
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient amide bond formation via tropylium ion organocatalysis†
Mohanad A. Hussein*ab,
Karrar Al-Ameed *cd,
Ali K. Almansorie and
Naeemah Jabbar Owaidf
*cd,
Ali K. Almansorie and
Naeemah Jabbar Owaidf
aDepartment of Biochemistry, College of Medicine, Jabir Ibn Hayyan University for Medical and Pharmaceutical Sciences, Iraq
bDepartment of Chemistry, College of Science, The University of New South Wales, Australia. E-mail: z5032126@zmail.unsw.edu.au
cDepartment of Chemistry, University of Kufa, 54001, Iraq. E-mail: karrar.alameed@uokufa.edu.iq
dCollege of Engineering, University of Warith AL-Anbiyaa, Karbala, Iraq
eCollege of Pharmacy, University of Al-Ameed, Karbala, Iraq
fDepartment of Chemistry, College of Science, University of Baghdad, Iraq
First published on 29th October 2024
Abstract
Amide bond formation is a fundamental reaction in organic synthesis, essential for the construction of peptides and various bioactive molecules. Traditional methods for amide bond formation often require harsh conditions, expensive reagents, or generate undesirable by-products. Herein, we report a novel and efficient method for amide bond formation utilizing tropylium ion organocatalysis. The tropylium ion, known for its stability and unique electronic properties, facilitates the activation of carboxylic acids and amines, promoting their coupling under mild conditions. This method demonstrates high yields and broad substrate scope, including sterically hindered and functionalized reactants. The mechanistic studies suggest that the tropylium ion acts as an electrophilic catalyst, enhancing the nucleophilicity of the amine and the electrophilicity of the carboxylic acid, thereby accelerating the formation of the amide bond. This work opens new avenues for the development of green and sustainable catalytic processes in organic synthesis. In order to explore the effect of the different substituents on the thermodynamics of the condensation reactions of the synthesized compounds, we have conducted a series of calculations using density functional theory to compute the reaction energies. The calculations also investigated the effect of the substitutions on the hydrogen migration in the tautomerism effect and the energy splitting of the frontier orbitals.
Introduction
The amide bond is a structural unit of fundamental importance in both nature and the field of synthetic organic chemistry.1–4 In nature, amide bonds form the backbones of all peptides and proteins. Additionally, they are present in a variety of natural products, making them one of the most central building blocks found in biomolecules. Furthermore, amide bonds are prevalent in a wide range of chemical products such as agrochemicals, pharmaceuticals, and polymers, emphasizing their major importance to the chemical industry as well.1–8 Thus, it is not surprising that amide bond formation has been an intensively investigated research area over the past decades. Recent efforts have focused on developing new and efficient catalytic protocols to replace traditional methods based on coupling reagents.1,2,4,9–11Coupling reagents have long been the go-to method for amide bond formation due to their high efficiency and broad substrate scope. This is especially true in the area of solid-phase peptide synthesis, where multiple amide bond-forming reactions need to be executed consistently with near-quantitative yields.6,12,13 However, these reagents are associated with high costs and poor atom economy. Consequently, the pharmaceutical industry has frequently called for the development of more cost-effective methods.14,15
The catalytic condensation of carboxylic acids and amines represents a highly attractive strategy with the potential to meet the rigorous standards of the pharmaceutical industry. This transformation is inherently atom-economic and sustainable, as the only byproduct formed in an ideal condensation reaction is water. If a non-toxic catalytic system is used, the overall reaction protocol should exhibit a highly favorable green chemistry profile. Unfortunately, the number of catalytic protocols available for this transformation is still limited despite intensive research efforts over the past decades. So far, the catalytic condensation of carboxylic acids and amines has predominantly been achieved using various kinds of Lewis acid catalysts. Of these, catalysts based on boron4,16–21 and group IV metals such as Al,22 Hf,23,24 Ti,22,25 Zr24,26–29 have by far been the most intensively studied over the past decade.
Separate from the impressive prior art in catalytic amide bond formation, the tropylium ion has emerged as a highly competent Lewis acid catalyst that has found use in a wide range of organic transformations, including, for example C–C bond in 1,3-diketone compounds,30 Ritter reactions,31 tropylium ion undergoes noncatalyzed, regioselective additions to a large variety of Michael acceptors,32 hydroboration reactions,33 hydration reactions of alkynes34 and cyclization reactions.35 Coupling reagents like TBTU and HBTU are widely used in peptide synthesis to activate carboxylic acids, typically yielding good to high results. HBTU is often paired with 1-hydroxybenzotriazole (HOBt) to minimize racemization, which is crucial for maintaining peptide bond stereochemistry. However, these reagents can be less effective with bulky substrates due to steric hindrance, and they produce toxic byproducts, raising environmental concerns and potentially affecting the desired product.1,2,10,11
In contrast, the tropylium ion has emerged as a more efficient and eco-friendly alternative. It operates under milder conditions, eliminating the need for traditional coupling reagents, thus simplifying the reaction and reducing waste. Reactions involving tropylium ions not only deliver comparable or even better yields than TBTU/HBTU, but they are also less impacted by steric hindrance, making them ideal for bulkier substrates. Environmentally, tropylium ion reactions are more aligned with green chemistry principles, as they avoid the production of harmful byproducts typical of conventional coupling reagents.30,32
To the best of our knowledge, despite its numerous applications, the tropylium ion has never been studied as a catalyst for the condensation of carboxylic acids and amines. However, as demonstrated herein, the tropylium ion is indeed a highly efficient Lewis acid catalyst for this transformation, ushering in a new paradigm in catalytic amide bond formation. Remarkably, efficient condensation was observed between equimolar amounts of carboxylic acids and amines. Furthermore, the tropylium ion enabled the use of anilines as condensation partners, which are a highly elusive substrate class in many catalytic protocols. In this work, we will present the optimization of the tropylium-based amidation protocol, describe its scope and limitations, and demonstrate its practical utility in synthesizing some pharmaceutical agents.
Results and discussion
Optimization of reaction conditions
Our inspiration to use the tropylium ion as a catalyst for the direct formation of amides through the condensation of carboxylic acids and amines stems from its unique non-benzenoid aromatic cation structure and its balance between stability and reactivity. This idea is derived from our earlier works in the area of tropylium salt-mediated chemical transformations1,6 based on our recent and previous reports on tropylium ion-promoted chemistry1,7 we believed that the tropylium ion could serve as an efficient organic Lewis acid catalyst to activate carboxylic acids for direct amidation under mild conditions.Therefore, phenylacetic acid 2a and benzylamine 3a were selected as model substrate to investigate the catalytic activity of tropylium tetrafluoroborate 1 in the direct amidation (Table 1). 4 Å pore size of activated molecular sieves were used to scavenge water formed during the course of the reaction and thereby not effected the desired yield.
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Cat. | mol (%) | Solvent | T (°C) | t (h) | 4ab (%) |
| a Reaction conditions: 1 mmol 2a in the indicated solvent, 3a 1 mmol and catalyst 1 in microwave vial at the indicated temperature for the indicated time.b Yield of the isolated 4a.c Reaction was run with 150 °C.d 10 mol% of a Brønsted acid catalyst was used instead of tropylium catalyst 1. | ||||||
| 1 | 1 | No cat. | THF | 70 °C | 15 | <10% |
| 2 | 1 | 5 mol% | DCM | rt | 15 | 30 |
| 3 | 1 | 5 mol% | DCM | 50 °C | 15 | 37 |
| 4 | 1 | 5 mol% | DCM | 50 °C | 15 | 46 |
| 5 | 1 | 10 mol% | THF | 45 °C | 7 | 52 |
| 6 | 1 | 10 mol% | THF | 70 °C | 15 | 89 |
| 7 | 1 | 10 mol% | DCE | 70 °C | 15 | 55 |
| 8 | 1 | 10 mol% | DMF | 70 °C | 15 | 62 |
| 9 | 1 | 10 mol% | Toluene | 70 °C | 15 | 37 |
| 10 | 1 | 10 mol% | MeCN | 70 °C | 15 | 55 |
| 11 | 1 | 10 mol% | 1,4-Dioxane | 70 °C | 15 | 45 |
| 12 | 1 | 10 mol% | MeOH | 70 °C | 15 | 61 |
| 13c | 1 | 10 mol% | THF | 150 °C | 1 h mw | 51 |
| 14 | 1 | 5 mol% | THF | 70 °C | 15 | 69 |
| 15 | 1 | 1 mol% | THF | 70 °C | 15 | 35 |
| 16d | HBF4 | 10 mol% | THF | 70 °C | 15 | 15 |
| 17d | TfOH | 10 mol% | THF | 70 °C | 15 | 18 |
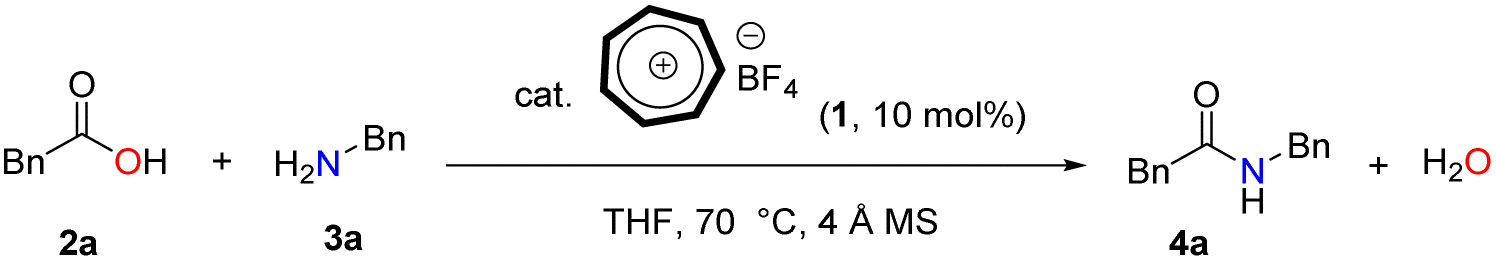
Initial optimization of the amidation reaction using 5 mol% of catalyst 1 at room temperature resulted in superior outcomes (Table 1, entry 2). It is important to note that the final product is produced only in trace amounts in the absence of the catalyst, as confirmed by NMR (entry 1). Screening over 8 different solvents revealed that only THF provided the highest efficiency for amide product 4a. This is attributed to the higher solubility of tropylium and the substrates in THF compared to the other solvents. It's worth noting that the reaction efficiency significantly decreased when increasing the catalytic loading and decreasing the reaction time and temperature (entry 5). However, increasing the reaction temperature and using 10 mol% tropylium catalyst led to an increase in reaction efficiency, yielding 89% of product 4a (entry 6). Utilizing tropylium BF4, the catalyst loading could be reduced to 5 mol% with minimal impact on reaction efficiency (entry 14). However, employing the same reaction conditions with a decrease in catalytic loading to 1 mol% showed reduced efficiency in the tropylium-catalyzed system (entry 15). This clearly demonstrates that decreasing the concentration of active catalyst in the reaction is sensitive to product inhibition, leading to a decrease in the reaction rate, and vice versa.
Only a limited number of methods have been reported that utilize Brønsted acids to catalyze direct amidation. To confirm this, a series of experiments were conducted to further optimize using strong Brønsted acids such as HBF4 and TfOH. However, these reaction conditions led to lower product yields and a more complicated reaction mixture (entries 16 and 17). They also confirm that the catalytic ability of tropylium is dominant in this reaction. Under these optimized reaction conditions, we subsequently used the conditions outlined in (Table 1, entry 6) to perform tropylium-promoted reactions with a range of non-activated carboxylic acids and amine derivatives.36
Notably, the tropylium ion can promote direct amidation for carboxylic acids and amines with bulky or electronically not active structures, however, the reaction proves to be sluggish, which generate the corresponding products in moderate efficiency. Phenylacetic acid 2a and benzylamine 3a, carrying both electron-withdrawing and electron-donating groups, generate smoothly the corresponding amides in good to excellent yields (Scheme 1 4a–4d). Electron-deficient aryl, cyclic, and heterocyclic benzoic acids behave similarly to their phenylacetic acid analogues. Consequently, they reacted smoothly with benzylamine to yield the corresponding products in moderate to high yields using 10 mol% of tropylium ion (Scheme 1, 4e–4i, 4k, 4l). Utilizing cinnamic acid as a versatile substrate in organic synthesis enables the formation of various derivatives with diverse applications. This is due to its possession of a double bond in its structure, which can be used for further reactions (Scheme 1, 4j).37 When treating fatty acids with different chain lengths as substrates with benzylamine, the amidation yield was high without being effected with length of carbon chain (Scheme 1. 4m, 4n).
 | ||
| Scheme 1 Substrate scope of tropylium-promoted direct amidation. | ||
We also expanded the applicability of our protocol by exploring the use of chiral starting materials such as methyl-L-proline and protected proline derivatives in tropylium-promoted direct amidation. Protected L-amino acid moieties are structurally important for many valuable synthetic precursors and biologically relevant organic compounds. Gratifyingly, we were able to access the desired product in high yield using benzylamine as the nucleophile (Scheme 1, 4o and 4p).
In contrast, the amidation reaction with electron-withdrawing and electron-donating substituents on the aromatic ring of benzylamine derivatives was favorable, and did not affect the amidation yield (Scheme 1, 4q–4u).
Furthermore, heterocyclic compounds, which are prevalent in many pharmaceuticals on the market today, are considered fundamental in medicinal chemistry due to their versatility and unique properties.38,39 Beyond drugs already available, ongoing research is exploring their potential in treating various cancers. Specifically, anticancer studies are leveraging the adaptable nature and dynamic structure of these compounds.40 So, we focused on using benzylamine substrates that contain N-heterocyclic systems like homopiperonylamine, furfurylamine, and morpholine rings in our reaction conditions. This led to the production of compounds (Scheme 1, 4v–4y) with good yields.
Notably, cyclic and aliphatic amines as nucleophiles behave similarly to their benzylamine analogues to give the corresponding products smooth reaction profiles (Scheme 1 4z–4zb). Using the optimized conditions, we then explored the scope of our reaction by testing structurally hindered cyclic and acyclic secondary amines, which resulted in the corresponding products being produced in varying yields, from low to high (Scheme 1 4zc–4zg).
Mechanism
Although clarifying the mechanistic details of this reaction was not the main focus of this work, based on our previous research in tropylium-promoted chemistry,1,12 we assumed that the tropylium ion acts as a Lewis acid catalyst in this reaction, facilitating the nucleophilic attack of amine 3 on carboxylic acid 2. The tropylium ion 1 can coordinate with the oxygen center of carboxylic acid 2, rendering the adjacent carbon electron-deficient. This facilitates a nucleophilic substitution reaction involving the nitrogen center of the amide substrate (Scheme 2) through either the SN1 or SN2 pathway. A subsequent hydrolysis reaction converts the iminium intermediate 6 into the amide product 4. Additionally, the tropylium ion might coordinate with water in the reaction medium, increasing its Brønsted acidity and potentially catalyzing the reaction through a hidden Brønsted acid pathway. However, by testing strong Brønsted acids (entries 16 and 17 in the optimization table), we demonstrated that the tropylium ion is the primary catalyst in this study.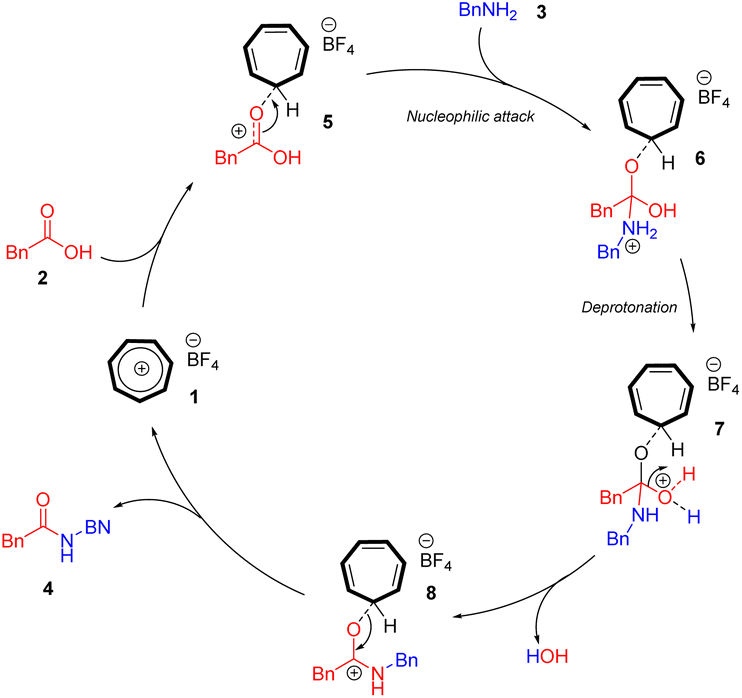 | ||
| Scheme 2 Proposed mechanism. | ||
Computational studies
Density functional theory calculations have been employed in this paper to gain more detailed molecular insights into the uncatalyzed condensation of benzylamine with different carboxylic acids, ranging from a–p, as illustrated in Scheme 1. Fig. 1 shows the relative fluctuations in energies of different properties. The energies of the calculated parameters were scaled to units where the correlation could be traceable. The reaction energies of the amide direct condensation showed positive variations in energy above the unsubstituted benzylamide, a. While the substitution effect caused the frequencies to shift toward lower energies (with exceptions c and e), this means the substitution effect tends to weaken the amide bond. Furthermore, the well-known electron-withdrawn group CN, NO2 in g and c respectively, contribute equality in stabilization of the amide formation with energy of ∼15 kcal mol−1, while prominent the electron-donating group (–OMe) in compound h destabilize the formation of amide bond.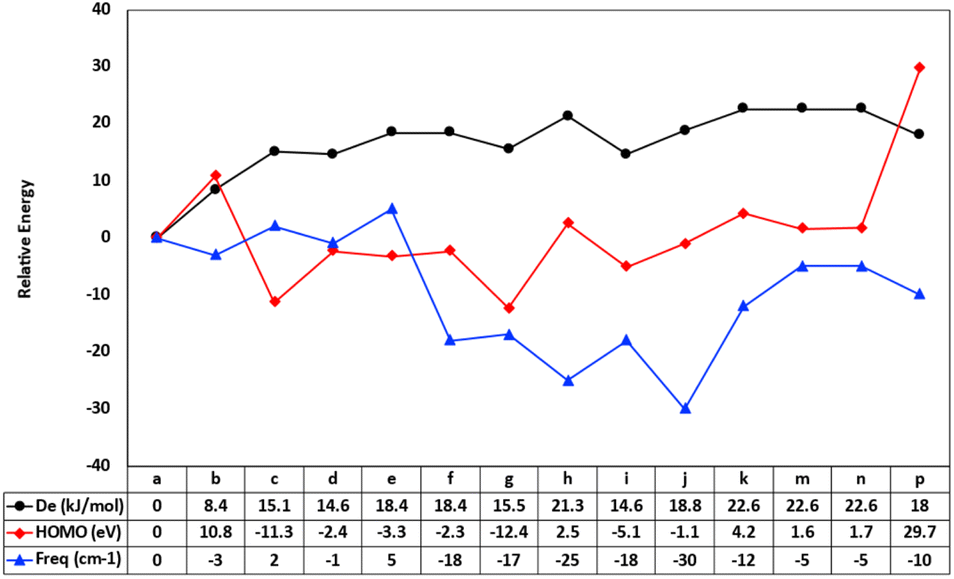 | ||
| Fig. 1 The relative changes in the synthesized species of amide scope: the frequencies of amide bonds (blue line) in (cm−1), the amide dissociation energies (black line) in kcal mol−1 and the HOMOs (red line) in eV. | ||
We also considered the highest occupied molecular orbitals (HOMOs), as they cover the area of the amide bonds. Therefore, changes in the amide bond could be reflected in the eigenvalues of HOMOs. Compounds f, g, h, and i reflect the effect of aromatic ring substitution on the selected amide bond parameters. However, there is no consistent pattern in the substitution effects on the rings. This inconsistency can be interpreted due to the different nature of substituted groups that affect resonance. For example, the cyano group is known to be more deactivating than the carboethoxy group.
At first glance, one can clearly see a matching pattern in the behavior of these parameters as we move from compound a to p in the carboxylic scope scheme. Regardless of the geometry relaxation of synthesized species, the small shifts in bond frequencies of the amide bond can be reflected in bond strength. This results in a comparative trend with both dissociation energies of the amide bond. The condensing reaction energies also show patterns related to the frontier orbitals. Although Kohn–Sham DFT orbitals cannot be fully trusted to compare to the reaction energies, the extension of these orbitals over the amide bond contributes to the bond order and hence its strength.
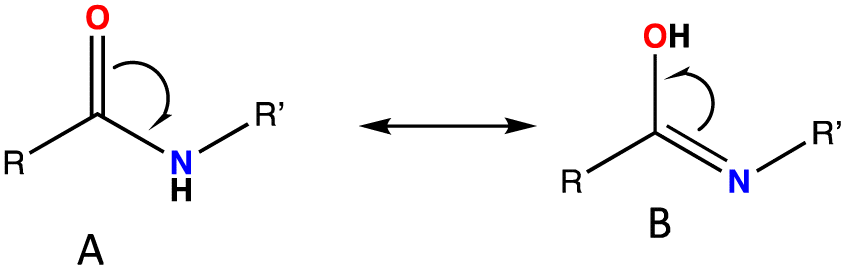
The tautomerization energy of shifting the proton between the nitrogen and oxygen atoms in the amide bond was calculated by taking the energy difference between the two isoelectronic structures (ETaut = EB − EA), A and B, as shown in Fig. 2. The positive values indicate that the keto form is more energetically favoured. The corresponding highest occupied molecular orbital also represented to show the effect on the hydrogen migration on the electronic distribution of the valence electrons. We have selected eight compounds that could be sufficient to demonstrate the preference for switching between the two forms. The energies of the tautomerism reaction shows that maximum change between the enol and keto forms before and after substitution can affect the tautomerism penalty is less than 2 kcal mol−1.
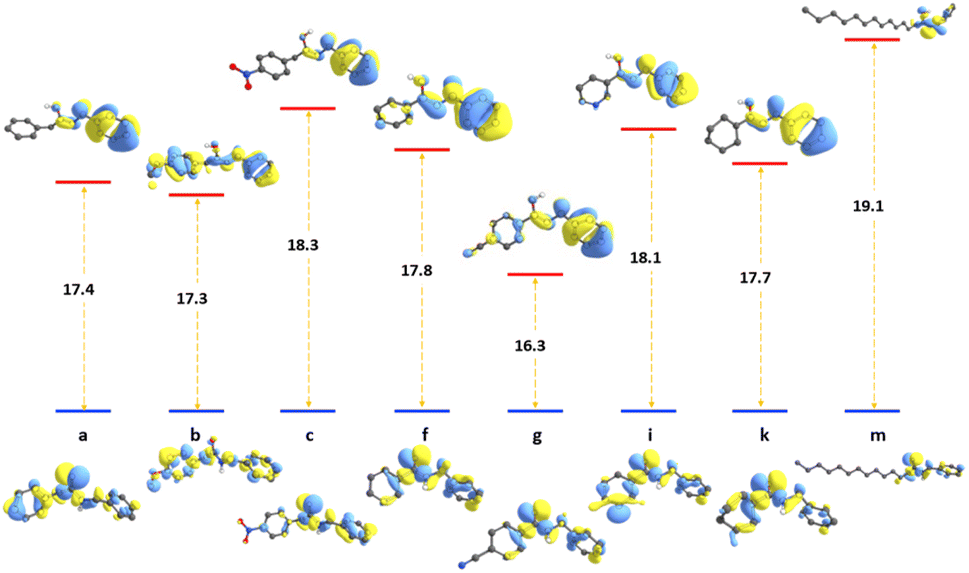 | ||
| Fig. 2 The tautomerism energy of switching from keto to enol form in kcal mol−1 for selected compounds listed in carboxylic scope, with corresponding HOMO orbitals of both forms. | ||
The computational details
The calculation for the condensation reaction of all the synthesized compounds were performed using Kohn–Sham density functional theory. We have performed geometry optimization using well-known Becke's hybrid B3LYP functional41,42 to the equilibrium geometries. The Ahlrichs' triple zeta polarized (def2-TZVP) basis set43 for all atoms supplemented with the auxiliary basis sets of Weigend (def2/J).44 To simulate the reaction conditions, the all optimization calculations the presence Conductor-like Polarizable Continuum Model (CPCM)45 of tetrahydrofuran (THF) solvent with dielectric constant ζ = 7.25. Subsequent vibrational calculations to check that all the eigenvalues of Hessian matrix contain no imaginary frequencies, series of single point frequency calculations were performed on the calculated compounds. The reaction energies were calculated for all the species that involved in the uncatalysed direct condensation reaction| RR′NH + R′′COOH → RR′NCOR′′ + H2O |
The reaction energy extracted by
| ΔE = (ERR′NCOR′′ + EH2O) − (ERR′NH + ER′′COOH) |
While tautomerism energies were performed by taking the difference between the keto and enol form on the amide bond of the synthesized compounds.
| ΔE = Eketo − Eenol |
All the calculations were performed using the open-source computational chemistry software ORCA, version 4.1.2 was developed by Neese group at Max-Planck Institute.46 The frontier orbitals are well-known for their crucial factor to understand the tautomerism effect on compounds.47,48 The frontier orbital also have been discussed to number of tautomer's to interpret the effect of tautomerism on the frontier orbitals. The results show (Fig. 3) there is only slight effect on the Kohn–Sham occupied orbitals with obvious consistency while the empty orbitals experience some deviation from consistency. The difference in the latter is reflected in some of the values of the HOMO–LUMO energy gaps between different compounds in both, enol and keto forms, taking the keto form as reference, we can notice the gaps of p and k compounds decrease significantly (27% and 14% respectively) upon the re-hydrogenation of enol form, in contrast, compound p frontier orbitals gap increases by 10% as the hydrogen shift from keto and enol form. These changes are mainly a direct result of the changes in the LUMO eigenvalues. The HOMO energies for both forms are changing uniformly. The rest of data show less pronounced differences (<4%) in the magnetite of the energy splitting of the frontier orbitals. The nitro and cyano groups in compound c stabilize both frontier orbitals, such trend reported in different studies49–52 yet, the results reported here offer clearer depiction to the behavior of the frontier orbitals toward the hydrogen migration between keto and enol forms.
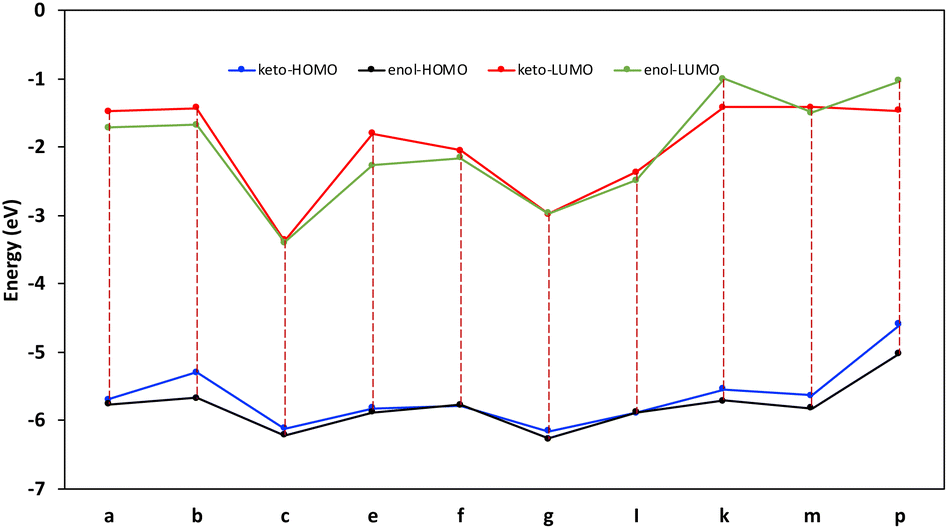 | ||
| Fig. 3 The frontier orbitals eigenvalues of the keto and enol forms upon the tautomerism shift. | ||
Experimental section
General procedure
All reagents and solvents were purchased and used as received from commercial suppliers or synthesized according to cited procedures. Oxygen and moisture sensitive reactions were carried out in dried glassware under nitrogen atmosphere. Flash chromatography was performed using 15–45 μm silica gel cartridges (60 Å mesh) on a Teledyne ISCO Combiflash Rf SiliaSep. SiO2 cartridges used for all the purifications were provided by SiliCycle. Analytical thin layer chromatography (TLC) was performed on E. Merck silica gel 60 F254 plates and the spots were visualized by UV light (254 nm). NMR spectra for the characterization of compounds were recorded at room temperature on a Bruker instrument 400 MHz (1H) and at 101 MHz (13C), or 500 MHz (1H) and at 126 MHz (13C). All the NMR data were collected for CDCl3 and DMSO-d6 solutions at ambient temperature. Chemical shifts (δ) are reported in parts per million (ppm) relative to CHCl3 (δ(H) = 7.26 ppm and δ(C) = 77.2 ppm) and DMSO solvent peaks (δ(H) = 2.50 ppm and δ(C) = 39.5 ppm). Data for 1H-NMR are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constants (Hz), and integration. High resolution mass spectroscopy (HRMS) measurements were carried out on a Bruker micro TOF/ESI mass spectrometer, and HRMS data analysis was performed using the software “Bruker Daltonics Data Analysis”, version 4.0 SP5.General procedure – substrate scope Scheme 1
To a 5 mL Schlenk tube loaded with a stirring bar containing the phenylacetic acid or benzoic acid derivatives (1 mmol, 1 equiv.) and activated molecular sieves 4 Å pellets (0.5 g) tropylium tetrafluoroborate (catalyst) 1 (0.2 mmol, 10 mol%) and dry THF (5 mL) was added through the septa, primary or secondary amine 3a (1 mmol, 1 equiv.) was added slowly through the septa and the resulting mixture was then heated to 70–90 °C for 15 hours. The crude reaction mixture was filtered through a pad of silica eluted with 100 mL EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et3N (100:1) and the target product was isolated from the crude mixture by column chromatography (silica-gel, hexane/ethyl acetate).
:Et3N (100:1) and the target product was isolated from the crude mixture by column chromatography (silica-gel, hexane/ethyl acetate).
Conclusion
We conducted a comprehensive mechanistic study of tropylium-catalyzed amidation reactions involving carboxylic acids and amines, using NMR spectroscopy and computational calculations. Additionally, we demonstrated the effectiveness of tetrafluoroborate as an organic Lewis acid in promoting these amidation reactions, resulting in good to excellent yields of the corresponding amide products. Drawing on our previous research in tropylium-promoted chemistry, this reaction protocol is adaptable to microwave and continuous flow conditions, suggesting promising applications in future organic synthesis endeavors. To investigate how different substituents affect the thermodynamics of condensation reactions in the synthesized compounds, a series of calculations were conducted using DFT to compute reaction energies. These calculations also examined the impact of substitutions on hydrogen migration during tautomerism and the energy splitting of the frontier orbitals. The calculations show that the tautomerism of the synthesized compounds effects the reaction energies only by slight margin of energy, while frontier orbital splitting changes mainly influenced the LUMO orbitals.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† The datasets generated and/or analyzed during the current study include experimental raw data: NMR and 13C measurement and geometrical coordinates. No special access permissions are required. For further information, please contact the corresponding author at E-mail: karrar.alameed@uokufa.edu.iq.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MH would like to thank Department of Chemistry at the University of New South Wales and Uppsala University for the research facilities and Jabir Ibn Hayyan University for medical and pharmaceutical sciences for the financial support. KA thanks the University of Kufa the financial support.References
- E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 30, 4641–4651 CrossRef.
- A. Ojeda-Porras and D. Gamba-Sánchez, J. Org. Chem., 2016, 81, 11548–11555 CrossRef.
- J. Pitzer and K. Steiner, J. Biotechnol., 2016, 235, 32–46 CrossRef PubMed.
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC.
- A. Tomberg and J. Boström, Drug Discovery Today, 2020, 25, 2174–2181 CrossRef PubMed.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef.
- M. Winnacker and B. Riegera, Polym. Chem., 2016, 7, 7039–7046 RSC.
- J. W. Bode, Curr. Opin. Drug Discovery Dev., 2006, 9, 765–775 Search PubMed.
- M. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef.
- M. Todorovic and D. M. Perrin, Chem. Rec., 2020, 112, 24210 Search PubMed.
- N. Martín and F. G. Cirujano, Catal. Commun., 2022, 164, 106420 CrossRef.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef PubMed.
- D. Constable, P. Dunn, J. Hayler, G. Humphrey, J. L. Leazer Jr, R. Linderman, K. Lorenz, J. Manley, B. Pearlman, A. Wells, A. Zaksh and T. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- M. Bryan, O. logo, P. Dunn, D. Entwistle, O. Gallou, S. Koenig, J. Hayler, M. Hickey, S. Hughes, M. Kopach, G. Moine, P. Richardson and R. Franz, Green Chem., 2018, 20, 5082–5103 RSC.
- Y. Du, T. Barber, S. Lim, H. Rzepa, I. Baxendale and A. Whiting, Chem. Commun., 2019, 55, 2916–2919 RSC.
- D. Sawant, D. Bagal, S. Ogawa, K. Selvam and S. Saito, Org. Lett., 2018, 20, 4397–4400 CrossRef CAS PubMed.
- M. Sabatini, L. Boulton and T. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed.
- H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 571–577 CrossRef CAS.
- T. M. El Dine, W. Erb, Y. Berhault, J. Rouden and J. Blanchet, J. Org. Chem., 2015, 80, 4532–4544 CrossRef.
- R. Lanigan, P. Starkov and T. Sheppard, J. Org. Chem., 2013, 78, 4512–4523 CrossRef CAS PubMed.
- Å. Nordahl and R. Carlson, Acta Chem. Scand., Ser. B, 1988, 42, 28–34 CrossRef.
- H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271–3277 CrossRef CAS.
- F. Azambuja and T. Parac-Vogt, ACS Catal., 2019, 9, 10245–10252 CrossRef.
- H. Lundberg, F. Tinnis and H. Adolfsson, Synlett, 2012, 23, 2201–2204 CrossRef CAS.
- C. Allen, A. Chhatwala and J. Williams, Chem. Commun., 2012, 48, 666–668 RSC.
- H. Lundberg, F. Tinnis, J. Zhang, A. Algarra, F. Himo and H. Adolfsson, J. Am. Chem. Soc., 2017, 139, 2286–2295 CrossRef CAS PubMed.
- F. Azambuja, A. Loosen, D. Conic, M. Besselaar, J. Harvey and T. Vogt, Chem.–Eur. J., 2012, 18, 3822–3826 CrossRef PubMed.
- H. Lundberg, F. Tinnis and H. Adolfsson, Organomet. Chem., 2019, 33, 5062 CrossRef.
- S. Doan, M. A. Hussein and T. V. Nguyen, Chem. Commun., 2021, 57, 8901–8904 RSC.
- M. A. Hussein, U. Tran, T. V. Huynh, J. Ho, M. Bhadbhade, H. Mayr and T. V. Nguyen, Angew. Chem., Int. Ed. Engl., 2020, 59, 1455–1459 CrossRef CAS PubMed.
- S. Doana and T. V. Nguyen, Green Chem., 2022, 24, 7382–7387 RSC.
- G. Oss, J. Ho and T. V Nguyen, Eur. J. Org Chem., 2018, 29, 3974–3981 CrossRef.
- D. Lyons, A. Dinh, N. Ton, R. D. Crocker, B. K. Mai and T. V. Nguyen, Org. Lett., 2022, 24, 2520–2525 CrossRef PubMed.
- D. J. M. Lyons, R. D. Crocker, M. Blümel and T. V. Nguyen, Angew. Chem., Int. Ed., 2017, 56, 1466–1484 CrossRef PubMed.
- M. A. Hussein, V. T. Huynh, R. Hommelsheim, R. M. Koenigs and T. V. Nguyen, Chem. Commun., 2018, 54, 12970–12973 RSC.
- N. Ruwizhi and B. A. Aderibigbe, Int. J. Mol. Sci., 2020, 21, 5712 CrossRef.
- X. Xu, et al., Bioorg. Med. Chem., 2023, 82, 117234 CrossRef.
- D. Izuchukwu and J. Conradie, J. Inorg. Biochem., 2023, 246, 112268 CrossRef PubMed.
- A. Ahmed, A. H. Gaber, A. M. Bayoumi, F. F. El-morsy, A. B. M. Sherbiny and I. H. E. Mehany, Bioorg. Chem., 2022, 120, 105591 CrossRef PubMed.
- M. A. Hussein, V. T. Huynh, R. Hommelsheim, R. M. Koenigs and T. V. Nguyen, Chem. Commun., 2018, 54, 12970–12973 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev., 1988, 37, 785–789 Search PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- V. arone and M. Cossi, J. Phys. Chem., 1998, 102, 1995–2001 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Phys. Chem., 2020, 22, 152 Search PubMed.
- Y. Shee, T.-L. Yeh, J.-Y. Hsiao, A. Yang, Y.-C. Lin and M.-H. Hsieh, npj Quantum Inf., 2023, 9, 102 CrossRef.
- M. W. Schmidt, E. A. Hull and T. L. Windus, J. Phys. Chem. A, 2015, 119, 10408–10427 CrossRef.
- B. Ośmiałowski, E. D. Raczyńsk and T. M. Krygowski, J. Org. Chem., 2006, 71, 3727–3736 Search PubMed.
- Y. Mao, M. H. Gordon and Y. Shao, Chem. Sci., 2018, 9, 8598–8607 RSC.
- A. H. Mageed and K. Al-Ameed, New J. Chem., 2021, 45(39), 18433–18442 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04534c |
| This journal is © The Royal Society of Chemistry 2024 |