
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile, catalyst- and additive-free, and scalable approach to the photochemical preparation of disulfides from organosulfenyl chlorides†
Wei Liuab,
Jiayi Wang *a and
Gonghua Song*a
*a and
Gonghua Song*a
aShanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology, Shanghai, 200237, P. R. China. E-mail: jiayi.wang@ecust.edu.cn; ghsong@ecust.edu.cn
bState Key Laboratory of Fluorine-containing Functional Membrane Materials, Shandong 256400, P. R. China
First published on 11th October 2024
Abstract
A novel, clean and efficient protocol for the preparation of disulfides has been developed through the photochemical radical homo- and cross-coupling reaction of sulfenyl chlorides under LED irradiation and without the use of any catalyst and additive. The representative photochemical homo-coupling of trichloromethyl sulfenyl chloride has been successfully conducted on kilogram-scale in a continuous flow mode. The solvent and the main byproduct can be recovered in high yields, which makes the approach be highly atom economical.
Introduction
Disulfides are widely used in biological studies and material science. For example, the disulfide unit often plays a key role for the formation and stabilization of the 3D structure of polypeptides and proteins in diverse biochemical processes.1 A disulfide derivate can also be promisingly served as the linker to form an antibody-drug conjugate by attaching a chemical probe, an imaging agent or a drag to a biomolecule.2 In fact, many biologically active natural products, pharmaceuticals and agrochemicals contain disulfide moieties.3,4 In addition, disulfides are often known as popular rubber additives (Fig. 1).5 Due to its wide application, a plethora of synthetic methods to access disulfides have been reported.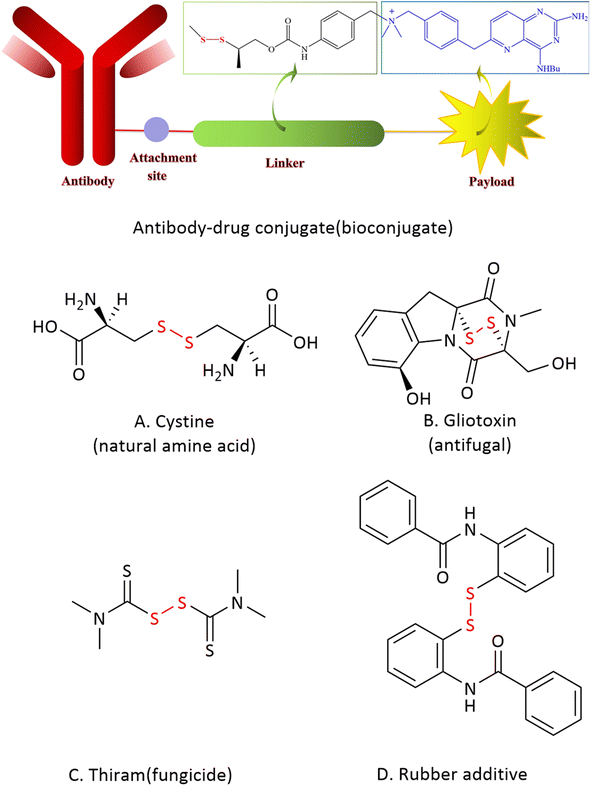 | ||
| Fig. 1 Representative disulfides and their applications. | ||
The most common method for the preparation of symmetrical disulfides is through the oxidative coupling of thiols. Various oxidizing agents such as air, iodine, hydrogen peroxide and chromates have been successfully applied to this transformation with the help of a metal or metal-free catalyst (Scheme 1a).6 The nucleophilic substitution of S–H bonds (Scheme 1b),7 the reductive coupling of sulfonyl chlorides, thiosulfonates and thiosulfates, and the reductive dimerization of thiocyanates and ethoxythio have also been utilized for the preparation of symmetrical disulfides (Scheme 1c).8 More recently, Wu & Tang et al. reported a radical pathway towards the synthesis of symmetrical disulfides from thiols catalyzed by an environmentally friendly catalyst zeolite ETS-10 in the absence of oxidants and additives (Scheme 1d).9 Although in some special cases, an unsymmetrical disulfide can become the main product of coupling reactions between two different thiols, the chemoselective approaches for the synthesis of unsymmetrical disulfides have recently received extensive attention since more diverse disulfide products could be expected. The metal-catalyzed cross-coupling or nucleophilic substitution approaches with prefunctionalized/activated disulfurating reagents (RSS-LG) have proven effective for constructing unsymmetrical disulfides (Scheme 1e).10 The unsymmetrical disulfides have also been obtained via a catalytic disulfide exchange reaction (Scheme 1f).11 Lei et al. reported a facile oxidant- and catalyst-free electrochemical method for the cross-coupling of an aryl mercaptan with an alkyl mercaptan by utilizing n-Bu4NBF4 as the electrolyte (Scheme 1g).12 On the other hand, as an environmentally friendly alternative to the conventional synthetic chemistry, photochemistry has recently attracted much attention.13 Photochemical protocols have also been successfully employed for the preparation of disulfides. Different metal and metal-free photocatalysts have been used for the synthesis of both symmetrical and unsymmetrical disulfides from different thiols (Scheme 1h).14 In addition, Wu & Parrt reported a simple and effective approach for the preparation of unsymmetrical disulfides through the radical substitution on tetrasulfides with the help of a photocatalyst and blue LED irradiation (Scheme 1i).15 A broad range of unsymmetrical disulfides were obtained in acceptable yields. Although great success has been achieved in the synthesis of disulfides, they are usually associated with some drawbacks such as the use of metal catalysts, photocatalysts and/or environmentally harmful additives.16 Herein, we report the synthesis of disulfides through a photochemical homo/cross-radical coupling reaction of organosulfenyl chlorides without the use of any catalyst, oxidant/reductant and other additives (Scheme 1j). The reaction can be performed both in a batch and a continuous-flow mode. Both the solvent and main byproduct can be effectively recovered by distillation. Although the synthesis of organosulfenyl chlorides is not usually so “green”, this new protocol provides an alternative choice for researchers in related fields.
 | ||
| Scheme 1 Approaches for the synthesis of disulfides. | ||
Results and discussion
Initially, the photochemical reaction of trichloromethyl sulfenyl chloride 1a in water was conducted under LED irradiation at 365 nm. Gratifyingly, the reaction proceeded smoothly and the desired product 1,2-bis(trichloromethyl)disulfide 2a was obtained in a moderate yield of 46% (Table 1, entry 1). Encouraged by this result, various solvents were then screened. Methanol, ethanol and isopropanol improved the disulfide yields to 82%, 80% and 74%, respectively (Table 1, entries 2–4). Whereas acetic acid (CH3COOH) was inferior and gave a relatively lower yield of 50% (Table 1, entry 5). No product was detected when the reaction was conducted in chloroform (CHCl3) (Table 1, entry 6). Acetonitrile (CH3CN), benzene and toluene were also applicable for this reaction, affording the desired product in a yield of 76%, 79% and 75%, respectively (Table 1, entries 7–9). The employment of n-pentane gave 2a in an excellent yield of 90% (Table 1, entry 10), while cyclohexane was found to be the best one among the solvents screened with the highest yield of 92% at 365 nm (Table 1, entry 11).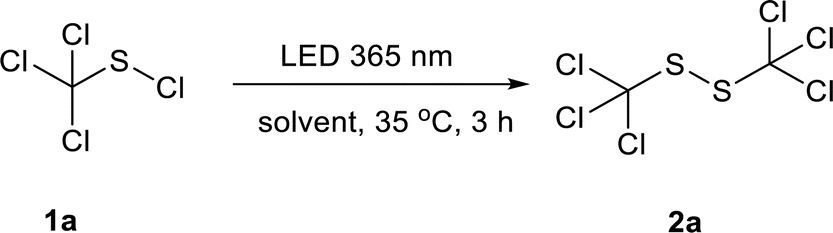
A short reaction time of 15 minutes was found to be enough to furnish the reaction (Table 2, entries 1–5). The effect of irradiation wavelength in the range of 350 nm to 455 nm was then investigated. The disulfide yield increased firstly with increasing wavelength, and an excellent isolated yield of 93% was achieved at 405 nm (Table 2, entries 6–8). The light irradiation at the wavelengths of 420 and 455 nm led to decreased yields of 81% and 75% respectively (Table 2, entries 9–10). Further screening on the reaction temperature revealed that either decreasing the temperature to 25 °C (Table 2, entry 11) or elevating it to 45 °C (Table 2, entry 12) did not improve the reaction yield. Therefore, the best reaction conditions were established as follows: irradiating 1a in cyclohexane with a LED of 405 nm at 35 °C for 15 minutes (Table 2, entry 8).
|
||||
|---|---|---|---|---|
| Entry | Wavelength (nm) | Temp. (°C) | Time (min) | Yield b (%) |
| a Reaction conditions: 50 mmol 1a, cyclohexane (53 mL), LED.b Determined by GC area normalization method.c Isolated yield. | ||||
| 1 | 365 | 35 | 120 | 92 |
| 2 | 365 | 35 | 60 | 92 |
| 3 | 365 | 35 | 30 | 92 |
| 4 | 365 | 35 | 15 | 92 |
| 5 | 365 | 35 | 10 | 90 |
| 6 | 350 | 35 | 15 | 62 |
| 7 | 385 | 35 | 15 | 93 |
| 8 | 405 | 35 | 15 | 96 (93c) |
| 9 | 420 | 35 | 15 | 81 |
| 10 | 455 | 35 | 15 | 75 |
| 11 | 405 | 25 | 15 | 90 |
| 12 | 405 | 45 | 15 | 92 |
With the optimized conditions in hand, the scope of various sulfenyl chlorides was investigated (Scheme 2). To our delight, expected symmetrical disulfides were obtained in good to excellent yields for various substituted phenyl sulfenyl chlorides. 1,2-bis(phenyl)disulfide (2b) was produced from phenyl sulfenyl chloride with a good yield of 73%. 4-Bromo-substituted phenyl sulfenyl chloride gave the corresponding product (2c) in a similar yield. All of the phenyl sulfenyl chlorides with a strong electron-withdrawing group of trifluoromethyl at the ortho-, meta- or para-position of the benzene ring generated the corresponding products with excellent yields (2d: 88%, 2e: 96% and 2f: 91%, respectively). While the 4-cyanophenyl sulfenyl chloride delivered the product smoothly with a good yield of 81% (2g), the 2-nitrophenyl sulfenyl chloride failed in the reaction (2h), which might due to the existence of the photosensitive nitro group.17 The substrates with an electron-donating substitute (–Me and –OMe) on the benzene ring were applicable to this reaction with moderate product yields (2i and 2j). 4,4′-disulfanediyldibenzoic acid (2k) and dimethyl 4,4′-disulfanediyldibenzoate (2l) were prepared from corresponding sulfenyl chlorides with a yield of 79% and 31% respectively. When 4-amine phenylsulfenyl chloride was applied, a salt-like solid mixture containing 24% of 4,4′-disulfanediyldianiline (2m) according to HPLC analysis (area normalization method) was obtained. Both naphthalen-1-yl sulfenyl chloride and naphthalen-2-yl sulfenyl chloride could give corresponding products in good yields (2n & 2o). Furthermore, the heterocyclic compound thiazol-2-yl sulfenyl chloride reacted smoothly to generate 1,2-di(thiazol-2-yl)disulfide (2p) in a moderate yield of 60%. In contrast, pyridyl sulfenyl chloride produced a salt-like solid containing 46% of 1,2-bis(4-pyridyl)disulfide (2q). When isopropyl sulfenyl chloride was used, a mixture of 1,3-bis(isopropyl)trisulfide (main product), 1,2-bis(isopropyl)disulfide (2r) and isopropyl sulfide was obtained. Benzylic sulfenyl chloride gave a mixture of 1,2-bis(benzyl)disulfide (2s) with other impurities. S-chlorocysteine also gave a salt-like solid containing corresponding disulfide product (2t) of 22%.
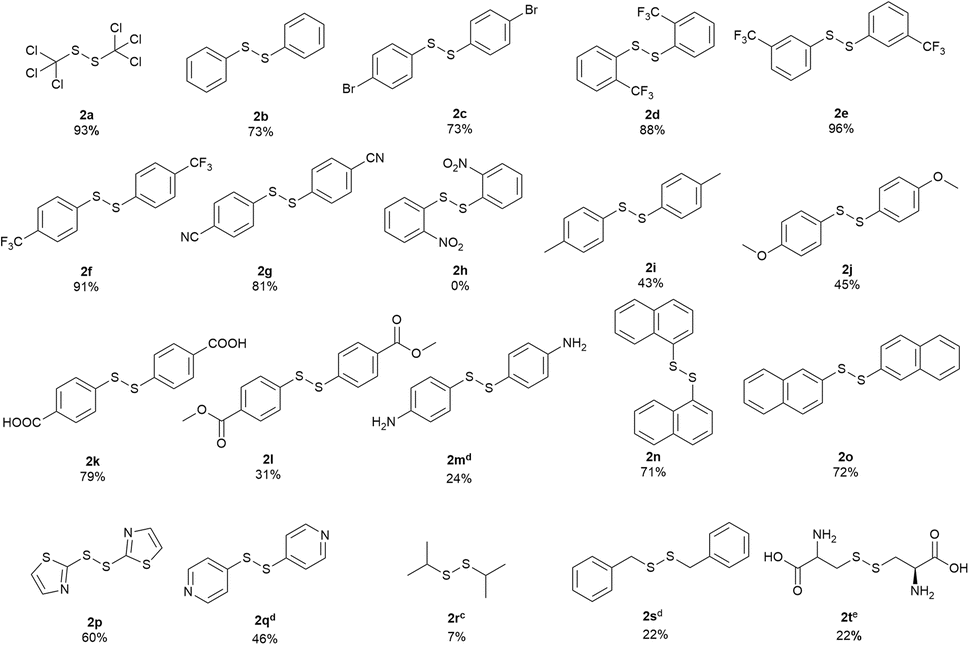 | ||
| Scheme 2 Substrate scope of symmetrical disulfides. a Reaction conditions: substrate (5 mmol), cyclohexane (5.3 mL), LED 405 nm, 35 °C, 15 minutes. b Isolated yield. c Determined by GCMS. d Determined by HPLC (254 nm) area normalization method. e Determined by HPLC (210 nm) area normalization method. | ||
Encouraged by the success in the homo-coupling of sulfenyl chlorides, the cross-coupling reaction between two different sulfenyl chlorides was then investigated. The reaction conditions were screened by the model reaction between trichloromethyl sulfenyl chloride (1a) and phenyl sulfenyl chloride (1b). The impact of different molar ratios and irradiation wavelength in the range of 350–455 nm was investigated (Table 3). To our delight, 1-phenyl-2-(trichloromethyl)disulfide (2ab) was obtained in a moderate yield of 49% with a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (1a:1b) under the 405 nm LED irradiation (Table 3, entry 3).
:1 (1a:1b) under the 405 nm LED irradiation (Table 3, entry 3).

Subsequently, the cross-coupling of trichloromethyl sulfenyl chloride (1a) with different aryl sulfenyl chlorides and heteroaryl sulfenyl chlorides were investigated (Scheme 3). The aryl sulfenyl chlorides bearing both an electron-withdrawing group (1d, 1f & 1g) and an electron-donating group (1i) on the benzene ring were applicable in this transformation. 1-(Naphthalen-2-yl)-2-(trichloromethyl)disulfide (2ao) and 2-((trichloromethyl)disulfaneyl)benzo[d]oxazole (2au) were also obtained in a moderate yield of 45% and 39%, respectively.
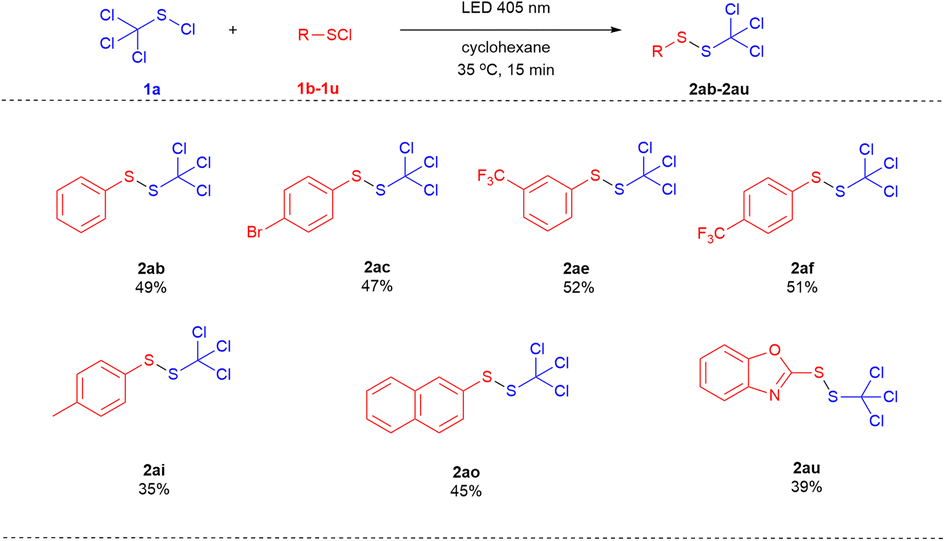 | ||
| Scheme 3 Cross coupling between trichloromethyl sulfenyl chloride (1a) and different sulfenyl chlorides.a,b a Reaction conditions: substrate (1 mmol), 1a (2 mmol), cyclohexane (3.3 mL), LED 405 nm, 35 °C, 15 minutes. b Isolated yield base on the aryl sulfenyl chloride (R–SCl). | ||
Next, the cross-coupling between phenyl sulfenyl chloride (1b) and other aryl sulfenyl chlorides was explored (Scheme 4). Several unsymmetrical diaryl disulfides, such as 2bc, 2bf and 2bi were achieved in moderate yields of 40–50%.
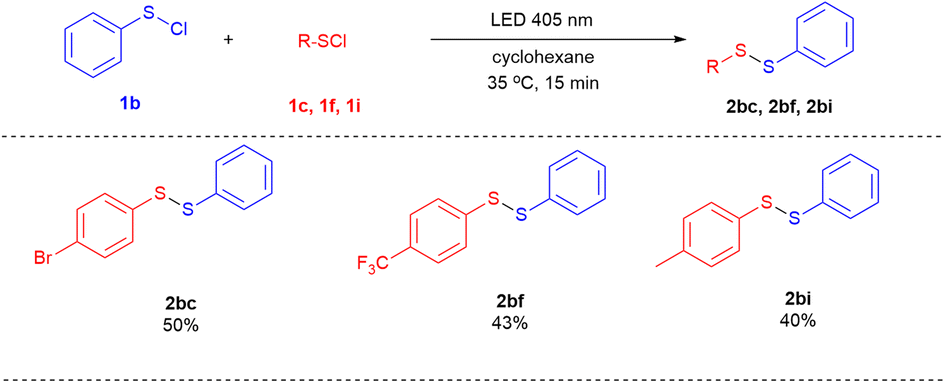 | ||
| Scheme 4 Cross coupling between phenyl sulfenyl chloride (1b) and other aryl sulfenyl chloridesa,b. a Reaction conditions: 1b (2 mmol), aryl sulfenyl chloride (1 mmol), cyclohexane (3.3 mL), LED 405 nm, 35 °C, 15 minutes. b Isolated yield base on R–SCl. | ||
To establish the mechanism of the reaction, a series of control experiments were performed (Scheme 5). It was found that the product yield was not affected when the reaction was conducted under a nitrogen or argon atmosphere (Scheme 5a). When the oxygen free radical scavenger benzoquinone was added, the reaction went smoothly and a product yield of 92% was obtained (Scheme 5b), implying that oxygen did not play a dominant role in this transformation. No product was detected without the LED irradiation (Scheme 5c). While the addition of 2,2′-azobis(2-methylpropionitrile) (AIBN) under dark condition made the reaction proceed successfully with a product yield of 85% (Scheme 5d), the presence of free radical inhibitor TEMPO upon LED irradiation could completely block the reaction (Scheme 5e), which indicating that a radical pathway was involved in this transformation. In addition, the GC-MS analysis of the model reaction mixture under optimized conditions showed the presence of chlorocyclohexane (main byproduct), cyclohexyl(trichloromethyl)sulfide (minor byproduct) and trace of chloroform and 1-cyclohexyl-2-(trichloromethyl)-disulfide. Persistent emission of hydrogen chloride with slight chlorine gas was also detected with a pH test paper and a potassium iodide starch test paper.
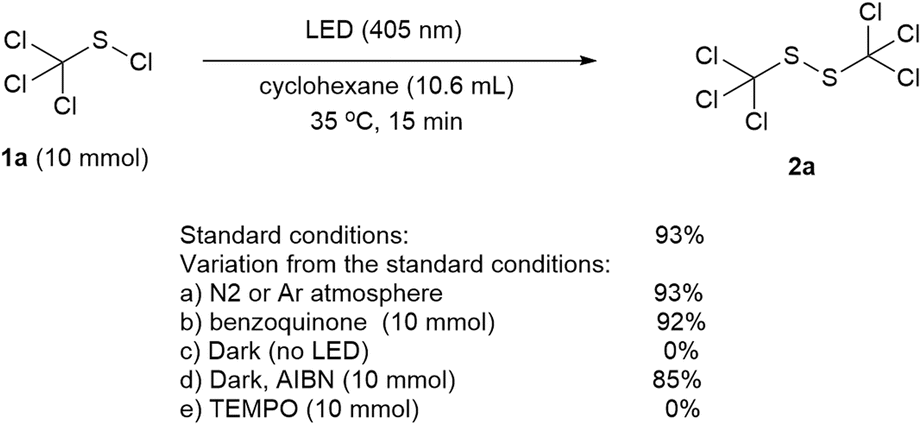 | ||
| Scheme 5 Control experiments. | ||
Based on the experimental evidences and the literature report,18 a plausible reaction mechanism was proposed as shown in Scheme 6. The photochemical cleavage of trichloromethyl sulfenyl chloride 1a under 405 nm LED irradiation generates the thiyl radical E and a chlorine radical. The homo-coupling of thiyl radical E produces the desired product 1,2-bis(trichloromethyl)disulfide 2a. The hydrogen atom transfer (HAT) process between cyclohexane and chlorine radical gives the cyclohexyl radical F,19 which further undergoes a cross-coupling with chlorine radical to produce the main byproduct chlorocyclohexane 4, indicating that the solvent cyclohexane simultaneously plays the role of a sacrificial reagent. A minor amount of chlorine radical is annihilated to form the byproduct chlorine gas. Cyclohexyl(trichloromethyl)sulfide 5 is generated by E and F. The photochemical cleavage of 5 affords the radical G and H, which can further produce chloroform 6, and 1-cyclohexyl-2-(trichloromethyl)disulfide 7 through a HAT process and a radical cross-coupling reaction, respectively.
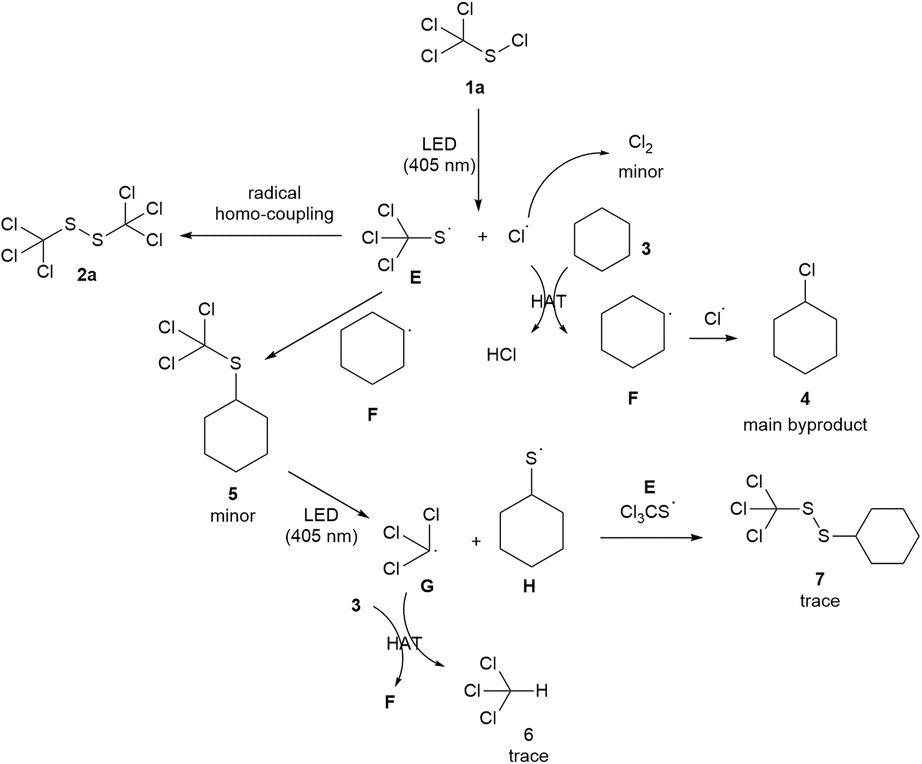 | ||
| Scheme 6 A plausible mechanism. | ||
On the other hand, over the last decade, the continuous flow photochemistry has increasingly attracted attention from researchers both in academia and industry.20 As the distribution of photons becomes more problematic with increasing reactor dimensions due to the attenuation effect of photon transport, photochemical transformation in flow mode can facilitate the photochemical processes and their subsequent scale-up due to the uniformly irradiation on the reaction mixture.21 The photochemical homo-coupling of trichloromethyl sulfenyl chloride 1a was therefore investigated in flow mode with a Corning G1 reactor equipped with a tail gas absorber using water as the absorbent. After the optimization on the residence time and reaction temperature (see ESI, Table S1†), the best continuous flow reaction conditions were obtained as follows: 1a in cyclohexane (0.85 M, 13.7 mL min−1), 85 °C, 405 nm LED, residence time 3 minutes.
The photochemical homo-coupling of trichloromethyl sulfenyl chloride 1a was then continuously run for more than 12 hours. After separating both the solvent cyclohexane and the main byproduct chlorocyclohexane out from the reaction mixture by distillation, 1410 g homo-coupling product 2a (93.7% in yield) was obtained in a purity of 99.0% (GC area normalization method). Meanwhile, through an atmospheric and then vacuum distillation process, 7276 g cyclohexane in a purity of 99.6% (GC area normalization method) was recovered in a recovery of 96.1% by the conventional distillation, and the main byproduct chlorocyclo-hexane, which is a useful organic intermediate for the synthesis of rubber additives, was then obtained in a yield of 99.5% (1200 g) and in a purity of 98.3% (GC area normalization method) by the vacuum distillation. The recovered cyclohexane was reused in the next run and 2a was achieved in the same yield (93.8%) (see ESI†, solvent reuse experiment).
Conclusions
In summary, we have demonstrated a facile and clean protocol for the synthesis of both symmetrical and unsymmetrical disulfides from sulfenyl chlorides in moderate to excellent yields under the LED irradiation. The photochemical radical coupling reaction of sulfenyl chlorides was performable for a variety of aryl sulfenyl chlorides bearing both electron-donating and withdrawing substituents in the absence of any catalyst, oxidant/reductant and other additive in 15 minutes. The reaction can be scaled up to kilogram scale in a continuous-flow mode. The solvent as well as the main byproduct can be recovered in high yields, which makes the process be highly atomic economical. The recovered solvent can be reused without any negative effect on the reaction. Thus, the present work provides an efficient, eco-friendly, and scalable method for the preparation of disulfides.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts.Notes and references
- (a) E. Gross, C. S. Sevier, A. Vala, C. A. Kaiser and D. Fass, Nat. Struct. Mol. Biol., 2002, 9, 61–67 CrossRef PubMed; (b) M. Gongora-Bernitez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef PubMed.
- L. He, L. L. Wang, Z. S. Wang, T. T. Li, H. Chen, Y. N. Zhang, Z. P. Hu, D. S. Dimitrov, J. J. Du and X. B. Liao, J. Med. Chem., 2021, 64, 15716–15726 CrossRef PubMed.
- Y.-M. Go and D. P. Jones, Free Radic. Biol. Med., 2011, 50, 495–509 CrossRef PubMed.
- S. S. Kumar, M. S. S. Bharati and S. V. Rao, J. Phys. D Appl. Phys., 2023, 37, 56 Search PubMed.
- J. I. Cunneen and F. W. Shipley, J. Polym. Sci., 1959, 36, 77–90 CrossRef.
- (a) J. B. Arterburn, M. C. Perry, S. L. Nelson, B. R. Dible and M. S. Holguin, J. Am. Chem. Soc., 1997, 119, 9309–9310 CrossRef; (b) A. R. Hajipour, S. E. Mallakpour and H. Adibi, J. Org. Chem., 2002, 67, 8666–8668 CrossRef; (c) J. K. Vandavasi, W. P. Hu, C. Y. Chen and J. J. Wang, Tetrahedron, 2011, 67, 8895–8901 CrossRef CAS; (d) H. M. Wang, Q. Q. Lu, C. H. Qian, C. Liu, W. Liu, K. Chen and A. W. Lei, Angew. Chem., Int. Ed., 2016, 55, 1094–1097 CrossRef CAS; (e) H. H. Cui, W. Wei, D. S. Yang, Y. L. Zhang, H. J. Zhao, L. L. Wang and H. Wang, Green Chem., 2017, 3520–3524 RSC; (f) H. Xu, Y.-F. Zhang and X. Lang, Chin. Chem. Lett., 2020, 1520–1524 CrossRef; (g) J. R. L. Sousa, M. S. Franco, L. D. Mendes, L. A. Araújo, J. S. S. Neto, T. E. A. Frizon, V. B. dos Santos, E. Carasek, S. Saba, J. Rafique and A. L. Braga, Org. Biomol. Chem., 2024, 22, 2175–2181 RSC.
- M. Bao and M. Shimizu, Tetrahedron, 2003, 59, 9655–9659 CrossRef.
- (a) Y. Zheng, F. L. Qing, Y. G. Huang and X. H. Xu, Adv. Synth. Catal., 2016, 358, 3477–3481 CrossRef; (b) G. A. Olah, S. C. Narang, L. S. Field and G. F. Salem, J. Org. Chem., 1980, 4792–4793 CrossRef; (c) G. A. Olah, S. C. Narang, L. D. Field and R. Karpeles, J. Org. Chem., 1981, 46, 2408–2410 CrossRef CAS; (d) Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 9188–9190 RSC; (e) H. Kumar, A. Dubey, G. Prajapati, R. Kant, R. S. Ampapathibd and P. K. Mandal, New J. Chem., 2022, 46, 3426–3430 RSC; (f) K. Taira, W. L. Mock and D. G. Gorenstein, J. Am. Chem. Soc., 1984, 106, 7831–7835 CrossRef.
- C. J. Zhu, D. F. Wu, H. L. Liu, C. W. Meng and T. D. Tang, Green Chem., 2022, 24, 9033–9039 RSC.
- (a) C. M. Park, B. A. Johnson, J. C. Duan, J. J. Park, J. J. Day, D. Gang, W. J. Qian and M. Xian, Org. Lett., 2016, 18, 904–907 CrossRef; (b) X. Xiao, M. H. Feng and X. F. Jiang, Angew. Chem., Int. Ed., 2016, 55, 14121–14125 CrossRef PubMed; (c) W. G. Wang, Y. Z. Lin, Y. D. Ma, C.-H. Tung and Z. H. Xu, Org. Lett., 2018, 20, 3829–3832 CrossRef PubMed; (d) J. X. Zou, J. H. Chen, T. Shi, Y. S. Hou, F. Cao, Y. Q. Wang, X. D. Wang, Z. Jia, Q. Y. Zhao and Z. Wang, ACS Catal., 2019, 9, 11426–11430 CrossRef CAS; (e) J. H. Xue and X. F. Jiang, Nat. Commun., 2020, 4170–4177 CrossRef CAS; (f) W. C. Gao, J. Tian, Y.-Z. Shang and X. F. Jiang, Chem. Sci., 2020, 11, 3903–3908 RSC; (g) J. H. Xue and X. F. Jiang, Org. Lett., 2020, 8044–8048 CrossRef PubMed; (h) E. Zysman-Colman and D. N. Harpp, J. Org. Chem., 2005, 5964–5973 CrossRef PubMed; (i) N. Eghbali, D. S. Bohle and D. N. Harpp, J. Org. Chem., 2006, 71, 6659–6661 CrossRef PubMed.
- (a) M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 6624–6625 CrossRef; (b) J. M. Guo, J. J. Zha, C.-H. Ding, Q. T. Tan and B. Xu, Org. Lett., 2021, 23, 3167–3172 CrossRef PubMed; (c) A. A. Ukuwela, A. I. Bush, A. G. Wedd and Z. G. Xiao, Chem. Sci., 2018,(9), 1173–1183 RSC; (d) M. Ohtani and M. Narisada, J. Org. Chem., 1991, 56, 5475–5478 CrossRef; (e) M. J. Song, Q. Y. Hu, Z.-Y. Li, X. Q. Sun and K. Yang, Chin. Chem. Lett., 2022, 33, 4269–4272 CrossRef.
- P. F. Huang, P. Wang, S. Tang, Z. J. Fu and A. W. Lei, Angew. Chem., Int. Ed., 2018, 57, 8115–8119 CrossRef.
- (a) R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Chem. Rev., 2023, 123, 5225–5261 CrossRef; (b) H. J. Kim, J. H. Yoon and S. Yoon, J. Phys. Chem. A, 2010, 114, 12010–12015 CrossRef PubMed; (c) M. Oba, K. Tanaka, K. Nishiyama and W. Ando, Org. Chem., 2011, 76, 4173–4177 CrossRef; (d) K. Y. D. Tan, G. F. Teng and W. Y. Fan, Organometallics, 2011, 4136–4143 CrossRef; (e) P. H. Huang, P. Wang, S. Tang, Z. J. Fu and A. W. Lei, Angew. Chem., Int. Ed., 2018, 27, 8247–8251 CrossRef; (f) D. G. Wang, Q. He, K. Q. Shi, M. T. Xiong, Y. F. Zhou and Y. J. Pan, Adv. Synth. Catal., 2021, 1–7 CrossRef; (g) D. H. Dethe, A. Srivastava, B. D. Dherange and B. V. Kumar, Adv. Synth. Catal., 2018, 360, 3020–3025 CrossRef.
- (a) X.-B. Xu, Z. J. Li, Y.-J. Gao, Q. Y. Meng, S. Yu, R. G. Weiss, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2014, 53, 2085–2089 CrossRef; (b) N. Spiliopoulou and C. G. Kokotos, Green Chem., 2021,(23), 546–551 RSC.
- Z. J. Wu and D. A. Pratt, J. Am. Chem. Soc., 2020, 142, 10284–10290 CrossRef PubMed.
- (a) D. Witt, Synthesis, 2008, 16, 2491–2509 CrossRef; (b) M. Wang and X. F. Jiang, Top. Curr. Chem., 2018, 376, 14 CrossRef.
- H.-R. Raquel, G.-G. Sara, R. P. María, S.-P. Samuel and S. Roberto, Org. Biomol. Chem., 2023, 21, 7791–7798 RSC.
- (a) A. Romieu, S. Bellon, D. Gasparutto and J. Cadet, Org. Lett., 2000, 2, 1085–1088 CrossRef; (b) L.-A. Chen and K. Sung, Org. Lett., 2009, 11, 3370–3373 CrossRef PubMed; (c) I. Suzuki, K. Kiyokawa, M. Yasuda and A. Baba, Org. Lett., 2013, 15, 1728–1731 CrossRef PubMed; (d) V. Prey, E. Gutschik and H. Berbalk, Monatsh. Chem., 1960, 91, 556–560 CrossRef.
- S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef.
- T. H. Rehm, Chem.–Eur. J., 2020, 26, 16952–16974 CrossRef PubMed.
- Y. H. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem.–Eur. J., 2014, 20, 10562–10589 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04568h |
| This journal is © The Royal Society of Chemistry 2024 |