DOI:
10.1039/D4RA03369H
(Paper)
RSC Adv., 2024,
14, 27799-27808
Bridging the size gap between experiment and theory: large-scale DFT calculations on realistic sized Pd particles for acetylene hydrogenation†
Received
7th May 2024
, Accepted 18th August 2024
First published on 2nd September 2024
Abstract
Metal nanoparticles, often supported on metal oxide promoters, are a cornerstone of heterogeneous catalysis. Experimentally, size effects are well-established and are manifested through changes to catalyst selectivity, activity and durability. Density Functional Theory (DFT) calculations have provided an attractive way to study these effects and rationalise the change in nanoparticle properties. However such computational studies are typically limited to smaller nanoparticles (approximately up to 50 atoms) due to the large computational cost of DFT. How well can such simulations describe the electronic properties of the much larger nanoparticles that are often used in practice? In this study, we use the ONETEP code, which is able to achieve more favourable computational scaling for metallic nanoparticles, to bridge this size gap. We present DFT calculations on entire Pd and Pd carbide nanoparticles of more than 300 atoms (approximately 2.5 nm diameter), and find major differences in the electronic structure of such large nanoparticles, in comparison to the commonly investigated smaller clusters. These differences are also manifested in the calculated chemical properties such as adsorption energies for C2H2, C2H4 and C2H6 on the pristine Pd and PdCx nanoparticles which are significantly larger (up to twice in value) for the ∼300 atoms structures. Furthermore, the adsorption of C2H2 and C2H4 on PdCx nanoparticles becomes weaker as more C is introduced in the Pd lattice whilst the impact of C concentration is also observed in the calculated reaction energies towards the hydrogenation of C2H2, where the formation of C2H6 is hindered. Our simulations show that PdCx nanoparticles of about 5% C per atom fraction and diameter of 2.5 nm could be potential candidate catalysts of high activity in hydrogenation reactions. The paradigm presented in this study will enable DFT to be applied on similar sized metal catalyst nanoparticles as in experimental investigations, strengthening the synergy between simulation and experiment in catalysis.
1. Introduction
The selective hydrogenation of acetylene has been extensively investigated as an important purification process of ethylene feedstocks in the production of polyethylene.1–4 During the exposure to hydrocarbons, the catalyst will adsorb acetylene, which strongly binds with Pd surface atoms. This will reduce the adsorption of ethylene whilst promoting the full hydrogenation of acetylene to ethane. Therefore, to increase selectivity towards ethylene via tuning the catalytic process, it is important to understand the mechanisms of ethylene and ethane formation as well as side-reactions like surface C–C oligomerization (that leads to green oil formation and deactivation of the catalyst).5,6
Supported Pd nanoparticles (NPs) are promising catalysts, widely used due to their high activity in a range of industrial applications7 such as the hydrogenation and oxidation of hydrocarbons and conversion of biomass;8 whilst being highly selective towards the partial hydrogenation of acetylene to ethylene. Additionally, the in situ formation of interstitial phases9 during catalysis, such as carbidic Pd,10 have attracted considerable interest due to their potential contribution in the increased selectivity towards the desirable products.11 Carbidic Pd is beneficial in blocking side-formation of phases such as hydrides,12,13 which otherwise would provide surface H that eventually hydrogenate ethylene to ethane. Insights on the formation mechanisms of PdC NPs are required, as well as the impact of NP size and shape with respect to the catalytic activity, aiming to tune the materials properties through controlled synthesis and achieve high stability, activity and selectivity.
The acetylene semi-hydrogenation over Pd/Al2O3 has been investigated via experimental and theoretical methods in the recent study of Gonçalves et al.14 where the reaction is modelled on a pyramidal Pd30 cluster. This study shows that full hydrogenation of C2H4 to C2H6 exhibits higher activation barriers as a first indication of Pd selectivity towards the production of ethylene. In the experimental and theoretical work of Liu et al.,15 DFT calculations show that desorption of ethylene is more favourable for the carbidic phase rather than the pristine high-coordinated Pd(111) surface. Furthermore, Vignola et al.16 investigated the C–C bond formation that leads to catalyst poisoning through the formation of oligomers. Oligomers block the active sites of the catalyst whilst consuming hydrogen that could hydrogenate acetylene to ethylene. In their study, they show that small Pd ensembles are considered as more appropriate catalysts to avoid oligomer formation, and that the particle size contribution should be further investigated as an important feature in catalytic activity. The role of subsurface C in alkyne hydrogenation has also attracted interest; in the experimental study of Teschner et al.,17 it is shown that the subsurface Pd sites filled with C or H, have a major role in the hydrogenation reactions taking place on the surface. The subsurface chemistry impact on the selective hydrogenation of ethylene has also been reported by Studt et al.18 In their theoretical study, DFT calculations have been performed showing that selectivity increases via weakening of the surface bond with adsorbates. In the case of Pd NPs, the size effect has been investigated by Sun et al.19 where experimental and computational work showed that C4 and green oil form on structures smaller than 2 nm, whilst within at a size of 2.6 nm, adsorption of ethylene becomes weaker. The C–C/C–H bonding has been investigated in the theoretical study of Zhao et al.,20 for a range of transition metal surfaces (where the most promising identified were the Pd(111) and Pt(111)), showing that the order in terms of acetylene hydrogenation activity is inverse to that of the selectivity towards ethylene. Yang et al.21 performed DFT calculations on a range of different Pd surfaces, examining the effect of subsurface C and H and showed that the close-packed Pd(111) exhibits the highest selectivity. The selective hydrogenation of acetylene in the presence of ethylene has been also investigated by Abdollahi et al.22 In their study, activation energies for the reaction process are reported for a range of Pdn (n = 2–15) nanoclusters. The Pd2 is reported to exhibit the best selectivity towards ethylene. Besides the synthesis method and characteristics such as the shape and size of the NPs, the support is also important towards activity and selectivity as reported in the experimental work by Benavidez et al.23 They showed that C supported Pd catalysts exhibit higher selectivity towards ethylene compared to oxide supported Pd catalysts. Additionally, supports such as gamma alumina may lead to green oil formation as reported by Asplund et al.24 The aforementioned works provided useful insights on the hydrogenation of acetylene on Pd catalysts as slabs and NPs, however the role of the PdCx formation on the hydrogenation reaction in realistic systems, comparable with experimental results is still required.
In this study, we address for the first time the challenge of the simulation system size in Pd based catalysis via performing large-scale DFT calculations on entire large Pd/PdCx NPs at different C concentrations. The structures used for our investigation were of more than 300 atoms and up to approximately 2.5 nm, going beyond the investigated system sizes reported so far in the literature by one order of magnitude. All geometries were fully relaxed, providing useful insights on the PdCx formation and the effect of C concentration on the hydrogenation of C2H2. The binding modes of adsorbed C2H2, C2H4 and C2H6 on the [100]/[111] facets of pristine and carbidised structures were firstly investigated and adsorption energies for the most stable configurations were obtained. Finally, we examined the impact of interstitial C at increasing (5% and 13% per atom fraction) concentrations on the reactants, intermediates, and products of the hydrogenation of C2H2 to C2H4 and C2H6.
2. Methodology
The linear-scaling DFT code ONETEP25 was used for the modelling of the pristine Pd and PdCx structures. For the construction of the density matrix, localized non-orthogonal Wannier functions (NGWFs) as expressed through a set of periodic sinc (p-sinc) functions26 were used. For these calculations, the p-sinc basis set was set to a kinetic energy cut-off of 800 eV. For the exchange and correlation interactions, the density functional of Perdew, Burke and Ernzerhof (PBE)27 was used together with the Grimme D2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 empirical correction for dispersion interactions. The core electrons were represented via norm-conserving pseudopotentials. The NGWFs and density matrix are concurrently optimized self-consistently via the Ensemble DFT (EDFT) method29 for metallic systems with a Fermi–Dirac smearing of 0.1 eV. The geometries were allowed to relax in the minimum energy configuration. For all atoms, an NGWF radius of 9.0 Bohr has been used, whilst geometry relaxations were performed at the Γ-point in cubic cells of 22–37 Å. The construction of Pd NPs was done using the ASE30 tool with the average Pd–Pd bond length of 2.74 Å. The schematic representation of the Pd and PdCx cells is generated using the CrystalMaker31 software, whilst for the reaction energies between different crystallographic configurations, the following formula is used:
28 empirical correction for dispersion interactions. The core electrons were represented via norm-conserving pseudopotentials. The NGWFs and density matrix are concurrently optimized self-consistently via the Ensemble DFT (EDFT) method29 for metallic systems with a Fermi–Dirac smearing of 0.1 eV. The geometries were allowed to relax in the minimum energy configuration. For all atoms, an NGWF radius of 9.0 Bohr has been used, whilst geometry relaxations were performed at the Γ-point in cubic cells of 22–37 Å. The construction of Pd NPs was done using the ASE30 tool with the average Pd–Pd bond length of 2.74 Å. The schematic representation of the Pd and PdCx cells is generated using the CrystalMaker31 software, whilst for the reaction energies between different crystallographic configurations, the following formula is used:
| EReaction = EProducts − EReactants |
where EProducts corresponds to the energy of the relaxed structures of products and EReactants to the energy of the relaxed structures of the reactants. For the adsorption energies of C2H2, C2H4, C2H6 and intermediates on the Pd surface the following formula is used:
| EAdsorption = EPd–adsorbate − (EPd/PdC + Eadsorbate) |
where EPd–adsorbate corresponds to the energy of the relaxed structure of Pd NP with the adsorbate, EPd/PdC to the energy of the relaxed structures for the Pd/PdCx NPs and Eadsorbate to the energy to the isolated molecule.
3. Results and discussion
3.1 Crystallography
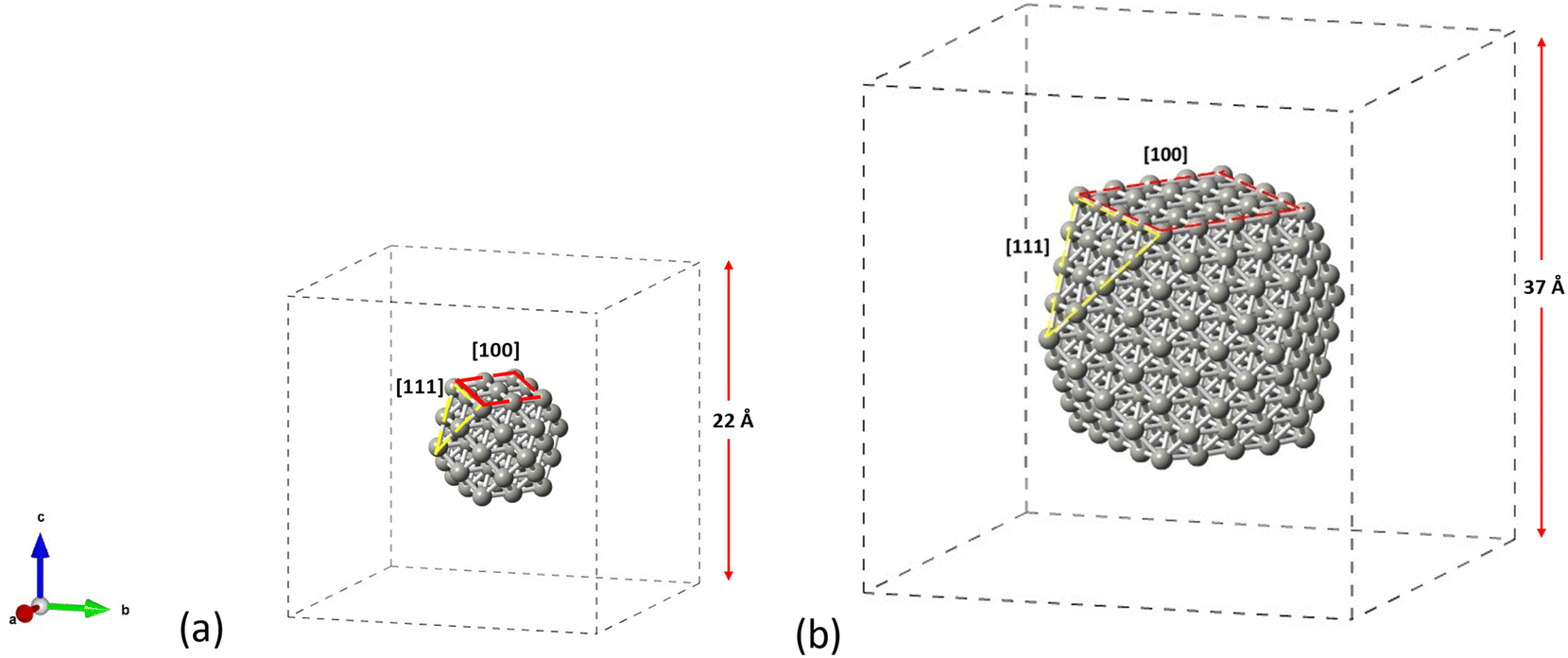
The structures considered as appropriate for this study are the cuboctahedral Pd55 and Pd309 NPs as shown in Fig. 1. We aim to account for both the [100] and [111] facets on the same particle in order to get insights on the materials performance under reaction conditions. The NP structures have been modelled in conditions of vacuum to avoid interactions with their periodic images, whilst allowing at least 5 Å of vacuum in each direction around the particle which was placed in the center of the simulation box. All structures were allowed to relax until optimized geometries were obtained. For the molecules of C2H2, C2H4 and C2H6, we performed separate geometry relaxations for the isolated structures prior to their introduction on the NP's surface.
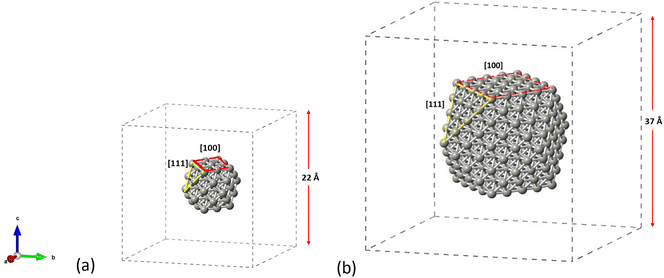 |
| | Fig. 1 Crystallographic arrangements of the NP structures showing the cuboctahedral (a) Pd55 and (b) Pd309, as placed in the centre of their simulation boxes. | |
3.2 Pd carbide NPs
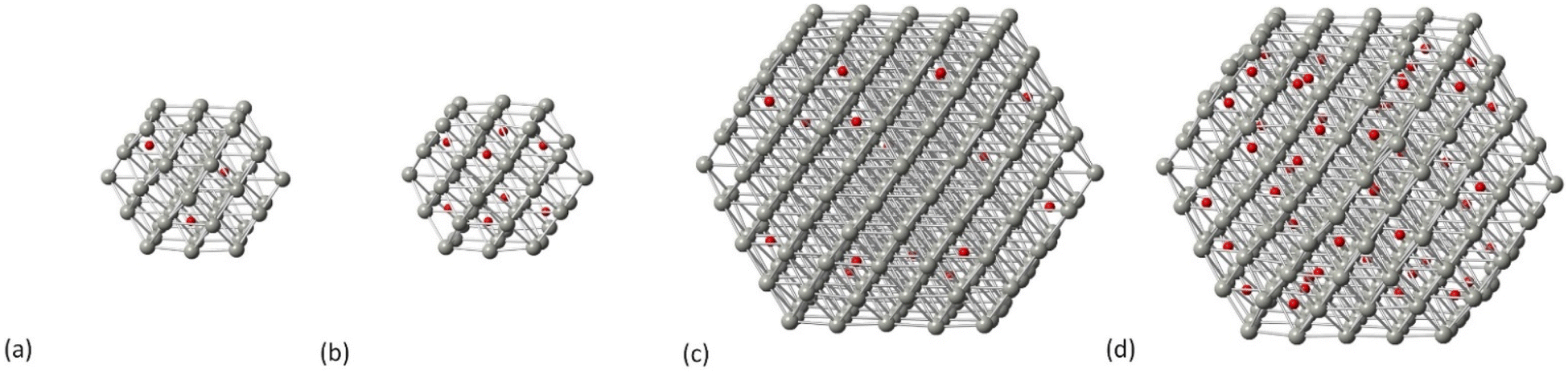
Carbidic Pd models were created through C incorporation corresponding to the occupation of the octahedral interstitial sites. In our previous work, we investigated the incorporation of C for different structures of Pd NPs,32 where we showed that there is a shape-dependent limitation of interstitial C concentration through the octahedral Pd sites; therefore, the carbidic phases considered in this work are up to a C concentration of 13% since we found that this is the maximum concentration that can be accommodated by all shapes. For the cuboctahedral structure, we have shown32 that the activation energy of carbidisation corresponds to 21.2 kJ mol−1 for the [111] facet and that, at increasing concentrations, the criterion is to allow at least one vacant site between C atoms that will preferentially occupy the subsurface area of the NP being long distance for low concentrations whilst aiming not to be in close neighboring sites at increasing amounts. In this study, two C concentrations (5% and 13%) have been considered. In Fig. 2 we show the carbidic structures for Pd55 and Pd309 whilst the average Pd–Pd bond distance for all structures is summarized in Table 1.
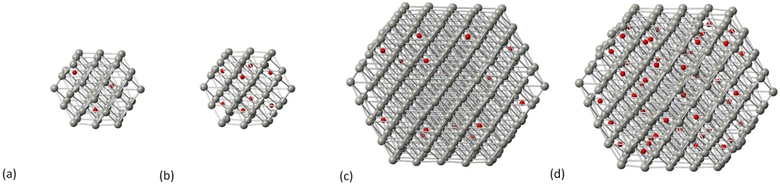 |
| | Fig. 2 The carbidic phases of Pd NPs at (a) Pd55 at 5% C concentration, (b) Pd55 at 13% C concentration, (c) Pd309 at 5% C concentration and (d) Pd309 at 13% C concentration. Grey spheres represent Pd atoms and red spheres represent C atoms. | |
Table 1 The average Pd–Pd bond length vs. C concentration in cuboctahedral Pd55 and Pd309
| C (%) |
Pd55 Pd–Pd (Å) |
Pd309 Pd–Pd (Å) |
| 0.0 |
2.73 |
2.75 |
| 5.0 |
2.74 |
2.77 |
| 13.0 |
2.75 |
2.80 |
3.3 C2H2, C2H4 and C2H6 adsorption on Pd NPs
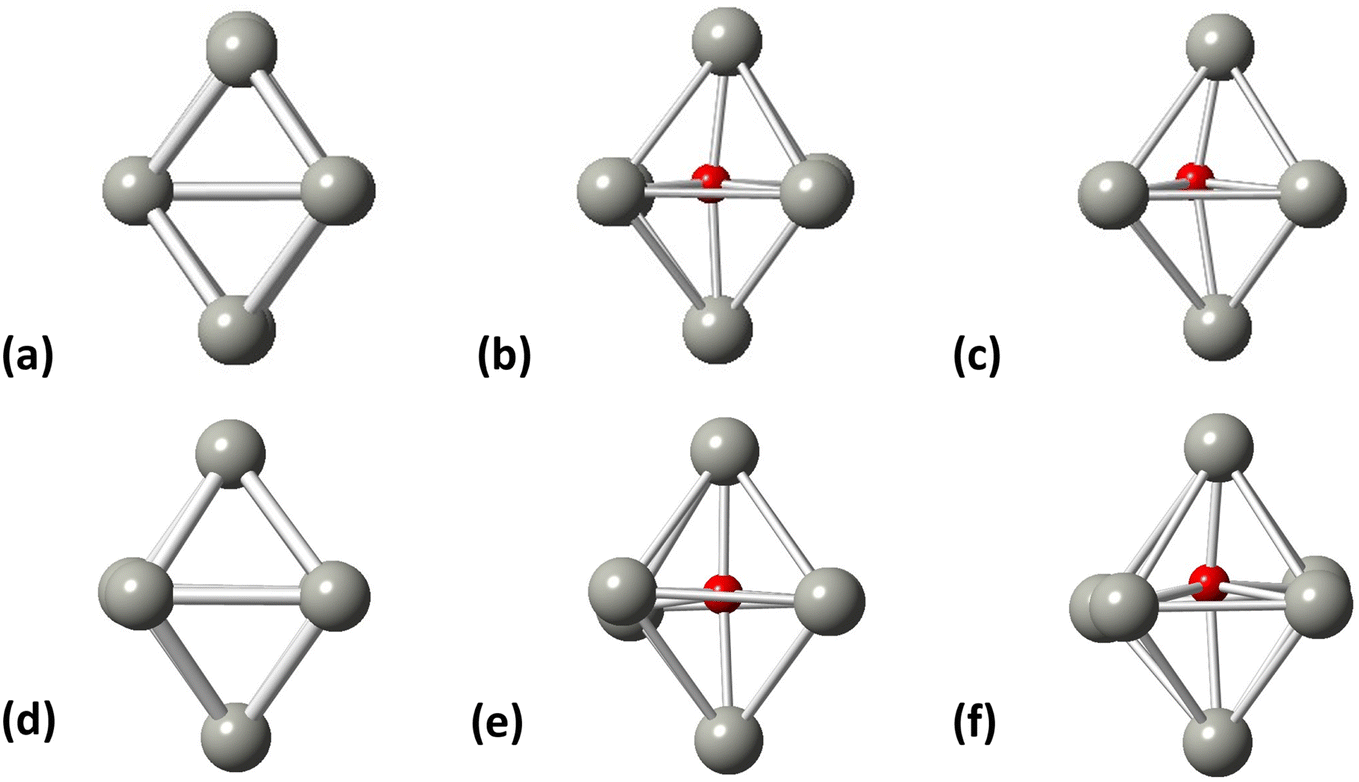
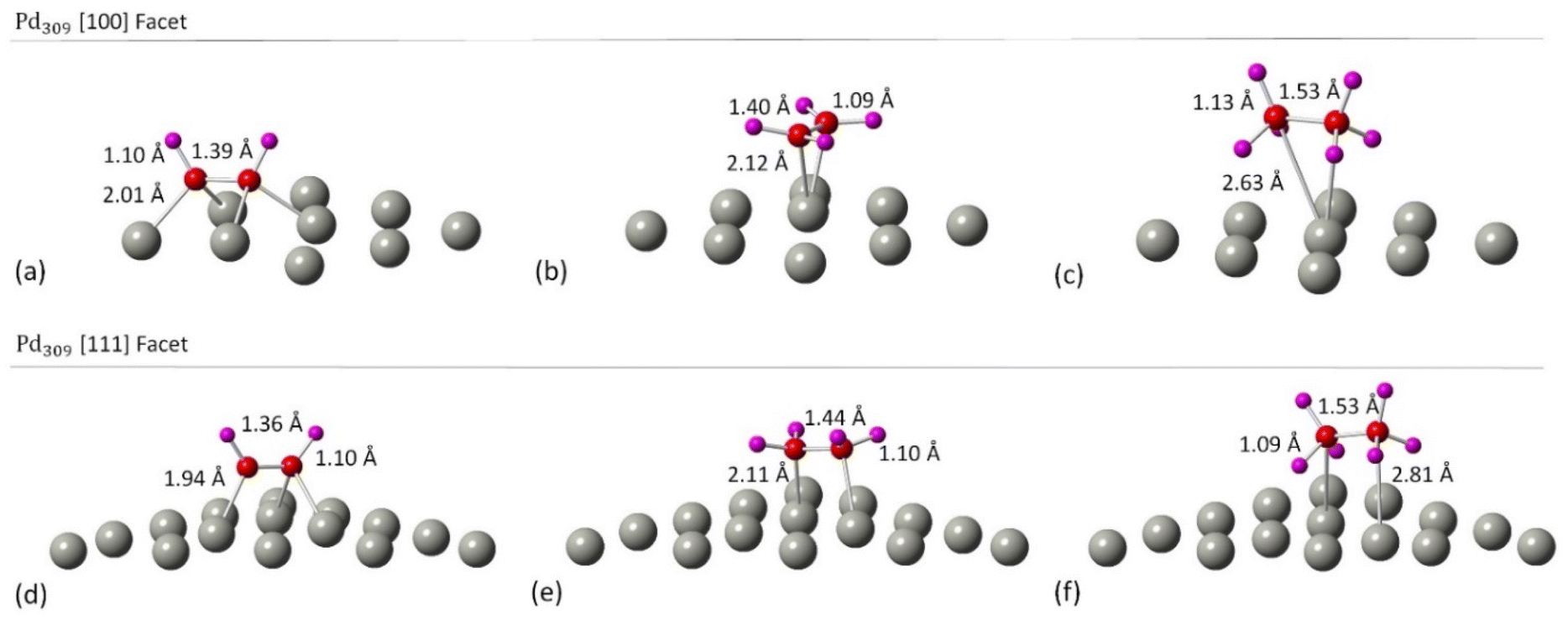
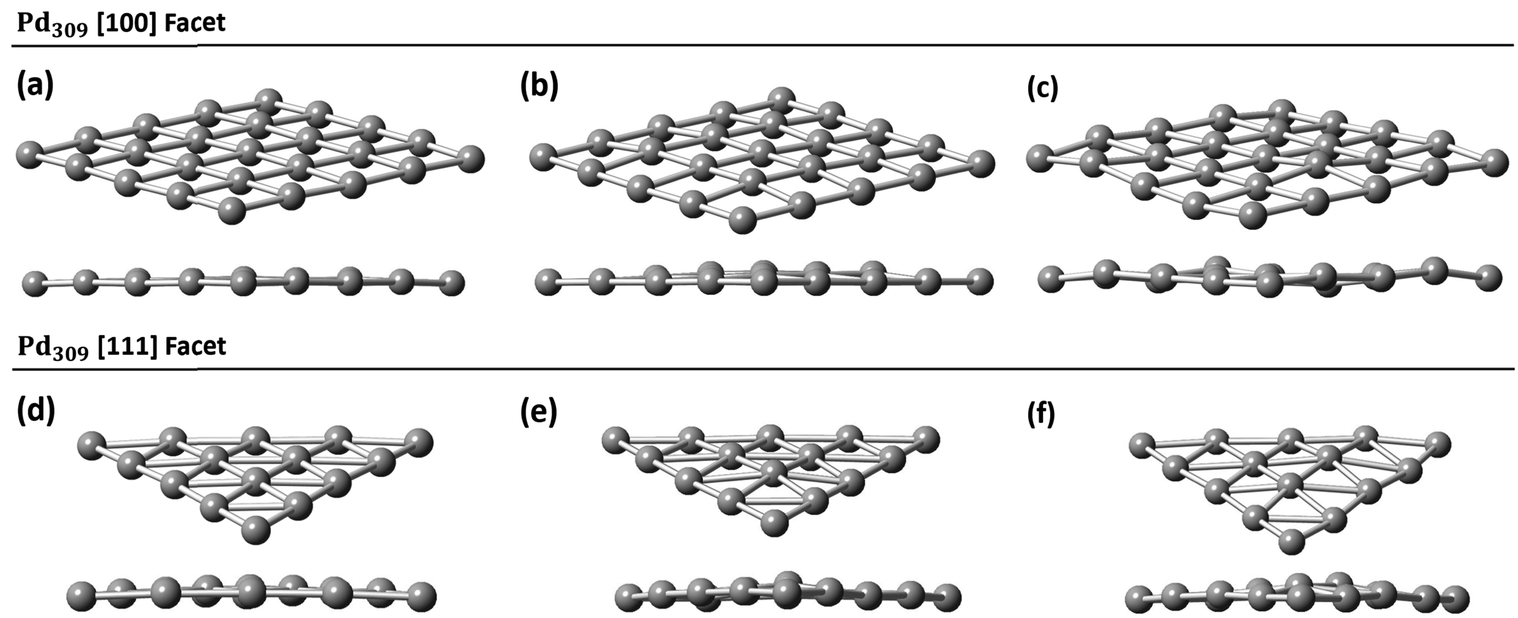
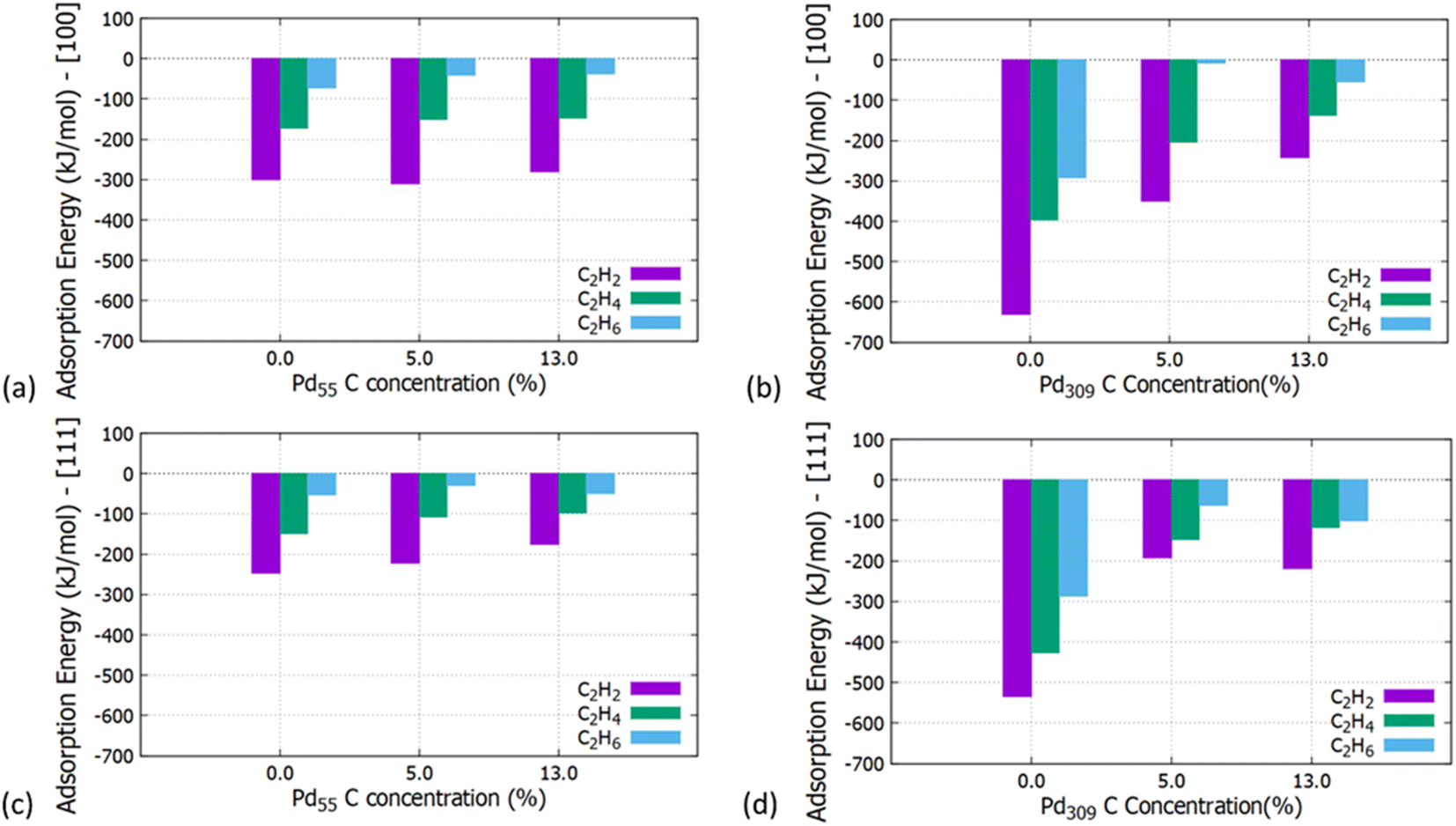
Initially, we studied the preferred adsorption and orientation of the molecules on the NP surface by doing full geometry relaxations of each NP-adsorbate complex. These geometries were used subsequently as configurations of reactants and products in the partial and full hydrogenation states of C2H2 to C2H4 and C2H6. The relaxation of C in the octahedral interstitial Pd sites at 0%, 5% and 13% concentration is shown in Fig. 3. The average Pd–C bond length for Fig. 3(b) and (c) is 2.04 Å and 2.07 Å respectively whilst for 3(e) and (f) is 2.01 Å and 2.06 Å respectively. Fig. 4 shows the relaxed structures of C2H2, C2H4 and C2H6 on the [100] and [111] facets of the pristine Pd309 surface. The Pd55 relaxed structures and NP–adsorbate bond lengths for all geometries used for this study are included in the ESI, Fig. S1–S6.† Additionally, the surface modifications for the [100] and [111] facets of the Pd309 are also shown in Fig. 5, whilst the geometries of C2H2, C2H4 and C2H6 on the [100] facet of pristine and carbidic Pd309 are shown in Fig. 6. It is evident that interstitial C will change the facet morphology leading to different binding arrangements between surface Pd and hydrocarbons. We compared the adsorption energies of the C2H2 to C2H4 and C2H6 molecules at different C concentrations, NP size and facet. The adsorption energies for both [100] and [111] facets of the cuboctahedral structures are shown in Fig. 7, whilst the obtained values are presented in Tables S1 and S2 in the ESI.† Our calculations showed that C2H2 adsorbs more strongly on the 4-fold site of the [100] facet and in agreement with the work of Crespo-Quesada et al.33 We also find that C2H2 is adsorbed more strongly (−300 kJ mol−1 for the [100] of Pd55 and −631.1 kJ mol−1 for the [100] of Pd309) on the surface of the NP than C2H4 (−172.9 kJ mol−1 for the [100] of Pd55 and −396.9 kJ mol−1 for the [100] of Pd309) and C2H6 (−72.0 kJ mol−1 for the [100] of Pd55 and −291.6 kJ mol−1 for the [100] of Pd309). We observed that the adsorption energies are considerably higher for the Pd309 structure, showing that the particle size is expected to affect the reaction. As we introduce interstitial C in the Pd lattice, the adsorption energies are reduced drastically for Pd309 but very little for Pd55. We also observed that the carbidic Pd has significantly lower adsorption energies for C2H4 on the [100] and [111] facets as compared to the pristine structure. This shows that ethylene will desorb more easily from the surface of the PdCx NP, hence it is a first indication of possible suppression of the full hydrogenation to ethane.
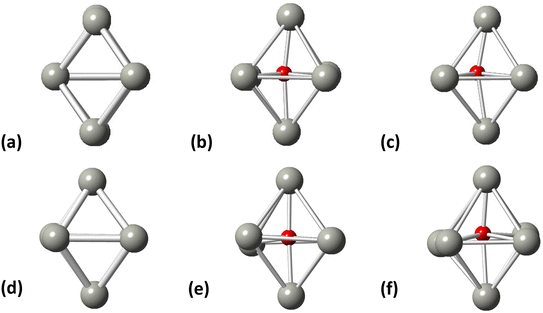 |
| | Fig. 3 Pd octahedral interstitial sites for (a) Pd55 at 0% C concentration, (b) Pd55 at 5% C concentration, (c) Pd55 at 13% C concentration, (d) Pd309 at 0% at C concentration, (e) Pd309 at 5% at C concentration, and (f) Pd309 at 13% at C concentration. Grey spheres represent Pd atoms and red spheres represent C atoms. | |
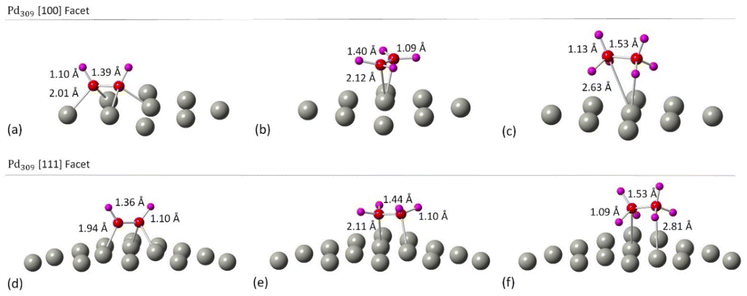 |
| | Fig. 4 The relaxed structures of (a) C2H2 on the [100] facet of Pd309 NP, (b) C2H4 on the [100] facet of Pd309 NP, (c) C2H6 on the [100] facet of Pd309 NP, (d) C2H2 on the [111] facet of Pd309 NP, (e) C2H4 on the [111] facet of Pd309 NP and (f) C2H6 on the [111] facet of Pd309 NP. Grey spheres represent Pd atoms, red spheres represent C atoms and purple spheres represent H atoms. Geometry relaxations were performed on the entire Pd NP–ligand complexes. | |
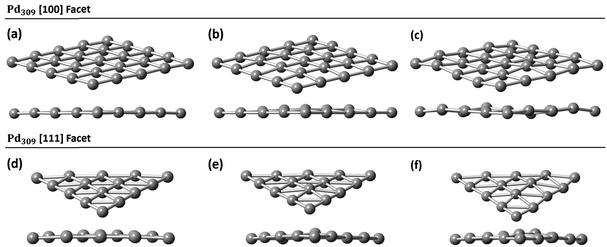 |
| | Fig. 5 Pd309 surface modifications for (a) the [100] facet at 0% C, (b) the [100] facet 5% C, (c) the [100] facet 13% C, (d) the [111] facet at 0% C, (e) the [111] facet at 5% C, and (f) the [111] facet at 13% C. | |
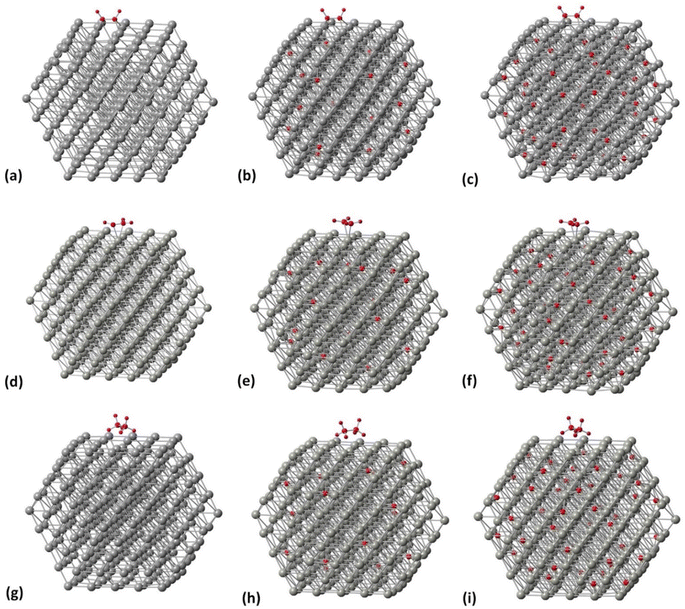 |
| | Fig. 6 Related geometries of molecule/PdCx NP complexes. (a)–(c) C2H2 on the [100] facet of Pd309 at 0, 5 and 13% C concentration, (d)–(f) C2H4 on the [100] facet of Pd309 at 0, 5 and 13% C concentration, and (g)–(i) C2H6 on the [100] facet of Pd309 at 0, 5 and 13% C concentration. | |
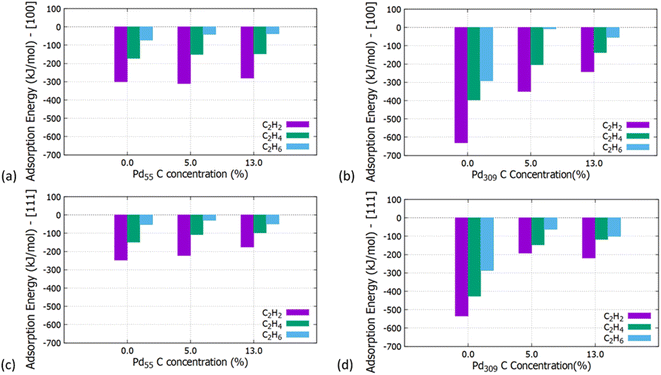 |
| | Fig. 7 Adsorption energies of C2H2, C2H4 and C2H6 on the (a) [100] facet of Pd55 at C concentration = 0%, 5% and 13%, (b) [100] facet of Pd309 at C concentration = 0%, 5% and 13%, (c) [111] facet of Pd55 at C concentration = 0%, 5% and 13% and (d) [111] facet of Pd309 at C concentration = 0%, 5% and 13%. | |
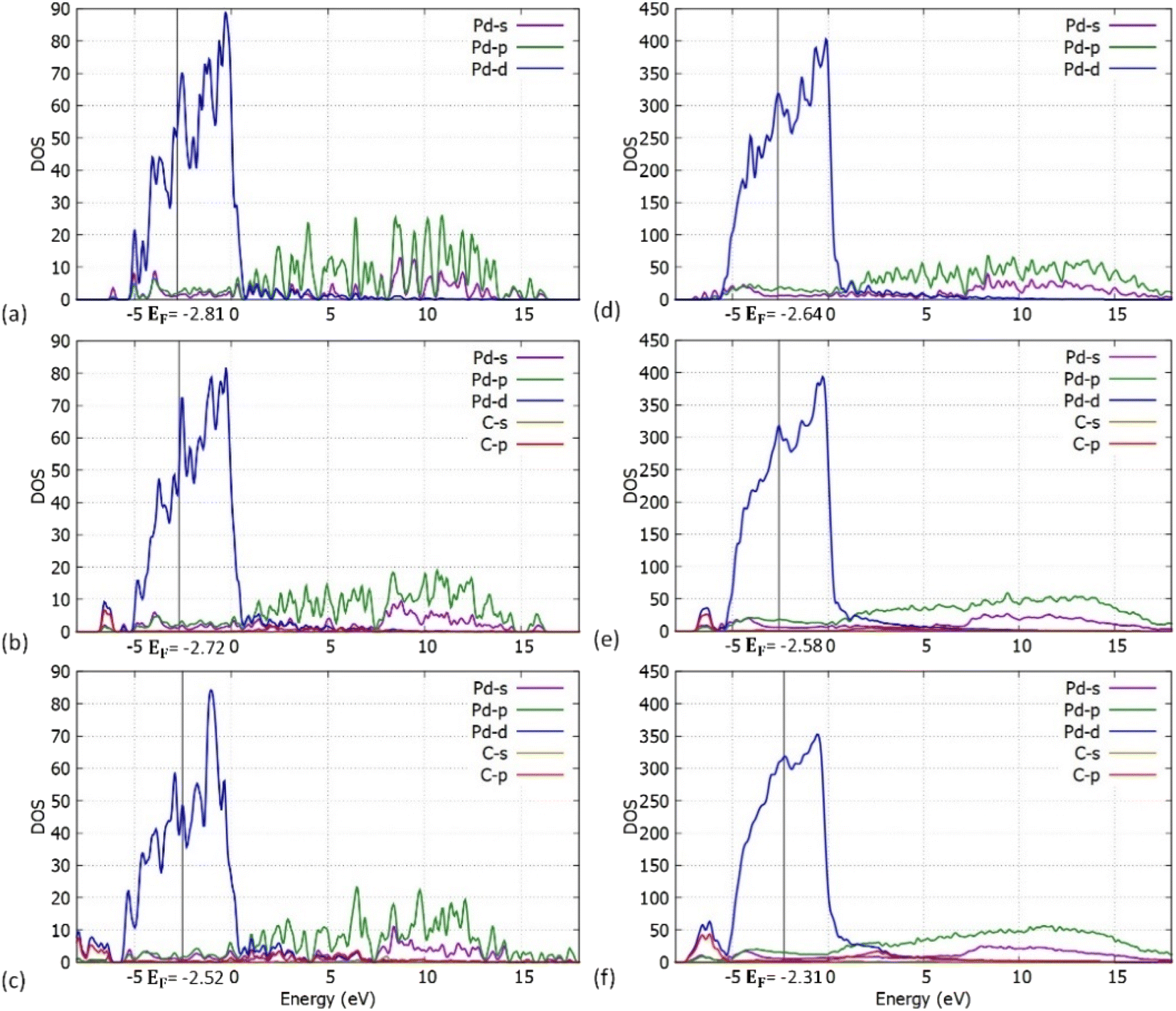
We have also performed density of states (DOS) investigations on the pristine and carbidic Pd55 and Pd309. As shown in Fig. 8, the metallic behaviour for both pristine structures is confirmed. Furthermore, Pd309 corresponds considerably more to bulk-like DOS when compared to Pd55. This behaviour is expected since the volume of the particle is larger, and its electronic structure is closer to bulk Pd.
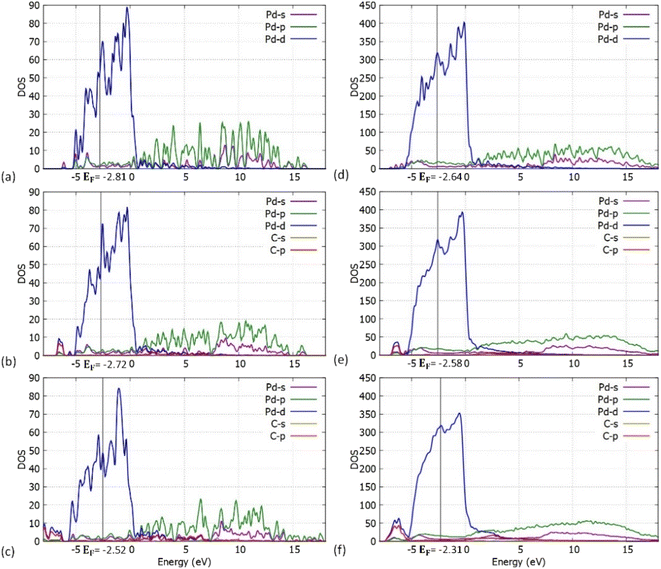 |
| | Fig. 8 Atom and orbital resolved DOS plots for the (a) Pd55 at C = 0% concentration, (b) Pd55 at C = 5% concentration, (c) Pd55 at C = 13% concentration, (d) Pd309 at C = 0% concentration, (e) Pd309 at C = 5% concentration and (f) Pd309 at C = 13% concentration. | |
To further explore the effect of C concentration on the hydrogenation of C2H2, we investigated the reaction energies of each stage of the formation of C2H4 and C2H6.
3.4 Hydrogenation reactions
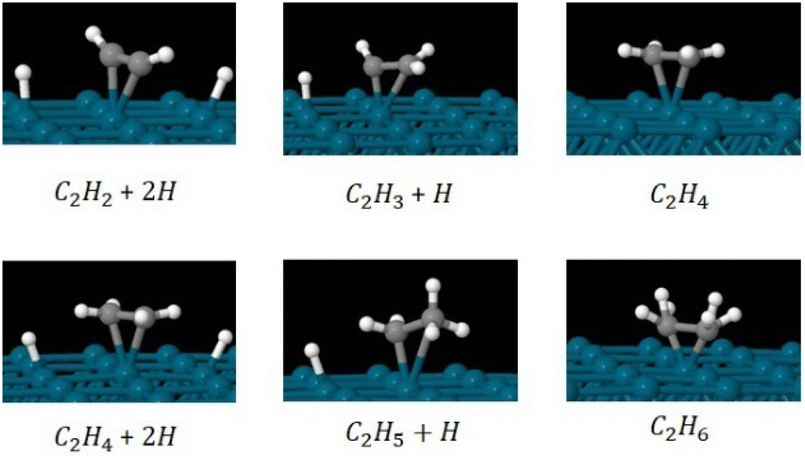
We investigated the partial and full hydrogenation of C2H2 on the pristine Pd55 and Pd309. Fig. 9 shows the reactants, products and intermediates on the Pd309 surface. The hydrogenation of acetylene on Pd NPs was investigated as a four-part process leading to partial and full hydrogenation corresponding to C2H4 and C2H6 as the two final products:
| (Part A) C2H2 + 2H → C2H3+H |
| (Part C) C2H4 + 2H → C2H5+H |
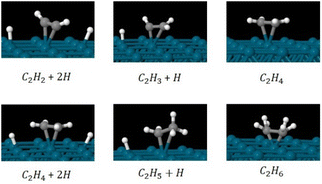 |
| | Fig. 9 Relaxed geometries of reactants, intermediates and products for the hydrogenation of C2H2 to C2H4 (part B) and C2H6 (part D) on the [100] facet of pristine Pd309. Geometry relaxations were performed on the entire Pd309–ligand complex. Blue spheres are Pd, grey spheres are C and white spheres are H. | |
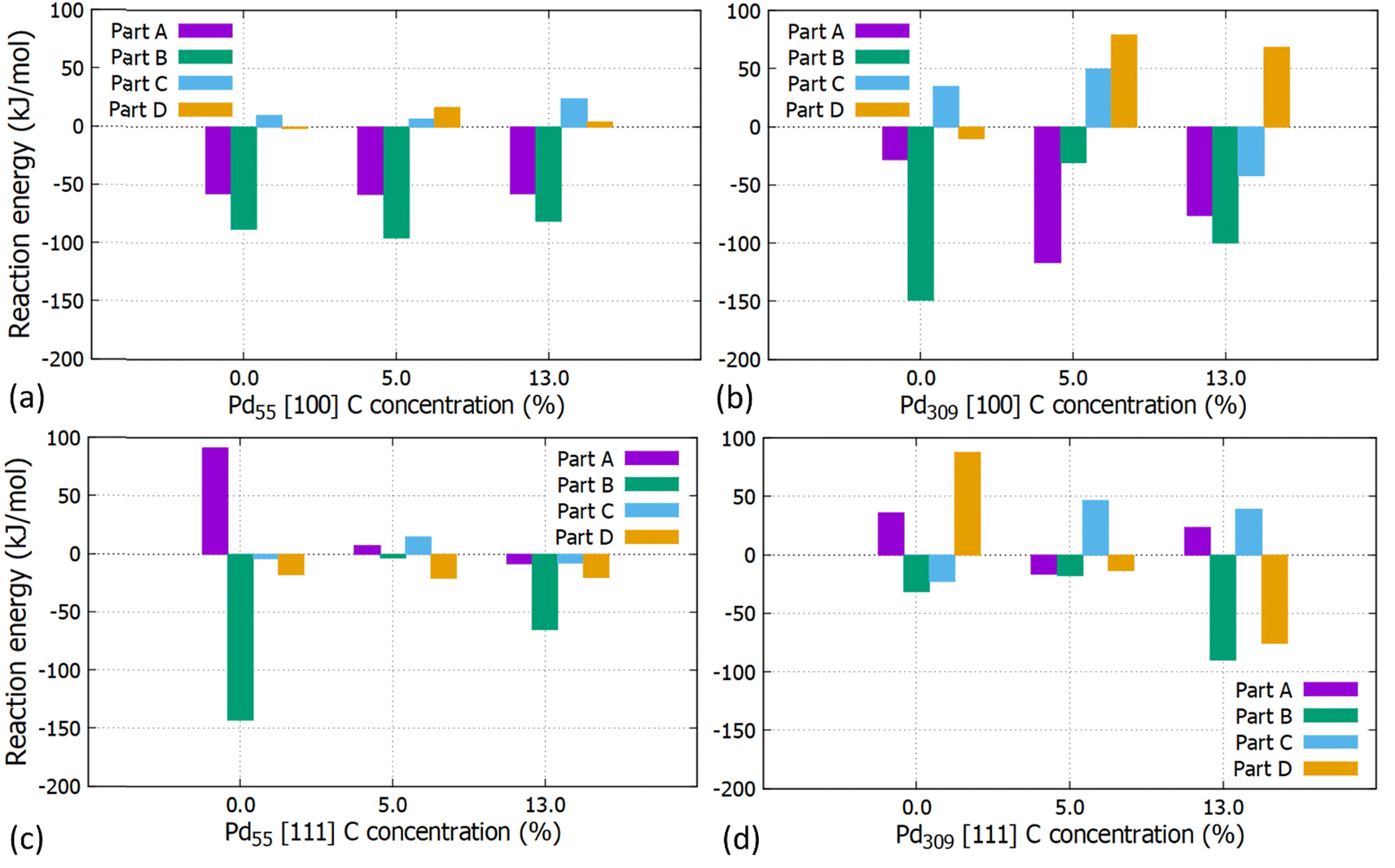
The reaction energies for the partial and full hydrogenation of C2H2 to C2H4 and C2H6 on the [100] and [111] facets of Pd55 and Pd309 at 0, 5 and 13% of interstitial C concentration are shown in Fig. 10. Our results show that information about the catalytic behaviour of PdCx NPs used in practical studies can be obtained by simulating entire large PdCx NPs, as done in this work.
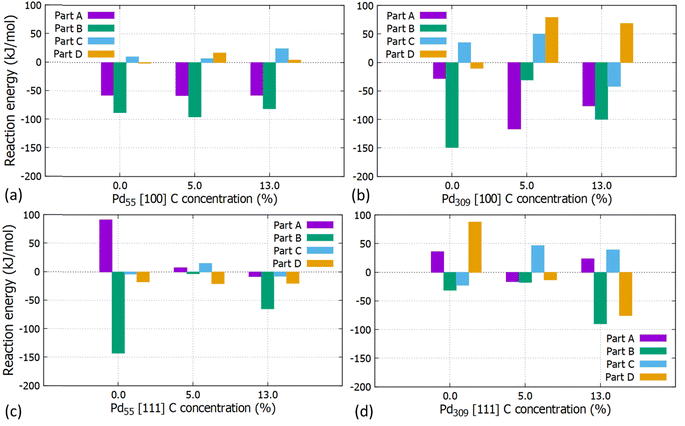 |
| | Fig. 10 Reaction energies for the partial and full acetylene hydrogenation on the (a) [100] facet of the Pd55 at C (=0%, 5% and 13%), (b) [100] facet of the Pd309 at C (=0%, 5% and 13%), (c) [111] facet of the Pd55 at C (=0%, 5% and 13%) and (d) [111] facet of Pd309 at C (=0%, 5% and 13%). | |
For Pd55, reaction energies for the [100] facet are exothermic for parts A (EReaction = −57.6 kJ mol−1) and B (EReaction = −88.1 kJ mol−1) whilst the introduction of interstitial C has a minor contribution. For the [111] facet, part A corresponds to endothermic reaction energies for pristine Pd and PdCx (5%). Here, interstitial C results to lower values and the behaviour is affected by the particle's distortion at the higher C concentration of 13%.
For the pristine Pd309 the partial hydrogenation on the [100] facet, part B is more exothermic (EReaction = −149.3 kJ mol−1) than part A (EReaction = −28.6 kJ mol−1) for the semi-hydrogenation to C2H4. As we introduce interstitial C in the Pd lattice, C2H3 forms more easily as the reaction energy becomes more exothermic, whilst when we reach the maximum considered C concentration of 13%, reaction energies correspond to similar values. For the [111] facet, part A is also endothermic. In contrast with Pd55, the reaction energy turns to endothermic for 5% C concentration showing that interstitial C, larger available surface area and reduced distortion of Pd309 will promote the reaction. Overall, we observe that parts A and B correspond to exothermic reactions showing that the formation of intermediate C2H3 and C2H4 as the final product on the [100] facet of Pd309 is energetically favourable. Furthermore, the carbidic phase has a considerable impact on the reaction energies. This is due to the stronger adsorption of intermediates such as C2H3 interacting with more surface Pd atoms compared to the pristine Pd309. This is due to the [100] facet getting distorted as a consequence of C doping. For the full hydrogenation to C2H6 (parts C and D), our calculations showed that the reaction to C2H5 is slightly endothermic (EReaction = 34.82 kJ mol−1) and full hydrogenation to ethane corresponds to a slightly exothermic energy of reaction (EReaction = −10.46 kJ mol−1). This behaviour is in agreement with previously reported computational work14 where the reaction energy for the formation of C2H5 is endothermic (approximately 25 kJ mol−1) and the reaction energy for the formation of C2H6 is exothermic (approximately −9.6 kJ mol−1). The same behaviour is observed for the increasing concentration of 5% C although here the full hydrogenation to ethane also turns to endothermic.
Our calculations unambiguously show that interstitial C at a concentration of around 5% promotes the formation of ethylene, since parts C and D that would lead to ethane are endothermic. Furthermore, at this concentration, the adsorption energy of C2H4 is reduced by more than 30% compared to the pristine Pd, whilst when increasing the interstitial C concentration close to saturation (at about 13%), part C becomes exothermic again. Therefore, more work is needed towards understanding the effect of the carbidic phase to increase selectivity in Pd catalysts.
4. Conclusions
We performed large-scale DFT calculations on entire cuboctahedral Pd55 and Pd309 NPs with different degrees of carbidisation to gain insights on the hydrogenation of C2H2 to C2H4 and C2H6. We investigated two phases of PdCx at concentrations of 5% and 13% (which is close to the maximum experimentally and computationally determined value) to examine how the particle size affects the adsorption and reaction energies during catalytic hydrogenation given that metal nanoparticle catalysts, often supported on metal oxide promoters, are essential to many applications in heterogeneous catalysis. Experimentally, size effects are well-established and are manifested through changes to catalyst selectivity, activity and durability and DFT calculations have provided an attractive way to study these effects and rationalise the change in nanoparticle properties. However, most DFT studies are typically limited to smaller nanoparticles (approximately up to 50 atoms) due to the large computational cost of DFT. Here we have used the ONETEP code, which is able to achieve more favourable computational scaling for metallic nanoparticles, to bridge this size gap and simulate nanoparticles of more than 300 atoms (approximately 2.5 nm diameter). In this study, we have used cuboctahedral Pd nanoparticles with 55 and 309 atoms and also their carbidised structures. We found that the adsorption energies of C2H2, C2H4 and C2H6 on the pristine Pd and PdCx NPs are considerably larger for the Pd309 structure. Furthermore, the carbidic phase for the Pd55 has minor impact on the adsorption energies whilst for the Pd309, a considerable decrease is observed as more C is introduced in the Pd interstitial sites. C2H2 adsorbs more strongly on the [100] facet, forming four C–Pd bonds, rather than on the [111]. However, the adsorption of C2H2 and C2H4 becomes weaker (on Pd309Cx) as more interstitial C is introduced with the latter being responsible for promoting desorption of ethylene and blocking full hydrogenation to ethane. Since the adsorption energies decrease for Pd309 with increasing C concentration, while Pd55 is insensitive to interstitial C, it is clear that catalytic selectivity towards partial hydrogenation can only be achieved with larger Pd NPs.
Overall, we see that the partial hydrogenation of C2H2 to C2H4 is favourable for both Pd55 and Pd309 with the exception of the [111] facet of Pd55 where monoatomic hydrogen relaxes towards the edges between facets making the hydrogenation reaction unfavourable. We showed that the size of the particle is expected to have a major impact on the reaction due to the available surface area where molecules adsorb as evidenced through the reaction energies. For the full hydrogenation of C2H2 we observed that the carbide phase has also an impact on the reaction towards the formation of C2H5 and C2H6, but only for Pd309, not Pd55. For a C concentration of 5%, the reaction is endothermic for both C2H5 and C2H6 for both facets preventing hydrogenation towards ethane. For a higher C concentration (13%) in Pd309, we found that hydrogenation to C2H5 becomes exothermic. This is an undesirable intermediate that could also lead to full hydrogenation to ethane. Therefore, the effect of the C concentration is complex, and more work is needed to tune it towards optimum selectivity and yield, but the present study provides starting points towards further optimization of the PdCx large nanoparticle catalyst.
This is the first time that simulations on entire large (>300 atoms) metallic NPs catalysts have been performed through DFT towards understanding catalytic hydrogenation reactions. We show that there is a dramatic difference in the behaviour of large Pd NPs as compared to small Pd NPs on the adsorption energies of hydrocarbons and the reaction energies between intermediates and products. Such DFT atomistic simulations of large realistic NPs are expected to act synergistically with experimental studies to provide detailed and valuable insights in Pd-based directed catalysis.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
AK, KM and RV are grateful to EPSRC (grant number EP/V000691/1) for postdoctoral funding. We are grateful to The Materials and Molecular Modelling MMM Hub (EPSRC grant number EP/T022213/1) for access to the Young supercomputer, the UKCP consortium (EPSRC grant number EP/X035956/1) for access to the ARCHER2 supercomputer and the University of Southampton for access to the Iridis5 supercomputer.
References
- A. Borodziński and G. C. Bond, Selective Hydrogenation of Ethyne in Ethene-Rich Streams on Palladium Catalysts. Part 1. Effect of Changes to the Catalyst during Reaction, Catal. Rev., 2006, 48(2), 91–144, DOI:10.1080/01614940500364909
 .
. - M. Armbrüster, M. Behrens, F. Cinquini, K. Föttinger, Y. Grin, A. Haghofer, B. Klötzer, A. Knop-Gericke, H. Lorenz, A. Ota, S. Penner, J. Prinz, C. Rameshan, Z. Révay, D. Rosenthal, G. Rupprechter, P. Sautet, R. Schlögl, L. Shao, L. Szentmiklósi, D. Teschner, D. Torres, R. Wagner, R. Widmer and G. Wowsnick, How to Control the Selectivity of Palladium-based Catalysts in Hydrogenation Reactions: The Role of Subsurface Chemistry, ChemCatChem, 2012, 4, 1048–1063, DOI:10.1002/cctc.201200100 .
- W. J. Kim and S. H. Moon, Modified Pd Catalysts for the Selective Hydrogenation of Acetylene, Catal. Today, 2012, 185, 2–16, DOI:10.1016/j.cattod.2011.09.037 .
- W. Huang, J. R. McCormick, R. F. Lobo and J. G. Chen, Selective hydrogenation of acetylene in the presence of ethylene on zeolite-supported bimetallic catalysts, J. Catal., 2007, 246(1), 40–45, DOI:10.1016/j.jcat.2006.11.013 .
- A. N. R. Bos and K. R. Westerterp, Mechanism and kinetics of the selective hydrogenation of ethyne and ethene, Chem. Eng. Process., 1993, 32(1), 1–7, DOI:10.1016/0255-2701(93)87001-B .
- I. Y. Ahn, J. H. Lee, S. S. Kum and S. H. Moon, Formation of C4 species in the deactivation of a Pd/SiO2 catalyst during the selective hydrogenation of acetylene, Catal. Today, 2007, 123(1–4), 151–157, DOI:10.1016/j.cattod.2007.02.011 .
- H. A. Aleksandrov, L. V. Moskaleva, Z.-J. Zhao, D. Basaran, Z.-X. Chen, D. Mei and N. Rösch, Ethylene conversion to ethylidyne on Pd(111) and Pt(111): a first-principles-based kinetic Monte Carlo study, J. Catal., 2012, 285(1), 187–195, DOI:10.1016/j.jcat.2011.09.035 .
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, A. Chutia, N. Jian, R. E. Palmer, M. Perdjon, A. Thetford, N. Dimitratos, A. Villa and P. P. Wells, Tandem Site- and Size-Controlled Pd Nanoparticles for the Directed Hydrogenation of Furfural, ACS Catal., 2017, 7(4), 2266–2274, DOI:10.1021/acscatal.6b03190 .
- M. Maciejewski and A. Baker, Incorporation and reactivity of carbon in palladium, Pure Appl. Chem., 1995, 67(11), 1879–1884, DOI:10.1351/pac199567111879 .
- N. Seriani, F. Mittendorfer and G. Kresse, Carbon in palladium catalysts: a metastable carbide, J. Chem. Phys., 2010, 132, 024711, DOI:10.1063/1.3290813 .
- D. Torres, F. Cinquini and P. Sautet, Pressure and Temperature Effects on the Formation of a Pd/C Surface Carbide: Insights into the Role of Pd/C as a Selective Catalytic State for the Partial Hydrogenation of Acetylene, J. Phys. Chem. C, 2013, 117(21), 11059–11065 CrossRef CAS .
- N. K. Nag, A Study on the Formation of Palladium Hydride in a Carbon-Supported Palladium Catalyst, J. Phys. Chem. B, 2001, 105(25), 5945–5949, DOI:10.1021/jp004535q .
- Y. Zhou, W. Baaziz, O. Ersen, P. A. Kots, E. I. Vovk, X. Zhou, Y. Yang and V. V. Ordomsky, Decomposition of Supported Pd Hydride Nanoparticles for the Synthesis of Highly Dispersed Metallic Catalyst, Chem. Mater., 2018, 30(22), 8116–8120, DOI:10.1021/acs.chemmater.8b02192 .
- L. P. L. Gonçalves, J. Wang, S. Vinati, E. Barborini, X.-K. Wei, M. Heggen, M. Franco, J. P. S. Sousa, D. Y. Petrovykh, O. S. G. P. Soares, K. Kovnir, J. Akola and Y. V. Kolen'ko, Combined experimental and theoretical study of acetylene semi-hydrogenation over Pd/Al2O3, Int. J. Hydrogen Energy, 2020, 45(2), 1283–1296, DOI:10.1016/j.ijhydene.2019.04.086 .
- Y. Liu, F. Fu, A. McCue, W. Jones, D. Rao, J. Feng, Y. He and D. Li, Adsorbate-Induced Structural Evolution of Pd Catalyst for Selective Hydrogenation of Acetylene, ACS Catal., 2020, 10(24), 15048–15059, DOI:10.1021/acscatal.0c03897 .
- E. Vignola, S. N. Steinmann, A. A. Farra, B. D. Vandegehuchte, D. Curulla and P. Sautet, Evaluating the Risk of C–C Bond Formation during Selective Hydrogenation of Acetylene on Palladium, ACS Catal., 2018, 8(3), 1662–1671, DOI:10.1021/acscatal.7b03752 .
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, The Roles of Subsurface Carbon and Hydrogen in Palladium-Catalyzed Alkyne Hydrogenation, Science, 2008, 320(5872), 86–89, DOI:10.1126/science.1155200 .
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Sørensen, C. Christensen and J. Nørskov, On the Role of Surface Modifications of Palladium Catalysts in the Selective Hydrogenation of Acetylene, Angew. Chem., Int. Ed., 2008, 47(48), 9299–9302 CrossRef CAS PubMed .
- M. Sun, F. Wang, G. Lv and X. Zhang, Size effect of PdC nanoparticles synthesized by S-containing silane coupling agents on semi-hydrogenation of acetylene, Appl. Surf. Sci., 2022, 153021, DOI:10.1016/j.apsusc.2022.153021 .
- Z.-J. Zhao, J. Zhao, X. Chang, S. Zha, L. Zeng and J. Gong, Competition of C-C bond formation and C-H bond formation for acetylene hydrogenation on transition metals: a density functional theory study, AIChE J., 2019, 65(3), 1059–1066, DOI:10.1002/aic.16492 .
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, Influence of surface structures, subsurface carbon and hydrogen, and surface alloying on the activity and selectivity of acetylene hydrogenation on Pd surfaces: a density functional theory study, J. Catal., 2013, 305, 264–276, DOI:10.1016/j.jcat.2013.05.027 .
- T. Abdollahi and D. Farmanzadeh, Selective hydrogenation of acetylene in the presence of ethylene on palladium nanocluster surfaces: a DFT study, Appl. Surf. Sci., 2018, 433, 513–529, DOI:10.1016/j.apsusc.2017.10.085 .
- A. D. Benavidez, P. D. Burton, J. L. Nogales, A. R. Jenkins, S. A. Ivanov, J. T. Miller, A. M. Karim and A. K. Datye, Improved selectivity of carbon-supported palladium catalysts for the hydrogenation of acetylene in excess ethylene, Appl. Catal., 2014, 482, 108–115, DOI:10.1016/j.apcata.2014.05.027 .
- S. Asplund, Coke Formation and Its Effect on Internal Mass Transfer and Selectivity in Pd-Catalysed Acetylene Hydrogenation, J. Catal., 1996, 158(1), 267–278, DOI:10.1006/jcat.1996.0026 .
- J. C. A. Prentice, J. Aarons, J. C. Womack, A. E. A. Allen, L. Andrinopoulos, L. Anton, R. A. Bell, A. Bhandari, G. A. Bramley, R. J. Charlton, R. J. Clements, D. J. Cole, G. Constantinescu, F. Corsetti, S. M.-M. Dubois, K. K. B. Duff, J. M. Escartín, A. Greco, Q. Hill, L. P. Lee, E. Linscott, D. D. O'Regan, M. J. S. Phipps, L. E. Ratcliff, Á. Ruiz Serrano, E. W. Tait, G. Teobaldi, V. Vitale, N. Yeung, T. J. Zuehlsdorff, J. Dziedzic, P. D. Haynes, N. D. M. Hine, A. A. Mostofi, M. C. Payne and C.-K. Skylaris, The ONETEP
linear-scaling density functional theory program, J. Chem. Phys., 2020, 152, 174111, DOI:10.1063/5.0004445 .
- C.-K. Skylaris, A. A. Mostofi, P. D. Haynes, O. Diéguez and M. C. Payne, Nonorthogonal generalized Wannier function pseudopotential plane-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 035119, DOI:10.1103/PhysRevB.66.035119 .
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865, DOI:10.1103/PhysRevLett.77.3865 .
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799, DOI:10.1002/jcc.20495 .
- Á. Ruiz-Serrano and C.-K. Skylaris, A variational method for density functional theory calculations on metallic systems with thousands of atoms, J. Chem. Phys., 2013, 139, 054107, DOI:10.1063/1.4817001 .
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The Atomic Simulation Environment-A Python Library for Working with Atoms, J. Phys.: Condens. Matter, 2017, 29, 273002, DOI:10.1088/1361-648X/aa680e .
- CrystalMaker®: a crystal and molecular structures program for Mac and Windows, 9.0.3, CrystalMaker Software Ltd, Oxford, England, 2014, https://www.crystalmaker.com Search PubMed .
- A. Kordatos, K. Mohammed, R. Vakili, A. Goguet, H. Manyar, E. Gibson, M. Carravetta, P. Wells and C.-K. Skylaris, Atomistic simulations on the carbidisation processes in Pd nanoparticles, RSC Adv., 2023, 13, 5619–5626, 10.1039/D2RA07462A .
- M. Crespo-Quesada, S. Yoon, M. Jin, A. Prestianni, R. Cortese, F. Cárdenas-Lizana, D. Duca, A. Weidenkaff and L. Kiwi-Minsker, Shape-Dependence of Pd Nanocrystal Carburization during Acetylene Hydrogenation, J. Phys. Chem. C, 2015, 119, 1101, DOI:10.1021/jp510347r .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Khaled Mohammed
a,
Khaled Mohammed