
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, biological evaluation, and in silico studies of novel N-substituted-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamides as promising anticancer agents†
Navid Dastyafteh‡
a,
Manica Negahdaripour‡ bc,
Mohammad Hosein Sayahid,
Mina Emamib,
Younes Ghasemibc,
Elham Safaeie,
Homa Azizianf,
Zahra Pakrouh Jahromibc,
Mehdi Asadif,
Mohammad Reza Mohajeri-Tehrania,
Fateme Zareb,
Minoo Shahidig,
Zahra Pooraskarig,
Sayed Mahmoud Sajjadi-Jazia,
Bagher Larijania,
Mohammad Mahdavi*a and
Sara Ranjbar*b
bc,
Mohammad Hosein Sayahid,
Mina Emamib,
Younes Ghasemibc,
Elham Safaeie,
Homa Azizianf,
Zahra Pakrouh Jahromibc,
Mehdi Asadif,
Mohammad Reza Mohajeri-Tehrania,
Fateme Zareb,
Minoo Shahidig,
Zahra Pooraskarig,
Sayed Mahmoud Sajjadi-Jazia,
Bagher Larijania,
Mohammad Mahdavi*a and
Sara Ranjbar*b
aEndocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran. E-mail: momahdavi@tums.ac.ir
bPharmaceutical Sciences Research Center, Shiraz University of Medical Sciences, Shiraz, Iran. E-mail: ranjbar_sa@sums.ac.ir; ranjbar90156@gmail.com
cDepartment of Pharmaceutical Biotechnology, School of Pharmacy, Shiraz University of Medical Sciences, Iran
dDepartment of Chemistry, Payame Noor University, Tehran, Iran
eSchool of Chemistry, College of Science, University of Tehran, Tehran, Iran
fDepartment of Medicinal Chemistry, School of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
gDepartment of Hematology and Blood Transfusion, School of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran
First published on 5th November 2024
Abstract
Novel benzimidazole-based derivatives were synthesized and their cytotoxic activities were evaluated against two human cancer cells, SW480 and A549, and the normal human MRC-5 cells, using the MTT assay. N-(2,4-Dimethoxyphenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5o) showed excellent cytotoxicity with IC50 values of 0.15 ± 0.01 and 3.68 ± 0.59 μM against A549 and SW480. Compound 5o had 38.5-, 62.9- and 3.1-fold superior cytotoxicity than cisplatin (IC50 = 5.77 ± 1.60 μM), etoposide (IC50 = 9.44 ± 1.98 μM), and doxorubicin (IC50 = 0.46 ± 0.02 μM), respectively against A549 cells. Moreover, 5o exhibited high selectivity towards A549 (SI = 794.6) and SW480 (SI = 32.4) cancer cells compared with the normal MRC-5. Further studies revealed the ability of 5o to induce apoptosis and arrest the cell cycle at the S phase in A549 cells. Molecular docking studies revealed 5o was well accommodated within the pocket of topoisomerase IIα-DNA, as a possible target. Molecular dynamics simulation studies confirmed the stability of the 5o-IIα-DNA complex. Compound 5o was predicted to have appropriate drug-likeness and pharmacokinetic properties.
1. Introduction
Cancer is the second biggest public health problem worldwide and accounts for one of every five deaths. The annual number of cancer-related deaths is estimated to increase to 11.4 million in 2030.1 Despite various cancer treatments, there is an urgent need for innovative chemotherapeutic agents due to the resistance and toxicity associated with the currently available drugs. Therefore, many studies have been devoted to the discovery of novel anticancer compounds in our research group.2–7Benzimidazole is a bicyclic nitrogen-containing molecule that has emerged as a privileged pharmacophore in medicinal chemistry due to its diverse biological activities such as antibacterial,8,9 antifungal,10 anti-inflammatory,11 antiviral,12 analgesic,13 anti-oxidant,14 anticonvulsant,15 antitubercular,16 antidiabetic,17 and antimalarial18 activities. Anticancer activity has also been reported for benzimidazoles.19–22 The structural similarity of benzimidazole with natural nitrogenous bases such as purine makes it an indispensable anchor for developing anticancer agents. Besides, benzimidazole provides both hydrogen bond donor and acceptor capacities in one nucleus; consequently, it can bind to different cancer-related targets through hydrogen bonding interactions.23,24 Benzimidazole derivatives exhibit anticancer properties through different mechanisms including the disruption of microtubule polymerization,25 the induction of apoptosis,26 cell cycle arrest,27 anti-angiogenesis,28,29 and inhibition of receptors involved in cancer such as topoisomerases,30 dihydrofolate reductase enzyme,31 tubulin,32 cyclin-dependent kinases,33 epidermal growth factor receptor (EGFR),34 vascular endothelial growth factor receptor (VEGFR),35–38 and so on.
The benzimidazole core is present in the structure of some anticancer drugs, e.g. nocodazole (an antimitotic agent), veliparib (a PARP inhibitor), and dovitinib (a VEGFR inhibitor) as illustrated in Fig. 1. Moreover, the cytotoxic potential of a series of 2-(substituted-phenyl) benzimidazole derivatives had been evaluated by Huynh et al. The results revealed that the presence of electron-donating groups on C-2-phenyl ring caused significant increase of the anticancer activity. The derivative containing a 3,4,5-trimethoxyphenyl group on position 2 of the benzimidazole core (Fig. 1, compound I), displayed superior cytotoxicity over other derivatives against A549 cells with an IC50 value of 11.75 μg mL−1.39 So far, most studies have been devoted to investigating the anticancer potential of benzimidazoles with diverse substitutions on different positions of the imidazole moiety, and C-6-substituted derivatives have been the subject of less studies. Therefore, we aimed to explore the anticancer effect of 2-(substituted-3,4,5-trimethoxyphenyl) benzimidazole derivatives bearing different carboxamides on the C-6 position (Fig. 1).
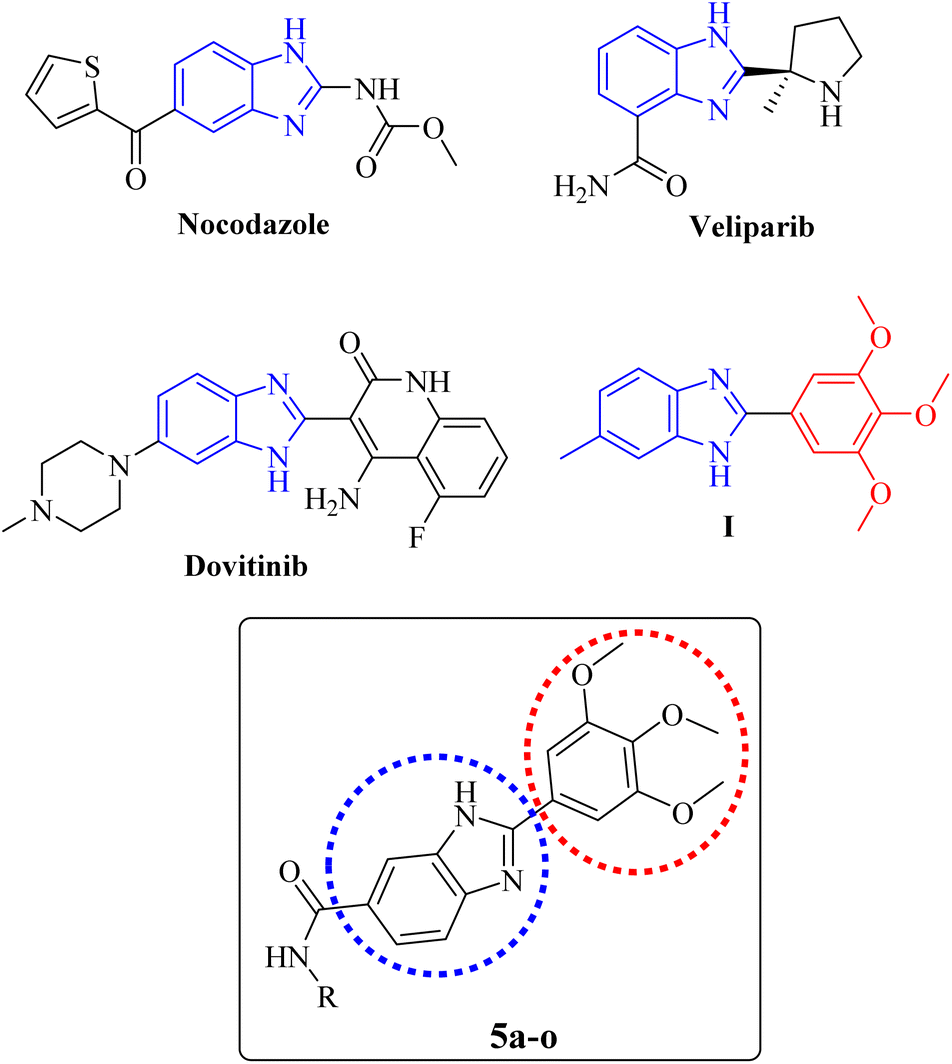 | ||
| Fig. 1 Rational study design, illustrating the structures of benzimidazole-containing anticancer agents and the general structure of the designed 2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamides (5a–o). | ||
In this study, the design and synthesis of a novel series of 2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamides (Fig. 1, 5a–o) as anticancer agents were reported. The compounds were screened for their cytotoxicity against cancer cell lines. Further biological assessments including, cell cycle analysis and apoptosis investigation were conducted for the most potent derivative. Subsequently, in silico studies, including molecular docking, molecular dynamics simulations, and also prediction of drug-likeness and pharmacokinetic properties were carried out.
2. Results and discussion
2.1. Synthesis
The synthetic pathway for new 2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide derivatives (5a–o) are outlined in Scheme 1. The required 2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxylic acid (3) was prepared by the reaction of 3,4,5-trimethoxybenzaldehyde (1) and 3,4-diaminobenzoic acid (2) in the presence of catalytic amount of Na2S2O5 in DMF. The target products (5a–o), were prepared via the reaction of the acquired carboxylic acid (3) with the corresponding substituted amine (4a–o) in DMF in the presence of DIPEA and TBTU as the base and coupling reagent, respectively. The structures of the synthesized derivatives were elucidated by 1HNMR, 13CNMR, as well as IR spectroscopy. The purity of the compounds was confirmed by elemental analyses.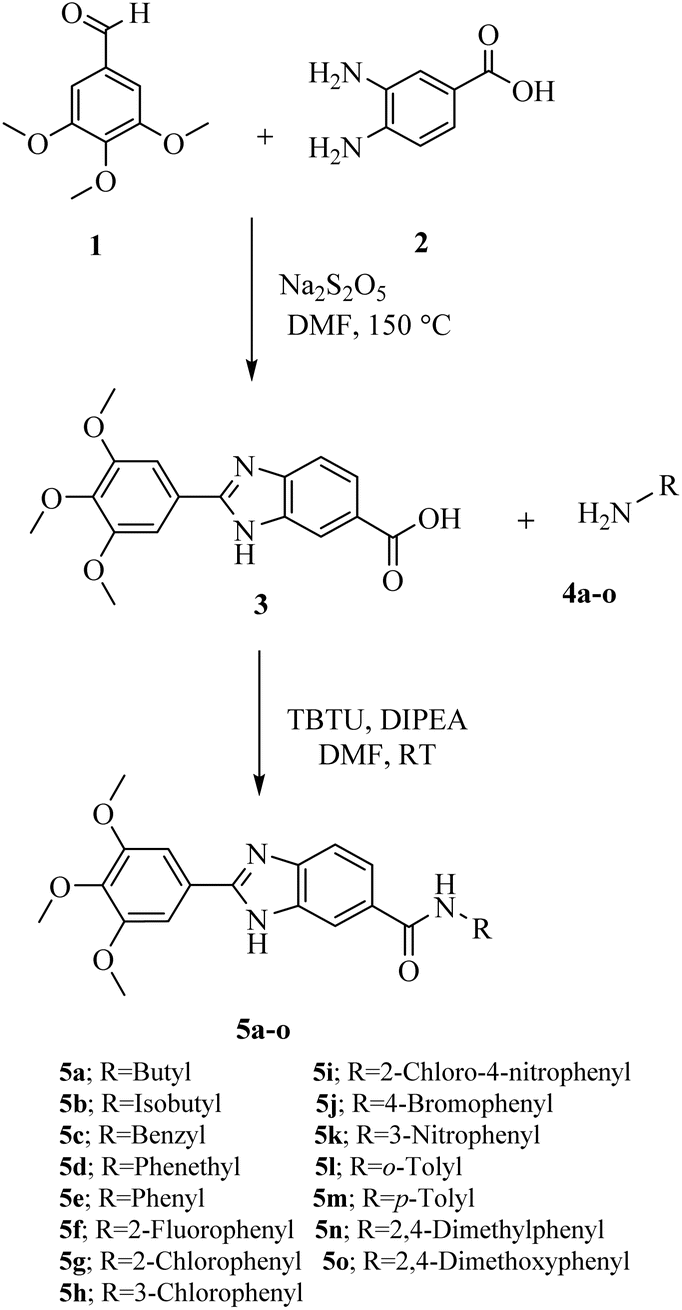 | ||
| Scheme 1 Synthesis of N-substituted-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamides (5a–o). | ||
2.2. In vitro cytotoxic activity
The cytotoxic effect of the synthesized N-substituted-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide derivatives (5a–o) were assessed against SW480 and A549 cancer cell lines and the results are presented in terms of IC50 values in Table 1. Cisplatin, doxorubicin, and etoposide were used as the positive controls.
|
|||
|---|---|---|---|
| Compound | R | IC50 (μM)a | |
| SW480b | A549c | ||
| a Mean ± SD of 3–4 independent replicates.b SW480; primary colon cancer.c A549; adenocarcinomic human alveolar basal epithelial cells. | |||
| 5a | Butyl | 46.12 ± 9.23 | 24.80 ± 5.21 |
| 5b | Isobutyl | 18.75 ± 3.25 | 0.82 ± 0.20 |
| 5c | Benzyl | 34.84 ± 5.37 | 16.33 ± 5.73 |
| 5d | Phenethyl | 18.20 ± 2.69 | 0.66 ± 0.13 |
| 5e | Phenyl | 5.45 ± 0.67 | 0.23 ± 0.05 |
| 5f | 2-Fluorphenyl | 6.38 ± 1.95 | 0.67 ± 0.15 |
| 5g | 2-Chlorophenyl | 49.08 ± 2.84 | 17.26 ± 3.50 |
| 5h | 3-Chlorophenyl | 52.73 ± 2.96 | 27.49 ± 6.78 |
| 5i | 2-Chloro-4-nitrophenyl | 20.21 ± 7.21 | 5.78 ± 4.85 |
| 5j | 4-Bromophenyl | 15.21 ± 2.58 | 13.07 ± 1.27 |
| 5k | 3-Nitrophenyl | 16.75 ± 1.41 | 14.38 ± 3.53 |
| 5l | o-Tolyl | 9.31 ± 0.21 | 0.92 ± 0.39 |
| 5m | p-Tolyl | 12.95 ± 1.20 | 0.52 ± 0.12 |
| 5n | 2,4-Dimethylphenyl | 14.82 ± 1.63 | 1.38 ± 0.23 |
| 5o | 2,4-Dimethoxyphenyl | 3.68 ± 0.59 | 0.15 ± 0.01 |
| Cisplatin | — | 15.21 ± 0.35 | 5.77 ± 1.60 |
| Doxorubicin | — | 0.59 ± 0.07 | 0.46 ± 0.02 |
| Etoposide | — | 16.62 ± 2.30 | 9.44 ± 1.98 |
The derivatives (5a–o) showed IC50 ranges of 0.15–27.49 μM, and 3.68–52.73 μM against A549 and SW480 cell lines, respectively. The majority of compounds had superior antiproliferative activities over cisplatin in both cancer cell lines. Derivatives 5e and 5o showed better IC50 values than doxorubicin against A549 cells. Moreover, the cytotoxic activities of 5f and 5m were comparable to doxorubicin against A549 cells. Compound 5o, bearing a 2,4-dimethoxyphenyl substitution, was the most potent cytotoxic agent with IC50 values of 0.15 ± 0.01 and 3.68 ± 0.59 μM against A549 and SW480, respectively. Compound 5o showed 38.5-, 62.9-, and 3.1-fold higher cytotoxic activities than cisplatin (IC50 = 5.77 ± 1.60 μM), etoposide (IC50 = 9.44 ± 1.98 μM), and doxorubicin (IC50 = 0.46 ± 0.02 μM), respectively against A549 cells. In the case of the SW480 cell line, the cytotoxic activity of 5o was 4.1-and 4.5-fold stronger than cisplatin (IC50 = 15.21 ± 0.35 μM) and etoposide (IC50 = 16.62 ± 2.30). Compound 5e, having a phenyl substitution, was the second-best cytotoxic derivative with IC50 values of 0.23 ± 0.05 and 5.45 ± 0.67 μM against A549 and SW480, respectively.
According to the results, a detailed structure–activity relationship is presented (Fig. 2). Changing linear butyl substitution in 5a to branched isobutyl substitution (as in 5b) significantly enhanced the cytotoxicity against both cancer cells. Insertion of a phenyl substitution (as in 5e) led to a noticeable increase in the activity compared to the aliphatic substituted derivatives 5a and 5b. However, increasing the distance between the amine function and the phenyl ring (as in 5c and 5d) would decrease the cytotoxic effect. Any substituent introduction, except methoxy, on the phenyl ring reduced the activity. Therefore, compound 5o, bearing the methoxy groups at the ortho and para positions of the phenyl ring, was the most cytotoxic compound against the two cancer cell lines. The lipophilic electron-donating methoxy substitutions not only may provide the formation of several hydrophobic interactions with the target but may also participate in the formation of hydrogen bonding interactions through the electronegative oxygen atom as the acceptor. Inserting a chlorine atom on the phenyl ring caused 5g and 5h to have the lowest cytotoxicity compared to other derivatives. Generally, it seems that electronegativity and lipophilicity were the determining factors for the cytotoxic activity of these compounds. In the case of halogen-substituted derivatives, introducing the most electronegative fluorine atom (as in 5f) and the most lipophilic bromine atom (as in 5j) improved the IC50 values compared with the chlorine substitution. Inserting a nitro moiety at the meta position (compound 5k) resulted in superior activity compared to chlorine substitution. In methyl-substituted derivatives (5i, 5m, and 5n), the insertion of two methyl substitutions on the phenyl ring would reduce the activity. Compounds 5h and 5k substituted with 3-Cl and 3-NO2 were among the least active derivatives, therefore, it can be stated that the meta position of phenyl ring was less favorable than the ortho and para positions.
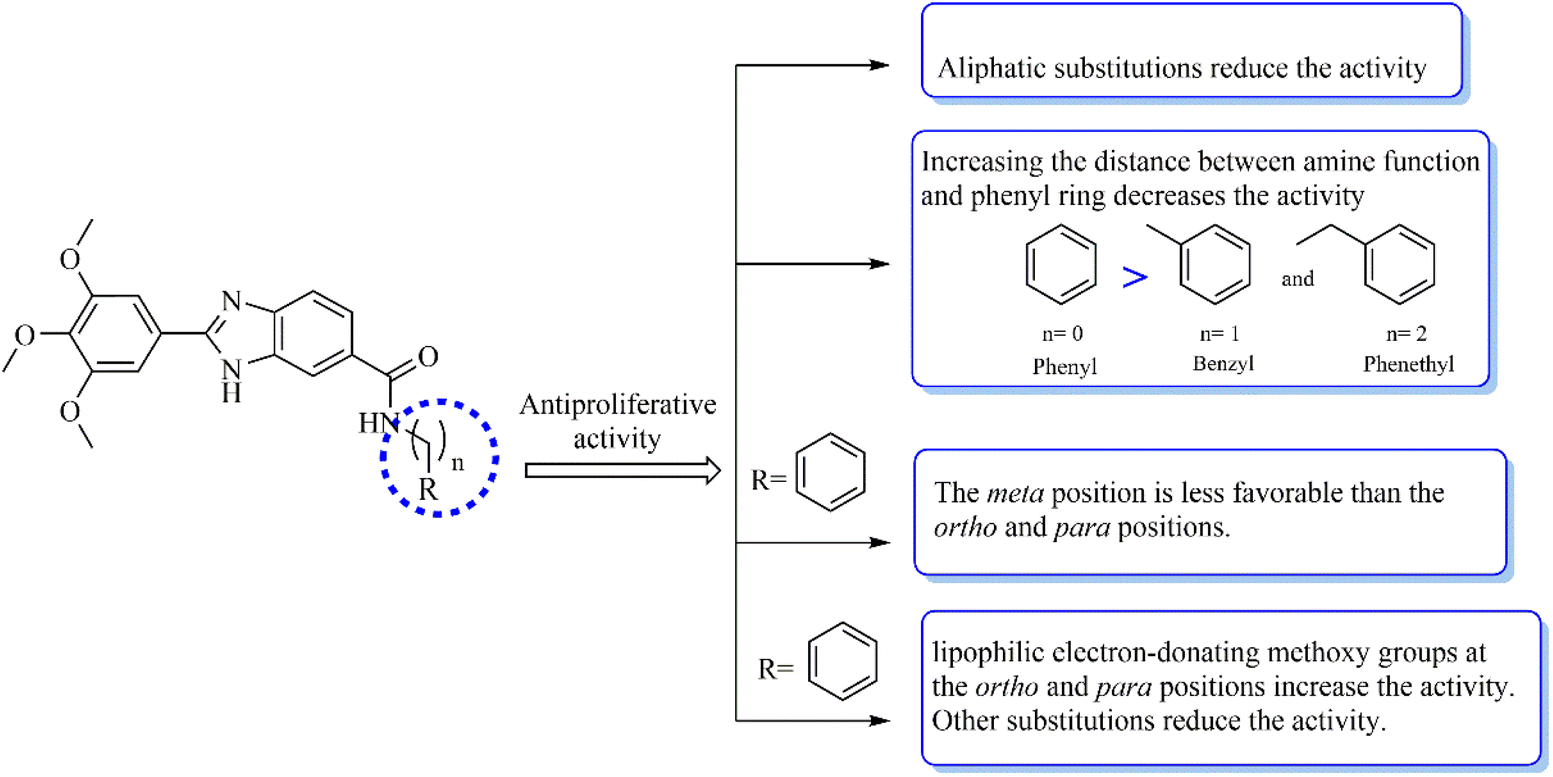 | ||
| Fig. 2 The structure–activity relationship of the synthesized compounds. | ||
The cytotoxic effects of four derivatives showing the most promising cytotoxicity potential against the cancer cell lines, 5e, 5f, 5m, and 5o, were evaluated against the normal human cell line MRC-5, and their selectivity index (SI) was calculated. Cisplatin and doxorubicin were used as the reference drugs. As shown in Table 2, the selected compounds showed greater SI toward A549 cancer cells. Compound 5o was remarkably the most selective derivative toward A549 cells (SI = 794.6) and SW480 (SI = 32.4) cells. The compound had significantly higher cytotoxic selectivity than the positive controls cisplatin and doxorubicin toward cancerous cells over normal cell lines. Compounds 5e, 5f, and 5m presented considerable cytotoxicity against normal MRC-5 cells with IC50 values of 11.55 ± 1.30, 10.58 ± 1.73, and 15.0 ± 1.62 μM, respectively, which are 50.2-, 15.8-, and 28.9-fold higher than those of A549 cells. However, 5e, 5f, and 5m demonstrated poor selectivity toward SW480 cells with SI values of 2.1, 1.7, and 1.2, respectively.
| Compound | aIC50 (μM) MRC-5b | SIc | |
|---|---|---|---|
| SW480 | A549 | ||
| a Values are expressed as mean ± SD of three independent replicates.b MRC-5: human fetal lung fibroblast cells.c SI: selectivity index = IC50 of a compound in a normal cell line/IC50 of the same compound in a cancerous cell line. | |||
| 5e | 11.55 ± 1.30 | 2.1 | 50.2 |
| 5f | 10.58 ± 1.73 | 1.7 | 15.8 |
| 5m | 15.05 ± 1.62 | 1.2 | 28.9 |
| 5o | 119.20 ± 2.46 | 32.4 | 794.6 |
| Cisplatin | 17.16 ± 1.07 | 1.1 | 3.0 |
| Doxorubicin | 0.39 ± 0.06 | 0.7 | 0.8 |
2.3. Cell cycle arrest
The cell cycle analysis was performed against A549 cells by flow cytometry in the presence of compound 5o for 24 h using propidium iodide (PI). Treatment of A549 cells with compound 5o caused significant increases in cell population in the S phase from 18.89% in control to 43.58% and 60.31% at 0.15 and 0.30 μM concentrations of 5o, respectively, with the corresponding reductions in G0/G1 (67.87% in control to 46.44% and 35.16% for 0.15 and 0.30 μM of 5o) and G2/M (13.24% in control to 7.98% and 4.53% for 0.15 and 0.30 μM of 5o) phases (Fig. 3). These results revealed that 5o induced the growth arrest of A549 cells at the S phase of the cell cycle in a dose-dependent manner.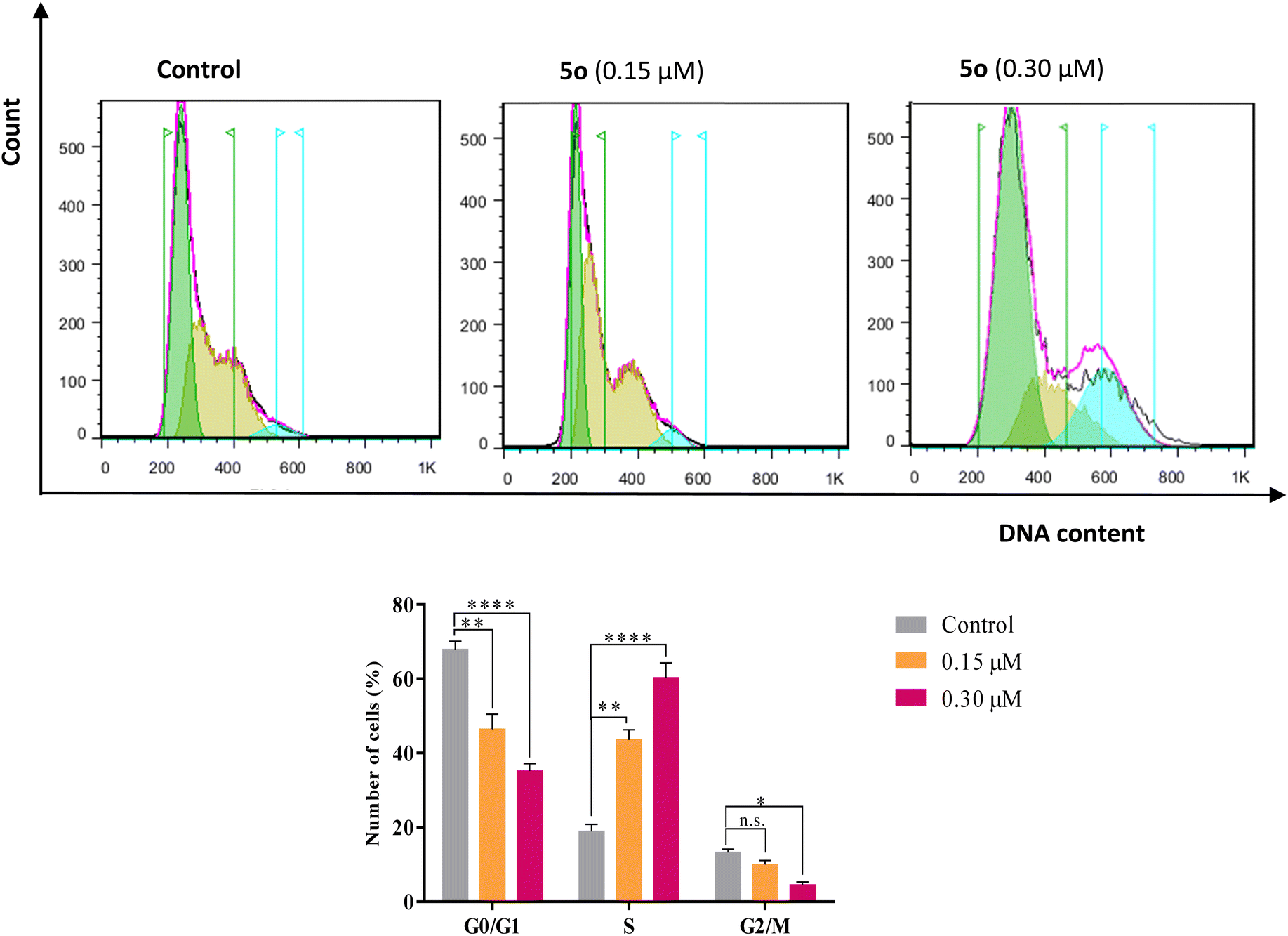 | ||
| Fig. 3 Flow cytometry analysis of cell-cycle progression of A549 cells after treatment with 0.15 and 0.30 μM concentrations of compound 5o for 24 h. Data are the mean ± SD of three independent experiments. n.s.: non-significant. *p < 0.05; **p < 0.01; and ****p < 0.0001 by one-way ANOVA and Tukey's post hoc test to compare the percentages of apoptotic cells with the control cells. | ||
2.4. Apoptosis induction
Apoptosis rates of compound 5o on A549 cells were determined for 24 h by flow cytometry. The Annexin V-fluorescein isothiocyanate (FITC) along with PI were applied as reagents for the detection of apoptosis.40 The staining of A549 cells with Annexin V-FITC and PI is represented by flow cytometry diagrams that indicate the cell death induction pathway.41 The resulting diagrams are shown in Fig. 4. In these diagrams, Q1 (annexin−/PI+), Q2 (annexin+/PI+), Q3 (annexin+/PI-), and Q4 (annexin−/PI−) were defined as necrotic, late apoptotic, early apoptotic, and intact, respectively.42 In the case of DMSO-treated cells (control), the percent of late and early apoptotic cells were 2.4% and 1.8%, respectively. After treatment with 0.15 and 0.30 μM concentrations of 5o, the percentage of late apoptotic cells increased to 5.4% and 39.9%, respectively, while the values of early apoptotic cells were 9.8% and 34.4%. The total apoptosis rates (Q2 + Q3) induced by compound 5o at 0.15 and 0.30 μM concentrations were 15.2% and 74.3%, respectively. Thus, the results support induced apoptosis in A549 cells by compound 5o in a dose-dependent manner. | ||
| Fig. 4 Graphical representation of the effect of compound 5o treatment on induction of apoptosis. Scatter plots were obtained by the flow cytometry analysis of A549 cells treated with 0.15 and 0.30 μM concentrations of 5o for 24 h after double staining with Annexin V-FITC and PI. | ||
2.5. Molecular docking
DNA topoisomerases are a group of enzymes that regulate the topology of DNA and are vital for biological processes, including DNA replication, transcription, recombination, and repair. Topoisomerase inhibitors have become potential anti-cancer medications.43 There are two types of topoisomerases: topoisomerase I (Topo I), which cleaves one of the DNA strands and topoisomerase II (Topo II), which induces breaks in double-stranded DNA. Topo II consists of two functionally different isoforms: Topo IIα and Topo IIβ.44 Topo IIα has a key role in cell division. It is highly expressed in proliferating and cancer cells, consequently, has appeared as a prominent target for anticancer drugs. Topo IIβ plays a key function in transcriptional regulation, cell development, and differentiation. Several topoisomerase inhibitors have been introduced for cancer treatment, such as camptothecin (Topo I inhibitor), doxorubicin (Topo II inhibitor), etoposide (Topo II inhibitor), and mitoxantrone (Topo II inhibitor).45A literature review revealed that topoisomerases can be proposed as possible targets for the anti-cancer benzimidazole derivatives. Bielawski, et al. reported bis-benzimidazoles as Topo I and Topo II inhibitors that interact with the GC base pair at the DNA minor groove.46 A benzimidazole-acridine derivative was reported to inhibit Topo I and promote cell death in K562 cells through the intrinsic apoptotic pathway.47 Li et al. introduced two benzimidazole–rhodanine conjugates as non-intercalative Topo II inhibitors that bind to the ATP-binding site of the Topo II enzyme.48 An antiproliferative benzimidazole hybrid was discovered to inhibit Topo II, arrest the cell cycle at the S phase, and induce apoptosis.49
The molecular docking simulations were performed to predict the binding affinity of 5o with the ATPase domain of Topo II (PDB IDs: 1ZXM and 1ZXN) and the central domain of Topo I-DNA (PDB ID: 1T8I), Topo IIα-DNA (PDB ID: 5GWK), and Topo IIβ-DNA (PDB ID: 4G0V). To validate the docking method, the internal ligands were docked in the corresponding binding site, and the obtained binding energies and root-mean-square deviation (RMSD) values are listed in Table 3. RMSD values (<2.0 Å) proved that the top-ranked conformer of ligands superimposed well over the corresponding X-ray crystallographic one into the binding site of the enzymes. According to the predicted binding energies, derivative 5o preferentially targeted Topo IIα-DNA (ΔG = −9.97 kcal mol−1), and Topo IIβ-DNA (ΔG = −9.06 kcal mol−1) rather than Topo I-DNA (ΔG = −8.73 kcal mol−1), and the ATPase domain of Topo IIα (ΔGs = −7.89 and −7.51 kcal mol−1) (Table 3).
| Topoisomerase | PDB | Co-crystallized ligand | RMSD (Å) | Binding energies (kcal mol−1) | |
|---|---|---|---|---|---|
| Co-crystallized ligand | 5o | ||||
| Human Topo I-DNA | 1T8I | Camptothecin | 0.66 | −10.63 | −8.73 |
| Human Topo IIα ATPase-no DNA | 1ZXM | ANP | 1.63 | −10.64 | −7.89 |
| Human Topo IIα ATPase-no DNA | 1ZXN | ADP | 1.56 | −10.19 | −7.51 |
| Human Topo IIα-DNA | 5GWK | Etoposide | 1.10 | −10.54 | −9.97 |
| Human Topo IIβ-DNA | 4 G0V | Mitoxantrone | 1.14 | −10.40 | −9.06 |
The docking interactions and binding modes of compound 5o and etoposide in complex with Topo IIα-DNA are shown in Fig. 5.
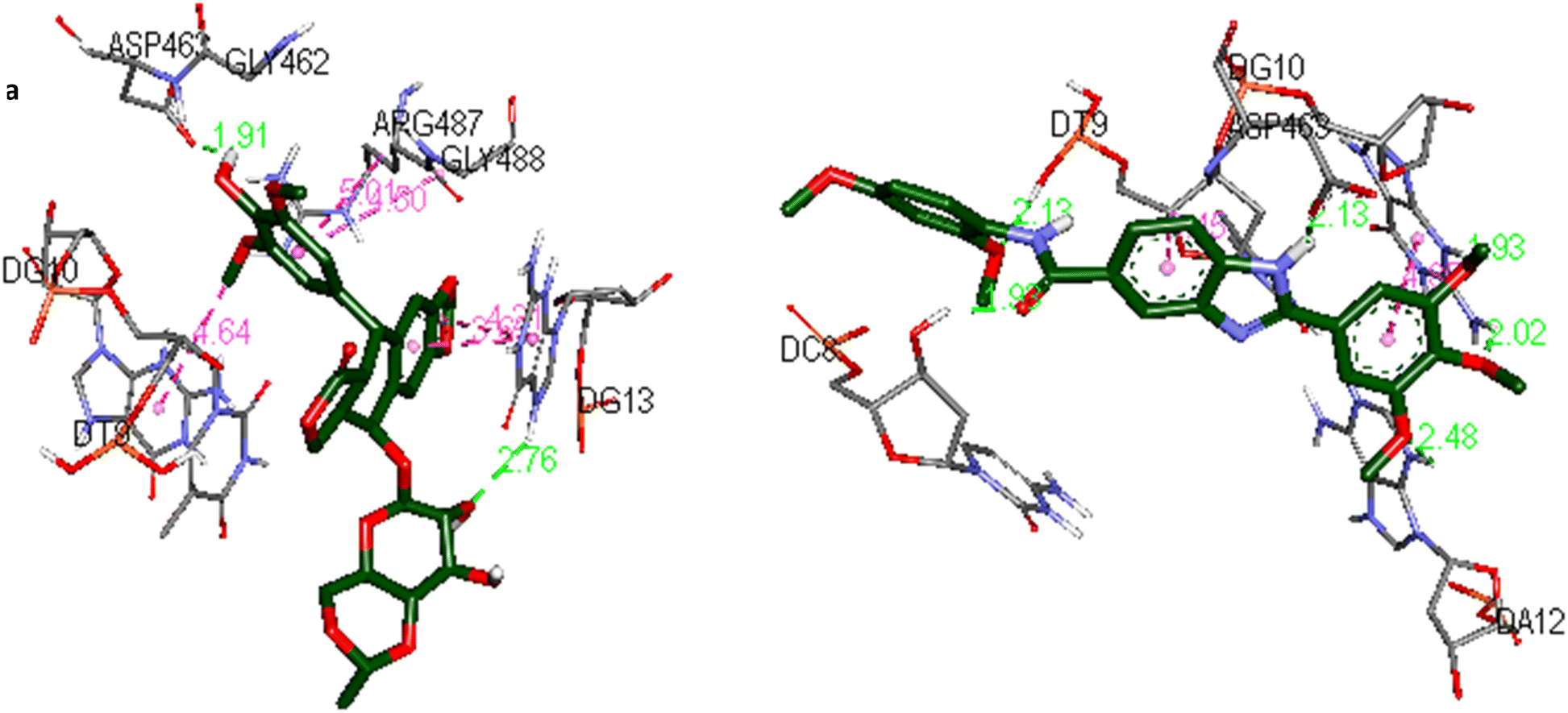 | ||
| Fig. 5 The docking interactions of (a) etoposide and (b) 5o in the active site of the Topo IIα-DNA (PDB ID: 5GWK) by using Discovery Studio 2021 Client software (http://accelrys.com/). | ||
Docking analysis of etoposide (Fig. 5a) represented the hydrogen bonding interactions with ASP463 and guanine DG13. Moreover, the compound established hydrophobic interactions including amide–Pi stacked, Pi–Pi stacked, and Pi–alkyl interactions with ARG487, guanine DG13, and guanine DG10.
In the case of compound 5o (Fig. 5b) the 3,4,5-trimethoxyphenyl moiety was intercalated in double-stranded DNA by establishing hydrogen bonding interactions with guanine DG10 and adenine DA12. The phenyl ring was also involved in a Pi–Pi T-shaped interaction with guanine DG10. The 2,4-dimethoxyphenyl moiety and the amide group were involved in hydrogen bonding interactions with thymine DT9 and cytosine DC8, respectively. The benzimidazole core was stabilized by hydrogen bonding and Pi–sigma interactions with ASP463 and thymine DT9, respectively.
2.6. Molecular dynamics simulations
Molecular dynamics (MD) simulations were performed to predict the dynamic interactions and stability of compound 5o in the active site of the Topo IIα-DNA. The RMSD values represent the deviation of each residue's position over time. An RMSD value lower than 2.5 Å indicates a stable interaction between the ligand and the protein.50 The backbone RMSD values for compound 5o throughout the 100 ns simulation duration are illustrated in Fig. 6. The compound demonstrated a consistently low RMSD, averaging 1.86 Å, indicative of its robust structural stability throughout the simulation process.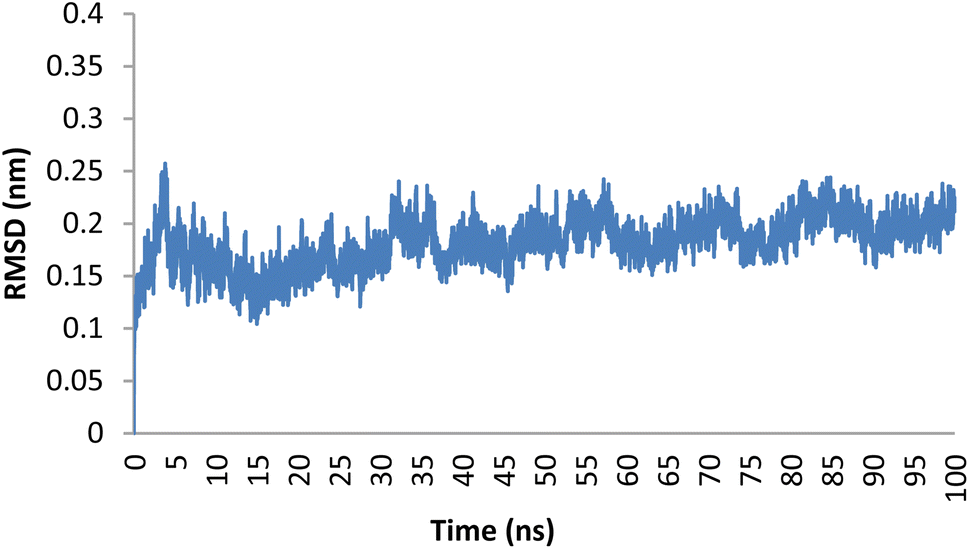 | ||
| Fig. 6 Plot of RMSD for compound 5o within the binding pocket of the Topo IIα-DNA (PDB ID: 5GWK) over a 100 ns duration. | ||
Hydrogen binding interactions are crucial in describing the stability of ligand–protein complexes. The number of hydrogen bonds established between compound 5o and the residues in the active site of Topo IIα-DNA over time are shown in Fig. 7. The findings indicate that, over a 100 ns simulation, compound 5o showed zero to three hydrogen bonds. Table 4 provides an overview of the specific hydrogen bonds (occupancy more than 1%) formed between the ligands and active site residues, along with details on their occupancy. The results indicate that 5o formed three stable hydrogen bonds with ASP463, DT9, and LYS614, showing an occupancy of 18.62%, 7.99%, and 1.60%, respectively. This aligns with our earlier observations in the molecular docking results.
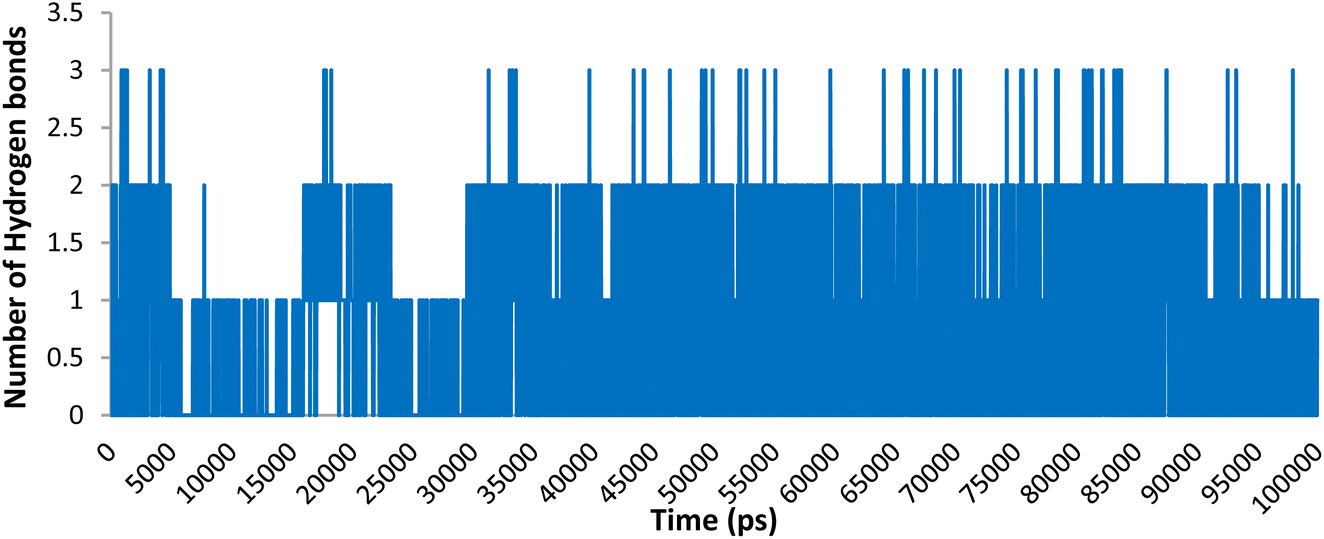 | ||
| Fig. 7 Number of hydrogen bonding interactions of compound 5o within the active site of Topo IIα-DNA. | ||
| Donor | Acceptor | Occupancy |
|---|---|---|
| 5o | ASP463-side | 18.69% |
| DT9-side | 5o | 7.99% |
| LYS614-side | 5o | 1.60% |
We utilized trajectory clustering to minimize the abundance of frames in a typical trajectory file, resulting in a representative set of distinctive frames. Fig. 8 shows the structure representative of the most populated cluster for the 5o-Topo IIα-DNA complex. Our calculations exposed several key interactions between the ligand and nearby residues that confer the system a stable conformation and endors the action of 5o as a topo II poison. The topo II aids the ligand's anchoring within the binding site by a tough hydrogen bonding interaction formed between ASP463 and the benzimidazole ring at a distance of 1.96 Å. Another hydrogen bond was also observed between LYS614 and the oxygen atom of the 2,4-dimethoxyphenyl group at a distance of 2.65 Å. The neighboring DNA bases were also involved in the stabilization of the complex. Specifically, the DT9 established a hydrogen bonding interaction with the 2-methoxy of 2,4-dimethoxyphenyl at a distance of 1.95 Å. The DT9 and DC8 bases displayed Pi-sigma interactions with the benzimidazole ring (3.82 Å), and 3-methoxy of 3,4,5-trimethoxyphenyl moiety (3.59 Å), respectively. The 3,4,5-trimethoxyphenyl formed several hydrophobic interactions with DA12 and DG13. The hydrogen bonding interactions described above were tracked during the simulations. The simulations corroborate the role of ASP463 and DT9 in stabilizing 5o at the binding site. Indeed, we monitored the distance between the atoms of amino acids (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif)
![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) , O
, O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) , and N atoms of ASP463, DT9, and LYS614) and 5o (N of benzimidazole core, O of 2-methoxy, and O of 4-methoxy) involved in hydrogen bonding and found that the distances remained under 3.1 Å during the simulation (Fig. 9).
, and N atoms of ASP463, DT9, and LYS614) and 5o (N of benzimidazole core, O of 2-methoxy, and O of 4-methoxy) involved in hydrogen bonding and found that the distances remained under 3.1 Å during the simulation (Fig. 9).
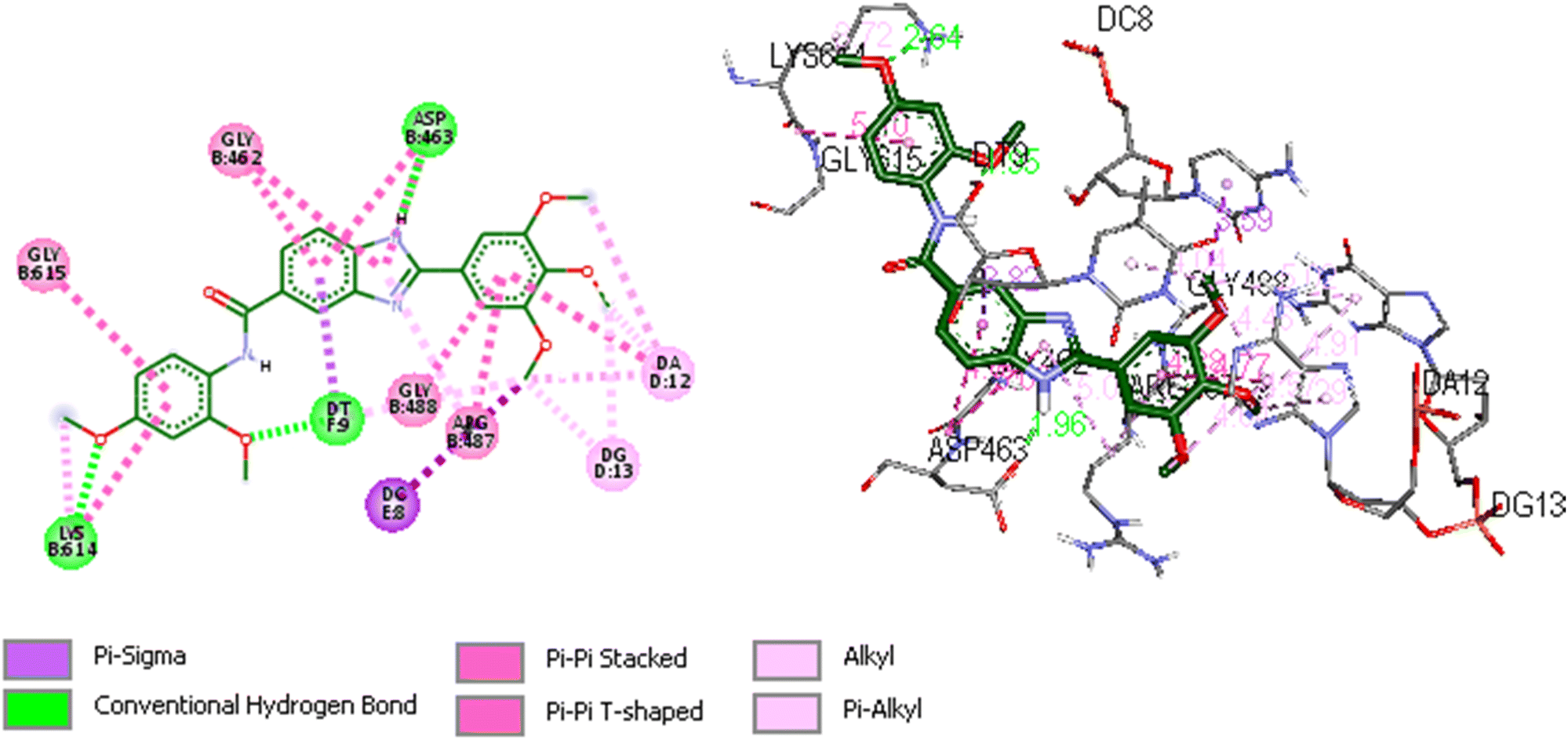 | ||
| Fig. 8 Representative image of 2D and 3D binding mode and interactions of 5o within the binding site of Topo IIα-DNA (PDB ID: 5GWK) obtained through cluster analysis. The image was generated by using Discovery Studio 2021 Client software (http://accelrys.com/). | ||
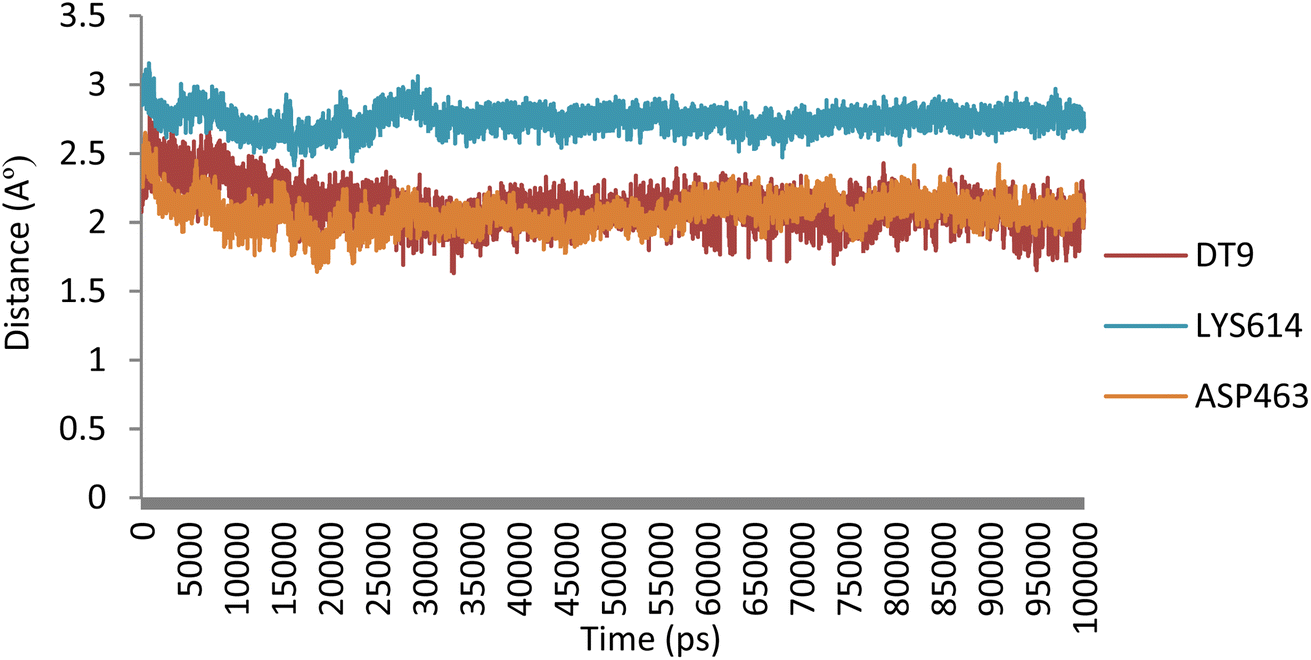 | ||
| Fig. 9 Evolution of the distances of hydrogen bonding interactions between 5o and Topo IIα-DNA over time. | ||
The Molecular Mechanics Poisson Boltzmann Surface Area (MM-PBSA) method was used to assess the binding free energy of 5o. Decomposition energy analysis per residue was calculated to determine the contribution of each residue to the binding energy of the ligand in the Topo IIα-DNA complex. The binding energy terms and their contribution to the total binding energy of 5o at the active site of the Topo IIα-DNA are listed in Table 5. According to the analysis, 5o exhibited a binding free energy of −91.29 kJ mol−1. The favorable energy term for 5o binding energy was to be driven by the van der Waals (VDW) energy rather than the electrostatic energy. This justifies the low number of hydrogen bonds and their low occupancies obtained from hydrogen bonding analysis. Additionally, according to the per residue decomposition analysis (Fig. 10),GLY462, ASP463, GLY488, LYS614, GLY615, HIS759, DC8, DT9, DA12, DG10, and DC11 of Topo IIα-DNA played pivotal roles in the binding stabilization of 5o with the receptor.
| Compound | ΔEvdw (kJ mol−1) | ΔEelec (kJ mol−1) | ΔGpolar (kJ mol−1) | ΔGnonpolar (kJ mol−1) | ΔGbind (kJ mol−1) |
|---|---|---|---|---|---|
| 5o | −158.87 ± 1.05 | −49.15 ± 1.12 | 134.72 ± 3.64 | −18.19 ± 0.15 | −91.29 ± 3.22 |
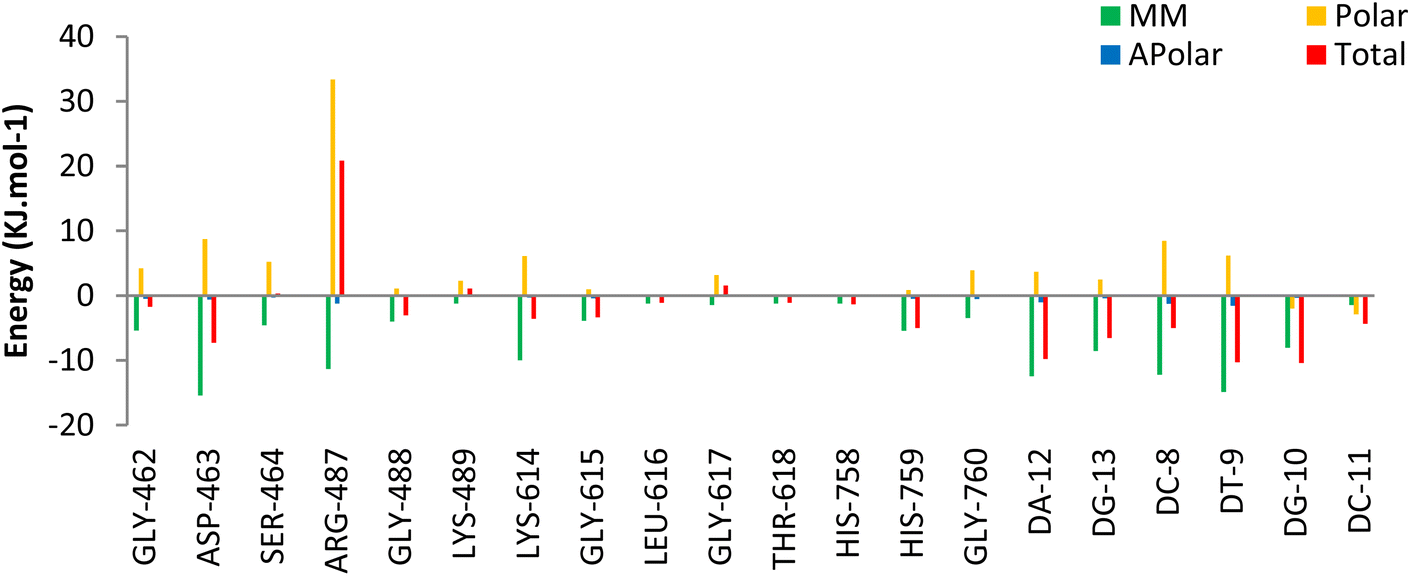 | ||
| Fig. 10 The per-residue decomposition energy analysis results for 5o-Topo IIα-DNA complex. These energies are delineated on a per-residue basis, illustrating contributions from total (red), molecular mechanical MM (green), polar (orange), and apolar (blue) energies. | ||
Overall, hydrogen bond, cluster, and decomposition energy analyses revealed robust interactions, predominantly driven by hydrophobic contacts of 5o within the active site of Topo IIα-DNA. Moreover, the role of hydrogen bond interactions (especially with ASP463 and DT9) in the stabilization of 5o was confirmed.
2.7. In silico prediction of drug-likeness and pharmacokinetics
The drug-likeness profile and pharmacokinetic properties of compound 5o are shown in Table 6. The results showed that compound 5o fulfilled the drug-likeness rules, including Lipinski, Ghose, Veber, Egan, and Muegge. Moreover, high human intestine absorption, low blood–brain barrier permeation (may not cause neurotoxicity), and moderate in vitro human colorectal carcinoma cell permeability were predicted for compound 5o. According to the results, compound 5o can be proposed as a drug-like candidate with promising pharmacokinetic properties.| ID | Drug-likeness | Pharmacokinetics | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Lipinski | Ghose | Veber | Egan | Muegge | HIAa | BBBb | Caco2c | PPBd | |
| a Human intestinal absorption (percentage); 0–20 (poor), 20–70 (moderate), 70–100 (well).b Blood–brain barrier penetration.c In vitro Caco2 (human colorectal carcinoma) cell permeability (nm s−1); <4 (low), 4–70 (moderate), >70 (high).d In vitro plasma protein binding (percentage); >90 (strongly), <90 (weakly). | |||||||||
| 5o | Yes | Yes | Yes | Yes | Yes | 92.99 | No | 27.56 | 88.00 |
3. Conclusions
Novel N-substituted-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamides (5a–o) were designed and synthesized as anticancer agents. The derivatives were screened for their cytotoxicity against cancerous (A549 and SW480) and normal (MRC-5) cells. All the tested compounds showed notable cytotoxic activity against the two cancer cell lines with an IC50 range of 0.15–52.73 μM. However, the compounds were more effective against A549 cells. As biological evaluation results revealed, the antiproliferative potential depends on the nature of carboxamide substitutions. Compound 5o, bearing a 2,4-dimethoxyphenyl, was the most potent derivative of all the tested compounds. It was more effective than cisplatin and etoposide towards the two cancer cells, and doxorubicin towards A549. Compound 5o exhibited low toxicity against MRC-5 normal cells. This compound effectively arrested A549 cells at the S phase and induced apoptosis at early and late stages. Compound 5o was well accommodated in the Topo IIα-DNA binding pocket. Moreover, MD simulation experiments confirmed the stability of compound 5o, and revealed the crucial role of the 3,4,5-trimethoxyphenyl, benzimidazole, and 2,4-dimethoxyphenyl moieties in establishing hydrogen bonding and hydrophobic interactions with the adjacent amino acids and DNA nucleotides. Finally, appropriate pharmacokinetic properties were predicted for the compound. This research identified new benzimidazole analogs and diversified the scope of anticancer agents. Further investigations on the enzyme inhibitory activity of the compounds may provide one step forward for the discovery of a novel chemotherapeutic drug.4. Materials and methods
4.1. Apparatus
All chemicals and reagents were of commercial grade. The progress of the reactions was followed using pre-coated aluminum sheet thin layer chromatography (TLC) plates (MERCK, silica gel 60-F254). The IR spectra were collected with a Nicolet FT-IR Magna 550 spectrometer (KBr disks). The nuclear magnetic resonance (NMR) spectra were recorded with a Bruker DRX-500 NMR spectrometer (1H: 301 MHz, 13C: 76 MHz) using DMSO-d6 as solvents. Chemical shifts (δ) are expressed in parts per million (ppm), and J values are given in hertz (Hz). Melting points were determined with a Barnstead IA9300 system (Electrothermal, UK), and are uncorrected. Elemental analyses were carried out by a PerkinElmer 2400 CHN Elemental Analyzer and were within ±0.4% of the theoretical values for C, H, and N.4.2. Chemistry
Brown solid; yield: 2626.6 mg, 80%; mp: 132–134 °C; Rf = 0.66 (ethyl acetate); 1H NMR (400 MHz, DMSO-d6) δ 8.25 (s, 1H), 7.99 (dd, J = 8.5, 1.5 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.60 (s, 2H), 3.93 (s, 6H), 3.79 (s, 3H) ppm; 13C NMR (100 MHz, DMSO-d6) δ 168.3, (154.4, 154.4),142.9, 140.7, 139.7, 124.1, 117.2, 115.3, (104.8, 104.8), 60.9, (56.9, 56.9) ppm; anal. calcd for C17H16N2O5: C, 62.19; H, 4.91; N, 8.53. Found: C, 62.34; H, 5.06; N, 8.69.
4.2.2.1 N-Butyl-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5a). Cream solid; yield: 433.5 mg, 93%; mp: 167–169 °C; Rf = 0.54 (ethyl acetate); IR (KBr, vmax): 3340 (NH), 3030 (CH aromatic), 2910 (CH aliphatic), 1680 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.08 (s, 1H, NH), 8.45 (t, J = 5.6 Hz, 1H, NH), 8.12 (s, 1H, H Ar), 7.77 (d, J = 8.2 Hz, 1H, H Ar), 7.64 (d, J = 8.5 Hz, 1H, H Ar), 7.57 (s, 2H, H Ar), 3.93 (s, 6H, 2× OCH3), 3.77 (s, 3H, OCH3), 3.32 (q, J = 6.5 Hz, 2H, CH2), 1.56 (p, 2H, CH2), 1.46–1.30 (m, 2H, CH2), 0.94 (t, J = 7.3 Hz, 3H, CH3) ppm: δ 13C NMR (76 MHz, DMSO-d6) δ 167.1, (153.7, 153.7), 153.4, 139.7, 129.4, 125.6, (104.5, 104.5), 60.6, (56.5, 56.5), 31.9, 20.2, 14.2 ppm; anal. calcd for C21H25N3O4: C, 65.78; H, 6.57; N, 10.96. Found: C, 65.94; H, 6.72; N, 11.12.
4.2.2.2 N-Isobutyl-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5b). Brown solid; yield: 328.7 mg, 89%; mp: 158–160 °C; Rf = 0.53 (ethyl acetate); IR (KBr, vmax): 3325 (NH), 3010 (CH aromatic), 2915 (CH aliphatic),1652 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.09 (s, 1H, NH), 8.48 (s, 1H, H Ar), 8.15 (m, 1H, NH), 7.78 (d, J = 8.3 Hz, 1H, H Ar), 7.66 (s, 1H, H Ar), 7.57 (s, 2H, H Ar), 3.93 (s, 6H, 2× OCH3), 3.77 (s, 3H, OCH3), 3.15 (t, J = 6.4 Hz, 2H, CH2), 1.91 (h, J = 13.4, 6.7 Hz, 1H, CH), 0.93 (d, J = 6.6 Hz, 6H, 2× CH3) ppm: 13C NMR (76 MHz, DMSO-d6) δ 167.2, (153.7, 153.7), 153.5, 139.7, 125.6, (104.5, 104.5), 60.6, (56.5, 56.5), 47.3, 28.67, 20.7 ppm; anal. calcd for C21H25N3O4: C, 65.78; H, 6.57; N, 10.96. Found: C, 65.91; H, 6.76; N, 11.09.
4.2.2.3 N-Benzyl-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5c). Brown solid; yield: 388.2 mg, 93%; mp:170–172 °C; Rf = 0.33 (ethyl acetate); IR (KBr, vmax): 3320 (NH), 3035 (CH aromatic), 2980 (CH aliphatic), 1675 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.16 (s, 1H, NH), 9.31–8.79 (m, 1H, NH), 7.92–7.69 (m, 2H, H Ar), 7.68–7.55 (m, 3H, H Ar), 7.46–7.22 (m, 3H, H Ar), 4.57 (d, J = 4.9 Hz, 2H, CH2), 3.94 (s, 6H, 2× OCH3), 3.77 (s, 3H, OCH3) ppm; 13C NMR (76 MHz, DMSO-d6) δ 167.1, (153.7, 153.7), 153.3, 146.4, 140.5, 139.7, (128.7, 128.7), 127.6, (127.1, 127.1), 125.6, 122.7, 121.6, 118.5, 111.3, (104.5, 104.5), 60.6, (56.5, 56.5), 43.2 ppm; anal. calcd for C24H23N3O4: C, 69.05; H, 5.55; N, 10.07. Found: C, 69.26; H, 5.69; N, 10.27.
4.2.2.4 N-Phenethyl-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5d). Brown solid; yield: 384.0 mg, 89%; mp: 161–163 °C; Rf = 0.39 (ethyl acetate); IR (KBr, vmax): 3320 (NH), 3030 (CH aromatic), 2875 (CH aliphatic), 1659 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.12 (s, 1H, NH), 8.62 (s, 1H, H Ar), 8.15 (m, 1H, NH), 7.79 (d, J = 8.0 Hz, 1H, H Ar), 7.68 (s, 1H, H Ar), 7.59 (s, 2H, H Ar), 7.42–7.26 (m, 4H, H Ar), 7.26–7.16 (m, 1H, H Ar), 3.94 (s, 6H, 2× OCH3), 3.77 (s, 3H, OCH3), 3.57 (q, J = 6.7 Hz, 2H, CH2), 2.92 (t, J = 7.3 Hz, 2H, CH2) ppm: 13C NMR (76 MHz, DMSO-d6) δ 167.2, (153.7, 153.7), 140.2, 139.7, (129.2, 129.2), (128.8, 128.8), 126.5, 125.6, (104.5, 104.5), 60.6, (56.5, 56.5), 41.5, 35.7 ppm; anal. calcd for C25H25N3O4: C, 69.59; H, 5.84; N, 9.74. Found: C, 69.74; H, 6.01; N, 9.89.
4.2.2.5 N-Phenyl-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5e). Brown solid; yield: 359.0 mg, 89%; mp: 166–168 °C; Rf = 0.33 (ethyl acetate); IR (KBr, vmax): 3320 (NH), 3025 (CH aromatic), 2885 (CH aliphatic), 1662 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 10.29 (s, 1H, NH), 8.27 (s, 1H, H Ar), 7.94–7.80 (m, 3H, H Ar), 7.73 (d, J = 8.4 Hz, 1H, H Ar), 7.59 (s, 2H, H Ar), 7.38 (t, J = 7.7 Hz, 2H, H Ar), 7.11 (t, J = 7.3 Hz, 1H, H Ar), 3.94 (s, 6H, 2× OCH3), 3.77 (s, 3H, OCH3): 13C NMR (76 MHz, DMSO-d6) δ 166.4, (153.8, 153.8), 153.7, 139.9, 139.8, 129.5, (129.1, 129.1), 125.2, 123.9, 122.7, (120.8, 120.8), (104.6, 104.6), 60.7, 56.8 ppm: anal. calcd for C23H21N3O4: C, 68.47; H, 5.25; N, 10.42. Found: C, 68.68; H, 5.49; N, 10.59.
4.2.2.6 N-(2-Fluorophenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5f). Brown solid; yield: 396.1 mg, 94%; mp: 177–179 °C; Rf = 0.32 (ethyl acetate); IR (KBr, vmax): 3325 (NH), 3045 (CH aromatic), 2970 (CH aliphatic), 1664 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.21 (s, 1H, NH), 10.13 (s, 1H, NH), 8.30 (s, 1H, H Ar), 7.92 (d, J = 8.4 Hz, 1H, H Ar), 7.78–7.68 (m, 2H, H Ar), 7.60 (s, 2H, H Ar), 7.39–7.18 (m, 3H, H Ar), 3.95 (s, 6H, 2× OCH3), 3.78 (s, 3H, OCH3) ppm: 13C NMR (76 MHz, DMSO-d6) δ 166.3, 156.32 (d, Jc–f = 246.5 Hz), (153.8, 153.8), 139.8, 128.4, 127.67, 127.2, 127.1, 126.5, 125.4, 124.8, 124.7, 122.7, 116.4, 116.1, (104.6, 104.6), 60.7, (56.6, 56.6) ppm; anal. calcd for C23H20FN3O4: C, 65.55; H, 4.78; N, 9.97. Found: C, 65.74; H, 4.93; N, 10.13.
4.2.2.7 N-(2-Chlorophenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5g). Brown solid; yield: 332.8 mg, 76%; mp: 191–193 °C; Rf = 0.30 (ethyl acetate); IR (KBr, vmax): 3330 (NH), 3020 (CH aromatic), 2970 (CH aliphatic), 1667 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.22 (s, 1H, NH), 10.04 (s, 1H, NH), 7.86 (d, J = 10.2 Hz, 1H), 7.80–7.64 (m, 3H, H Ar), 7.64–7.47 (m, 5H, H Ar), 7.41 (t, J = 7.6 Hz, 1H, H Ar), 3.97 (s, 6H, 2× OCH3), 3.79 (s, 3H, OCH3) ppm; 13C NMR (76 MHz, DMSO-d6) δ 168.4, 163.9, (153.7, 153.7), 143.3, 139.8, 129.0, 127.8, 125.8, 125.3, 119.6, 117.5, 110.1, (104.6, 104.6), 60.6, (56.5, 56.5) ppm; anal. calcd for C23H20ClN3O4: C, 72.77; H, 4.63; N, 10.29. Found: C, 72.98; H, 4.84; N, 10.51.
4.2.2.8 N-(3-Chlorophenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5h). Brown solid; yield: 376.6 mg, 86%; mp: 180–182 °C; Rf = 0.29 (ethyl acetate); IR (KBr, vmax): 3310 (NH), 3050 (CH aromatic), 2980 (CH aliphatic), 1660 (C
O) cm−1; 1H NMR ((301 MHz, DMSO-d6) δ 12.47 (s, 1H, NH)), 10.05 (s, 1H, NH), 8.23 (s, 1H, H Ar), 8.05–7.95 (m, 1H, H Ar), 7.90 (d, J = 8.5 Hz, 1H, H Ar), 7.79–7.66 (m, 2H, H Ar), 7.61–7.49 (m, 3H, H Ar), 7.41 (t, J = 7.6 Hz, 1H, H Ar), 3.93 (s, 6H, 2× OCH3), 3.77 (s, 3H, OCH3) ppm; δ 13C NMR (76 MHz, DMSO-d6) δ 168.3, 162.8, (153.7, 153.7), 143.3, 139.9, 128.3, 127.8, 125.2, 125.1, 125.0124.2, 119.6, 110.1, (104.7, 104.7), 60.6, (56.5, 56.5) ppm; anal. calcd for C23H20ClN3O4: C, 72.77; H, 4.63; N, 10.29. Found: C, 72.95; H, 4.78; N, 10.49.
4.2.2.9 N-(2-Chloro-4-nitrophenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5i). Brown solid; yield: 415.3 mg, 86%; mp: 198–200 °C; Rf = 0.25 (ethyl acetate); IR (KBr, vmax): 3320 (NH), 3035 (CH aromatic), 2960 (CH aliphatic), 1675 (C
O), 1560–1345 (NO2) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.24 (s, 2H, NH), 8.21 (s, 1H, H Ar), 8.00 (d, J = 8.4 Hz, 1H, H Ar), 7.87 (d, J = 8.2 Hz, 1H, H Ar), 7.79–7.65 (m, 2H, H Ar), 7.57 (s, 2H, H Ar), 7.42 (t, J = 7.4 Hz, 1H, H Ar), 3.94 (s, 6H, 2× OCH3), 3.77 (s, 3H, OCH3) ppm; 13C NMR (76 MHz, DMSO-d6) δ 168.4, (153.8, 153.8), 143.3, 139.81, 128.3, 127.8, 125.3, 125.0, 124.1, 119.6, 110.1, (104.6, 104.6), 60.6, (56.5, 56.5) ppm; anal. calcd for C23H19ClN4O6: C, 57.21; H, 3.97; N, 11.60. Found: C, 57.42; H, 4.11; N, 11.78.
4.2.2.10 N-(4-Bromophenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5j). Brown solid; yield: 414.8 mg, 86%; mp: 186–188 °C; Rf = 0.32 (ethyl acetate); IR (KBr, vmax): 3335 (NH), 3020 (CH aromatic), 2965 (CH aliphatic), 1671 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 10.68 (s, 1H, NH), 8.36 (s, 1H, H Ar), 8.09 (d, J = 8.6 Hz, 1H, H Ar), 7.94–7.79 (m, 5H, H Ar), 7.57 (d, J = 8.7 Hz, 2H, H Ar), 3.97 (s, 6H, 2× OCH3), 3.82 (s, 3H, OCH3) ppm. 13C NMR (76 MHz, DMSO-d6) δ 165.4, (153.9, 153.9), 151.5, 141.8, 139.0, 135.8, 133.5, 131.9, 125.4, 122.8, 119.5, 115.9, 114.5, 114.2, (106.4, 106.4), 60.8, (57.0, 57.0) ppm; anal. calcd for C23H20BrN3O4: C, 57.27; H, 4.18; N, 8.71. Found: C, 57.42; H, 4.39; N, 8.88.
4.2.2.11 N-(3-Nitrophenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5k). Brown solid; yield: 417.0 mg, 93%; mp: 195–197 °C; Rf = 0.27 (ethyl acetate); IR (KBr, vmax): 3325 (NH), 3030 (CH aromatic), 2980 (CH aliphatic), 1665 (C
O), 1565–1350 (NO2) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 10.83 (s, 1H, NH), 8.90 (s, 1H, NH), 8.35 (s, 1H, H Ar), 8.26 (d, J = 8.0 Hz, 1H, H Ar), 7.99 (d, J = 8.1 Hz, 2H, H Ar), 7.82 (d, J = 8.2 Hz, 1H, H Ar), 7.71 (d, J = 8.2 Hz, 1H, H Ar), 7.66 (s, 2H, H Ar), 3.96 (s, 6H, 2× OCH3), 3.79 (s, 3H, OCH3) ppm; 13C NMR (76 MHz, DMSO-d6) δ 166.6, (153.8, 153.8), 148.41, 141.1, 130.5, 129.5, 126.7, 123.7, 118.5, 114.8, (105.1, 105.1), 60.7, (56.7, 56.7) ppm; anal. calcd for C23H20N4O6: C, 61.60; H, 4.50; N, 12.49. Found: C, 61.80; H, 4.68; N, 12.71.
4.2.2.12 N-(o-Tolyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5l). Brown solid; yield: 342.3 mg, 82%; mp: 183–185 °C; Rf = 0.32 (ethyl acetate); IR (KBr, vmax): 3335 (NH), 3015 (CH aromatic), 2970 (CH aliphatic), 1658 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.16 (s, 1H, NH), 9.90 (s, 1H, NH), 8.31 (s, 1H, H Ar), 7.94 (d, J = 8.4 Hz, 1H, H Ar), 7.75 (d, J = 8.4 Hz, 1H, H Ar), 7.62 (s, 2H, H Ar), 7.46 (d, J = 7.6 Hz, 1H, H Ar), 7.34–7.13 (m, 3H, H Ar), 3.95 (s, 6H, 2× OCH3), 3.79 (s, 3H, OCH3), 2.33 (s, 3H, CH3) ppm; 13C NMR (76 MHz, DMSO-d6) δ 166.2, (153.8, 153.8), 139.8, 137.2, 134.1, 130.8, 129.1, 127.1, 126.5, 126.3, 125.5, 122.6, (104.6, 104.6), 60.7, (56.6, 56.6), 18.4 ppm; anal. calcd for C24H23N3O4: C, 69.05; H, 5.55; N, 10.07. Found: C, 69.24; H, 5.72; N, 10.23.
4.2.2.13 N-(p-Tolyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5m). Brown solid; yield: 392.4 mg, 94%; mp: 175–177 °C; Rf = 0.32 (ethyl acetate); IR (KBr, vmax): 3325 (NH), 3030 (CH aromatic), 2965 (CH aliphatic), 1656 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.20 (s, 1H, NH), 10.22 (s, 1H, NH), 8.29 (s, 1H, H Ar), 7.90 (d, J = 8.5 Hz, 1H, H Ar), 7.75 (d, J = 6.9 Hz, 3H, H Ar), 7.61 (s, 2H, H Ar), 7.18 (d, J = 8.1 Hz, 2H, H Ar), 3.95 (s, 6H, 2× OCH3), 3.78 (s, 3H, OCH3), 2.30 (s, 3H, CH3) ppm: 13C NMR (76 MHz, DMSO-d6) δ 166.3, (153.8, 153.8), 139.8, 137.5, 132.8, 129.5, 125.5, 122.6, 120.8, (104.6, 104.6), 60.7, (56.5, 56.5), 21.0 ppm; anal. calcd for C24H23N3O4: C, 69.05; H, 5.55; N, 10.07. Found: C, 69.20; H, 5.74; N, 10.31.
4.2.2.14 N-(2,4-Dimethylphenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5n). Brown solid; yield: 405.6 mg, 94%; mp: 178–180 °C; Rf = 0.31 (ethyl acetate); IR (KBr, vmax): 3320 (NH), 3035 (CH aromatic), 2980 (CH aliphatic), 1660 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.19 (s, 1H, NH), 9.83 (s, 1H, NH), 8.31 (s, 1H, H Ar), 7.93 (d, J = 8.4 Hz, 1H, H Ar), 7.74 (d, J = 8.4 Hz, 1H, H Ar), 7.62 (s, 2H, H Ar), 7.31 (d, J = 7.9 Hz, 1H, H Ar), 7.10 (s, 1H, H Ar), 7.05 (d, J = 7.9 Hz, 1H, H Ar), 3.95 (s, 6H, 2× OCH3), 3.79 (s, 3H, OCH3), 2.29 (d, J = 10.3 Hz, 6H, 2× CH3) ppm: 13C NMR (76 MHz, DMSO-d6) δ 166.2, (153.8, 153.8), 153.7, 139.7, 135.3, 134.6, 133.9, 131.3, 129.1, 127.0, 127.0, 125.5, 122.6, (104.6, 104.6), 60.6, (56.5, 56.5), 21.0, 18.3 ppm; anal. calcd for C25H25N3O4: C, 69.59; H, 5.84; N, 9.74. Found: C, 69.79; H, 6.08; N, 9.91.
4.2.2.15 N-(2,4-Dimethoxyphenyl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-6-carboxamide (5o). Brown solid; yield: 412.5 mg, 89%; mp: 191–193 °C; Rf = 0.30 (ethyl acetate); IR (KBr, vmax): 3315 (NH), 3030 (CH aromatic), 2935 (CH aliphatic),1651 (C
O) cm−1; 1H NMR (301 MHz, DMSO-d6) δ 13.18 (s, 1H, NH), 9.38 (m, 1H, NH), 7.93–7.72 (m, 2H, H Ar), 7.71–7.54 (m, 4H, H Ar), 6.70 (s, 1H, H Ar), 6.58 (d, J = 8.3 Hz, 1H, H Ar), 3.94 (s, 6H, 2× OCH3), 3.85 (s, 3H, OCH3), 3.79 (d, J = 9.6 Hz, 6H, 2× OCH3) ppm: δ 13C NMR (76 MHz, DMSO-d6) δ 165.9, 158.1, (153.8, 153.8), 146.5, 143.8, 139.7, 137.7, 135.2, 128.9, 126.4, 125.5, 122.8, 120.7, 118.6, 111.4, (104.6, 104.6), 99.4, 60.7, (56.5, 56.5), 56.2, 55.8 ppm; anal. calcd for C25H25N3O6: C, 64.79; H, 5.44; N, 9.07. Found: C, 64.96; H, 5.67; N, 9.23.
4.3. MTT assay
The cytotoxic activity of the designed compounds (5a–o) was assessed by the MTT (3-(4,5-dimethylthiazol-yl)-2,5-diphenyl-tetrazolium bromide) assay according to our previous procedure.5,51 Two human cancer cell lines, SW480 (human colon adenocarcinoma) and A549 (human lung adenocarcinoma), and the normal MRC-5 (human fetal lung fibroblast cells) cells were obtained from the National Cell Bank of Iran (NCBI, Pasteur Institute, Tehran, Iran). RPMI 1640 culture media was used to culture cancer cell lines. The media was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Gibco, USA), and the cells were kept at 37 °C in a humidified CO2 incubator. Trypsin/EDTA 0.5% solution (Gibco/USA) was applied to harvest cells, and the cells were seeded in 96-well microplates at a density of 8 × 103 cells per well. The cells were treated with different concentrations of the designed compounds, cisplatin, etoposide, and doxorubicin in triplicate times. Three untreated wells were used as the negative control. After 72 h, the media was removed, and 100 μL fresh MTT solution was added to the wells. Then, the plates were incubated for 4 h at 37 °C to obtain formazan purple crystals. Finally, the media was removed, 150 μL DMSO was added, and incubated at 37 °C in the dark for 10 min to dissolve the crystals. The absorbance of individual wells was read at 490 nm using a microplate ELISA reader. Excel 2016 and Curve Expert 1.4 were used to analyze the data. The data are presented as mean ± SD of 3–4 independent experiments.4.4. Cell cycle
The total number of 5 × 104 A549 cells were pre-cultured in 24 well plates in high glucose DMEM culture medium for 16 h in standard culturing conditions. After treatment of cells with 0.18 and 0.36 μM of 5o for 24 h, cells were harvested with 0.25% trypsin, followed by washing with PBS and stained with propidium iodide (5 μL, 1 mg mL−1), and 1U of RNase A. Cell cycle analysis was performed using flow cytometry (BD FACSCalibur Flow Cytometer, BD, USA). The DNA content was analyzed using Flowjo 10.0 software.4.5. Apoptosis assay
1 × 105 of A549 cells were pre-cultured for 16 h followed by exposure to 0.18 and 0.36 μM of 5o for 24 h. Then, Annexin V/PI staining was performed using eBioscience™ Annexin V apoptosis detection kit (Invitrogen). Cells were washed once with phosphate-buffered saline (PBS), and once with 1000 μL 1× binding buffer. In the next step, the cells were suspended with 100 μL of binding buffer containing 5 μL of Annexin V-FITC for 15 min. Afterward, cells were washed again with 1000 μL binding buffer and resuspended in 200 μL of the same buffer containing 5 μL PI solution. The apoptosis rates were then determined by BD FACS Calibur™ flow cytometry (BD Biosciences, San Jose, CA, USA).4.6. Molecular docking analysis
The structures of ligands were sketched and optimized using CHEM 3D 16.0. The three dimensional crystal structures of different human topoisomerase proteins (PDB IDs: 1T8I, 1ZXM, 1ZXN, 5GWK, and 4G0V) were downloaded from protein data bank (https://www.rcsb.org). Preparation of the PDBQT formats was performed with AutoDock Tools 1.5.4 (ADT). Water molecules and the co-crystalized ligands were removed from the protein structure, and Gasteiger charges and polar hydrogens were added. PDBQT formats of the ligands were prepared by adding Gasteiger charges and setting the degree of torsions. Molecular docking study was conducted by AutoDock 4.2 software. The grid maps were constructed considering a grid box of 50 × 50 × 50 dimensions with 0.375 Å spacing centered at the ligands binding sites. For the docking process, the rigid macromolecule and Lamarckian genetic search algorithm were selected, and the number of runs was set at 100. The self-dock was performed to confirm the validity of the docking procedures. The orientations and interactions were visualized by the Discovery Studio 2021 Client.4.7. Molecular dynamics simulations
MD simulations were performed to explore the dynamic behavior of compound 5o in an aqueous environment. The simulations were conducted using the GROMACS 2016.3 software, employing the CHARMM27 force field for an accurate representation of the molecular interactions.52 The ligand parameters were generated using the SwissParam web server. The system, consisting of the ligand and explicit water molecules, was solvated in a cubic periodic box, and counter ions were added to neutralize the system. Following energy minimization, the system was equilibrated in two successive phases—NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature). Subsequently, a production MD run was performed for 100 ns. RMSD and hydrogen bond analysis were calculated to evaluate the stability and flexibility of the ligand within the binding pocket over the simulation period. Clustering analysis was performed to condense frames in a trajectory file using GROMACS's cluster module with the Gromos Clustering Algorithm based on a 0.35 nm cutoff. The MM-PBSA method was used to estimate binding free energies. All plots were generated using Excel software and Discovery Studio 2021 Client was utilized to illustrate the interaction analysis.4.8. Drug-likeness profile and pharmacokinetics prediction
The drug-likeness nature of 5o was predicted by SwissADME (https://www.swissadme.ch) free web tool. Pharmacokinetic properties were estimated by the preADMET online server (https://preadmet.bmdrc.org/).Data availability
The data supporting this article have been included in the ESI.†Author contributions
N. D.: methodology, investigation. M. N.: conceptualization, methodology, editing. M. H. S.: data curation. M. E.: investigation, formal analysis. Y. G.: resources, supervision. E. S.: formal analysis. H. A.: conceptualization. Z. P. J.: formal analysis. M. A.: data curation. M. R. M.-T.: visualization. F. Z.: original draft preparation. M. S.: methodology. Z. P.: visualization. S. M. S.-J.: validation. B. L.: resources, validation. M. M.: methodology, conceptualization, supervision, project administration. S. R.: methodology, conceptualization, supervision, project administration, writing – reviewing and editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Vice-Chancellor for Research, Shiraz University of Medical Sciences, Iran (grant number: 28745).References
- C. D. Mathers and D. Loncar, Updated projections of global mortality and burden of disease, World Health Organization, Geneva, 2005, p. 10 Search PubMed.
- M. Khoshneviszadeh, O. Firuzi, M. Aminsafaee, M. Kashefizadeh, S. Ranjbar, Z. Rezaei, H. Sadeghpour, F. Zargari, R. Miri and N. Edraki, Iran. J. Pharm. Res., 2021, 20, 161 CAS.
- S. Ranjbar, N. Edraki, M. Khoshneviszadeh, A. Foroumadi, R. Miri and M. Khoshneviszadeh, J. Res. Pharmaceut. Sci., 2018, 13, 1 CrossRef.
- S. Ranjbar, M. R. Shabanpoor, Z. Dehghani, O. Firuzi, N. Edraki and M. Khoshneviszadeh, Braz. J. Pharm. Sci., 2021, 57, e18074 CrossRef CAS.
- S. Ranjbar, M. Khoshneviszadeh, M. Tavakkoli, R. Miri, N. Edraki and O. Firuzi, Mol. Diversity, 2021, 1–20 Search PubMed.
- S. Ranjbar, F. F. Lashkarian, M. Khoshneviszadeh, F. Moosavi, A. Sakhteman, F. Zargari, L. Saso, O. Firuzi and N. Edraki, J. Mol. Struct., 2023, 1285, 135427 CrossRef CAS.
- S. Ranjbar, P. Sadeghian, S. Khademian, M. Emami, Z. P. Jahromi, S. H. Mirmajidi, F. Zare, M. Negahdaripour, Y. Ghasemi and M. Khoshneviszadeh, Heliyon, 2024, e29850 CrossRef CAS.
- E. C. Pham, T. V. Le Thi, H. H. L. Hong, B. N. V. Thi, L. B. Vong, T. T. Vu, D. D. Vo, N. V. T. Nguyen, K. N. B. Le and T. N. Truong, RSC Adv., 2023, 13, 399–420 RSC.
- E. C. Pham, T. V. T. Le and T. N. Truong, RSC Adv., 2022, 12, 21621–21646 RSC.
- R. Kumar and G. Singh, Pharmacophore, 2022, 13, 41–55 CrossRef.
- R. Veerasamy, A. Roy, R. Karunakaran and H. Rajak, Pharmaceuticals, 2021, 14, 663 CrossRef CAS PubMed.
- A. Kanwal, M. Ahmad, S. Aslam, S. A. R. Naqvi and M. J. Saif, Pharmaceut. Chem. J., 2019, 53, 179–187 CrossRef CAS.
- M. Gaba, S. Singh and C. Mohan, Eur. J. Med. Chem., 2014, 76, 494–505 CrossRef CAS PubMed.
- H. Hernández-López, C. J. Tejada-Rodríguez and S. Leyva-Ramos, Mini Rev. Med. Chem., 2022, 22, 1268–1280 CrossRef.
- K. Shabana, Salahuddin, A. Mazumder, H. Singh, R. Kumar, S. Tyagi, V. Datt, A. Shankar Sharma, M. Shahar Yar and M. Jawed Ahsan, ChemistrySelect, 2023, 8, e202300209 CrossRef CAS.
- K. Veena, M. S. Raghu, K. Y. Kumar, C. P. Kumar, F. A. Alharti, M. K. Prashanth and B.-H. Jeon, J. Mol. Struct., 2022, 1269, 133822 CrossRef CAS.
- N. Shayegan, A. Iraji, N. Bakhshi, A. Moazzam, M. A. Faramarzi, S. Mojtabavi, S. M. M. Pour, M. B. Tehrani, B. Larijani and Z. Rezaei, J. Mol. Struct., 2022, 1268, 133650 CrossRef CAS.
- H. D. Attram, S. Wittlin and K. Chibale, MedChemComm, 2019, 10, 450–455 RSC.
- Z. M. Alzhrani, M. M. Alam and S. Nazreen, Mini Rev. Med. Chem., 2022, 22, 365–386 CrossRef CAS.
- S. Venugopal, B. Kaur, A. Verma, P. Wadhwa, M. Magan, S. Hudda and V. Kakoty, Chem. Biol. Drug Des., 2023, 102, 357–376 CrossRef CAS.
- L. S. Feng, W. Q. Su, J. B. Cheng, T. Xiao, H. Z. Li, D. A. Chen and Z. L. Zhang, Arch. Pharmazie, 2022, 355, 2200051 CrossRef CAS.
- G. Satija, B. Sharma, A. Madan, A. Iqubal, M. Shaquiquzzaman, M. Akhter, S. Parvez, M. A. Khan and M. M. Alam, J. Heterocycl. Chem., 2022, 59, 22–66 CrossRef CAS.
- M. J. Akhtar, M. S. Yar, V. K. Sharma, A. A. Khan, Z. Ali, M. Haider and A. Pathak, Curr. Med. Chem., 2020, 27, 5970–6014 CrossRef CAS PubMed.
- Y. Bansal, R. Minhas, A. Singhal, R. K. Arora and G. Bansal, Curr. Org. Chem., 2021, 25, 669–694 CrossRef CAS.
- A. Kamal, A. B. Shaik, S. Polepalli, G. B. Kumar, V. S. Reddy, R. Mahesh, S. Garimella and N. Jain, Bioorg. Med. Chem., 2015, 23, 1082–1095 CrossRef CAS PubMed.
- P. Sharma, T. S. Reddy, D. Thummuri, K. R. Senwar, N. P. Kumar, V. Naidu, S. K. Bhargava and N. Shankaraiah, Eur. J. Med. Chem., 2016, 124, 608–621 CrossRef CAS.
- S. Nazreen, A. S. Almalki, S. E. I. Elbehairi, A. A. Shati, M. Y. Alfaifi, A. A. Elhenawy, N. I. Alsenani, A. Alfarsi, A. Alhadhrami and E. A. Alqurashi, Molecules, 2022, 27, 6899 CrossRef CAS.
- V. L. Ranganatha, B. V. Avin, P. Thirusangu, T. Prashanth, B. Prabhakar and S. A. Khanum, Life Sci., 2013, 93, 904–911 CrossRef CAS PubMed.
- J. Yellol, S. A. Perez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak and A. Espinosa, J. Med. Chem., 2015, 58, 7310–7327 CrossRef CAS.
- W. Zhou, W. Zhang, Y. Peng, Z.-H. Jiang, L. Zhang and Z. Du, Molecules, 2020, 25, 3180 CrossRef CAS PubMed.
- P. Singla, V. Luxami and K. Paul, Bioorg. Med. Chem., 2015, 23, 1691–1700 CrossRef CAS PubMed.
- Y. Ren, Y. Wang, G. Li, Z. Zhang, L. Ma, B. Cheng and J. Chen, J. Med. Chem., 2021, 64, 4498–4515 CrossRef CAS PubMed.
- R. H. El-Hameed, S. S. Fatahala and A. I. Sayed, Med. Chem., 2022, 18, 238–248 CrossRef CAS PubMed.
- M. J. Akhtar, A. A. Khan, Z. Ali, R. P. Dewangan, M. Rafi, M. Q. Hassan, M. S. Akhtar, A. A. Siddiqui, S. Partap and S. Pasha, Bioorg. Chem., 2018, 78, 158–169 CrossRef CAS.
- S. Ilhan, G. Dilekci, A. Guner and H. Bektas, Anti Cancer Agents Med. Chem., 2022, 22, 2109–2115 CrossRef CAS.
- X. Yuan, Q. Yang, T. Liu, K. Li, Y. Liu, C. Zhu, Z. Zhang, L. Li, C. Zhang and M. Xie, Eur. J. Med. Chem., 2019, 179, 147–165 CrossRef CAS PubMed.
- D. I. Othman, A. Hamdi, S. S. Tawfik, A. A. Elgazar and A. S. Mostafa, J. Enzyme Inhib. Med. Chem., 2023, 38, 2166037 CrossRef PubMed.
- H. M. Abd El-Lateef, M. A. Elbastawesy, T. M. Abdelghani Ibrahim, M. M. Khalaf, M. Gouda, M. G. Wahba, I. Zaki and M. M. Morcoss, Molecules, 2023, 28, 481 CrossRef CAS.
- T.-K.-C. Huynh, T.-H.-A. Nguyen, N.-H.-S. Tran, T.-D. Nguyen and T.-K.-D. Hoang, J. Chem. Sci., 2020, 132, 1–9 CrossRef.
- R. Kumar, A. Saneja and A. K. Panda, Lung Cancer Methods Protoc., 2021, 213–223 CAS.
- A. E. Evren, L. Yurttaş, B. Eksellı and G. Akalın-Cıftcı, Lett. Drug Des. Discovery, 2019, 16, 547–555 CrossRef CAS.
- J.-H. Lin, K.-T. Yang, W.-S. Lee, P.-C. Ting, Y.-P. Luo, D.-J. Lin, Y.-S. Wang and J.-C. Chang, Oxid. Med. Cell. Longev., 2022, 2022, 9523491 Search PubMed.
- P. A. Yakkala, N. R. Penumallu, S. Shafi and A. Kamal, Pharmaceuticals, 2023, 16, 1456 CrossRef CAS PubMed.
- K. Buzun, A. Bielawska, K. Bielawski and A. Gornowicz, J. Enzyme Inhib. Med. Chem., 2020, 35, 1781–1799 CrossRef CAS PubMed.
- H. K. Swedan, A. E. Kassab, E. M. Gedawy and S. E. Elmeligie, Bioorg. Chem., 2023, 136, 106548 CrossRef CAS.
- K. Bielawski, S. Wolczynski and A. Bielawska, Pol. J. Pharmacol., 2004, 56, 373–378 CAS.
- C. Gao, B. Li, B. Zhang, Q. Sun, L. Li, X. Li, C. Chen, C. Tan, H. Liu and Y. Jiang, Bioorg. Med. Chem., 2015, 23, 1800–1807 CrossRef CAS.
- P. Li, W. Zhang, H. Jiang, Y. Li, C. Dong, H. Chen, K. Zhang and Z. Du, MedChemComm, 2018, 9, 1194–1205 RSC.
- N. A. Nawareg, A. S. Mostafa, S. M. El-Messery and M. N. Nasr, Bioorg. Chem., 2022, 127, 106038 CrossRef CAS.
- S. Patel, A. D. Mackerell Jr and C. L. Brooks 3rd, J. Comput. Chem., 2004, 25, 1504–1514 CrossRef CAS PubMed.
- M. Gholampour, S. Ranjbar, N. Edraki, M. Mohabbati, O. Firuzi and M. Khoshneviszadeh, Bioorg. Chem., 2019, 88, 102967 CrossRef CAS PubMed.
- A. D. MacKerell Jr, N. Banavali and N. Foloppe, Biopolymers, 2000, 56, 257–265 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04492d |
| ‡ These two authors contributed equally to this work, and both are considered as the first authors. |
| This journal is © The Royal Society of Chemistry 2024 |