
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiodegradation of hydroxylated boron nitride nanoplatelets, their toxic effect and drug delivery application†
Kanwal Asifabc,
Md. Mahbubur Rahman d,
Andrea Augusto Sfrisoe,
Salvatore Parisia,
Vincenzo Canzonieriaf,
Isabella Caligiuria,
Flavio Rizzolio*ab and
Muhammad Adeel*bg
d,
Andrea Augusto Sfrisoe,
Salvatore Parisia,
Vincenzo Canzonieriaf,
Isabella Caligiuria,
Flavio Rizzolio*ab and
Muhammad Adeel*bg
aPathology Unit, Centro di Riferimento Oncologico di Aviano (C.R.O.) IRCCS, 33081, Aviano, Italy
bDepartment of Molecular Sciences and Nanosystems, Ca' Foscari University of Venice, 30172, Venice, Italy. E-mail: flavio.rizzolio@unive.it; Fax: +39-041 2348594; Tel: +39-041 2348910
cCentre for Endocrinology, Charterhouse Square, Queen Mary University of London, London EC1M 6BQ, UK
dDepartment of Energy Material Science & Engineering, Konkuk University, Chungju 27478, Republic of Korea
eDepartment of Chemical and Pharmaceutical Sciences, University of Ferrara, Italy
fDepartment of Medical, Surgical and Health Sciences, University of Trieste, 34149, Trieste, Italy
gDepartment of Bioengineering, Royal School of Mines, Imperial College London, London, SW7 2AZ, UK. E-mail: m.adeel@imperial.ac.uk; Fax: +39-041 2348594; Tel: +39-041 2348910
First published on 22nd August 2024
Abstract
Boron nitride is extensively used in various biomedical applications and often interacting with the blood circulatory system. However, the effect of its biotransformation in blood plasma, drug delivery applications, and antitumor effects remains unclear. Herein, we synthesized hydroxylated BN nanoplatelets (–OH/BNNPs) that are used to load doxorubicin (DOX) for cancer therapy. The stability of the –OH/BNNPs was tested in a lab-made, artificially developed, in vivo system for up to sixty days at two different pH values (pH 5.5 & 7.4). The results were compared thoroughly with pristine BN, and it is observed that –OH/BNNPs was very stable for up to two months compared to pristine BN that degraded during the next day. The –OH functionalization on the BNNP surface improves the DOX loading compared to the bulk BN since the –OH functional group facilitates drug absorption through hydrogen bonding. This causes the sustained release of the drug, which is an ideal requirement in drug delivery systems. The DOX-loaded –OH/BNNPs showed excellent therapeutic abilities on different cancer cell lines and organoids derived from colorectal cancer patients.
Introduction
Boron nitride (BN), also called white graphite, is drawing significant attention in various fields of science (e.g., medicine, chemistry, photovoltaics, electronics etc.) because of its versatile physicochemical properties.1–4 It is widely used in biomedical applications, including tissue engineering, bio-imaging, sensors, and specifically for cancer diagnostic and therapeutic applications.5–7 Several reports have been documented using BN of different shapes (rods, sheets) and sizes for cancer therapy. However, delivering BN into the blood circulatory system presents challenges related to their long-term stability (biotransformation), cytotoxic effects, drug delivery efficiency, and targeted therapy.Generally, upon entering the bloodstream, nanomaterials undergo alterations in their properties or undergo bio-transformation or biodegradation.8 These transformations happen when materials react with blood plasma containing small molecules like free radicals, peroxidase, small organic molecules, and other active components.9 This often affects the potency of the therapeutic system via the accumulation of nanobodies in vivo and causes uncertain health risks.8 Consequently, several reports have studied the stability of nanomaterials in blood plasma. For example, the stability of organic polymeric materials (poly-lactic acid (PLA) and poly-methyl-methacrylate (PMMA)) has been tested in blood plasma and other biosynthetic physiological solutions driving to the conclusion that PLA nanoparticles (aggregated in spleen homogenate and in gastric juice) are less stable compared to PMMA.10 In another study, McEnnis et al. utilized polystyrene nanoparticles that were incubated in goat blood plasma. They conducted an analysis using a nanoparticle tracking analysis (NTA) system.11 The research findings indicated that polystyrene nanoparticles coated with polyethylene glycol (PEG) exhibited reduced levels of multicomponent aggregation compared to uncoated polystyrene nanoparticles.11 As well as for organic nanomaterials, several stability tests of inorganic nanomaterials have also been tested. For example, Zhang et al. tested graphene-based nanomaterials stability in blood plasma, their biocompatibility, and antitumoral ability.8 Lim et al. studied the interaction of graphene oxide (GO) of different lateral size distributions with human blood plasma proteins. The authors found a correlation of lateral size dependent distributions and accumulation of nanoparticles with proteins.12 Several other stability tests of inorganic materials have also been done like on magnetite nanoparticles that are being used mostly for drug delivery and imaging applications. Garcia et al. synthesized different sizes of magnetite nanoparticles and incubated them into blood plasma at different time points. Several techniques were employed to observe the stability of the nanoparticles and it was observed that silica and chitosan-coated iron oxide (Fe3O4) nanoparticles were more stable compared to the uncovered magnetite nanoparticles.13 Apart from all these materials, boron-based compounds (e.g., boric acid, bulk BN, and their composites) have a special place in in vivo biology applications (therapy, sensing, and imaging applications)14 and the long-term stability of boron compounds have rarely been investigated. Boron-based compounds are recognized as self-therapeutic agents that demonstrate a preference for releasing boron specifically at tumoral sites.14 This property allows them to capture neutrons from these sites, a process utilized in the well-established standard neutron capture therapy.15 Previously, studies have indicated a notably rapid release of boron from the source materials such as boric acid and bulk BN.16 To improve the slow release of the boron from the source, BN is considered as the main source.16 It is because BN can be tuned into different sizes and shapes (boron nitride nano flasks, boron nitride nanotubes and boron nitride nanosheets etc.) to improve the stability and achieve the optimum outcomes for in vivo biology.17 These different shapes not only help for the sustained release of boron but also acquire predominant therapeutic outcomes.17 For example, Golberg et al. synthesized hollow BN nanospheres as a boron reservoir for prostate cancer therapy.16 The crystalline boron sphere releases boron in a controlled manner and decrease the tumor growth. The same group synthesized BN nanospheres with petal-like surfaces and loaded them with DOX for cancer therapy. They suggested the petal-like surface of the BN nanospheres can have the tendency to absorb more drugs on the surface and be able to have enhanced therapeutic outcomes.18 The two-dimensional (2D) nanomaterials are considered potential matrices to maximize the drug loading induced by their high surface-to-volume ratio.19–22 Recently, Weng et al. developed –OH functionalized 2D boron nitride nanosheets (BNNSs) for the Camptothecin (CPT) delivery. The authors claim that –OH groups enhanced the attachment of CPT and increased the dispersibility of the BNNSs. They have tested the stability of the materials for up to two days and claim that the materials are stable enough to use it as a cargo for CPT.23 Although –OH functionalization is generally effective in attaching drugs to cargo surfaces, there is still much to discover in terms of its long-term stability, cytotoxicity, and therapeutic outcomes. Herein, we synthesized –OH functionalized BN nanoplatelets (–OH/BNNPs) by a simple chemical oxidation of bulk BN. The synthesized –OH/BNNPs were employed for loading DOX and exhibited optimal therapeutic efficacy on different cancerous cell lines. The stability of the particles was tested up to 60 days at two distinct pH conditions (pH 5.5 & pH 7.4) and it was observed that –OH/BNNPs were stable compared to the bulk BN.
Materials and methods
Materials
Hexagonal BN (h-BN) powder, sulfuric acid (98%), potassium permanganate (KMnO4), hydrogen peroxide (H2O2), fetal bovine serum (FBS), ascorbic acid (AA), and phosphate buffer saline pH 7.4 tablets were bought from Sigma-Aldrich (St. Louis, Missouri, USA). Ultrapure water was used throughout the experiments using a Millipore Milli-Q Biocell A10 water purifying system. A2780, A2780cis (Sigma-Aldrich, St. Louis, MO, USA), and LNCaP (ATCC, Manassas, VA, USA) cells were seeded as per instruction provided by the manufacturers. CellTiter-Glo® was purchased from Promega (Madison, WI, USA).Instrumentations
To characterize the crystal structure of the synthesized –OH/BNNPs, an X-ray diffractometer (XRD, Philips) with the Cu Kα radiation (λ = 1.5406 Å) was used. Fourier transform infrared (FTIR) and Raman spectra were measured using an FTIR (MIDAC, M4000) and Raman spectrophotometer (Horiba Scientific, Xplora Plus, France), respectively, where the excitation wavelength was 532 nm for Raman spectra measurements. To observe the composition of chemicals, their oxidation states, and the bonding characteristics, X-ray photoelectron spectra (XPS) were measured by an XPS analyzer (XPS, Thermo Scientific K-Alpha, Thermo Fisher Scientific, UK). UV-Visible spectrophotometer (UV-Vis-31 Scan, ONDA, Modena, Italy) was used to quantify the optical absorption spectra of the samples. A field emission scanning electron microscope (FE-SEM, Carl Zeiss Sigma VP, Germany) and a high-resolution transmission electron microscope (HR-TEM, JEM-2100 (HRP)) were utilized for morphological characterization. Malvern Zetasizer machine was used to measure dynamic light scattering of pristine h-BN and –OH/BNNPs. The selected area diffraction pattern (SAED) was observed by HRTEM. To identify the IC50 values, luminescence measurements were conducted using the Tecan Infinite M1000 system (Tecan, Mannedorf, Switzerland). IHC staining was carried out using UltraVision LP Detection System HRP DAB kit (Thermo Scientific, Waltham).Synthesis of –OH/BNNPs
The –OH/BNNPs were synthesized according to our previously reported method.4 Briefly, 4 g of pristine h-BN powder was added into 100 mL of H2SO4 (98%) solution and stirred for 30 min at room temperature (RT). Then, 2 g of KMnO4 was slowly added into the solution at 0 °C and stirred for another 12 h at RT. Subsequently, 10 mL of H2O2 (30% w/w) was added and stirred for another 5 h at RT. Then, the suspended solid was collected by filtration using filter paper, and the filtrate was dispersed in water by sonication. After separating the large particles by settling quickly (around 2 minutes), the smaller dispersed particles were collected by centrifugation at 3000 rpm for 10 min. The obtained –OH/BNNPs were sequentially dispersed in water and centrifuged multiple times (for better washing) until the solution pH reached 7.0. Finally, the as-prepared –OH/BNNPs were dried in a vacuum oven at 40 °C for 24 h.Long-term stability tests of –OH/BNNPs
The long-term stability test of the –OH/BNNPs was performed by simulating the in vivo system.24 Briefly, 0.5 g of –OH/BNNP powder was dispersed into PBS solution with 5% FBS at two different pH (7.4 and 5.5). The –OH/BNNPs were dispersed properly by sonication and stored in an incubator at 37 °C with gentle shaking. On a weekly base, the samples were taken out from the incubator and spin down at 1000 rpm for 2 minutes to separate the powder from the supernatant, followed by drying in a vacuum oven at 40 °C for 24 h. All the dried samples were stored at −20 °C for further characterization. The stability of the samples at different day intervals (D0, D30 and D60) was examined at two different pH conditions (5.5 and 7.4) and characterized by XRD, Raman and FTIR spectroscopy.DOX loading in –OH/BNNPs
To load DOX into the –OH/BNNPs, 1 mL of DOX solution (2 mg mL−1) was mixed with 10 mg mL−1 of –OH/BNNPs. Then the mixture was kept overnight with rotation (on a gentle shaking) in the dark at RT. Afterwards, the samples were centrifuged (2000 rpm for 5 min) and washed with DI water to remove the free DOX.25 The loading efficiency of DOX on –OH/BNNPs was evaluated using a specific absorption wavelength at 480 nm with a UV-Vis spectrophotometer.Loading efficiency (LE) was calculated via eqn (1):
| LE (%) = [(total amount of drug − free drug in supernatant)/total amount of drug] × 100 | (1) |
DOX release test
In the DOX release experiments, 500 μL of DOX-OH/BNNPs and free DOX samples were added in 0.5 mL activated mini dialysis membrane (Cole-Parmer Instrument LTD, UK) in two different beakers containing PBS at respective pH values of 7.4 (representing normal physiological solution) and 5.0 (representing acidic tumor microenvironment), 100 rpm shaken at RT. At fixed time intervals, 20 μL of sample was collected (from inside the membrane), and the released DOX was evaluated by UV-spectrophotometer using a specific absorption wavelength at 480 nm.Drug release (DR) was calculated via eqn (2):26
| DR (%) = 100 − [(concentration at point T interval/concentration at point different T0)] × 100 | (2) |
Cell viability assay
To evaluate the cytotoxic effect of DOX-OH/BNNPs and free DOX, different cell lines, including A2780, A2780 cis and LNCaP were seeded in 96-well at the density of 1 × 103 per well. Afterwards, cells were treated with six concentrations (0.3–0.008 μM) of DOX-OH/BNNPs, only –OH/BNNPs, and free DOX. Next, cell viability was measured after 96 hours of treatment using the CellTiter-Glo® assay system in accordance with the manufacturer's instruction (Promega, Madison, WI, USA) using a Tecan M1000 instrument (Tecan, Mannedorf, Switzerland). IC50 values were analyzed from a non-linear regression analysis performed with GraphPad Prism software.Organoid isolation and culture
The patient-derived tumour organoids (PDTO) were obtained for research purposes from completely anonymous CRC specimens collected under a signed biobank informed consent at the National Cancer Institute (CRO) of Aviano, Italy. Organoids were produced following the protocol described by.27–29 Briefly, tumor tissues were digested with collagenase I at a concentration of 2 mg mL−1 for 30 min, the tissue suspension underwent centrifugation at 1500 rpm for 5 min. Following this, the resulting pellet was then resuspended in a Cultrex growth factor-reduced Basement Membrane Extract (BME), Type2 (R&D Systems Catalog no: 3533-001-02, Milan, Italy) and cultured in 24 well plates. Then, 450 μL of the organoid medium was added following the solidification of the hydrogel.27–29Toxicity assay on PDTO
To investigate the cytotoxic effect of DOX-OH/BNNPs, PDTO was seeded with 10 μl of Cultrex type 2 on a 96 multi-wells plate and treated with six different concentrations of DOX-OH/BNNPs and free DOX for 96 hours ranging concentrations from 34.5 μM to 0.011 μM with dilution factor 5. CellTiter-Glo®Luminescence assay (Promega, Madison, WI, US) was used to assessed cytotoxicity with a M1000 instrument (Tecan, Switzerland). IC50 values were analyzed by using GraphPad Prism software, a non-linear regression method was employed.Histopathological analysis of CRC PDTO
Formalin-fixed paraffin-embedded sections of tumor organoids were employed for histopathological analysis. The process involved collecting and fixing the organoids in phosphate-buffered 10% formalin, followed by embedding them in paraffin using a Micro NextGen Cell Blocking™ Kit (Cat no: M20; AV Bioinnovation) as per guidelines provided by the manufacturer. Haematoxylin and eosin (H&E) staining was performed on 5 μm sections using a Leica ST5020 multistainer. Additionally, sections of 2 μm were cut for immunohistochemistry (IHC) staining. A heat-induced antigen retrieval method and an UltraVision LP Detection System HRP DAB kit (ThermoFisher Scientific, Waltham, MA, USA) were used to perform IHC. To characterize PDTO, the following antibodies were utilized: Muc2 (Catalog no: ab76774, dilution 1: 100, Abcam, Cambridge, UK), CK20 (Catalog no: 790-4431, dilution 1.9 μg mL−1, Ventana Medical Systems, Arizona, US), CDX2 (Catalog no: ab76541, dilution 1.100, Abcam, Cambridge, UK). Immunohistochemistry images were taken using a light microscope at different magnifications.Statistical analysis
Statistical analysis was achieved with a two-tailed t-test conducted through GraphPad Prism 8.0 software. For all data, p < 0.05 were considered statistically significant.Results and discussion
Synthesis and mechanisms of reactions involving –OH/BNNPs
The chemical process of –OH/BNNPs formation, aimed at achieving enhanced –OH functionalization, is anticipated to occur through the following mechanism, as schematically shown in Fig. 1. Initially, when H2SO4 was added to the h-BN powder, the H+ ions intercalated into the h-BN layers through the naturally present N atoms in the h-BN structure.30,31 This causes an increase in the interlayer spacing between h-BN sheets (0.33 nm), which allows the intercalation of higher radius SO42− ions (0.258 nm) between the h-BN sheets and causes further expansion of the sheets.30 With the subsequent addition of KMnO4, the intercalation of K+ (rK+ = 0.138 nm) and MnO4− (rMnO4− = 0.229 nm) further enhanced the sheet expansion and exfoliated the h-BN. During this process, KMnO4 is easily oxidized and forms MnO2, which can be easily decorated onto the h-BN sheets.32 The following addition of H2O2 to remove MnO2, induced the formation of ˙OH radicals in solution. These reactive ˙OH radicals exhibit a propensity to readily target the B atom within the B–N bond located at the edges of BNNPs. This induces homolytic cleavage of the B–N bond, resulting in the formation of a covalent B–OH bond alongside a N˙ radical. Consequently, this process initiates the opening of the hexagonal ring structure of BNNPs and cutting the BN sheets into smaller sheets. Simultaneously, the layers of BNNPs undergo cleavage facilitated by –OH functionalization, driven by a subsequent ˙OH attack at the naturally occurring ˙B site on the basal plane.33 These also enabled additional cutting of h-BN sheets into smaller sheets and formed BNNPs with reduced size and thickness. Notably, the N˙ generated post –HO˙ attack at the B site within the B–N bond can be promptly deactivated by bonding with H+ in an aqueous solution.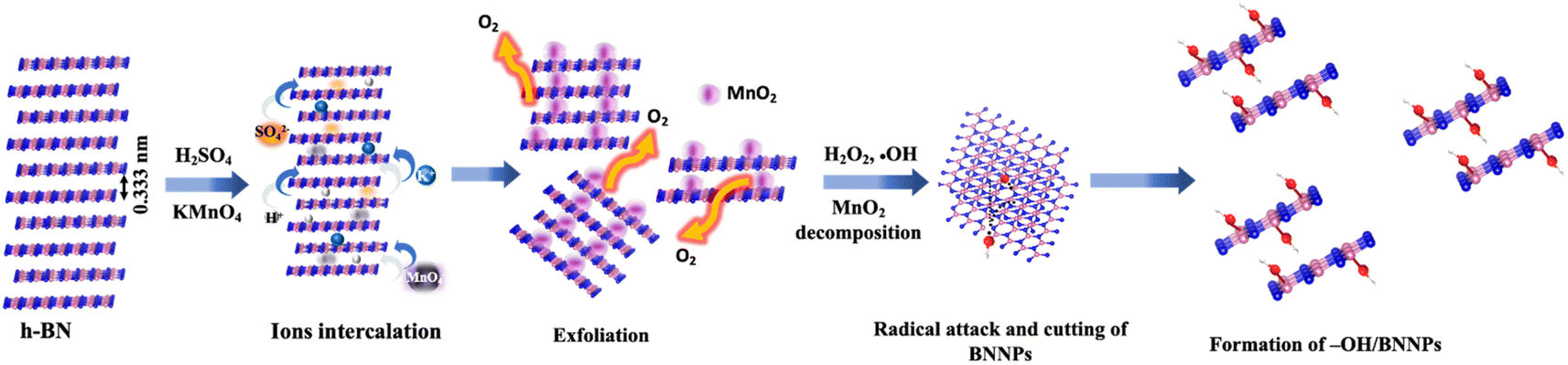 | ||
| Fig. 1 Schematic representation of the synthesis mechanism of –OH/BNNPs. | ||
Morphological analysis
Fig. 2a and b show the FE-SEM images of the pristine h-BN and –OH/BNNPs, respectively. It was clearly observed that bulk h-BN exhibited an agglomerated nature with higher particle size and thickness. In contrast, the particle size and thickness were reduced after exfoliation and functionalization of h-BN. The sizes of the h-BN and –OH/BNNPs were about 357.6 nm and 247.7 nm, respectively, which was measured by the dynamic light scattering (DLS) analysis (Fig. S1†). | ||
| Fig. 2 (a) and (b) are the FE-SEM images of the pristine h-BN and –OH/BNNPs, respectively. (c) HR-TEM image of the –OH/BNNPs and (d) the corresponding SAED pattern. | ||
The morphology and size of the –OH/BNNPs were further analyzed by HR-TEM measurements. The semi-transparency behaviour of –OH/BNNPs to the electron beam indicates the reduction in size and exfoliation of h-BN4 (Fig. 2c and S2†). The SAED pattern of the –OH/BNNPs exhibits the bright concentric rings of (002) and (100) planes (Fig. 1d), indicating the hexagonal phase of the BNNPs with high crystallinity.4,31
Structural analysis
Fig. 3(a) shows the XRD pattern in the 2θ angle range from 20° to 80° of bulk h-BN and as-synthesized –OH/BNNPs. The prominent high-intensity peak was observed for both bulk h-BN and –OH/BNNPs, demonstrating their high purity and crystallinity.4,31,34 The main diffraction peak of h-BN appears at 2θ of 26.72° along with other characteristics diffraction peaks at 41.68° and 54.97°. These diffraction peaks correspond to (002), (100), and (004) crystallographic planes of hexagonal BN, respectively.4,32,34 Due to the functionalization, exfoliation, and size reduction, the (002) diffraction peak of –OH/BNNPs was shifted to lower angles with 2θ values of 26.64° (Fig. 3(b)). This slight shifting in diffraction peak is because of the expansion of the unit cell volume induced by the –OH functionalization at the basal plane and edges of BN. The full width at half maximum (FWHM) was ca. 0.62° for –OH/BNNPs, and ca. 0.42° for bulk h-BN. This increase in FWHM of (002) diffraction plane for –OH/BNNPs can be attributed to the reduction of the thickness/size of bulk h-BN with successful –OH functionalization.32,35 To further characterize the structure and functional groups of –OH/BNNPs, Raman spectra was measured, as shown in Fig. 3(c). The magnified Raman spectra in the wavenumber range starting from 1300 to 1450 cm−1 exhibit the E2g phonon mode and B–N bond vibration. The E2g band of h-BN and –OH/BNNPs was located at 1383.82 cm−1 and 1378.26 cm−1, respectively. This shift in the E2g band of –OH/BNNPs indicates the reduction in size and successful –OH functionalization.36,37 Fig. 3(d) shows the FTIR spectra of the h-BN and –OH/BNNPs. Both spectra exhibited the same absorption bands at 1372 and 817 cm−1, corresponding to the B–N stretching and B–N bending vibrations, respectively. However, the –OH band of –OH/BNNPs exhibited a strong absorption at 3415.8 cm−1 compared to the bulk BN. This prominent decrease in transmittance of the –OH band in –OH/BNNPs confirms the successful –OH functionalization.38,39 Fig. S3† shows the UV-Visible absorption spectra of h-BN and –OH/BNNPs. The spectra clearly show the shifting of the absorption maxima (λmax) from 205.42 nm for h-BN to 211.58 nm for –OH/BNNPs, which is well-matched with the reported literature.35,40,41 Furthermore, upon exfoliation, cutting, and –OH functionalization of h-BN, the surface area (SABET) was increased from 23.24 m2 g−1 to 27.85 m2 g−1. The corresponding N2 adsorption isotherms of –OH/BNNPs and bulk h-BN are shown in Fig. S4.†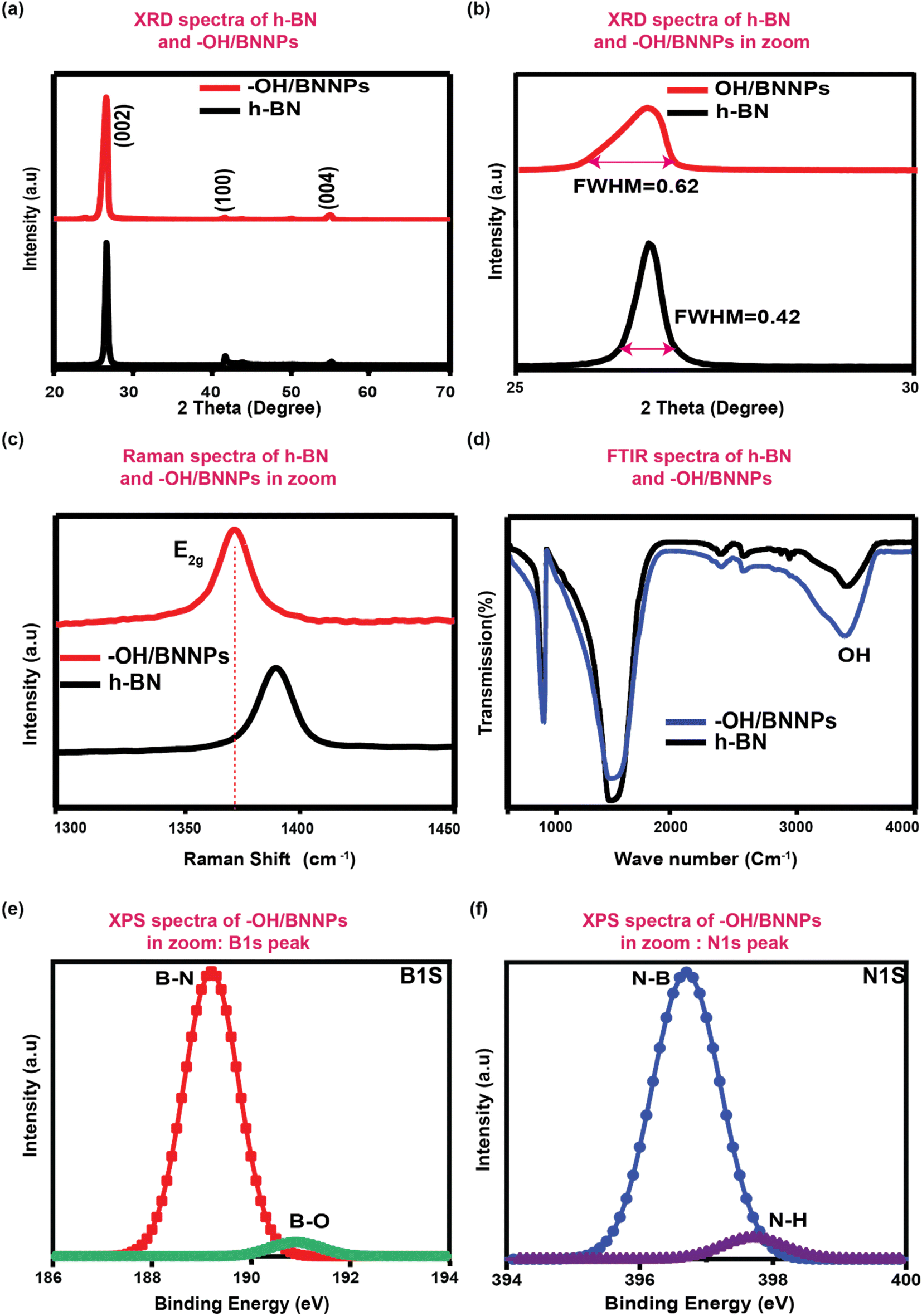 | ||
| Fig. 3 (a) XRD pattern of the bulk h-BN and –OH/BNNPs and (b) magnified XRD spectra. (c) Raman spectra of bulk h-BN and –OH/BNNPs. (d) FTIR spectra of bulk h-BN and –OH/BNNPs. (e) and (f) are the high-resolution XPS spectra of B 1s and N 1s in –OH/BNNPs, respectively. | ||
The XPS was measured to confirm the elemental composition and functional groups, and the corresponding survey XPS spectra of –OH/BNNPs are presented in Fig. S5.† The spectrum shows the B, N, and O element peaks in their reported binding energy positions. The additional Si and carbon peaks might have originated from the substrate/stage and ubiquitous carbon contamination, respectively. Fig. 3(e) and (f) show the high-resolution B 1s and N 1s spectra of –OH/BNNPs, respectively. The deconvoluted and fitted core-level B 1s spectra showed two peaks at 189.99 eV and 190.89 eV, which can be attributed to the B–N bonding and B–O binding, respectively. The appearance of the B–O bonding peak can be ascribed to the covalent grafting of oxygen functional groups in BNNPs. The fitted core-level O 1s spectra showed two peaks at the binding energies of 396.69 eV and 397.68 eV. The former peak can be ascribed to the B–O bond and later can be attributed to the N–H. The appearance of the N–H bonding indicates the HO˙ radicals attack at the B sites of BN and the deactivation of N˙ radical formation. These results confirmed the effective functionalization of BN with –OH functional groups.
Long-term stability at pH 7.4 and 5.5
The long-term stability of the –OH/BNNPs and h-BN was investigated in a solution similar to artificial body-like conditions at two different pHs (7.4 and 5.5), and the details of the development of body-like conditions are presented in the experimental section. For analyses, the samples were collected after treatment at each day interval for up to 60 days. Fig. 4(a)–(c) show the HR-TEM images of the –OH/BNNP samples after treatment at pH 7.4 with D0, D30, and D60, respectively, and Fig. 4 (d)–(f) display the HR-TEM images of the –OH/BNNP samples after treatment at pH 5.5 with D0, D30, and D60, respectively. It is evident that the –OH/BNNPs stay highly stable for up to 60 days without significant structural deterioration at both pH conditions. While bulk h-BN degraded completely after D30 and D60 when treated at both pH 7.4 and 5.5 (Fig. 4(g)–(i) and 4(j)–(l), respectively). | ||
| Fig. 4 HR-TEM images of –OH/BNNPs after treatment (a–c) at pH 7.4 (D0–D60) and (d–f) at pH 5.5 (D0–D60). HR-TEM images of h-BN after treatment (g–i) at pH 7.4 (D0–D60) and (j–l) at pH 5.5 (D0–D60). | ||
The structural stability of h-BN and –OH/BNNPs was assessed further by FTIR and XRD measurements. Fig. 5(a) and (b) show the FTIR spectra of the –OH/BNNPs after treatment at pH 7.4 and 5.5 at different day intervals. It was observed that the spectra were perfectly overlapped after treatment for 30 days and 60 days with the spectra of freshly prepared-OH/BNNPs without the appearance of any impurity peaks or decomposition products. This is consistent with the HR-TEM analyses. In contrast, the FTIR spectra of h-BN after treatment for 30 days and 60 days at pH 7.4 and 5.5 exhibited new IR bands by demising the prominent B–N stretching and bending vibrations at 1372 and 817 cm−1, respectively (Fig. 5(c) and (d), respectively), the comparison of the peaks of bulk h-BN and OH/BNNPs are highlighted in yellow colour. This result suggests the degradation or decomposition of h-BN, consistent with the HR-TEM analyses. However, the decomposed or degraded products of h-BN after the treatment are not identified clearly, which requires further depth spectroscopic analyses. Similar to the HR-TEM and FTIR analyses, the XRD pattern of –OH/BNNPs after treatment at pH 7.4 and 5.5 for 60 days showed that high intensity (002) plane peak remains unchanged considering the intensity and peak position (Fig. 5(e) and (f)). On the contrary, the XRD pattern of h-BN showed the complete disappearance of the (002) plane peak at 26.72° after 60 days at both pHs with a new high-intensity peak in the range between 17 and 19° (Fig. 5(e) and (f)). This further suggests the structural change of h-BN or its decomposition induced by the treatment in artificial body-like conditions at pH 7.4 and 5.5. Therefore, –OH/BNNPs can be considered as the best platform to deliver therapeutic compounds inside the body for sustainable drug delivery applications.
 | ||
| Fig. 5 FTIR spectra of –OH/BNNPs after treatment (a) at pH 7.4 (D0–D60) and (b) pH 5.5 (D0–D60). FTIR spectra of h-BN after treatment (c) at pH 7.4 (D0–D60) and (d) pH 5.5 (D0–D60). XRD pattern of –OH/BNNPs and h-BN after treatment (e) at pH 7.4 (D60) and (f) pH 5.5 (D60). | ||
–OH/BNNPs for sustained delivery of DOX
To investigate the loading efficiency of DOX onto the –OH/BNNPs, UV-Vis spectra of DOX were used to measure the supernatant after –OH/BNNPs -DOX overnight rotation. The encapsulation efficiency of DOX was 12% with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio of DOX and –OH/BNNPs formulation.
:5 ratio of DOX and –OH/BNNPs formulation.
The release profiles of DOX from –OH/BNNPs in PBS at different pH values are shown in Fig. 6(a). Compared to free DOX, the release from –OH/BNNPs is controlled over time. Although the profiles are similar, the release of DOX from –OH/BNNPs increased gradually to reach 99% at pH 5.0 while 94% at pH 7.4 after 72 hours. This result was due to the DOX protonation, DOX amino group underwent protonation at a lower pH, leading to an enhanced hydrophilicity of DOX. Consequently, the bond between DOX and the hydrophobic –OH/BNNPs surface was diminished, leading to the eventual release of DOX.25 Nevertheless, our results show the sustained release of DOX from –OH/BNNPs, which could be due to the OH functionalization of BNNP holding DOX due to its NH2 group and cause sustained release of DOX from BNNP over time.42–44
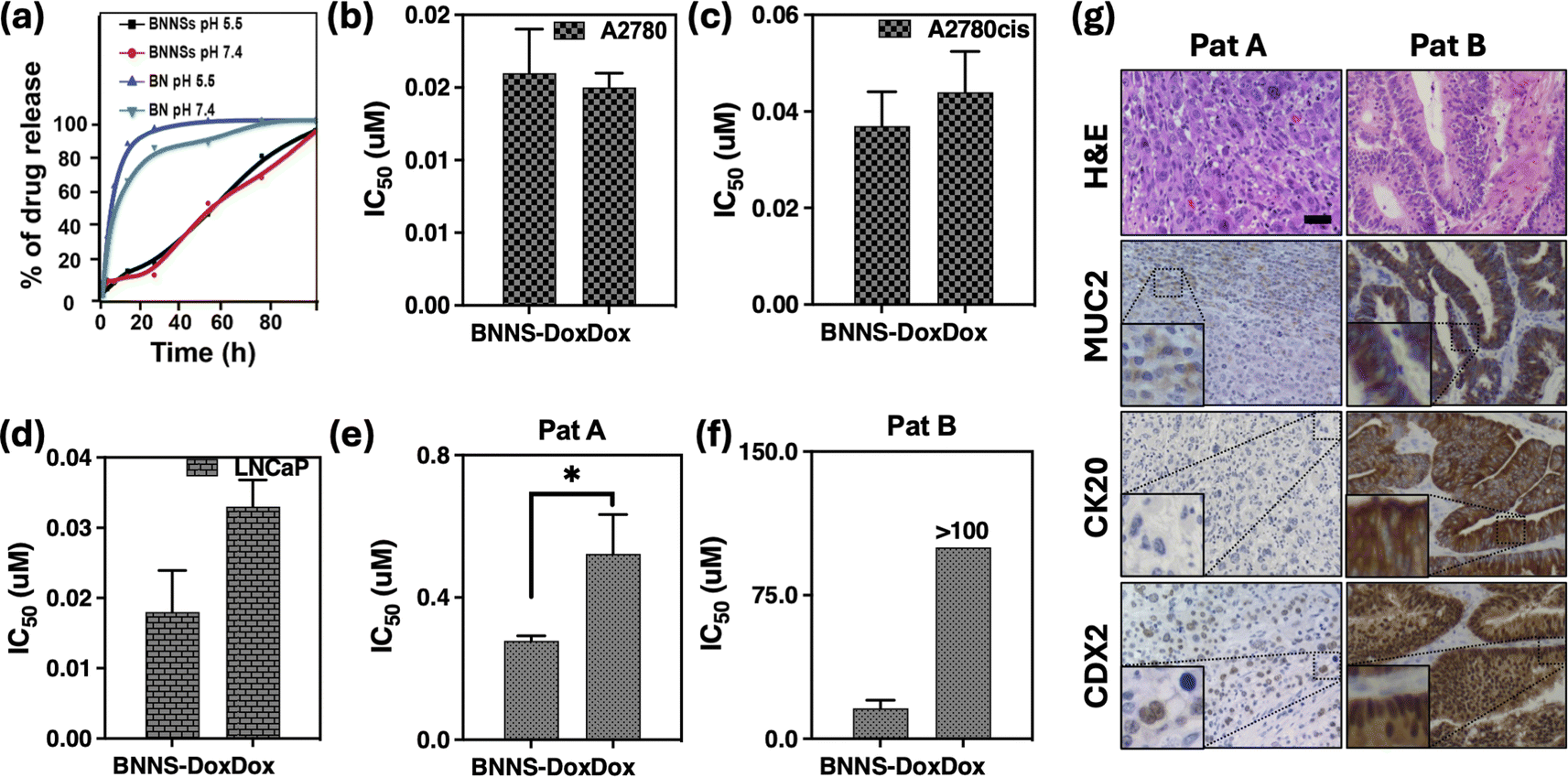 | ||
| Fig. 6 (a) Release test of –OH/BNNPs-DOX and free DOX at pH 5.5 and pH 7.4. (b–d) Cytotoxic evaluation. IC50 values of –OH/BNNPs-DOX and free DOX toward cancer cells. Values are expressed in μM. (e and f) Cytotoxicity of –OH/BNNPs-DOX and free DOX on CRC patients (Pat A and B). IC50 values are expressed as μM. (g) Immunohistochemistry and hematoxylin & eosin staining of tumor tissues from PAT A and B. Muc2 (Mucin 2), CK20 (cytokeratin 20), CdX2 (Caudal-type homeobox 2) markers of CRC. IHC images were at 20×, scale bar indicated 50 μm. | ||
Cytotoxicity assay of DOX –OH/BNNPs
Furthermore, to evaluate the cytotoxicity of DOX loaded in –OH/BNNPs, different cancerous cell lines (A2780, A2780cis, LNCaP) were treated with –OH/BNNPs-DOX and free DOX for 96 h. Both –OH/BNNPs-DOX and free DOX exhibit cytotoxicity on all cancer cell lines. Additionally, the DOX-loaded BNNPs exhibit a tendency of increased toxicity compared to free DOX, although this difference is not statistically significant in the A2780cis and LNCaP cell lines, as depicted in Fig. 6c and d. This result illustrates that incorporating smaller amounts of drug into these innovative –OH/BNNPs can result in increased potency for reducing cancer cell viability. One reasonable explanation for this phenomenon is that –OH/BNNPs facilitates the DOX accumulation inside cancer cells compared to free DOX taking advantage of the endocytosis process.43,45 Once taken up, the DOX would be activated and released within living cells in response to the acidic conditions of their aqueous environments in lysosomes, which typically have a pH lower than 5.Anticancer activity of –OH/BNNPs-DOX on human colorectal cancer organoids
Colorectal cancer (CRC) ranks as the third most diagnosed cancer worldwide, resulting in approximately one million deaths annually.46,47 The standard treatment options for CRC typically include chemotherapy, utilizing agents such as 5-fluorouracil, irinotecan, and oxaliplatin,48–50 alongside surgical intervention.51 Notably, advancements in chemotherapeutic regimens have contributed to an improvement in the overall survival rate among CRC patients over the past decade.52,53 However, the development of drug resistance remains a significant challenge, often resulting in chemotherapy failure54,55To assess the efficacy of –OH/BNNPs containing DOX in ex vivo models, we employed organoids derived from CRC patients. Numerous reports have demonstrated the application of organoids for evaluating the drug toxicity. This approach provides an alternative method to overcome the clinical challenges and achieve substantial results that replicate the observed patient response.24,27–29,46,47,56 We produced organoids originating from CRC patients to evaluate the efficacy of encapsulated DOX in –OH/BNNPs. As shown in Fig. 6(e) and (f), the IC50 values of –OH/BNNPs-DOX are in the range of 0.2–14 μM, in comparison to 0.4 – >100 μM of free DOX. These data demonstrate that encapsulated DOX in –OH/BNNPs exhibit higher potency and the potential to overcome the chemoresistance issue comparatively to the free DOX, which could be due to the –OH/BNNPs ability to retain DOX and facilitate gradual release over time.
To explore the molecular similarity between organoids and the parental tumor, an immunohistochemical analysis was conducted, revealing immunopositivity for Muc2 (Mucin 2), CK20 (cytokeratin 20) and CdX2 (Caudal-type homeobox 2) which are markers of CRC (Fig. 6g, S6†).
Conclusions
The long-term biotransformation study of nanoparticles is crucial for assessing risks in a clinical setting. It plays a vital role in bridging the gap between wet bench research and clinical application. Here in this research, we studied the long-term stability of the hydroxylated boron nitride nanoparticles for up to two months, at two different pH conditions (5.5 and 7.4) by simulating an in vivo system. The results were compared with pristine BN and it was observed that the hydroxylated BN is very stable for up to two months. The system was used to load the DOX and it was observed that the drug was absorbed on the surface due to the high surface-to-volume ratio of the material. Additionally, the drug interaction was facilitated through hydrogen bonding on the surface (through hydroxylated BN surface and naturally present OH functional groups in DOX structure) and showed a sustained release profile compared to the pristine BN. Furthermore, the system showed excellent therapeutic abilities on different cancer cell lines and CRC-PDTO. The findings of this study unveil an exciting prospect for evaluating the long-term risk assessment of cargos prior to their utilization in the healthcare system.Disclaimer
This manuscript has been edited for grammar and syntax using ChatGPT, an AI language model developed by OpenAI. While efforts have been made to improve the clarity and accuracy of the language, the final content and scientific interpretations are the sole responsibility of the authors and their collaborators. ChatGPT has been utilized solely to enhance readability and expression, without generating any data.Data availability
The data supporting the findings of this study are available within the paper, ESI† files and from the corresponding authors upon reasonable request. Source of data are provided with this paper.Author contributions
K. A.; M. A. involved in the conceptualization, experimental data and writing; Md. M. R.; I. C.; A. A. S., S. P. participated in the data analysis, review & editing; V. C. contributed in the biological samples, review & editing, funding acquisition; F. R. was participated in the data analysis, review & editing. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the Italian Ministry of Health, Ricerca Corrente.References
- V. Vatanpour, S. A. Naziri Mehrabani, B. Keskin, N. Arabi, B. Zeytuncu and I. Koyuncu, A Comprehensive Review on the Applications of Boron Nitride Nanomaterials in Membrane Fabrication and Modification, Ind. Eng. Chem. Res., 2021, 60, 13391–13424, DOI:10.1021/ACS.IECR.1C02102/ASSET/IMAGES/MEDIUM/IE1C02102_0021.GIF.
- A. Merlo, V. R. S. S. Mokkapati, S. Pandit and I. Mijakovic, Boron nitride nanomaterials: biocompatibility and bio-applications, Biomater. Sci., 2018, 6, 2298–2311, 10.1039/C8BM00516H.
- S. Roy, X. Zhang, A. B. Puthirath, A. Meiyazhagan, S. Bhattacharyya and M. M. Rahman, et al., Structure, Properties and Applications of Two-Dimensional Hexagonal Boron Nitride, Adv. Mater., 2021, 33, 2101589, DOI:10.1002/ADMA.202101589.
- M. Adeel, M. M. Rahman and J. J. Lee, Label-free aptasensor for the detection of cardiac biomarker myoglobin based on gold nanoparticles decorated boron nitride nanosheets, Biosens. Bioelectron., 2019, 126, 143–150, DOI:10.1016/J.BIOS.2018.10.060.
- A. E. Naclerio and P. R. Kidambi, A Review of Scalable Hexagonal Boron Nitride (h-BN) Synthesis for Present and Future Applications, Adv. Mater., 2023, 35, 2207374, DOI:10.1002/ADMA.202207374.
- X. F. Jiang, Q. Weng, X. B. Wang, X. Li, J. Zhang and D. Golberg, et al., Recent Progress on Fabrications and Applications of Boron Nitride Nanomaterials: A Review, J. Mater. Sci. Technol., 2015, 31, 589–598, DOI:10.1016/J.JMST.2014.12.008.
- P. Saxena, A. K. Gupta, V. Saharan and H. Harish, Toxicity of boron nitride nanoparticles influencing bio-physicochemical responses in freshwater green algae, Environ. Sci. Pollut. Res., 2023, 30, 23646–23654, DOI:10.1007/S11356-022-23912-X/FIGURES/4.
- D. Li, X. Hu and S. Zhang, Biodegradation of graphene-based nanomaterials in blood plasma affects their biocompatibility, drug delivery, targeted organs and antitumor ability, Biomaterials, 2019, 202, 12–25, DOI:10.1016/J.BIOMATERIALS.2019.02.020.
- I. Lynch, C. Weiss and E. Valsami-Jones, A strategy for grouping of nanomaterials based on key physico-chemical descriptors as a basis for safer-by-design NMs, Nano Today, 2014, 9, 266–270, DOI:10.1016/J.NANTOD.2014.05.001.
- S. Lazzari, D. Moscatelli, F. Codari, M. Salmona, M. Morbidelli and L. Diomede, Colloidal stability of polymeric nanoparticles in biological fluids, J. Nanopart. Res., 2012, 14, 1–10, DOI:10.1007/S11051-012-0920-7.
- M. S. Bannon, A. López Ruiz, K. Corrotea Reyes, M. Marquez, Z. Wallizadeh and M. Savarmand, et al., Nanoparticle Tracking Analysis of Polymer Nanoparticles in Blood Plasma, Part. Part. Syst. Charact., 2021, 38, 2100016, DOI:10.1002/PPSC.202100016.
- L. K. P. Kenry and C. T. Lim, Molecular interactions of graphene oxide with human blood plasma proteins, Nanoscale, 2016, 8, 9425–9441, 10.1039/C6NR01697A.
- S. E. Favela-Camacho, J. F. Pérez-Robles, P. E. García-Casillas and A. Godinez-Garcia, Stability of magnetite nanoparticles with different coatings in a simulated blood plasma, J. Nanopart. Res., 2016, 18, 1–9, DOI:10.1007/S11051-016-3482-2/FIGURES/6.
- M. Adeel, F. Duzagac, V. Canzonieri and F. Rizzolio, Self-Therapeutic Nanomaterials for Cancer Therapy: A Review, ACS Appl. Nano Mater., 2020, 3, 4962–4971, DOI:10.1021/ACSANM.0C00762/ASSET/IMAGES/LARGE/AN0C00762_0005.JPEG.
- K. Nedunchezhian, N. Aswath, M. Thiruppathy and S. Thirugnanamurthy, Boron Neutron Capture Therapy - A Literature Review, J. Clin. Diagn. Res., 2016, 10, ZE01–ZE04, DOI:10.7860/JCDR/2016/19890.9024.
- X. Li, X. Wang, J. Zhang, N. Hanagata, X. Wang and Q. Weng, et al., Hollow boron nitride nanospheres as boron reservoir for prostate cancer treatment, Nat. Commun., 2017, 8, 1–12, DOI:10.1038/ncomms13936.
- Q. Weng, X. Wang, X. Wang, Y. Bando and D. Golberg, Functionalized hexagonal boron nitride nanomaterials: emerging properties and applications, Chem. Soc. Rev., 2016, 45, 3989–4012, 10.1039/C5CS00869G.
- I. V. Sukhorukova, I. Y. Zhitnyak, A. M. Kovalskii, A. T. Matveev, O. I. Lebedev and X. Li, et al., Boron Nitride Nanoparticles with a Petal-Like Surface as Anticancer Drug-Delivery Systems, ACS Appl. Mater. Interfaces, 2015, 7, 17217–17225, DOI:10.1021/ACSAMI.5B04101/ASSET/IMAGES/MEDIUM/AM-2015-041013_0014.GIF.
- H. Zhang, T. Fan, W. Chen, Y. Li and B. Wang, Recent advances of two-dimensional materials in smart drug delivery nano-systems, Bioact. Mater., 2020, 5, 1071–1086, DOI:10.1016/J.BIOACTMAT.2020.06.012.
- J. Ouyang, S. Rao, R. Liu, L. Wang, W. Chen and W. Tao, et al., 2D materials-based nanomedicine: from discovery to applications, Adv. Drug Delivery Rev., 2022, 185, 114268, DOI:10.1016/J.ADDR.2022.114268.
- A. Jayakumar, S. Mathew, S. Radoor, J. T. Kim, J. W. Rhim and S. Siengchin, Recent advances in two-dimensional nanomaterials: properties, antimicrobial, and drug delivery application of nanocomposites, Mater. Today Chem., 2023, 30, 101492, DOI:10.1016/J.MTCHEM.2023.101492.
- M. J. Molaei, Two-dimensional (2D) materials beyond graphene in cancer drug delivery, photothermal and photodynamic therapy, recent advances and challenges ahead: a review, J. Drug Delivery Sci. Technol., 2021, 61, 101830, DOI:10.1016/J.JDDST.2020.101830.
- Y. Cheng, Y. Han, W. Zhang, L. Zeng, Y. Long and S. Wang, et al., Gram-scale synthesis of boron nitride nanosheets by salt-template method for anticancer drug delivery, Chem. Eng. J., 2022, 437, 135304, DOI:10.1016/J.CEJ.2022.135304.
- M. Adeel, S. Parisi, M. Mauceri, K. Asif, M. Bartoletti and F. Puglisi, et al., Self-Therapeutic Cobalt Hydroxide Nanosheets (Co(OH)2NS) for Ovarian Cancer Therapy, ACS Omega, 2021, 6, 28611–28619, DOI:10.1021/ACSOMEGA.1C03010/ASSET/IMAGES/LARGE/AO1C03010_0006.JPEG.
- S. Feng, H. Zhang, T. Yan, D. Huang, C. Zhi and H. Nakanishi, et al., Folate-conjugated boron nitride nanospheres for targeted delivery of anticancer drugs, Int. J. Nanomed., 2016, 11, 4573–4582, DOI:10.2147/IJN.S110689.
- S. Varghese, J. P. Chaudhary and C. Ghoroi, One-step dry synthesis of an iron based nano-biocomposite for controlled release of drugs, RSC Adv., 2020, 10, 13394–13404, 10.1039/D0RA01133A.
- K. Asif, M. Adeel, M. M. Rahman, I. Caligiuri, T. Perin and M. Cemazar, et al., Iron nitroprusside as a chemodynamic agent and inducer of ferroptosis for ovarian cancer therapy, J. Mater. Chem. B, 2023, 11, 3124–3135, 10.1039/D2TB02691K.
- K. Asif, M. Adeel, M. M. Rahman, A. A. Sfriso, M. Bartoletti and V. Canzonieri, et al., Silver nitroprusside as an efficient chemodynamic therapeutic agent and a peroxynitrite nanogenerator for targeted cancer therapies, J. Adv. Res., 2024, 56, 43–56, DOI:10.1016/J.JARE.2023.03.005.
- K. Asif, M. Adeel, M. Mahbubur Rahman, M. Bartoletti, S. K. Brezar and M. Cemazar, et al., Copper nitroprusside: an innovative approach for targeted cancer therapy via ROS modulation, Biomed. Pharmacother., 2024, 171, 116017, DOI:10.1016/J.BIOPHA.2023.116017.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun and A. S. Slesarev, et al., Correction to Improved Synthesis of Graphene Oxide, ACS Nano, 2018, 12, 2078, DOI:10.1021/ACSNANO.8B00128.
- W. Zhang, M. M. Rahman, F. Ahmed, N. S. Lopa, C. Ge and T. Ryu, et al., A two-step approach for improved exfoliation and cutting of boron nitride into boron nitride nanodisks with covalent functionalizations, Nanotechnology, 2020, 31, 425604, DOI:10.1088/1361-6528/AB9A76.
- H. R. Barai, M. M. Rahman, A. Rahim and S. W. Joo, α-MnO2 nanorod/boron nitride nanoplatelet composites for high-performance nanoscale dielectric pseudocapacitor applications, J. Ind. Eng. Chem., 2019, 79, 115–123, DOI:10.1016/J.JIEC.2019.06.009.
- T. Sainsbury, A. Satti, P. May, Z. Wang, I. McGovern and Y. K. Gun'ko, et al., Oxygen radical functionalization of boron nitride nanosheets, J. Am. Chem. Soc., 2012, 134, 18758–18771, DOI:10.1021/JA3080665/SUPPL_FILE/JA3080665_SI_001.PDF.
- M. Du, Y. Wu and X. Hao, A facile chemical exfoliation method to obtain large size boron nitride nanosheets, CrystEngComm, 2013, 15, 1782–1786, 10.1039/C2CE26446C.
- Z. Cui, A. J. Oyer, A. J. Glover, H. C. Schniepp and D. H. Adamson, Large Scale Thermal Exfoliation and Functionalization of Boron Nitride, Small, 2014, 10, 2352–2355, DOI:10.1002/SMLL.201303236.
- F. Xiao, S. Naficy, G. Casillas, M. H. Khan, T. Katkus and L. Jiang, et al., Edge-Hydroxylated Boron Nitride Nanosheets as an Effective Additive to Improve the Thermal Response of Hydrogels, Adv. Mater., 2015, 27, 7196–7203, DOI:10.1002/ADMA.201502803.
- Q. Weng, X. Wang, C. Zhi, Y. Bando and D. Golberg, Boron nitride porous microbelts for hydrogen storage, ACS Nano, 2013, 7, 1558–1565, DOI:10.1021/NN305320V/SUPPL_FILE/NN305320V_SI_001.PDF.
- D. Kong, D. Zhang, H. Guo, J. Zhao, Z. Wang and H. Hu, et al., Functionalized Boron Nitride Nanosheets/Poly(l-lactide) Nanocomposites and Their Crystallization Behavior, Polymers, 2019, 11, 440, DOI:10.3390/POLYM11030440.
- L. Zhai, Z. Liu, C. Li, X. Qu, Q. Zhang and G. Li, et al., Cyanate ester resin based composites with high toughness and thermal conductivity, RSC Adv., 2019, 9, 5722–5730, 10.1039/C8RA10244A.
- G. Gao, A. Mathkar, E. P. Martins, D. S. Galvão, D. Gao and P. Alves Da Silva Autreto, et al., Designing nanoscaled hybrids from atomic layered boron nitride with silver nanoparticle deposition, J. Mater. Chem. A, 2014, 2, 3148–3154, 10.1039/C3TA12892J.
- L. Wu, K. Wu, C. Lei, D. Liu, R. Du and F. Chen, et al., Surface modifications of boron nitride nanosheets for poly(vinylidene fluoride) based film capacitors: advantages of edge-hydroxylation, J. Mater. Chem. A, 2019, 7, 7664–7674, 10.1039/C9TA00616H.
- E. S. Permyakova, L. Y. Antipina, A. M. Kovalskii, I. Y. Zhitnyak, K. Y. Gudz and J. Polčak, et al., Experimental and Theoretical Study of Doxorubicin Physicochemical Interaction with BN(O) Drug Delivery Nanocarriers, J. Phys. Chem. C, 2018, 122, 26409–26418, DOI:10.1021/ACS.JPCC.8B07531/ASSET/IMAGES/MEDIUM/JP-2018-07531C_0009.GIF.
- Q. Weng, B. Wang, X. Wang, N. Hanagata, X. Li and D. Liu, et al., Highly water-soluble, porous, and biocompatible boron nitrides for anticancer drug delivery, ACS Nano, 2014, 8, 6123–6130, DOI:10.1021/NN5014808.
- J. Ren, L. Stagi and P. Innocenzi, Hydroxylated boron nitride materials: from structures to functional applications, J. Mater. Sci., 2020, 56, 4053–4079, DOI:10.1007/S10853-020-05513-6.
- A. Farahani, M. Jamshidi and M. Foroutan, Improving thermal/electrical properties of silicone rubber nanocomposite using exfoliated boron nitride nano sheets made by an effective/novel exfoliating agent, Mater. Des., 2023, 229, 111935, DOI:10.1016/J.MATDES.2023.111935.
- C. Su, K. A. Olsen, C. E. Bond and V. L. J. Whitehall, The Efficacy of Using Patient-Derived Organoids to Predict Treatment Response in Colorectal Cancer, Cancers, 2023, 15, 805, DOI:10.3390/CANCERS15030805/S1.
- T. Kiwaki and H. Kataoka, Patient-Derived Organoids of Colorectal Cancer: A Useful Tool for Personalized Medicine, J. Pers. Med., 2022, 12, 695, DOI:10.3390/JPM12050695.
- G. Argilés, J. Tabernero, R. Labianca, D. Hochhauser, R. Salazar and T. Iveson, et al., Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up, Ann. Oncol., 2020, 31, 1291–1305, DOI:10.1016/J.ANNONC.2020.06.022.
- S. Xiong and G. W. Xiao, Reverting doxorubicin resistance in colon cancer by targeting a key signaling protein, steroid receptor coactivator, Exp. Ther. Med., 2018, 15, 3751–3758, DOI:10.3892/ETM.2018.5912.
- T. A. Debele, C. K. Chen, L. Y. Yu and C. L. Lo, Lipopolyplex-Mediated Co-Delivery of Doxorubicin and FAK siRNA to Enhance Therapeutic Efficiency of Treating Colorectal Cancer, Pharmaceutics, 2023, 15, 596, DOI:10.3390/PHARMACEUTICS15020596/S1.
- T. Matsuda, K. Yamashita, H. Hasegawa, T. Oshikiri, M. Hosono and N. Higashino, et al., Recent updates in the surgical treatment of colorectal cancer, Ann. Gastroenterol. Surg., 2018, 2, 129–136, DOI:10.1002/AGS3.12061.
- M. Buunen, R. Veldkamp, W. C. Hop, E. Kuhry, J. Jeekel, E. Haglind, L. Påhlman, M. A. Cuesta, S. Msika, M. Morino, A. Lacy and H. J. Bonjer, Survival after laparoscopic surgery versus open surgery for colon cancer: long-term outcome of a randomised clinical trial, Lancet Oncol., 2009, 10, 44–52, DOI:10.1016/S1470-2045(08)70310-3.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Cancer drug resistance: an evolving paradigm, Nat. Rev. Cancer, 2013, 13, 714–726, DOI:10.1038/NRC3599.
- L. A. Diaz, R. T. Williams, J. Wu, I. Kinde, J. R. Hecht and J. Berlin, et al., The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers, Nature, 2012, 486, 537–540, DOI:10.1038/NATURE11219.
- J. A. Engelman, K. Zejnullahu, T. Mitsudomi, Y. Song, C. Hyland and O. P. Joon, et al., MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling, Science, 2007, 316, 1039–1043, DOI:10.1126/SCIENCE.1141478.
- T. Matsui and T. Shinozawa, Human Organoids for Predictive Toxicology Research and Drug Development, Front. Genet., 2021, 12, 767621, DOI:10.3389/FGENE.2021.767621/BIBTEX.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04433a |
| This journal is © The Royal Society of Chemistry 2024 |