyclic indolone NITD609 developed by Turner et al. has demonstrated remarkable antimalarial activity, inhibiting PfATP4, for which phase II clinical trials have been carried out on adults with uncomplicated malaria led by P. falciparum in sub-Saharan Africa.40 Similarly, spirocyclic chromane-based analogs (Fig. 1) have emerged as new pharmacophores with potent antimalarial activity.41
Sulfonamide-based derivatives exhibited diverse biological activities in medicinal chemistry which include fungicidal, nematicidal, and bactericidal activities.42 Several research groups have investigated the antimalarial activity of sulphonamide-containing compounds.43–45 Domínguez et al.46 synthesized the chalcone-based aryl sulfonamide derivatives (Fig. 1) for the inhibition of beta-hematin formation and evaluated their activity against the Plasmodium parasite. Similarly, Leeza Zaidi et al. 47 developed thienopyrimidine-based aryl sulphonamide derivatives (Fig. 1) which have antimalarial activity in the micromolar range. In addition, a combination of pyrimethamine-sulfadoxine which is a derivative of sulfonamide is still effective as a curative measure of malarial disease in pregnant women in Tanzania, indicating the importance of developing new sulphonamide-based derivatives to cure malaria.48 Additionally, recent reports49–51 described the antimalarial advancements and hydrophilicity that occur due to the incorporation of the piperazine ring in synthetic compounds and provided an easy way to understand the rationale behind the ring's introduction.
Thus, by keeping all these factors in mind, we disclosed the outcomes of our recent work comprising a series of new pyrimidine-tethered spirocyclic chromane-based sulphonamides and their antiplasmodial activity were examined toward drug-susceptible (3D7) strain and drug resistance (W2) strain of P. falciparum. Furthermore, the molecules with good antimalarial efficacy were tested for enzymatic assay (FP-2 and FP-3) followed by cytotoxicity and hemolysis assay. Our findings indicate that the newly synthesized compounds could be utilized as research models and are appropriate for use in future drug development.
2. Results and discussions
2.1. Synthetic chemistry
A linear synthetic method was applied to synthesize the design molecules (SZ1–SZ14), sketched in (Scheme 1). The synthesis began with preparing the intermediate (1) by treating the commercially available -ortho-hydroxy acetophenone and 1-boc-piperidone-4-one by known literature procedure. Then intermediate (1) was subjected to deprotection of the Boc group to gain the nucleophilic intermediate (2) using the standards deprotection conditions, TFA in the presence of solvent dichloromethane.41 Further, commercially available 4,6-dichloropyrimidine was reacted with the intermediate compound (2) along with triethylamine to synthesize the intermediate compound (3) which was subsequently treated with anhydrous piperazine and K2CO3 to get the final intermediate compound (4). Finally, the intermediate (4) was treated with various commercially available sulfonyl chlorides to furnish the target compounds (SZ1–SZ14) in good yield. | ||
| Scheme 1 Synthesis of sulfonamide derivatives based on pyrimidine-tethered spirochromane targeted compounds (SZ1–SZ14)a. aReagent and conditions: (a) pyrrolidine, methanol, reflux at 80 °C, 6–8 h, 77% yield; (b) trifluoroacetic acid (TFA), DCM, r.t., 2 h, 73% yield; (c) 4,6-dichloropyrimidine, triethylamine, dry THF, rt, overnight, 46% yield; (d) anhydrous piperazine, K2CO3, dioxane, reflux at 80 °C, 10 h, 70% yield; (e) various sulfonyl chlorides, dried methanol, 0 °C, 8 h, 54–70% yield. | ||
The final compounds were purified by column chromatography and nuclear magnetic resonance spectroscopy (NMR) was utilized to determine their structural elucidation. Correlated results are included in the supplemental data in Fig. S2–S17.† Moreover, the molar mass of the synthesized hybrids was validated using high-resolution mass spectrometry (HRMS), and it was discovered that the predicted mass and the molar mass matched well. The Fig. S18–S22 in the ESI† provides their projected molecular weight.
2.2. In vitro biological studies
| Compounds | Pf3D7 | PfW2 | Resistance index (RI)a |
|---|---|---|---|
| IC50 (μM) | IC50 (μM) | ||
| a ND = not determined.b Reported earlier.52 | |||
| SZ1 | 14.76 ± 0.24 | >15 | ND |
| SZ2 | 18.12 ± 0.43 | >15 | ND |
| SZ3 | 25.24 ± 0.35 | >15 | ND |
| SZ4 | 6.33 ± 0.29 | 14.36 ± 0.16 | 0.44 |
| SZ5 | 7.45 ± 0.37 | 13.28 ± 0.19 | 0.56 |
| SZ6 | 6.68 ± 0.09 | 4.76 ± 0.25 | 1.40 |
| SZ7 | 7.97 ± 0.33 | 5.64 ± 0.18 | 1.41 |
| SZ8 | 5.61 ± 0.42 | 8.12 ± 0.37 | 0.69 |
| SZ9 | 3.22 ± 0.07 | 3.89 ± 0.23 | 0.83 |
| SZ10 | 4.55 ± 0.34 | 6.67 ± 0.16 | 0.68 |
| SZ11 | 3.92 ± 0.25 | 5.58 ± 0.19 | 0.70 |
| SZ12 | 8.46 ± 0.28 | >15 | ND |
| SZ13 | 22.21 ± 0.36 | >15 | ND |
| SZ14 | 2.84 ± 0.21 | 3.12 ± 0.14 | 0.91 |
| CQb | 0.018 ± 0.002 | 0.051 ± 0.001 | 0.35 |
| PYRb | 0.015 ± 0.0002 | 129.0 ± 11.3 | 0.00012 |
The results demonstrated that molecules SZ10 (FP-2 = 7.2 μM and FP-3 = 8.8 μM) and SZ11 (FP-2 = 6.5 μM and FP-3 = 8.0 μM) were least potent against the two enzymes, FP-2 and FP-3 as they had the lowest effectiveness against the Pf3D7 strain while molecule SZ9 (FP-2 = 4.9 μM and FP-3 = 6.3 μM) and SZ14 (FP-2 = 4.1 μM and FP-3 = 5.4 μM) was most potent against both FP-2 and FP-3 enzymes among others. Additionally, the obtained IC50 values from this in vitro enzymatic assay supported the validation of the preliminary assay result against 3D7 and W2 strains. Consequently, the results indicated that the molecule was considered to be a potent inhibitor of falcipains belonging to enzyme cysteine proteases, and the SZ14 molecule may serve as a lead molecule against the plasmodium parasite.
| Compounds | Pf3D7 | PfW2 | Cytotoxicity (IC50 μM) | Selectivity index (SI) with Pf3D7 strain | Selectivity index (SI) with PfW2 strain | |||
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | Vero cell | A5489 | Vero cell | A5489 | Vero cell | A5489 | |
| a N.C. denotes no cytotoxicity noticed up to 1000 μM; SI (selectivity index) = ratio of cytotoxicity assay's IC50 value and the inhibitory concentration against the specific strains. | ||||||||
| SZ9 | 3.22 ± 0.07 | 3.89 ± 0.23 | N.C. | 78.47 ± 2.76 | 310.55 | 24.36 | 257.06 | 20.17 |
| SZ10 | 4.55 ± 0.34 | 6.67 ± 0.16 | N.C. | 122.46 ± 3.39 | 219.79 | 26.91 | 149.92 | 18.35 |
| SZ11 | 3.92 ± 0.25 | 5.58 ± 0.19 | N.C. | 94.15 ± 2.97 | 255.10 | 24.01 | 179.21 | 16.87 |
| SZ14 | 2.84 ± 0.21 | 3.12 ± 0.14 | N.C. | 66.22 ± 2.14 | 352.11 | 23.31 | 320.51 | 21.22 |
 | ||
| Fig. 2 Compound's impact on the growth and morphology of P. falciparum. | ||
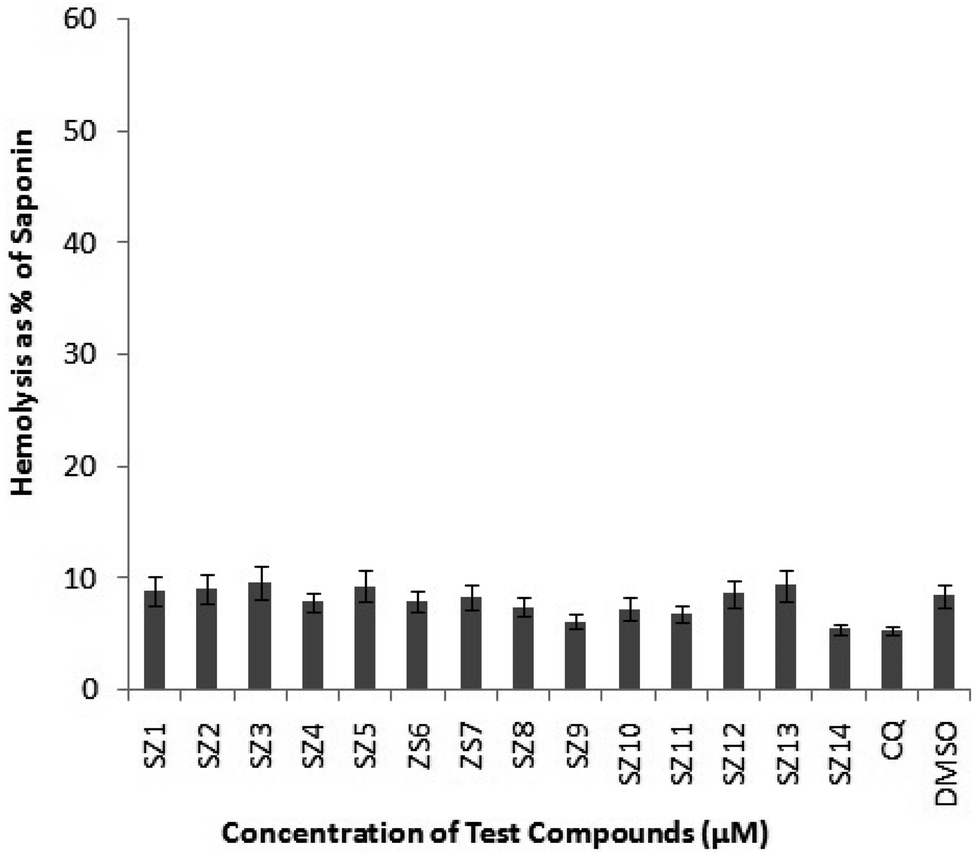 | ||
| Fig. 3 Hemolytic experiment for the synthesized substances (SZ1-QS14) at a dose of 100 μM, while the negative and positive visuals were controlled using 0.5% of DMSO and 0.05% of saponin, respectively. With a standard deviation, the data represents the mean of two studies, each of which was conducted twice. | ||
2.3. Computational study
The active pyrimidine-tethered spirochromane sulfonamide hybrids interacted with falcipain-2 and falcipains-3 target proteins with the formation of H-bonding along with π–π stacking interactions. The docking results are summarised in (Table 2) which displays the binding energy of the corresponding ligands. The calculated binding free energy for in vitro active compounds demonstrated that the entities SZ9 and SZ14 have shown a substantial binding affinity towards PfFP-2 with energy varied from −9.4 to −5.7 kcal mol−1 while −9.8 to −7.5 kcal mole−1 against PfFP-3 when compared with the standard inhibitors of PfFP-2(E64) and PfFP-3(K11017) (Table 4).
As shown in (Fig. 4) the catalytic site's amino acid residue of the enzyme falcipains-2 connected with the ligand SZ14 through hydrogen bonding between the carbonyl oxygen of the chromane ring of the ligand with the hydrogen of GLN36 and CYS39 at bond lengths of 2.95 Å and 2.37 Å correspondingly. The TRP206 residue interacted with the chromane benzene ring through π–π interactions for the ligands SZ14 and SZ9 (Fig. 4 and 5). Similarly, for both the ligands SZ14 and SZ9, the GLY83, SER149, and ASN173 amino acid residue was associated with the carbon–hydrogen bonding interaction to the carbons of the piperazine, oxygen atom of sulfonyl groups, and the carbons atom of piperidine ring of spirochromane respectively (Fig. 4 and 5) while LEU84 and ILE85 were involved with the π–σ along with π–alkyl interaction with the phenyl ring of the sulfonamide portion correspondingly. The ASP234 was visualized to have interacted with the fluorine atom forming a halogen-type interaction along with π–anion interaction with the phenyl ring for the compound SZ14 while for SZ9 it shows only the π–anion interaction. Additionally, a hydrogen bonding in the middle of the fluorine atom of the SZ9 ligand and the hydrogen atom of the ASN86 was visualized while such interaction was not displayed by entity SZ14.
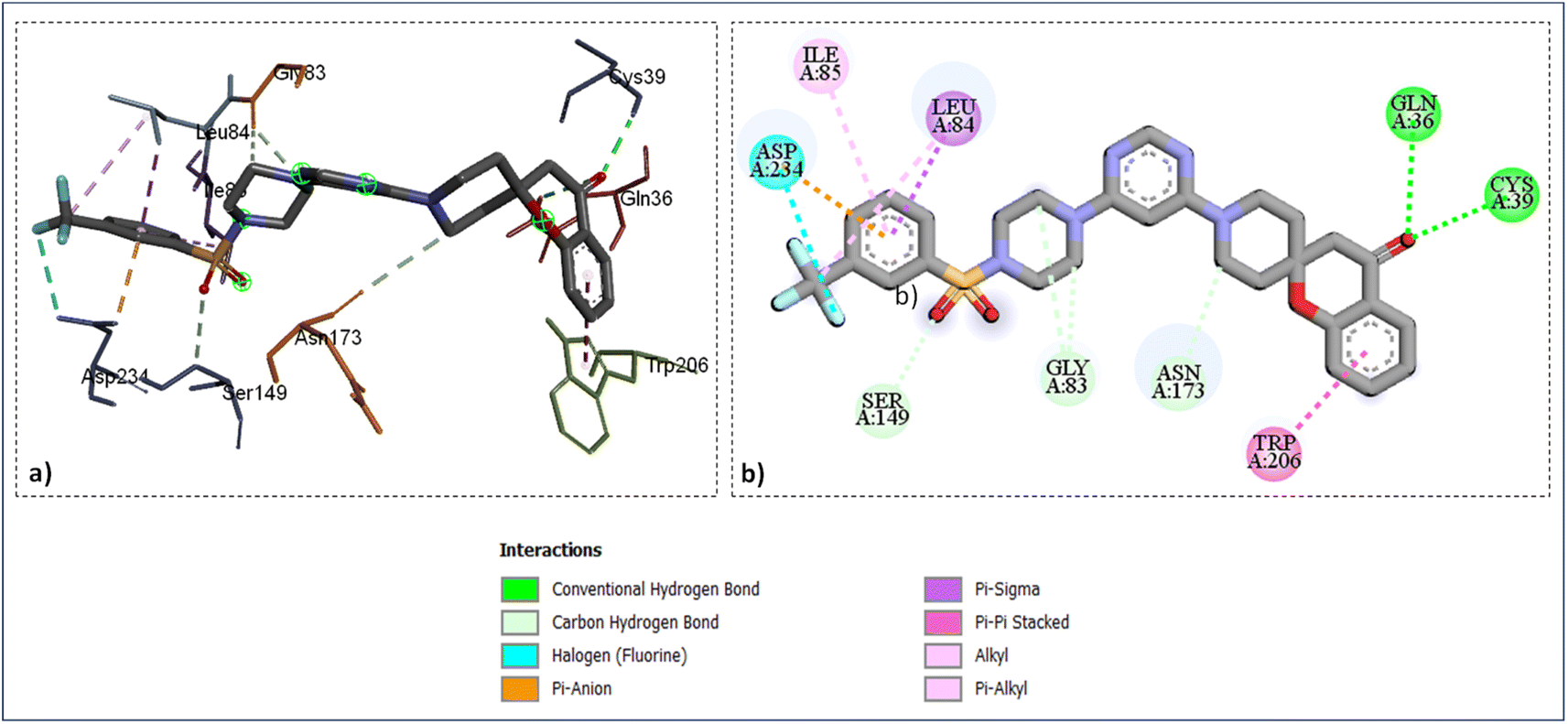 | ||
| Fig. 4 Ligand docked protein interactions: (a) 3D binding pose showing the interaction of SZ14 complexed with protein falcipain-2 (3BPF); (b) 2D binding pose showing the interaction of SZ14 complexed with protein falcipain-2 (3BPF). | ||
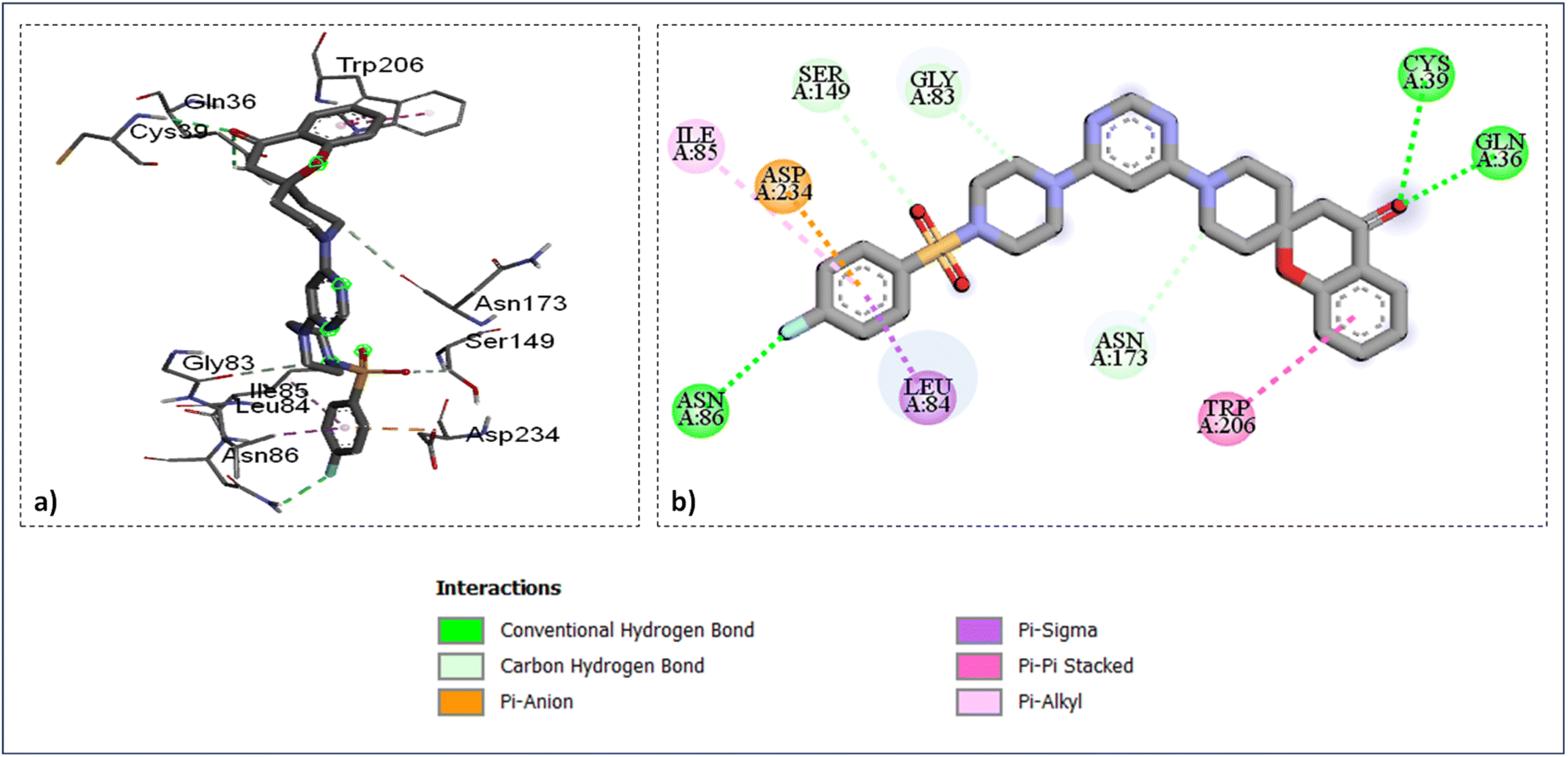 | ||
| Fig. 5 Ligand docked protein interactions: (a) 3D binding pose showing the interaction of SZ9 complexed with protein falcipain-2 (3BPF); (b) 2D binding pose showing the interaction of SZ9 complexed with protein falcipain-2 (3BPF). | ||
The docking study of the active compounds with PfFP-3 (3BWK) revealed that the compound SZ14 exhibited a π–π interaction between the TYR93 and the benzene ring, three hydrogen bonding between the three fluorine atoms of CF3 group with the GLY92 and HIS183 along with three carbon–hydrogen bond interaction: one within the GLY49 and nitrogen atom of the pyrimidine ring, the second between ASN182 and carbon of the piperazine ring, and third between TYR93 and one of the fluorine of the –CF3 group (Fig. 6). Furthermore, an alkyl interaction was observed between the ALA184 and carbon atoms of the CF3 group of the SZ14 ligand. The binding site of the targeted protein also interacted with the compound SZ9 with the formation of various interactions such as two π–π interactions between TRP215 and the phenyl ring of the ligand, the carbon–hydrogen bond interaction among the ASN182 and the carbon atoms of the piperidine ring. In addition, a π–alkyl interaction was viewed among the ALA166 and the benzene ring as well and another π–alkyl interaction was also noticed between PRO181 and the benzene ring of the spirochromane fragments in the binding site as imaged in Fig. 7.
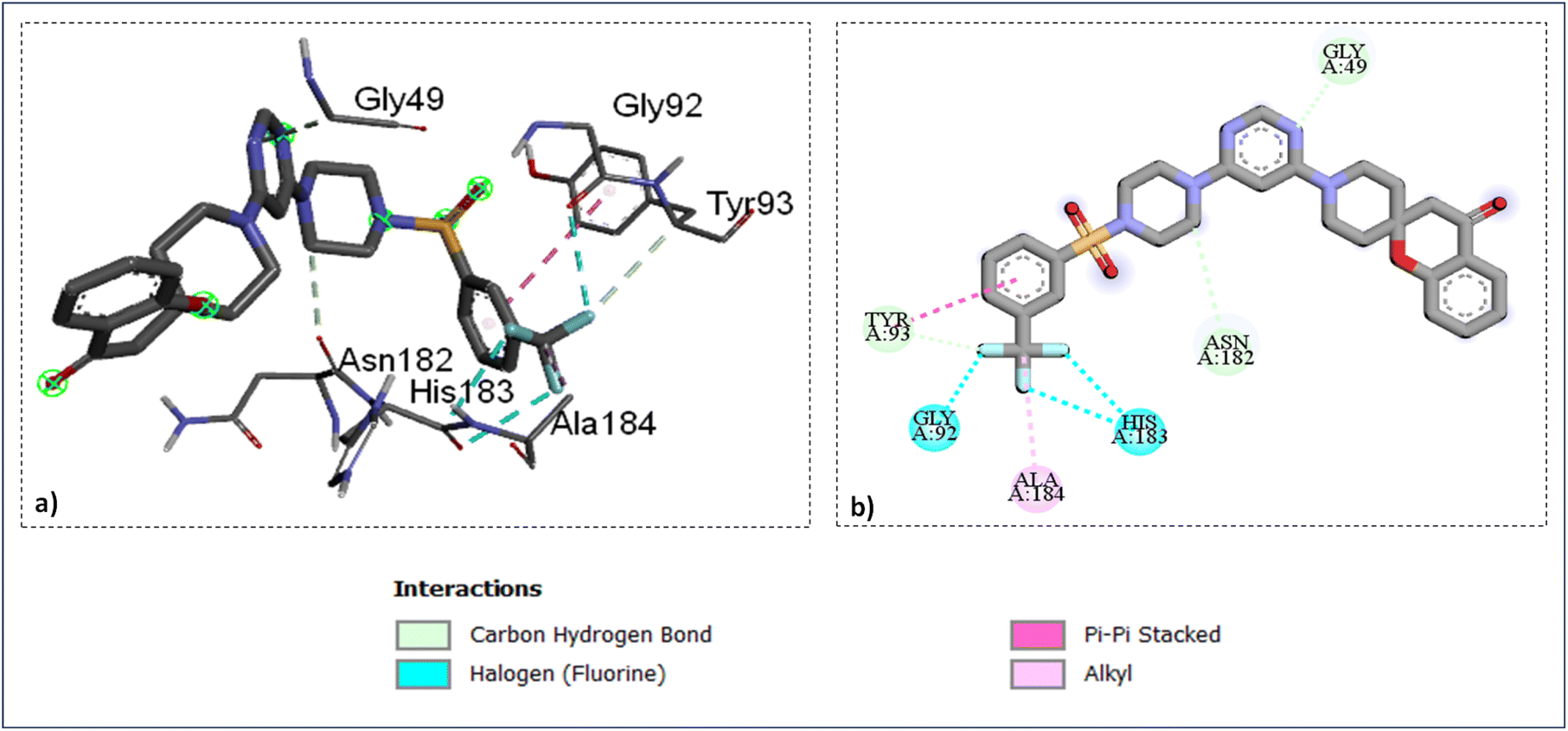 | ||
| Fig. 6 Ligand docked protein interactions: (a) 3D binding pose showing the interaction of SZ14 complexed with protein falcipain-3 (3BWK); (b) 2D binding pose showing the interaction of SZ14 complexed with protein falcipain-3 (3BWK). | ||
 | ||
| Fig. 7 Ligand docked protein interactions: (a) 3D binding pose showing the interaction of SZ9 complexed with protein falcipain-3 (3BWK); (b) 2D binding pose showing the interaction of SZ9 complexed with protein falcipain-3 (3BWK). | ||
To further investigate the aforementioned, the catalytic pockets of PfFP-2 and PfFP-3 were further docked with two reference inhibitors, E64 and K11017 respectively. The ligands SZ14 and SZ9 demonstrate a higher binding affinity and easily occupy the receptor pocket compared to the reference molecules, as seen in (Fig. 8), which reveals how it accommodates the reference inhibitor molecules and active synthesized ligands. The proximity of the active compounds to the catalytic sites of both target proteins (PfFP-2 and PfFP-3) indicates the effectiveness and active involvement of the hydrophobic effective molecules interacting with the enzyme's catalytic site and subsequently suppressing enzyme activity.
 | ||
| Fig. 8 Correlative visualizations of the inhibitors [E64(yellow) co-crystalized with PfFP-2(3BPF) and K11017(cyan) co-crystalized with PfFP-3(3BWK)] and the active entities SZ14 (blue) and SZ9 (red) bound to the active site of receptors: (a) ligands (SZ14, SZ9, and E64) bonded to the PfFP-2(3BPF) active site; (b) ligands (SZ14, SZ9, and K11017) bonded to the PfFP-3(3BWK) active site. | ||
| Compound | MW (<500 amu) | DonorHB (<5) | AccptHB (<10) | QPlogPo/w (<5) | N of violation (<2) | #Rotor (0–15) |
|---|---|---|---|---|---|---|
| SZ9 | 537.608 | 0 | 10.250 | 4.113 | 1 | 2 |
| SZ14 | 587.616 | 0 | 10.250 | 4.938 | 1 | 2 |
| CQ | 319.876 | 1 | 4 | 4.537 | 0 | 8 |
| PYM | 248.714 | 4 | 3 | 1.793 | 0 | 4 |
| Comp. | Percent human oral absorption (>80% – high & <25% – poor) | QPPCaco (<25 poor, >500 great) | QPlogBB (−3.0 to 1.5) | QPlogKhsa (−1.5 to 1.5) | QPlogHERG (below-5) | QPloS (−6.5 to 5) | PSA (70–200 Å) |
|---|---|---|---|---|---|---|---|
| SZ9 | 86.140 | 485.092 | −0.997 | 0.305 | −7.051 | −6.743 | 101.854 |
| SZ14 | 92.174 | 566.307 | −0.812 | 0.548 | −7.205 | −7.878 | 102.657 |
| CQ | 100.000 | 1460.226 | 0.407 | 0.591 | −6.329 | −4.550 | 25.049 |
| PYM | 84.401 | 420.427 | −0.768 | −0.251 | −4.286 | −2.938 | 73.489 |
3. Conclusion
The present investigation addressed the designing of a set of new fourteen pyrimidine-tethered spirochromane-based sulphonamide derivatives and their chemical synthesis followed by their biological evaluation against malarial parasitic disease. SZ14 displayed the strongest activity of all the synthesized compounds in vitro experiments against CQ-susceptible and CQ-resistant strains of P. falciparum, with IC50 values of 2.84 and 3.12 μM against both strains, respectively. Among all the tested compounds SZ14 and SZ9 were observed to possess antiplasmodial activity against PfFP-2 and PfFP-3 enzymes respectively. The selected compounds (SZ9, SZ10, SZ11, and SZ14) were also assessed for cytotoxicity in vitro and it was found that SZ14 possessed the highest anti-malarial activity while presenting the least cytotoxic impact against the lung cancer A5489 cell line, indicating a strong safety index and a strong preference for the Plasmodium pathogen in contrast to human cells. Further, in vitro hemolysis was carried out with uninfected human erythrocytes for all the active compounds to establish biocompatibility, confirming these compounds possess specific anti-malaria activity that is not due to lysis of the human erythrocytes. The docking study for the active compounds (SZ14 and SZ9) was performed with the FP-2 and FP-3 of the P. falciparum and stronger interaction was noticed with the amino acid unit at the active pocket of the targeted protein when compared to the known inhibitor of cysteine protease enzyme (FP-2 and FP-3). Moreover, the in silico physicochemical and ADME predictions described that these molecules possess good pharmacokinetic profiles, indicating that they might have a probability of being orally active lead molecules. Consequently, all the combined results from studies based on molecular docking, in vitro antimalarial assays, and predicted ADME profiling demonstrated that pyrimidine-tethered spirochromane-based sulphonamide derivatives could be further optimized for the development of potential antimalarial drugs.4. Experimental
4.1. Chemistry
All the required commercially available small building blocks, solvents, and reagents utilized in the reaction were procured via commercial suppliers like Bld pharm, sigma Aldrich, Avra, and Spectrochem and used in the chemical synthesis without further purification. To check the reaction progress, thin layer chromatography (TLC) was developed on precoated aluminum sheets (Merck silica gel 60 F254, 0.2 mm) by using a suitable system of solvents. For the visualization of developed chromatograms, UV light of a short wavelength was used. The Tanco melting point apparatus (ISO 9001: 2000 CO.) was used to obtain the melting of all the final products. Every synthetic intermediate and final product were isolated and pure through flash column chromatography by exploiting silica gel (200–240 Mesh) and the eluents were a binary mixture of ethyl acetate in petroleum ether and a binary mixture of methanol in chloroform. 1H NMR and 13C NMR of all the final compounds were reported on JEOL (JNM-ECZ 400S) 400 spectrometer. Relative chemical shifts (δ) in parts per million were measured in CDCl3 solvent to the internal reference standard tetramethyl silane (TMS, δ 0.0, 1H NMR) The molecular weight was confirmed by utilizing the mass spectra recorded on Agilent G6530AA (LC-HRMS-Q-TOF).4.1.5.1. 1′-(6-(4-((4-Nitrophenyl) sulfonyl) piperazin-1-yl)pyrimidin-4-yl)spiro[chromane-2,4′-piperidin]-4-one (SZ1). Yellow solid; melting point = 235–237 °C; yield: 0.96 g, 64.4%. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 8.8 Hz, 2H), 8.17 (s, 1H), 7.94 (d, J = 8.9 Hz, 2H), 7.87 (dd, J = 7.8, 1.8 Hz, 1H), 7.55–7.45 (m, 1H), 7.07–6.94 (m, 2H), 5.55 (s, 1H), 4.14–3.95 (m, 2H), 3.74–3.65 (m, 4H), 3.40–3.27 (m, 2H), 3.17–3.08 (m, 4H), 2.71 (s, 2H), 2.14–2.05 (m, 2H), 1.71–1.57 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.56, 163.02, 162.80, 159.02, 157.69, 150.45, 141.67, 136.57, 129.00 (2C), 126.79, 124.57 (2C), 121.45, 120.81, 118.36, 81.71, 78.01, 48.08, 45.67 (2C), 43.72 (2C), 39.92 (2C), 33.71 (2C). HRMS (ESI) in MeOH calcd for C27H28N6O6S [M + H]+: 565.1791, found: 565.1847.
4.1.5.2. 1′-(6-(4-((2-Nitrophenyl)sulfonyl)piperazin-1-yl) pyrimidin-4-yl)spiro [chromane-2,4′-piperidin]-4-one (SZ2). Greenish white solid; melting point = 142–144 °C; yield: 0.97 g, 65.1%. 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 7.98–7.93 (m, 1H), 7.88–7.83 (m, 1H), 7.76–7.66 (m, 2H), 7.63–7.58 (m, 1H), 7.54–7.46 (m, 1H), 7.07–6.94 (m, 2H), 5.62 (s, 1H), 4.12–4.01 (m, 2H), 3.74–3.62 (m, 6H), 3.39–3.30 (m, 4H), 2.72 (s, 2H), 2.09 (d, J = 14.3 Hz, 2H), 1.76–1.52 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.74, 163.00, 162.94, 159.06, 157.65, 148.49, 136.60, 134.13, 131.78, 131.08, 130.84, 126.74, 124.27, 121.42, 120.76, 118.41, 81.85, 63.76, 48.08 (2C), 45.60 (2C), 44.03 (2C), 39.94, 33.68 (2C). HRMS (ESI) in MeOH calcd for C27H28N6O6S [M + H]+: 565.1791, found: 565.1853.
4.1.5.3. 1′-(6-(4-(Naphthalen-2-ylsulfonyl)piperazin-1-yl)pyrimidin-4-yl)spiro[chromane-2,4′-piperidin]-4-one (SZ3). Light green; melting point = 196–198 °C; yield: 0.90 g, 60.0%. 1H NMR (400 MHz, CDCl3) δ 8.30 (br, 1H), 8.11 (s, 1H), 7.94 (d, J = 8.5 Hz, 2H), 7.89 (d, J = 8.4 Hz, 1H), 7.83 (dd, J = 7.8, 1.8 Hz, 1H), 7.70 (dd, J = 8.6, 1.8 Hz, 1H), 7.67–7.56 (m, 2H), 7.51–7.42 (m, 1H), 7.04–6.92 (m, 1H), 5.51 (s, 1H), 4.83–4.73 (m, 2H), 4.06–3.94 (m, 2H), 3.65 (t, J = 5.0 Hz, 2H), 3.34–3.22 (m, 2H), 3.10 (t, J = 5.1 Hz, 4H), 2.67 (s, 2H), 2.07–2.01 (m, 2H), 1.66–1.53 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.58, 162.85, 162.67, 158.95, 157.52, 136.48, 134.96, 132.33, 132.17, 129.42, 129.22, 129.17, 129.08, 127.97, 127.76, 126.64, 122.83, 121.31, 120.68, 118.30, 81.54, 77.96, 47.98, 45.77 (2C), 43.61 (2C), 39.82 (2C), 33.57 (2C). HRMS (ESI) in MeOH calcd for C31H31N5O4S [M + H]+: 570.2097, found: 570.2187.
4.1.5.4. 4-((4-(6-(4-Oxospiro[chromane-2,4′-piperidin]-1′-yl)pyrimidin-4-yl)piperazin-1-yl)sulfonyl)benzonitrile (SZ4). Light green solid; melting point = 170–172 °C; yield: 0.97 g, 67.8%. 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 7.91–7.80 (m, 5H), 7.54–7.46 (m, 1H), 7.07–6.93 (m, 2H), 5.56 (s, 1H), 4.13–3.98 (m, 2H), 3.72–3.65 (m, 4H), 3.40–3.29 (m, 2H), 3.11 (t, J = 5.1 Hz, 4H), 2.72 (s, 2H), 2.14–2.00 (m, 2H), 1.73–1.61 (m, 2H).13C NMR (101 MHz, CDCl3) δ 191.62, 163.00, 162.79, 159.01, 157.66, 140.03, 136.57, 133.11 (2C), 128.37 (2C), 126.75, 121.43, 120.77, 118.37, 117.19, 116.95, 81.72, 78.01, 48.06, 45.67 (2C), 43.69 (2C), 39.91 (2C), 33.69 (2C). HRMS (ESI) in MeOH calcd for C28H28N6O4S [M + H]+: 545.1893, found: 545.1954.
4.1.5.5. 1′-(6-(4-(Thiophen-2-ylsulfonyl)piperazin-1-yl) pyrimidin-4-yl) spiro[chromane-2,4′-piperidin]-4-one (SZ5). Greenish white solid; melting point = 163–165 °C; yield: 0.87 g, 63.6%. 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.85 (dd, J = 7.9, 1.8 Hz, 1H), 7.62 (dd, J = 5.0, 1.3 Hz, 1H), 7.55–7.45 (m, 2H), 7.13 (dd, J = 5.0, 3.7 Hz, 1H), 7.05–6.95 (m, 2H), 5.57 (s, 1H), 4.11–4.00 (m, 2H), 3.69 (t, J = 5.1 Hz, 4H), 3.39–3.27 (m, 2H), 3.10 (t, J = 5.1 Hz, 4H), 2.70 (s, 2H), 2.14–2.03 (m, 2H), 1.72–1.57 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.67, 162.95, 162.83, 159.04, 157.62, 136.58, 135.51, 132.87, 132.67, 127.94, 126.75, 121.42, 120.77, 118.38, 81.70, 78.04, 48.08, 45.80 (2C), 43.56 (2C), 39.96 (2C), 33.69 (2C). HRMS (ESI) in MeOH calcd for C25H27N5O4S2 [M + H]+: 526.1504, found: 526.1571.
4.1.5.6. 1′-(6-(4-((5-Chlorothiophen-2-yl) sulfonyl) piperazin-1-yl pyrimidin-4-yl) spiro [chromane-2,4′-piperidin]-4-one (SZ6). Light green solid; melting point 79–81 °C; yield: 0.78 g, 53.7%. 1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 7.86 (dd, J = 7.8, 1.8 Hz, 1H), 7.50 (ddd, J = 8.8, 7.3, 1.8 Hz, 1H), 7.32 (d, J = 4.0 Hz, 1H), 7.06–6.93 (m, 3H), 5.59 (s, 1H), 4.06 (d, J = 13.6 Hz, 2H), 3.74–3.67 (m, 4H), 3.43–3.29 (m, 2H), 3.12 (t, J = 5.2 Hz, 4H), 2.72 (s, 2H), 2.17–2.04 (m, 2H), 1.73–1.60 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.58, 163.02, 162.83, 159.07, 157.66, 138.01, 136.57, 133.77, 132.24, 127.37, 126.81, 121.46, 120.85, 118.40, 81.73, 78.07, 48.12, 45.78 (2C), 43.61 (2C), 40.01 (2C), 33.76 (2C). HRMS (ESI) in MeOH calcd for C25H26ClN5O4S2 [M + H]+: 560.1115, found: 560.1199.
4.1.5.7. 1′-(6-(4-(Pyridin-3-ylsulfonyl)piperazin-1-yl)pyrimidin-4-yl)spiro[chromane-2,4′-piperidin]-4-one (SZ7). Light green solid; melting point 144–146 °C; yield: 0.95 g, 69.3%.1H NMR (400 MHz, CDCl3) δ 8.97 (d, J = 1.5 Hz, 1H), 8.82 (dd, J = 4.9, 1.6 Hz, 1H), 8.16 (s, 1H), 8.03 (dt, J = 8.0, 1.7 Hz, 1H), 7.85 (dd, J = 7.9, 1.8 Hz, 1H), 7.54–7.45 (m, 2H), 7.05–6.92 (m, 2H), 5.56 (s, 1H), 4.22–3.89 (m, 2H), 3.69 (dd, J = 10.9, 6.4 Hz, 4H), 3.38–3.27 (m, 2H), 3.10 (t, J = 5.1 Hz, 4H), 2.71 (s, 2H), 2.12–2.04 (m, 2H), 1.69–1.59 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.64, 163.01, 162.82, 159.04, 157.68, 153.79, 148.54, 136.58, 135.51, 132.39, 126.77, 123.98, 121.44, 120.80, 118.39, 81.74, 78.03, 77.48, 48.09, 45.62 (2C), 43.69 (2C), 39.93 (2C), 33.71 (2C). HRMS (ESI) in MeOH calcd for C26H28N6O4S [M + H]+: 521.1893, found: 521.1980.
4.1.5.8. 1′-(6-(4-((2-Fluorophenyl)sulfonyl)piperazin-1-yl)pyrimidin-4-yl)spiro[chromane-2,4′-piperidin]-4-one (SZ8). Pale yellow solid; melting point 88–90 °C; yield:0.88 g, 62.4%.1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 7.88–7.80 (m, 2H), 7.62–7.53 (m, 1H), 7.53–7.46 (m, 1H), 7.29 (dd, J = 7.6, 1.2 Hz, 1H), 7.24–7.16 (m, 1H), 7.06–6.95 (m, 2H), 5.58 (s, 1H), 4.14–4.01 (m, 2H), 3.74–3.60 (m, 4H), 3.40–3.27 (m, 2H), 3.27–3.21 (m, 4H), 2.71 (s, 2H), 2.14–2.01 (m, 2H), 1.71–1.60 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.67, 163.02, 162.95, 159.05, 157.67, 136.58, 135.49 (d, J = 8.3 Hz), 131.42, 126.77, 124.75, 124.74, 121.42, 120.80, 118.40, 117.63, 117.41, 81.72, 78.06, 48.10, 45.49, 43.93 (2C), 39.94 (2C), 33.70 (2C). HRMS (ESI) in MeOH calcd for C27H28FN5O4S [M + H]+: 538.1846, found: 538.1913.
4.1.5.9. 1′-(6-(4-((4-Fluorophenyl)sulfonyl)piperazin-1-yl)pyrimidin-4-yl)spiro[chromane-2,4′-piperidin]-4-one (SZ9). White solid; melting point 160–162 °C; yield: 0.91 g, 64.1%. 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.85 (dt, J = 7.8, 1.6 Hz, 1H), 7.80–7.71 (m, 2H), 7.55–7.44 (m, 1H), 7.26–7.15 (m, 2H), 7.00 (q, J = 7.9 Hz, 2H), 5.56 (s, 1H), 4.09–3.99 (m, 2H), 3.67 (t, J = 5.2 Hz, 4H), 3.32 (t, J = 11.3 Hz, 2H), 3.04 (t, J = 5.1 Hz, 4H), 2.71 (s, 2H), 2.08 (d, J = 13.7 Hz, 2H), 1.73–1.56 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.63, 165.45 (d, J = 255.9 Hz), 162.96, 162.82, 159.02, 157.62, 136.56, 131.43, 130.58, 126.75, 121.42, 120.77, 118.37, 116.61 (d, J = 22.6 Hz) (2C), 81.65, 77.48, 77.16, 76.84, 48.06, 45.73 (2C), 43.62 (2C), 39.91 (2C), 33.67 (2C). HRMS (ESI) in MeOH calcd for C27H28FN5O4S [M + H]+: 538.1846, found: 538.1914. Elemental analysis: calculated C, 60.32; H, 5.25; N, 13.03; S, 5.96; found C, 60.23, H, 5.09; N, 12.75; S, 7.56.
4.1.5.10. 1′-(6-(4-((3,5-Difluorophenyl) sulfonyl) piperazin-1-yl) pyrimidin-4-yl) spiro [chromane-2,4′-piperidin]-4-one (SZ10). Off-white solid; melting point 196–198 °C; yield: 0.86 g, 58.6%. 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 7.97–7.77 (m, 1H), 7.59–7.44 (m, 1H), 7.34–7.26 (m, 2H), 7.13–6.91 (m, 3H), 5.58 (s, 1H), 4.23–3.86 (m, 2H), 3.80–3.58 (m, 4H), 3.34 (t, J = 11.3 Hz, 2H), 3.11 (t, J = 5.1 Hz, 4H), 2.72 (s, 2H), 2.25–1.93 (m, 2H), 1.73–1.57 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.62, 162.48 (dd, J = 267.4 Hz, 23.3 Hz), 159.02, 157.68, 138.88, 136.57, 126.77, 121.43, 120.79, 118.38, 111.29 (d, J = 27.8 Hz), 109.13, 108.89, 108.64, 81.68, 78.03, 48.08, 45.74 (2C), 43.64 (2C), 39.91 (2C), 33.69 (2C). HRMS (ESI) in MeOH calcd for C27H27F2N5O4S [M + H]+: 556.1752, found: 556.1818.
4.1.5.11. 1′-(6-(4-((3,4-Difluorophenyl)sulfonyl) piperazin-1-yl) pyrimidin-4-yl)spiro [chromane-2,4′-piperidin]-4-one (SZ11). Pale green solid; melting point 170–172 °C; yield: 0.92 g, 62.7%. 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 7.86 (d, J = 8.6 Hz, 1H), 7.55 (dq, J = 26.0, 8.5 Hz, 3H), 7.34 (q, J = 8.2 Hz, 1H), 7.07–6.91 (m, 2H), 5.57 (s, 1H), 4.14–3.99 (m, 2H), 3.81–3.59 (m, 4H), 3.40–3.26 (m, 2H), 3.11–3.04 (m, 4H), 2.72 (s, 2H), 2.33–1.87 (m, 2H), 1.80–1.46 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.61, 162.98, 162.77, 159.01, 157.65, 150.38 (d, J = 261.59 Hz), 150.20 (d, J = 278.76 Hz), 149.09, 148.96, 136.56, 132.32, 126.74, 124.92, 121.41, 120.77, 118.60, 118.37, 117.75, 117.55, 81.67, 78.01, 48.06, 45.70 (2C), 43.61 (2C), 39.89 (2C), 33.67 (2C). HRMS (ESI) in MeOH calcd for C27H27F2N5O4S [M + H]+: 556.1752, found: 556.1823.
4.1.5.12. 1′-(6-(4-((3,5-Dimethylisoxazol-4-yl)sulfonyl) piperazin-1-yl) pyrimidin-4-yl) spiro[chromane-2,4′-piperidin]-4-one (SZ12). Greenish white solid; melting point 182–184 °C; yield: 0.83 g, 58.4%. 1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 7.85 (dd, J = 7.8, 1.9 Hz, 1H), 7.56–7.44 (m, 1H), 7.00 (dd, J = 13.5, 7.8 Hz, 2H), 5.59 (s, 1H), 4.18–3.97 (m, 2H), 3.74–3.61 (m, 4H), 3.43–3.23 (m, 2H), 3.16 (t, J = 5.1 Hz, 4H), 2.72 (s, 2H), 2.63 (s, 3H), 2.39 (s, 3H), 2.21–1.94 (m, 2H), 1.80–1.49 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.59, 174.09, 162.86, 162.69, 159.01, 158.00, 157.52, 136.58, 126.77, 121.45, 120.78, 118.38, 112.95, 81.71, 78.00, 77.48, 77.16, 76.84, 48.07, 45.14 (2C), 43.70 (2C), 39.96 (2C), 33.70 (2C), 13.14, 11.51. HRMS (ESI) in MeOH calcd for C26H30N6O5S [M + H]+: 539.1998, found: 539.2064.
4.1.5.13. 1′-(6-(4-(Morpholinosulfonyl) piperazin-1-yl) pyrimidin-4-yl) spiro[chromane-2,4′-piperidin]-4-one (SZ13). Light yellow solid; melting point 146–148 °C; yield: 0.95 g, 68.3%. 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 7.86 (dd, J = 7.8, 1.8 Hz, 1H), 7.50 (ddd, J = 8.8, 7.2, 1.8 Hz, 1H), 7.06–6.96 (m, 2H), 5.61 (s, 1H), 4.14–4.04 (m, 2H), 3.77–3.68 (m, 4H), 3.67–3.60 (m, 4H), 3.42–3.29 (m, 6H), 3.27–3.20 (m, 4H), 2.72 (s, 2H), 2.16–2.06 (m, 2H), 1.74–1.61 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.60, 162.96, 162.80, 159.03, 157.37, 136.58, 126.78, 121.45, 120.81, 118.39, 81.77, 78.03, 66.44 (2C), 48.10, 46.68 (2C), 46.18 (2C), 44.12 (2C), 40.05 (2C), 33.73 (2C). HRMS (ESI) in MeOH calcd for C25H32N6O5S [M + H]+: 529.2155, found: 529.2250.
4.1.5.14 1′-(6-(4-((3-(Trifluoromethyl)phenyl)sulfonyl)piperazin-1-yl) pyrimidin-4-yl)spiro [chromane-2,4′-piperidin]-4-one (SZ14). Off-white solid; melting point 138–140 °C; yield: 0.98 g, 63.2%. 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 8.00 (s, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.90–7.80 (m, 2H), 7.70 (t, J = 7.8 Hz, 1H), 7.54–7.44 (m, 1H), 7.05–6.94 (m, 2H), 5.56 (s, 1H), 4.11–3.96 (m, 2H), 3.75–3.62 (m, 4H), 3.42–3.24 (m, 2H), 3.09 (t, J = 5.1 Hz, 4H), 2.71 (s, 2H), 2.14–2.00 (m, 2H), 1.73–1.56 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.55, 163.06, 162.87, 159.05, 157.71, 137.03, 136.55, 132.61, 132.27, 131.94, 131.61, 131.00, 130.18, 129.85 (q, J = 3.03 Hz), 126.78, 124.73 (q, J = 4.04 Hz), 121.44, 120.85, 118.39, 81.71, 78.05, 48.11, 45.75 (2C), 43.69 (2C), 39.96 (2C), 33.74 (2C). HRMS (ESI) in MeOH calcd for C28H28F3N5O4S [M + H]+: 588.1814, found: 588.1909. Elemental analysis: calculated C, 57.23; H, 4.80; N, 11.92; S, 5.46; found: C, 58.01; H, 4.36; N, 11.67; S, 7.07.
4.2. Biological study
An investigation was executed to investigate the impact of a few compounds on the parasitic proliferation. The early ring stage 3D7 parasite of P. falciparum was dosed for 48 hours with substances (at 3 × IC50 concentration) or DMSO (Con, at 0.1%). Giemsa-stained smears were created from the test and control cultured before and following the treatment (48 hours). Photographs of the smears were observed and recorded using a 100× objective light microscope, Axio-Imager Z2.
whereas: Ao stands for absorbance of cells treated by 0.1% DMSO media, At stands for absorbance of cells treated among diverse concentrations of the test.
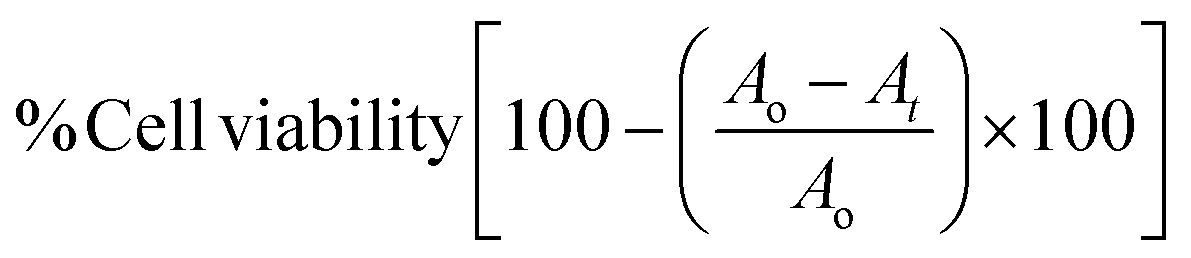
Furthermore, the corresponding cytotoxicity was assessed with the lung cancer A5489 cell line using the previously described standard procedure.67 10% foetal bovine serum supplemented Dulbecco's modified Eagle's medium (DMEM, Gibco, USA) was used to cultivate the cells. The test chemical compounds were administered at different dosages to cells seeded in a 160 μL medium at a density of 1 × 104 cells per well in a 96-well plate, which then underwent incubation for 24 hours at 37 °C. After being incubated for 24 hours, the medium was extracted, and MTT (25 μL) in 4 mmgg mmLL−1−1 was introduced. This was accompanied by an additional 4 hour incubation period. After discarding the medium, DMSO (200 μL) was applied to liquidate the formazan crystals. Triplicates of the experiment were conducted, and the control wells were compared to evaluate the vitality of the cells. The Software GraphPad Prism, Version 3 (San Diego, CA) was applied for the determination of every test molecule's half-maximal inhibitory concentration (IC50).
4.3. Computational study
Abbreviations
| P. falciparum | Plasmodium falciparum |
| FP-2 | Falcipains-2 |
| FP-3 | Falcipains-3 |
| ADME | Absorption, distribution, metabolism, and excretion |
| CQ | Chloroquine |
| FDA | Food and Drug Administration |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
AR: docking studies, designing of the molecule, standardization of synthetic protocol, synthesis of compounds, analyzing NMR, compiling the results, and writing the original draft of the manuscript. SA: standardization of synthetic protocol, synthesis of compounds, reviewing, and editing the original manuscript. AS: characterizations, reviewing, and editing the original manuscript. SK: methodology. SF: reviewing and editing the original manuscript. TSP, JBD, and BCD: performing the antimalarial assay, enzyme inhibition assay, cytotoxicity, hemolysis, compilation of the results, analysis of the results, and writing related sections of the antimalarial activity. NH: conceptualization of the study, supervising the synthetic work, editing the manuscript, and arranging the funds for this project.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The author A. R. wishes to acknowledge the Ministry of Minority Affairs, Govt. of India for the research fellowship [No. f.82-27/2019(SA-III), UGC-Ref. No. 191620050863]. NH acknowledges SPARC (File No. SPARC/2019-2020/P2269/SL), the Ministry of Education, Govt. of India for funding this project. TSP, JBD, and BCD are grateful to the principal and management of V. P. and R. P. T. P. Science College, Vallabh Vidyanagar for providing all the necessary research facilities and financial support for this research work.References
- M. A. Phillips, J. N. Burrows, C. Manyando, R. H. van Huijsduijnen, W. C. Van Voorhis and T. N. C. Wells, Nat. Rev. Dis. Primers, 2017, 3, 17050 CrossRef PubMed.
- A. F. Cowman, J. Healer, D. Marapana and K. Marsh, Cell, 2016, 167, 610–624 CrossRef CAS PubMed.
- B. M. Greenwood, D. A. Fidock, D. E. Kyle, S. H. I. Kappe, P. L. Alonso, F. H. Collins and P. E. Duffy, J. Clin. Invest., 2008, 118, 1266–1276 CrossRef CAS PubMed.
- World Health Organization, World Malaria Report 2023, Geneva, 2023 Search PubMed.
- E. A. Ashley, M. Dhorda, R. M. Fairhurst, C. Amaratunga, P. Lim, S. Suon, S. Sreng, J. M. Anderson, S. Mao, B. Sam, C. Sopha, C. M. Chuor, C. Nguon, S. Sovannaroth, S. Pukrittayakamee, P. Jittamala, K. Chotivanich, K. Chutasmit, C. Suchatsoonthorn, R. Runcharoen, T. T. Hien, N. T. Thuy-Nhien, N. V. Thanh, N. H. Phu, Y. Htut, K.-T. Han, K. H. Aye, O. A. Mokuolu, R. R. Olaosebikan, O. O. Folaranmi, M. Mayxay, M. Khanthavong, B. Hongvanthong, P. N. Newton, M. A. Onyamboko, C. I. Fanello, A. K. Tshefu, N. Mishra, N. Valecha, A. P. Phyo, F. Nosten, P. Yi, R. Tripura, S. Borrmann, M. Bashraheil, J. Peshu, M. A. Faiz, A. Ghose, M. A. Hossain, R. Samad, M. R. Rahman, M. M. Hasan, A. Islam, O. Miotto, R. Amato, B. MacInnis, J. Stalker, D. P. Kwiatkowski, Z. Bozdech, A. Jeeyapant, P. Y. Cheah, T. Sakulthaew, J. Chalk, B. Intharabut, K. Silamut, S. J. Lee, B. Vihokhern, C. Kunasol, M. Imwong, J. Tarning, W. J. Taylor, S. Yeung, C. J. Woodrow, J. A. Flegg, D. Das, J. Smith, M. Venkatesan, C. V. Plowe, K. Stepniewska, P. J. Guerin, A. M. Dondorp, N. P. Day and N. J. White, N. Engl. J. Med., 2014, 371, 411–423 CrossRef PubMed.
- A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. J. Day, N. Lindegardh, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455–467 CrossRef CAS PubMed.
- A. Uwimana, E. Legrand, B. H. Stokes, J.-L. M. Ndikumana, M. Warsame, N. Umulisa, D. Ngamije, T. Munyaneza, J.-B. Mazarati, K. Munguti, P. Campagne, A. Criscuolo, F. Ariey, M. Murindahabi, P. Ringwald, D. A. Fidock, A. Mbituyumuremyi and D. Menard, Nat. Med., 2020, 26, 1602–1608 CrossRef CAS PubMed.
- R. Leung-Toung, Y. Zhao, W. Li, T. Tam, K. Karimian and M. Spino, Curr. Med. Chem., 2006, 13, 547–581 CrossRef CAS PubMed.
- J. McKerrow, Bioorg. Med. Chem., 1999, 7, 639–644 CrossRef CAS PubMed.
- P. L. Olliaro and Y. Yuthavong, Pharmacol. Ther., 1999, 81, 91–110 CrossRef CAS PubMed.
- J. L. Siqueira-Neto, A. Debnath, L.-I. McCall, J. A. Bernatchez, M. Ndao, S. L. Reed and P. J. Rosenthal, PLoS Neglected Trop. Dis., 2018, 12, e0006512 CrossRef PubMed.
- C. Teixeira, J. R. B. Gomes and P. Gomes, Curr. Med. Chem., 2011, 18, 1555–1572 CrossRef CAS PubMed.
- P. J. Rosenthal, J. Exp. Biol., 2003, 206, 3735–3744 CrossRef CAS PubMed.
- M. Marco and J. Miguel Coteron, Curr. Top. Med. Chem., 2012, 12, 408–444 CrossRef CAS PubMed.
- P. S. Sijwali, K. Kato, K. B. Seydel, J. Gut, J. Lehman, M. Klemba, D. E. Goldberg, L. H. Miller and P. J. Rosenthal, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8721–8726 CrossRef CAS PubMed.
- P. S. Sijwali, J. Koo, N. Singh and P. J. Rosenthal, Mol. Biochem. Parasitol., 2006, 150, 96–106 CrossRef CAS PubMed.
- T. Conroy, J. T. Guo, N. Elias, K. M. Cergol, J. Gut, J. Legac, L. Khatoon, Y. Liu, S. McGowan, P. J. Rosenthal, N. H. Hunt and R. J. Payne, J. Med. Chem., 2014, 57, 10557–10563 CrossRef CAS PubMed.
- F. Shah, J. Gut, J. Legac, D. Shivakumar, W. Sherman, P. J. Rosenthal and M. A. Avery, J. Chem. Inf. Model., 2012, 52, 696–710 CrossRef CAS PubMed.
- T. M. Musyoka, A. M. Kanzi, K. A. Lobb and Ö. Tastan Bishop, J. Biomol. Struct. Dyn., 2016, 34, 2084–2101 CrossRef CAS PubMed.
- H. Li, J. Huang, L. Chen, X. Liu, T. Chen, J. Zhu, W. Lu, X. Shen, J. Li, R. Hilgenfeld and H. Jiang, J. Med. Chem., 2009, 52, 4936–4940 CrossRef CAS PubMed.
- P. M. Njogu, E. M. Guantai, E. Pavadai and K. Chibale, ACS Infect. Dis., 2016, 2, 8–31 CrossRef CAS PubMed.
- D. Rana, M. Kalamuddin, S. Sundriyal, V. Jaiswal, G. Sharma, K. Das Sarma, P. S. Sijwali, A. Mohmmed, P. Malhotra and N. Mahindroo, Bioorg. Med. Chem., 2020, 28, 115155 CrossRef CAS PubMed.
- A. Singh, M. Kalamuddin, M. Maqbool, A. Mohmmed, P. Malhotra and N. Hoda, Bioorg. Chem., 2021, 108, 104514 CrossRef CAS PubMed.
- H. Madhav, T. S. Patel, Z. Rizvi, G. S. Reddy, A. Rahman, M. A. Rahman, S. Ahmedi, S. Fatima, K. Saxena, N. Manzoor, S. Bhattacharjee, B. C. Dixit, P. S. Sijwali and N. Hoda, Eur. J. Med. Chem., 2023, 258, 115564 CrossRef CAS PubMed.
- F. W. Muregi and A. Ishih, Drug Dev. Res., 2010, 71, 20–32 CrossRef CAS PubMed.
- F. Simon, Nat. Rev. Drug Discovery, 2006, 5, 881–882 CrossRef CAS PubMed.
- J. Walsh and A. Bell, Curr. Pharm. Des., 2009, 15, 2970–2985 CrossRef CAS PubMed.
- M. Jain, S. Vangapandu, S. Sachdeva, S. Singh, P. P. Singh, G. B. Jena, K. Tikoo, P. Ramarao, C. L. Kaul and R. Jain, J. Med. Chem., 2004, 47, 285–287 CrossRef CAS PubMed.
- S. B. Patil, Heliyon, 2023, 9, e16773 CrossRef CAS PubMed.
- K. Singh and T. Kaur, Medchemcomm, 2016, 7, 749–768 RSC.
- R. T. Delfino, O. A. Santos-Filho and J. D. Figueroa-Villar, J. Braz. Chem. Soc., 2002, 13, 727–741 CrossRef CAS.
- K. A. Galal, A. Truong, F. Kwarcinski, C. de Silva, K. Avalani, T. M. Havener, M. E. Chirgwin, E. Merten, H. W. Ong, C. Willis, A. Abdelwaly, M. A. Helal, E. R. Derbyshire, R. Zutshi and D. H. Drewry, J. Med. Chem., 2022, 65, 13172–13197 CrossRef CAS PubMed.
- J. M. Coterón, D. Catterick, J. Castro, M. J. Chaparro, B. Díaz, E. Fernández, S. Ferrer, F. J. Gamo, M. Gordo, J. Gut, L. de las Heras, J. Legac, M. Marco, J. Miguel, V. Muñoz, E. Porras, J. C. de la Rosa, J. R. Ruiz, E. Sandoval, P. Ventosa, P. J. Rosenthal and J. M. Fiandor, J. Med. Chem., 2010, 53, 6129–6152 CrossRef PubMed.
- S. Chowdhary, Shalini, J. Mosnier, I. Fonta, B. Pradines, N. Cele, P. Seboletswe, P. Singh and V. Kumar, ACS Med. Chem. Lett., 2022, 13, 1068–1076 CrossRef CAS PubMed.
- M. Wang, T. Tang, R. Li, Z. Huang, D. Ling, L. Zheng, Y. Ding, T. Liu, W. Xu, F. Zhu, H. Min, R. Boonhok, F. Mao, J. Zhu, X. Li, L. Jiang and J. Li, J. Med. Chem., 2022, 65, 4156–4181 CrossRef CAS PubMed.
- R. Oliveira, R. C. Guedes, P. Meireles, I. S. Albuquerque, L. M. Gonçalves, E. Pires, M. R. Bronze, J. Gut, P. J. Rosenthal, M. Prudêncio, R. Moreira, P. M. O'Neill and F. Lopes, J. Med. Chem., 2014, 57, 4916–4923 CrossRef CAS PubMed.
- K. Hiesinger, D. Dar'in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150–183 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- M. Brindisi, S. Gemma, S. Kunjir, L. Di Cerbo, S. Brogi, S. Parapini, S. D'Alessandro, D. Taramelli, A. Habluetzel, S. Tapanelli, S. Lamponi, E. Novellino, G. Campiani and S. Butini, Medchemcomm, 2015, 6, 357–362 RSC.
- E. K. Schmitt, G. Ndayisaba, A. Yeka, K. P. Asante, M. P. Grobusch, E. Karita, H. Mugerwa, S. Asiimwe, A. Oduro, B. Fofana, S. Doumbia, G. Su, K. Csermak Renner, V. K. Venishetty, S. Sayyed, J. Straimer, I. Demin, S. Barsainya, C. Boulton and P. Gandhi, Clin. Infect. Dis., 2022, 74, 1831–1839 CrossRef CAS PubMed.
- B. F. Roberts, I. D. Iyamu, S. Lee, E. Lee, L. Ayong, D. E. Kyle, Y. Yuan, R. Manetsch and D. Chakrabarti, Int. J. Parasitol.: Drugs Drug Resist., 2016, 6, 85–92 Search PubMed.
- M. Jain, S. Nehra, P. C. Trivedi and R. V. Singh, Heterocycl. Commun., 2003, 9, 1 Search PubMed.
- T. S. Patel, J. D. Bhatt, R. B. Dixit, C. J. Chudasama, B. D. Patel and B. C. Dixit, Arch. Pharm., 2019, 352, 1900099 CrossRef PubMed.
- V. R. Karpina, S. S. Kovalenko, S. M. Kovalenko, O. G. Drushlyak, N. D. Bunyatyan, V. A. Georgiyants, V. V. Ivanov, T. Langer and L. Maes, Molecules, 2020, 25, 4485 CrossRef CAS PubMed.
- T. F. Mabasa, B. Awe, D. Laming and H. H. Kinfe, Med. Chem., 2019, 15, 685–692 CrossRef CAS PubMed.
- J. N. Domínguez, C. León, J. Rodrigues, N. Gamboa de Domínguez, J. Gut and P. J. Rosenthal, Farm., 2005, 60, 307–311 CrossRef PubMed.
- S. Leeza Zaidi, S. M. Agarwal, P. Chavalitshewinkoon-Petmitr, T. Suksangpleng, K. Ahmad, F. Avecilla and A. Azam, RSC Adv., 2016, 6, 90371–90383 RSC.
- A. I. Marealle, D. P. Mbwambo, W. P. Mikomangwa, M. Kilonzi, H. J. Mlyuka and R. F. Mutagonda, Malar. J., 2018, 17, 409 CrossRef PubMed.
- M. Sinha, V. R. Dola, P. Agarwal, K. Srivastava, W. Haq, S. K. Puri and S. B. Katti, Bioorg. Med. Chem., 2014, 22, 3573–3586 CrossRef CAS PubMed.
- M. V. Papadopoulou, W. D. Bloomer, H. S. Rosenzweig, S. R. Wilkinson, J. Szular and M. Kaiser, Eur. J. Med. Chem., 2016, 117, 179–186 CrossRef CAS PubMed.
- D. J. Lowes, W. A. Guiguemde, M. C. Connelly, F. Zhu, M. S. Sigal, J. A. Clark, A. S. Lemoff, J. L. Derisi, E. B. Wilson and R. K. Guy, J. Med. Chem., 2011, 54, 7477–7485 CrossRef CAS PubMed.
- R. Sudhakar, N. Adhikari, S. Pamnani, A. Panda, M. Bhattacharjee, Z. Rizvi, S. Shehzad, D. Gupta and P. S. Sijwali, Microbiol. Spectrum, 2022, 10, 3 Search PubMed.
- C. L. Yeates, J. F. Batchelor, E. C. Capon, N. J. Cheesman, M. Fry, A. T. Hudson, M. Pudney, H. Trimming, J. Woolven, J. M. Bueno, J. Chicharro, E. Fernández, J. M. Fiandor, D. Gargallo-Viola, F. Gómez de las Heras, E. Herreros and M. L. León, J. Med. Chem., 2008, 51, 2845–2852 CrossRef CAS PubMed.
- R. P. Brueckner, B. G. Schuster, K. C. Lasseter and E. T. Lin, Am. J. Trop. Med. Hyg., 1998, 58, 645–649 CrossRef CAS PubMed.
- F. Salas, J. Fichmann, G. K. Lee, M. D. Scott and P. J. Rosenthal, Infect. Immun., 1995, 63, 2120–2125 CrossRef CAS PubMed.
- P. Rosenthal, Emerging Infect. Dis., 1998, 4, 49–57 CrossRef CAS PubMed.
- S. C. Stolze, E. Deu, F. Kaschani, N. Li, B. I. Florea, K. H. Richau, T. Colby, R. A. L. van der Hoorn, H. S. Overkleeft, M. Bogyo and M. Kaiser, Chem. Biol., 2012, 19, 1546–1555 CrossRef CAS PubMed.
- J. Olson, Bioorg. Med. Chem., 1999, 7, 633–638 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- T. Huang, J. Sun, Q. Wang, J. Gao and Y. Liu, Molecules, 2015, 20, 16221–16234 CrossRef CAS PubMed.
- C. Lambros and J. P. Vanderberg, J. Parasitol., 1979, 65, 418 CrossRef CAS PubMed.
- P. S. Sijwali and P. J. Rosenthal, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4384–4389 CrossRef CAS PubMed.
- W. Chen, Z. Huang, W. Wang, F. Mao, L. Guan, Y. Tang, H. Jiang, J. Li, J. Huang, L. Jiang and J. Zhu, Bioorg. Med. Chem., 2017, 25, 6467–6478 CrossRef CAS PubMed.
- R. Pignatello, G. Spampinato, V. Sorrenti, L. Vicari, C. Di Giacomo, A. Vanella and G. Puglisi, Pharm. Pharmacol. Commun., 1999, 5, 299–305 CrossRef CAS.
- J. D. Bhatt, C. J. Chudasama and K. D. Patel, Arch. Pharm., 2017, 350, 1700088 CrossRef PubMed.
- K. Pal, M. K. Raza, J. Legac, A. Rahman, S. Manzoor, S. Bhattacharjee, P. J. Rosenthal and N. Hoda, Eur. J. Med. Chem., 2023, 248, 115055 CrossRef CAS PubMed.
- M. F. A. do Nascimento, T. F. Borgati, L. C. R. de Souza, C. A. Tagliati and A. B. de Oliveira, Toxicol. Appl. Pharmacol., 2020, 401, 115074 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04370g |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |