
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular motifs formed by CH3/Cl-substituted arene groups: evidence for structural differences in thiophosphoramides and similarities in their complexes†
Saeed Hosseinpoor a,
Mehrdad Pourayoubi*a,
Eliška Zmeškalováb and
Morgane Pouponb
a,
Mehrdad Pourayoubi*a,
Eliška Zmeškalováb and
Morgane Pouponb
aDepartment of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran. E-mail: pourayoubi@um.ac.ir
bInstitute of Physics of the Czech Academy of Sciences, Na Slovance 2, Prague 8, 182 21, Czech Republic
First published on 11th October 2024
Abstract
Differences/similarities of supramolecular motifs are discussed in two new thiophosphoramide structures and their Ni molecular complexes: (C2H5O)2P(S)(NHC(S)NHCH2C6H4X) and [{(C2H5O)2P(S)(NC(S)NHCH2C6H4X)}2Ni] (X = Cl/CH3 I/II and III/IV). The structures have equal numbers of donor/acceptor sites contributing to classical hydrogen bonds (PS/CS and 2 × NH in ligands and 2 × PS and 2 × NH in the complexes). However, these donor and acceptor sites contribute to inter/intramolecular hydrogen bonding in ligands and intramolecular hydrogen bonding in complexes. In the supramolecular assemblies of the ligands, the classic hydrogen bonds (N–H⋯S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) are restricted in dimer synthons, and the weaker interactions (formed by Cl/CH3 substituents) compete against each other. In the complexes, despite the lack of classic intermolecular hydrogen bond, numerous weak interactions, e.g., C–H⋯Y (Y = S, O, Ni, N, and π), contribute to the molecular assemblies, which do not include the participation of Cl/CH3. Thus, different packing features of ligands, but similar in complexes are observed. Each ligand and the associated complex show nearly equal supramolecular motifs in the slice of the substituted benzyl groups, related to the formation of C–H⋯Cl/π⋯π for the 4-Cl-C6H4CH2 groups in I/III and C–H⋯π for the 4-CH3-C6H4CH2 groups in II/IV. The repeatabilities of the motifs made by 4-Cl-C6H4CH2/4-CH3-C6H4CH2 were checked by surveying 142/844 structures with 178/1482 segments in the CSD, which show that 17% and 12% of the structures exhibited similarities with the title structures. The methods X-ray crystallography, 2D fingerprint plots, electrostatic potential surfaces, QTAIM, and energy framework calculations were applied to present the discussion.
C) are restricted in dimer synthons, and the weaker interactions (formed by Cl/CH3 substituents) compete against each other. In the complexes, despite the lack of classic intermolecular hydrogen bond, numerous weak interactions, e.g., C–H⋯Y (Y = S, O, Ni, N, and π), contribute to the molecular assemblies, which do not include the participation of Cl/CH3. Thus, different packing features of ligands, but similar in complexes are observed. Each ligand and the associated complex show nearly equal supramolecular motifs in the slice of the substituted benzyl groups, related to the formation of C–H⋯Cl/π⋯π for the 4-Cl-C6H4CH2 groups in I/III and C–H⋯π for the 4-CH3-C6H4CH2 groups in II/IV. The repeatabilities of the motifs made by 4-Cl-C6H4CH2/4-CH3-C6H4CH2 were checked by surveying 142/844 structures with 178/1482 segments in the CSD, which show that 17% and 12% of the structures exhibited similarities with the title structures. The methods X-ray crystallography, 2D fingerprint plots, electrostatic potential surfaces, QTAIM, and energy framework calculations were applied to present the discussion.
Introduction
The prediction and design of crystalline structures have been a topic of interest, due to various applications in material sciences and pharmaceutics.1–4 In this context, strong to moderate interactions, notably hydrogen bonds, play a significant role in forming molecular assemblies and are extensively addressed.3,5–7 Although weak interactions often have less impact on the formation of supramolecular assemblies, their role is still questionable, and there are reports on the study of such interactions in molecular/ionic crystal structures5,8 and macromolecule materials.9,10The importance of weak interactions can become clearer when two almost the same molecules with just a difference in the substituent (influential for weak interactions) form two crystal structures with different packing features.11,12 In organic compounds, Cl/CH3 units—which have about the same volume—are frequently utilized as substituents. Their disparate electrical characteristics lead to different actions as donor/acceptor sites in contacts and to form different molecular assemblies. The electron density of chlorine, bonded to one other atom, is anisotropically distributed, resulting in the formation of (i) a negative belt orthogonal to the covalent bond (a possible electron donor site) and (ii) a σ-hole upward of the chlorine bond with lower electron density and generally positive electrostatic potential (a possible electron acceptor site).5,13
However, the expected behavior of organic chlorine is its hydrogen acceptor capability in weak hydrogen bonds. Methyl groups, with positive electrostatic potential on their hydrogens, usually take part in weak hydrogen bonds and dihydrogen interactions. Nevertheless, a negative region on the associated carbon atom was calculated, where it is bonded to an electropositive atom, proposing the potential electron donor behavior.14,15 In fact, the electron density around methyl is practically distributed in opposition to chlorine.
In addition to direct interactions of substituents, their influence on the electrostatic potential of neighboring groups can impact the molecular assembly. Regarding the substitutions of the aryl groups, Wheeler showed the through-space effect of substituents on the electrostatic potential,16 although he explained that the main effect of substituents is due to their direct and local interaction.17,18
Some researchers show that exchanging non-polar substituents with the same sizes in molecules19 (e.g. Cl/CH3) or even with different sizes20 (e.g. CF3/CH3) may cause the production of isostructures. Further research showed that in some cases, the directional interactions of Cl atoms may alter the structure.21 An old CSD study indicated that about 30% of the crystal structures with Cl/CH3-substitution that had been documented up to that time were isostructure.22 Earlier investigations affirm the isostructurality in Cl/CH3-exchanged crystal structures, in which interactions with no contribution of the Cl atom dominate.23–27 In some structures where the DH⋯Cl (D = hydrogen donor atom) and Cl⋯Cl interactions are dominant with respect to other weak interactions, the structures are changed by exchanging the Cl atom with the CH3 group.24,28,29 Moreover, there are examples of structural similarity of hydrogen bond pattern, based on stronger interactions, but with differences in crystal packing related to the contribution of Cl/CH3.30 Nonetheless, the prediction of the possible isostructurality is challenging, and it is still up for discussion how weak to moderate interactions play a role in discriminating or not in structures with exchanged methyl and chloro substituents.
The (R1O)2P(S)(NHC(S)R2) thiourea-based thiophosphoramides (R1 = aryl/alkyl, and R2 = amine groups) are studied in complexation processes,31,32 manufacturing of antibacterial agents,33,34 membranes,35 as sulfide donor precursors in the fabrication of photoluminescence thin-films,36,37 and in the concepts related to crystal engineering.32,38,39
Some attempts have been made to look into the structural features of the thiophosphoramide family affected by the methyl and chlorine substituents, individually. However, the comparison of motifs and interactions created by chlorine/methyl substituted aryl groups and their effects on supramolecular assemblies is limited to a pair of Hg complexes.40 Herein, we report on two new thiourea-based thiophosphoramide structures and their nickel complexes, including CH3/Cl substituents attached to an arene ring (I–IV, Chart 1). The differences between these structures are probed by closely looking at the crystal structures using computational methods. The role of weak interactions is examined in the effectiveness of Cl/CH3 exchange on structural changes. Additionally, the structures with 4-Cl-C6H4CH2 and 4-CH3-C6H4CH2 fragments were retrieved from the Cambridge Structural Database to find the probability of the creation of the aryl⋯aryl motifs similar to I/III and II/IV.
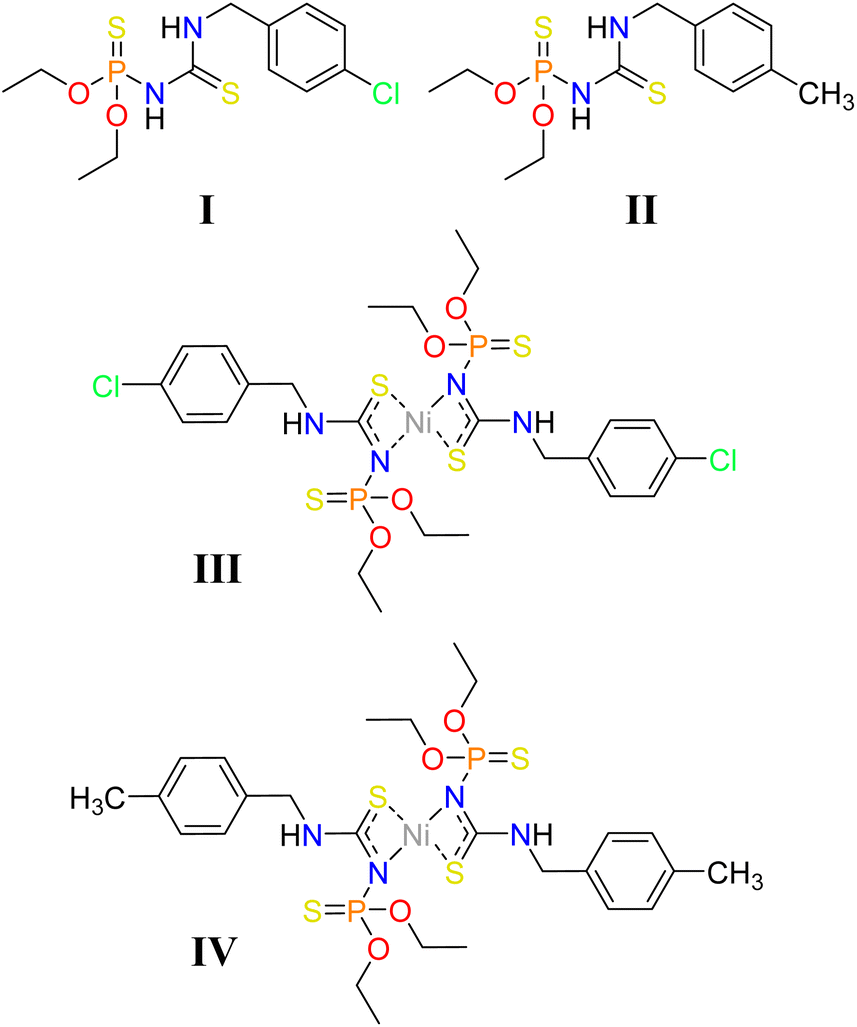 | ||
| Chart 1 Structural formulas of I–IV. | ||
Experimental and theoretical methods
Diffraction data were collected on a Rigaku/Oxford Diffraction diffractometer using Cu Kα radiation at 95 K (I) and 120 K (II–IV). The data were registered with an Atlas S2 CCD area detector. CrysAlis PRO41 was used to control the experiment and process data. Shelxt42 was used for the structure solution, and Jana2020![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 was used to refine and finalize the structure model. The structures were solved by the direct method and refined by the full-matrix least-squares method on F2. All non-hydrogen atoms were refined anisotropically, and the hydrogen atoms were all located on a difference-Fourier map, but those attached to carbon atoms were repositioned geometrically. The H atoms were initially refined with soft restraints on the bond lengths and angles to regularize their geometry (C–H in the range 0.93–0.98 Å, N–H in the range 0.86–0.89 Å and Uiso(H) in the range 1.2–1.5 times Ueq of the parent atom), after which the positions were refined with riding constraints.
43 was used to refine and finalize the structure model. The structures were solved by the direct method and refined by the full-matrix least-squares method on F2. All non-hydrogen atoms were refined anisotropically, and the hydrogen atoms were all located on a difference-Fourier map, but those attached to carbon atoms were repositioned geometrically. The H atoms were initially refined with soft restraints on the bond lengths and angles to regularize their geometry (C–H in the range 0.93–0.98 Å, N–H in the range 0.86–0.89 Å and Uiso(H) in the range 1.2–1.5 times Ueq of the parent atom), after which the positions were refined with riding constraints.
X-ray crystallographic parameters of I–IV are gathered in Table 1, and selected bond lengths and angles and hydrogen bond parameters are given in Tables S1–S5.† The molecular structures are represented in Fig. S1.†
| I | II | III | IV | |
|---|---|---|---|---|
| Chemical formula | C12H18ClN2O2PS2 | C13H21N2O2PS2 | C12H17ClN2Ni0.50O2PS2 | C13H20N2Ni0.50O2PS2 |
| Mr | 352.85 | 332.43 | 381.19 | 360.77 |
| Crystal system, space group | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Triclinic, P |
Triclinic, P |
Triclinic, P |
| Temperature (K) | 95 | 120 | 120 | 120 |
| a, b, c (Å) | 7.6290 (1), 7.6702 (1), 14.1025 (4) | 5.8046 (2), 10.7878 (2), 13.3907 (3) | 7.8502 (2), 10.1868 (4), 11.6914 (4) | 8.2171 (3), 9.9942 (4), 11.1612 (4) |
| α, β, γ (°) | 94.2128 (19), 99.531 (2), 98.0165 (16) | 81.0002 (18), 83.334 (2), 89.276 (2) | 113.275 (3), 97.462 (3), 97.123 (3) | 113.611 (3), 93.655 (3), 92.926 (3) |
| V (Å3) | 801.93 (3) | 822.58 (4) | 835.66 (5) | 835.21 (6) |
| Z | 2 | 2 | 2 | 2 |
| μ (mm−1) | 5.51 | 3.88 | 5.86 | 4.39 |
| Crystal size (mm) | 0.49 × 0.27 × 0.14 | 0.79 × 0.55 × 0.38 | 0.31 × 0.27 × 0.12 | 0.76 × 0.28 × 0.22 |
| Tmin, Tmax | 0.11, 0.46 | 0.10, 0.23 | 0.15, 0.49 | 0.03, 0.38 |
| No. of measured, independent, and observed [I > 2.0σ(I)] reflections | 11539, 3210, 3153 |
13323, 2929, 2793 |
12744, 2996, 2716 |
13759, 2985, 2692 |
| Rint | 0.023 | 0.038 | 0.096 | 0.068 |
| (Sinθ/λ)max (Å−1) |
0.624 | 0.600 | 0.600 | 0.600 |
| R[F2 > 2σ(F2)] | 0.026 | 0.031 | 0.050 | 0.045 |
| wR(F2) | 0.071 | 0.097 | 0.141 | 0.139 |
| S | 0.97 | 1.04 | 1.00 | 0.99 |
| No. of reflections | 3210 | 2929 | 2996 | 2985 |
| No. of parameters | 182 | 190 | 188 | 187 |
| Δρmax, Δρmin (e Å−3) | 0.54, −0.28 | 0.36, −0.37 | 0.76, −0.81 | 0.64, −0.91 |
All the chemical substances utilized in this investigation were procured from commercially available sources and employed in their original state. 1H, 13C{1H}, and 31P{1H} NMR spectra were recorded using a Bruker Avance III 300 MHz spectrometer. As external standards, chemical shifts were determined relative to TMS for 1H and 13C and 85% H3PO4 for 31P. IR spectra were recorded on a Buck 500 Scientific spectrometer using KBr pellets. The mass spectra were recorded with a Varian Mat CH-7 spectrometer at 70 eV.
The molecular electrostatic potential (ESP) surfaces were calculated using the Gaussian 09 software44 at the B3LYP/cc-pVDZ level of theory, for molecules extracted from crystal structures after neutron-normalization of hydrogen positions.
Crystal lattice energies of structures I and II were calculated using CrystalExplorer 21.5 with the CE-B3LYP method and 6-31G(d,p) basis set.45 In this approach, the interaction energies of a molecule with the first sphere of the neighboring molecules are computed, and the effects of the further molecules on the lattice energy are ignored. The interaction energies (Etot) are calculated using the equation:
| Etot = keleEele + kpolEpol + kdisEdis + krepErep |
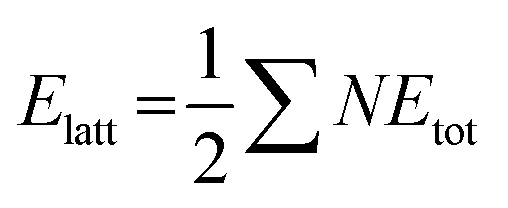 formula,47 in which N is the number of molecular pairs in the molecular shell, and Etot is the total energy of each molecular pair. Crystal lattice energies, interaction energies of molecular pairs, and their components (electrostatic, dispersion, polarization, and repulsion) are given in Tables S7 and S8.†
formula,47 in which N is the number of molecular pairs in the molecular shell, and Etot is the total energy of each molecular pair. Crystal lattice energies, interaction energies of molecular pairs, and their components (electrostatic, dispersion, polarization, and repulsion) are given in Tables S7 and S8.†
Intermolecular interactions of molecular pairs with the most prominent energies in the crystal structure were studied using the QTAIM method.48 In structures I and II, the molecular pairs with total interaction energies ≤ −15 kJ mol−1 (obtained from crystal lattice energy calculations) were selected. The wavefunction files were obtained using Gaussian 09 software44 with the D3-B3LYP/cc-pVDZ level of theory, and the QTAIM diagrams were plotted with the MultiWFN program package.49 Selected topological parameters including potential energy density (V(r)), kinetic energy density (G(r)), total energy density (H(r)), total electronic density (ρ(r)), and its corresponding Laplacian (∇2ρ(r)) at bond critical points (BCPs) are given in Table 2. The V(r) values can be applied in the formula E = V(r)/2 for estimating the hydrogen bond strength of molecular systems in the gas phase.50 In the crystal structures, reporting the interaction energy values obtained from this estimation can be misguiding due to some observed deviations from the results of other computational methods.51 Nevertheless, the V(r) values can still be utilized as a criterion for the approximate comparison of the interaction strengths that are employed in this study. The ρ(r) is another parameter that is used for approximate comparison of the interaction strength.52 Based on Koch–Popelier criteria if the ρ(r) value at a BCP of a CH⋯X contact ranges at 0.002–0.035 a.u. a hydrogen bond is established.53
| Pair code | Interaction | ρ(r) | ∇2ρ(r) | G(r) | V(r) | H(r) |
|---|---|---|---|---|---|---|
| Intramolecular (I) | NH⋯SP |
0.0194 | 0.0473 | 0.0111 | −0.0104 | 0.0007 |
| Ia | NH⋯SC |
0.0224 | 0.0473 | 0.0115 | −0.0112 | 0.0003 |
| CH⋯SC |
0.0061 | 0.0185 | 0.0039 | −0.0031 | 0.0008 | |
| Ib | CH⋯O | 0.0089 | 0.0274 | 0.0065 | −0.0062 | 0.0003 |
| Ic | CH⋯SC |
0.0048 | 0.0150 | 0.0030 | −0.0022 | 0.0008 |
| CH⋯N | 0.0063 | 0.0215 | 0.0045 | −0.0037 | 0.0008 | |
| CH⋯O | 0.0041 | 0.0180 | 0.0035 | −0.0025 | 0.0010 | |
| Id | CH⋯SP |
0.0060 | 0.0152 | 0.0033 | −0.0028 | 0.0005 |
| CH⋯N | 0.0044 | 0.0165 | 0.0033 | −0.0024 | 0.0009 | |
| CH⋯SC |
0.0043 | 0.0128 | 0.0026 | −0.0020 | 0.0006 | |
| CH⋯SC |
0.0042 | 0.0127 | 0.0026 | −0.0020 | 0.0006 | |
| CH⋯π | 0.0066 | 0.0200 | 0.0042 | −0.0033 | 0.0009 | |
| NH⋯Cl | 0.0038 | 0.0137 | 0.0028 | −0.0021 | 0.0007 | |
| CH⋯Cl | 0.0048 | 0.0156 | 0.0031 | −0.0022 | 0.0009 | |
| CH⋯Cl | 0.0050 | 0.0162 | 0.0033 | −0.0025 | 0.0008 | |
| CH⋯HC | 0.0048 | 0.0177 | 0.0035 | −0.0026 | 0.0009 | |
| Ie | CH⋯SC |
0.0035 | 0.0112 | 0.0022 | −0.0015 | 0.0007 |
| CH⋯SC |
0.0041 | 0.0114 | 0.0023 | −0.0018 | 0.0005 | |
| CH⋯SP |
0.0077 | 0.0211 | 0.0046 | −0.0038 | 0.0008 | |
| If | π⋯π | 0.0052 | 0.0122 | 0.0026 | −0.0022 | 0.0004 |
| CH⋯Cl | 0.0064 | 0.0219 | 0.0044 | −0.0034 | 0.0010 | |
| C(π)⋯Cl | 0.0055 | 0.0161 | 0.0033 | −0.0027 | 0.0006 | |
| Ig | CH⋯SP |
0.0056 | 0.0173 | 0.0035 | −0.0026 | 0.0009 |
| CH⋯HC | 0.0047 | 0.0162 | 0.0033 | −0.0025 | 0.0008 | |
| Intramolecular (II) | NH⋯SP |
0.0202 | 0.0474 | 0.0113 | −0.0108 | 0.0005 |
| IIa | NH⋯SC |
0.0219 | 0.0462 | 0.0112 | −0.0109 | 0.0003 |
| CH⋯SC |
0.0061 | 0.0206 | 0.0041 | −0.0032 | 0.0009 | |
| CH⋯SC |
0.0039 | 0.0128 | 0.0025 | −0.0018 | 0.0007 | |
| IIb | CH⋯SC |
0.0037 | 0.0102 | 0.0021 | −0.0016 | 0.0005 |
| O⋯SP |
0.0033 | 0.0117 | 0.0023 | −0.0018 | 0.0005 | |
| N⋯SP |
0.0044 | 0.0120 | 0.0026 | −0.0023 | 0.0003 | |
| CH⋯O | 0.0101 | 0.0311 | 0.0075 | −0.0073 | 0.0002 | |
| CH⋯HC | 0.0033 | 0.0129 | 0.0024 | −0.0015 | 0.0009 | |
| CH⋯HC | 0.0030 | 0.0119 | 0.0021 | −0.0013 | 0.0008 | |
| CH⋯HC | 0.0040 | 0.0143 | 0.0026 | −0.0015 | 0.0011 | |
| CH⋯HC | 0.0040 | 0.0149 | 0.0028 | −0.0019 | 0.0009 | |
| IIc | CH⋯SP |
0.0091 | 0.0247 | 0.0054 | −0.0047 | 0.0007 |
| NH⋯π | 0.0050 | 0.0174 | 0.0034 | −0.0025 | 0.0009 | |
| CH⋯π | 0.0073 | 0.0223 | 0.0047 | −0.0038 | 0.0009 | |
| π⋯π | 0.0043 | 0.0109 | 0.0023 | −0.0019 | 0.0004 | |
| IId | CH⋯SC |
0.0066 | 0.0173 | 0.0037 | −0.0031 | 0.0006 |
| CH⋯HC | 0.0032 | 0.0122 | 0.0022 | −0.0013 | 0.0009 | |
| IIe | CH⋯SP |
0.0040 | 0.0112 | 0.0023 | −0.0017 | 0.0006 |
| CH⋯π | 0.0049 | 0.0162 | 0.0032 | −0.0025 | 0.0007 | |
| CH⋯π | 0.0040 | 0.0126 | 0.0026 | −0.0020 | 0.0006 | |
| IIf | CH⋯HC | 0.0042 | 0.0173 | 0.0031 | −0.0020 | 0.0011 |
| CH⋯HC | 0.0045 | 0.0180 | 0.0034 | −0.0022 | 0.0012 | |
| Intramolecular (III) | NH⋯SP |
0.0201 | 0.0536 | 0.0128 | −0.0122 | 0.0006 |
| IIIa | CH⋯O | 0.0050 | 0.0193 | 0.0040 | −0.0032 | 0.0008 |
| CH⋯SP |
0.0068 | 0.0211 | 0.0044 | −0.0035 | 0.0009 | |
| CH⋯N | 0.0049 | 0.0166 | 0.0035 | −0.0029 | 0.0006 | |
| CH⋯π | 0.0044 | 0.0142 | 0.0028 | −0.0022 | 0.0006 | |
| CH⋯Ni | 0.0062 | 0.0187 | 0.0039 | −0.0032 | 0.0007 | |
| IIIb | CH⋯SP |
0.0078 | 0.0227 | 0.0049 | −0.0041 | 0.0008 |
| IIIc | CH⋯SP |
0.0040 | 0.0128 | 0.0025 | −0.0017 | 0.0008 |
| CH⋯HC | 0.0034 | 0.0140 | 0.0026 | −0.0017 | 0.0009 | |
| CH⋯HC | 0.0024 | 0.0123 | 0.0022 | −0.0013 | 0.0009 | |
| IIId | CH⋯SP |
0.0042 | 0.0136 | 0.0027 | −0.0019 | 0.0008 |
| CH⋯Cl | 0.0038 | 0.0125 | 0.0025 | −0.0020 | 0.0005 | |
| Cl⋯S | 0.0048 | 0.0122 | 0.0026 | −0.0022 | 0.0004 | |
| IIIe | CH⋯S–C | 0.0050 | 0.0152 | 0.0031 | −0.0024 | 0.0007 |
| CH⋯O | 0.0028 | 0.0132 | 0.0024 | −0.0015 | 0.0009 | |
| CH⋯HC | 0.0045 | 0.0182 | 0.0034 | −0.0023 | 0.0011 | |
| CH⋯HC | 0.0035 | 0.0142 | 0.0027 | −0.0018 | 0.0009 | |
| IIIf | CH⋯Cl | 0.0050 | 0.0162 | 0.0033 | −0.0026 | 0.0007 |
| π⋯π | 0.0048 | 0.0116 | 0.0025 | −0.0021 | 0.0004 | |
| Intramolecular (IV) | NH⋯SP |
0.0183 | 0.0502 | 0.0118 | −0.0111 | 0.0007 |
| IVa | CH⋯O | 0.0066 | 0.0227 | 0.0051 | −0.0045 | 0.0006 |
| CH⋯O | 0.0032 | 0.0133 | 0.0026 | −0.0018 | 0.0008 | |
| CH⋯SP |
0.0055 | 0.0187 | 0.0036 | −0.0025 | 0.0011 | |
| CH⋯π | 0.0066 | 0.0217 | 0.0045 | −0.0035 | 0.0010 | |
| CH⋯Ni | 0.0057 | 0.0184 | 0.0037 | −0.0027 | 0.0010 | |
| IVb | CH⋯SP |
0.0042 | 0.0119 | 0.0024 | −0.0017 | 0.0007 |
| CH⋯SP |
0.0041 | 0.0129 | 0.0025 | −0.0018 | 0.0007 | |
| IVc | CH⋯HC | 0.0034 | 0.0166 | 0.0028 | −0.0015 | 0.0013 |
| IVd | CH⋯SP |
0.0044 | 0.0123 | 0.0025 | −0.0020 | 0.0005 |
| CH⋯SP |
0.0041 | 0.0128 | 0.0025 | −0.0018 | 0.0007 | |
| IVe | CH⋯O | 0.0020 | 0.0092 | 0.0016 | −0.0010 | 0.0006 |
| CH⋯HC | 0.0045 | 0.0211 | 0.0037 | −0.0022 | 0.0015 | |
| IVf | CH⋯S–C | 0.0033 | 0.0095 | 0.0019 | −0.0014 | 0.0005 |
| CH⋯π | 0.0032 | 0.0090 | 0.0018 | −0.0014 | 0.0004 |
The 2D fingerprint plot represents the distance of each point on the Hirshfeld surface to the nearest internal atom (di) and external atom (de). Accordingly, the term di + de can be used as a criterion for estimating interaction strengths. The fragment patches of the generated Hirshfeld surface show the regions in contact with neighbor molecules and their area.
Synthesis and crystallization
:1 mole ratio) for 18 hours. The potassium chloride salt was filtered off, and the filtrate was reacted with 4-chlorobenzylamine for I or 4-methyl benzylamine for II (1:1 mole ratio). After 4 hours, the solution was filtered off and left for slow evaporation. The obtained crystals, suitable for X-ray analysis, were separated, washed with acetonitrile, and dried.I: m.p.: 84 °C. IR (KBr disc, ν, cm−1): 3258, 3099, 2920, 1552, 1492, 1417, 1330, 1284, 1181, 1098, 1026, 879, 806. MS (70 eV, EI): m/z (%) = 353 (2) [M + 1]+, 352 (22) [M]+, 292 (23) [M–2C2H6]+, 259 (34) [M–2C2H5OH–1]+, 139 (98) [C7H6ClN]+, 121 (97) [CNPS2]+, 77 (65) [C6H5]+, 29 (100) [C2H5]+. 31P{1H} NMR (121 MHz, DMSO-d6): δ (ppm) = 60.57 (s). 1H NMR (301 MHz, DMSO-d6): δ (ppm) = 9.33 (s, 1H), 8.55 (t, 3JH–H = 5.6 Hz, 1H), 7.43 (d, 3JH–H = 8.5 Hz, 2H), 7.34 (d, 3JH–H = 8.5 Hz, 2H), 4.70 (apparent d, J = 5.6 Hz, 2H), 4.12 (m, 4H), 1.27 (t, 3JH–H = 7.0 Hz, 6H). 13C{1H} NMR (75 MHz, DMSO-d6): δ (ppm) = 181.28 (d, 2JC–P = 4.1 Hz), 137.41 (s), 132.19 (s), 129.78 (s), 128.82 (s), 64.12 (d, 2JC–P = 5.3 Hz), 47.16 (s), 16.13 (d, 3JC–P = 7.9 Hz).
II: m.p.: 97 °C. IR (KBr disc, ν, cm−1): 3264, 3094, 2913, 1768, 1550, 1501, 1420, 1332, 1286, 1184, 1107, 1024, 973, 882, 803, 654. MS (70 eV, EI): m/z (%) = 333 (72) [M + 1]+, 332 (75) [M]+, 331 (72) [M–1]+, 122 (72) [CHNPS2]+, 120 (24) [NHCH2C6H4CH3]+, 28 (100) [C2H4]+. 31P{1H} NMR (121 MHz, CDCl3): δ (ppm) = 50.80 (s). 1H NMR (301 MHz, CDCl3): δ (ppm) = 8.20 (t, J = 2.7 Hz, 1H), 7.76 (d, 2JH–P = 9.7 Hz, 1H), 7.35 (d, 3JH–H = 8.4 Hz, 2H), 7.25 (d, 3JH–H = 8.4 Hz, 2H), 4.82 (apparent d, J = 5.1 Hz, 2H), 4.17 (m, 4H), 2.37 (s, 3H), 1.32 (t, 3JH–H = 7.1 Hz, 6H). 13C{1H} NMR (75 MHz, CDCl3): δ (ppm) = 180.84 (d, 2JC–P = 6.3 Hz), 137.62 (s), 133.31 (s), 129.50 (s), 127.71 (s), 64.68 (d, 2JC–P = 5.1 Hz), 50.07 (s), 21.19 (s), 15.80 (d, 3JC–P = 7.9 Hz).
:1 mole ratio). Then, a solution of NiCl2·6H2O in methanol was added dropwise (ligand:metal mole ratio = 2:1), and the solution was stirred for 3 hours. The potassium chloride salt was filtered off, and the filtrate was left until the solvent evaporated. Violet crystals, suitable for X-ray analysis, were obtained from recrystallizing residual precipitate in a mixture of methanol/acetonitrile (1:1 v/v).III: m.p.: 133 °C. IR (KBr disc, ν, cm−1): 3168, 2978, 2914, 1565, 1494, 1394, 1340, 1262, 1093, 1027, 969, 907, 814. 31P{1H} NMR (121 MHz, CDCl3): δ (ppm) = 57.32 (s). 1H NMR (301 MHz, CDCl3): δ (ppm) = 9.42 (s, 2H), 7.25 (d, 3JH–H = 8.1 Hz, 4H), 7.18 (d, 3JH–H = 8.0 Hz, 4H), 4.46 (apparent d, J = 6.0 Hz, 4H), 4.36–3.93 (m, 8H), 1.42 (t, 3JH–H = 7.0 Hz, 12H). 13C{1H} NMR (75 MHz, CDCl3): δ (ppm) = 187.13 (weak signal), 134.41 (s), 133.78 (s), 128.99 (s), 128.93 (s), 63.17 (d, 2JC–P = 4.7 Hz), 45.21 (s), 15.98 (d, 3JC–P = 8.1 Hz).
IV: m.p.: 134 °C. IR (KBr disc, ν, cm−1): 3180, 2982, 2905, 2861, 1555, 1461, 1404, 1339, 1251, 1162, 1101, 1045, 959, 906, 820, 801, 669. 31P{1H} NMR (121 MHz, CDCl3): δ (ppm) = 56.77 (s). 1H NMR (301 MHz, CDCl3): δ (ppm) = 9.36 (s, 2H), 7.25–7.07 (m, 8H), 4.45 (apparent d, J = 5.8 Hz, 4H), 4.38–3.95 (m, 8H), 2.38 (s, 6H), 1.44 (t, 3JH–H = 7.1 Hz, 12H). 13C{1H} NMR (75 MHz, CDCl3): δ (ppm) = 186.79 (weak signal), 137.78 (s), 132.74 (s), 129.51 (s), 127.58 (s), 63.08 (d, 2JC–P = 4.8 Hz), 45.77 (s), 21.18 (s), 16.01 (d, 3JC–P = 8.4 Hz).
Results and discussion
Crystal structure description
Single crystals of ligands I/II, and complexes III/IV were obtained from CH3CN, and CH3CN/CH3OH (1:1 v/v) solutions, respectively, in the ambient conditions. All four compounds crystallize in the triclinic P space group, with one molecule in the asymmetric units of I/II and one-half of the molecule in the asymmetric units of III/IV (Fig. S1†). The complete molecules of III and IV are generated by an inversion element.
In these structures, the P atoms have a distorted tetrahedral P(S)(N)(O)2 environment, and the N atom attached to phosphorus is also linked to the carbon atom of a C(S)N moiety. Both these nitrogen atoms in all four structures show almost a planar configuration with the NHC2, NHPC, or NPCNi environments. The bond lengths and angles are in the normal range of analogous structures and according to the atoms involved.54,55 In the coordination compounds III and IV, the nickel atom has a square planar NiS2N2 geometry.
After the deprotonation of the P(S)NHC(S) segment (from ligands to complexes), the phosphorus–nitrogen and carbon–nitrogen bonds are strengthened, while the carbon–sulfur bonds are weakened. The related values in I/II and III/IV (dP–N = 1.6731 (12)/1.6714 (15) Å & 1.647 (2)/1.646 (2) Å, dC–N = 1.3772 (18)/1.376 (2) Å & 1.343 (4)/1.348 (4) Å, dC–S = 1.6839 (14)/1.6807 (18) Å & 1.727 (3)/1.729 (3) Å) show the changes in carbon–sulfur bonds are higher.
In all structures, the intramolecular N–H⋯SP hydrogen bond is made by the NH unit of the X-C6H4CH2NH segment (X = Cl/CH3), forming a 6-membered hydrogen-bonded ring (an S6 graph-set, graph set notations are described in the ref. 56). Typically, this hydrogen bond in structure III is slightly stronger than the associated ligand (I) (according to the V(r) = −0.0122/−0.0104 a.u. and ρ(r) = 0.0201/0.0194 a.u. for III/I). The sulfur atom of the thiocarbonyl group cooperates in the intermolecular N–H⋯S hydrogen bonds in I/II or coordination Ni–S bonds in III/IV. These structural features are also observed in the reported constitutional isomers,33,34 despite the different packing characteristics due to their different molecular conformations.
The conformations of the P(S)NC(S)N segment are described by the S–P–N–C/P–N–C–N/P–N–C–S torsion angles. These torsion angles show ±sc/+sp/+ap conformations in I/II and +sp/+sp/+ap conformations in III/IV (sp = synperiplanar, ap = antiperiplanar, and sc = synclinal). The differences of conformations in ligands and complexes are related to the SP–N–C torsion angles (I/II: 48.44°/−50.77°, III/IV: 13.28°/24.41°), which correspond to an almost flat hydrogen-bonded S6 ring in complexes and higher twisting of the similar ring in ligands. This trend to planarity is a result of the deprotonation of NH units in complexes.
The superposition of coordination structures (III and IV, Fig. 1A) shows a relative mirror image relation (if the difference of Cl/CH3 substituents is ignored). This feature does not appear in the ligand structures (I and II, Fig. 1B), where the orientations of amine and ethyl groups are different.
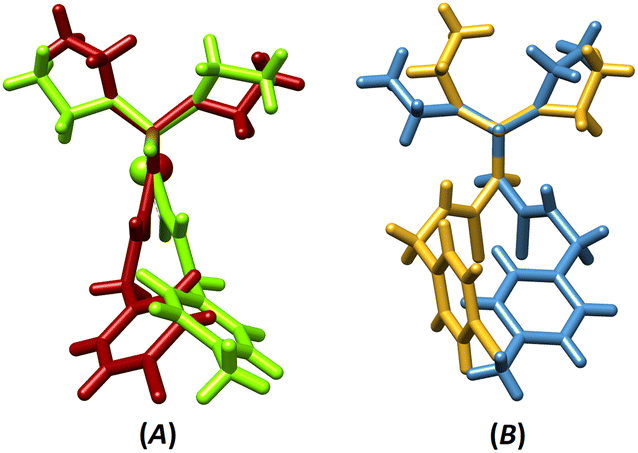 | ||
| Fig. 1 The overlays of (A) III/IV (RMSD = 0.002) and (B) I/II (RMSD = 0.010) with fixing the PSN moieties on each other (for III/IV, one-half of the molecule was considered). | ||
The ESP maps of molecules in crystal structures were generated to make a better image of the potential of each part of molecules for establishing interactions (Fig. 2). Accordingly, in the ligand structures, the highest accumulation of negative ESP is around the CS sulfur atom (the surface energy, Esur = −26.7 and −26.0 kcal mol−1 for I and II). The involvement of the PS sulfur atom in the intramolecular hydrogen bond decreases the negative ESP accumulation (−14.0 and −14.4 kcal mol−1 for I and II). The positive ESP is accumulated on the P-bonded NH groups (the highest Esur = +29.0 and +27.8 kcal mol−1 for I and II), creating a high potential for establishing intermolecular hydrogen bonds.
 | ||
| Fig. 2 ESP surfaces of I–IV with the isovalue 0.0004 a.u. The energies (in kcal mol−1) for the selected points of the surfaces are given at the B3LYP/cc-pVDZ level of theory. | ||
In complexes, coordinating the C–S and P-bonded nitrogen atom to the Ni atom reduces the negative ESP of this sulfur atom, and removes the positive ESP around the nitrogen atom. The most negative ESP is located on one of the ethoxy O atoms (the lowest Esur = −23.6 and −28.2 kcal mol−1 for III and IV), while positive ESP is almost uniformly expanded on all CH units. Thus, interactions are weak in the crystals and molecules do not include suitable sites to form prominent interactions.
The ESP surfaces visualize the differences in the Cl/CH3 behaviors. Negative ESP is distributed as a belt around the Cl atom, and on the σ-hole region, and the absolute value of the negative ESP decreases. This trend is almost reversed for the methyl group. The belt around the CH3 group contains a positive ESP, while by relocating to the region along the σ-bond, the value of the positive ESP decreases.
A remarkable feature in ESP surfaces is the effect of electronegative atoms on the surrounding environment. In all structures, the ESP on the surface of the side of the aryl ring closer to the sulfur atom is more negative than the other side. In I and II, the Esur of near/far faces of the aryl ring to the CS group are equal to −17.3/–10.9, and −22.0/–17.0 kcal mol−1. In III and IV, unlike I and II, the PS group is oriented towards the aryl ring, and the Esur of near/far faces of the aryl ring is equal to −7.7/–4.8, and −13.1/–11.5 kcal mol−1, respectively. In complexes, the cooperation of the electronegative N and S atoms with an oxygen atom situated sterically above them creates a vast region with high negative ESP, which is maximized on the oxygen atom. This region even covers the surroundings of the Ni atom, which individually has a positive charge.
Based on Rozas classification, all of the hydrogen bonds are weak, and positive ∇2ρ(r) and H(r) values of all BCPs indicate the absence of any strong/moderate hydrogen bond.57
In the molecular assembly of I, a dimer synthon (R22(8)) is formed through a pair of N–H⋯SC hydrogen bonds (the most important interaction, V(r) = −0.0112 a.u., ρ(r) = 0.0224 a.u.), which is strengthened by the C–H⋯SC interactions (pair Ia, Fig. 3). Such dimers are connected through two equal C–H⋯O hydrogen bonds (V(r) = −0.0062 a.u., ρ(r) = 0.0089 a.u., pair Ib), and construct a tape in the direction of [110]. Weak C–H⋯N hydrogen bond (V(r) = −0.0037 a.u., ρ(r) = 0.0063 a.u.), C–H⋯O, and C–H⋯SC contacts in pair Ic, C–H⋯π interactions and some weak hydrogen bonds in pair Id, and three C–H⋯S hydrogen bonds in pair Ie (Table 2) link the tapes together to make sheets parallel to the ab-plane (ab-assembly, Fig. 3). The sheets are stacked through π⋯π interaction, along with the C–H⋯Cl, and C⋯Cl in pair If, and C–H⋯SP, and H⋯H interactions in pair Ig, to form a three-dimensional network (Fig. 3B).
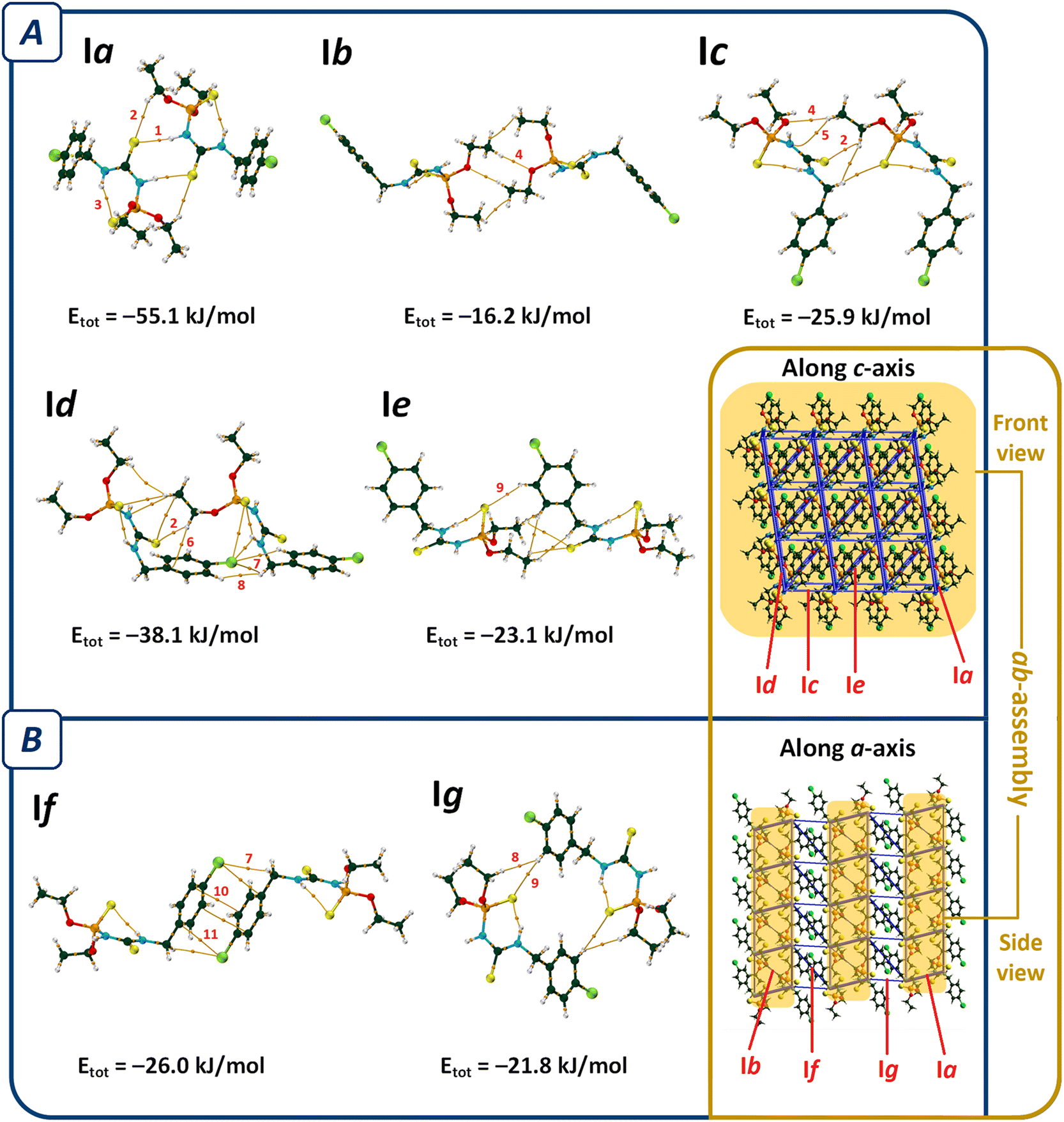 | ||
| Fig. 3 QTAIM diagrams of the most influential molecular pairs (cutoff energy value = −15 kJ mol−1) in the packing of structure I, and two views of the energy framework considering these molecular pairs along the c- and a-axes. The most prominent interactions in each pair are labeled: NH⋯SC (1), CH⋯SC (2), NH⋯SP (3), CH⋯O (4), CH⋯N (5), CH⋯π (6), CH⋯Cl (7), H⋯H (8), CH⋯SP (9), π⋯π (10), and Cl⋯C (11). Total interaction energies (Etot) are given at the bottom of each pair. Section (A) shows pairs contributing to form ab-assembly and section (B) represents pairs that stack ab-assemblies together (atom color codes: P = orange, O = red, N = cyan, C = dark green, Cl = light green, S = yellow, and H = white). | ||
The most important molecular pair in II (pair IIa, Fig. 4) is similar to I, which includes N–H⋯SC hydrogen bonds (V(r) = −0.0109 a.u., ρ(r) = 0.0219 a.u., R22(8) motif), and C–H⋯SC interactions. However, the number of the latter type of interactions is different (in comparison with pair Ia, Table 2).
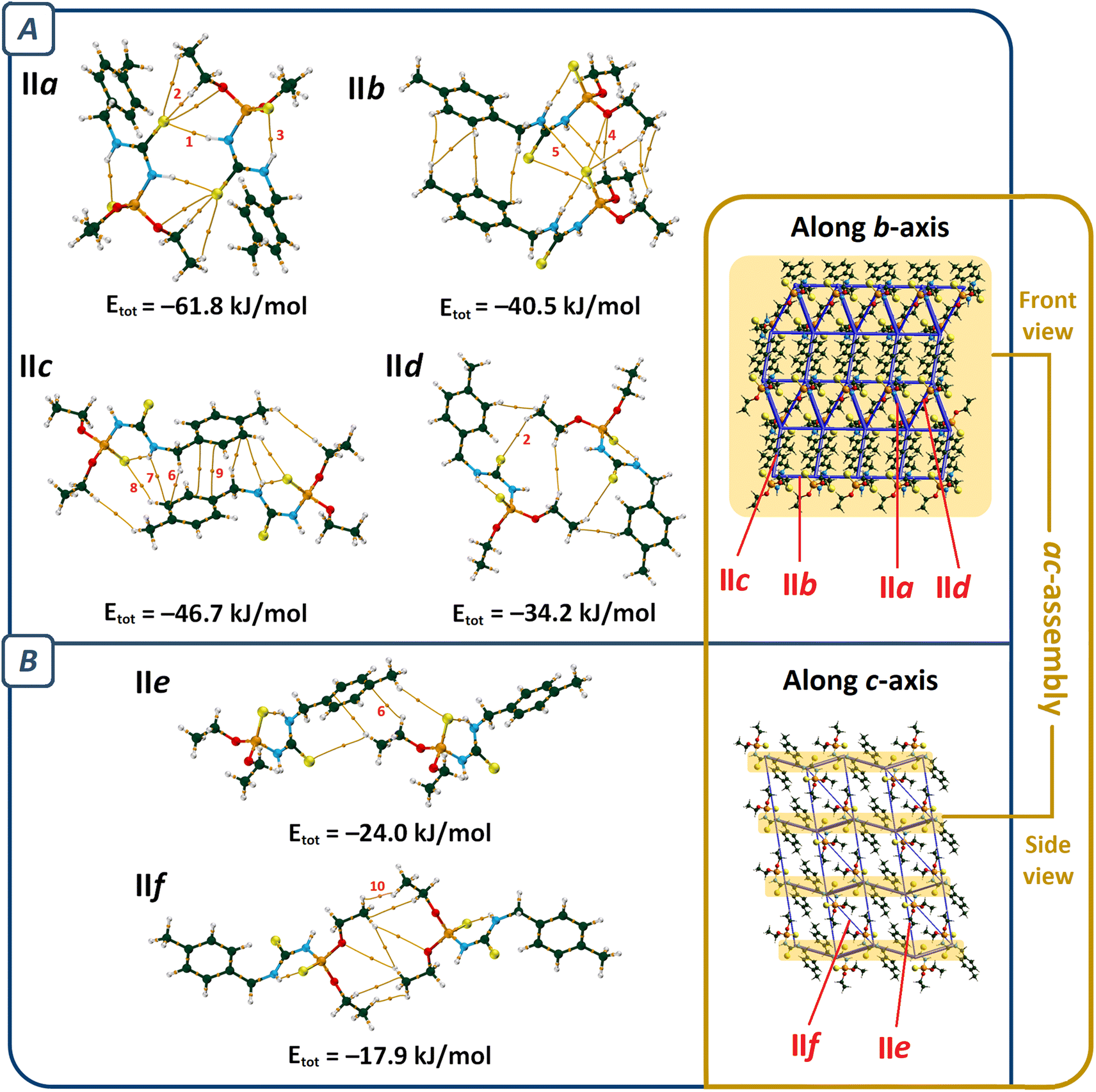 | ||
| Fig. 4 QTAIM diagrams of the most influential molecular pairs (cutoff energy value = −15 kJ mol−1) in the packing of structure II, and two views of the energy framework considering these molecular pairs along the b- and c-axes. Total interaction energies (Etot) are given at the bottom of each pair. The most prominent interactions in each pair are labeled: NH⋯SC (1), CH⋯SC (2), NH⋯SP (3), CH⋯O (4), S⋯N (5), CH⋯π (6), NH⋯π (7), CH⋯SP (8), π⋯π (9), and H⋯H (10). Section (A) shows pairs contributing to form ac-assembly, and section (B) represents pairs that stack ac-assemblies together (atom color codes: P = orange, O = red, N = cyan, C = dark green, S = yellow, and H = white). | ||
Regardless of pair IIa, the molecular assembly in II is significantly different from I. The C–H⋯O hydrogen bond of pair IIb holds the dimers IIa together (V(r) = −0.0073 a.u., ρ(r) = 0.0101 a.u.) to make a one-dimensional assembly along the a-axis, which is reinforced by PS⋯N, PS⋯O, C–H⋯SC, and dihydrogen contacts. The C–H⋯SP hydrogen bond (V(r) = −0.0047 a.u., ρ(r) = 0.0091 a.u.) and the interactions related to the aryl fragment (C–H⋯π, N–H⋯π, and π⋯π) in pair IIc and the C–H⋯SC interactions in pair IId extend the supramolecular architecture to a two-dimensional assembly parallel to the ac-plane (ac-assembly, Fig. 4A). Such symmetry-related sheets are connected through C–H⋯SP, C–H⋯π, and H⋯H interactions (pairs IIe and IIf) to form a three-dimensional supramolecular architecture (Fig. 4B).
The molecular packing maps of structures III and IV have a close similarity (Fig. 5 and 6). In both structures, the most numbers of intermolecular interactions are seen along the a-axis, which include C–H⋯SP, C–H⋯O, C–H⋯Ni, C–H⋯N, and C–H⋯π interactions (pairs IIIa and IVa, Fig. 5). The planar environments at N and Ni atoms and the high electrostatic potential of the [(CNHC(S)NP(S))2Ni] part (Fig. 2) influence the formation of ribbon arrangements shown as green and purple in Fig. 5A. In these pairs, the strongest interactions (based on potential energy density) are the C–H⋯SP (IIIa) and C–H⋯O (IVa) hydrogen bonds with the V(r) values of −0.0035 and −0.0045 a.u., respectively. The intermolecular anagostic C–H⋯Ni interactions are seen in III/IV (V(r) = −0.0032/−0.0027 a.u. and ρ(r) = 0.0062/0.0057 a.u.). The lengths and angles of the C–H⋯Ni interactions in III/IV stand 2.946/3.010 Å and 122.31°/113.05°, respectively, which are in the normal range of such rarely occurred interactions (2.3–3.0 Å and 110–170°).58,59 The ESP surfaces show that the formation of C–H⋯Ni interactions is related to the electrostatic nature caused by the electronegative atoms neighbor to Ni, which make a vast region with negative electrostatic potential and attracts hydrogens.
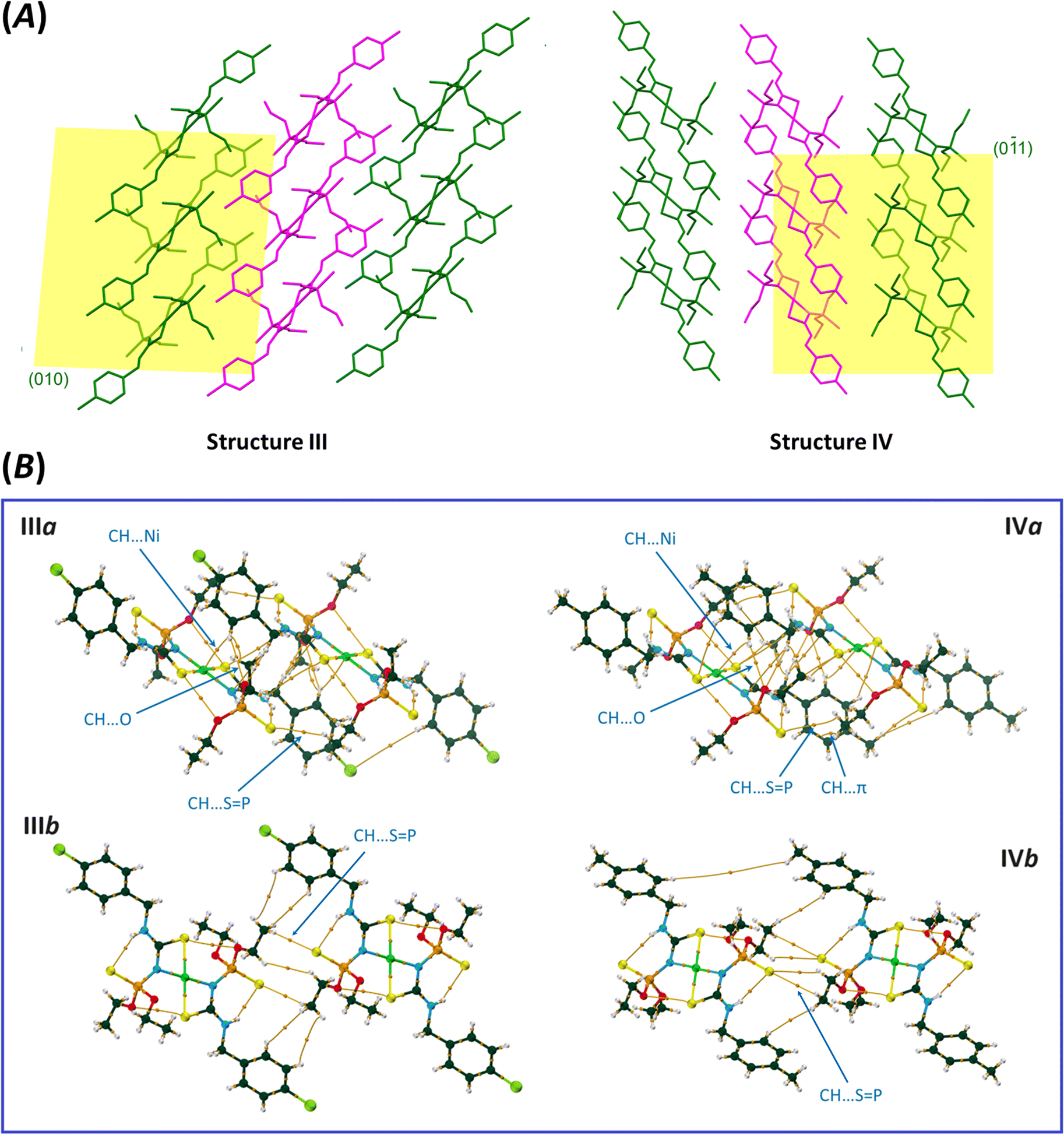 | ||
| Fig. 5 (A) Two-dimensional assemblies formed by the molecular pairs IIIa/IIIb and IVa/IVb. Ribbons, represented by green and purple colors, constructed through the consequence of pairs IIIa and IVa; molecules in the ribbons with different colors interacted together as shown in the pairs IIIb and IVb. (B) QTAIM diagrams of the molecular pairs IIIa/IVa and IIIb/IVb (atom color codes: Ni = green, P = orange, O = red, N = cyan, C = dark green, S = yellow, Cl = light green, and H = white). | ||
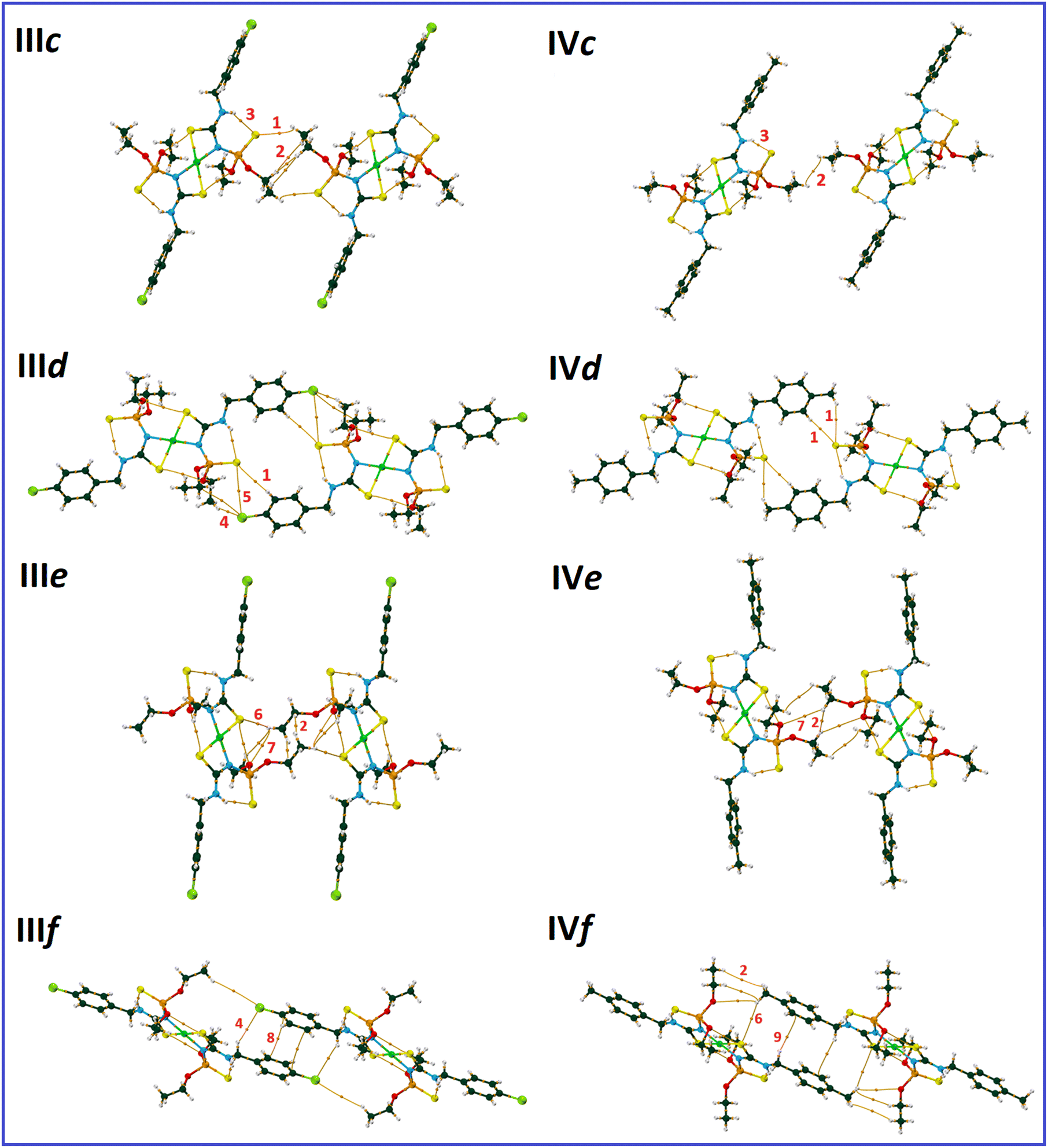 | ||
| Fig. 6 QTAIM diagrams of the molecular pairs IIIc–IIIf and IVc–IVf (atom color codes: Ni = green, P = orange, O = red, N = cyan, C = dark green, S = yellow, Cl = light green, and H = white). The most prominent interactions in each pair are labeled: CH⋯SP (1), H⋯H (2), NH⋯SP (3), CH⋯Cl (4), S⋯Cl (5), CH⋯S–C (6), CH⋯O (7), π⋯π (8), and CH⋯π (9). | ||
The noted assemblies are extended to two-dimensional architectures by the C–H⋯SP hydrogen bonds in the directions parallel to the ac plane for III and (01) plane for IV [Fig. 5, V(r) = −0.0041, −0.0017 (pair IIIb), −0.0018 and −0.0017 a.u. (pair IVb)]. The thiophosphoryl group contributes to some other C–H⋯SP interactions, as represented in pairs IIIc, IIId, and IVd (Fig. 6). The stabilization of III in the b-direction and IV in the c-direction is happened by C–H⋯O and CH⋯HC interactions between two ethoxy groups (pairs IIIe and IVe, V(r) values are given in Table 2) to form three-dimensional networks altogether with the other noted interactions.
The main difference in the structures originates from the interactions related to the 4-chlorobenzyl and 4-methylbenzyl moieties (IIIf and IVf). The CH2C6H4Cl and CH2C6H4CH3 moieties in III and IV take part in similar contacts to the associated ligands, i.e. C–H⋯Cl/π⋯π and two equal C–H⋯π interactions, respectively (Fig. 6).
In pair IVf, there is a slippage between two adjacent arene rings compared to IIIf. This slippage also changes the orientation of the ethoxy groups. The conformation of the P–O–C–C segments are +ap/+ap for III and +ap/+ac (ac = anticlinal) for IV. The different conformations cause a larger distance of chlorine and ethoxy with respect to the methyl and ethoxy groups in associated compounds. However, the chlorine atom attracts one of the terminal hydrogen atoms of the ethoxy group and makes a C–H⋯Cl hydrogen bond (Fig. 6, V(r) = −0.0026 a.u., ρ(r) = 0.0050 a.u.). In the lack of the chlorine atom (in IV), the CH2 unit of benzyl takes part in the C–H⋯π interaction; thus, the closeness of the ethoxy and methyl groups forces the ethoxy group backward.
Considerable differences in the crystal packings of I and II are reflected in the pronounced differences in the volumes of unit cells (801.93 (3) and 822.58 (4) Å3). While III and IV (with more similarities) show comparable volumes (835.66 (5) and 835.21 (6) Å3).
Analysis of contacts using fingerprint plots
The 2D fingerprint plots illustrate the contribution rates of contacts in molecular packing, as shown in Fig. 7. In all structures, H⋯H contacts have more share relative to the other contacts and are developed in a vast region of plots. These contacts relative to total contacts have 41.8% in I and 46.5% in III, with the Cl substituent, and increase to 60.9% in II and 61.2% in IV, with the CH3 group. The smallest corresponding di + de values are marked with label 1 in the figure.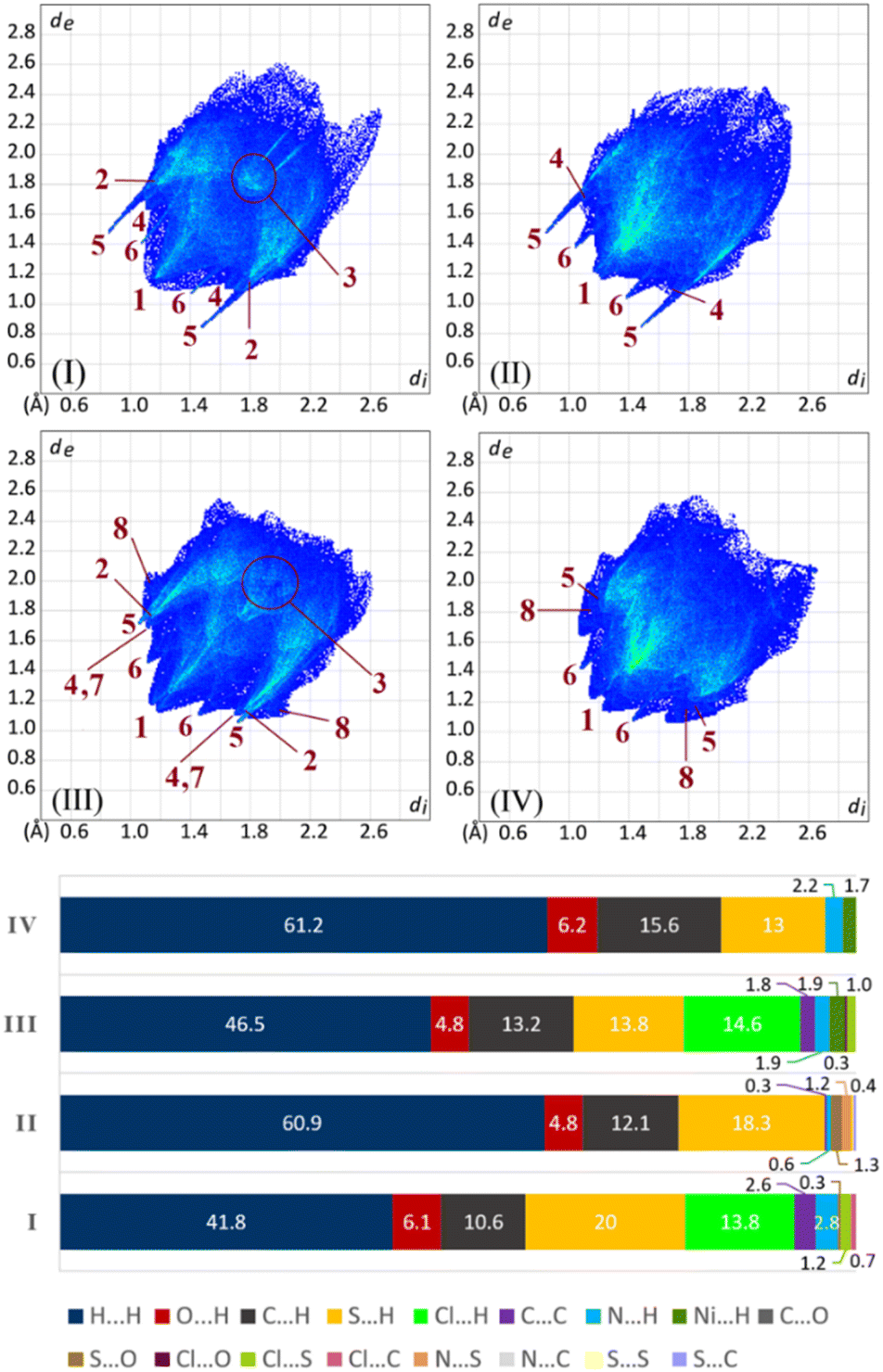 | ||
| Fig. 7 2D fingerprint plots and percentages of contacts in structures I–IV. Locations of corresponding minima di + de values for contacts are shown by the numbers (contact codes: 1 = H⋯H, 2 = Cl⋯H, 3 = C⋯C, 4 = C⋯H, 5 = S⋯H, 6 = O⋯H, 7 = N⋯H, and 8 = Ni⋯H). | ||
The sum of H⋯H% and Cl⋯H% contacts in I and III are comparable with the percentages of H⋯H contacts in II and IV. This value for the structure I (H⋯H% + Cl⋯H% = 55.6%) is remarkably lower than H⋯H% in the structure II (H⋯H% = 60.9%). Part of this difference is due to the π⋯π interactions, which are fewer in II with respect to I (C⋯C% = 0.3% and 2.6%). The environment around the chlorine atom in III does not have any notable difference relative to the methyl group in IV (III: H⋯H% + Cl⋯H% = 61.1%, IV: H⋯H% = 61.2%), which is a result of the almost similar molecular packing.
The contribution of the arene rings in the molecular packing (π⋯π/C–H⋯π) appears as C⋯C and C⋯H contacts (labels 3 and 4). In two structures having chlorine atoms (I and III), the arene rings take part in π⋯π interaction, while in the analogous structures having methyl groups (II and IV), the π⋯π interactions do not matter (C⋯C is absent in IV). This scarcity/absence of π⋯π interactions in II/IV is compensated by raising the C–H⋯π interactions.
In I/II, the H⋯S contacts appear as the tallest spikes (with di + de ≈ 2.3–2.5, label 5) due to the N–H⋯SC hydrogen bonds, while in III/IV, the weak C–H⋯SP hydrogen bonds appear as shorter spikes (with di + de ≈ 2.8–3.0). The H⋯O contacts manifest two small symmetric spikes in all structures (label 6). The C–H⋯N contacts in III, and C–H⋯Ni contacts in III/IV have small contributions, which are introduced with labels 7 and 8, respectively (Fig. 7).
Study of the origin of similarities/differences in packing features
Scrutinizing intermolecular interactions in analogous structures helps to understand the origin of changes in the packing features. Structures III and IV have very slight variances, while I and II differ significantly. On the other hand, the supramolecular networks of III and IV are negligibly affected by the Cl/CH3 substituents. Notably, in pairs IIIa and IVa, with the most contact surfaces (covering 43.2% and 35.9% of the Hirshfeld surfaces of III and IV, respectively) and the majority of interactions, the Cl/CH3 substituents do not have a significant role in the formation of the assemblies. Structures I and II have completely different packing features. Hence, it stands to reason that the change of substituent (Cl/CH3) will determine the difference between the crystal packing features.A common characteristic of the structures reported here (I–IV) is the intramolecular N–H⋯SP hydrogen bonds, similar to most of the analogous structures retrieved from the Cambridge Structural Database.60 In structures I and II, the centrosymmetric hydrogen-bonded dimers are formed through a pair of N–H⋯SC hydrogen bonds. Other packing features are different and heavily depend on the Cl/CH3 substituents.
The results of crystal lattice energy calculations demonstrate that structure I has a much bigger amount of attraction force (Eatt) than structure II (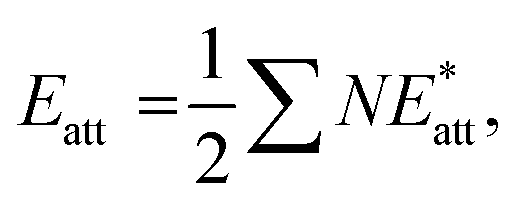 in which
in which 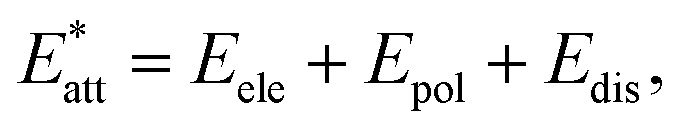 Eatt(I) = −301.2 kJ mol−1, Eatt(II) = −279.4 kJ mol−1), counteracting with repulsion force, despite the crystal lattice energy of II is slightly larger (Elatt(I) = −164.8 kJ mol−1, and Elatt(II) = −166.8 kJ mol−1). The greater attraction force of I (arising from interactions of Cl substituent) results in denser molecular packing (the volumes within Hirshfeld surfaces for I and II equal 392.83 and 403.22 Å3, respectively). In such conditions, investigating the interactions of the altered fragments (aryl groups) in these two structures can shed light on factors affecting changes in molecular packing features.
Eatt(I) = −301.2 kJ mol−1, Eatt(II) = −279.4 kJ mol−1), counteracting with repulsion force, despite the crystal lattice energy of II is slightly larger (Elatt(I) = −164.8 kJ mol−1, and Elatt(II) = −166.8 kJ mol−1). The greater attraction force of I (arising from interactions of Cl substituent) results in denser molecular packing (the volumes within Hirshfeld surfaces for I and II equal 392.83 and 403.22 Å3, respectively). In such conditions, investigating the interactions of the altered fragments (aryl groups) in these two structures can shed light on factors affecting changes in molecular packing features.
In structure I, the most important interactions around the aryl fragment include π⋯π with two near C⋯C bond paths (V(r) = −0.0022 a.u.), a pair of equal C–H⋯Cl interactions (V(r) = −0.0034 a.u., pair If, Fig. 3), and a C–H⋯π interaction (pair Id, V(r) = −0.0033 a.u., Fig. 3). Moreover, the chlorine atom is involved in hydrogen bonding with three near hydrogen atoms (pair Id, Fig. 3).
In structure II, the environment around the aryl fragment exhibits some variations. Despite the presence of the C–H⋯π contacts formed by two hydrogens of the ethyl fragment (pair IIe, V(r) = −0.0025 and −0.0020 a.u., Fig. 4), the behavior of the aryl fragment in interacting with one another differs from the structure I. In this structure, the aryl rings interact by a pair of equal C–H⋯π interactions (pair IIc, Fig. 4), along with a π⋯π interaction with fewer potential energy density (V(r) = −0.0019 a.u. for two observed bond paths).
Thus, the chlorine atom in I with higher electrostatic potential has the propensity to establish weak hydrogen bonds (C–H⋯Cl with V(r) = −0.0034, −0.0025, and −0.0022 a.u. and N–H⋯Cl with V(r) = −0.0021 a.u., pairs Id and If). Especially in pair If, it creates a motif that is stabilized by the cooperation of a π⋯π interaction; while in II, the CH3-substituent with lower electrostatic potential is not involved in such interactions. The interactions of the CH3 substituent are weaker and in lack of Cl-involved hydrogen bonds, the molecules have more freedom to form different competing interactions. Especially in pair IIc, aryl fragments are stacked through C–H⋯π and π⋯π interactions. Hence, the electrostatic differences of the CH3/Cl substituents lead to the different packing features of I and II.
Structures III and IV have nearly identical molecular packing; however, similar to structures I and II, the mutual interactions of aryl fragments are different. These fragments in structures III and IV make contacts almost resembling I and II, respectively. In structure III, dimers are formed through π⋯π and C–H⋯Cl interactions (pair IIIf), while in structure IV, analogous dimers (pair IVf) are constructed through C–H⋯π and H2C–H⋯S–C interactions, and the π⋯π interactions are absent.
Similar supramolecular motifs related to the CH2C6H4Cl fragment (involving C–H⋯Cl and π⋯π interactions) were also observed in the structures with the refcodes HELXAB and HELXEF (with the formulas P(S)(NHCH2C6H4Cl)3 and [P(S)(NHCH2C6H4Cl)3]2Hg2Cl4, respectively).40
As a result, the role of the electrostatic potential is crucial in these variations. Hydrogens in all structures tend to interact with groups having high negative ESP values. In the aryl fragment of structures I and III, the Cl atom has a high negative ESP, but in the absence of the Cl atom in II and IV, the π-electron cloud is more negative, which attracts the hydrogen atoms.
To make a more comprehensive evaluation of the repeatability of these patterns, a survey in the CSD was conducted based on the structures having CH2C6H4-4-Cl and CH2C6H4-4-CH3 fragments. The first search was carried out based on the motifs containing two CH2C6H4-4-Cl moieties interacted with two C⋯C contacts (between two carbons in positions of 2 or 3 of aryl rings) within the specified distance ≤ 3.8 Å. The second search was done based on the motifs including two CH2C6H4-4-CH3 moieties, which have at least two reciprocal C⋯H contacts (between a carbon atom in any position of aryl ring and a hydrogen of CH2 unit) with a distance ≤ 3.3 Å.
Among structures that possess CH2C6H4-4-Cl fragment, 142 queries out of 844 have patterns similar to those in the structures I and III (containing π⋯π interactions with C⋯C distances up to 3.8 Å and C–H⋯Cl hydrogen bond, Fig. 8), which shows 17% probability of repetition of these motifs. Fig. 8 shows the frequency of π⋯π interaction lengths of the specified patterns. Accordingly, the lengths of π⋯π interactions in structures I and III are within the normal range (Cg⋯Cg = 3.640 and 3.644 Å, respectively).
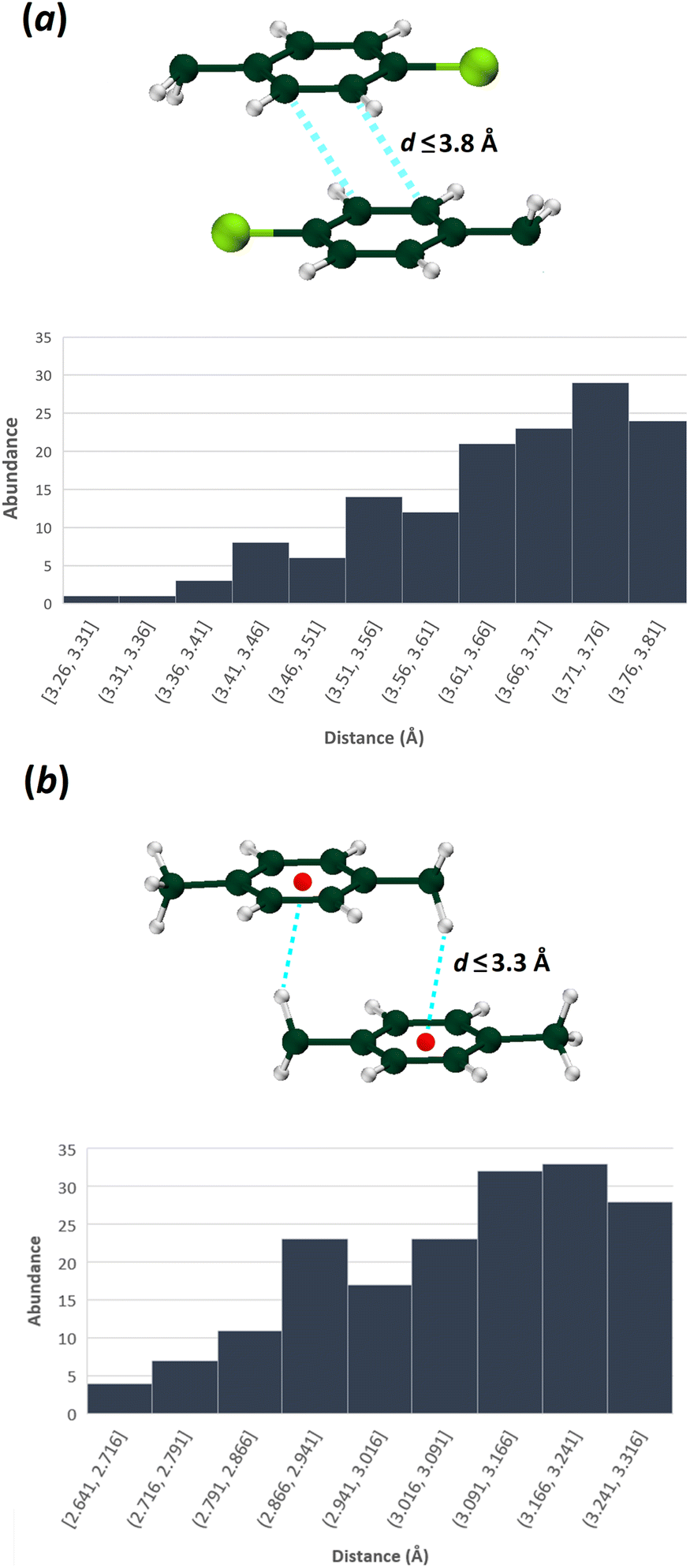 | ||
| Fig. 8 The distribution of indicated interaction lengths between (a) two CH2C6H4Cl, and (b) two CH2C6H4CH3 fragments (CSD version 5.42 updated at September 2021 was used). | ||
For the structures with the CH2C6H4-4-CH3 fragment, 178 queries out of 1482 have patterns similar to those in structures II and IV (i.e., a 12% probability of repeating, applying a criterion of C⋯H distances up to 3.3 Å). As was noted earlier, this motif includes a pair of equal C–H⋯π interactions, with the CH2 moiety as a hydrogen bond donor (Fig. 8). According to the abundance histogram, the C–H⋯π interaction length in the structures II and IV is in the normal range (C–H⋯C = 2.841 and 3.198 Å).
Spectroscopy
In the IR spectra, the bands centered at 3258/3099 cm−1 for I and 3264/3094 cm−1 for II are associated with the NH vibrations. For the related nickel complexes, only one band appears in this region, centered at 3168 cm−1 for III and 3180 cm−1 for IV (due to the deprotonation of one NH unit in the coordinated ligand). For I and II, the bands at 1552 and 1550 cm−1 are related to the SC–N vibration that for III and IV are shifted to 1565 and 1555 cm−1, similar to those reported for analogous ligands/complexes.32
The phosphorus signals appear as a singlet (in 31P{1H} NMR) at 60.57 and 50.80 ppm for I and II and 57.32 and 56.77 ppm for III and IV, respectively (solvent: DMSO-d6 for I and CDCl3 for II, III, and IV).
In the 1H NMR spectra, triplets in the range of 1.27–1.44 ppm and multiplets in the range of 3.93–4.38 ppm correspond to the CH3 and CH2 moieties of ethyl groups. The CH2 moiety of the benzyl group appears as a doublet (within 4.45–4.82 ppm). A typical pattern of the para-substituted arene ring (two doublets) is observed for I–III in the 7.18–7.43 ppm range, but in IV, an overlap signal appears at 7.25–7.07 ppm.
The shapes of NH signals are surprising, as they show the effects of hydrogen bonding, complexation, and solvent. For I (in DMSO-d6), the triplet at 8.55 ppm corresponds to the NH unit that bonded to the CH2 moiety, and the broad peak at 9.33 ppm corresponds to the NH unit of the P(S)NHC(S) segment. For II (in CDCl3), the NH unit bonded to the CH2 moiety appears a triplet, slightly shifted with regard to I (at 8.20 ppm); however, the shift and pattern of the other NH unit are meaningful (at 7.76 ppm as a doublet with J = 9.7 Hz). These differences are related to the solvent effect and different hydrogen bonding in the two solvents. The pattern of the noted triplet in CDCl3 is also different from the triplet in DMSO-d6, where in CDCl3, it tends to broaden, probably due to the remaining intramolecular NH⋯S hydrogen bonding, and the through space phosphorus nucleus effect to this NH unit. In both complexes, only one broad NH signal appears at 9.42 ppm for III and 9.36 ppm for IV (complexes lack the other NH). The broadening is related to the remaining NH⋯S hydrogen bonds in solution, which are stronger in complexes with respect to the corresponding ligands.
In the 13C{1H} NMR spectra of all four compounds, the CH3 and CH2 groups (of ethyl moiety) and the CS group appear as doublets, due to 3J coupling with phosphorus for methyl and 2J couplings for two others. Typically, for I, the related signals appear at 16.13 (3JCP = 7.9 Hz), 64.12 (2JCP = 5.3 Hz), and 181.28 ppm (2JCP = 4.1 Hz). The chemical shifts and coupling constants related to similar carbon atoms in three other compounds show no significant differences, except for low-intensity signals of CS units in complexes. The other aliphatic/aromatic signals appear as singlets at the expected regions, as noted in the Experimental section.
Conclusions
The (C2H5O)2P(S)(NHC(S)NHCH2C6H4X) thiophosphoramides and the [{(C2H5O)2P(S)(NC(S)NHCH2C6H4X)}2Ni] complexes (X = Cl/CH3, I/II and III/IV) were synthesized to study the effect of the Cl/CH3 substituents in the crystal packing. Despite the same crystallization process, the molecular packing features are different in the ligands but are similar in the complexes. These characteristics can be monitored by comparing the degrees of participation of the Cl⋯H and H⋯H contacts with the H⋯H contacts, respectively in compounds including the chlorine substituents (I/III) with analogous CH3-substituted compounds (II/IV) by the Hirshfeld surface analyses. The sum of Cl⋯H% and H⋯H% percentages in I is different from the H⋯H% in II, but nearly equal sum values are observed in III and IV. The other feature is different unit cell volumes of the ligands and similar ones of the complexes.By analyzing crystal structures, the origins of the similarities/differences were investigated, and the following conclusions were drawn: in the ligands, the dimer synthons are formed through classical N–H⋯SC hydrogen bonds, and to connect the dimers some weak interactions compete with each other. Thus, the variations in the molecular packing features are brought about by the Cl and CH3 substituents, which have different capacities for establishing interactions. In structure I, the chlorine atom takes part in the C–H⋯Cl hydrogen bonds, which drives the aryl rings to stack through π⋯π interactions. In structure II with the CH3 substituent, the molecules take part in C–H⋯π and π⋯π interactions.
In the molecular complexes, the planar environment around N and Ni atoms, the electron delocalization in the NCS segment, and intramolecular N–H⋯SP hydrogen bonds cause the planarity and rigidity of the middle part of the molecule (i.e. the [(CNHC(S)NP(S))2Ni] fragment). These geometry requirements and high negative electrostatic potential in this region cause to attract hydrogens and make CH⋯O, CH⋯N, CH⋯Ni, and CH⋯S interactions, which facilitate molecular stacking and the creation of ribbon assemblies. A large part of the interactions is established in these ribbons without the contribution of the Cl/CH3 substituents. Consequently, despite the lack of moderate interactions in III and IV, several numbers of weak interactions contribute to constructing the main feature of supramolecular assembly. Thus, the structures become strikingly alike. The slight differences in the structures arise from the slippage of the CH2C6H4CH3 moieties in IV relative to the CH2C6H4Cl moieties in III, which causes the different conformations of ethoxy groups.
Altogether, it can be concluded that as much as the role of stronger directional interactions (than those established by Cl/CH3 groups) in the expansion of the structure in different dimensions decreases, the influence of the Cl/CH3 exchange on the structural change increases. From the viewpoint of motifs made by aryl groups, the structures III and IV are similar to the corresponding ligands I and II.
The variations observed in the aryl⋯aryl motifs in structures possessing Cl substituent (I and III) in comparison to structures with CH3 substituent (II and IV) may be attributed to the higher negative ESP of the Cl atom relative to the CH3 group. In the presence of the Cl atom, the hydrogens are preferentially attracted to the Cl, as a part of the negative charge of the π-system is transferred to the Cl atom. Thus, the motifs are constructed through CH⋯Cl and π⋯π interactions. In the absence of Cl, the hydrogens are attracted to the π-cloud of the aryl ring to form CH⋯π interactions.
The analysis of structures possessing CH2C6H4Cl and CH2C6H4CH3 moieties retrieved from the CSD show around 17% and 12% repeatability of aryl⋯aryl motifs similar to those observed in I/III and II/IV, respectively.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for I–IV have been deposited at the CCDC under CCDC numbers 2311920–2311923.†Author contributions
Saeed Hosseinpoor (investigation, formal analysis, software, writing – original draft), Mehrdad Pourayoubi (project administration, writing – review & editing), Eliška Zmeškalová (formal analysis (X-ray crystallography), review & editing), Morgane Poupon (formal analysis (X-ray crystallography)).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work by the Ferdowsi University of Mashhad is gratefully acknowledged (Project No. 56293/3). This research was supported by the project TERAFIT – CZ.02.01.01/00/22_008/0004594 and by the Czech Science Foundation project 24-10558S.References
- Q. Zhu and S. Hattori, J. Mater. Res., 2023, 38, 19–36 CrossRef.
- S. M. Woodley and R. Catlow, Nat. Mater., 2008, 7, 937–946 CrossRef PubMed.
- M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19, 1426–1453 CrossRef.
- M. A. Neumann, J. van de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef PubMed.
- C. B. Aakeröy, S. Panikkattu, P. D. Chopade and J. Desper, CrystEngComm, 2013, 15, 3125–3136 RSC.
- M. Pourayoubi, M. Toghraee, J. Zhu, M. Dušek, P. J. Bereciartua and V. Eigner, CrystEngComm, 2014, 16, 10870–10887 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef PubMed.
- Y. Lu, J. Lin, L. Wang, L. Zhang and C. Cai, Chem. Rev., 2020, 120, 4111–4140 CrossRef PubMed.
- W.-L. Guan, J.-F. Chen, J. Liu, B. Shi, H. Yao, Y.-M. Zhang, T.-B. Wei and Q. Lin, Coord. Chem. Rev., 2024, 507, 215717 CrossRef CAS.
- I. Bouabdallah, T. Harit, M. Rahal, F. Malek, M. Tillard and D. Eddike, Acta Chim. Slov., 2021, 68, 718–727 CrossRef CAS.
- C.-H. Kuo, D.-C. Huang, W.-T. Peng, K. Goto, I. Chao and Y.-T. Tao, J. Mater. Chem. C, 2014, 2, 3928–3935 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- O. Loveday and J. Echeverría, Nat. Commun., 2021, 12, 5030 CrossRef CAS PubMed.
- H.-Y. Zhuo, L.-X. Jiang, Q.-Z. Li, W.-Z. Li and J.-B. Cheng, Chem. Phys. Lett., 2014, 608, 90–94 CrossRef CAS.
- S. E. Wheeler and K. N. Houk, J. Chem. Theory Comput., 2009, 5, 2301–2312 CrossRef CAS.
- S. E. Wheeler, Acc. Chem. Res., 2013, 46, 1029–1038 CrossRef CAS PubMed.
- S. E. Wheeler, J. Am. Chem. Soc., 2011, 133, 10262–10274 CrossRef CAS.
- A. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1st edn, 1973 Search PubMed.
- A. Nangia, New J. Chem., 2000, 24, 1049–1055 RSC.
- G. R. Desiraju and J. A. R. P. Sarma, J. Chem. Sci., 1986, 96, 599–605 CrossRef CAS.
- M. R. Edwards, W. Jones, W. D. S. Motherwell and G. P. Shields, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2001, 356, 337–353 CrossRef CAS.
- S. Hosseinpoor, M. Pourayoubi, M. Abrishami, M. Sobati, F. Karimi Ahmadabad, F. Sabbaghi, M. Nečas, M. Dušek, M. Kučeráková and M. Kaur, CrystEngComm, 2023, 25, 2557–2569 RSC.
- A. Singh, A. Ramanan and D. Bandyopadhyay, Cryst. Growth Des., 2011, 11, 2743–2754 CrossRef CAS.
- A. K. S. Romasanta, D. Braga, M. T. Duarte and F. Grepioni, CrystEngComm, 2017, 19, 653–660 RSC.
- S. Ranjan, R. Devarapalli, S. Kundu, S. Saha, S. Deolka, V. R. Vangala and C. M. Reddy, IUCrJ, 2020, 7, 173–183 CrossRef CAS PubMed.
- S. L. Tan and E. R. T. Tiekink, CrystEngComm, 2021, 23, 1723–1743 RSC.
- M. R. Edwards, W. Jones and W. D. S. Motherwell, CrystEngComm, 2006, 8, 545–551 RSC.
- N. K. Nath and A. Nangia, Cryst. Growth Des., 2012, 12, 5411–5425 CrossRef.
- M. Polito, E. D'Oria, L. Maini, P. G. Karamertzanis, F. Grepioni, D. Braga and S. L. Price, CrystEngComm, 2008, 10, 1848–1854 RSC.
- D. A. Safin, M. G. Babashkina, M. Bolte and Y. Garcia, CrystEngComm, 2012, 14, 774–778 RSC.
- D. A. Safin, M. G. Babashkina, M. P. Mitoraj, P. Kubisiak, K. Robeyns, M. Bolte and Y. Garcia, Inorg. Chem. Front., 2016, 3, 1419–1431 RSC.
- K. E. Metlushka, D. N. Sadkova, L. N. Shaimardanova, K. A. Nikitina, K. A. Ivshin, D. R. Islamov, O. N. Kataeva, A. V. Alfonsov, V. E. Kataev, A. D. Voloshina, L. N. Punegova and V. A. Alfonsov, Inorg. Chem. Commun., 2016, 66, 11–14 CrossRef.
- O. N. Kataeva, K. E. Metlushka, Z. R. Yamaleeva, K. A. Ivshin, A. G. Kiiamov, O. A. Lodochnikova, K. A. Nikitina, D. N. Sadkova, L. N. Punegova, A. D. Voloshina, A. P. Lyubina, A. S. Sapunova, O. G. Sinyashin and V. A. Alfonsov, Cryst. Growth Des., 2019, 19, 4044–4056 CrossRef.
- R. C. Luckay, X. Sheng, C. E. Strasser, H. G. Raubenheimer, D. A. Safin, M. G. Babashkina and A. Klein, Dalton Trans., 2009, 4646–4652 RSC.
- I. D. Rojas-Montoya, A. Santana-Silva, V. García-Montalvo, M.-Á. Muñoz-Hernández and M. Rivera, New J. Chem., 2014, 38, 4702–4710 RSC.
- V. Flores-Romero, O. L. García-Guzmán, A. Aguirre-Bautista, I. D. Rojas-Montoya, V. García-Montalvo, M. Rivera, O. Jiménez-Sandoval, M.-Á. Muñoz-Hernández and S. Hernández-Ortega, New J. Chem., 2020, 44, 10367–10379 RSC.
- M. P. Mitoraj, M. G. Babashkina, A. Y. Isaev, Y. M. Chichigina, K. Robeyns, Y. Garcia and D. A. Safin, Cryst. Growth Des., 2018, 18, 5385–5397 CrossRef.
- M. P. Mitoraj, F. Sagan, M. G. Babashkina, A. Y. Isaev, Y. M. Chichigina and D. A. Safin, Eur. J. Org. Chem., 2019, 2019, 493–503 CrossRef.
- M. Khorramaki, M. Abad, V. Darugar, M. Pourayoubi, M. Vakili, M. Nečas, D. Choquesillo-Lazarte, P. V. Andreev and E. S. Shchegravina, Polyhedron, 2022, 228, 116157 CrossRef.
- Agilent, CrysAlis PRO, Agil. Technol. Ltd, Yarnton, Oxfordshire, UK, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- V. Petříček, L. Palatinus, J. Plášil and M. Dušek, Z. Kristallogr. - Cryst. Mater., 2023, 238, 271–282 CrossRef.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef PubMed.
- M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, J. Phys. Chem. Lett., 2014, 5, 4249–4255 CrossRef PubMed.
- M. A. Spackman, CrystEngComm, 2018, 20, 5340–5347 RSC.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1994 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef.
- M. A. Spackman, Cryst. Growth Des., 2015, 15, 5624–5628 CrossRef.
- B. Bankiewicz, P. Matczak and M. Palusiak, J. Phys. Chem. A, 2012, 116, 452–459 CrossRef PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef.
- M. G. Babashkina, D. A. Safin, M. Bolte, M. Srebro, M. Mitoraj, A. Uthe, A. Klein and M. Köckerling, Dalton Trans., 2011, 40, 3142–3153 RSC.
- K. Metlushka, A. Tufatullin, L. Shaimardanova, D. Sadkova, K. Nikitina, O. Lodochnikova, O. Kataeva and V. Alfonsov, Heteroat. Chem., 2014, 25, 636–643 CrossRef.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef PubMed.
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154–11161 CrossRef.
- G. Rajput, V. Singh, A. N. Gupta, M. K. Yadav, V. Kumar, S. K. Singh, A. Prasad, M. G. B. Drew and N. Singh, CrystEngComm, 2013, 15, 4676–4683 RSC.
- M. P. Mitoraj, M. G. Babashkina, K. Robeyns, F. Sagan, D. W. Szczepanik, Y. V. Seredina, Y. Garcia and D. A. Safin, Organometallics, 2019, 38, 1973–1981 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2311920–2311923. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05281a |
| This journal is © The Royal Society of Chemistry 2024 |