DOI:
10.1039/D4RA04553J
(Paper)
RSC Adv., 2024,
14, 26239-26245
Theoretical investigation of the excited-state intramolecular double proton transfer process of 2,2′-(benzo[1,2-d:4,5-d′]bis(thiazole)-2,6-diyl)diphenol†
Received
21st June 2024
, Accepted 30th July 2024
First published on 29th August 2024
Abstract
In this work, the excited state intramolecular double proton transfer (ESIDPT) mechanism of 2,2′-(benzo[1,2-d:4,5-d′]bis(thiazole)-2,6-diyl)diphenol (BTAP) is proposed using density functional theory (DFT) and time-dependent DFT (TDDFT). The changes in bond lengths, bond angles and IR vibrational spectra associated with two intramolecular hydrogen bonds of BTAP upon photoexcitation indicate that the hydrogen bonds are strengthened in the excited state, facilitating the ESIDPT process. Investigation of the constructed S1-state potential energy surface proposes that BTAP prefers a stepwise ESIDPT mechanism. Electronic spectra and frontier molecular orbitals (FMOs) are also presented to illustrate the luminescent properties of BTAP.
1. Introduction
Proton transfer is one of the fundamental processes in the fields of chemistry, biology and physics. Since Weller discovered the dual-emission property in salicylic acid and attributed the large Stokes-shifted fluorescent emission to the excited state intramolecular proton transfer (ESIPT) mechanism, the ESIPT process has been elaborated systematically using experimental and theoretical methods.1–12 It is widely accepted that the ESIPT process occurs via a four-step photophysical scheme: (1) transition to the excited state upon photoexcitation, (2) proton transfer with an intramolecular hydrogen bond to form a phototautomer, (3) radiative relaxation to produce a large Stokes-shifted fluorescence to the ground state, and (4) reverse proton transfer. Owing to the unique feature of ESIPT, this mechanism has been applied to the design of fluorescent probes,13–19 bio-imaging techniques,20–26 white-light OLEDs27–30 and so on.
Biological systems typically involve two or more proton transfers, whereas most excited state intramolecular proton transfer processes involve only a single proton transfer. In order to mimic the proton relay in biochemical processes, there is an urgent need to design and investigate molecular models with excited state double or multiple proton transfer properties. In recent years, excited-state intramolecular double proton transfer (ESIDPT) has gradually received widespread attention. G. Krishnamoorthy et al.31 performed a comprehensive experimental and theoretical analysis on the ESIDPT characteristics of a fluorescent molecule, 2,2′-(1,4-phenylenebis(1H-benzo[d]imidazole-1,2-diyl))diphenol, which contained two 2-(2′-hydroxyphenyl)benzimidazole unit. Zhao et al.32 presented 2,5-bis(4,5-diphenyl-1H-imidazole-2-yl)benzene-1,4-diol derivatives bearing double intramolecular hydrogen bonds and further elucidated the effects of chalcogen substitution on hydrogen bond interactions and the ESIPT mechanism. Yongqing Li and co-workers33 confirmed a stepwise ESIDPT mechanism of 2,2′-bipyridine-3,3′-diol-5,5′-dicarboxylic acid ethyl ester and proposed a method to regulate the stepwise ESIDPT process through solvent polarity and external electric field. Apart from these examples, some different types of ESIDPT compounds have been reported;34–42 however, the number of ESIDPT model molecules is still limited.
In this work, we theoretically investigated the ESIDPT mechanism of a new model molecule 2,2′-(benzo[1,2-d:4,5-d′]bis(thiazole)-2,6-diyl)diphenol (BTAP), which bears the phenol group as the hydrogen bond donor and benzobisthiazole group as the hydrogen bond acceptor. The geometric structures of BTAP and its ESIPT isomers in the ground and excited states are optimized using density-functional theory (DFT) and time-dependent density-functional theory (TDDFT), as shown in Fig. 1. The bond lengths, bond angles and infrared vibration spectrum related to intramolecular hydrogen bonds are analyzed in detail to reveal the effect of photoexcitation on intramolecular hydrogen bonds. The potential energy surfaces in the S0 and S1 states are constructed to elaborate the ESIDPT mechanism of BTAP. We also provide electronic spectra and frontier molecular orbitals (FMOs) to illustrate the luminescent properties of BTAP.
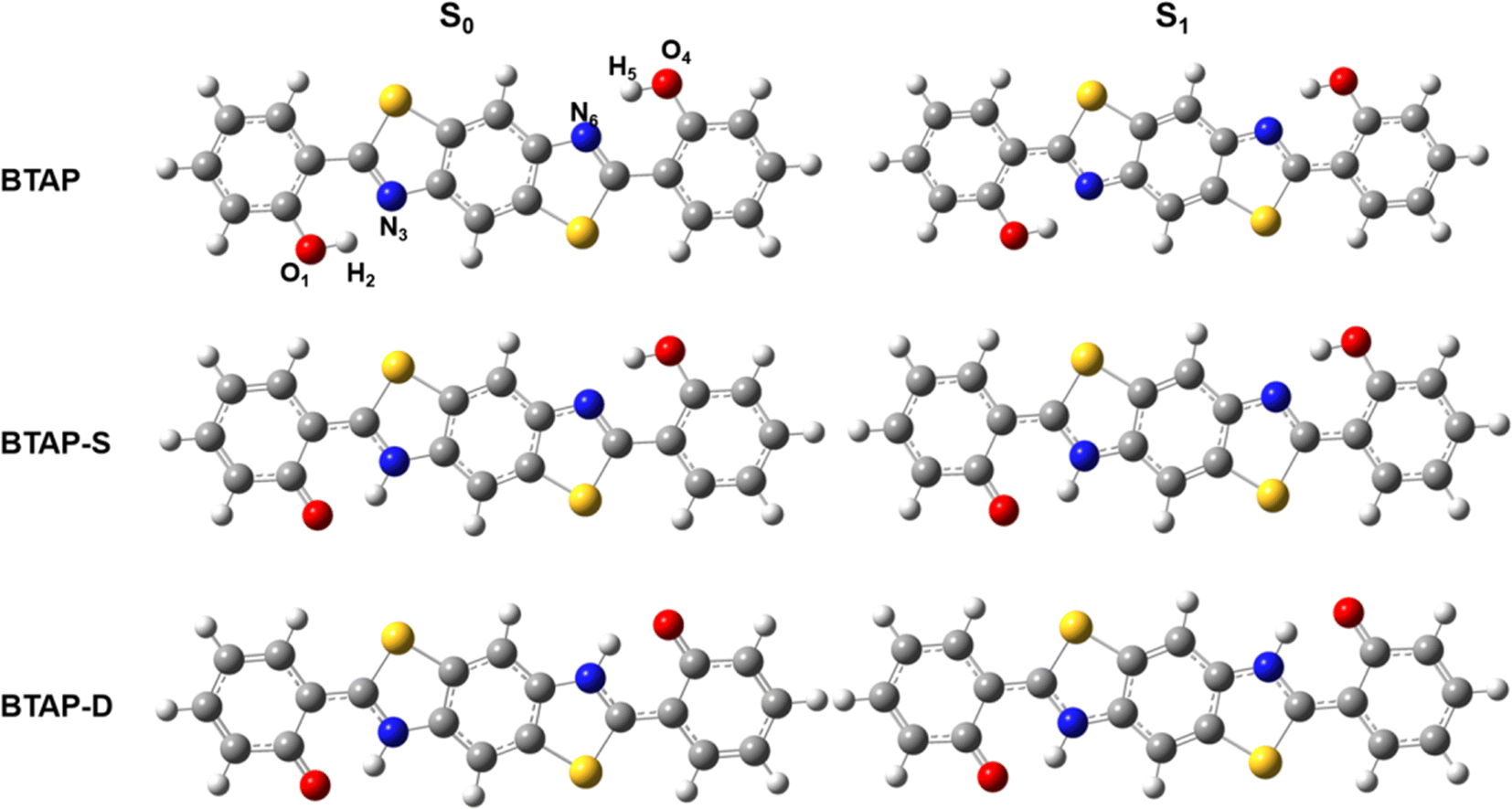
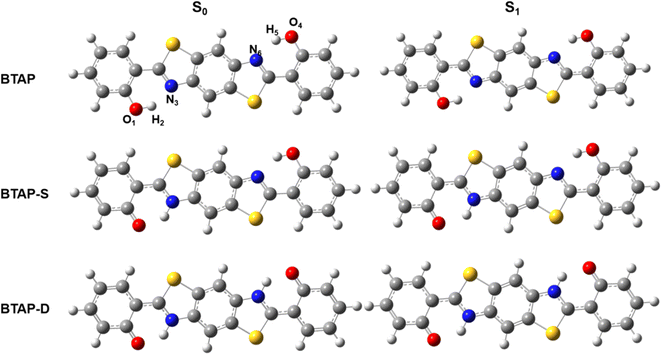 |
| | Fig. 1 Optimized geometry structures of BTAP, its single proton transfer tautomer BTAP-S and double proton transfer tautomer BTAP-D. | |
2. Computational details
All theoretical calculations presented were carried out using Gaussian 09 software.43 Geometry optimizations of the ground state and excited state were performed via density functional theory (DFT) and time-depend density functional theory (TDDFT) methods, respectively, using Becke's three-parameter hybrid exchange functional with the Lee–Yang–Parr gradient-corrected correlation functional (B3LYP)44,45 and 6-31++G(d, p) basis set. The incorporation of Grimme's D3 version of dispersion in all calculations ensures the comprehensive consideration of the dispersion forces. To be consistent with the experimental result, dichloromethane was used as the solvent with the polarizable continuum model (PCM) with the integral equation formalism variant (IEFPCM).46 The geometry optimizations were performed without constraints on bond lengths, angles, or dihedral angles. All the local minima were determined without the imaginary modes by vibrational analysis calculations. Based on the optimized structures, the absorption and fluorescent spectra were calculated at the B3LYP/6-31++G(d, p) level. In addition, to study the proton transfer behavior of the molecule, the potential energy surface surfaces (PESs) were scanned at S0 and S1 states by gradually extending the bond lengths of O1–H2 and O4–H5, respectively, without structural constraints.
3. Results and discussion
3.1. Geometry structure and hydrogen bond
Geometry optimization of the ESIPT isomers of BTAP molecule in S0 and S1 states was conducted using the B3LYP functional and 6-31++G(d, p) basis set. The optimized ESIPT isomer structures of BTAP, BTAP-S (single proton transfer form) and BTAP-D (double proton transfer form) in the S0 and S1 states are shown in Fig. 1. Since photoexcited ESIPT behaviors are closely related to intramolecular double hydrogen bonding interactions, the effects of photoexcitation on hydrogen bonding effects were elaborated first. All atoms involved in intramolecular hydrogen bonding were numbered as O1–H2⋯N3 and O4–H5⋯N6 for description. Considering the symmetry of the molecular structure, the structural changes on both sides of the molecule were consistent.
The change of hydrogen bond strength before and after photo-excitation was illustrated by comparing the bond lengths, bond angles and vibrational frequency of intramolecular hydrogen bonds in ground and excited states. Some important structural parameters associated with hydrogen bonds are shown in Table 1. For the BTAP form, the bond lengths of O1–H2 and O4–H5 were elongated from 0.995 Å (S0) to 1.000 Å (S1). The hydrogen bond lengths of H2⋯N3 and H5⋯N6 are shortened from 1.758 Å (S0) to 1.739 Å (S1), which indicated that the intramolecular hydrogen bond in the S1 state was strengthened. The increase in the bond angles of O1–H2⋯N3 and O4–H5⋯N6 from 146.0° to 147.0° confirmed the enhancement of hydrogen bonds in the S1 state. For the BTAP-S form, compared with the S0 state, the bond length of hydroxyl (O4–H5) is lengthened, the distance of hydrogen bond (H5⋯N6) is shortened, and the bond angle (O4–H5⋯N6) of the hydrogen bond is enlarged in the S1 state. The changes in bond length and angle of the BTAP and BTAP-S indicate that the intramolecular hydrogen bonds of enol segment tautomers are enhanced in the S1 state, meaning that the ESIPT reaction is more likely to occur in the S1 state. In contrast, the structural changes of keto's segments in BTAP-S (O1–H2⋯N3) and BTAP-D (O1–H2⋯N3 and O4–H5⋯N6) show that the intramolecular hydrogen bonds are weakened in the S1 state, indicating that the reverse ESIPT prefer to occur in the S0 state.
Table 1 The primary bond lengths (Å) and bond angles δ (°) of BTAP, BTAP-S and BTAP-D structures in the S0 and S1 states
| |
BTAP |
BTAP-S |
BTAP-D |
| S0 |
S1 |
S0 |
S1 |
S0 |
S1 |
| O1–H2 |
0.995 |
1.000 |
0.994 |
0.995 |
1.676 |
1.763 |
| H2–N3 |
1.758 |
1.739 |
1.763 |
1.754 |
1.049 |
1.039 |
| O4–H5 |
0.995 |
1.000 |
1.680 |
1.836 |
1.676 |
1.763 |
| H5–N6 |
1.758 |
1.739 |
1.049 |
1.032 |
1.049 |
1.039 |
| δ(O1–H2–N3) |
146.0° |
147.0° |
145.9° |
146.8° |
137.8° |
135.0° |
| δ(O4–H5–N6) |
146.0° |
147.0° |
137.3° |
132.6° |
137.8° |
135.0° |
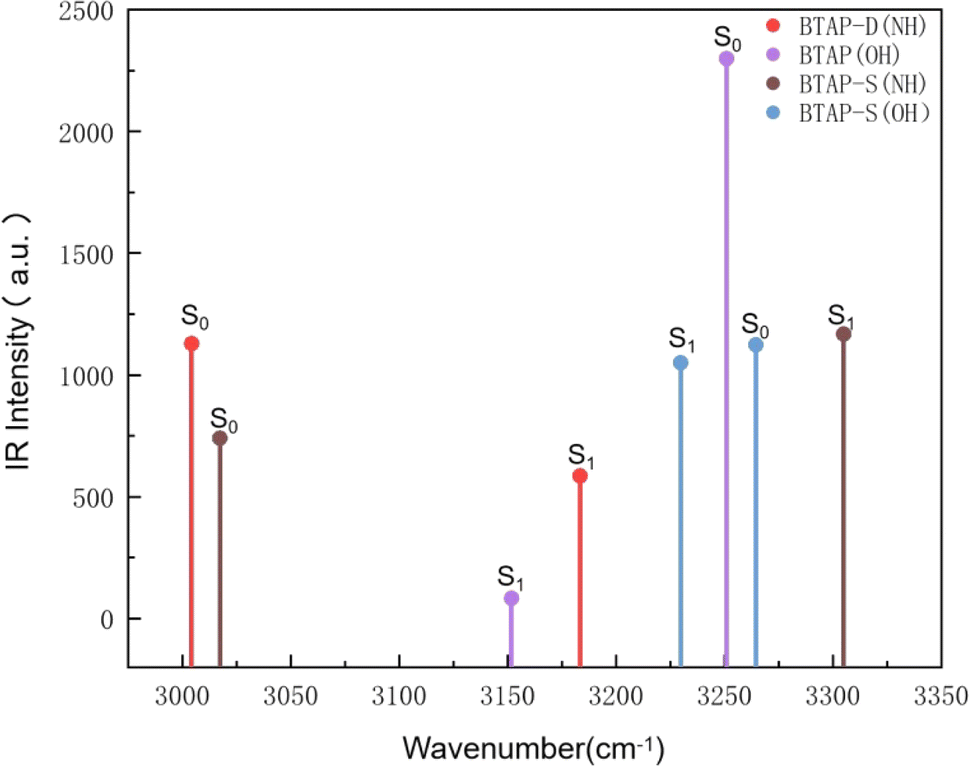
The infrared vibration spectra analysis at the hydrogen bond sites is also used to elaborate on the changes in the hydrogen bond strength between the ground state and the excited state. Herein, the theoretical IR results involved in stretching vibration of hydrogen bond sites in BTAP, BTAP-S and BTAP-D are presented in Fig. 2. As shown in Fig. 2, the vibration frequencies of O1–H2 (O4–H5) for BTAP are located at 3251 cm−1 in the S0 state and turn to 3152 cm−1 after photoexcitation to the S1 state. The vibration frequencies are red-shifted by 99 cm−1 when BTAP is photoexcited from the S0 state to the S1 state. It means that hydrogen bonds are enhanced, facilitating the process of intramolecular proton transfer in the excited state. For the BTAP-S form, the vibration frequency of O4–H5 is red-shifted by 34 cm−1 from 3264 cm−1 in the S0 state to 3230 cm−1 in the S1 state, which indicates that the hydrogen bond O4–H5⋯N6 is enhanced in the S1 state. However, the blue-shift of 288 cm−1 occurs for H2–N3 from 3017 cm−1 in the S0 state to 3305 cm−1 in the S1 state, and the hydrogen bond strength O1–H2⋯N3 is superior in the S0 state than in the S1 state. Similarly, the stretch vibrational frequency of N3–H2 (N6–H5) in BTAP-D is also blue-shifted by 179 cm−1 from 3004 cm−1 in the S0 state to 3183 cm−1 in the S1 state, which illustrates that the intensity of the hydrogen bond O1–H2⋯N3 (O4–H5⋯N6) is stronger in the S0 state than in the S1 state. In summary, the infrared vibration analysis indicates that the proton transfer process is more likely to occur in the excited state while the inverse proton transfer process is more likely to occur in the ground state, which is consistent with the analysis of changes in geometry structure parameters.
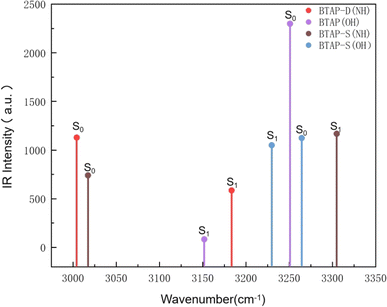 |
| | Fig. 2 Calculated infrared vibration spectra of BTAP, BTAP-S and BTAP-D in the spectral region of OH and NH stretching bands in S0 and S1 states. | |
3.2. Potential energy surfaces and proton transfer process
To illustrate the ESIPT mechanism further and to explore whether the double proton transfer occurs stepwise or synchronously, and the calculations of potential energy surfaces (PESs) were performed using the constrained optimizations in the S0 state and S1 state geometrical structures of BTAP along with the fixed O1–H2 bond length and O4–H5 bond length. As shown in Fig. 3, the PESs of the S0 state is scanned with varying the O1–H2 and O4–H5 bond lengths from 0.995 to 2.15 Å in steps of 0.05 Å. There exist four local minimum points whose coordinates are located at A (1.00 Å, 1.00 Å), B (1.00 Å, 1.70 Å), C (1.70 Å, 1.00 Å) and D (1.70 Å, 1.70 Å). Owing to the symmetry of the molecular structure, point B and point C exhibit significant symmetrical characteristics and the energy values of the two points are equal. It is not difficult to find that points A, B (C) and D can approximatively represent BTAP, BTAP-S and BTAP-D, respectively. The calculated results show that the energies of the four local minimum points from large to small are ED > EC = EB > EA in the S0 state. Thus, the BTAP prefers to exist in the form of an enol tautomer in the S0 state. Similarly, there also exist four stable points on the potential energy surface of the S1 state, whose coordinates located A* (1.00 Å, 1.00 Å), B* (1.00 Å, 1.80 Å), C* (1.80 Å, 1.00 Å) and D* (1.75 Å, 1.75 Å). The potential energy barriers among these stable configurations of the S0 and S1 states are also listed in Table 2. As shown in Table 2, the potential energy barriers in the S1 state are relatively lower than those in the S0 state, indicating that the proton transfer process is easier to occur in the S1 state than in the S0 state. The synchronous double proton transfer (BTAP → BTAP-D) and the sequential single proton transfer (BTAP → BTAP-S → BTAP-D) are the two primary pathways for the excited state intramolecular double proton transfer of BTAP. The potential energy barrier of the pathway (BTAP → BTAP-D) is 17.8 kcal mol−1 and is too high to overcome the energy barrier, hence the double proton transfer is difficult to happen spontaneously. However, the acceptable barrier of 7.9 kcal mol−1 (BTAP → BTAP-S) implies the possibility of proton transfer, leading to the generation of BTAP-S. Subsequently, another proton would transfer to form BTAP-D across a mild barrier of 11.4 kcal mol−1 (BTAP-S → BTAP-D). Therefore, it is very likely that the double proton transfer undergoes a stepwise mechanism. Because the energies of the four local minimum points from large to small follow ED* > EA* > EC* = EB* in the S1 state, the energy barrier of the reverse proton transfer process for BTAP-D → BTAP-S (2 kcal mol−1) is lower than the one of the forward proton transfer process and BTAP-S → BTAP (8.1 kcal mol−1) is very close to the one of the forward proton transfer process, suggesting that the proton transfer process is reversible and the tautomers BTAP, BTAP-S, and BTAP-D coexist in the S1 state.
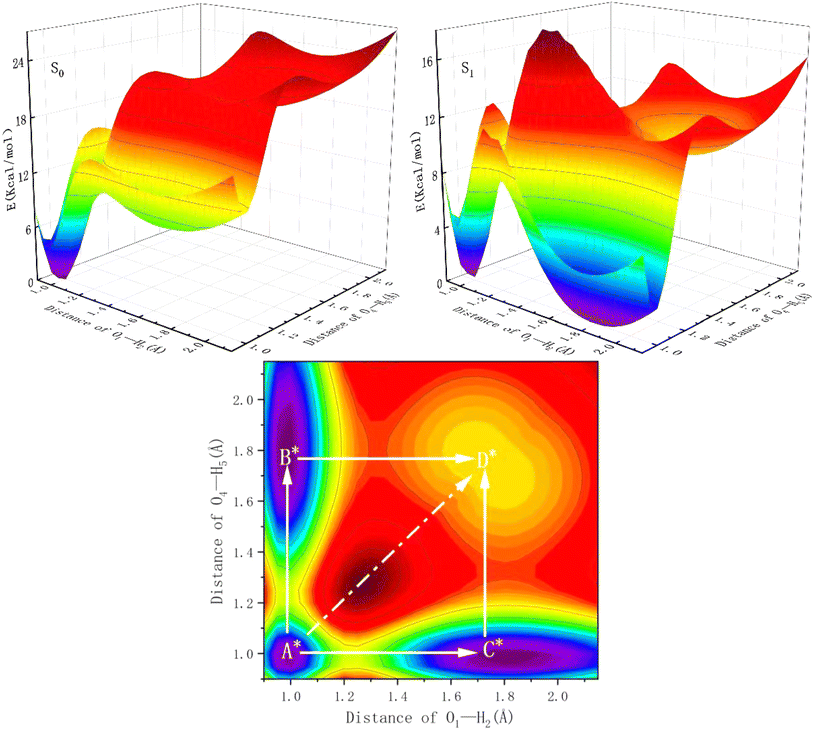 |
| | Fig. 3 The potential energy surfaces of the BTAP system in the S0 and S1 states and the projective plane in the S1 state as functions of O1–H2 and O4–H5 bond lengths. | |
Table 2 The potential energy barriers (kcal mol−1) in S0 and S1 states (E': reverse processes)
| |
EA→B |
EB→D |
EA→D |
EA*→B* |
EB*→D* |
EA*→D* |
| E |
11.2 |
11.5 |
22.6 |
7.9 |
11.4 |
17.8 |
| E′ |
1.9 |
1.8 |
3.6 |
8.1 |
2 |
8.6 |
3.3. Electronic spectra and frontier molecular orbitals
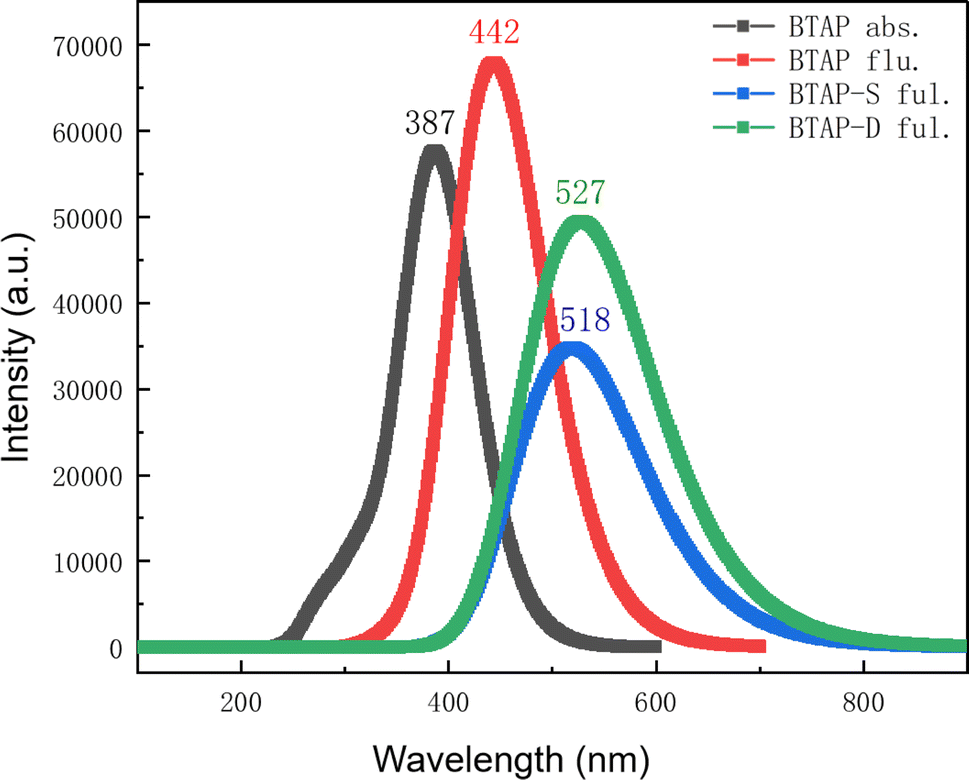
As the result of PESs shows, there exist three tautomers BTAP, BTAP-S, and BTAP-D in the S1 state, which would emit fluorescence upon photo-excitation. Therefore, the absorption and fluorescence spectra of the three molecules are simulated at the B3LYP/6-31++G(d, p) level. The absorption spectrum is shown in Fig. 4. The computational absorption peak value of BTAP in dichloromethane is 388 nm and coincides well with the experimental value of 386 nm (Fig. S1†). In addition, the fluorescence spectra in dichloromethane are shown in Fig. 4. The calculated fluorescence peak values of BTAP, BTAP-S and BTAP-D are 442 nm, 518 nm and 527 nm, respectively. The normal emission of BTAP with a Stokes shift of 57 nm is in good agreement with the experimental value of 403 nm. The two fluorescent emission peaks of BTAP-S and BTAP-D are very close and overlap to form one peak so that only one large stokes-shifted fluorescent emission peak of keto-forms at 523 nm appears in the experimental spectrum. The perfect agreement of the spectral data demonstrates the rationality of the simulation using the DFT/TD-DFT methods with B3LYP functional and 6-31++G(d, p) basis set.
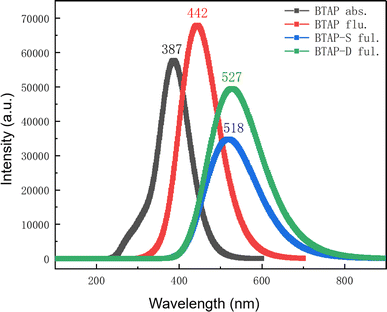 |
| | Fig. 4 Theoretical electronic spectra of BTAP, BTAP-S and BTAP-D in DCM. | |
The charge distribution and charge transfer induced by photo-excitation can depict qualitatively the properties of the excited state and frontier molecular orbitals (FMOs) mainly contributing to the S0–S1 transition, hence, the frontier MOs of BTAP are analyzed in dichloromethane solvent to explore the nature of the excited state. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) orbital are calculated and shown in Fig. 5. Both HOMO and LUMO orbitals exhibit a π character, showing that the S0 → S1 transition is an allowed ππ*-type transition. Mulliken's charge analysis is adopted for a detailed investigation of the charge distribution over the atoms involved in intramolecular hydrogen bonds and is used to explore the influence of charge transfer on the excited state proton transfer process. As shown in Table 3, the electron density on the O atom and N atom decreases from the S0 state to the S1 state, and it decreases more on the O atom for the BTAP tautomer. The negative charge of the O1 atom (O4 atom) decreases from −0.679 in the S0 state to −0.670 in the S1 state, meanwhile, a slight decrease from −0.253 to −0.251 in the negative charge of the N4 atom (N5 atom) occurs. This evidence indicates that the intensity of the hydroxyl group weakens and the intensity of the hydrogen bond strengthens in the S1 state for BTAP, which facilitates the ESIPT reaction.
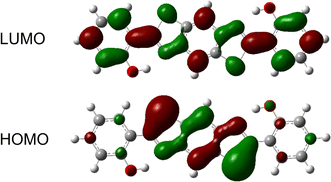 |
| | Fig. 5 Frontier molecular orbitals (HOMO and LUMO) of the BTAP form. | |
Table 3 The Mulliken's charge of O1, H2, N3, O4, H5 and N6 atoms for BTAP in both S0 and S1 states
| Atoms |
S0 |
S1 |
| O1 |
−0.679 |
−0.670 |
| H2 |
0.560 |
0.560 |
| N3 |
−0.253 |
−0.251 |
| O4 |
−0.679 |
−0.670 |
| H5 |
0.560 |
0.560 |
| N6 |
−0.253 |
−0.251 |
4. Conclusions
In this work, the ESIPT mechanism of 2,2′-(benzo[1,2-d:4,5-d′]bis(thiazole)-2,6-diyl)diphenol was systematically investigated based on the DFT/TD-DFT methods. By comparing the changes in bond length, bond angle, and infrared vibration spectrum between the ground state and excited state, we observed a significant enhancement of the intramolecular hydrogen bond in the excited state, which promotes the occurrence of proton transfer reactions. The scanned PESs show that the potential energy barrier of the synchronous double proton transfer is so high that the two protons cannot transfer simultaneously in the excited state, therefore, the BTAP follows a stepwise ESIPT process which has a lower barrier that is easily overcome. The spectral data obtained from theoretical calculations is consistent with the experimental ones, validating the rationality of the theoretical simulation methods used in this work. The FMOs and Mulliken's charge analysis exhibit that the charge transfer upon photoexcitation enhances the proton transfer process.
Data availability
The data supporting this article have been included within the manuscript and its ESI.†
Author contributions
Yongchao Hao: Writing – original draft; theoretical calculation; methodology; funding acquisition; resources; supervision. Xiaoran Li: data curation; formal analysis. Hongfang Li: data curation. Shanyan Chang: data curation. Jiangyu Zhang: formal analysis; Lili Dong: funding acquisition; resources; supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the financial support of the S & T Program of Xingtai (2023ZZ089, 2022ZZ118), Hebei Province Foundation of Returned Scholars (C20200105), Science Research Project of Hebei Education Department (QN2020512). Further, the author thanks other co-authors for their contributions, and we are also grateful to the other members of the research team for their help.
References
- H. Gu, W. J. Wang, W. Y. Wu, M. L. Wang, Y. R. Liu, Y. J. Jiao, F. Wang, F. Wang and X. Q. Chen, Chem. Commun., 2023, 59, 2056–2071 RSC.
- J. F. Zhao, P. Song, L. Feng, X. Wang and Z. Tang, J. Mol. Liq., 2023, 380, 121763 CrossRef CAS.
- O. Anitha, M. Mathivanan, B. Tharmalingam, T. Thiruppathiraja, S. Ghorai, R. Natarajan, V. Thiagarajan, S. Lakshmipathi and B. Murugesapandian, Dyes Pigments, 2023, 212, 111091 CrossRef CAS.
- K. Liu, J. Zhang, Q. Y. Shi, L. P. Ding, T. H. Liu and Y. Fang, J. Am. Chem. Soc., 2023, 145, 7408–7415 CrossRef CAS PubMed.
- T. Frizon, C. Salla, F. Grillo, F. S. Rodembusch, V. S. Câmara, H. C. Silva, E. Zapp, E. Junca, F. Z. Galetto, A. M. de Costa, G. J. Pedroso, A. A. Chepluki, S. Saba and J. Rafique, Spectrochim. Acta, Part A, 2023, 288, 122050 CrossRef CAS PubMed.
- J. F. Zhao, B. Jin and Z. Tang, Phys. Chem. Chem. Phys., 2022, 24, 27660–27669 RSC.
- S. Kaya, H. G. Aydin, S. Keskin, Z. Ekmekci and N. Menges, J. Photochem. Photobiol., A, 2021, 420, 113487 CrossRef CAS.
- S. Ding, A. X. Xu, A. K. Sun, Y. Xia and Y. J. Liu, Spectrochim. Acta, Part A, 2021, 245, 118937 CrossRef CAS PubMed.
- Y. T. Qi, Z. Tang, H. B. Zhan, Y. Wang, Y. L. Zhao, X. Fei, J. Tian, L. Yu and J. Y. Liu, Spectrochim. Acta, Part A, 2020, 224, 117359 CrossRef CAS PubMed.
- B. Kuzu, M. Tan, Z. Ekmekci and N. Menges, J. Photochem. Photobiol., A, 2019, 381, 111874 CrossRef CAS.
- S. Y. Yin, S. S. Sun, M. Pan, L. Chen, Z. Wang, Y. J. Hou, Y. N. Fan, H. P. Wang and C. Y. Su, J. Photochem. Photobiol., A, 2018, 355, 377–381 CrossRef CAS.
- N. Suzuki, A. Fukazawa, K. Nagura, S. Saito, H. Kitoh-Nishioka, D. Yokogawa, S. Irle and S. Yamaguchi, Angew. Chem. Int. Ed., 2014, 53, 8231–8235 CrossRef CAS PubMed.
- A. I. Said, N. I. Georgiev and V. B. Bojinov, J. Photochem. Photobiol., A, 2024, 446, 115176 CrossRef CAS.
- Y. R. Huang, Y. Li, Y. Li, K. L. Zhong and L. J. Tang, New J. Chem., 2023, 47, 6916–6923 RSC.
- H. X. Ren, F. J. Huo, W. Wen and C. X. Yin, Dyes Pigments, 2022, 199, 110111 CrossRef CAS.
- A. Y. Bi, M. Liu, S. Huang, F. Zheng, J. P. Ding, J. Y. Wu, G. Tang and W. B. Zeng, Chem. Commun., 2021, 57, 3496–3499 RSC.
- N. Nehra, R. Kaushik, D. G. Vikas and R. K. Tittal, J. Mol. Struct., 2020, 1207, 127839 CrossRef CAS.
- X. X. Tang, X. G. Zhu, H. L. Xu, H. Sun, X. Han, Q. Li, B. B. Zhou and Z. H. Ni, Spectrochim. Acta, Part A, 2022, 281, 121567 CrossRef CAS PubMed.
- H. N. Peng, X. M. Peng, J. Q. Huang, A. Huang, S. J. Xu, J. J. Zhou, S. S. Huang and X. P. Cai, J. Mol. Struct., 2020, 1212, 128138 CrossRef CAS.
- T. X. Xiao, C. Bao, L. L. Zhang, K. Diao, D. X. Ren, C. X. Wei, Z. Y. Li and X. Q. Sun, J. Mater. Chem. A, 2022, 10, 8528–8534 RSC.
- S. Bhowal and A. Ghosh, RSC Adv., 2021, 11, 27787–27800 RSC.
- H. Y. Xu, W. Chen, L. X. Ju and H. F. Lu, Spectrochim. Acta, Part A, 2021, 247, 119074 CrossRef CAS PubMed.
- D. Aydin, I. B. Gunay, S. Elmas, T. Savran, F. N. Arslan, G. Sadi and I. Yilmaz, New J. Chem., 2020, 44, 12079–12089 RSC.
- P. Zhang, Y. Z. Xiao, Q. Zhang, Z. X. Zhang, H. W. Yu and C. F. Ding, New J. Chem., 2019, 43, 7620–7627 RSC.
- C. Balakrishnan, M. A. Neelakantan and S. Banerjee, Sens. Actuators, B, 2017, 253, 1012–1025 CrossRef CAS.
- R. Alam, T. Mistri, R. Bhowmick, A. Katarkar, K. Chaudhuri and M. Ali, RSC Adv., 2016, 6, 1268–1278 RSC.
- L. Duarte, J. C. Germino, J. F. Berbigier, C. A. Barboza, M. M. Faleiros, D. D. Simoni, M. T. Galante, M. S. de Holanda, F. S. Rodembusch and T. Atvars, Phys. Chem. Chem. Phys., 2019, 21, 1172–1182 RSC.
- L. Duarte, J. C. Germino, R. A. Mendes, J. F. Berbigier, M. M. Faleiros, F. S. Rodembusch and T. Atvars, Dyes Pigments, 2019, 171, 107671 CrossRef CAS.
- M. Mohan, M. N. Satyanarayan and D. R. Trivedi, New J. Chem., 2019, 43, 10413–10428 RSC.
- W. J. Yang and X. B. Chen, Phys. Chem. Chem. Phys., 2014, 16, 4242–4250 RSC.
- A. Das Kanungo, Ila, S. Panda, U. Phukon, M. Sathiyendran and G. Krishnamoorthy, J. Photochem. Photobiol., A, 2024, 450, 115412 CrossRef CAS.
- C. Liu, J. Zhao, J. Chen, M. Wang, M. Hou and L. Yang, Phys. Chem. Chem. Phys., 2024, 26, 6335–6344 RSC.
- H. Zhuang, W. Shi, G. Zhao and Y. Li, Phys. Chem. Chem. Phys., 2024, 26, 12016–12026 RSC.
- X. Tang, Y. Zhang and C. Sun, Phys. Chem. Chem. Phys., 2024, 26, 10439–10448 RSC.
- J. Zhao and C. Liu, Molecules, 2023, 28, 5951 CrossRef CAS PubMed.
- J. Zhao, H. Zhang, L. Fan, F. Li and P. Song, Spectrochim. Acta, Part A, 2023, 299, 122831 CrossRef CAS PubMed.
- X. Xin, W. Shi, Y. Zhao, G. Zhao and Y. Li, Chem. Phys., 2023, 570, 111882 CrossRef CAS.
- D. Ushakou, J. Lumin., 2023, 263, 120032 CrossRef CAS.
- H. Mu, D. Li, J. Gao, Y. Wang, Y. Zhang, G. Jin and H. Li, J. Mol. Struct., 2023, 1294, 136385 CrossRef CAS.
- G. Kumar, K. Paul and V. Luxami, New J. Chem., 2020, 44, 12866–12874 RSC.
- J. Zhao, H. Dong and Y. Zheng, J. Phys. Chem. A, 2018, 122, 1200–1208 CrossRef CAS PubMed.
- Y. Zhao, Y. Yang, Y. Ma and Y. Li, J. Lumin., 2018, 201, 189–195 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- J. T. R. Cammi, J. Comput. Chem., 1995, 16, 1449–1458 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Xiaoran Lia,
Hongfang Lia,
Shanyan Changa,
Jiangyu Zhanga and
Lili Dong*a
*ab,
Xiaoran Lia,
Hongfang Lia,
Shanyan Changa,
Jiangyu Zhanga and
Lili Dong*a