DOI:
10.1039/D4RA04633A
(Paper)
RSC Adv., 2024,
14, 26494-26504
An efficient and simple approach for synthesizing indazole compounds using palladium-catalyzed Suzuki–Miyaura cross-coupling†
Received
25th June 2024
, Accepted 15th August 2024
First published on 22nd August 2024
Abstract
A series of indazole derivatives (6a–6i and 7a–7i) has been synthesized using Suzuki Miyaura cross-coupling with a palladium catalyst from readily available starting materials. An efficient and reliable methodology was employed for the synthesis, and the compounds were thoroughly characterized using 1H NMR, 13C NMR, FT-IR, and HRMS analysis to confirm their structural integrity and purity. Density function theory (DFT) computation identified four compounds (6g, 6h, 7g, and 7h) with significant energy band gaps. Additionally, the molecular electrostatic potential study highlighted the distinct electrical characteristics of these indazole molecules. Subsequent molecular docking investigations were carried out using the AUTODOCK method with two separate protein data bank (PDB) structures (6FEW, 4WA9) involved in renal cancer pathways. The results showed that eight substances PDB: 6FEW (6g, 6h, 7g, and 7h) and PDB: 4WA9 (6a, 6c, and 7c, 7g) had the highest binding energies, indicating their potential as therapeutic agents for treating kidney cancer.
Introduction
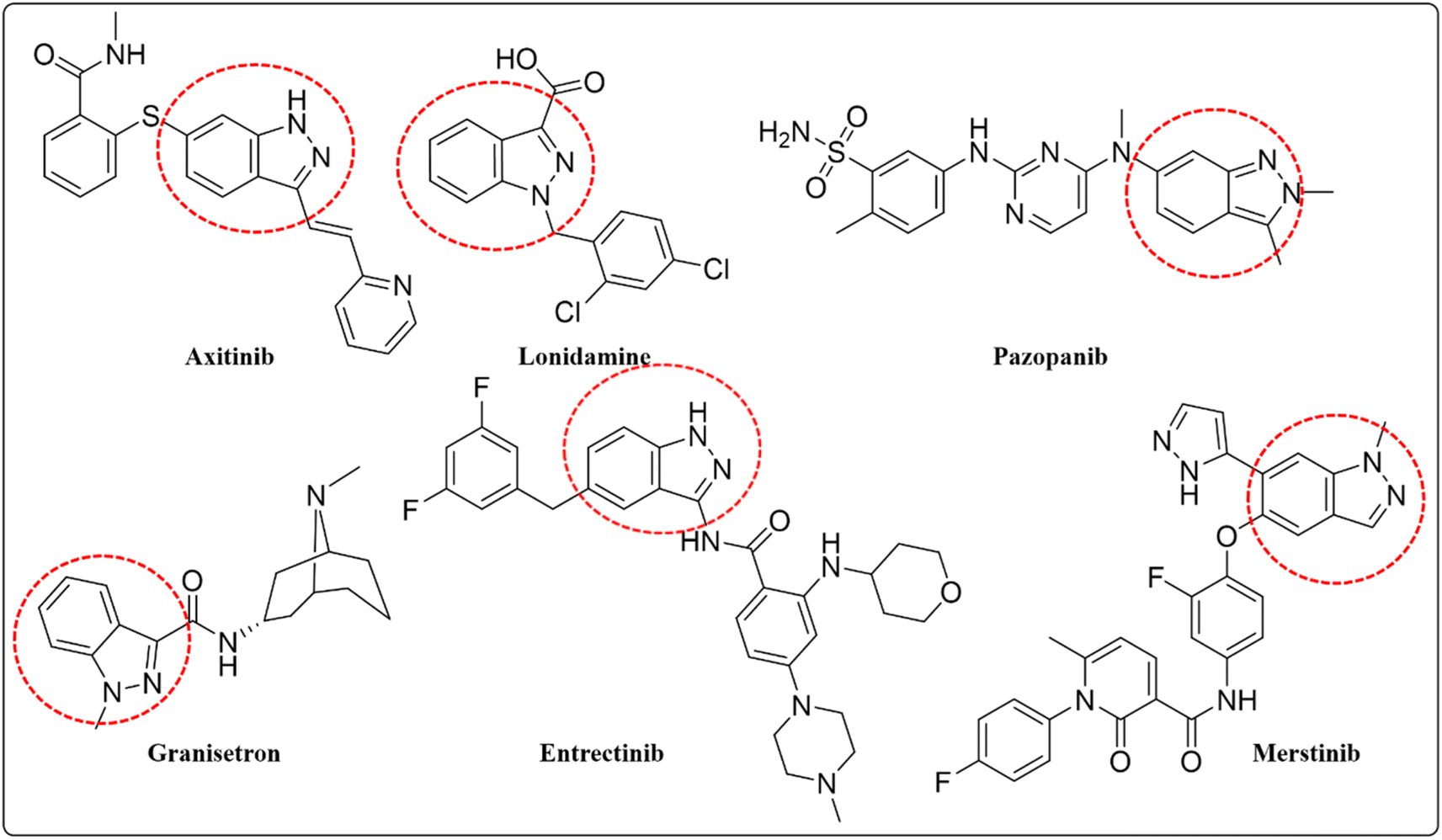
Indazoles are important nitrogen-containing heterocyclic compounds; consisting of a ten-electron aromatic molecule with a pyrazole and benzene ring.1 Direct radical addition is effective at the C3 position of indazole such as alkylation, arylation, benzylation, amination, sulfonation, trifluoromethylation, and phosphorylation.2 Indazole is a suitable pharmacophore with distinct physiological and pharmacological activities and it can be used as a bio-isosteric alternative for other heterocycle structures such as indole and benzimidazole.3 Numerous natural and synthetic medicinal chemicals are produced from indazoles, including axitinib lonidamine, granisetron, and benzydamine (Fig. 1). Axitinib is a powerful tyrosine kinase inhibitor used to treat renal cell cancer. Lonidamine is a hexokinase inhibitor, commonly used as a chemotherapeutic drug renowned for its antitumor characteristics. Granisetron is a specific 5-HT-3 antagonist, is used as an antiemetic in cancer radiotherapy. Benzydamine is also used for its anti-inflammatory and analgesic properties.4 The in vitro and in vivo anti-cancer effectiveness of indazole compounds produced as kinase inhibitors has been intensively studied. For example, the multikinase inhibitor pazopanib (Fig. 1), the c-met inhibitor merestinib (Fig. 1), and the tropomyosin receptor kinase/proto-oncogene tyrosine-protein kinase ROS1/anaplastic lymphoma kinase inhibitor entrectinib (Fig. 1) are all authorized for the treatment of various cancer types.5–8 Cancer is characterized as unexpected or aberrant cell growth (neoplasia) produced by a variety of metabolic abnormalities in cell physiology. Metabolic problems have existed for many decades, and scientists are working to create perfect management or entire irradiation therapy remains a difficulty and is still being investigated today.9 Globocan, the worldwide cancer research agency, estimated 19.3 million new cancer cases and 10 million cancer deaths in 2020. Cancer is one of the leading causes of mortality globally. Females account for more breast cancer patients (11.7%), whereas males account for the bulk of lung cancer cases (18%). Other common cancers include colorectal (9.4%), liver (8.3%), and prostate (7.3%). In 2020, a high number of new cases of kidney cancer were projected. Renal cell carcinoma is responsible for 90% of all kidney cancer occurrences globally. Therefore there is a need to develop novel anticancer medications with low side effects and good bioavailability.10,11 Heterocyclic organic chemistry has spurred interest in its potential biological, agricultural, and industrial applications. Indazole heterocycle molecule is a functional substituent and scaffold that has been investigated for its potential as a kinase inhibitor in cancer treatment. It possesses pharmacological properties12 such as including anti-tumor,13 anti-HIV,14 anti-inflammatory,15 anti-depressant,16 anti-leukemia,17 anti-convulsant,18 anti-arthritic,19 anthelmintic,20 anti-diabetic,21 and antipyretic. Due to its effectiveness, many anticancer drugs utilize indazole as a key structural component.22 The Suzuki–Miyaura cross-coupling technique is used to synthesize indazole compounds, which is a highly effective method of forming the C–C bond. This technique involves, reacting an aryl halide with organoboronic acid using palladium as a catalyst. The synthesis process uses palladium (Pd) precursors (salts).23 The most efficient method of creating new indazole compounds is by using the Pd (0) to Pd(II) cycle. This involves treating an aryl halide with Pd (0) in an oxidative manner to create a Pd(II) intermediate. This intermediate is then trans-metalated and withdrawn reductively to produce the active Pd (0) and the corresponding product.24
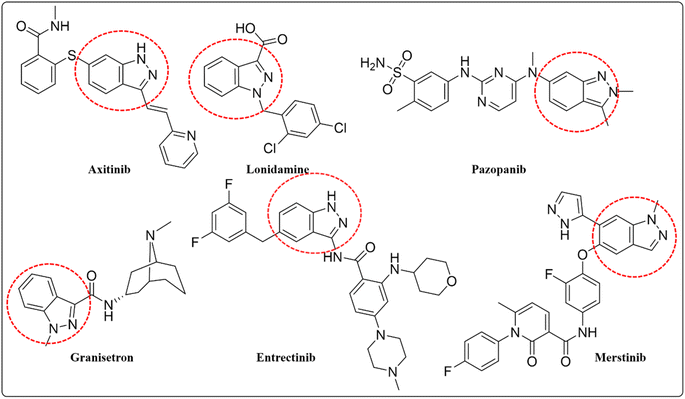 |
| | Fig. 1 Commercially available indazole drugs. | |
Results and discussion
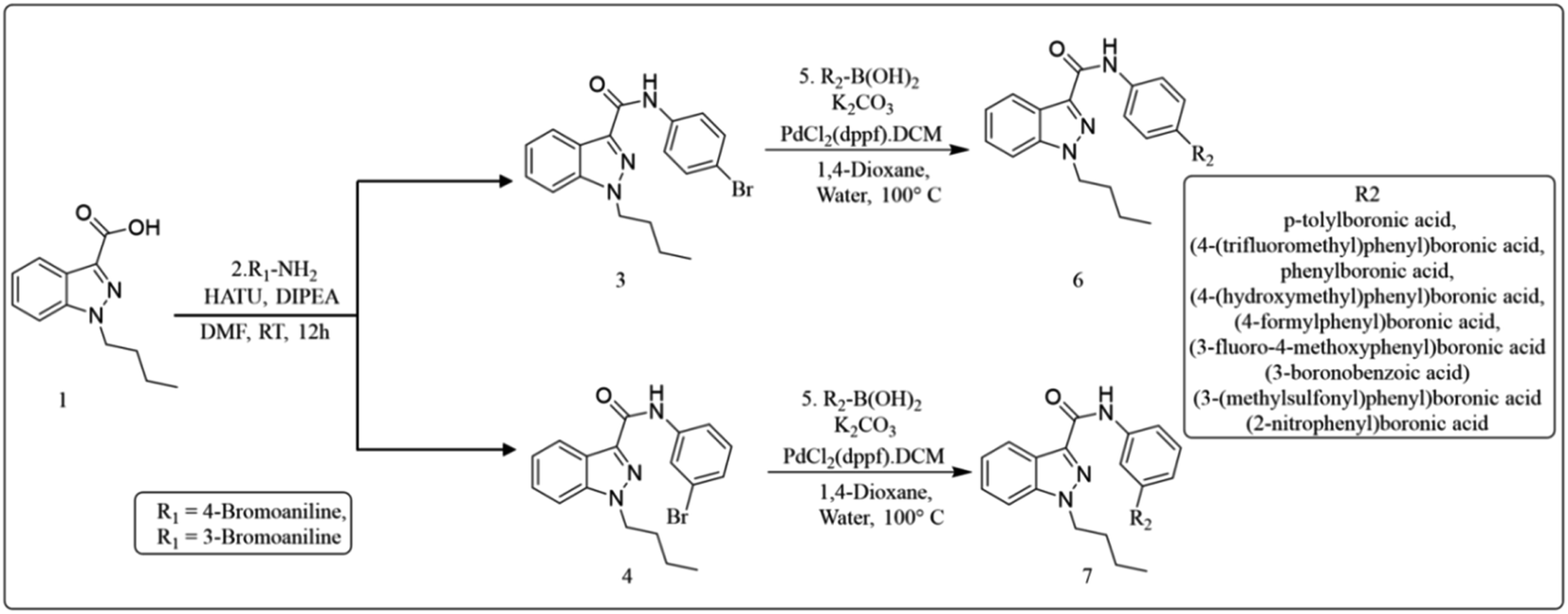
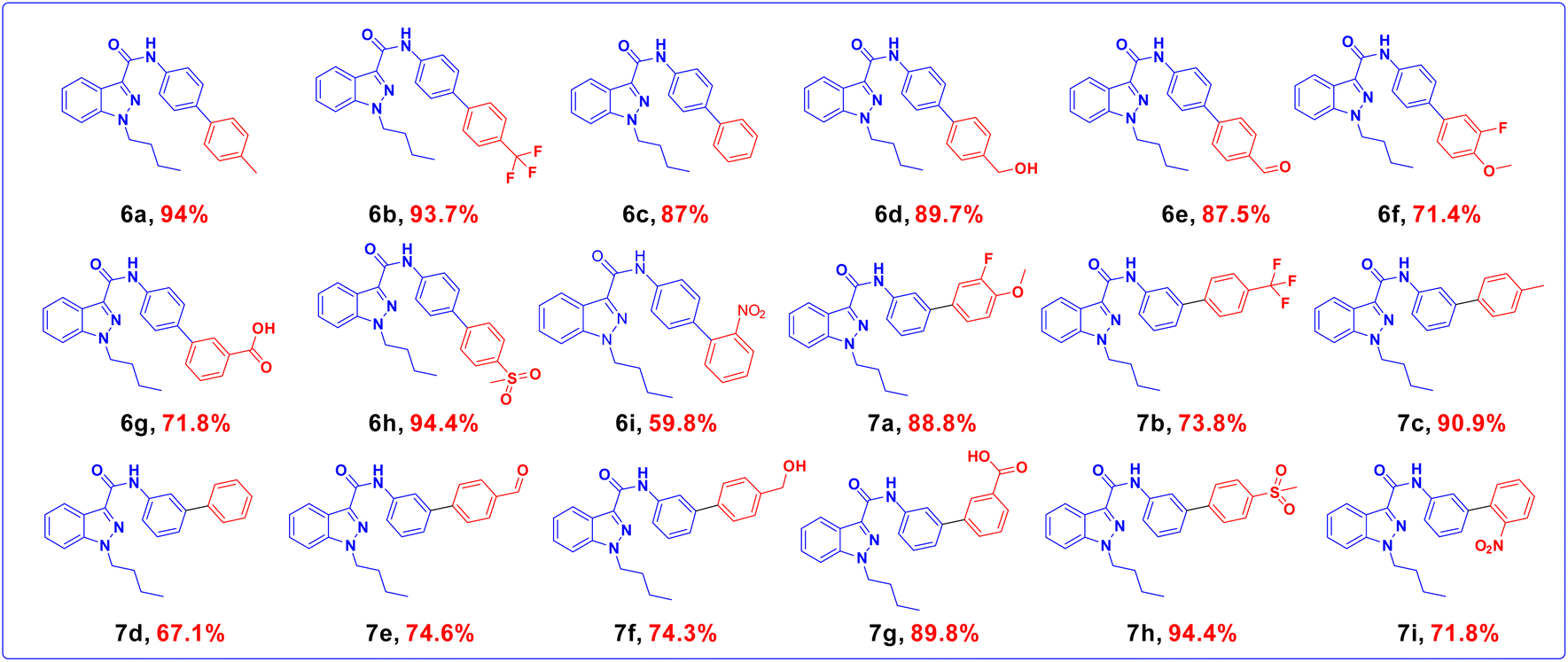
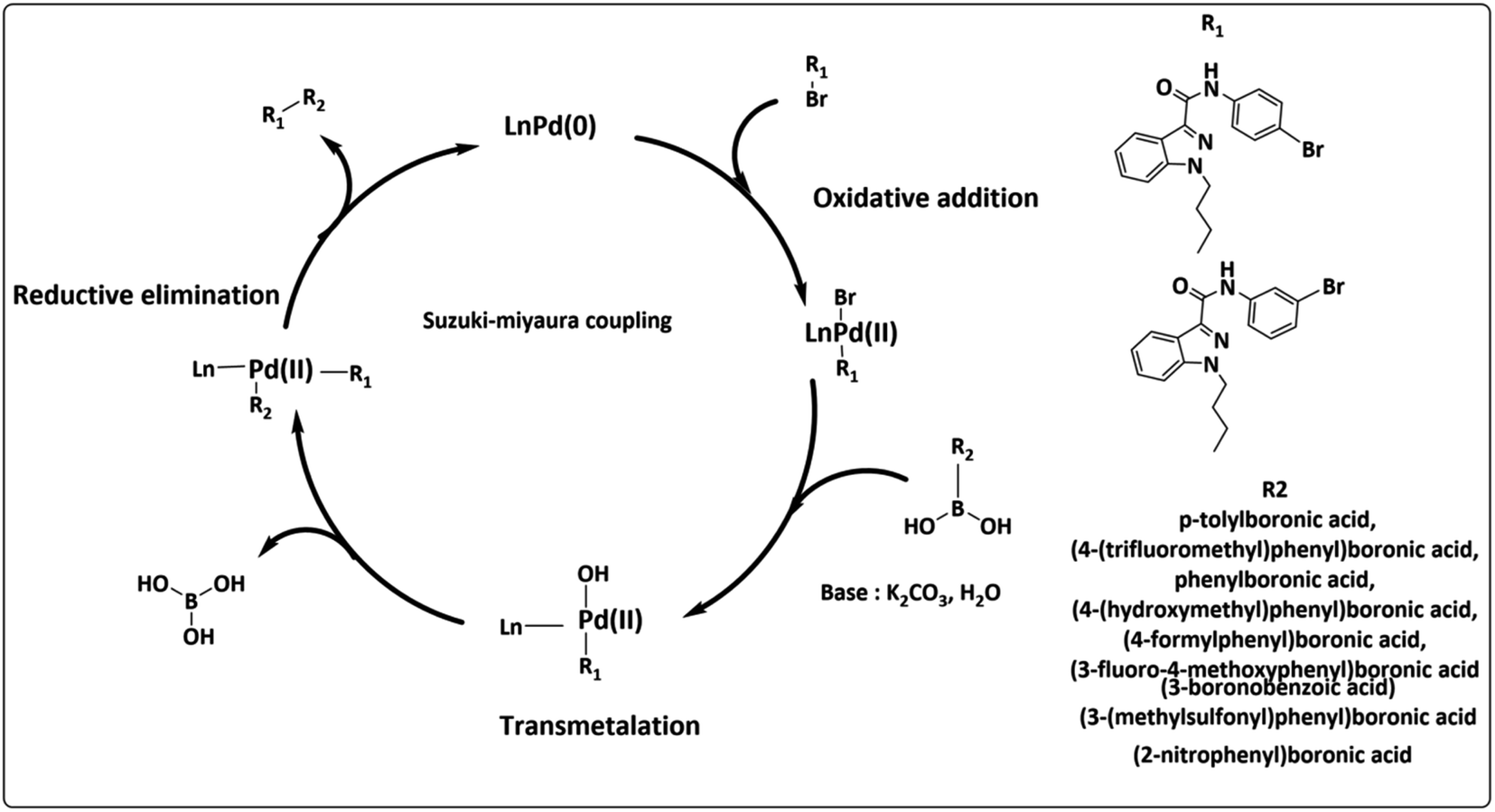
The widely accessible desired target molecules N-(4-bromophenyl)-1-butyl-1H-indazole-3-carboxamide (6) and N-(3-bromophenyl)-1-butyl-1H-indazole-3-carboxamide (7) were synthesized using acid amide cross-coupling. The synthesis involved employing HATU (hexafluorophosphate azabenzotriazole tetramethyl uronium) amide coupling reagent and DIPEA (N,N-diisopropylethylamine) in DMF at room temperature for 12–16 hours. After the reaction was completed, as confirmed by TLC using 20% ethyl acetate/hexane as eluent, the reaction mixture was quenched with ice-cold water to precipitate solid, which was then dried under a hot-air oven at 60–70 °C for 2 hours to obtain off-white solids of (6) and (7). Suzuki–Miyaura coupling of 6 and 7 with various organoboronic acids was employed to develop novel indazole compounds (Fig. 2). Optimization studies are detailed in Table 1. The reaction involved using Bromo-indazole carboxamide (0.404 mmol), boronic acid (1.212 mmol), and K2CO3 (1.212 mmol) as bases in PdCl2(dppf)·(DCM) (0.020 mmol), water, and 1,4-dioxane/water were used as solvents at 100 °C for 12 hours. TLC (20% ethyl acetate/hexane) confirmed the completion of the reaction, and the reaction mixture was filtered through a Celite pad under a high vacuum. The filtrate was then dissolved in ethyl acetate, and extracted (2 × 50 mL) with water (50 mL). The organic layer was dried over sodium sulphate under decreased pressure to produce crude, which was refined using column chromatography (Rf = 0.5, 15% ethyl acetate/hexane). This method was used to generate all of the derivatives given in (Fig. 3), and (Fig. 4) depicts a plausible mechanism for the Suzuki–Miyaura coupling.
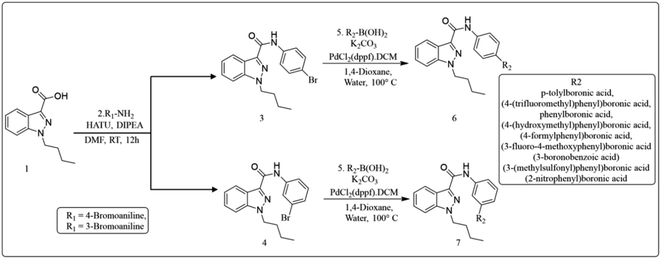 |
| | Fig. 2 Procedure for preparation of indazole derivatives by Suzuki reaction. | |
Table 1 Optimization studies
| Entry |
Catalyst |
Base |
Solvent |
Yield % |
| 1 |
PdCl2(dppf)·DCM |
K2CO3 |
THF/water |
35% |
| 2 |
PdCl2(dppf)·DCM |
Cs2CO3 |
THF/water |
30% |
| 3 |
PdCl2(dppf)·DCM |
Na2CO3 |
THF/water |
40% |
| 4 |
PdCl2(dppf)·DCM |
DIPEA |
THF/water |
Trace |
| 5 |
PdCl2(dppf)·DCM |
K2CO3 |
Toluene/water |
30% |
| 6 |
PdCl2(dppf)·DCM |
Cs2CO3 |
Toluene/water |
25% |
| 7 |
PdCl2(dppf)·DCM |
Na2CO3 |
Toluene/water |
30% |
| 8 |
PdCl2(dppf)·DCM |
DIPEA |
Toluene/water |
Trace |
| 9 |
PdCl2(dppf)·DCM |
K2CO3 |
1,4-Dioxane/water |
94.4% |
| 10 |
PdCl2(dppf)·DCM |
Cs2CO3 |
1,4-Dioxane/water |
60% |
| 11 |
PdCl2(dppf)·DCM |
Na2CO3 |
1,4-Dioxane/water |
65% |
| 12 |
PdCl2(dppf)·DCM |
DIPEA |
1,4-Dioxane/water |
Trace |
| 13 |
PdCl2(dppf)·DCM |
K2CO3 |
DMF/water |
27% |
| 14 |
PdCl2(dppf)·DCM |
Cs2CO3 |
DMF/water |
22% |
| 15 |
PdCl2(dppf)·DCM |
Na2CO3 |
DMF/water |
30% |
| 16 |
PdCl2(dppf)·DCM |
DIPEA |
DMF/water |
Trace |
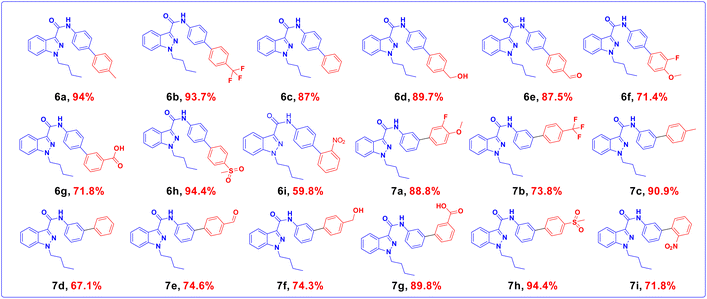 |
| | Fig. 3 Indazole derivatives from 6a–6i, and 7a–7i. | |
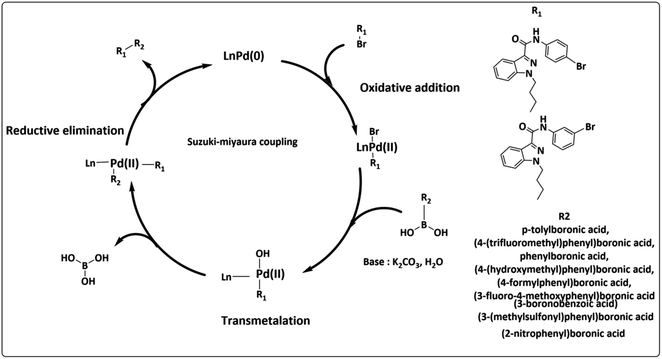 |
| | Fig. 4 Plausible mechanism for Suzuki–Miyaura coupling. | |
DFT studies
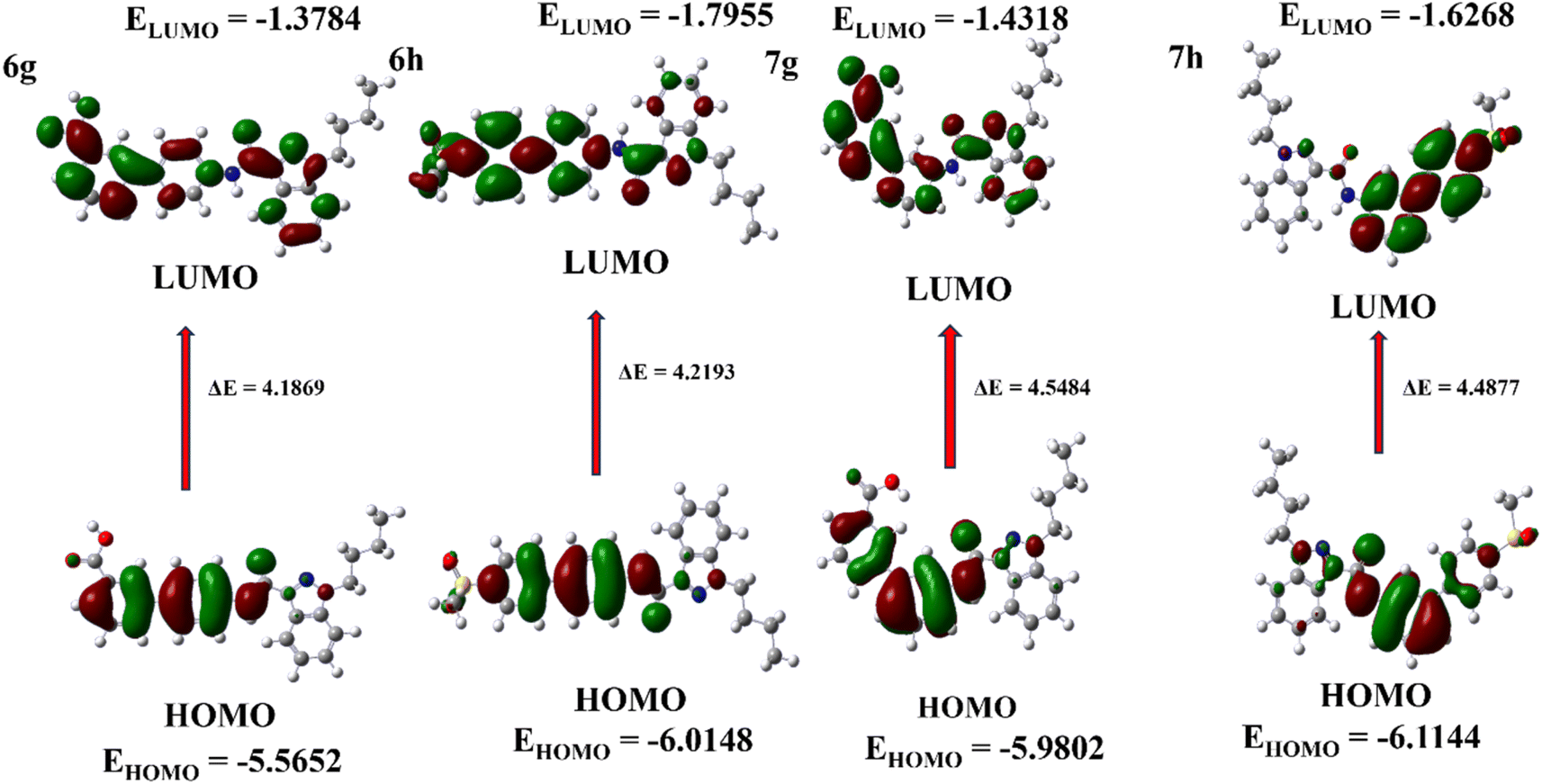
Theoretical investigation was conducted using density functional theory (DFT) calculations, and indazole derivatives were modelled in the Gaussian 6.1 software using the base set B3LYP/6-31G(d,p). The Frontier Molecular Orbital (FMO) is a type of molecular orbital described in ESI.†25–27 The HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) were calculated using the DFT technique. The EHOMO (HOMO energy) values indicate that the molecule donates electrons to the acceptor molecule, while the ELUMO (LUMO energy) values suggest that the molecule readily accepts electrons from the donor molecule.28 All derivatives were calculated using the DFT technique. The four compounds with the highest energy values are 6g, 6h, 7g, and 7h, and (Fig. 5) displays the frontier orbital (FMO) of these compounds. To calculate the energy difference between HOMO and LUMO values, the formula ΔE = ELUMO − EHOMO can be applied.29 The ESI† contains estimated DFT values for other indazole derivatives. HOMO–LUMO energies were used to calculate electronic parameters, including ionization potential (I), electron affinity (A), and global descriptors such as absolute electronegativity (χ), chemical potential (μ), hardness (η), softness (S), and electrophilicity index (ω) of electron-donating power (ω−) and electron-accepting power (ω+),30,31 at the 6-31G(d,p) level, with the parameters listed in Table 2. The ESI† demonstrates the electronic characteristics of the indazole compounds.
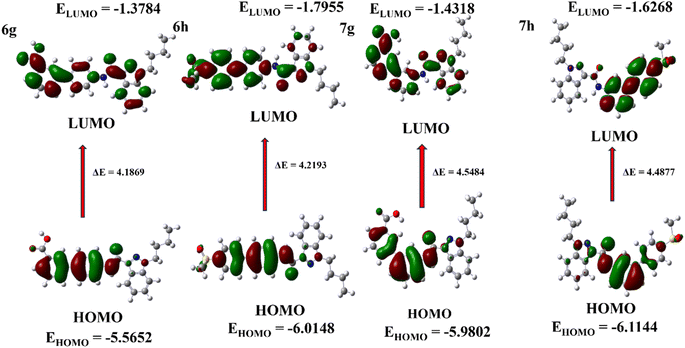 |
| | Fig. 5 Frontier molecular orbitals and HOMO–LUMO energy level diagram of 6g, 6h, 7g, and 7h. | |
Table 2 Electronic parameters of indazole derivatives for 6g, 6h, and 7g, 7ha
| S. no. |
HOMO (eV) |
LUMO (eV) |
ΔE = ELUMO − EHOMO (eV) |
I = −EHOMO (eV) |
A = – ELUMO (eV) |
χ = (I + A)/2 (eV) |
μ = –χ (eV) |
η = (I − A)/2 (eV) |
S = 1/η (eV) |
ω = μ2/2 η |
ω− = (3I + A)2/16(I − A) |
ω+ = (I + 3A)2/16(I − A) |
| HOMO = highest occupied molecular orbital, LUMO = lowest unoccupied molecular orbital, I = ionisation, A = electron affinity, χ = electronegativity, μ = potential efficiency, η = hardness, S = softness, ω = electrophilicity, ω− = e donating power, ω+ = e accepting power. |
| 6g |
−5.5652 |
−1.3784 |
4.1869 |
5.5652 |
1.3784 |
3.4718 |
−3.4718 |
2.0934 |
0.4777 |
12.6165 |
85.4837 |
24.6230 |
| 6h |
−6.0148 |
−1.7955 |
4.2193 |
6.0148 |
1.7955 |
3.9051 |
−3.9051 |
2.1096 |
0.4740 |
16.0862 |
103.7999 |
34.2789 |
| 7g |
−5.9802 |
−1.4318 |
4.5484 |
5.9802 |
1.4318 |
3.7060 |
−3.7060 |
2.2742 |
0.4397 |
15.6177 |
106.6871 |
30.0165 |
| 7h |
−6.1144 |
−1.6268 |
4.4877 |
6.1144 |
1.6268 |
3.8706 |
−3.8706 |
2.2438 |
0.4457 |
16.8079 |
111.8555 |
33.9051 |
Molecular electrostatic potential
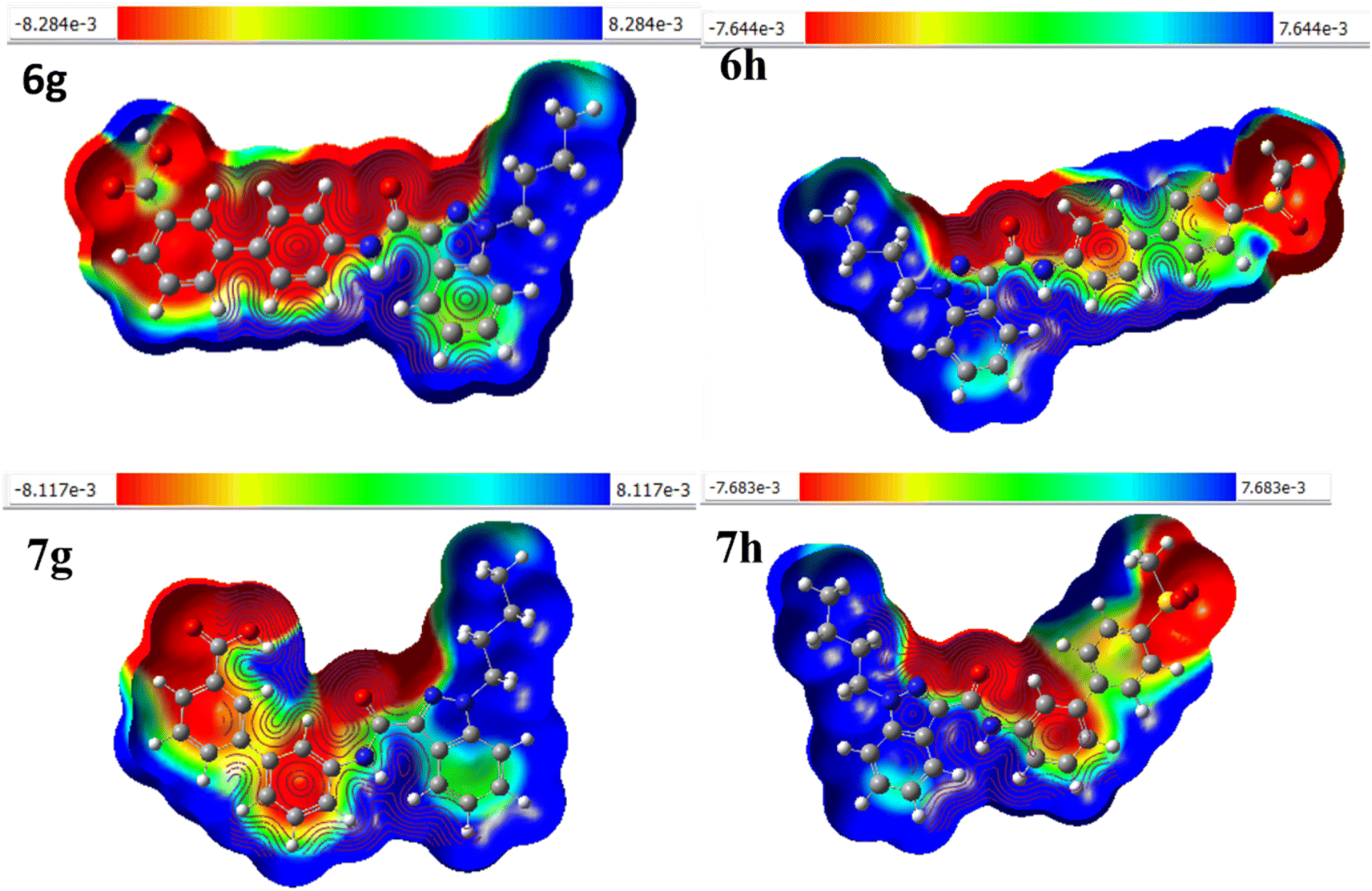
The molecular electrostatic potential (MEP) can be used to predict a molecule's reactivity to nucleophilic and electrophilic attacks. It is based on the molecule's electrical properties such as dipole moment, electronegativity, atomic charges, and so on. MEP is particularly useful for understanding electrophilic and nucleophilic reactions and hydrogen bonding. In this study, the MEP surface analysis of the compound was performed using DFT calculations with the optimized structure and the B3LYP/6-31G(d,p) basis set.32–34 The analysis revealed the electrophilic (positive) and nucleophilic (negative) attack regions on molecules 6g, 6h, 7g, and 7h (Fig. 6). shows the electron densities of 6g (−8.284e negative and 8.284e positive site), 6h (−7.664e negative and 7.664e positive site), 7g (−8.117e negative site and +8.117e positive site), and 7h (−7.683e negative site and 7.683e positive site), representing the compounds' mapped electrostatic potential surface. Different colours represent different electrostatic potential levels, as well as zones of positive, negative, and neutral potential. Blue signifies an electrophilic site, red symbolizes a nucleophilic site, and green indicates an area with a neutral electrostatic potential. Nucleophilic sites are found on oxygen, nitrogen, and sulfur atoms, while electrophilic sites are on hydrogen atoms. As a result, nucleophilic and electrophilic molecules tend to prefer locations with lower electronegative potential and higher electrostatic potential.
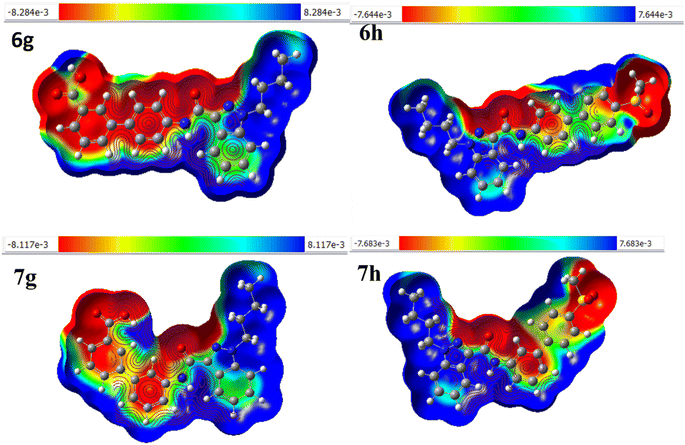 |
| | Fig. 6 The molecular electrostatic potential surface of 6g, 6h, 7g, and 7h. | |
Molecular docking
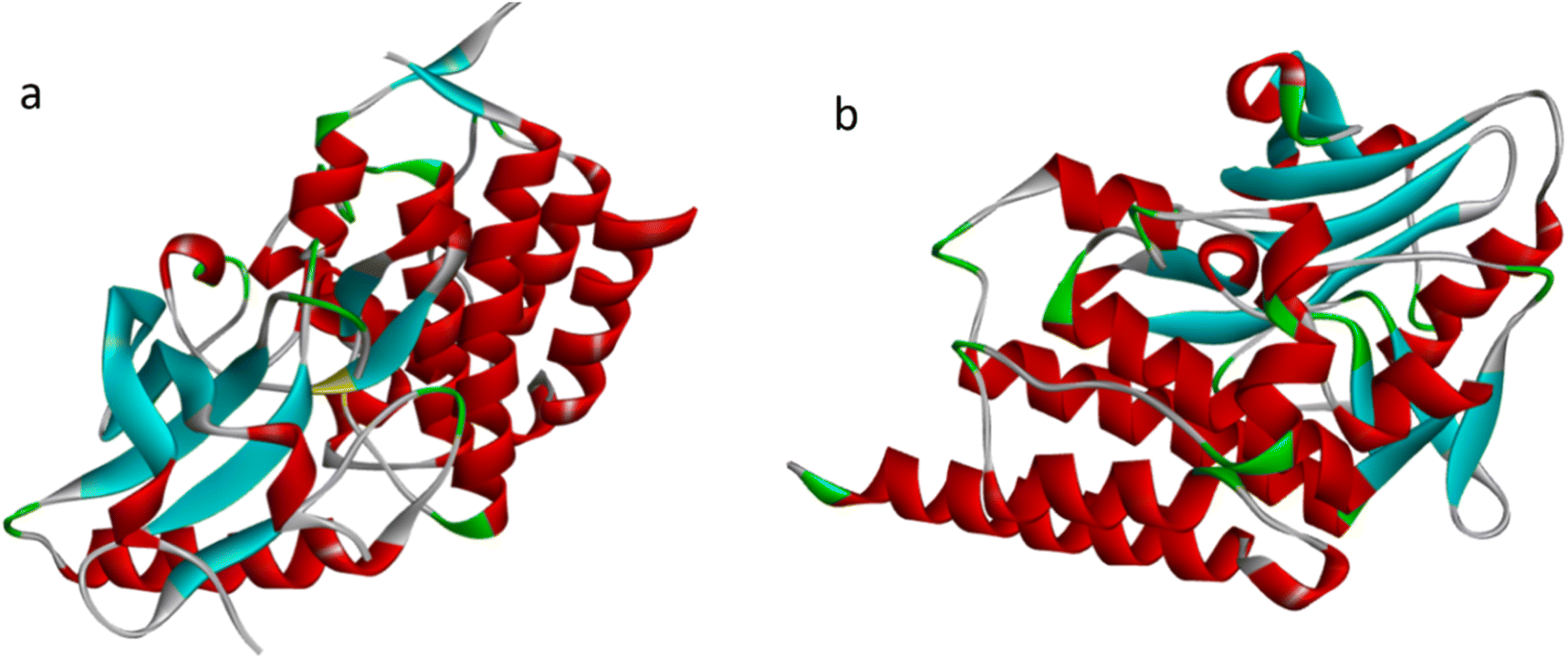
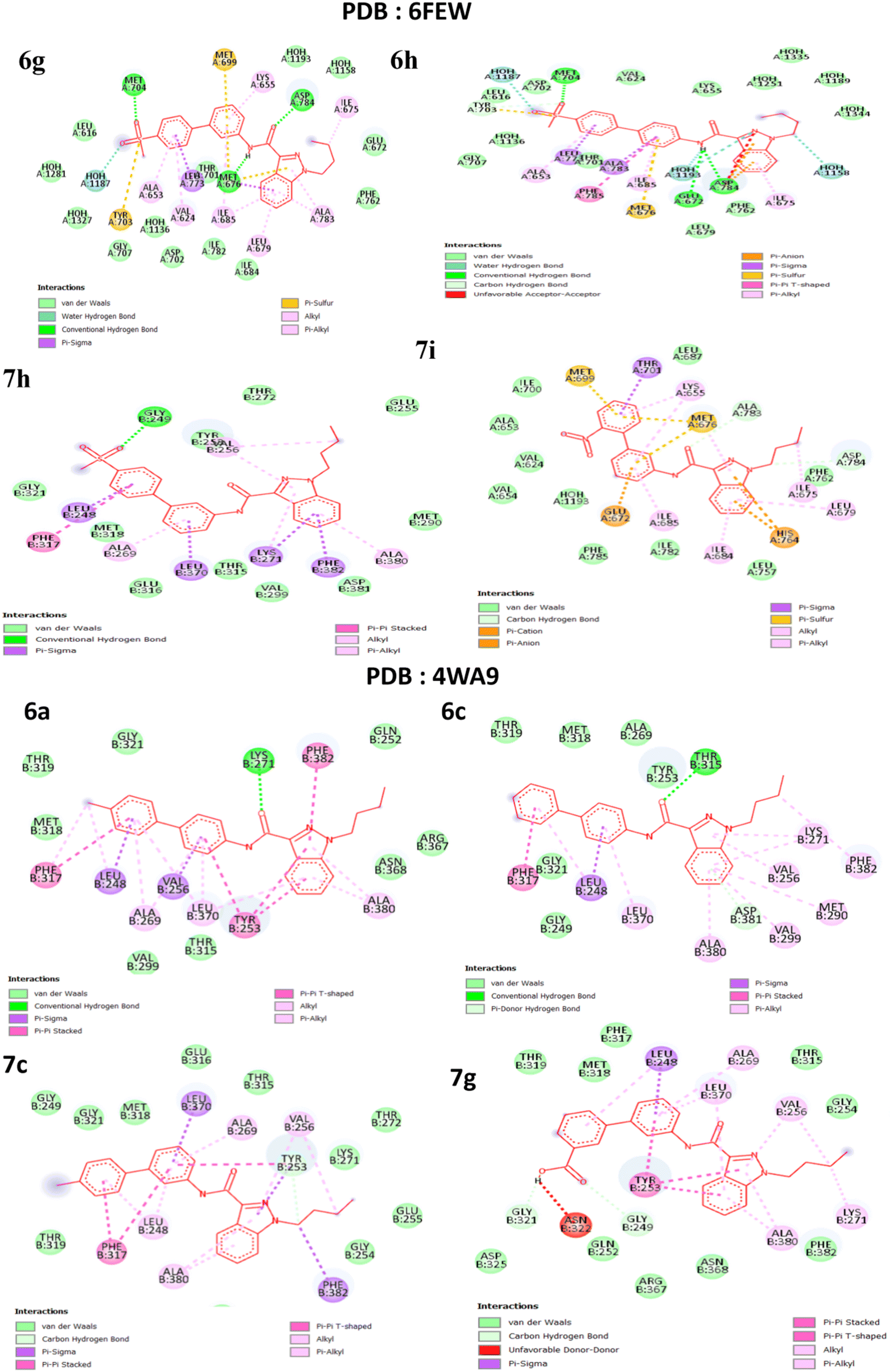
The literature has identified entrectinib, pazopanib, and axitinib as the three most commonly available indazole-containing medicines with potent anti-renal cancer activity. To explore the efficacy of these newly synthesised molecules, we utilized axitinib-related PDB 4WA9 and another renal cancer-related PDB 6few. To explore the binding interaction of the indazole derivative, we conducted in silico studies. Auto Dock tools (version 1.5.6) were used to generate receptor grid maps for the docking investigation.35 We searched the protein databank for the protein crystallographic structures (PDB: 4WA9 and PDB: 6few) and utilized them in our studies (PDB: 4WA9, the crystal structure of human abl1 wild-type kinase domain in association with axitinib and PDB: 6FEW, DDR1: 2-[8-(1H-idazole-5-carbonyl) (4-oxo-1phenyl-1,3,8-triazspiro [4.5] decan-3-yl]-N-methylacetamide) (Fig. 7). The protein was synthesized using the Bioviva Discovery Studio software program (http://www.discover.3ds.com/discovery-studio-visualizer-download).36 Following that, we eliminated the protein's water molecules, binding ligands, and other components. After removing water molecules, binding ligands, and other components, the atoms were filled by polar hydrogen atoms and allocated Gasteiger charges within the receptor. The ligand was then charged with Gasteiger in the Avogadro program and the corresponding protein structure was used as an input file for the auto grid method after the Kollman charges were assigned [maps were created using auto grid with 0.375 Å spacing between grid points for each atom type in the ligand,37 the grid box's center was set to x = 9.47003, y = 154.99967, and z = 35.72421. The active site box has dimensions of 40.40-40 (ref. 38)]. Flexible ligand docking was performed on the selected molecules. Each docked system received ten cycles of the Auto Dock search using the Lamarckian genetic method. After doing all of the derivative docking experiments, we compared the results to the axitinib-related PDB: 4wa9 and the renal cancer-related PDB: 6few. All of the derivatives showed almost identical binding energies displayed in Table 3, (1) final intermolecular energy + vander Waal + H-bond + desolv energy, (2) final total internal energy, (3) torsional free energy, and (4) unbound system's energy, which were obtained using the formula [(1) + (2) + (3) − (4)] in kcal mol−1. Furthermore, these docking studies illustrated the interaction of amino acids, using the 2D diagram-related structure displayed in Fig. 8. The Discovery Studio visualizer was used to analyze 2D ligand–receptor interactions and all derivatives was highlighted in the ESI.†
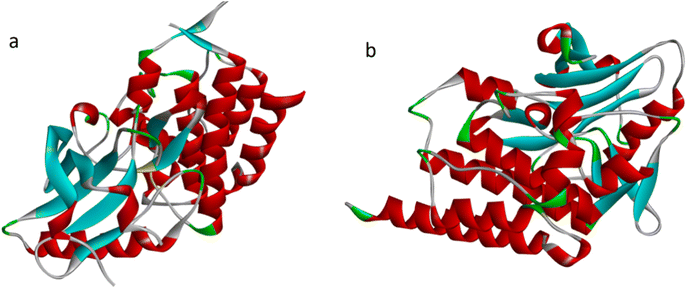 |
| | Fig. 7 (a) PDB: 6FEW, DDR1, 2-[8-(1H-idazole-5-carbonyl)-4-oxo-1phenyl-1,3,8-triazspiro[4.5]decan-3-yl]-N-methylacetamide (b) PDB: 4WA9, the crystal structure of human abl1 wild-type kinase domain in complex with axitinib. | |
Table 3 Docking results in terms of binding energy (kcal mol−1), most efficient anticancer (renal cancer) 6a, 6c, 6g, 6h, and 7c, 7g, 7h,7i derivatives
| Entry |
Receptor |
(1) Final intermolecular energy + van der Waal + H-bond + desolv energy (kcal mol−1) |
(2) Final total internal energy (kcal mol−1) |
(3) Torsional free energy (kcal mol−1) |
(4) Unbound system's energy (kcal mol−1) |
(ΔG) binding energy = [(1) + (2) + (3) − (4)] (kcal mol−1) |
| 6a |
4wa9 |
−12.21 |
−1.64 |
+1.79 |
−1.89 |
−10.42 |
| 6c |
4wa9 |
−10.87 |
−1.87 |
+1.79 |
−1.87 |
−9.08 |
| 7c |
4wa9 |
−12.06 |
−0.98 |
+1.79 |
−0.98 |
−10.27 |
| 7g |
4wa9 |
−12.95 |
−1.01 |
+2.39 |
−1.01 |
−10.57 |
| 6g |
6few |
−14.40 |
−1.71 |
+2.39 |
−1.71 |
−12.02 |
| 6h |
6few |
−13.88 |
−1.89 |
+2.09 |
−1.89 |
−11.79 |
| 7h |
6few |
−15.28 |
−1.48 |
+2.09 |
−1.48 |
−13.19 |
| 7i |
6few |
−15.08 |
−2.14 |
+2.09 |
−2.14 |
−12.99 |
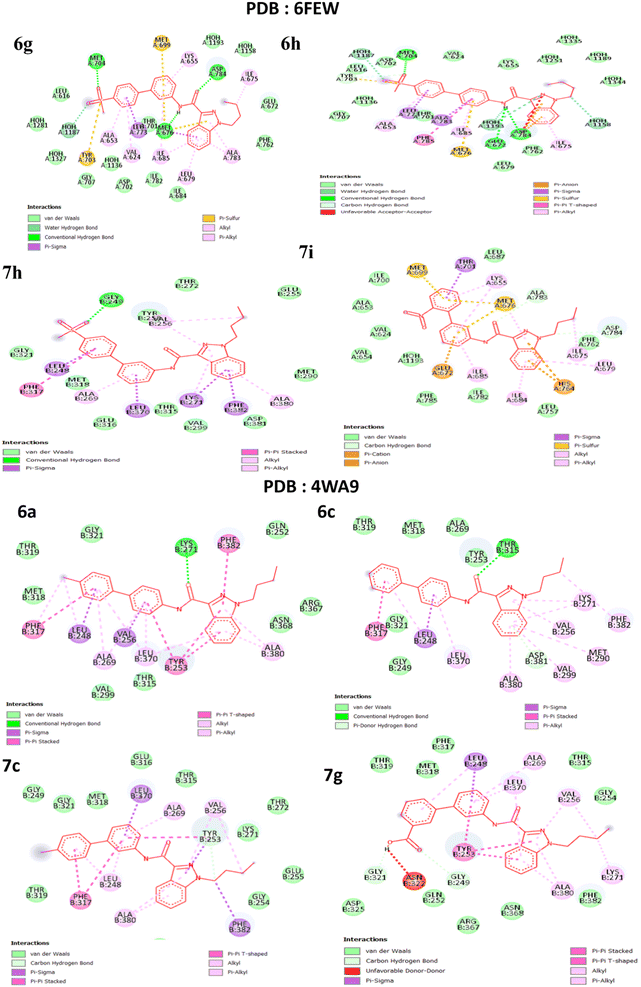 |
| | Fig. 8 2D predicted binding mode of compounds PDB: 6FEW (6g, 6h and 7h, 7i) and PDB: 4wa9 (6a, 6c, and 7c, 7g). | |
Conclusions
In summary, the study has discovered a new method of combining indazole derivatives and various boronic acids using the Suzuki–Miyaura coupling mechanism. The synthesized derivatives were thoroughly validated using FT-IR, HRMS, and 1H and 13C NMR spectroscopy. Using Density Functional Theory (DFT) calculations, four compounds with high energy states were identified and analysed through molecular electrostatic potential surface investigations. Docking studies were conducted using AutoDock software version 1.5.6 and Protein Data Bank entries 4WA9 and 6FEW. The results showed that compounds (6g, 6h, and 7h, 7i) and (6a, 6c, and 7c, 7g) exhibited strong binding affinities.
Experimental section
All the necessary chemicals and solvents used in the experiment were purchased from commercially available chemical suppliers and used as received without further purification. The solvents DMF and 1,4-dioxane were obtained from Finar chemical suppliers, while the remaining chemicals were purchased from Avra Chemicals and Sigma-Aldrich. All the reactions were carried out in round bottom flasks. The infrared absorption spectra were recorded on an affinity-1 (4000–200 cm−1). The 1-butyl-1H-indazole-3-carboxamide was prepared according to the reported protocols. The 1H-NMR and 13C-NMR spectra were recorded at room temperature on a Bruker Avance III (400 MHz) spectrometer using CDCl3 as a solvent and TMS as an internal standard and are referred to as the residual solvent signal CDCl3: (7.26) for 1H and (77.16) for 13C NMR: dimethyl sulfoxide-d6 (2.50) for 1H and (39.50) for 13C NMR: chemical shift (δ) is given in ppm and coupling constant (J) were measured in Hz. The following abbreviations are used: s-singlet, d-doublet, dd-doublet of doublet, t-triplet, td-triplet of doublet, dt-doublet of triplet, q-quartet, qd-quartet of doublet, qn-quintet, br-broad, m-multiplet. HRMS ESI-MS was recorded using Xeo G2 XS OT of (water) and values are given m/z. Column chromatography uses a glass column of silica gel (100–200 mesh). Analytical TLC was carried out on Macherey-Nagel 60 F245 aluminium-backed silica gel plates.
General procedure for the synthesis of 3 and 4
To a stirred solution of 1-butyl-1H-indazole-3-carboxylic acid (250 mg, 1.146 mmol) was dissolved in DMF (10 mL), HATU (2 equivalents), and DIPEA (3 equivalents) was added to the reaction mixture, then commercial amines (2 equivalents) were added. The reaction mixture was stirred at room temperature for 8–16 hours. After completion of the reaction, the resultant reaction mixture was poured into ice-cold water, and solid precipitated and filtered through the Buchner funnel under a high vacuum dried to obtain pure compound 3 and 4.
General procedure for Suzuki reaction (6a–6i, 7a–7i)
To a stirred solution of N-(3-bromophenyl)-1-butyl-1H-indazole-3-carboxamide (150 mg) was dissolved in 1,4-dioxane/water (6/2 mL), K2CO3 (3 eq.), and Pd(dppf)Cl2·DCM (0.05 eq.) was added to the reaction mixture, and commercial boronic acids (3 eq.) were added. The reaction mixture was stirred at room temperature for 8–12 hours. After completion of the reaction, the resultant reaction mixture was filtered through a Celite pad with a Buchner funnel under a high vacuum, the filtrate was extracted with water and ethyl acetate (4 × 20 mL). The organic layer was dried with anhydrous sodium sulphate and the solvent was removed under reduced pressure to afford crude product. The crude was purified by silica gel chromatography to obtain a pure compound (6a–6i, 7a–7i).
Characterization of indazole derivatives (3, 4) and (6a–6i, 7a–7i)
1-Butyl-1H-indazole-3-carboxylic acid to N-(4-bromophenyl)-1-butyl-1H-indazole-3-carboxamide (3). Appearance: brown solid (melting point = 96–99 °C, yield = 82.5%, weight = 210.6 mg). 1H NMR [400 MHz, DMSO-d6]: δ 10.41 (s, 1H), 8.23 (d, J = 8.40 Hz, 1H), 7.90 (d, J = 8.80 Hz, 2H), 7.83 (d, J = 8.40 Hz, 1H), 7.55–7.52 (m, 3H), 7.32 (t, J = 7.60 Hz, 1H), 4.55 (t, J = 7.20 Hz, 2H), 1.90 (q, J = 7.20 Hz, 2H), 1.34–1.32 (m, 2H), 0.91 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, DMSO-d6]: δ 161.16, 141.18, 138.68, 137.23, 131.82, 127.24, 123.18, 122.74, 122.18, 111.04, 55.37, 31.99, 19.92, 14.01. IR STRECHING (cm−1): 3272 (N–H), 1655 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). HRMS (ESI) m/z: [M + H] calculated C18H18BrN3O 372.0711 + found 372.0707 [M + H].
O). HRMS (ESI) m/z: [M + H] calculated C18H18BrN3O 372.0711 + found 372.0707 [M + H].
1-Butyl-1H-indazole-3-carboxylic acid to N-(3-bromophenyl)-1-butyl-1H-indazole-3-carboxamide (4). Appearance: brown solid melting point = 96–99 °C, yield = 78.4%, weight = 200 mg). 1H NMR [400 MHz, CDCl3]: δ 8.78 (s, 1H), 8.32 (d, J = 8.40 Hz, 1H), 7.95 (d, J = 1.60 Hz, 1H), 7.60–7.59 (m, 1H), 7.37–7.36 (m, 2H), 7.16 (t, J = 7.20 Hz, 2H), 4.34 (t, J = 7.20 Hz, 2H), 1.87 (q, J = 7.60 Hz, 2H), 1.34–1.32 (m, 2H), 0.89 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.51, 141.11, 139.38, 136.73, 126.99, 126.83, 123.03, 122.94, 122.73, 122.44, 118.03, 109.48, 49.38, 31.38, 20.07, 13.66. IR STRECHING (cm−1): 3272 (N–H), 1655 (CO). HRMS (ESI) m/z: [M + H] calculated C18H18BrN3O 372.0711 + found 372.0707 [M + H].
1-Butyl-N-(4-methyl-[1,1-biphenyl]-4-yl)-1H-indazole-3-carboxamide (6a). Appearance: off white solid, yield = 94%, weight = 145 mg, melting point = 94–97 °C, (eluent = 15% ethyl acetate/hexane). 1H NMR [400 MHz, CDCl3]: δ 8.83 (s, 1H), 8.37 (d, J = 8.00 Hz, 1H), 7.75 (d, J = 8.40 Hz, 2H), 7.52 (d, J = 8.80 Hz, 2H), 7.43 (t, J = 8.40 Hz, 2H), 7.36 (t, J = 2.00 Hz, 2H), 7.17 (t, J = 5.60 Hz, 2H), 4.35 (t, J = 7.20 Hz, 3H), 2.31 (s, 2H), 1.88 (q, J = 7.60 Hz, 2H), 1.26–1.27 (m, 2H), 0.90 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.54, 141.11, 137.78, 137.16, 137.14, 136.79, 136.71, 129.51, 127.45, 126.91, 126.68, 123.00, 122.91, 122.88, 119, 109.41, 49.34, 31.85, 21.11, 20.09, 13.68. DEPT-135 NMR [100 MHz, CDCl3]: δ 129.51, 127.45, 126.91, 122.91, 122.88, 119.94, 109.42, 49.35, 31.85, 21.11, 20.09, 13.69. FT-IR (cm−1): 3358 (N–H), 1659 (CO). HRMS (ESI) m/z: [M + H] calculated C25H25N3O 384.2076 + found 384.2078 [M + H].
1-Butyl-N-(4-(trifluoromethyl)-[1,1-biphenyl]-4-yl)-1H-indazole-3-carboxamide (6b). Appearance: off white solid, yield = 93.7%, weight = 165 mg, melting point = 101–105 °C, (eluent = 15% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.88 (s, 1H), 8.37 (d, J = 8.00 Hz, 1H), 7.80 (d, J = 8.80 Hz, 2H), 7.59–7.61 (m, 4H), 7.55 (d, J = 8.40 Hz, 2H), 7.37 (t, J = 1.60 Hz, 2H), 4.36 (t, J = 7.20 Hz, 2H), 1.89 (q, J = 7.20 Hz, 2H), 1.26–1.28 (m, 2H), 0.90 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.60, 144.11, 141.12, 138.29, 136.98, 135.06, 129.18, 128.85, 127.85, 127.02, 126.98, 125.70, 122.98, 122.83, 120.00, 109.46, 49.38, 31.84, 20.08, 13.67. DEPT-135 NMR [100 MHz, CDCl3]: δ 127.085, 127.02, 126.98, 125.70, 122.98, 120.00, 109.46, 109.46, 49.38, 31.84, 20.08, 13.67. FT-IR (cm−1): 3351 (N–H), 1664 (CO). HRMS (ESI) m/z: [M + H] calculated C25H22F3N3O 438.1793 + found 438.1794 [M + H].
N-([1,1-Biphenyl]-4-yl)-1-butyl-1H-indazole-3-carboxamide (6c). Appearance: off white solid, yield = 87%, weight = 130 mg, melting point = 83–88 °C, (eluent = 10% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.85 (s, 1H), 8.37 (d, J = 8.00 Hz, 1H), 7.77 (d, J = 8.40 Hz, 2H), 7.53–7.54 (m, 4H), 7.34–7.36 (m, 4H), 4.35 (t, J = 6.80 Hz, 2H), 1.89 (q, J = 7.60 Hz, 2H), 1.36–1.34 (m, 2H), 0.90 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.55, 141.12, 140.66, 137.43, 137.12, 136.75, 128.78, 127.03, 126.92, 126.85, 123.01, 122.91, 119.94, 109.41, 49.35, 31.84, 20.08, 13.67. DEPT-135 NMR [100 MHz, CDCl3]: δ 128.78, 127.67, 127.03, 126.86, 122.91, 122.89 119.93, 109.41, 49.35, 31.85, 20.09, 13.68. FT-IR (cm−1): N–H (3363), CO (1660). HRMS (ESI) m/z: [M + H] calculated C24H22N3O 370.1919 + found 370.1924 [M + H].
1-Butyl-N-(4-(hydroxymethyl)-[1,1-biphenyl]-4-yl)-1H-indazole-3-carboxamide (6d). Appearance: off white solid (yield = 89.7%, weight = 145 mg, melting point = 81–86 °C, (eluent = 25% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.86 (s, 1H), 8.37 (d, J = 7.60 Hz, 1H), 7.76 (d, J = 7.20 Hz, 2H), 7.53 (s, 3H), 7.46 (d, J = 6.40 Hz, 1H), 7.36 (d, J = 9.20 Hz, 3H), 7.25 (d, J = 5.60 Hz, 2H), 4.69 (s, 2H), 4.35 (s, 2H), 1.88 (s, 2H), 1.31 (d, J = 6.80 Hz, 2H), 0.90 (t, J = 6.80 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.59, 141.50, 141.09, 137.48, 137.06, 136.46, 136.46, 129.02, 127.66, 126.94, 126.10, 125.64, 125.42, 122.99, 122.93, 122.87, 119.94, 109.44, 65.37, 49.35, 31.85, 20.08, 13.69. DEPT-135 NMR [100 MHz, CDCl3]: δ 129.02, 127.66, 126.94, 126.10, 125.65, 125.42, 122.93, 122.86, 119.94, 109.44, 65.37, 49.35, 31.85, 20.09, 13.69. FT-IR (cm−1): 3403 (OH), 3250 (N–H), 1645 (CO). HRMS (ESI) m/z: [M + H] calculated C25H25N3O2 400.2025 + found 400.2037 [M + H].
1-Butyl-N-(4-formyl-[1,1-biphenyl]-4-yl)-1H-indazole-3-carboxamide (6e). Appearance: off white solid, yield = 87.5%, weight = 140 mg, melting point = 93–97 °C, (eluent = 25% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 10.01 (s, 1H), 8.89 (s, 1H), 8.37 (d, J = 8.00 Hz, 1H), 8.03 (s, 1H), 7.75–7.77 (m, 4H), 7.58 (d, J = 8.64 Hz, 2H), 7.52 (t, J = 8.00 Hz, 1H), 7.37–7.38 (m, 2H), 4.36 (t, J = 7.20 Hz, 2H), 1.89 (q, J = 7.20 Hz, 2H), 1.26–1.28 (m, 2H), 0.00 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 192.45, 160.61, 141.56, 138.12, 136.98, 136.94, 135.04, 129.52, 128.38, 127.81, 127.70, 126.97, 122.97, 122.83, 122.05, 109.46, 49.38, 31.84, 20.08, 13.67. DEPT-135 NMR [100 MHz, CDCl3]: δ 132.74, 129.52, 128.39, 127.81, 127.70, 126.97, 122.97, 122.82, 120.05, 109.46, 49.38, 31.84, 20.08, 13.68. FT-IR (cm−1): 2955 (CHO), 3301 (N–H), 1693 (CO). HRMS (ESI) m/z: [M + H] calculated C25H23N3O2 398.1868 + found 398.1868 [M + H].
1-Butyl-N-(3-fluoro-4-methoxy-[1,1-biphenyl]-4-yl)-1H-indazole-3-carboxamide (6f). Appearance: off white solid, yield = 71.4%, weight = 120 mg, melting point = 80–85 °C, (eluent = 15% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.84 (s, 1H), 8.37 (d, J = 8.00 Hz, 1H), 7.76 (d, J = 8.00 Hz, 2H), 7.48 (d, J = 7.60 Hz, 2H), 7.39 (s, 2H), 7.19–7.26 (m, 4H), 6.95 (t, J = 8.00 Hz, 1H), 4.37 (t, J = 6.80 Hz, 2H), 3.86 (s, 3H), 1.90 (t, J = 7.20 Hz, 2H), 1.32 (q, J = 6.80 Hz, 2H), 0.912 (t, J = 6.80 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.65, 141.11, 138.62, 137.04, 129.51, 127.20, 126.39, 122.97, 122.91, 122.85, 122.26, 119.98, 118.41, 117.92, 115.00, 114.81, 113.58, 109.43, 56.37, 49.36, 31.84, 20.08, 13.68. DEPT-135 NMR [100 MHz, CDCl3]: δ 129.51, 127.20, 126.93, 122.91, 122.86, 122.26, 119.98, 118.41, 117.92, 115.00, 114.81, 113.56, 109.44, 56.37, 49.36, 31.85, 20.08, 13.68. FT-IR (cm−1): 3380 (N–H), 2955 (–OCH3), 1681 (CO). HRMS (ESI) m/z: [M + H] calculated C25H24FN3O2 418.1931 + found 418.1933 [M + H].
4-(1-Butyl-1H-indazole-3-carboxamido)-[1,1-biphenyl]-3-carboxylic acid (6g). Appearance: off white solid, yield = 71.8%, weight = 120 mg, melting point = 159–164 °C, (eluent = 30% ethyl acetate/hexane) 1H NMR [400 MHz, DMSO-d6]: δ 10.39 (s, 1H), 8.26 (d, J = −8.40 Hz, 1H), 8.22 (s, 1H), 8.05 (d, J = 8.40 Hz, 2H), 7.92 (t, J = 8.40 Hz, 2H), 7.84 (d, J = 8.40 Hz, 1H), 7.73 (d, J = 8.80 Hz, 2H), 7.60 (t, J = 7.60 Hz, 1H), 7.51 (t, J = 8.00 Hz, 1H), 7.34 (t, J = 7.60 Hz, 1H), 4.57 (t, J = 7.20 Hz, 2H), 0.00 (q, J = 7.20 Hz, 2H), 1.27–1.28 (m, 2H), 0.92 (t, J = 7.60 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 167.18, 161.14, 141.9, 140.54, 139.14, 137.37, 134.62, 132.00, 131.14, 129.77, 128.32, 127.39, 127.24, 123.16, 122.91, 122.22, 121.22, 111.02, 49.11, 31.99, 19.93, 14.02. DEPT-135 NMR [100 MHz, CDCl3]: δ 131.14, 129.77, 128.33, 127.39, 127.36, 127.25, 123.17, 122.22, 121.22, 111.03, 49.11, 31.99, 19.93, 14.02. FT-IR (cm−1): 3281 (N–H), 1715 (CO). HRMS (ESI) m/z: [M + H] calculated C25H23N3O3 414.1817 + found 414.1818 [M + H].
1-Butyl-N-(4-(methylsulfonyl)-[1,1-biphenylyl]-4-yl)-1H-indazole-3-carboxamide (6h). Appearance: off white solid, yield = 94.4%, weight = 170 mg, melting point = 79–84 °C, (eluent = 15% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.98 (s, 1H), 8.44 (d, J = −8.00 Hz, 1H), 8.00 (d, J = 8.00 Hz, 2H), 7.91 (d, J = 8.40 Hz, 2H), 7.79 (d, J = 8.40 Hz, 2H), 7.65 (d, J = 8.40 Hz, 2H), 7.47 (d, J = 4.80 Hz, 2H), 7.32–7.32 (m, 1H), 4.45 (t, J = 7.20 Hz, 2H), 3.10 (s, 3H), 1.98 (q, J = 7.20 Hz, 2H), 1.35–1.37 (m, 2H), 0.99 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.61, 1466.10, 141.15, 138.79, 138.75, 136.93, 134.33, 128.02, 127.94, 127.54, 127.01, 123.02, 122.99, 122.80, 120.06, 109.47, 49.40, 44.67, 31.83, 20.07, 13.65. DEPT-135 NMR [100 MHz, CDCl3]: δ 128.28, 126.96, 126.86, 126.46, 125.97, 122.00, 121.73, 119.91, 119.01, 116.73, 48.32, 43.53, 30.78, 28.66, 19.02, 12.63. FT-IR (cm−1): 3075 (N–H), 1676 (CO). HRMS (ESI) m/z: [M + H] calculated C25H23N3O3S 448.1695 + found 448.1697 [M + H].
1-Butyl-N-(2-nitro[1,1-biphenyl]-4-1H-indazole-3-carboxamide) (6i). Appearance: off white solid, yield = 59.8%, weight = 100 mg, melting point = 88–94 °C, (eluent = 15% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.86 (s, 1H), 8.37 (d, J = 8.00 Hz, 1H), 7.77 (d, J = 7.60 Hz, 3H), 7.54 (t, J = 7.60 Hz, 1H), 7.40 (d, J = 9.20 Hz, 4H), 7.28 (s, 1H), 4.37 (t, J = 7.20 Hz, 2H), 1.90 (q, J = 7.20 Hz, 2H), 1.30 (d, J = 35.60 Hz, 2H), 0.91 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.57, 149.36, 141.14, 138.27, 137, 135.87, 132.69, 132.24, 132, 128.7, 126.94, 124.1, 123, 122.93, 122.87, 119.76, 109.43, 49.36, 31.81, 20.06, 13.64. DEPT-135 NMR [100 MHz, CDCl3]: δ 132.24, 132, 128.7, 127.97, 126.94, 124.1, 122.93, 122.87, 119.76, 109.43, 49.36, 31.81, 20.06, 13.64. FT-IR (cm−1): 3390 (N–H), 1671 (CO), 1521 (NO2). HRMS (ESI) m/z: [M + H] calculated C25H22N4O3 415.1770 + found 415.1773 [M + H].
1-Butyl-N-(3-fluoro-4-methoxy-[1,1-biphenyl]-3-yl)-1H-indazole-3-carboxamide (7a). Appearance: off white solid, yield = 88.8%, weight = 150 mg, melting point = 96–100 °C, (eluent = 15% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.86 (s, 1H), 8.37 (d, J = 8.00 Hz, 1H), 7.93 (s, 1H), 7.63 (d, J = 8.00 Hz, 1H), 7.30–7.31 (m, 5H), 7.18–7.21 (m, 2H), 6.95 (t, J = 8.80 Hz, 1H), 4.36 (t, J = 6.80 Hz, 2H), 3.86 (s, 3H), 1.90 (q, J = 7.20 Hz, 2H), 1.27–1.29 (m, 2H), 0.904 (t, J = 7.60 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.66, 153.77, 147.24, 141.11, 140.66, 138.62, 137.35, 137.04, 129.51, 127.19, 126.93,122.97, 122.83, 122.26, 119.99, 118.42, 117.92, 114.99, 109.44, 56.37, 49.36, 31.84, 20.08, 13.67. DEPT-135 NMR [100 MHz, CDCl3]: δ 129.51, 127.20, 126.93, 122.91, 122.83, 122.26, 119.99, 118.42, 117.92, 114.99, 114.80, 113.59, 113.57, 109.44 56.37, 49.36, 31.85, 20.08, 13.68. FT-IR (cm−1): 3277 (N–H), 2767 (OCH3), 1664 (CO). HRMS (ESI) m/z: [M + H] calculated C25H24FN3O2 418.1931+ found 418.1931 [M + H].
1-Butyl-N-(4-(trifluoromethyl)-[1,1-biphenyl]-3-yl)-1H-indazole-3-carboxamide (7b). Appearance: off white solid, yield = 73.8%, weight = 130 mg, melting point = 66–70 °C, (eluent = 15% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.89 (s, 1H), 8.37 (d, J = 8.00 Hz, 1H), 8.05 (s, 1H), 7.61–7.64 (m, 5H), 7.38–7.39 (m, 3H), 7.18 (d, J = 3.60 Hz, 1H), 4.37 (t, J = 6.80 Hz, 2H), 1.90 (q, J = 7.20 Hz, 2H), 1.27–1.29 (m, 2H), 0.91 (t, J = 7.60 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.69, 144.40, 141.14, 140.71, 138.77, 136.99, 129.64, 127.58, 126.97, 125.69, 125.65, 122.97, 122.77, 119.30, 118.50, 109.45, 49.38, 31.84, 20.08, 13.66. DEPT-135 NMR [100 MHz, CDCl3]: δ 129.64, 127.58, 126.97, 125.69, 122.97, 122.83, 122.78, 119.30, 118.50, 109.45, 49.38, 31.84, 20.08, 13.66. FT-IR (cm−1): 3289 (N–H), 1668 (CO). HRMS (ESI) m/z: [M + H] calculated C25H22F3N3O 438.1793 + found 438.1795 [M + H].
1-Butyl-N-(4-methyl-[1,1-biphenyl]-3-yl)-1H-indazole-3-carboxamide (7c). Appearance: off white solid, yield = 90.9%, weight = 140 mg, melting point = 107–110 °C, (eluent = 15% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.86 (s, 1H), 8.38 (d, J = 8.00 Hz, 1H), 7.92 (s, 1H), 7.68 (d, J = 8.00 Hz, 1H), 7.49 (d, J = 8.00 Hz, 2H), 7.34–7.36 (m, 3H), 7.29 (s, 1H), 7.18 (t, J = 4.40 Hz, 2H), 4.37 (t, J = 7.20 Hz, 2H), 2.33 (s, 3H), 1.90 (q, J = 7.60 Hz, 2H), 1.27–1.29 (m, 2H), 0.91 (t, J = 7.60 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.61, 142.11, 141.11, 138.50, 137.94, 137.24, 137.15, 129.45, 129.39, 127.10, 126.90, 122.99, 122.91, 122.85, 122.57, 118.27, 118.17, 109.40, 49.34, 31.85, 21.14, 20.09, 13.68. DEPT-135 NMR [100 MHz, CDCl3]: δ 129.46, 129.40, 127.10, 126.90, 122.91, 122.86, 122.57, 118.27, 118.17, 109.40, 49.34, 31.85, 21.14, 20.09, 13.68. FT-IR (cm−1): 3289 (N–H), 1668 (CO). HRMS (ESI) m/z: [M + H] calculated C25H25N3O 384.2076 + found 384.2080 [M + H].
N-([1,1-Biphenyl]-3-yl)-1-butyl-1H-indazole-3-carboxamide (7d). Appearance: off white solid, yield = 67.1%, weight = 100 mg, melting point = 65–68 °C, (eluent = 10% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.94 (s, 1H), 8.45 (d, J = 6.80 Hz, 1H), 8.02 (s, 1H), 7.76 (d, J = 6.00 Hz, 1H), 7.66 (d, J = 5.60 Hz, 2H), 7.33–7.36 (m, 8H), 4.43 (s, 2H), 1.97 (s, 2H), 1.38 (d, J = 6.00 Hz, 2H), 0.97 (s, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.63, 142.21, 141.12, 140.86, 138.55, 137.12, 129.44, 128.74, 127.46, 127.29, 123.00, 122.90, 122.88, 118.52, 118.41, 109.42, 49.35, 31.85, 20.09, 13.68. DEPT-135 NMR [100 MHz, CDCl3]: δ 129.44, 128.74, 127.29, 126.91, 122.88, 122.75, 118.52, 118.41, 109.42, 49.35, 31.85, 20.09, 13.68. FT-IR (cm−1): 3014 (N–H), 1670 (CO). HRMS (ESI) m/z: [M + H] calculated C24H24N3O 370.1919 + found 370.1920 [M + H].
1-Butyl-N-(4-formyl-[1,1-biphenyl]-3-yl)-1H-indazole-3-carboxamide (7e). Appearance: off white solid, yield = 74.6%, weight = 120 mg, melting point = 67–72 °C, (eluent = 25% ethyl acetate/hexane) 1H NMR [400 MHz, CDCl3]: δ 10.10 (s, 1H), 9.00 (s, 1H), 8.44 (d, J = 6.40 Hz, 1H), 8.13 (d, J = 14.40 Hz, 2H), 7.76–7.78 (m, 3H), 7.61 (s, 1H), 7.26–7.33 (m, 5H), 4.45 (s, 2H), 1.98 (s, 2H), 1.39 (d, J = 5.20 Hz, 2H), 0.98 (s, 3H). 13C NMR [100 MHz, CDCl3]: δ 192.47, 160.76, 141.82, 140.66, 138.73, 136.93, 136.87, 133.29, 129.71, 129.51, 128.72, 128.44, 126.98, 122.96, 122.78, 122.72, 119.21, 118.40, 109.48, 49.40, 31.85, 20.09, 13.68. DEPT-135 NMR [100 MHz, CDCl3]: δ 133.28, 129.71, 129.51, 128.72, 128.44, 126.98, 122.99, 122.71, 119.21, 118.39, 109.48, 49.40, 31.85, 20.09, 13.68. FT-IR (cm−1): 3257 (N–H), 2952 (CHO), 1690 (CO). HRMS (ESI) m/z: [M + H] calculated C25H23N3O2 398.1868 + found 398.1873 [M + H].
1-Butyl-N-(4-(hydroxymethyl)-[1,1-biphenyl]-3-yl)-1H-indazole-3-carboxamide (7f). Appearance: off white solid, yield = 74.3%, weight = 120 mg, melting point = 77–82 °C, (eluent = 25% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.97 (s, 1H), 8.43 (d, J = 7.60 Hz, 1H), 8.01 (s, 1H), 7.74 (d, J = 7.20 Hz, 1H), 7.64 (s, 1H), 7.57 (d, J = 7.60 Hz, 1H), 7.42–7.44 (m, 4H), 7.26–7.31 (m, 3H), 4.76 (s, 2H), 4.58 (s, 1H), 4.43 (t, J = 6.40 Hz, 2H), 1.95–1.97 (m, 2H), 1.36–1.37 (m, 2H), 0.97 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.71, 141.94, 141.39, 141.12, 138.47, 137.03, 129.76, 129.46, 128.99, 126.95, 126.57, 126.10, 125.87, 122.97, 122.94, 122.84, 122.81, 118.67, 118.44, 114.65, 113.84, 109.44, 49.37, 31.85, 20.08, 13.68. DEPT-135 NMR [100 MHz, CDCl3]: δ 129.76, 129.47, 128.99, 126.95, 126.57, 126.10, 125.87, 122.94, 122.84, 122.81, 118.82, 118.67, 118.44, 114.66, 113.84, 109.44, 65.04, 49.38, 31.85, 20.09, 13.68. FT-IR (cm−1): 3383 (O–H), 1663 (CO). HRMS (ESI) m/z: [M + H] calculated C25H25N3O2 400.2025 + found 400.2027 [M + H].
3-(1-Butyl-1H-indazole-3-carboxamido)-[1,1-biphenyl]-3-carboxylic acid (7g). Appearance: off white solid, yield = 89.8%, weight = 150 mg, melting point = 145–148 °C, (eluent = 30% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.95 (s, 1H), 8.38 (d, J = 8.00 Hz, 1H), 8.33 (s, 1H), 8.04 (d, J = 7.60 Hz, 1H), 7.95 (s, 1H), 7.84 (d, J = 7.60 Hz, 1H), 7.78 (d, J = 8.00 Hz, 1H), 7.48 (t, J = 7.60 Hz, 1H), 7.32–7.34 (m, 4H), 4.37 (t, J = 7.20 Hz, 2H), 1.90 (q, J = 7.20 Hz, 2H), 1.27–1.29 (m, 2H), 0.90 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 175.23, 160.68, 141.29, 141.12, 140.91, 138.71, 132.56, 129.67, 129.17, 129.00, 128.90, 126.94, 122.99, 122.93, 122.88, 122.75, 119.08, 118.37, 109.43, 49.38, 31.86, 20.09, 13.68. DEPT-135 NMR [100 MHz, CDCl3]: δ 135.56, 129.67, 129.17, 129.00, 128.89, 126.95, 122.94, 122.88, 122.76, 119.08, 118.37, 109.43, 49.38, 31.86, 20.09, 13.68. FT-IR (cm−1): 3281 (N–H), 1715 (CO). HRMS (ESI) m/z: [M + H] calculated C25H23N3O3 414.1817 + found 414.1818 [M + H].
1-Butyl-N-(4-(methylsulfonyl)-[1,1-biphenyl]-3-yl)-1H-indazole-3-carboxamide (7h). Appearance: off white solid, yield = 94.4%, weight = 170 mg, melting point = 110–106 °C, (eluent = 25% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.91 (s, 1H), 8.36 (d, J = 8.00 Hz, 1H), 8.12 (d, J = Hz, 1H), 7.94 (d, J = 8.40 Hz, 2H), 7.77 (d, J = 8.00 Hz, 2H), 7.65 (d, J = 7.60 Hz, 1H), 7.38–7.40 (m, 3H), 7.30 (d, J = 7.60 Hz, 1H), 7.19 (s, 1H), 4.38 (t, J = 7.20 Hz, 2H), 3.03 (s, 3H), 0.91 (q, J = 7.20 Hz, 2H), 1.27–1.29 (m, 2H), 0.91 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 159.70, 145.41, 140.13, 139.08, 138.27, 137.88, 135.90, 128.73, 127.16, 125.98, 121.99, 121.96, 121.82, 121.76, 118.71, 108.45, 48.37, 43.65, 30.81, 19.05, 12.63. DEPT-135 NMR [100 MHz, CDCl3]: δ 128.72, 127.15, 126.85, 125.97, 121.98, 121.81, 121.72, 118.68, 117.57, 108.46, 48.36, 43.64, 30.81, 19.04, 12.64. FT-IR (cm−1): N–H (3336), 1669 (CO). HRMS (ESI) m/z: [M + H] calculated C25H25N3O3S 448.1695 + found 448.1876 [M + H].
1-Butyl-N-(2-nitro-[1,1-biphenyl]-3-yl)-1H-indazole-3-carboxamide (7i). Appearance: off white solid, yield = 71.8%, weight = 120 mg, melting point = 99–104 °C, (eluent = 15% ethyl acetate/hexane), 1H NMR [400 MHz, CDCl3]: δ 8.93 (s, 1H), 8.42 (d, J = 8.00 Hz, 1H), 7.89 (d, J = 8.00 Hz, 1H), 7.80 (d, J = 9.20 Hz, 1H), 7.62 (d, J = 7.20 Hz, 1H), 7.52 (d, J = 6.00 Hz, 1H), 7.43–7.44 (m, 3H), 7.32 (t, J = 6.00 Hz, 1H), 7.26 (s, 1H), 7.07 (d, J = 7.60 Hz, 1H), 4.44 (t, J = 7.20 Hz, 2H), 0.00 (q, J = 7.60 Hz, 2H), 1.33–1.35 (m, 2H), 0.98 (t, J = 7.20 Hz, 3H). 13C NMR [100 MHz, CDCl3]: δ 160.61, 149.20, 141.11, 138.43, 138.39, 136.92, 136.16, 132.35, 129.36, 128.30, 126.91, 124.10, 123.39, 122.96, 122.82, 119.37, 119.12, 109.44, 49.36, 31.82, 20.07, 13.67. DEPT-135 NMR [100 MHz, CDCl3]: δ 132.35, 132.15, 129.37, 128.30, 126.92, 124.11, 123.39, 122.92, 122.82, 119.37, 119.12, 109.14. FT-IR (cm−1): N–H (3384), CO (1676), NO2 (1524). HRMS (ESI) m/z: [M + H] calculated C25H22N3O3 415.1770 + found 415.1771 [M + H].
Data availability
Detailed experimental procedures and characterization data for all novel compounds such as 1H and 13C NMR, Dept-135, COSY, HSQC, IR, and HRMS spectra are submitted as ESI.† The above content also included in the appropriate section of the manuscript.
Author contributions
The corresponding author Vijayaparthasarathi Vijayakumar has designed and executed the work, while the first author Bandaru Gopi has synthesized and characterized the target molecules and carried out the studies.
Conflicts of interest
No conflict of interest.
Acknowledgements
The VIT University administration in Vellore, India, is acknowledged by the authors for the seed grant (SG20230086) and for providing research facilities; and SIF-Chemistry at VIT University for providing NMR facilities. We sincerely thank Dr S. Sarveswari for her perceptive feedback and valuable insights on how to wrap up the research. The author acknowledges Ms. Rebecca Susan Philip's suggestion to finish the assignment.
References
- Z. Yang, J. T. Yu and C. Pan, Org. Biomol. Chem., 2022, 20, 7746–7764 RSC.
- L. Zhang, H. Yan, Y. Fan, X. Luo, Y. Pan, Y. Liu, Y. Cai and Q. Xia, J. Org. Chem., 2023, 88, 5731–5744 CrossRef CAS PubMed.
- T. Y. Wu, S. Dhole, M. Selvaraju and C. M. Sun, ACS Comb. Sci., 2018, 20, 156–163 CrossRef CAS PubMed.
- H. Zhang, Z. Ye, N. Chen, Z. Chen and F. Zhang, Green Chem., 2022, 24, 1463–1468 RSC.
- W. Wei, Z. Liu, X. Wu, C. Gan, X. Su, H. Liu, H. Que, Q. Zhang, Q. Xue, L. Yue, L. Yu and T. Ye, RSC Adv., 2021, 11, 15675–15687 RSC.
- S. Miyamoto, S. Kakutani, Y. Sato, A. Hanashi, Y. Kinoshita and A. Ishikawa, Jpn. J. Clin. Oncol., 2018, 48, 503–513 CrossRef PubMed.
- A. R. He, R. B. Cohen, C. S. Denlinger, A. Sama, A. Birnbaum, J. Hwang, T. Sato, N. Lewis, M. Mynderse, M. Niland, J. Giles, J. Wallin, B. Moser, W. Zhang, R. Walgren and E. R. Plimack, Oncologist, 2019, 24, e930–e942 CrossRef CAS.
- Z. T. Al-Salama and S. J. Keam, Drugs, 2019, 79, 1477–1483 CrossRef PubMed.
- S. Puri and K. Juvale, J. Mol. Struct., 2022, 1269, 133727 CrossRef CAS.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef.
- L. Bukavina, K. Bensalah, F. Bray, M. Carlo, B. Challacombe, J. A. Karam, W. Kassouf, T. Mitchell, R. Montironi, T. O'Brien, V. Panebianco, G. Scelo, B. Shuch, H. van Poppel, C. D. Blosser and S. P. Psutka, Eur. Urol., 2022, 82, 529–542 CrossRef PubMed.
- Y. Wan, S. He, W. Li and Z. Tang, Anticancer Agents Med. Chem., 2018, 18, 1228–1234 CrossRef CAS PubMed.
- M. R. Tammana, V. Vijayakumar and S. Sarveswari, J. Indian Chem. Soc., 2024, 101168 CrossRef CAS.
- H. J. Zhang, G. T. Tan, V. D. Hoang, N. Van Hung, N. M. Cuong, D. D. Soejarto, J. M. Pezzuto and H. H. S. Fong, J. Nat. Prod., 2003, 66, 263–268 CrossRef CAS.
- B. Gopi and V. Vijayakumar, RSC Adv., 2024, 14(19), 13218–13226 RSC.
- D. R. Kerzare, S. S. Menghani and P. B. Khedekar, Indian J. Pharm. Educ. Res., 2018, 52, 110–121 CrossRef CAS.
- B. Ko, Y. Jang, M. H. Kim, T. T. Lam, H. K. Seo, P. Jeong, M. Choi, K. W. Kang, S. D. Lee, J. H. Park, M. Kim, S. Y. Han and Y. C. Kim, Eur. J. Med. Chem., 2023, 262, 115860 CrossRef CAS.
- J. M. Honnanayakanavar, O. Obulesu and S. Suresh, Org. Biomol. Chem., 2022, 20, 2993–3028 RSC.
- Y. H. Khan, S. Qasim, A. M. Uttra, N. H. Alotaibi, A. S. Alanazi, A. I. Alzarea, A. D. Alatawi and T. Hussain Mallhi, Life Sci., 2023, 324, 121742 CrossRef CAS PubMed.
- X. P. Kang, W. Lian, L. Xu, J. Wang, H. Jia, B. Zhang, A. L. Liu and G. H. Du, RSC Adv., 2018, 8, 5286–5297 RSC.
- R. Rafique, K. M. Khan, K. Arshia, S. Chigurupati, A. Wadood, A. U. Rehman, A. Karunanidhi, S. Hameed, M. Taha and M. al-Rashida, Bioorg. Chem., 2020, 94, 103195 CrossRef CAS PubMed.
- S. Bhattacharjee, S. Laru and A. Hajra, Org. Biomol. Chem., 2022, 20, 8893–8897 RSC.
- Q. Pan, Y. Wu, A. Zheng, X. Wang, X. Li, W. Wang, M. Gao, Z. Bibi, S. Chaudhary and Y. Sun, Molecules, 2023, 28, 7190 CrossRef CAS PubMed.
- J. Yu, A. Zheng, L. Jin, Y. Wu, Q. Pan, X. Wang, X. Li, W. Wang, M. Gao and Y. Sun, Appl. Sci., 2023, 13, 4095 CrossRef CAS.
- V. Hemalatha and V. Vijayakumar, Inorg. Chem. Commun., 2022, 144, 109894 CrossRef CAS.
- M. S. Sunitha and S. Sarveswari, Inorg. Chim. Acta, 2024, 562, 121893 CrossRef CAS.
- M. S. Sunitha and S. Sarveswari, Inorg. Chem. Commun., 2024, 160, 111973 CrossRef CAS.
- R. H. Waghchaure and V. A. Adole, J. Mol. Struct., 2024, 1296, 136724 CrossRef CAS.
- T. Uelisson da Silva, E. Tomaz da Silva, K. de Carvalho Pougy, C. Henrique da Silva Lima and S. de Paula Machado, Inorg. Chem. Commun., 2022, 135, 109120 CrossRef CAS.
- C. Kucuk, S. Celik, S. Yurdakul, E. Cotelı and B. Erdem, Polyhedron, 2023,(241), 116469 CrossRef CAS.
- J. K. Ojha, G. Ramesh and B. V. Reddy, Chem. Phys. Impact, 2023,(7), 100280 CrossRef.
- M. A. Mumit, T. K. Pal, M. A. Alam, M. A. A. A. A. Islam, S. Paul and M. C. Sheikh, J. Mol. Struct., 2020, 1220, 128715 CrossRef CAS.
- A. Jumabaev, U. Holikulov, H. Hushvaktov, N. ISSAOUI and A. Absanov, J. Mol. Liq., 2023, 377, 121552 CrossRef CAS.
- V. Hemalatha and V. Vijayakumar, J. Mol. Struct., 2024, 1311, 138322 CrossRef CAS.
- S. S. Bushra, K. M. Khan, N. Ullah, M. Mahdavi, M. A. Faramarzi, B. Larijani, U. Salar, R. Rafique, M. Taha and S. Perveen, J. Mol. Struct., 2021, 1242, 130826 CrossRef CAS.
- D. Veeranna, L. Ramdas, G. Ravi, S. Bujji, V. Thumma and J. Ramchander, ChemistrySelect, 2022, e202201758 CrossRef CAS.
- M. Adib, F. Peytam, M. Rahmanian-Jazi, M. Mohammadi-Khanaposhtani, S. Mahernia, H. R. Bijanzadeh, M. Jahani, S. Imanparast, M. A. Faramarzi, M. Mahdavi and B. Larijani, New J. Chem., 2018, 42, 17268–17278 RSC.
- V. H. Hoang, N. T. K. Trang, T. C. Minh, L. T. B. Long, T. H. Lan, N. T. Hue, L. Q. Tien, T. X. Nguyen, Y. T. K. Nguyen, H. Yoo and P. T. Tran, Bioorg. Med. Chem., 2023, 90, 117377 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Vijayaparthasarathi Vijayakumar
and
Vijayaparthasarathi Vijayakumar