
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClose-packed nitronyl nitroxide radicals by Au–S self-assembly: strong ferromagnetic coupling†
Zhipeng Xua,
Yongliang Qinb,
Dongdong Weia,
Jie Jin a,
Long Zhenga,
Jie Xua,
Hui Liua,
Ranran Chena and
Di Wang*a
a,
Long Zhenga,
Jie Xua,
Hui Liua,
Ranran Chena and
Di Wang*a
aSchool of Materials Science and Chemical Engineering, Anhui Jianzhu University, Hefei 230601, China. E-mail: wangdi@ahjzu.edu.cn
bAnhui Province Key Laboratory of Condensed Matter Physics at Extreme Conditions, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei, Anhui 230031, China
First published on 2nd September 2024
Abstract
The study of the magnetism of tightly arranged nitronyl nitroxide (NN) radicals via Au–S self-assembly is interesting. In this study, a series of radicals (S-NN, D-NN, BS-NN, BD-NN) along with two types of nanomaterials (S-NPs, D-NPs) were synthesized. NN was chosen for the magnetic units. Their structures have been successfully synthesized and analyzed. The spin magnetic properties were characterized by electron paramagnetic resonance (EPR) and superconducting quantum interference device (SQUID) measurement. The analysis revealed that the self-assembled NN formed via Au–S bonds exhibits high packing density. Furthermore, it was gratifying to observe that the AuNPs exhibit ferromagnetism after the surface modification by NN. This results in strong ferromagnetic exchange interactions of S-NPs and D-NPs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :JS-NPs = +279.715 K and JD-NPs = +254.913 K, respectively.
:JS-NPs = +279.715 K and JD-NPs = +254.913 K, respectively.
Introduction
In recent years, organic magnetic hybrid materials have attracted a lot of interest from scientists. These materials combine the predictable low density, ease of processing,1,2 structural tunability and diversity of organic materials with the magnetic functionality found in magnetic materials. An important task in the design of these organic magnetic hybrid materials is the integration of organic structure with magnetic properties. Many studies have focused on organic–inorganic hybrid magnets, which integrate inorganic components with organic molecules within a unified structure, have been synthesized to control the magnetic properties of materials.3–5 However, the magnetic properties of such materials rely on the inorganic components, thereby somewhat limiting the combination of organic structure with magnetic properties. Organic intrinsic magnets6 are a class of organic magnetic materials in which the aggregation of unpaired electrons7 within organic structures leads to the emergence of magnetic properties.8 Current research on organic intrinsic magnets predominantly focuses on the field of molecules. Different stable radicals, such as TEMPO,9 NN,10–12 PTM,13 etc., were used in the research. For single radicals, their crystal arrangement, and magnetic characteristics have been extensively characterized and investigated by introducing hydrogen bonds, ligand groups or diverse bridging structures. Some progress has been made in studying the structural properties and functionalization of organic intrinsic magnets. However, compared to traditional iron–cobalt–nickel series magnets,14,15 the aggregated state magnetism of organic intrinsic magnets is relatively weak. Therefore, it is desirable to employ the self-assembly strategy to design organic magnetic hybrid materials with good aggregated state magnetism.NN, a stable neutral organic radical with good spin delocalization properties and large value of spin polarization at room temperature16–21 has been extensively studied for its excellent spin magnetic properties and serves as a promising candidate for magnetic units. Self-assembly13,22–25 is a technique wherein fundamental units spontaneously organize into an ordered and dense structure. Gold nanoparticles26–28 possess advantages of adjustable shape and size, large specific surface area, facile synthesis, surface modification capabilities, and excellent biocompatibility, which have attracted more and more attention. The technology of functionalization on the surface of gold nanoparticles by Au–S self-assembly has been quite mature.
V. Loveras et al. proposed a one-pot reaction method for obtaining monodisperse22 gold nanoparticles, which can be utilized to synthesize organic radical-functionalized gold nanoparticles with controllable size, uniform morphology, and stable properties. However, this study did not conduct qualitative and quantitative analysis of the aggregated magnetic properties of the synthesised magnetic materials.
Herein, we present the synthesis and characterization of several novel self-assembled agglomerated organic materials. NN was selected as the magnetic unit, and the Au–S self-assembly strategy was employed to enhance the close packing of organic spin units. The synthesis of NN section is based on the previously reported reference, combined with the general synthesis steps18 developed by our group. Long flexible organic chains were chosen for self-assembly on the gold surface to facilitate vertical alignment and dense packing.24,29 The specific synthesis pathway is shown in Fig. 1a. Subsequently, monodisperse gold nanoparticles (AuNPs) with uniform particle size were selected and synthesized according to reported procedures. At room temperature, the synthesized bi-single nitronyl nitroxide (BS-NN) and bi-double nitronyl nitroxide (BD-NN) are added to the nanoparticles. Based on the Au–S interaction, the disulfide bonds were broken, and self-assembled on the surface of the AuNPs. Single radical self-assembled gold nanoparticles (S-NPs) and double radical self-assembled gold nanoparticles (D-NPs) were obtained, respectively. The synthesis diagram is shown in Fig. 1b. The products were characterized and analyzed through UV-vis, IR, CV, TEM, SEM, EDS, DLS measurements. Furthermore, RT-EPR, VT-EPR and SQUID measurements revealed the strong ferromagnetic interactions in both S-NPs and D-NPs, and DFT calculations were performed to collect information about the electronic structure of them (Fig. S9†).
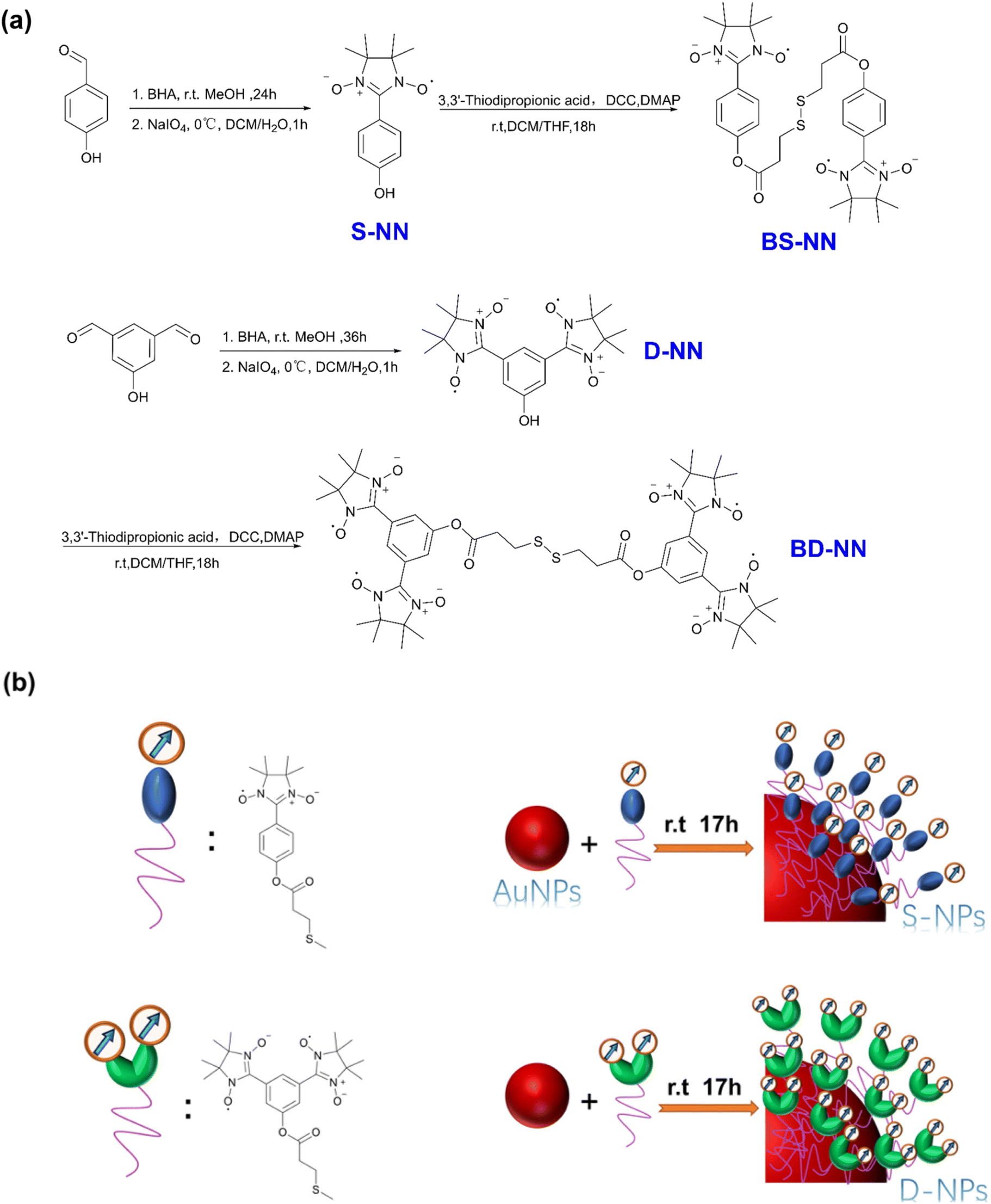 | ||
| Fig. 1 (a) Synthesis flow chart of S-NN, BS-NN, D-NN and BD-NN; (b) composite concept diagram of S-NPs and D-NPs. The red ball refers to trioctylamine stabilized gold nanoparticles. | ||
Experimental
Materials
All products used were purchased from aladdin and have not been further purified unless otherwise specified. The compound 2,3-bis(hydroxy-amino)-2,3-dimethylbutane (BHA)19 was synthesized as described in the literature. All reaction units were dried in a blast dryer and synthesized in a dry argon atmosphere.General considerations
Unless otherwise noted, CW X-band EPR spectra are recorded in toluene diluted and oxygen-free solutions at concentrations of 10−4 moles. G-factor correction using 2,2-diphenyl-1-picrohydrazine (g = 2.00370) as standard, equipped with a frequency counter and liquid helium flow temperature control, simulated with WINEPR SimFonia software. All electrochemical measurements were performed using CHI760E (Shanghai Chenhua, China). Elemental analysis was performed by scanning electron microscopy (SEM JSM-7500F) and mapping. The phase structure of the samples was ascertained by X-ray diffraction (XRD, Bruker, D8-ADVANCE) and Raman spectra (Renishaw InVia Raman spectrometer). The organic structure was verified using the liquid-mass spectrometer (Waters xevo TQD). Transmission electron microscope (Hitachi HITACHI-HT7700) was used to observe the morphology of the sample, and the average particle size of the sample was calculated by imageJ. The particle size of the sample was analyzed using a particle size analyzer (Malvern Instr, UK). Fourier transform infrared spectroscopy (Nicolet6700) was used to determine the functional groups of the samples. The UV-vis absorption spectra were recorded with Shanghai Jingke L7 spectrophotometer at room temperature. The spectroscopic experiments were carried out in toluene as solvent at room temperature. VT and VM susceptibility were measured using the Quantum Design MPS-XL-7 SQUID magnetization meter. The sample was wrapped with about 10 mg of Teflon tape and measured in a measuring tube, minus the background signal and diamagnetic correction of the gold nanoparticle sample.Results and discussion
Optical properties
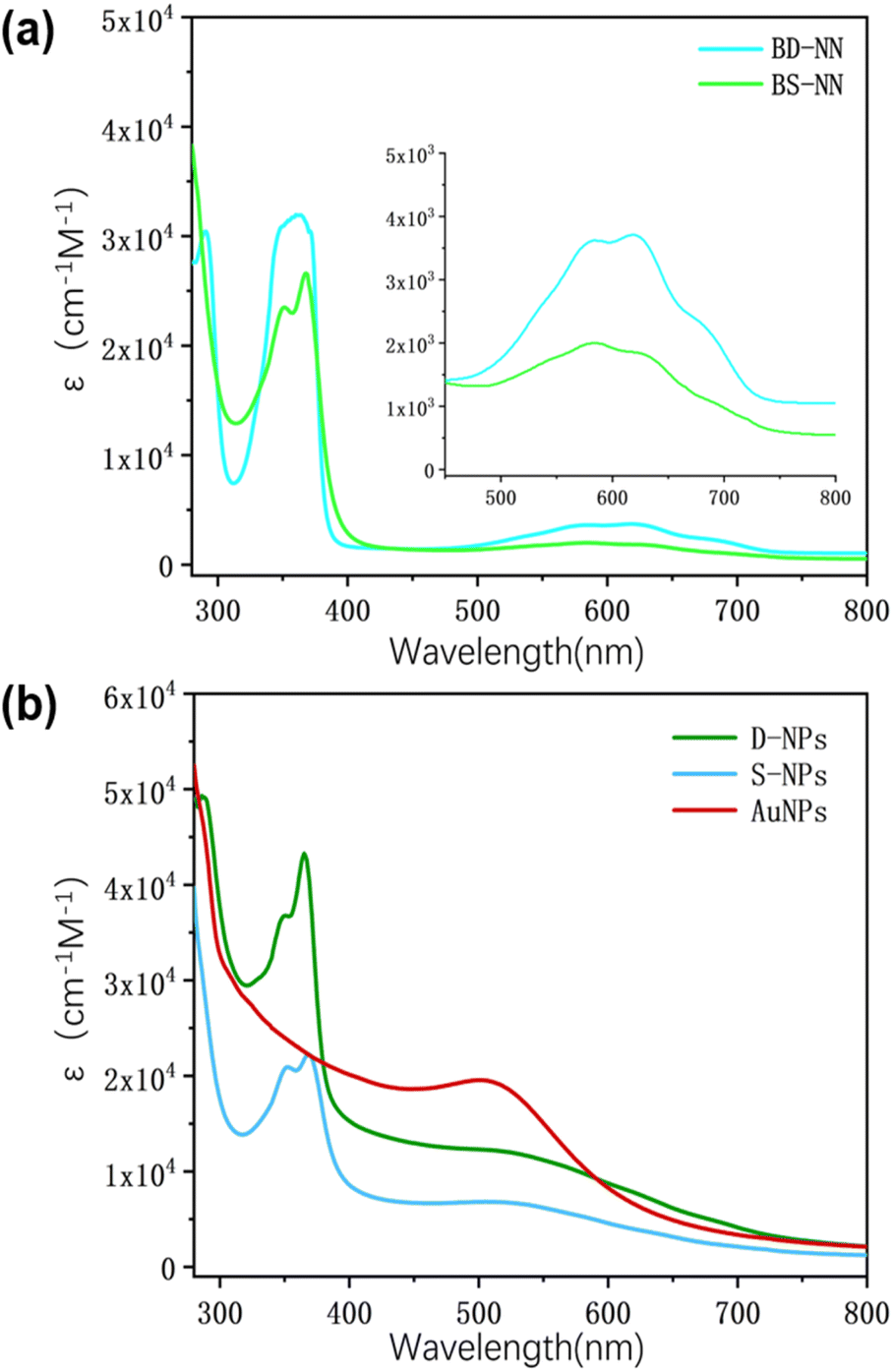 | ||
| Fig. 2 (a) UV spectra of BS-NN and BD-NN; (b) UV spectra of AuNPs, S-NPs and D-NPs. Tests were performed at the same concentration. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1720 cm−1, and a stretching vibrational peak of C–O–C at 1200 cm−1. The appearance of the characteristic vibrational peak of S–S, indicating the successful synthesis of the ester bond. As shown in Fig. 3b, the characteristic peaks of ester bond were retained near 1720 cm−1 and 1200 cm−1 for S-NPs and D-NPs. Additionally, the characteristic peaks of S–S disappeared between 500–600 cm−1, signifying the successful synthesis of NPs based on gold–sulfur self-assembly, where the disulfide bond had been broken and self-assembly with gold had occurred.
O at 1720 cm−1, and a stretching vibrational peak of C–O–C at 1200 cm−1. The appearance of the characteristic vibrational peak of S–S, indicating the successful synthesis of the ester bond. As shown in Fig. 3b, the characteristic peaks of ester bond were retained near 1720 cm−1 and 1200 cm−1 for S-NPs and D-NPs. Additionally, the characteristic peaks of S–S disappeared between 500–600 cm−1, signifying the successful synthesis of NPs based on gold–sulfur self-assembly, where the disulfide bond had been broken and self-assembly with gold had occurred.
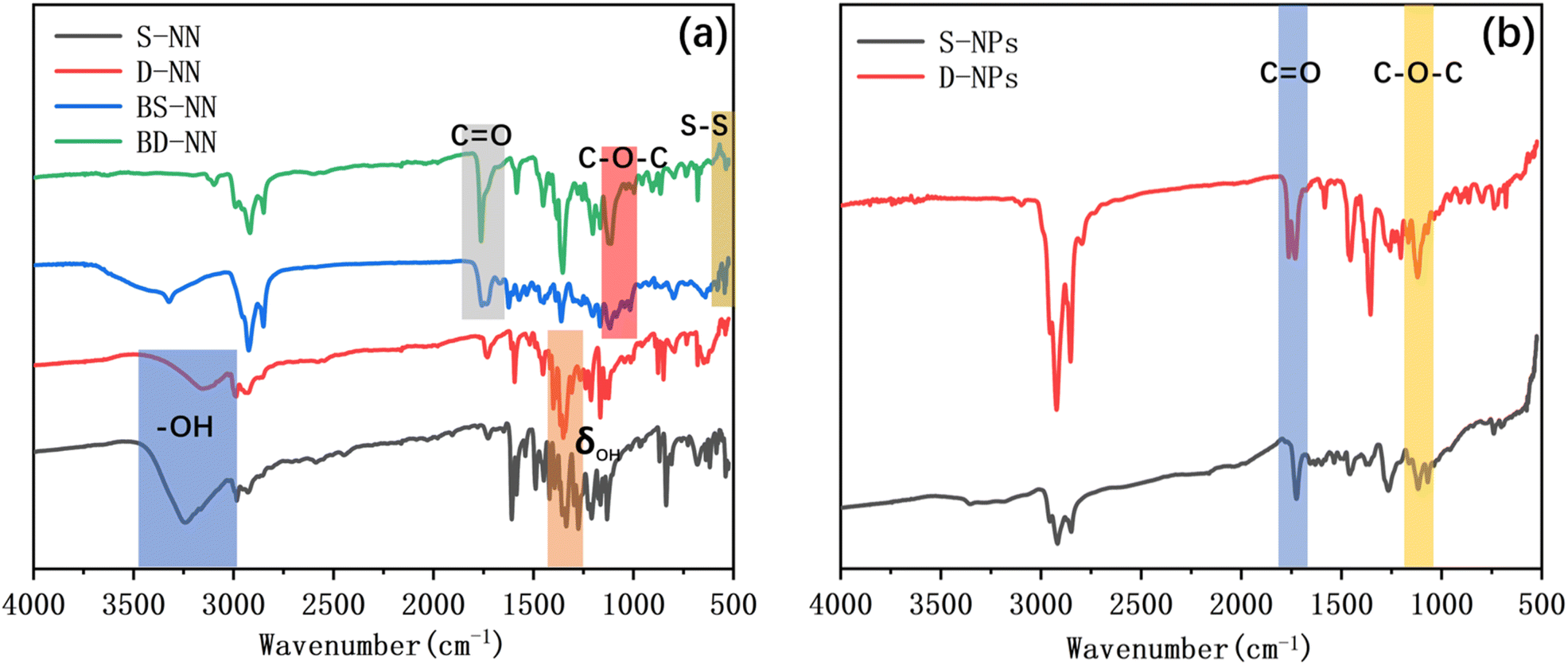 | ||
| Fig. 3 (a) IR spectrum of S-NN, D-NN, BS-NN and BD-NN; (b) IR spectrum of S-NPs and D-NPs. | ||
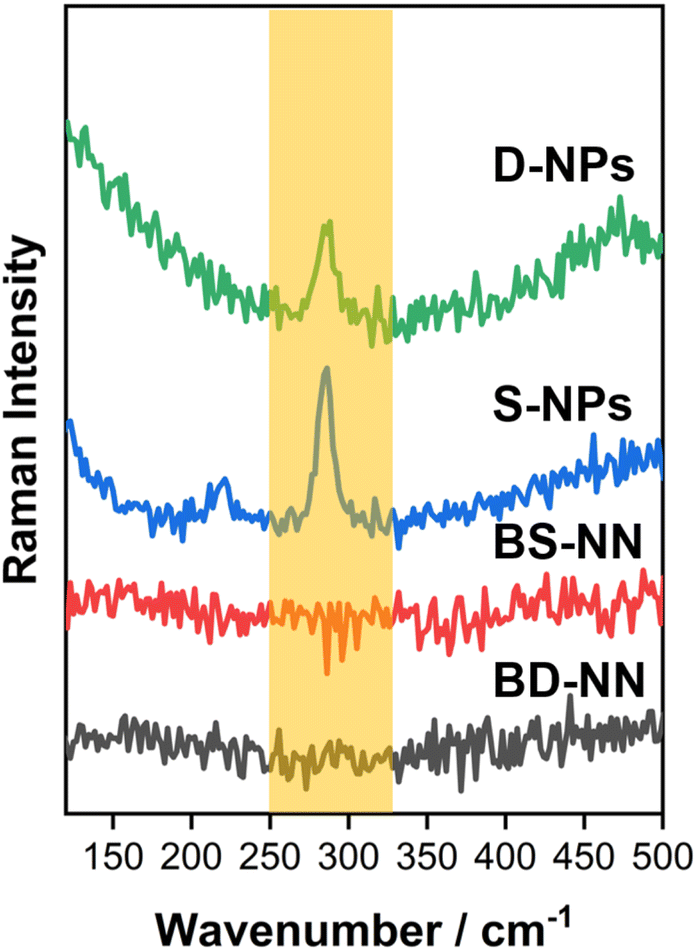 | ||
| Fig. 4 Raman spectrum of BS-NN, BD-NN, S-NPs and D-NPs. | ||
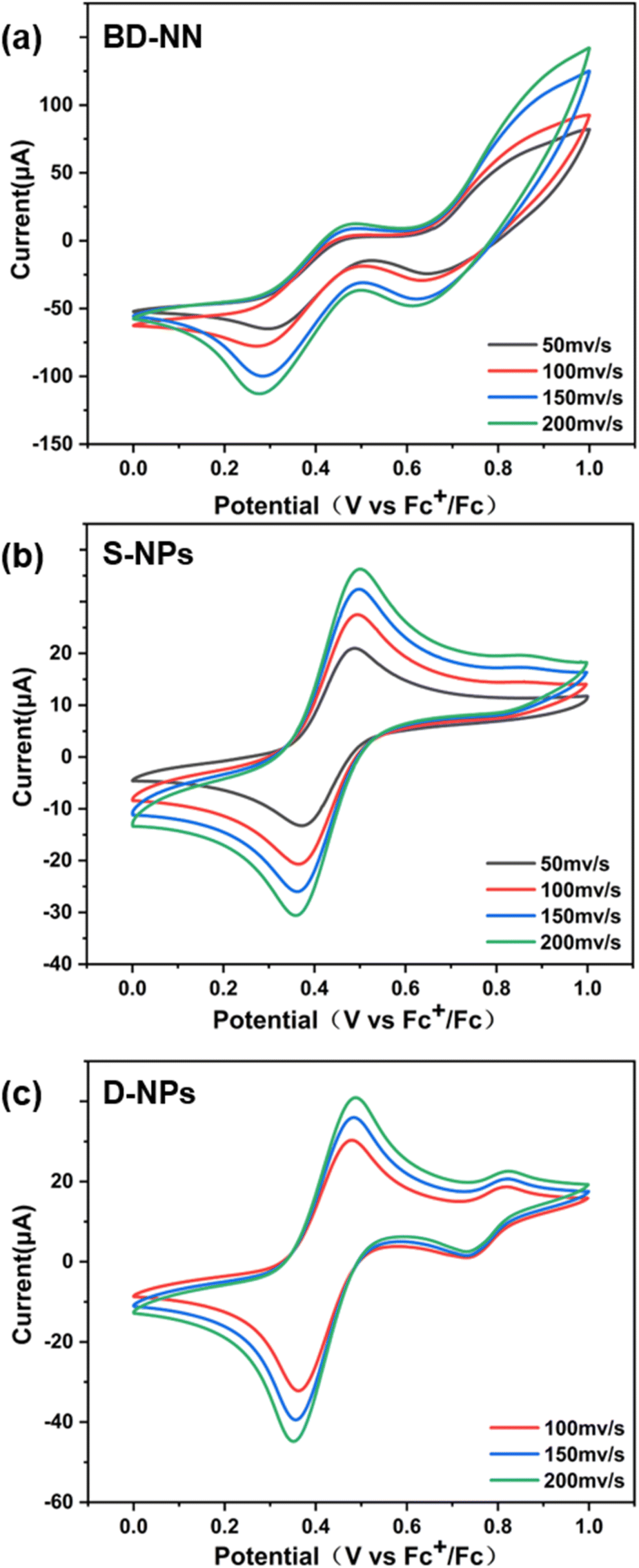 | ||
| Fig. 5 (a) CV spectrum of BD-NN; (b) CV spectrum of S-NPs; (c) CV spectrum of D-NPs. Each set of tests adopts 50 mV s−1, 100 mV s−1, 150 mV s−1, and 200 mV s−1 sweep speeds. The CVs were all tested in CH3CN with 0.02 M tetrabutylammonium hexafluorophosphate as electrolyte and Pt as working electrode. | ||
In addition, an increase in the scanning rate led to higher peak intensities, as observed from the CV data. This result can be attributed to surface-restricted electroactive substances, further confirming the successful preparation of radical-grafted nanoparticles. Furthermore, multiple groups of tests under the same conditions yielded nearly identical redox waves and current intensities, enhancing the accuracy of this dataset.
Discussion of radicals packing density
TEM. We conducted transmission electron microscopy (TEM) analysis to examine the samples. The test results (Fig. 6a and c) revealed that the S-NPs exhibited excellent dispersion, and nearly all of them appeared spherical in shape. The sample data were statistically analyzed using ImageJ software (Fig. 6b and d), which calculated the average particle size of S-NPs was 2.57 nm, primarily distributed within the range of 1.5–3 nm. Similarly, the average particle size of D-NPs was determined to be 2.54 nm, mainly distributed within the range of 1.5–2.5 nm. Particle size was also validated by DLS data (Fig. S6†).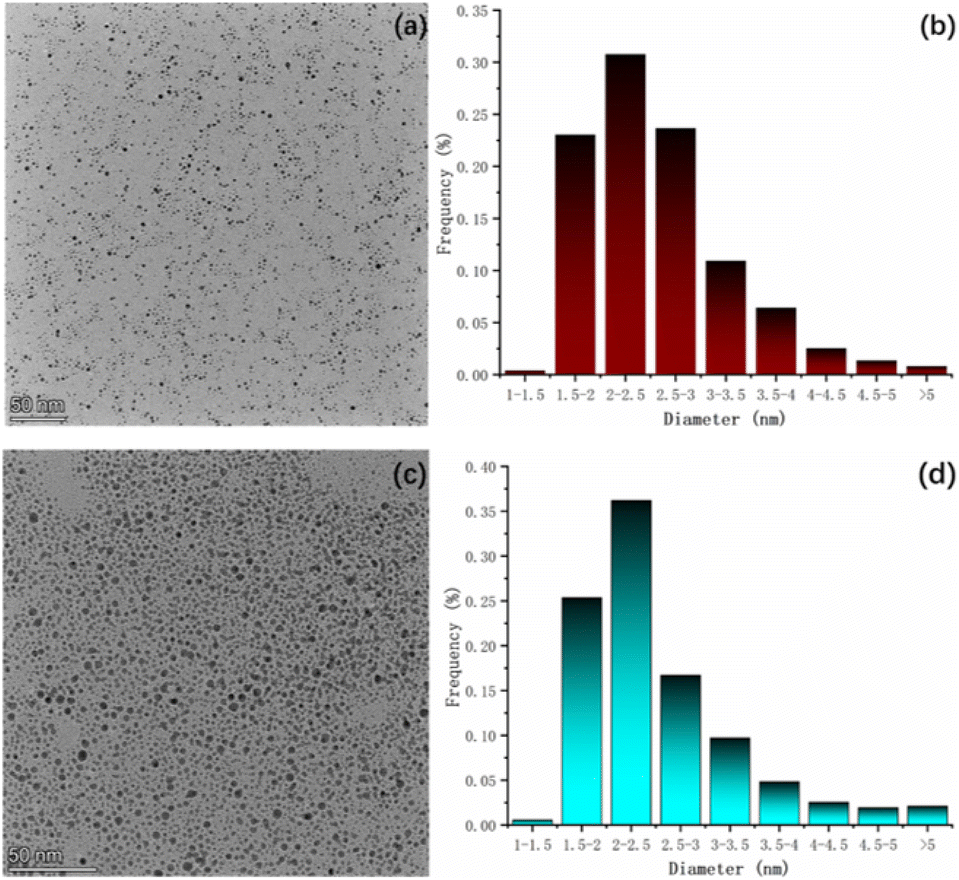 | ||
| Fig. 6 (a) TEM spectrum of S-NPs; (b) statistical graph of particle size of S-NPs; (c) TEM spectrum of D-NPs; (d) statistical graph of particle size of D-NPs. | ||
The slightly smaller particle size of D-NPs compared to S-NPs can be attributed to the structural characteristics of the radicals involved. Firstly, the linear structure of the double radicals is not as long as that of the single radicals. Secondly, the linear structure of the double radicals is not as bulky as that of the single radicals. These factors contribute to the smaller size of D-NPs. The stereo-hindrance effect leads to an increase in the packing density of the NN moiety but a decrease in the packing density of the flexible chain moiety. Consequently, the organic portion's content is reduced, resulting in smaller particle size for D-NPs.
EDS. In order to determine the elemental distribution of the samples, EDS tests were carried out on several synthesized samples, respectively (Fig. S2–S5†). The test spectra are presented in Fig. S2–S5.† The data were averaged over the four groups tested and statistically presented in Table 1, and the counting calculation data are shown in Table 2. The calculation reveal that the self-assembled density of S-NPs is 13.2 Å2, which is different from the previous reports. This discrepancy can be attributed to the following three reasons: (i) different elements selected for counting radicals; (ii) variances in the radii of prepared nanoparticles; (iii) variations in specific surface area. The self-assembled density of D-NPs is 2.8 Å2, indicating a significant increase in the packing density compared to S-NPs.
| Product | Element | Line type | Weight% | Atomic% |
|---|---|---|---|---|
| AuNPs | Au | M series | 97.38 | 72.58 |
| N | K series | 2.62 | 27.42 | |
| S-NPs | Au | M series | 81.77 | 27.70 |
| S | K series | 3.67 | 7.65 | |
| N | K series | 6.59 | 31.41 | |
| O | K series | 7.97 | 33.24 | |
| D-NPs | Au | M series | 55.91 | 10.39 |
| S | K series | 13.08 | 14.94 | |
| N | K series | 11.42 | 29.85 | |
| O | K series | 19.58 | 44.82 |
| Product | Particle diameter (nm) | AuNPs area (Å2) | The average number of NN per AuNP | Maximum packing density S0 (Å2) |
|---|---|---|---|---|
| S-NPs | 2.57 | 2075 | 157 | 13.2 |
| D-NPs | 2.54 | 2027 | 728 | 2.8 |
Both S-NPs and D-NPs were synthesized from the same batch of AuNPs, the influence of the AuNPs size can be excluded. The radical packing density of D-NPs is approximately 4.75 times higher than that of S-NPs, which can be attributed to three factors: (i) the BD-NN structure contains twice as many NN as BS-NN; (ii) interactions between the two radical groups in the m-phenylene diradical on the D-NPs may reduce the steric hindrance between the radical groups, thus contributing to a more ordered arrangement of the radicals on the AuNPs; (iii) the strong magnetic interactions between the radicals on the gold surface may facilitate their close packing. In summary, it can be concluded that self-assembly using multi-radicals is indeed an effective strategy for enhancing the compact packing of radicals.
Magnetic properties study
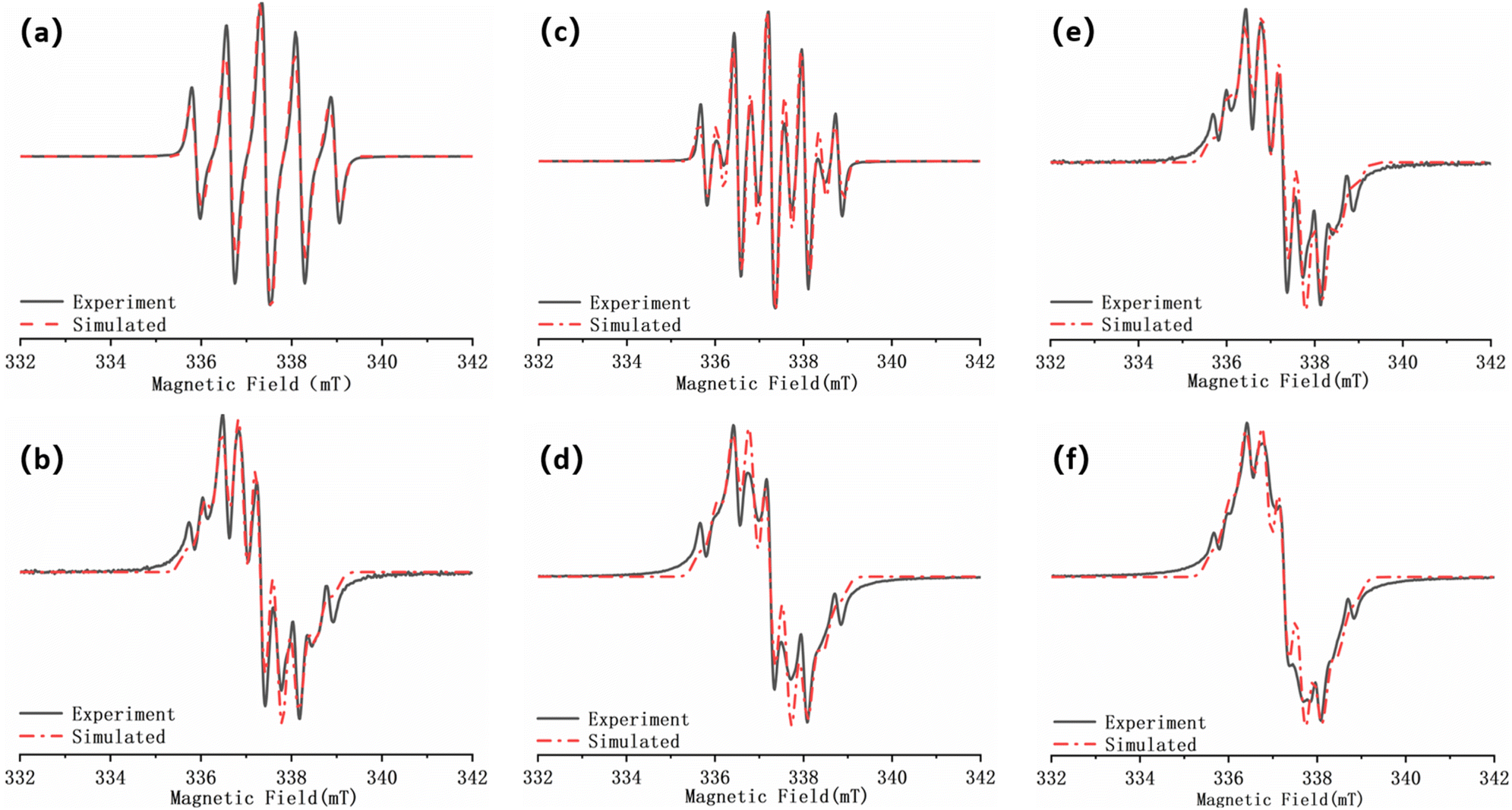 | ||
| Fig. 7 (a) EPR spectrum of S-NN; (b) EPR spectrum of D-NN; (c) EPR spectrum of BS-NN; (d) EPR spectrum of BD-NN; (e) EPR spectrum of S-NPs; (f) EPR spectrum of D-NPs. The black curves are experimental data and the red curves are fitted data. (a), (b), (c), (d) were obtained by testing in DCM and (e), (f) were obtained by testing in toluene. All spectra were obtained at room temperature. | ||
| Sample | λ (nm) | ε (cm−1 M−1) | g | aN/2 (G) | |D/hc| (cm−1) | r (Å) |
|---|---|---|---|---|---|---|
| S-NN | — | — | 2.00650 | 7.50 | — | — |
| D-NN | — | — | 2.00675 | 3.55 | — | — |
| BS-NN | 578619 |
36853744 |
2.00685 | 7.50 | — | — |
| BD-NN | 581634 |
20061822 |
2.00690 | 3.65 | 0.00541 | 7.88 |
| S-NPs | 623 | — | 2.00690 | 3.70 | 0.00531 | 7.93 |
| D-NPs | 628 | — | 2.00690 | 3.55 | 0.00550 | 7.84 |
The packing molar ratio (HAuCl4:B-NN = 5:1) was selected,22 and the gradient test plots are shown in Fig. S7 and S8.† According to the previous reports,33 at room temperature, the single radical of NN are five-line spectra, whereas the well-coupled double radicals present nine-line spectra. Fig. 7a illustrates S-NN, which shows a standard five-line spectra. The fitting results yields a g value of 2.00650 and aN = 7.5 G, consistent with previous reports, which is consistent with typical NN-type single radical hyperfine splitting features.
Due to the flexible nature of the bi-radical skeleton in BS-NN, the coupling between the bi-radicals is primarily spatial, and the coupling strength is weak. As a result, the spectra differ from the standard nine-line spectra of strongly interacting two-radicals, as verified in Fig. 7c. For D-NN, the fit yields the g value of 2.00675 and aN = 3.55 G, which is about half of the hyperfine splitting in a typical NN single radical. However, the overall spectral line splitting is poorer (Fig. 7b). This can be attributed to the close proximity of the interstitial double radical on benzene, leading to strong intramolecular spin–spin interactions. When esterification is performed to synthesize BD-NN (Fig. 7d), the radicals within the molecule strongly interact with each other. Consequently, the uniformity of line splitting is further compromised. The EPR spectra of both S-NPs (Fig. 7e) and D-NPs (Fig. 7f) resemble the spectra of their precursor B-radicals when the AuNPs with maximum packing density are prepared at the suitable ratio. However, their linewidths closely approach their aN value, resulting in poorer linewidth uniformity and signal intensity. This can be attributed to the close packing of radicals on the surface of AuNPs, which further strengthens the spatial coupling between the radicals.
When radicals are spatially close to the gold surface, spin conduction and coupling at the gold surface is achieved through complex spin–spin interactions (including spatial, chemical bonding, and spin off-domains on gold surface).22 These interactions may contribute to the deterioration of the ultrafine cleavage fraction of EPR signals in the gold-nanoscale self-assembled samples.
In order to further investigate the magnetic coupling mechanism between the densely packed radicals on AuNPs, the cryogenic solution EPR spectra of all the samples in 10–100 K DCM were recorded, and the J values were obtained from the fitting of IT-T diagrams:18 Fitting the plot of IT versus T gives ΔES-T = +130.69 cal mol−1, 2J kB−1 = +66.89 K for S-NPs and ΔES-T = +81.16 cal mol−1, 2J kB−1 = +41.54 K for D-NPs (Fig. 8) (Table 4). Among them, BD-NN and D-NPs exhibit clear |ΔMs| = 2 signals in the half-field (Fig. S8†). From the test spectra, the experimental parameter D value can be obtained. In general, a larger D value implies a closer intramolecular spin–spin distance. Using the point approximation, 2D = 3g2 μB/r3, the dipole–dipole distance can be derived approximately and used to analyze the distances of neighboring spins within the molecule. The value of D depends on the dipole–dipole interactions in the spin–spin system and not directly on the exchange interactions. Consequently, the value of D reflects the spatial distance. Theoretically, the radical portions on S-NPs should be isolated from each other without intramolecular interactions. However, the disulfide bond-breaking self-assembly is a dynamically reversible process, and under high-density packing conditions, it may partially convert back to BS-NN. On the other hand, it has been reported that the self-assembled radicals on the gold surface can result in a spin–spin interaction similar to the intramolecular spin–spin interactions. This explains the existence of certain intramolecular interactions in S-NPs. All tested and calculated data are shown in Fig. S8† and Table 3. The plot of IT vs. T was fitted with the transformed Bleaney–Bowers eqn (1).34 Where BS-NN consistent with previously reported spectra,22 exhibit a well-defined zero-field splitting (zfs) pattern, this is a typical EPR pattern for mono-radical phenyl nitronyl nitroxide. BS-NN shows a weak peak at |ΔMs| = 2, suggesting very weak dipole interactions within the molecule, mainly due to the excessive distances along the bond between the spin units. For S-NPs, |D/hc| = 0.00531 cm−1, which calculates to an average spin–spin distance of 7.93 Å. For D-NPs, |D/hc| = 0.00550 cm−1, which calculates to an average spin–spin distance of 7.84 Å. It can be clearly observed that D-NPs achieve smaller spin–spin distances relative to S-NPs. This implied that the D-NPs achieve higher radical packing densities.
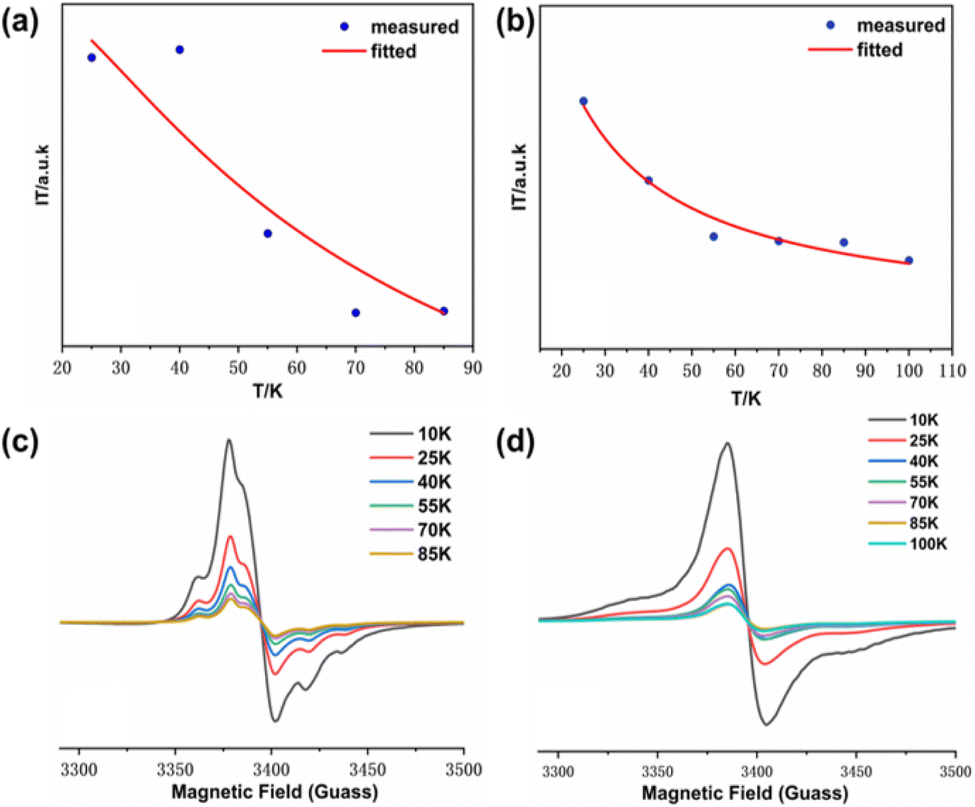 | ||
| Fig. 8 (a) IT-T fitted images of S-NPs; (b) IT-T fitted images of D-NPs; (c) EPR spectra of S-NPs measured over a temperature gradient; (d) EPR spectra of D-NPs measured over a temperature gradient. All samples are solved in DCM for test. | ||
The magnetic properties analyzed by the variable-temperature EPR test spectra are different from previously reported.22 We believe that this difference is due to the lack of formula fitting and only empirical judgment in previous work. In fact, the thiol-modified gold nanoparticles are ferromagnetic,35 and the radicals used for self-assembly are also ferromagnetic,36 so the NN modified gold nanoparticles should also be ferromagnetic. This was confirmed by subsequent SQUID analysis as well.
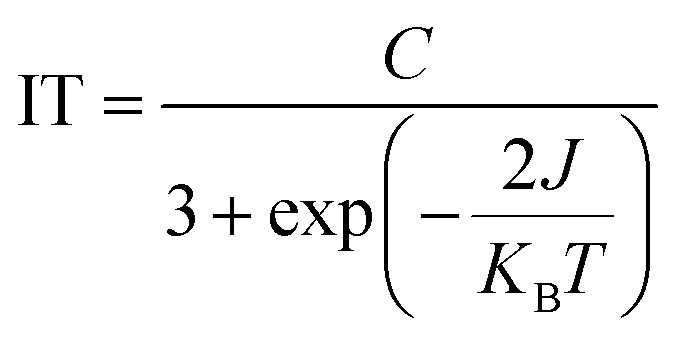 | (1) |
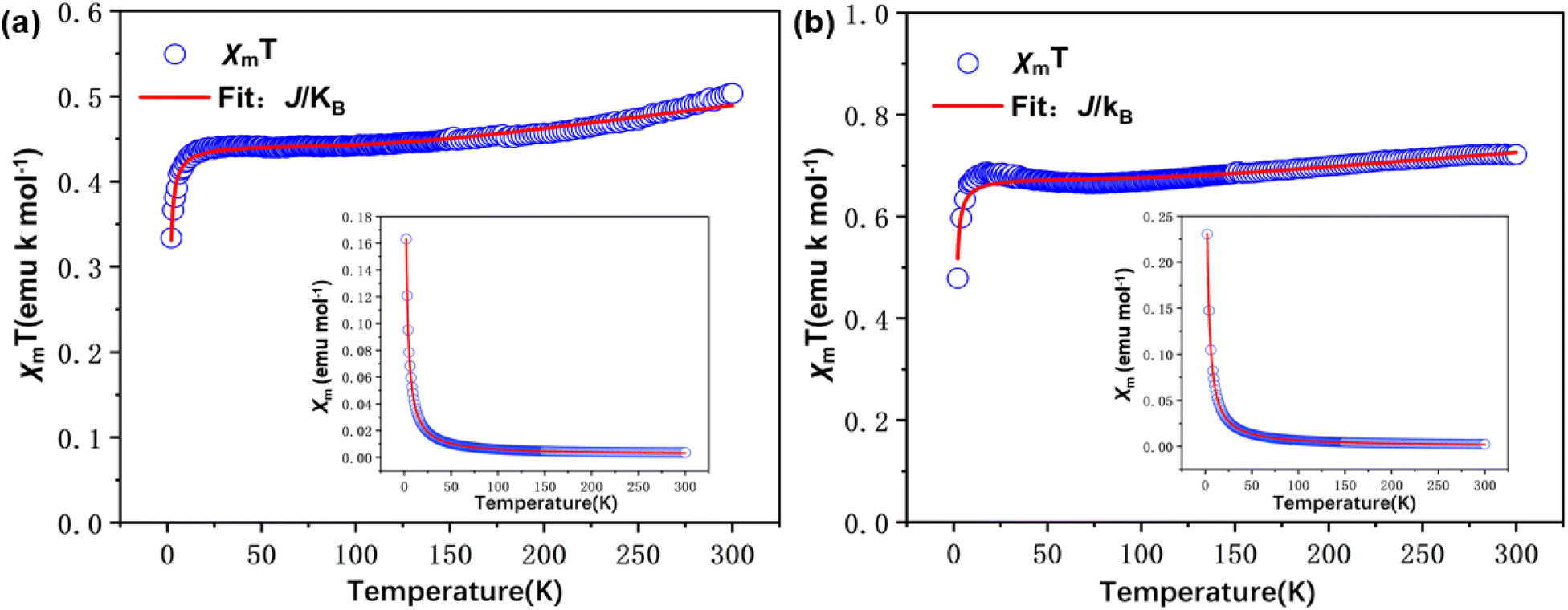 | ||
| Fig. 9 SQUID magnetometry of solid samples: plots of χmT vs. T at H = 10000.31543 Oe in the warming mode (blue circle). Inset plots: χm vs. T in the warming mode and fitting (solid red line) curves of (a) S-NPs; (b) D-NPs. The drawing data is subtracted from the background. | ||
In order to obtain the relevant parameters of the sample by fitting the test curve, we used the Hamiltonian H = −2JS1S2 (where S1 = S2 = 1/2). The processed data were corrected and calculated as χm vs. T and χmT vs. T, and fitted using the Bleaney–Bowers eqn (2).38
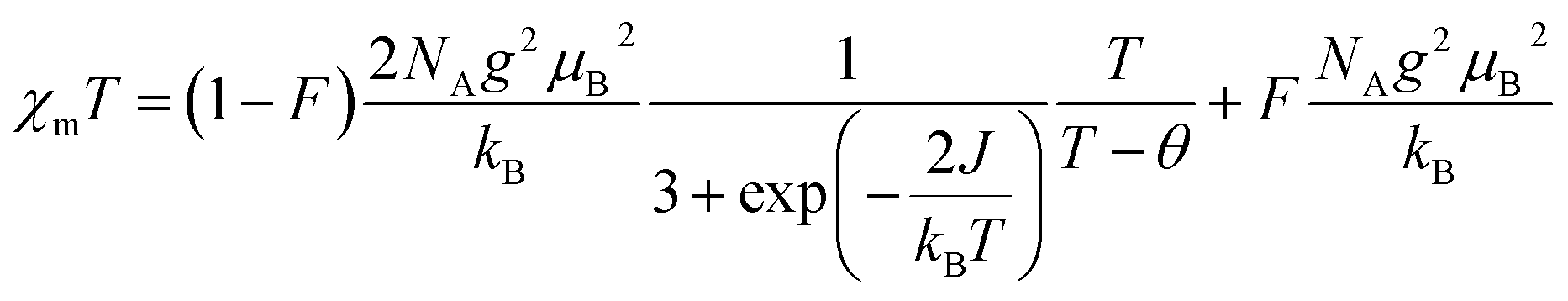 | (2) |
Measurements were performed over the temperature range of 2–300 K at a fixed field strength of 10000 Oe. According to the results fitted by eqn (2), both S-NPs and D-NPs exhibit strong ferromagnetism: JS-NPs = +279.715 K and JD-NPs = +254.913 K, the results with the J obtained from VT-EPR were shown in Table 4. The χmT − T curve of the S-NPs and D-NPs exhibited temperature dependence during the warming process. At 300 K, the χmT of S-NPs reaches approximately 0.490 cm3 K mol−1, which exceeds the theoretical value of 0.375 cm3 K mol−1 for single radicals. This can be attributed to the strong ferromagnetic coupling interaction. When the temperature is cooled down from 300 K to 58 K, the value of χmT decreases slowly and uniformly. This can be explained to the slightly intermolecular antiferromagnetic coupling. As the temperature decreases from 58 K to 23 K, the χmT value exhibits a slight increase, illustrating the ferromagnetism in S-NPs. At 300 K, the χmT of D-NPs reaches approximately 0.770 cm3 K mol−1, which is basically consistent with the theoretical value of 0.750 cm3 K mol−1 for the diradical. The χmT gradually decreased during cooling from 300 K to 73 K. This is mainly due to the slightly intermolecular antiferromagnetic coupling caused by the spatial motion of the radical-connected soft chains in the high-temperature region. Upon further cooling from 73 K to 25 K, the χmT of D-NPs shows a relatively obvious increase. This illustrates the ferromagnetism in D-NPs.39,40
The first striking feature to be observed in the M-H magnetization reversal loops is the presence of hysteresis and a remnant magnetization, all indicative of ferromagnetic behavior (Fig. 10) at both room temperature 1.9 K and 300 K. The M value is higher than that of the previously reported alkyl mercaptan modified gold nanoparticles,35 indicating the presence of better ferromagnetism. Among them, the M of D-NPs is higher than S-NPs, which is consistent with magnetic analysis from χmT vs. T and VT-EPR.
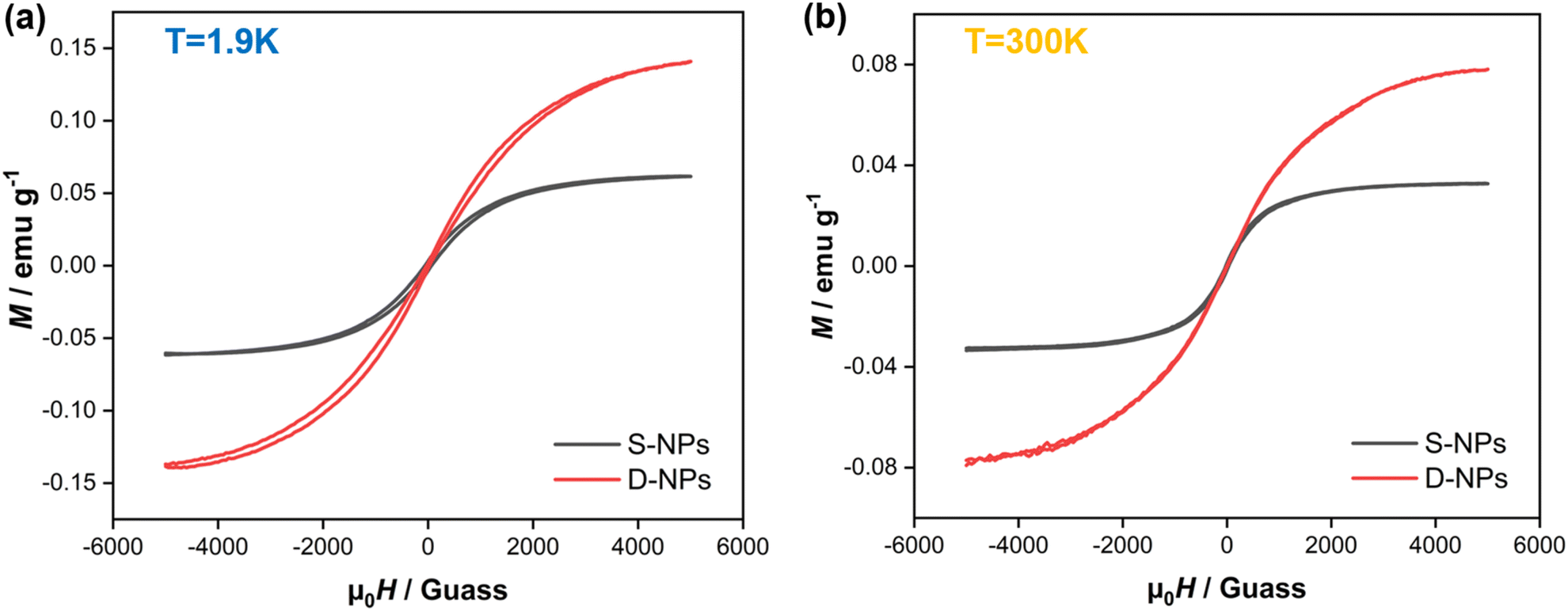 | ||
| Fig. 10 Magnetization reversal loops measured at (a) 1.9 K and (b) 300 K. | ||
We compared the magnetic properties of S-NPs with D-NPs. The J value of S-NPs is slightly larger than D-NPs. The two NN groups which are meta-connected on the benzene have an intramolecular ferromagnetism of 23 K.36 The intramolecular interactions are present in D-NPs while absent in S-NPs. Besides, intermolecular spin interactions through space are generally antiferromagnetic.41 The radical packing density of D-NPs is 4.75 times higher than S-NPs, resulting in a stronger intermolecular antiferromagnetism through space directly. The combined effect of intramolecular interactions and spin interactions through space results in the J value of S-NPs being slightly larger than D-NPs. The occurrence of ferromagnetism at Au–S interfaces has been observed, and the ferromagnetism has been associated with Au 5d localized holes that are the result of charge transfer from the Au surface atoms to the S atoms of the organic ligands when forming the Au–S bonds.35,42 There are A–S bond on Au surface in both S-NPs and D-NPs, and we refer to the resulting ferromagnetism as spin interactions through the gold surface. It can be deduced that the considerable ferromagnetism in S-NPs and D-NPs should be attributed to the spin interactions through Au surface primarily. This analysis also suggests that the strategy of achieving close-packing through Au–S self-assembly to obtain a material with strong magnetic coupling is a feasible approach.
Conclusions
Herein, a series of self-assembled organic spin hybrid samples were prepared based on the AuNPs by utilizing the binding of Au–S bonds and innovatively choosing NN-type double radicals. The accuracy of the structural synthesis of the samples was verified by UV, IR, MS, Raman spectrum and CV tests. The morphology and radical packing density of the samples were discussed by TEM and EDS. The magnetic properties of the samples were investigated and analyzed by use EPR and SQUID. The results of packing density analysis indicate that the self-assembled strategy employing two radicals achieved higher packing densities on the gold surface and that the D-NPs achieved higher packing densities on the AuNPs surface than those previously reported for NN type radicals. Magnetic analysis showed that S-NPs and D-NPs exhibited a higher ferromagnetism than previously reported alkyl mercaptan modified gold nanoparticles and most of NN type organic radicals, which is the superposition of the ferromagnetism due to the Au 5d localized holes on the gold surface and the spin magnetism of NN. Besides, it shows that the use of the radical self-assembly strategy on the gold surface is a promising approach for synthesizing macroscopic ferromagnets in the aggregated state. In addition, AuNPs with magnetic properties have a wide range of applications in the biomaterials, such as drug targeting, magnetic imaging. This study provides a valuable strategy for the preparation of organic magnetic hybrid magnets with tightly ordered organic spin structures.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Zhipeng Xu: conceptualization, investigation, writing – original draft. Dongdong Wei: investigation, validation. Yongliang Qin: formal analysis. Jie Jin: supervision. Long Zheng: resources. Jie Xu: software. Hui Liu: methodology. Ranran Chen: resources. Di Wang: funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the following funds: National Natural Science Foundation of China (No. 52003004), Anhui Provincial Outstanding Youth Scientific Research Project (No. 2023AH030036), China Railway Scientific Research Project (HYB20200183). Natural Science Foundation of Anhui Province (2108085QE216), Natural Science Foundation of the Anhui Higher Education Institutions (2022AH050243).Notes and references
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 CrossRef CAS PubMed.
- R. A. Layfield, Organometallics, 2014, 33, 1084–1099 CrossRef CAS.
- I.-J. Baek and W.-J. Cho, Solid-State Electron., 2018, 140, 129–133 CrossRef CAS.
- W. Canon-Mancisidor, M. Zapata-Lizama, P. Hermosilla-Ibanez, C. Cruz, D. Venegas-Yazigi and G. Minguez Espallargas, Chem. Commun., 2019, 55, 14992–14995 RSC.
- K. Taniguchi, M. Nishio, N. Abe, P. J. Huang, S. Kimura, T. H. Arima and H. Miyasaka, Angew. Chem., Int. Ed. Engl., 2021, 60, 14350–14354 CrossRef CAS PubMed.
- F. Liu, X. Hou, B. Hu and R. Li, Molecules, 2021, 26, 6130 CrossRef CAS PubMed.
- K. Kolanji, P. Ravat, A. S. Bogomyakov, V. I. Ovcharenko, D. Schollmeyer and M. Baumgarten, J. Org. Chem., 2017, 82, 7764–7773 CrossRef CAS PubMed.
- M. Mas-Torrent, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2012, 112, 2506–2527 CrossRef CAS PubMed.
- A. C. Petre Ionita, B. C. Gilbert and V. Chechik, J. Am. Chem. Soc., 2002, 124, 9048–9049 CrossRef PubMed.
- E. V. Tretyakov, P. A. Fedyushin, E. V. Panteleeva, D. V. Stass, I. Y. Bagryanskaya, I. V. Beregovaya and A. S. Bogomyakov, J. Org. Chem., 2017, 82, 4179–4185 CrossRef CAS PubMed.
- P. Ravat, Y. Borozdina, Y. Ito, V. Enkelmann and M. Baumgarten, Cryst. Growth Des., 2014, 14, 5840–5846 CrossRef CAS.
- R. Rausch, A. M. Krause, I. Krummenacher, H. Braunschweig and F. Wurthner, J. Org. Chem., 2021, 86, 2447–2457 CrossRef CAS PubMed.
- V. Lloveras, P. Elias-Rodriguez, L. Bursi, E. Shirdel, A. R. Go Experimental assistanceni, A. Calzolari and J. Vidal-Gancedo, Nano Lett., 2022, 22, 768–774 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- D. Ling, N. Lee and T. Hyeon, Acc. Chem. Res., 2015, 48, 1276–1285 CrossRef CAS PubMed.
- T. Furui, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui, K. Okada, E. V. Tretyakov, S. E. Tolstikov, G. V. Romanenko and V. I. Ovcharenko, Inorg. Chem., 2014, 53, 802–809 CrossRef CAS PubMed.
- R. Tanimoto, T. Wada, K. Okada, D. Shiomi, K. Sato, T. Takui, S. Suzuki, T. Naota and M. Kozaki, Inorg. Chem., 2022, 61, 3018–3023 CrossRef CAS PubMed.
- Z. Yue, J. Liu, M. Baumgarten and D. Wang, J. Phys. Chem. A, 2023, 127, 1565–1575 CrossRef CAS PubMed.
- D. Wang, Y. Ma, B. Wolf, A. I. Kokorin and M. Baumgarten, J. Phys. Chem. A, 2018, 122, 574–581 CrossRef CAS PubMed.
- B. Mladenova-Kattnig, G. Grampp and A. I. Kokorin, Appl. Magn. Reson., 2015, 46, 1359–1366 CrossRef CAS.
- Y. Gao, J. Hu and Y. Ju, Acta Chim. Sin., 2016, 74, 312–329 CrossRef CAS.
- V. Loveras, E. Badetti, J. Veciana and J. Vidal-Gancedo, Nanoscale, 2016, 8, 5049–5058 RSC.
- Y. Wang, N. S. Hush and J. R. Reimers, J. Am. Chem. Soc., 2007, 129, 14532–14533 CrossRef CAS PubMed.
- M. S. Inkpen, Z. F. Liu, H. Li, L. M. Campos, J. B. Neaton and L. Venkataraman, Nat. Chem., 2019, 11, 351–358 CrossRef CAS PubMed.
- Y. Ohya, N. Miyoshi, M. Hashizume, T. Tamaki, T. Uehara, S. Shingubara and A. Kuzuya, Small, 2012, 8, 2335–2340 CrossRef CAS PubMed.
- J. Hu, R. Jiang, H. Zhang, Y. Guo, J. Wang and J. Wang, Nanoscale, 2018, 10, 18473–18481 RSC.
- S. Rautiainen, J. Chen, M. Vehkamäki and T. Repo, Top. Catal., 2016, 59, 1138–1142 CrossRef CAS.
- P. D. Ortiz, J. Castillo-Rodriguez, X. Zarate, R. Martin-Trasanco, M. Benito, I. Mata, E. Molins and E. Schott, Langmuir, 2018, 34, 9402–9409 CrossRef CAS PubMed.
- D. J. Lavrich, S. M. Wetterer, S. L. Bernasek and G. Scoles, J. Phys. Chem. B, 1998, 102, 3456–3465 CrossRef CAS.
- C. K. A. Nyamekye, S. C. Weibel and E. A. Smith, J. Raman Spectrosc., 2021, 52, 1246–1255 CrossRef CAS.
- X. Feng, J. Yang, X. Duan, Y. Cao, B. Chen, W. Chen, D. Lin, G. Qian, D. Chen, C. Yang and X. Zhou, ACS Catal., 2018, 8, 7799–7808 CrossRef CAS.
- C. Qi, Y. Cheng, Z. Yang, T. Ishida, H. Su, J. Zhang, X. Sun, L. Sun, L. Zhao and T. Murayama, J. Catal., 2024, 436, 11560 CrossRef.
- U. Eichhoff and P. Höfer, Appl. Magn. Reson., 2020, 51, 1723–1737 CrossRef CAS.
- S. Hase, D. Shiomi, K. Sato and T. Takui, J. Mater. Chem., 2001, 11, 756–760 RSC.
- P. Dong, E. A. Fisher, M.-V. Meli and S. Trudel, Nanoscale, 2020, 12, 19797–19803 RSC.
- M. E. Ali and S. N. Datta, J. Phys. Chem. A, 2006, 110, 2776–2784 CrossRef CAS PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- C. Shi, L. Gao, M. Baumgarten, D. Wei, Z. Xu, W. Wang and D. Wang, Magnetochemistry, 2023, 9, 178 CrossRef CAS.
- K. Tanaka, K. Furuichi, M. Kozaki, S. Suzuki, D. Shiomi, K. Sato, T. Takui and K. Okada, Polyhedron, 2007, 26, 2021–2026 CrossRef CAS.
- L. Catala, J. Le Moigne, N. Kyritsakas, P. Rey, J. J. Novoa and P. Turek, Chem. - Eur. J., 2001, 7, 2466–2480 CrossRef CAS PubMed.
- T. Mitsumori, K. Inoue, N. Koga and H. Iwamura, J. Am. Chem. Soc., 1995, 117, 2467–2478 CrossRef CAS.
- M. Suda, N. Kameyama, M. Suzuki, N. Kawamura and Y. Einaga, Angew. Chem., 2008, 120, 166–169 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04506h |
| This journal is © The Royal Society of Chemistry 2024 |