
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColour tuneability of heteroleptic iridium complexes through second-sphere coordination†
Barbora Balónová ,
T. Harri Jones,
Allison E. True,
Sydney M. Hetherington and
Barry A. Blight*
,
T. Harri Jones,
Allison E. True,
Sydney M. Hetherington and
Barry A. Blight*
Department of Chemistry, University of New Brunswick, Fredericton, NB E3B 5A3, Canada. E-mail: b.blight@unb.ca
First published on 29th October 2024
Abstract
Novel H-bond-rich iridium(III) complexes with different systems of cyclometalating ligands bearing the general formula [Ir(C^N)2(N^N)] were synthesised, characterised and their photophysical properties determined. The incorporated guanidine moieties allow us to introduce a hydrogen-bonding array for the formation of self-assembled iridium-bound hydrogen-bonded systems. Through a series of experimental and computational studies we demonstrated the host–guest chemistry of these systems. These hydrogen-bonding interactions were shown to have significant impacts on chromaticity, quantum yields and lifetimes with emphasis on energy transfer studies based on this second-sphere coordination approach.
Introduction
Iridium complexes have catalysed significant research interest due to their various applications such as bioimaging,1–6 organic photovoltaic cells,7–10 and catalysis.11–14 Particularly, the implementation of iridium complexes as emitters in phosphorescent organic light-emitting diodes (PhOLEDs) has attracted great attention.15–17 Cyclometallated iridium complexes have exhibited short lifetimes in excited states and high photoluminescence efficiencies which makes them particularly interesting when exploited in PhOLEDs.18–20 Phosphorescent iridium emitters can harvest both the generated singlet and triplet excitation pathways which theoretically makes them able to realise a 100% external quantum efficiency giving them an advantage in comparison to fluorescent systems, which can only reach a maximum of 25%.21 To fulfil the requirements of full-colour OLED displays, the colour regulation toward efficient red, green and blue emissions is highly desirable. Colour tunability is one of the key features of cyclometallated iridium complexes,22–27 and can be achieved through synthetic modifications of C^N ligands; these modifications change the energy of the frontier orbitals that take part in the photoexcitation process. The major triplet excited states observed in phosphorescent iridium complexes, according to Shakirova et al.,28 are (a) metal perturbed intra-ligand (3IL) transition (or ligand centred transition), (b) metal to ligands charge transfer (3MLCT) and (c) inter-ligand charge transfer (3LLCT). The energy of the excited states can be changed in multiple ways, but the most common approach is the synthetic modification of C^N ligands through incorporation of either donor or acceptor substituents.29–32 Other methodologies that can shift the emission colour of iridium complexes include size variations with increase of rigidity of the cyclometallating ligands, for instance, dibenzo[f,h]quinoxaline ligands that are able to bathochromically shift the phosphorescence colour.33To date, iridium complexes emitting in green and yellow regions have reported high quantum efficiencies.34,35 On the other hand, orange and red emitting iridium complexes typically suffer from slightly lower quantum yields since the radiative rate constant (kr) has a dependence on the excited state energy10 and it usually decreases with longer emission wavelengths.36 Blue iridium complexes are still under intense investigation for their application in the display and lighting industry as they also have relatively low efficiencies compared to standard Ir(III) yellow and green devices. According to Pal et al.,37 the criteria for deep blue emitters are: (i) possess the required chromaticity standards defined by the National Television System Committee (NTSC) and European Broadcasting Union (EBU) with Commission International de l’Éclairage (CIE)38 coordinates of (0.14, 0.08) and (0.15, 0.06), respectively; (ii) possess high photoluminescence quantum yields (ΦPL) that translate into high external quantum efficiencies in OLED and (iii) exhibit competitive device stabilities to fluorescent systems.38
Despite the efforts of various research groups,39–41 the design and synthesis of stable, efficient, and pure blue iridium complexes remains challenging. This study presents the design and synthesis of neutral iridium complex 1 (Fig. 1), with cyan-blue emission in CHCl3 solution with CIE coordinates: (0.18, 0.31). The chromaticity of complex 1 can be modified and reach deep blue emission (CIE coordinates: (0.15, 0.11)) when combined with compound 5 (Fig. 2). Furthermore, we prepared neutral yellow complex 2 and red/orange emissive iridium complexes 3 and 4 (Fig. 1), which are also desirable for their application in the display industry, but moreover the iridium complexes with red emission are often exploited in bioimaging and anticancer studies.42,43 The reasoning behind the synthesis of yellow complex 2 was to examine if white emission can be achieved by combining it with guest 5 (blue fluorescence in solution), as white emission is a crucial component in white OLEDs (WOLEDs).44,45
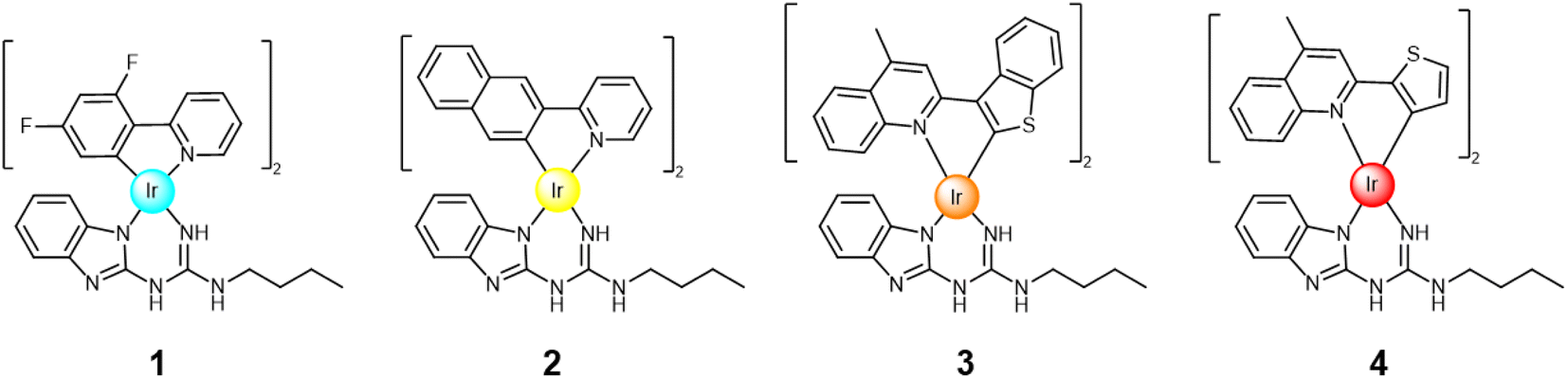 | ||
| Fig. 1 Cyclometallated Ir(III) complexes 1–4 studied herein. | ||
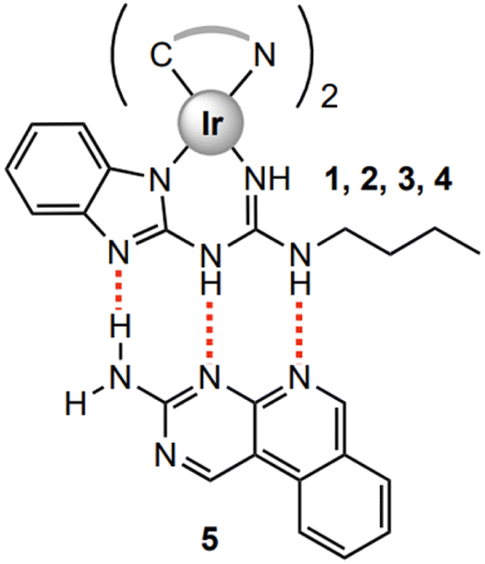 | ||
| Fig. 2 Proposed triple H-bonded systems, with preorganised arrangement that contributes to the stability of 1·5 to 4·5 systems. | ||
Usually, WOLEDs are made up of the three primary colours (RGB), so there is a broad gap (up to 100 nm) in the white-light spectrum, which means that the emission ranging from 520 to 620 nm is missing. White emission can be obtained based on the principle of additive colour mixing. Combination of blue and complementary yellow emissions is an attractive approach to reach white emission.46 Concomitantly, guest molecule 5 represents a suitable binding partner for yellow iridium complex 2, creating self-assembled hydrogen bonding systems, with preorganised arrangement that contributes to the stability of the 2·5 system. As defined by the CIE chromaticity system, an ideal white light has coordinates of x = 0.33, y = 0.33 and can be realised by additive colour mixing.47,48 Separately, through the synthesis of heteroleptic iridium complexes 3 and 4, based on thiophenephenylquinoline derivatives as C^N ligands, red emission was targeted.
Our team has reported a series of iridium complexes with ancillary ligands based on guanidine and thiourea moieties, i.e., inherently H-bond rich motifs (Fig. 2).49 Through second-sphere coordination via H-bonding, we introduced a de novo strategy to alter photophysical properties of emitters, accessing a linear colour scale with a discretely interacting two component system, reaching into colour regions obviating the need of synthetic modifications around the metal centre. We have further demonstrated the effectiveness of this approach through electrochemiluminescence in which this supramolecular system exhibits unique photoemissive processes upon electrochemical excitation.50,51
Here, we present the design, synthesis, and both an experimental and computational investigation of the photoluminescent properties of yellow, orange/red and blue iridium complexes 1–4 (Fig. 1). This expands the library of H-bond capable emitters towards colour tuning in phosphorescent materials (Fig. 3). All complexes were examined as single species and as co-complexes in combination with previously reported guest molecule 5.49 Compound 5 represents a suitable binding partner for the guanidine ancillary ligand that was combined with analogues iridium complexes49,52 due to its ability to form triple H-bonded systems, modifying the photophysical properties of each complex.
 | ||
| Fig. 3 Vials from emission studies of iridium complexes 1–4 with added aliquots of compound 5. Vials are illuminated with long wave UV-light (365 nm). Values in % corresponding to the molar equivalents of guest content (5) in solution. | ||
Results
Synthesis
Details for the synthesis of compounds 1–5 are presented in the experimental section S1-2 (see ESI†).Photo-physical measurements
Absorption–emission spectra of iridium complexes 1–4 were measured in CHCl3 solution at the same concentration (1.0 × 10−5 M).As shown in Fig. 4 above and S26 (ESI; Section S4†), the absorption spectra of the four complexes are mainly comprised of three parts. Complexes 1–4 show relatively strong bands in the range of 250 to 350 nm, which can be attributed to spin-allowed singlet state ligand centred (1LC) π–π* transitions. Absorptions from 350 to 450 nm can be ascribed to spin-allowed metal-to-ligand charge transfer singlet state and ligand-to-ligand charge transfer 1MLCT/1LLCT. The lowest energy component, extending beyond 450 nm arises primarily from spin-forbidden 3MLCT/3LLCT transitions.53 These types of electronic transitions are consistent with pertinent literature.54,55 The photoluminescence data, including emission maxima, lifetimes and quantum yields of complexes 1–4 are summarised in Table 1.
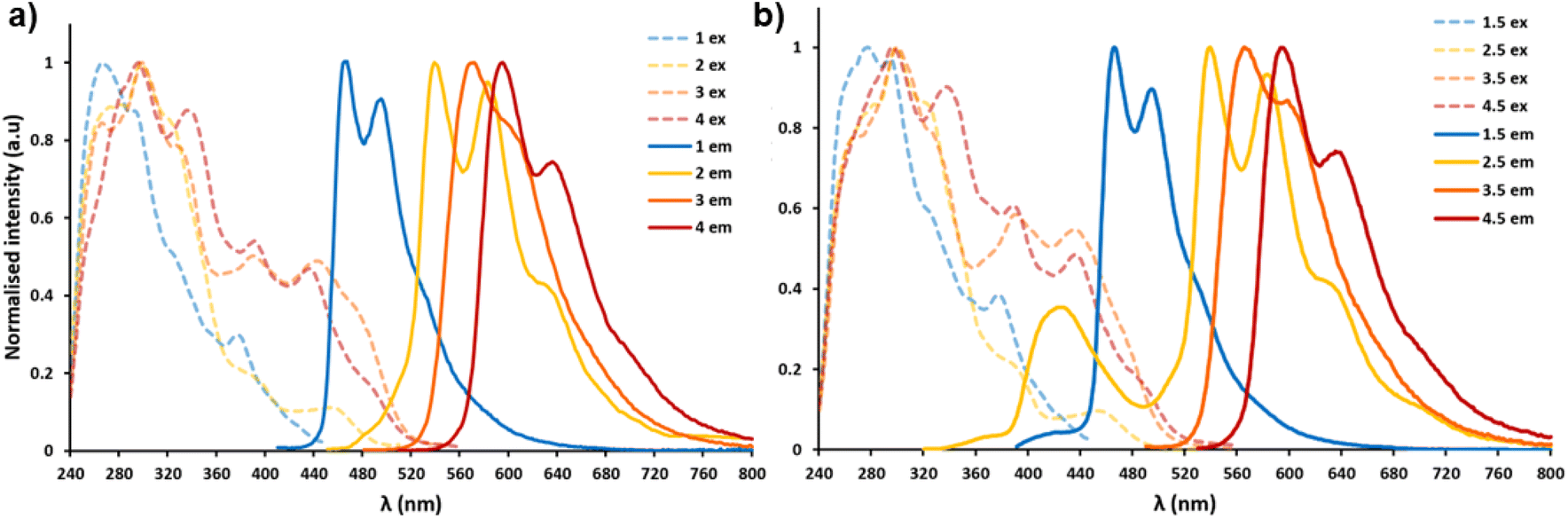 | ||
Fig. 4 (a) The normalised absorption–emission spectra of complex 1–4, dashed line representing the excitation of each complex and solid line representing the emission, all spectra were measured in CHCl3 (1.0 × 10−5 M). (b) The normalised absorption–emission spectra of complex 1–4 with compound 5 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in CHCl3 (1.0 × 10−5 M). :1 molar ratio in CHCl3 (1.0 × 10−5 M). | ||
| System | λexca (nm) | λema (nm) | ΦPLb | τc (μs) | krd (×105 s−1) | knre (×105 s−1) |
|---|---|---|---|---|---|---|
| a All data was collected in CHCl3 and degassed for 1 min (argon) at 298 K.b PLQYs for Ir(III) complexes 1–4, determined using the relative method. Further details of standard in the ESI S1 and S5.c Decay lifetimes using TCSPC excited at 365 nm and their corresponding chi-squared values for Ir(III) complexes 1–4, in CHCl3 degassed for 20 min (Ar) at 298 K. Complexes 1, 2, 3, and 4 decay biexponentially.d The radiative (kr) rate constant was calculated as kr = ΦPL/τPL.e The nonradiative (knr) rate constant was calculated as knr = (1 − ΦPL)/τPL. | ||||||
| 1 | 264, 378 | 467, 493 | 86.7 | 2.3 | 3.8 | 0.6 |
| 1·5 (1:1) |
280, 378 | 462, 489 | 82.2 | 2.1 | 0.4 | 0.9 |
| 2 | 296, 320 | 545, 588 | 8.7 | 0.0019 | 448.5 | 4706.2 |
| 2·5 (1:1) |
298, 325 | 538, 584 | 15.6 | 0.0036 | 430.9 | 2331.5 |
| 2·5 (1:1.3) |
298, 326 | 538, 584 | 33.2 | 0.0034 | 985.2 | 1982.2 |
| 3 | 298, 332 | 570, 610 | 23.4 | 1.7 | 1.4 | 4.5 |
| 3·5 (1:1) |
297, 340 | 569, 607 | 43.3 | 4.3 | 1.0 | 1.3 |
| 4 | 296, 338 | 594, 640 | 36.1 | 3.6 | 1.0 | 1.8 |
| 4·5 (1:1) |
308, 391 | 593, 635 | 41.4 | 3.6 | 1.2 | 1.6 |
All iridium compounds studied herein exhibit dual emission to some extent, complex 1 emits a cyan-blue colour (467, 493 nm) with a high quantum yield (ΦPL = 86.7%) and a microsecond lifetime (τ = 2.3 μs) showing clear phosphorescent character. When in the presence of compound 5 forming co-complex 1·5, we see a decrease in both quantum yield and lifetime in the co-complex as well as a minor blue shift ∼4 nm in the peak of the iridium complex's emission, this ‘blue shift’ is a common trend of all complexes in this study when compound 5 is present in solution. Complex 2 emits yellow (545, 588 nm) with a relatively low quantum yield (ΦPL = 8.7%), lifetime measurements showed an uncharacteristically short lifetime of 19 ns (short for a cyclometallated Ir(III) complex). Upon addition of compound 5 an enhancement in the lifetime of up to 36 ns was recorded, it was also possible to achieve near white light emission (CIE coordinates: 0.34, 0.33) when 1.3 molar equivalents of 5 were added to 2, as well as an increase to the overall quantum yield of this co-complex. Complexes 3 and 4 emitted orange and red respectively, both have long lifetimes which could be enhanced through equimolar additions of compound 5. Both compounds also saw enhancement in quantum yields when forming a co-complex with compound 5 in solution. The overall blue shifting in the colour of the bimolecular systems can be ascribed to a combination of the presence of the blue emitting compound 5 and the H-bonding interactions of compound 5 with each of iridium complexes 1–4.
Solid state photophysical measurements were also collected by preparing polymer films of 2 wt% (calculated based on weight of complex) dispersion in PMMA from argon degassed CHCl3 using a spin coating method. Films with equimolar host–guest complexes were then made and the photophysical properties measured. In PMMA films, the blue shift in emission was weakened compared to solution when one molar equivalent of compound 5 was added to complexes 2, 3, and 4. The emission profile of 1 saw a minor red shift of 2 nm when compound 5 was added to form the co-complex in film. In all solid-state (PMMA) co-complexes, except 2·5, the presence of compound 5 also led to a decrease in quantum yield. The table summarising these results can be found in ESI; S7.†
Host–guest chemistry
All complexes were studied as single species in CHCl3 solutions and in combination with guest molecule 5. Stacked emission spectra from the host–guest studies of complexes 1–4 in combination with 5 are presented in the ESI (Section S4†) and their association constants (Ka; M−1) summarised in Table 2. Surprisingly, the trend in association constants' relative strengths did not correlate with well-established Hammett parameters, which can be used to benchmark the electron-withdrawing (EWD) or donating (EDG) ability of a substituent or ligand.56 However, since the H-bonding interactions are occurring between the guest molecule 5 and the same ancillary ligand of the iridium complexes, the cyclometalating ligands do exhibit a noteworthy influence on the Ka values of each HG complex.Molecular recognition imparted by the H-bond sites enacts directed assemblies of the heterodimers with measurable association constants. Further, the stoichiometric addition of 5 can tune the colour of the overall visible light emissions of our host–guest systems. These effects on the photophysical properties by equimolar additions of compound 5 are summarised in Table 1.
Electrochemical studies
The electrochemical properties of our Ir-complexes and co-systems were investigated using cyclic voltammetry. Relevant data is summarised in Table 3 below, and all potentials reported are referenced to a ferrocene standard. Complexes 1–4 all exhibit irreversible reductions between (−1.52 to −1.48 V), these irreversible reductions are consistent with other cyclometallated iridium complexes in the literature.57 Upon additions of one molar equivalent of compound 5, no consistent trends were seen in the irreversible reduction peaks; from this and previous literature, we can infer these reduction peaks are centred on the various C^N ligands.57 Each complex also has a reversible oxidation, the values of which vary significantly from 1.17 V in complex 1 to 0.54 V in complex 4 with a clear decreasing trend of the Eox value with the red shifting of the iridium complex. These reversible oxidations are often ascribed to the Ir(III)/Ir(IV) redox couple with contributions from an N^N− ligand.49,58 Upon addition of compound 5 to complexes 1–4 forming their respective assemblies in solution, the Eox values anodically shift by a range of values (0.10 to 0.03 V). The largest change is complex 1 in 1·5 where the anode shift is 0.1 V, leading to a ΔE value increase of 0.11 V. Overall, these anode shifts in the presence of compound 5 agree with our previous study into the H-bonding properties of the benzimidazolyl-guanidine ligands H-bonding interactions with compound 5 as well as electrochemical studies into frontier orbitals.49,59| System | Eoxa (V) | Ereda (V) | ΔEa (V) | (com)E0–0b (eV) | E0–0c (eV) | E(PS*/PS−)d (V) | E(PS*/PS+)e (V) |
|---|---|---|---|---|---|---|---|
| a All samples were prepped and studied in an argon flushed glove box in degassed, dry CHCl3. Further details of electrodes and apparatus can be found in ESI S1.b (com)E0–0 calculated from the computationally obtained HOMO–LUMO gap.c E0–0 was calculated from the crossover of normalised absorption and emission spectra.d E(PS*/PS−) = Ered + E0–0.e E(PS*/PS+) = Eox − E0–0. | |||||||
| 1 | 1.17 | −1.51 | 2.68 | 3.16 | 2.81 | 1.30 | −1.64 |
| 1·5 (1:1) |
1.27 | −1.50 | 2.77 | 3.26 | 2.84 | 1.34 | −1.57 |
| 2 | 0.82 | −1.49 | 2.31 | 2.95 | 2.58 | 1.09 | −1.76 |
| 2·5 (1:1) |
0.85 | −1.51 | 2.36 | 3.07 | 2.59 | 1.08 | −1.74 |
| 3 | 0.67 | −1.52 | 2.19 | 2.83 | 2.36 | 0.84 | −1.69 |
| 3·5 (1:1) |
0.71 | −1.50 | 2.21 | 2.96 | 2.39 | 0.89 | −1.68 |
| 4 | 0.54 | −1.51 | 2.05 | 2.79 | 2.26 | 0.75 | −1.72 |
| 4·5 (1:1) |
0.61 | −1.48 | 2.09 | 2.84 | 2.28 | 0.80 | −1.67 |
Computational studies
To further understand the findings of the photophysical and electrochemical data, DFT and TD-DFT studies were performed at the B3LYP/(6-31G**/LANL2DZ) level on complexes 1–4 and their respective assemblies with 5. Information on software packages used, structural inputs, specific orbital energies, and visualisations of molecular orbitals can be found in ESI; S9.†In iridium complexes 1–4 (Fig. 5), the HOMO resides upon the ancillary ligand's π-orbitals along with partial residuals on the iridium metal's d and π-orbitals. This is due to the large concentration of nitrogen within the ancillary benzimidazolyl-linked guanidine ligand. The HOMO energies of the complexes range between −4.75 and −4.57 eV. Although the HOMOs in complexes 1–4 reside mostly on the guanidine ligand, there is still variation in these energies. As there are residuals upon the iridium centre, the variation in the HOMO energies between 1–4 can be attributed to the changes in the other two cyclometallating ligands of the complex.
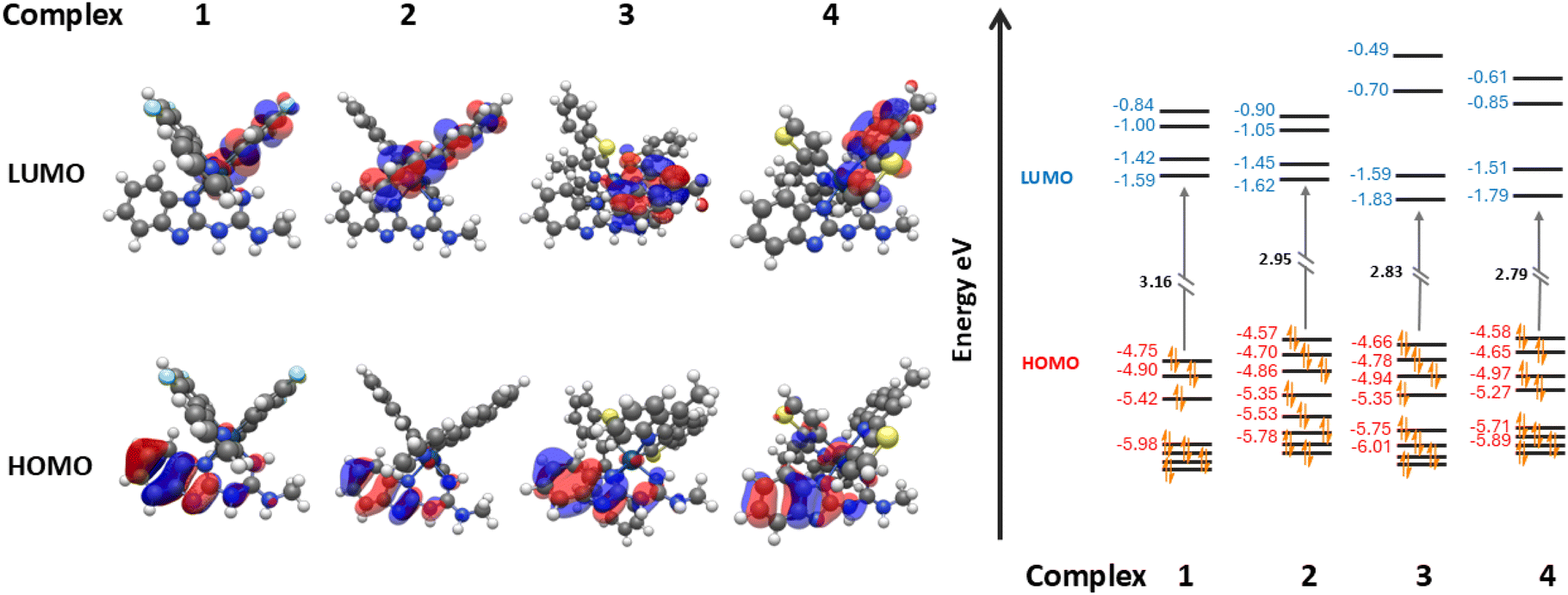 | ||
| Fig. 5 (left) TD-DFT predicted HOMO and LUMO orbital distributions of complex 1–4 and (right) visualisation of the energy levels of frontier orbitals with computationally obtained HOMO–LUMO gap. | ||
When compound 5 is modelled in the presence of complexes 1–4, a change in the HOMO level is observed in the co-complex. The HOMO continues to reside on the guanidine ligand of the iridium complex, but the HOMOs of the assemblies see an increase in energy, with the HOMO energies now ranging from −4.65 to −4.48 eV. This increased HOMO stability is due to frontier HOMO–LUMO interactions between the HOMO on the guanidine ligand of the complexes and the LUMO residing on compound 5.59
Studies show LUMO levels are affected by the introduction of compound 5 differently. In complexes 1–4 the LUMO resides on the π-orbitals of one of the two varying C^N cyclometallating ligands depending on the compound. Rather predictably, the LUMO energies tend to decrease from complex 1–4 from a peak of −1.59 eV in complex 1 to the lowest of −1.79 eV in complex 4. This lowering of the LUMO energy is due to the introduction of extended π-conjugation in the C^N cyclometallating ligands and the removal of electron withdrawing fluoride groups, leading to increased LUMO stability.60 When compound 5 is introduced to form their co-complexes, the LUMO of each assembly resides on compound 5. This artificially lowers the HOMO–LUMO gap of the co-complex, and the former LUMO of the iridium complex becomes the LUMO+1 of the new HG complex. In every case despite the stabilisation of the HOMO, this reassignment of the LUMO leads to an increased HOMO–LUMO+1 (formerly HOMO–LUMO) gap of orbitals residing on the iridium complexes. For example, in the case of complex 3 and assembly 3·5 the LUMO energy of complex 3 is −1.83 eV, but the LUMO+1 energy of 3·5 is −1.64 eV leading to a large increase in the Ir-centred HOMO–LUMO+1 gap and a calculated blue shift in the emission profile of the complex (Fig. 6).
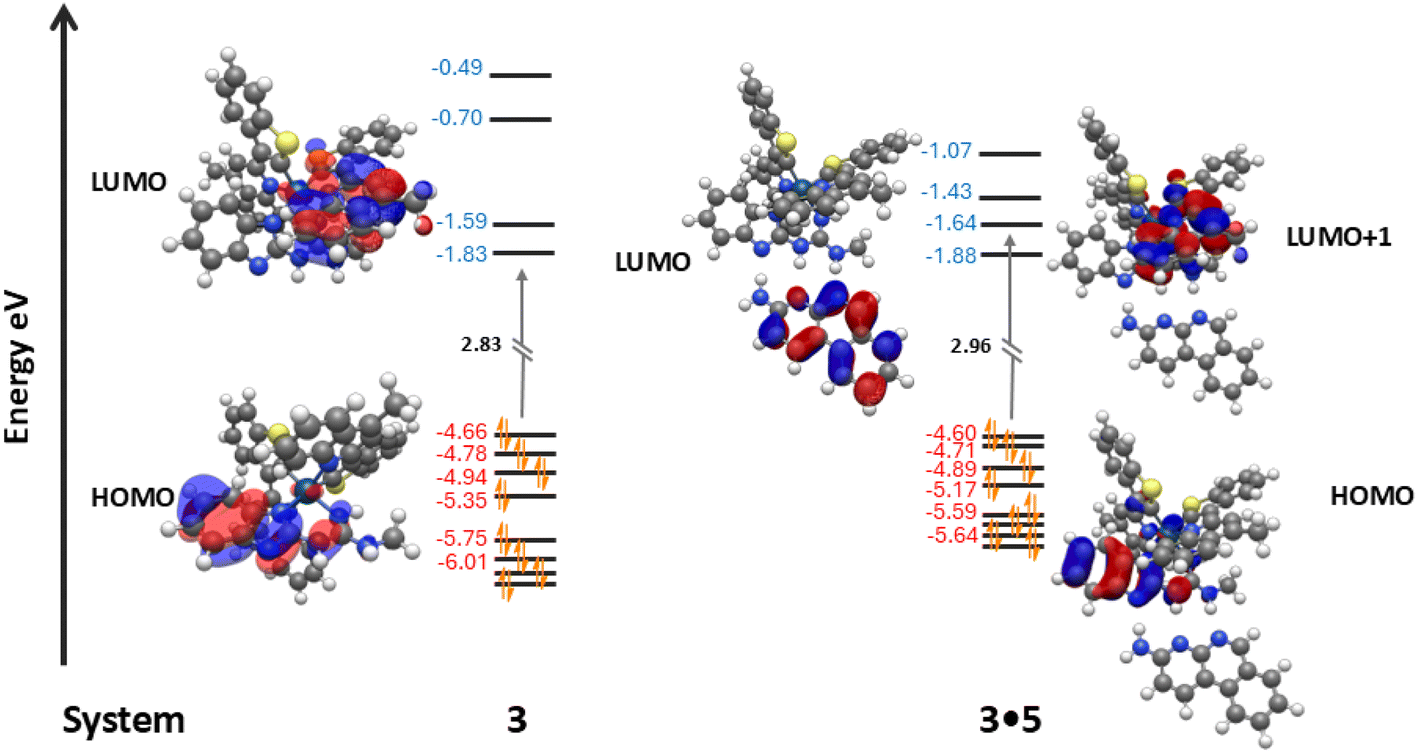 | ||
| Fig. 6 Comparison of the TD-DFT computationally modelled frontier orbitals of complex 3 and assembly 3·5. | ||
Continuing to use the example of complex 3 and HG assembly 3·5; during the first emission T1 excited state of complex 3, the transition contributions typically follow HOMO−1/HOMO−2/HOMO−3 to the LUMO, resulting in the predicted emission of 566 nm. Major contributions during the first T1 excited state in the co-system 3·5 are from HOMO−1/HOMO−2/HOMO−3 to LUMO+1, indicating the LUMO+1 is supplanting the position of the LUMO (located on 5) in triplet state excitations within the iridium complexes. Furthermore, the predicted T1 emission of 3·5 has blue-shifted by 1 nm to 565 nm indicating the increase in the HOMO → LUMO+1 gap from the introduction of 5 has resulted in a change in the emission profile of the iridium complex.
Discussion
The cyan-blue emission of complex 1 can be ascribed to the introduction of fluorine atoms on the C^N ligand, which generally stabilises the HOMO orbital and leads to an increased HOMO–LUMO gap.30 In this case, computational investigations revealed that the LUMO resides on the C^N ligand while the HOMO is residing on the guanidine ligand, due to the large electron density of the benzimidazole moiety of the ligand. It may then be assumed that the electron withdrawing fluorine atoms are destabilising the LUMO on the C^N ligands, therefore increasing the HOMO–LUMO gap as evidenced by the largest LUMO value of the complex (−1.59 eV). Upon excitation, emission of cyan-blue iridium 1 (CIE coordinates: (0.18, 0.31) and (0.16, 0.32), respectively) was modified by adding aliquots of 5 dissolved in a solvent system of CHCl3 with 1% DMSO accessing a linear colour change reaching into deep blue region for 1:1 systems (CIE coordinates: (0.15, 0.11)) without the need to synthetically modify the structure of complex 1. Additionally, changes in ΦPL and lifetime values for 1·5 in a 1:1 ratio in CHCl3 would also serve as a preliminary proof of a mutual communication between the two species (host and guest) that are designed to form stable self-assembled H-bonded systems. For the co-system 1·5, a decrease in quantum yield was observed (1·5 in CHCl3 solution ΦPL = 82.2%) when compared with compound 1 (ΦPL = 86.7%). The lifetime decay of complex 1 was measured to be 2.3 μs for the major component and decreased upon introduction of complement 5. From the study of the 1·5 system in CHCl3 solution, the longer lifetime component with value 2.1 μs was ascribed to the photoexcitation of complex 1 as the major component with a 97% contribution. In addition, a fluorescence resonance energy transfer (FRET) experiment was conducted for 1·5 system in CHCl3 solution.61 As presented in Fig. 7, the emission intensity of compound 5 decreased and the emission intensity of complex 1 slightly increased after these two components were combined in 1:1 solution mixture. This study illustrates that compound 5 represents a donor molecule that can modestly transfer its energy to complex 1 in the excited state.
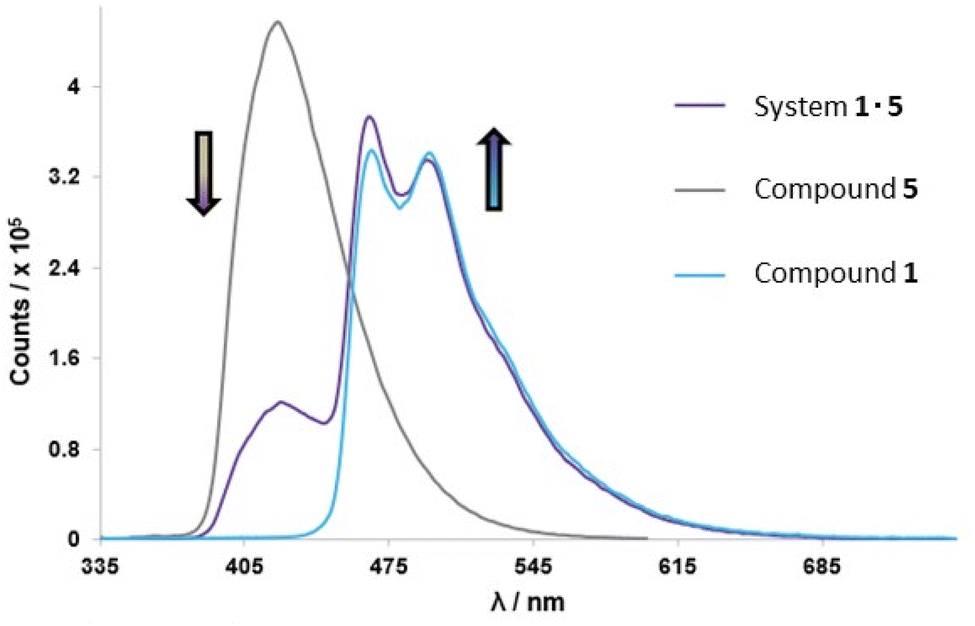 | ||
| Fig. 7 Overlaid emission spectra of complex 1 (blue), compound 5 (grey) and co-complex 1·5 (purple) from FRET studies in CHCl3 solution. All samples excited at λex = 350 nm. | ||
The blue shift of the complex was further investigated using through TD-DFT studies where we identified that the HOMO–LUMO gap of complex 1 had been increased by the presence of compound 5 by almost 10 eV. Here, H-bonding with compound 5 stabilises the HOMO localised on the benzimidazole moiety, increasing it by almost 10 eV. Due to the HOMO–LUMO frontier orbital interaction between 1 and 5, the LUMO+1 in the co-complex (residing on the C^N ligand; the LUMO of the iridium complex) was subsequently destabilised by 20 eV. Electrochemical studies confirmed that introducing 5 to complex 1 increases the ΔE by 0.11 V, with the Eox value anodically shifting and the Ered value having little change. We posit that H-bonding between compound 5 and the benzimidazolyl-guanidine ligand of the iridium complex leads to increased HOMO stability as the HOMO–LUMO interaction between frontier HOMO orbitals of complex 1 and the LUMO of compound 5 share the electron density residing in complex 1's HOMO.59
Iridium complex 2 emits yellow light (at much lower relative intensity) while exhibiting a very low quantum yield (ΦPL = 3.4%) and a short lifetime (τ = 1.94 ns), especially in solution. When complex 2 is dispersed in a 2 wt% load in PMMA films there is a dramatic increase in the quantum yield (ΦPL = 15.1%) despite maintaining a short lifetime. Upon adding aliquots of compound 5 to 2 in solution, we see a gradual increase in quantum yield while maintaining its relatively short lifetimes leading to very high kr and knr values for 2·5. Though these photophysical properties are not particularly noteworthy, addition of 1.3 equivalents of compound 5 to complex 2 during an emission titration resulted in white emission with CIE coordinates: (0.34, 0.33) (Fig. 8). Since ideal white light has coordinates of (0.33, 0.33),34 this was an exciting result. The titration with compound 5 also improved complex 2's uncharacteristically low quantum yield, and lifetime. The new quantum yield of the 1:1 co-complex 2·5 increased to ΦPL = 15.6% in CHCl3 solution. Increasing the ratio of 2·5 slightly to 1:1.3, produced pure white light emission with an observable doubling in quantum yield (ΦPL = 33.2%). We mainly ascribed this increase to the blue emitting compound 5, as compound 5 is known to have efficient fluorescent emission properties (ΦPL = 40%, τ = 3 ns).49 This ideal ratio of host–guest for white light was also determined through a recently reported theoretical calculation (eqn (1)) to estimate the overall contributions of 2 and 5 in the co-system.
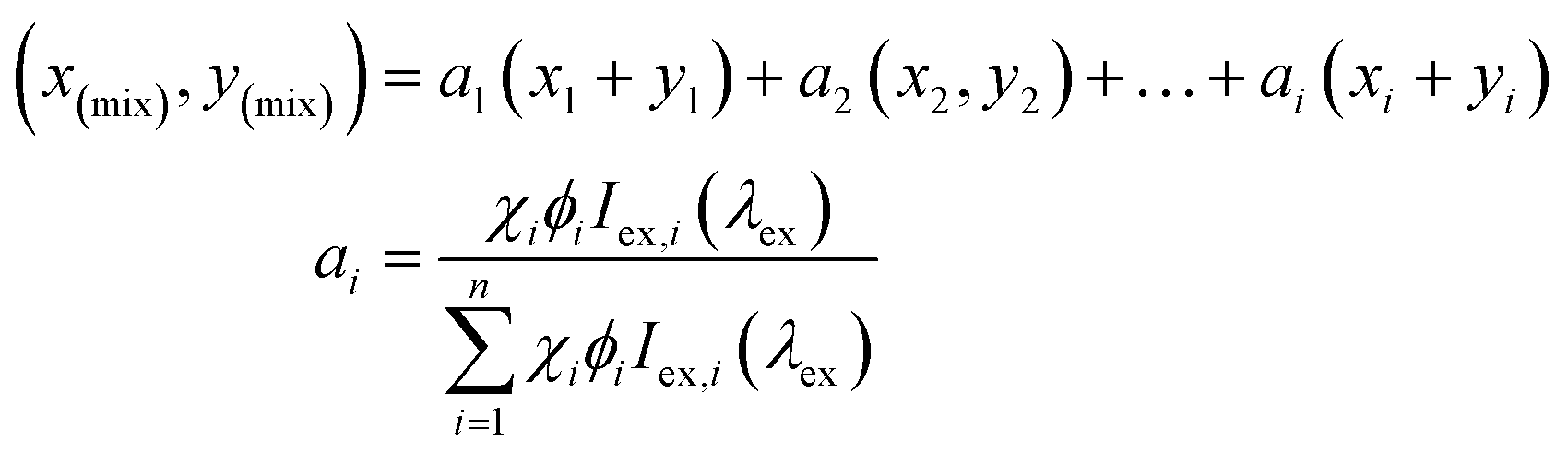 | (1) |
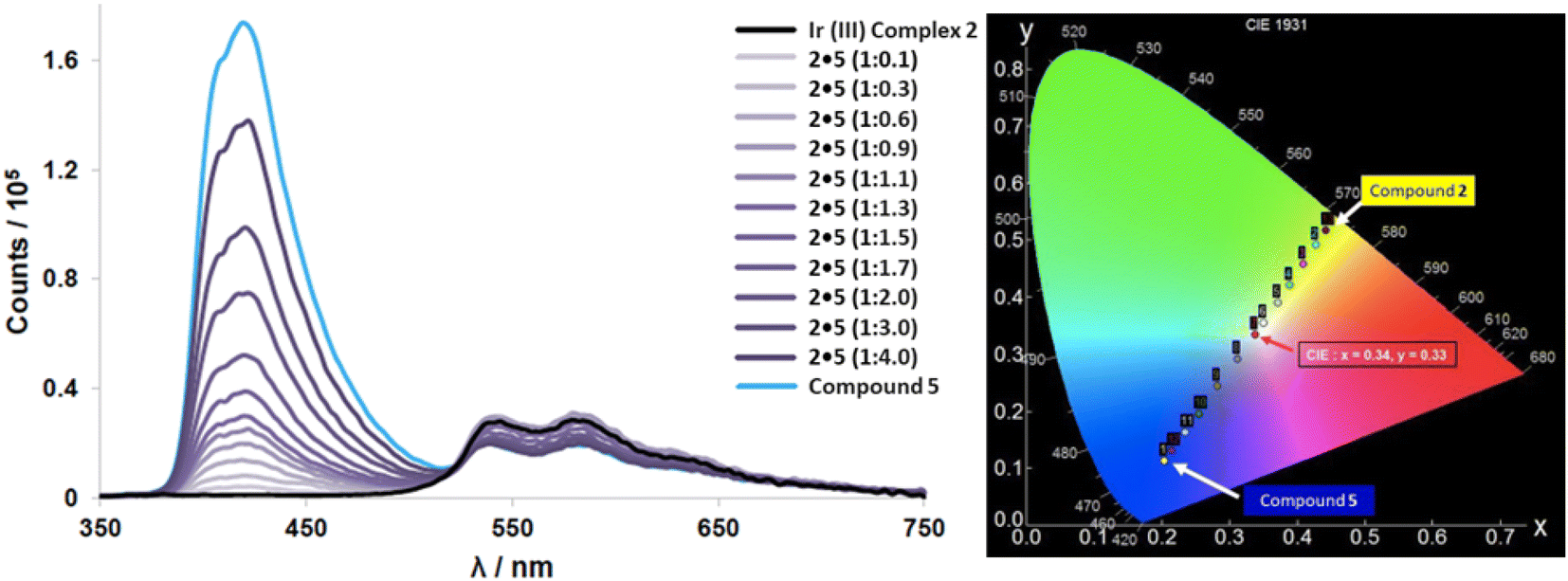 | ||
| Fig. 8 (left) Emission spectra from titration studies of complex 2 with additions of compound 5 measured in CHCl3 solution. Black line represents emission of iridium complex 2 without any additions, blue line represents compound 5 with no complex 2 present. (right) CIE diagram from emission titration experiment of host complex 2 and guest compound 5. | ||
This equation, developed by Price et al.,62 allows for the molar equivalents required in specific colour tuned emissions to be estimated to a high degree of accuracy. The equation takes into account the PL contributions of the multiple components into a linear combination or relative brightness (ai) as the product of the ith components mole fraction (χi), photoluminescent quantum yield (Φi), and excitation intensity (Iex,i) at a common wavelength.
In this instance, the equation can only be used to give a close estimation of the correct constituent ratio of the co-complex for white light emission because the equation does not account for energy transfer within assemblies such as those presented herein. Upon use of the equation, we determined the theoretical molar ratio for white light emission was 1:1.41, which is notably different from our obtained molar ratio of 1:1.3, further indicating energy transfer exists within co-complex 2·5. When attempting this ‘ideal’ white light ratio in PMMA films, the system did not achieve near white light emission, rather a more efficient yellow system, leading us to believe that the hydrogen bond association in the solution phase is necessary to achieve the white light emission.
The computational data for 2·5 again shows increased HOMO stability in the presence of compound 5 due to the frontier orbital HOMO–LUMO interactions as well as the destabilisation of the LUMO+1 residing on the iridium explaining the blue shift of the emission of the iridium complex. This is further supported by the electrochemical data that shows also shows an anode shift of 0.03 V in the Eox. This value is the smallest shift of all Eox values reported herein, showing the weakest H-bonding and frontier HOMO–LUMO interactions between iridium complex and its complement. This weaker frontier orbital interaction aligns with our UV-vis titration studies where it was found that complex 2 had the lowest ΔGa value of −15.9 kJ mol−1 and a Ka with compound 5 of only 6.1 × 102, proving that complex 2 had the weakest affinity for compound 5.
Iridium complexes 3 and 4 were synthesised to study orange/red emissive complexes and their communication with compound 5 via energy transfer. The energy transfer (ET) values were calculated from the ratio between the corrected excitation spectrum of host–guest pairs and their absorption spectra. Emission spectra of systems 3·5 and 4·5 in CHCl3 solution were collected at a higher wavelength λex = 640 nm, which is beyond the emission window of guest compound 5. We observed that the ET efficiency for 3·5 was approximately 36% from the ratio between the corrected excitation spectrum of 3·5 (Fig. 9).
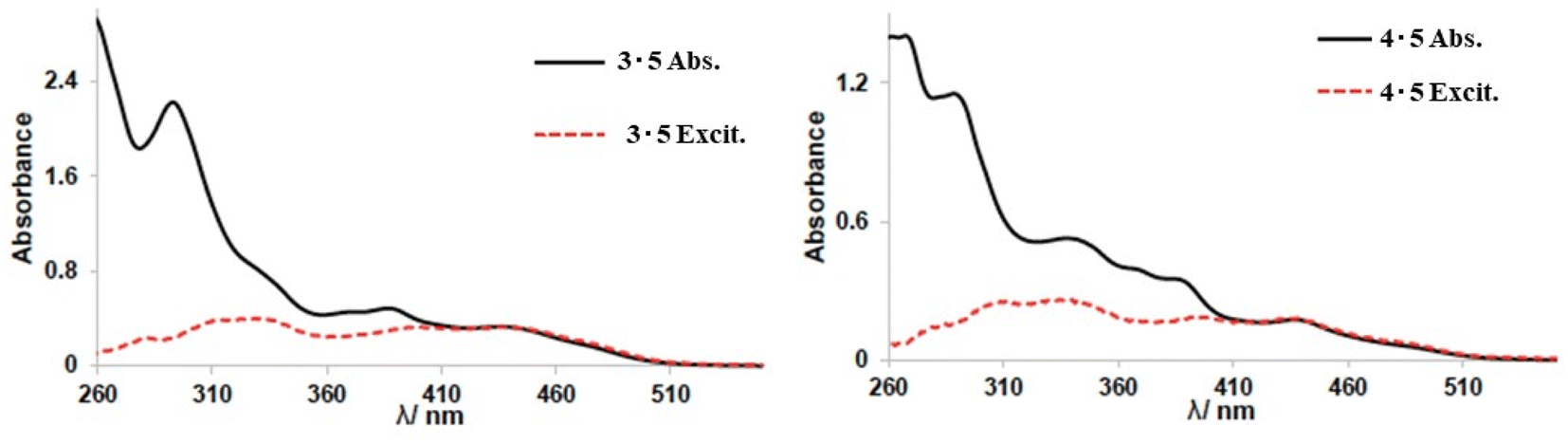 | ||
| Fig. 9 Absorption (solid black line) and corrected PL excitation (red dashed line measured at 570 nm) for co-complexes 3·5 (left) and 4·5 (right) (1:1 ratio of Ir complex and compound 5). Spectra normalised to absorbance at 440 nm. | ||
The ET efficiency for 4·5 was calculated to be 35% which represents an almost identical value to system 3·5. From this study, it can be concluded that even if the emission properties of complexes 3 and 4 were slightly different (shift in emission maxima ∼ 20 nm), their energy transfer efficiency values when combined with compound 5 are comparable. Furthermore, these experiments demonstrated that there is a sufficient distance for “communication” between complexes 3/4 and guest compound 5 in 1:1 solution. To further examine the interaction of complexes 3/4 with 5, emission spectra of 1:1 mixture in CHCl3 solutions were collected for both systems 3·5 and 4·5.
As shown in Table 1 there was an increase in the photoluminescence quantum yields measured for assemblies 3·5 and 4·5 when compared to the values of the complexes 3 and 4. Complex 3 had a more significant change when introduced to compound 5 with the quantum yield increasing from ΦPL = 23.4% to 43.3%. As presented in Table 1, the lifetime decays for both 3·5 and 4·5 increased when compared to their respective parent Ir-complexes. The longer lifetimes (4.3 μs and 3.6 μs), which are ascribed to the direct photoexcitation of the complexes 3 and 4, represent the major components with contributions of 90% and 99%, respectively.
Both complexes 3 and 4 were also investigated electrochemically. Anodic shifts of 0.04 and 0.07 V, respectively, were observed when bound to compound 5. These shifts are consistent with the interactions observed for the other studied complexes, and further supported by Ka values established in the UV-Vis binding studies. Similarly as for complexes 1 and 2, the TD-DFT studies exhibited large changes in the HOMO–LUMO gap with the introduction of compound 5 to both 3 and 4. Computational studies of both 3·5 and 4·5 demonstrate increased HOMO stability through HOMO–LUMO interactions, and the LUMO+1 being subsequently destabilised leading to an increased HOMO–LUMO+1 gap and a minor blue shift in the iridium complex's emission. What is not reflected in the computational or electrochemical data is the enhancement in the quantum yields. Complex 1 saw a minor quenching when introduced to compound 5. Complex 2 demonstrated an increase in the co-system quantum yield when 5 is introduced, which has a significantly higher quantum yield than complex 2 itself.49 Complexes 3 and 4 in combination with 5 exceed all their respective unimolecular quantum yields, the major components of the emission within 3·5 and 4·5 are ascribed to the iridium complexes directly (both computationally and experimentally). These results along with the efficient ET leads us to hypothesise that the presence of compound 5 directly led to an enhancement of the emission properties of complexes 3 and 4 through their H-bonding interactions.
Conclusions
In conclusion, we have demonstrated the design, synthesis and characterisation of four novel heteroleptic Ir(III) complexes all incorporating a benzimidazole-linked guanidine ligand allowing for a high degree of colour tuneability. Each iridium complex (1–4) was further studied to demonstrate a unique property when combined with compound 5. Emission of cyan-blue complex 1 can be modified to access a deep blue emission39 through addition of compound 5. Electrochemical and computational data of 1 also revealed the complex has an appropriate energetic profile to act as a photosensitiser for photocatalytic water splitting; an area we are currently exploring.63 Yellow complex 2 demonstrated white light emission, CIE coordinates: (0.33, 0.34), after controlled addition of compound 5 in solution along with an increased quantum yield. Future studies into more yellow emitting Ir(III) complexes integrating our benzimidazolyl-guanidine ligand along with compound 5 could lead to highly efficient white light emission assuming the yellow iridium components efficiency can be increased. Complexes 3 and 4 showed tuneable orange/red emission, a desirable property for PhOLED applications as well as bio-imaging.43,44 In particular, complex 4 showed red emission with λem = 593 nm and a shoulder peak at 640 nm in CHCl3 solution. Energy transfer efficiency was calculated for complexes 3 and 4 when mixed with compound 5 in a 1:1 ratio in CHCl3 (35% for system 3·5 and 36% for system 4·5). Complexes 3 and 4 would be suitable for further studies on biological applications due to the biologically active guanidine moieties, which represent an available binding site for appropriate guest molecules in supramolecular hydrogen bonded systems.
Data availability
The raw data that underpins this work can be accessed through the University of New Brunswick's data repository: https://doi.org/10.25545/AIBVAX.Author contributions
BB and THJ contributed equally to the completion of this work and are attributed co-first authorship. BB: conceptualisation, experimental methodology, investigation, formal analysis, data curation, writing – original draft. THJ: experimental methodology, computational methodology, investigation, validation, formal analysis, writing. AET investigation, formal analysis, validation. SMH: investigation, validation. BAB: funding procurement, conceptualisation, writing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge the University of New Brunswick, and its Department of Chemistry for financial and technical support. This work was fully supported by Natural Science and Engineering Council of Canada (NSERC; DG RGPIN-2018-04021) and New Brunswick Foundation for Innovation (NBIF; RAI-2019-023).References
- Z. Lu, M. Shangguan, X. Jiang, P. Xu, L. Hou and T. Wang, Dyes Pigm., 2019, 171, 107715 CrossRef CAS.
- K. Y. Zhang, P. Gao, G. Sun, T. Zhang, X. Li, S. Liu, Q. Zhao, K. K.-W. Lo and W. Huang, J. Am. Chem. Soc., 2018, 140, 7827–7834 CrossRef CAS.
- K. Fan, S.-S. Bao, W.-X. Nie, C.-H. Liao and L.-M. Zheng, Inorg. Chem., 2018, 57, 1079–1089 CrossRef CAS.
- J. Shen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2021, 443, 214016 CrossRef CAS.
- S. Abbas, I. Din, A. Raheel and A. Tameez ud Din, Appl. Organomet. Chem., 2020, 34, e5413 CrossRef CAS.
- L. Cho-Cheung Lee and K. Kam-Wing Lo, Chem.–Asian J., 2022, 17, e202200840 CrossRef.
- T. Yang, X. Gao, Y. He, H. Wang and Y. Tao, J. Mater. Chem. C, 2020, 8, 5761–5768 RSC.
- T. Yang, B. Wang, Y. He, A. Zhou, Z. Yao, G. Xing and Y. Tao, Inorg. Chem., 2023, 62, 5920–5930 CrossRef CAS PubMed.
- Q. Wu, Y. Cheng, Z. Xue, X. Gao, M. Wang, W. Yuan, S. Huettner, S. Wan, X. Cao, Y. Tao and W. Huang, Chem. Commun., 2019, 55, 2640–2643 RSC.
- E. Baranoff and P. Kumar, in Iridium (III) in Optoelectronic and Photonics Applications, John Wiley & Sons, Ltd, Chichester, UK, 2017, pp. 655–681 Search PubMed.
- X.-S. Gu, N. Yu, X.-H. Yang, A.-T. Zhu, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2019, 21, 4111–4115 CrossRef CAS PubMed.
- S.-M. Lu, Z. Wang, J. Wang, J. Li and C. Li, Green Chem., 2018, 20, 1835–1840 RSC.
- H. Wu, Y. Wang, Z. Shi, X. Wang, J. Yang, M. Xiao, J. Ge, W. Xing and C. Liu, J. Mater. Chem. A, 2022, 10, 13170–13189 RSC.
- W. J. Kerr, G. J. Knox and L. C. Paterson, J. Labelled Compd. Radiopharm., 2020, 63, 281–295 CrossRef CAS PubMed.
- (a) I. Omae, Coord. Chem. Rev., 2016, 310, 154–169 CrossRef CAS; (b) T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Coord. Chem. Rev., 2018, 374, 55–92 CrossRef CAS.
- J. Kang, K.-M. Park, K. H. Lee, J. Y. Lee and Y. Kang, Dyes Pigm., 2021, 190, 109334 CrossRef CAS.
- J. Jayabharathi, V. Thanikachalam and S. Thilagavathy, Coord. Chem. Rev., 2023, 483, 215100 CrossRef CAS.
- S. Guo, T. Huang, S. Liu, K. Y. Zhang, H. Yang, J. Han, Q. Zhao and W. Huang, Chem. Sci., 2017, 8, 348–360 RSC.
- X. Liu, B. Yao, Z. Zhang, X. Zhao, B. Zhang, W.-Y. Wong, Y. Cheng and Z. Xie, J. Mater. Chem. C, 2016, 4, 5787–5794 RSC.
- H. Benjamin, M. A. Fox, A. S. Batsanov, H. A. Al-Attar, C. Li, Z. Ren, A. P. Monkman and M. R. Bryce, Dalton Trans., 2017, 46, 10996–11007 RSC.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- N.-Y. Chau, P.-Y. Ho, C.-L. Ho, D. Ma and W.-Y. Wong, J. Organomet. Chem., 2017, 829, 92–100 CrossRef CAS.
- A. F. Henwood and E. Zysman-Colman, Chem. Commun., 2017, 53, 807–826 RSC.
- C. Hierlinger, D. B. Cordes, A. M. Z. Slawin, D. Jacquemin, V. Guerchais and E. Zysman-Colman, Dalton Trans., 2018, 47, 10569–10577 RSC.
- K.-R. Kim, J. Oh and J.-I. Hong, Analyst, 2023, 148, 5619–5626 RSC.
- Y. Liu, Y. Li, T. Pu, Y. Pei, Y. Fan, C. Xu and F. Li, New J. Chem., 2023, 47, 16794–16798 RSC.
- J. R. Shakirova, O. A. Tomashenko, E. E. Galenko, A. F. Khlebnikov, P. Hirva, G. L. Starova, S.-H. Su, P.-T. Chou and S. P. Tunik, Inorg. Chem., 2018, 57, 6853–6864 CrossRef CAS PubMed.
- I. R. Laskar and T.-M. Chen, Chem. Mater., 2004, 16, 111–117 CrossRef CAS.
- S. Takizawa, J. Nishida, T. Tsuzuki, S. Tokito and Y. Yamashita, Inorg. Chem., 2007, 46, 4308–4319 CrossRef CAS PubMed.
- Y. Wang, N. Sun, B. F. E. Curchod, L. Male, D. Ma, J. Fan, Y. Liu, W. Zhu and E. Baranoff, J. Mater. Chem. C, 2016, 4, 3738–3746 RSC.
- C.-J. Li, S.-Y. Yin, H.-P. Wang, Z.-W. Wei and M. Pan, J. Photochem. Photobiol., A, 2019, 379, 99–104 CrossRef CAS.
- (a) F.-M. Hwang, H.-Y. Chen, P.-S. Chen, C.-S. Liu, Y. Chi, C.-F. Shu, F.-I. Wu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Inorg. Chem., 2005, 44, 1344–1353 CrossRef CAS; (b) G. Li, D. G. Congrave, D. Zhu, Z. Su and M. R. Bryce, Polyhedron, 2018, 140, 146–157 CrossRef CAS.
- (a) W.-K. Hu, S.-H. Li, X.-F. Ma, S.-X. Zhou, Q. Zhang, J.-Y. Xu, P. Shi, B.-H. Tong, M.-K. Fung and L. Fu, Dyes Pigm., 2018, 150, 284–292 CrossRef CAS; (b) J. Yao, S. Ying, Q. Sun, Y. Dai, X. Qiao, D. Yang, J. Chen and D. Ma, J. Mater. Chem. C, 2019, 7, 11293–11302 RSC.
- Z. Chen, H. Zhang, D. Wen, W. Wu, Q. Zeng, S. Chen and W. Y. Wong, Chem. Sci., 2020, 11, 2342–2349 RSC.
- P.-N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198–10207 CrossRef CAS.
- A. K. Pal, S. Krotkus, M. Fontani, C. F. R. Mackenzie, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Adv. Mater., 2018, 30, 1804231 CrossRef PubMed.
- R. Bai, X. Meng, X. Wang and L. He, Adv. Funct. Mater., 2020, 30, 1907169 CrossRef CAS.
- T. Smith and J. Guild, Trans. Opt. Soc., 1931, 33, 73–134 CrossRef.
- X. Yang, X. Xu and G. Zhou, J. Mater. Chem. C, 2015, 3, 913–944 RSC.
- J.-H. Lee, G. Sarada, C.-K. Moon, W. Cho, K.-H. Kim, Y. G. Park, J. Y. Lee, S.-H. Jin and J.-J. Kim, Adv. Opt. Mater., 2015, 3, 211–220 CrossRef CAS.
- Y. Miao, P. Tao, L. Gao, X. Li, L. Wei, S. Liu, H. Wang, B. Xu and Q. Zhao, J. Mater. Chem. C, 2018, 6, 6656–6665 RSC.
- J. Zhu, B. Z. Tang and K. K. Lo, Chem.–Eur. J., 2019, 25, 10633–10641 CrossRef CAS.
- V. Venkatesh, R. Berrocal-Martin, C. J. Wedge, I. Romero-Canelon, C. Sanchez-Cano, J.-I. Song, J. P. C. Coverdale, P. Zhang, G. J. Clarkson, A. Habtemariam, S. W. Magennis, R. J. Deeth and P. J. Sadler, Chem. Sci., 2017, 8, 8271–8278 RSC.
- S. Wang, Q. Yang, B. Zhang, L. Zhao, D. Xia, J. Ding, Z. Xie and L. Wang, Adv. Opt. Mater., 2017, 5, 1700514 CrossRef.
- H. Wu, L. Ying, W. Yang and Y. Cao, Chem. Soc. Rev., 2009, 38, 3391–3400 RSC.
- P. Tao, W.-L. Li, J. Zhang, S. Guo, Q. Zhao, H. Wang, B. Wei, S.-J. Liu, X.-H. Zhou, Q. Yu, B.-S. Xu and W. Huang, Adv. Funct. Mater., 2016, 26, 881–894 CrossRef CAS.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS.
- B. Balónová, D. Rota Martir, E. R. Clark, H. J. Shepherd, E. Zysman-Colman and B. A. Blight, Inorg. Chem., 2018, 57(14), 8581–8587 CrossRef PubMed.
- L. Yang, J. R. Adsetts, R. Zhang, B. Balónová, M. T. Piqueras, K. Chu, C. Zhang, E. Zysman-Colman, B. A. Blight and Z. Ding, J. Electroanal. Chem., 2022, 906, 115891 CrossRef CAS.
- L. Yang, R. Zhang, B. Balónová, A. E. True, K. Chu, J. R. Adsetts, C. Zhang, X. Qin, E. Zysman-Colman, B. A. Blight and Z. Ding, J. Electroanal. Chem., 2022, 920, 116594 CrossRef CAS.
- B. Balónová and B. A. Blight, Front. Chem., 2021, 9, 712698 CrossRef.
- (a) P.-N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198–10207 CrossRef CAS; (b) Y.-J. Cho, S.-Y. Kim, C. M. Choi, N. J. Kim, C. H. Kim, D. W. Cho, H.-J. Son, C. Pac and S. O. Kang, Inorg. Chem., 2017, 56, 5305–5315 CrossRef CAS PubMed.
- H. Zhen, C. Luo, W. Yang, W. Song, B. Du, J. Jiang, Y. Zhang and Y. Cao, Macromolecules, 2016, 39, 1693–1700 CrossRef.
- L. Zhao, S. Wang, L. Lu, J. Ding and L. Wang, J. Mater. Chem. C, 2017, 5, 9753–9760 RSC.
- C. A. Hollingsworth, P. G. Seybold and C. M. Hadad, Int. J. Quantum Chem., 2002, 90(4–5), 1396–1403 CrossRef CAS.
- H. J. Bae, J. Chung, H. Kim, J. Park, K. M. Lee, T.-W. Koh, Y. S. Lee, S. Yoo, Y. Do and M. H. Lee, Inorg. Chem., 2014, 53, 128–138 CrossRef CAS PubMed.
- M. Hebda, G. Stochel, K. Szaciłowski and W. Macyk, J. Phys. Chem. B, 2006, 110, 15275–15283 CrossRef CAS PubMed.
- T. Polat, F. Bulut, I. Arıcan, F. Kandemirli and G. Yildirim, J. Mol. Struct., 2015, 1101, 189–211 CrossRef CAS.
- C. You, D. Liu, M. Zhu, J. Yu, B. Zhang, Y. Liu, Y. Wang and W. Zhu, J. Mater. Chem. C, 2020, 8, 7079–7088 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40(3), 1305–1323 RSC.
- J. Price, B. Balónová, B. A. Blight and S. Eisler, Chem. Sci., 2021, 12(36), 12092–12097 RSC.
- Y. Zhou, P. He, X.-F. Mo, C. Liu, Z.-L. Gan, H.-X. Tong and X.-Y. Yi, Inorg. Chem., 2021, 60, 6266–6275 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04535a |
| This journal is © The Royal Society of Chemistry 2024 |