
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering the defects of UiO-66 MOF for an improved catalytic detoxification of CWA simulant: methyl paraoxon†
Selva Balasubramanianab,
Arockia Jayalatha Kulandaisamy b and
John Bosco Balaguru Rayappan*ab
b and
John Bosco Balaguru Rayappan*ab
aCentre for Nanotechnology & Advanced Biomaterials (CeNTAB), SASTRA Deemed University, Thanjavur, Tamil Nadu – 613 401, India. E-mail: rjbosco@ece.sastra.edu; Fax: +91 4362 264 120; Tel: +91 4362 350 009. ext. 2255
bSchool of Electrical & Electronics Engineering (SEEE), SASTRA Deemed University, Thanjavur, Tamil Nadu – 613 401, India
First published on 4th October 2024
Abstract
Exigency in search of an ideal candidate for an effective detoxification of chemical warfare agents is still continuing. Zirconium-based Metal–organic Framework (MOF) UiO-66 has shown a significant detoxification of such toxic chemicals owing to its tunable physio-chemical properties and profuse catalytic sites. In this context, a series of UiO-66 MOFs synthesized by tuning the acidity constant (pKa) and concentration of the modulator, synthesis temperature and water molecules was tested for their detoxification efficiency against the simulant ‘methyl-paraoxon’ at room temperature. Amongst, HCl modulated UiO-66 across the considered synthesis temperature have shown competent catalytic performance in virtue of defects generation within its structure. In addition, the role of catalytic features of UiO-66 obtained by tailoring its defects in enhancing the degradation efficiency has been systematically investigated. The detoxification efficiency of 98.5% with a half-life time of 0.23 min has confirmed the effectiveness of engineered defects in enhancing the catalytic activity of UiO-66 in detoxifying the identified simulant.
1. Introduction
In view of providing a safe environment to civilians and military personnel against the exposure of Chemical Warfare Agents (CWAs), it is imperative to develop effective filtration and/or detoxification material interfaces.1 The inevitability of robust defence against targeted CWAs has been an active area of research for not only their detection but detoxification.2 In this context, Metal–organic Frameworks (MOFs) have shown a notable performance towards the broad spectrum of nerve and blistering agents.3,4 The attractive detoxification features of MOFs in absorbing the toxic agents and converting the same to a harmless product within a short span of time has unbolted more findings in the area of MOFs catalysis.Amongst MOFs, Zirconium MOFs (Zr-MOFs) in particular UiO-66, UiO-67 and UiO-68 (ref. 5) have received astounding attention due to their assorted functional tunability with formidable thermal, chemical and mechanical stability. The strong Lewis acidic nature of Zr with higher oxygen affinity results in robust Zr–O bond formation, which lead to the formation of stable MOFs that arises from the strong backbone of organic and inorganic components.6,7 The building structure of these MOFs consists of hexanuclear Zr6O4(OH)412+ secondary building units (SBUs), wherein the 12 connected nodes bridged together with organic linkers giving rise to diverse geometrical features, topologies and crystal structures.8 The stability of MOF is decisively contributed by the SBUs due to their reversible characteristics between Zr6O4(OH)4 (hydroxylated) and Zr6O6 (dehydroxylated) structure.6
Among UiO MOFs, UiO-66 has been explored more for a variety of applications because of its unprecedented stability, porosity, specific surface area and catalytic properties.9,10 The UiO-66 structure comprises of two cages, tetrahedron and octahedron with typical cage size of 7.5 Å and 12 Å, wherein the face of octahedral cages partaking with 8 tetrahedral cages and the edge partaking with additional 8 octahedral pores.11 The core of UiO-66, Zr6(μ3-O)4(μ3-OH)4, contains six Zr atoms occupying the octahedron vertices, in which four of its faces are hydroxylated and remaining four are in oxide state.12,13 This structural arrangement has inherent detoxification characteristics through the prevalent reaction mechanism, which involves the direct coordination of nerve agents to the Zr atoms and formation of P–OH bond via nucleophilic addition.14 In addition, studies have shown that those nerve agents containing organophosphorus compounds likely to have more than one adsorption sites in UiO-66, but the same reaction is not quite possible in the pristine structure of UiO, since all the Zr atoms in SBUs are completely coordinated. Yet, the establishment of defective SBUs by removal of one linker molecule leave the two octacoordinated Zr atoms to be uncoordinated thereby facilitates the enhanced reactive nature of UiO-66.
It is desired that physiochemical properties of UiO-66 can be fine-tuned towards various applications due to the versatility of various synthesis approaches such as solvothermal,15 modulated synthesis,8 isoreticular synthesis16 and mixed ligand approach.17 As further insight, reaction temperature,18 molar concentration,19 choice of solvent and modulator20,21 have their own influence in re-designing the resultant MOFs characteristics. Notably, the suitable choice of modulators typically, (i) monocarboxylic acids (coordination modulators) or (ii) inorganic acid species (protonation modulators) in the due course of synthesis process greatly influence the crystallite size of MOFs.22,23 In addition, modulators play a major role in the formation of catalytic sites in terms of creating missing-linker and missing-cluster defects.24 It is known that missing linker defects denote the removal of organic linkers from the structure and thereby leaving coordination vacancies between two adjacent metal clusters.25 On the other hand, missing cluster defects are generated due to the elimination of Zr6O4(OH)412+ SBUs that coordinate with the twelve linker molecules.26 Meticulously, the acidity constant (pKa) and optimal molar ratio of modulators are the significant parameters that have dominance towards the crystallization process and defect formation.27 The creation of such defects either would be a missing linker or missing cluster that obviously engendering larger cavities within the crystal structure and allows with guest molecules interaction.28 Also, the role of synthesis temperature and an inversely proportional relation between the temperature and density of defect sites was inferred and it is ascribed due to upsurge in the lability of strong Zr–O (carboxylate) bonds.29
Although, many upcoming research progresses about UiO-66 were documented, most of them pay particular attention either to their material characteristics or catalytic degradation performance. The insights on the pathway of material characteristics towards CWAs detoxification are yet to be explored. In this background, a series of UiO-66 MOF catalysts was synthesized with respect to its synthesis parameters namely acidity constant of modulator, HCl concentration, water molecules and synthesis temperature. The extent to which these considered parameters contributed to the physicochemical properties of the final product by means of its analytical results altogether were explored cumulatively, which has not yet been demonstrated to certain extent. In addition, the effects of those resultant structural reconstructions on their catalytic degradation performance were estimated. The detoxification artifice of nerve agents was chosen due to its highest in vivo toxicity. Since the use of nerve agents are perilous, paraoxon-methyl (an equivalent simulant to nerve agents) has been considered as the use of corresponding simulants which greatly abridges the experimental procedure.
2. Experimental section
2.1 Materials
Zirconium(IV) chloride (ZrCl4, M.W.: 233.04 g mol−1) and terephthalic acid (H2BDC, 98%, M.W.: 166.13 g mol−1), paraoxon-methyl PESTANAL™ analytical standard (DMNP, M.W.: 247.14 g mol−1), 4-ethylmorpholine (N-EM, M.W.: 115.17 g mol−1) were purchased from Sigma Aldrich, USA. N,N-Dimethylformamide (dry DMF, 99%, M.W.: 73.09 g mol−1) and acetone (99%, M.W.: 58 g mol−1) were purchased from AVRA, India. Acetic acid (AA, glacial 100%, M.W.: 60.05 g mol−1) and hydrochloric acid (HCl, 37%, EMPLURA®) were purchased from MERCK, India. Formic acid (FA, M.W.: 46.03 g mol−1) was procured from SPECTRUM, India. 4-Nitrophenol/p-nitrophenoxide (C6H5O3N, M.W.: 139.11 g mol−1) extra pure AR was procured from SISCO Research Laboratories Pvt. Ltd, India. UV treated de-ionized water was supplied by MS Technologists (India).2.2 UiO-66 synthesis
UiO-66 was synthesized via solvothermal technique with the precursors of ZrCl4/H2BDC in an equivalent ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Two different precursors were prepared with the following description; solution set #1: ZrCl4 (6 mmol) was dissolved in DMF in a borosilicate reagent bottle and corresponding modulators (M = AA (8 eq.), FA (8 eq.), HCl (X = 4 to 12 eq.) in relative to the H2BDC precursor concentration and water (63 & 100%)) of appropriate amount was added into it separately for each individual synthesis reactions under constant stirring for 20 min and subsequent sonication for another 20 min at room temperature was performed. In terms of MOFs synthesis via modulator assisted pathway, the reaction will be done under acidic conditions to evade the metal oxides/hydroxides formation as well as to keep adequate concentration of metal complexes for the hybrid framework formation. Thus, the role of modulators in the growth process of MOF crystals were analyzed by varying different modulators in terms of their acidity constant (pKa). Solution #2: H2BDC (6 mmol) was separately dissolved in DMF in a beaker under stirring and kept under sonication for 20 min. Again, under stirring, solution #2 was mixed with the samples of solution set #1 under stirring and the same environment is maintained for the next 20 min. Then, the reagent bottle was tightly sealed with screw cap and placed in a hot air oven for 24 h at different synthesis temperatures (T = 80, 100 and 120 °C). After 24 h, the reaction gets completed and the solution was allowed to cool down to room temperature. Afterwards, the samples were centrifuged to decan the mother solvent and washed with fresh DMF, and acetone (3× times) each to remove the unreacted solvent molecules. Finally, the samples were dried at 80 °C for 24 h under vacuum (600 mm. Hg) condition. The appropriate synthesis conditions are listed in Table S1.†
:1. Two different precursors were prepared with the following description; solution set #1: ZrCl4 (6 mmol) was dissolved in DMF in a borosilicate reagent bottle and corresponding modulators (M = AA (8 eq.), FA (8 eq.), HCl (X = 4 to 12 eq.) in relative to the H2BDC precursor concentration and water (63 & 100%)) of appropriate amount was added into it separately for each individual synthesis reactions under constant stirring for 20 min and subsequent sonication for another 20 min at room temperature was performed. In terms of MOFs synthesis via modulator assisted pathway, the reaction will be done under acidic conditions to evade the metal oxides/hydroxides formation as well as to keep adequate concentration of metal complexes for the hybrid framework formation. Thus, the role of modulators in the growth process of MOF crystals were analyzed by varying different modulators in terms of their acidity constant (pKa). Solution #2: H2BDC (6 mmol) was separately dissolved in DMF in a beaker under stirring and kept under sonication for 20 min. Again, under stirring, solution #2 was mixed with the samples of solution set #1 under stirring and the same environment is maintained for the next 20 min. Then, the reagent bottle was tightly sealed with screw cap and placed in a hot air oven for 24 h at different synthesis temperatures (T = 80, 100 and 120 °C). After 24 h, the reaction gets completed and the solution was allowed to cool down to room temperature. Afterwards, the samples were centrifuged to decan the mother solvent and washed with fresh DMF, and acetone (3× times) each to remove the unreacted solvent molecules. Finally, the samples were dried at 80 °C for 24 h under vacuum (600 mm. Hg) condition. The appropriate synthesis conditions are listed in Table S1.†
2.3 Buffer and stock solution
To execute the detoxification process, 0.45 M of N-EM buffer solution (pH = ∼10.2) was prepared and utilized as it is throughout the studies. An aqueous solution of DMNP (0.025 mmol) stock was separately prepared using 0.45 M N-EM and stored. Typically, 6.7 mg of as-synthesized UiO-66 powder was added to three 1.5 mL centrifuge tube, each containing 0.5 mL of N-EM aqueous solution (0.45 M) at room temperature and subjected to sonication for about 20 min. Then, the prepared analyte DMNP solution was added to the above suspension and ten microliters of aliquot was taken from the same at regular time intervals, diluted with 5 mL of N-EM buffer to find out the extent of reaction. The progressive rate of reaction was observed with UV spectrometer and the changes in absorbance intensity were monitored between the wavelength range of 200 and 500 nm. The changes in peak intensity at 403 nm, an indicator of 4-nitrophenol/p-nitrophenoxide (4-NP) formation was focused to evaluate the catalytic degradation performance.2.4 Instrumental techniques
Powder X-ray diffraction (PXRD) patterns for the synthesized UiO-66 sample was recorded using the Bruker D8 Focus X-Ray Diffractometer (Cu Kα source λ = 1.5406 Å, 40 kV, 40 mA Japan). High resolution-scanning electron microscope (TESCAN, Czech Republic) and Transmission Electron Microscope (TEM, JEOL 2100F, Japan) were used to evaluate the morphological features. X-ray photoelectron spectrometer (XPS, K-ALPHA, Thermo Scientific, UK) having monochromate Al Kα radiation of 1486.6 eV energy was utilized to determine and quantify the chemical states of elements. Functional group analysis spectra were acquired from Bruker Alpha – T Fourier Transform Infrared Spectrometer (FTIR), Germany. The spectra were recorded between 4000 and 500 cm−1 in transmission mode and accumulating 48 scans with a spectral resolution of 4 cm−1. The Brunauer–Emmett–Teller (BET) surface area and porosity of the samples were probed using ASAP 2020 porosity analyser (Micromeritics, Australia). All the samples were degassed prior to analysis and N2 adsorption–desorption isotherms were carried out at 77 K. Thermogravimetric analysis (TGA) was carried out using TA Instruments (Model: SDT Q600), USA. The analysis has been carried out between the temperature range of 25 and 950 °C with a temperature ramp of 20 °C min−1. Finally, the catalytic degradation studies have been performed using UV-vis spectrophotometer (Evolution 201, Thermo Scientific).3. Results and discussion
3.1 Crystal and structural studies
PXRD patterns of as-synthesized UiO-66 samples shown in Fig. 1 confirmed the crystalline nature through the characteristic diffraction peaks indexed to (1 1 1), (2 0 0), (6 0 0), (7 1 1), and (6 6 4) planes, which are in well agreement with the isostructural UiO-66 cubic crystal structure (information card no.: 4512072).30 The calculated lattice parameters are enumerated in Table S2.† The HCl modulated samples exhibited greater crystallinity in relative to the AA and FA modulated samples as shown in Fig. 1(a). In this study, modulators with decrease in pKa values (AA = 4.76 < FA = 3.74 < HCl = −6.3) lead to greater crystal growth. The introduction of HCl to the reaction medium accelerated the solubility of ZrCl4 (ref. 31) and the counterpart of HCl, i.e., water as a source of protons provided the bridging O2− and OH− ligands,32 which in-turn facilitated the formation of Zr4+ oxo/hydroxo SBU clusters.33 The addition of H2BDC linker to this reaction mixture resulted in the formation of multinuclear clusters and UiO-66 crystal structure in the elevated temperature environment.34 This controlled nucleation was due to the presence of stronger HCl acidic condition, which favoured the protonation of H2BDC linker molecules, as it retarded the dissociation of H2BDC to BDC.35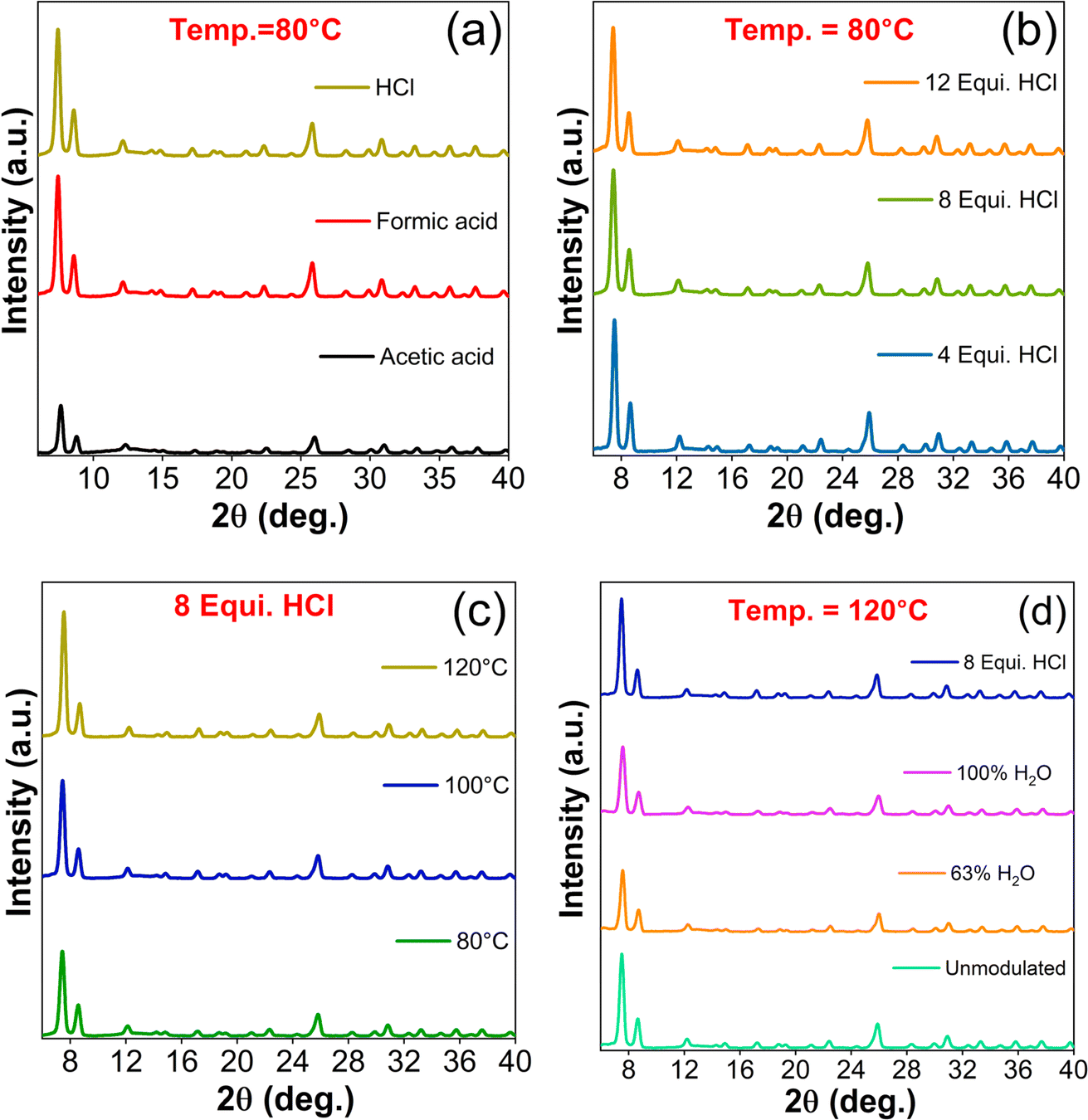 | ||
| Fig. 1 PXRD patterns of UiO-66 samples resulting from various synthesis conditions; (a) modulators with different pKa values (acetic, formic and HCl), (b) different equivalents of HCl at 80 °C, (c) various synthesis temperatures for 8 equivalence of HCl, and (d) comparison of unmodulated UiO-66 with water and HCl modulated samples. | ||
On the other hand, the monocarboxylic nature of AA and FA tend to bind with metal nodes competing linker molecules resulted in lesser crystallinity.35 Also, it has been observed that HCl modulated samples get precipitated completely as the end product whereas, unmodulated and organic acids modulated samples remained in colloidal state in accordance with the earlier claim.11 Therefore, it is confirmed that the addition of HCl hustles the reaction towards product formation either by conditioning the solvent DMF and/or supporting the Zr6 clusters preformation, leading to the formation of highly crystallized UiO-66.
Further, the role HCl equivalence in the formation of UiO-66 was investigated by varying it from 4 to 12 equivalence in steps of 4. The escalation of HCl concentration from 4 to 12 equivalence decreased the relative intensity of characteristic peaks (Fig. 1(b), S1(a & e)†). The increase in deprotonation of carboxylic linker (i.e., partial deprotonation) with an increase in HCl concentration at 80 and 100 °C, might have attributed to lower crystallinity. In other words, diminutive of the reaction between Zr clusters and ligands might have concomitantly induced the formation of defects, which were observed in the (2 0 0) reflection intensity.11 When the temperature was increased to 120 °C, the above observed phenomena got spurned wherein the (1 1 1) reflection started to grow and (2 0 0) reflection endured with minimal intensity variation (Fig. S1(b)†).
The PXRD patterns of synthesized UiO-66-T (T from 80 to 120 °C) samples with HCl modulator followed an increase in (1 1 1) reflection intensity in proportion to the synthesis temperature and vice versa for (2 0 0) plane, which accounted for the missing linker. In line with defects generation, the increase in temperature favoured the crystallization irrespective of HCl concentration. In specific, the intensity of (1 1 1) plane increased to a greater proportion when the synthesis temperature was changed from 80 to 100 °C (Fig. 1(c) & S1(c)†). For 12 equivalence of HCl, (2 0 0) plane intensity was immune to change with respect to synthesis temperature and turned up to the dormant phase (Fig. S1(d & f)†). In overall, the synthesis of UiO-66 at low temperature with higher HCl concentration and/or lower HCl concentration at high temperature may beneficial to fine-tune the concession between the crystallinity and defect states.
In order to further elucidate the acidic environment of reaction medium, set of experiments were carried out without the addition of HCl. The samples synthesized at 80 and 100 °C without HCl showed a negligible yield due to the deficiency of thermal activation and inadequacy of water source for the formation of Zr4+ oxo/hydroxo clusters.33 Hence, it is proven that the role of water molecules presents in HCl aqueous solution have an irreplaceable impact in structuring the MOF crystals. To explicate the need of water content, one sample was added with 0.89 mL of water (63% of water in 8 equiv. of 37% aqueous HCl) and another with 1.39 mL (to mimic the previous situation with 0% of aqueous HCl). Among them, the absence of HCl favoured the UiO-66 formation, nevertheless, the intensities of characteristic reflections were much lower than that of unmodulated and HCl modulated samples (Fig. 1(d)). This trend to a greater extent supported the proposed hypothesis that water molecules as proton source alone is not sufficient in the formulation of crystalline UiO-66 but the harmonious action between water and HCl protonation features plays a crucial role in achieving the desired structural diversity of UiO-66 MOFs. In addition, suitable choice of modulator pKa values, their concentration and the synthesis temperature essentially decide the UiO-66 crystallinity and its structural defects.
3.2 Topology studies
The morphologies of as-synthesized samples were observed using SEM. In Fig. 2(a–c), HCl modulated UiO-66 MOFs unveil the large number of asymmetrical agglomerated spheroidal particles rather than regular octahedral morphology.36 It could be presumed that the agglomeration of each individual UiO-66 particles with other has led to the bigger complexes. In case of other organic acids (AA, FA), the formation of spherical like structures continues with a greater number of ordered smaller grains (Fig. 2(d & e)) and the crystal growth remains in intermittent stage to octahedron crystal structure. In spite of the increased synthesis temperature (80 to 120 °C), the obtained irregular spheroidal structure (Fig. 2(f & g)) remains invariant thereby emphasized the role of HCl concentration dominance over temperature. TEM analysis was done to further confirm the internal structural arrangement of samples. From micrographs (Fig. S2†), it was found that all HCl equivalence (4, 8, 12) samples have exhibited the irregular spheroidal structure. Also, for the samples synthesized with respect to synthesis temperature, it was found that irregular spheroidal continues to aggregate to form bigger particles. | ||
| Fig. 2 SEM images of UiO-66 samples: HCl modulated samples at 80 °C of 4, 8, 12 eq. (a–c), acetic and formic acid modulated samples (d & e), 8 eq. of HCl modulated samples at 100 °C and 120 °C (f & g) and unmodulated & water modulated samples at 120 °C (h & i). | ||
In the due process of synthesizing unmodulated samples, the regular octahedron morphology is scarcely obtained at 120 °C (Fig. 2(h)) and no yield was obtained at lower temperatures (80 & 100 °C). In addition, UiO-66 synthesised with water modulated exhibited the truncated pebble structure (Fig. 2(i)) thereby denoted the incomplete crystal formation. Succinctly, it is discernible that, the aggregation of multi-clusters has been occurred in greater extent in the modulated UiO-66 samples before the occurrence of complete crystallization.
3.3 Elemental composition
XPS analysis was carried out for the synthesized samples to confirm the chemical states as well as the chemical composition of UiO-66 and evaluated the changes in composition with respect to their synthesis conditions. Since the survey spectra and their corresponding high-resolution spectra follows same trend except with the change in their composition ratio. The trend of survey and the deconvoluted spectra has been given in Fig. S3.† The exact variation in composition has been picturized in Fig. 3 and S3.† The survey spectrum of each synthesized sample has revealed the presence of Zr, C, and O elements (Fig. S3(a)†). The high-resolution spectra of each element have been deconvoluted into multiple peaks to discern the transition in their chemical composition and the explanation is as follows. The characteristic peak of Zr valence states is attributed to Zr 3d5/2 and Zr 3d3/2, which possess the binding energy difference of 2.3 eV (Fig. S3(b)†) and confirmed the presence of zirconium-oxo cluster (Zr4+). Likewise, deconvoluted O 1s spectra (Fig. S3(c)†) showed three peaks in the order of COOH/Zr–O–H, Zr–O–C and Zr–O–Zr binding. The C 1s spectra (Fig. S3(d)†) were split into three peaks, which are ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O and C–C. An ensemble of all the samples and elemental composition with their corresponding atomic ratio percentage are listed in Table S3.† Among them, changes in the elemental composition of O 1s spectra were estimated to evaluate the defects formed in the series of UiO-66 samples synthesized in different experimental conditions. Three distinct peaks in the O 1s spectra aforementioned are assigned to the oxygen containing species namely coordinated free carboxylate group/hydroxy group, coordinated carboxylate group and the bridging ligand groups at the Zr6 node.
O, C–O and C–C. An ensemble of all the samples and elemental composition with their corresponding atomic ratio percentage are listed in Table S3.† Among them, changes in the elemental composition of O 1s spectra were estimated to evaluate the defects formed in the series of UiO-66 samples synthesized in different experimental conditions. Three distinct peaks in the O 1s spectra aforementioned are assigned to the oxygen containing species namely coordinated free carboxylate group/hydroxy group, coordinated carboxylate group and the bridging ligand groups at the Zr6 node.
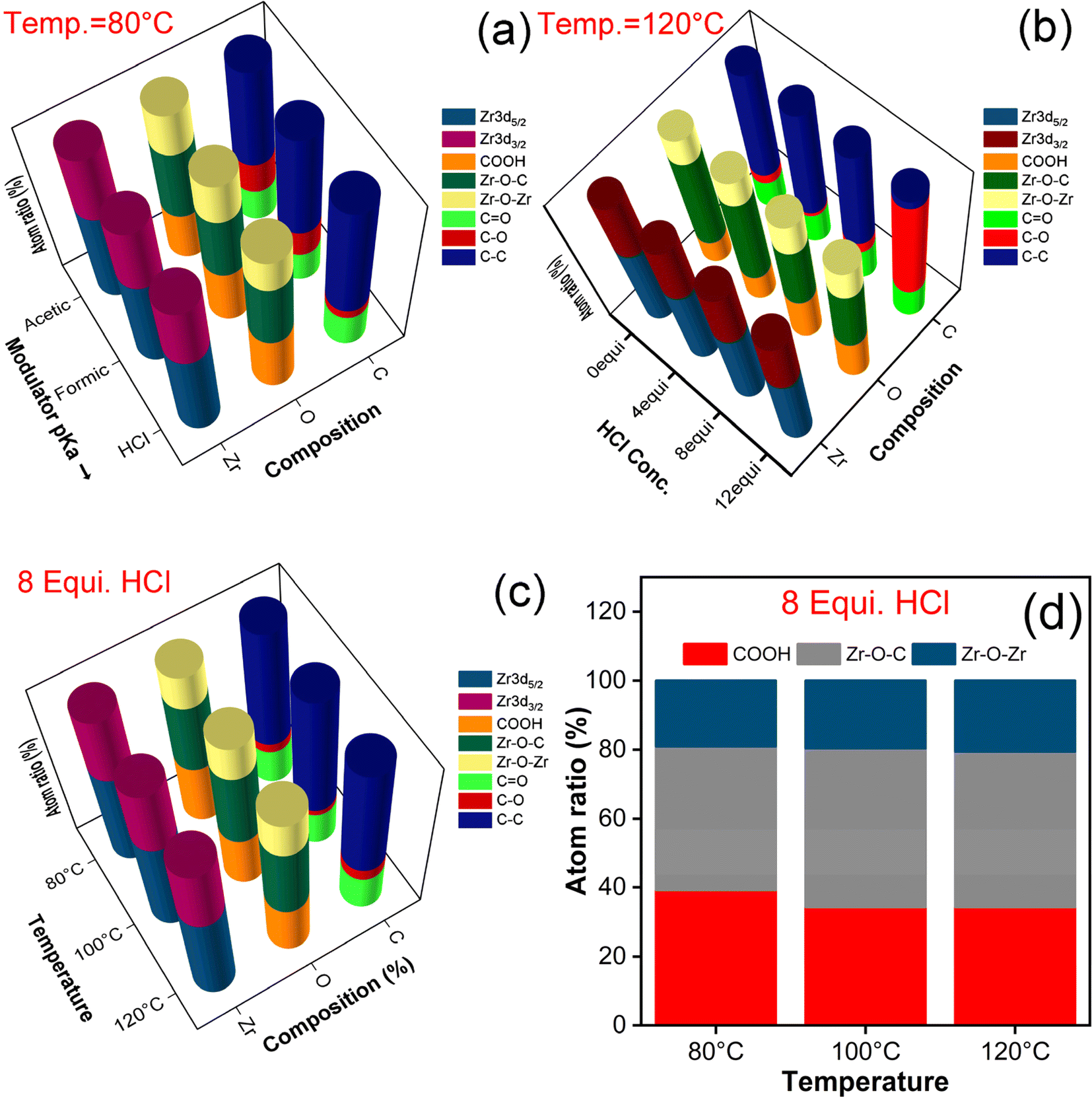 | ||
| Fig. 3 The elemental composition of Zr, C, and O in UiO-66 samples of different batches characterized by XPS: (a) effect of modulators pKa, (b) increase of HCl equivalence, (c) increase of synthesis temperature, and (d) estimation of uncoordinated carboxyl group. | ||
The existence of coordinated free carboxylate groups designate the presence of missing cluster/linker defects in the samples. Even in case of missing cluster defects, the elimination of metal node is accompanied with the elimination of bridged ligands associated with them, which in-turn can be pronounced cohesively as missing linker defects.37 Hence, the estimation of COOH composition will be directly correlated to the relative density of defects in the corresponding samples.38 In, an increased amount of COOH with the decrease of modulator pKa values (AA < FA < HCl) confirmed the dominance of HCl in creating defects in UiO-66. The proportionality of defects formation is in direct variation with HCl equivalence for each synthesis temperature (80, 100, & 120 °C) as evidenced in Fig. 3(b), S4(a–b & f).† Also, it is to be noted that the unmodulated sample exhibited the low level of COOH presence than modulated samples, which resembled the complete coordination of ligand to the inorganic moiety depicting the near defect free sample (Fig. S4(c)†). In addition, the availability of COOH concentration for water modulated samples was varied incrementally upon the stoichiometric addition of water molecules and the COOH range was nearly same with HCl modulated samples at 120 °C. However, the concentration of COOH is inversely corelated to synthesis temperature; increase in temperature leads to decline in defect formation as seen in Fig. 3(c, d), S4(d & e)† and is in good agreement with the PXRD results. Again, it is proven that increase in HCl concentration leads to generation of defects but the synthesis temperature hinders the formation of defects.29
3.4 Functional group analysis
FTIR spectra of UiO-66 samples were observed for different combinations of synthesis parameters as illustrated in Fig. 4(a–d), which shows similar characteristic peak patterns except the variation in their intensity. The strong, broad peak at 3404 cm−1 represents the stretching vibration of O–H bond, arises due to the adsorbed water and deprotonated carboxylic acids.36 The peaks positioned at 1583 and 1506 cm−1 attributes to CC stretching vibration of phenyl ring in the H2BDC linker molecules.39,40 The peaks at 1657 and 1400 cm−1 ascribes the asymmetrical and symmetrical stretching vibrations of CO bond respectively. In specific, the strong band of about 1400 cm−1 confirmed the existence of reaction between Zr4+ and –COOH that relates to free carboxylic acid group in the framework of υas (COO−) mode.41 The bands at 748 and 665 cm−1 relate to the formation of corresponding symmetric and asymmetric vibration of O–Zr–O bonding42 further confirmed the formation of Zr6(OH)4O4 clusters.43 Besides the shape/intensity of unmodulated UiO-66 spectrum is meagre than the modulated one in the same region depict its defect free characteristics. For instance, the peaks at 3404, 1657 and 1400 cm−1 which represent the O–H and CO stretching vibrations are very broad and less intense unlike the modulated samples.
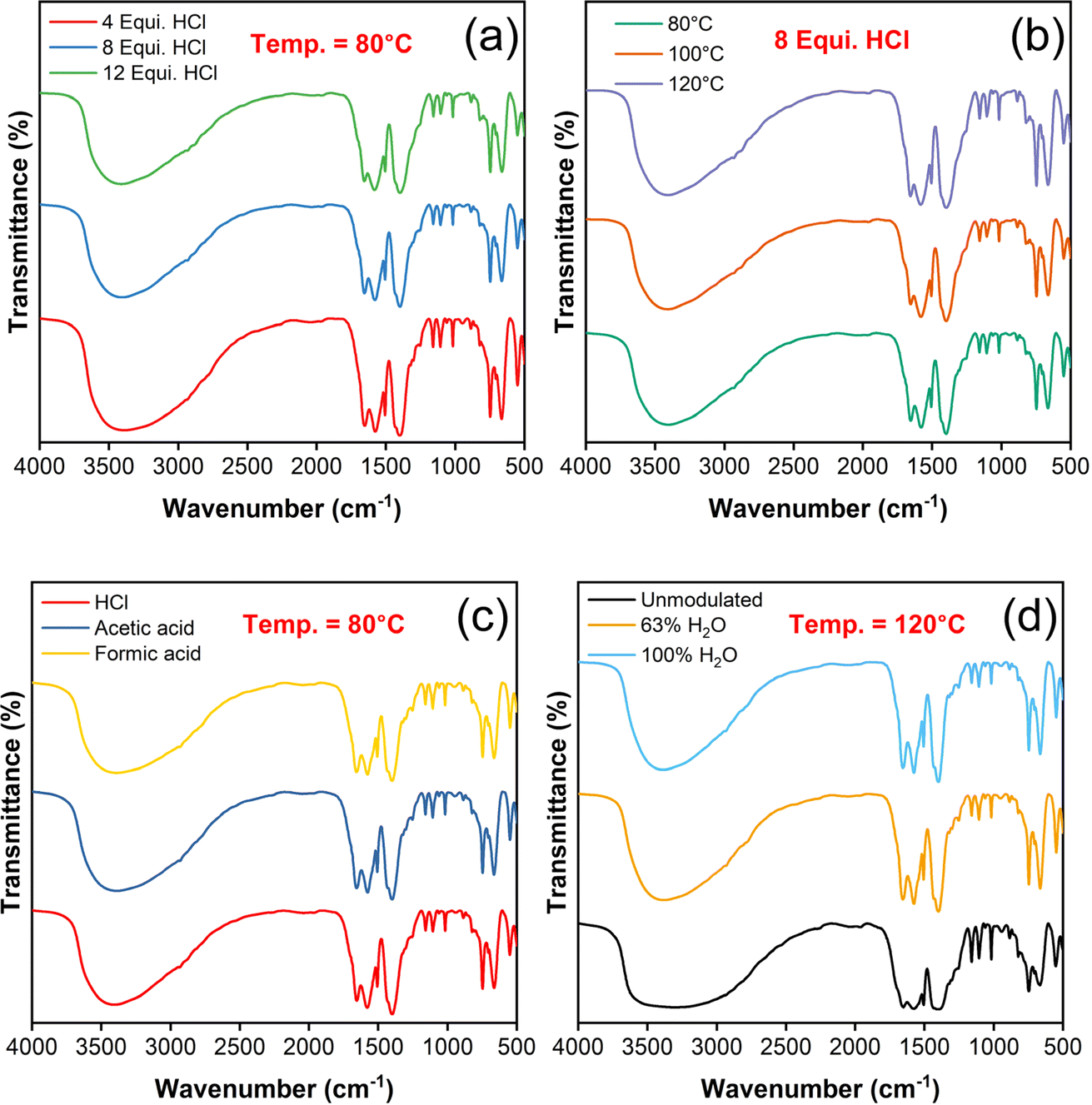 | ||
| Fig. 4 FTIR spectra of the UiO-66 samples: change in (a) HCl equivalence, (b) synthesis temperature, (c) modulators and (d) unmodulated & water modulated samples. | ||
3.5 Texture analysis
The N2 adsorption–desorption behaviour of as-synthesized UiO-66 samples were carried out to probe the BET surface area and pore structures. The estimated range of surface area of the samples were between 597.47 and 882.27 m2 g−1 and all the samples exhibited type I isotherm behaviour as shown in Fig. 5(a–c) and S5(a–d).† The high nitrogen uptake at relatively low pressure confirmed the presence of micropores,44 and convexity of adsorption curve at P/Po > 0.8 indicates the presence of mesopores wherein, the adsorption capacity has been increased slenderly at P/Po = 1.45 Thus, cumulatively accounted for the coexistence of micro- and mesopores structure. During desorption, pore evaporation and multi-layer film formation might have occurred.45 The declining trend in desorption curve and narrow down of difference between adsorption–desorption curve indicated the existence of narrow slit like pores including micropore region that offers large pore spaces.46 The change in surface area, pore volume and pore size are listed in Table S4.†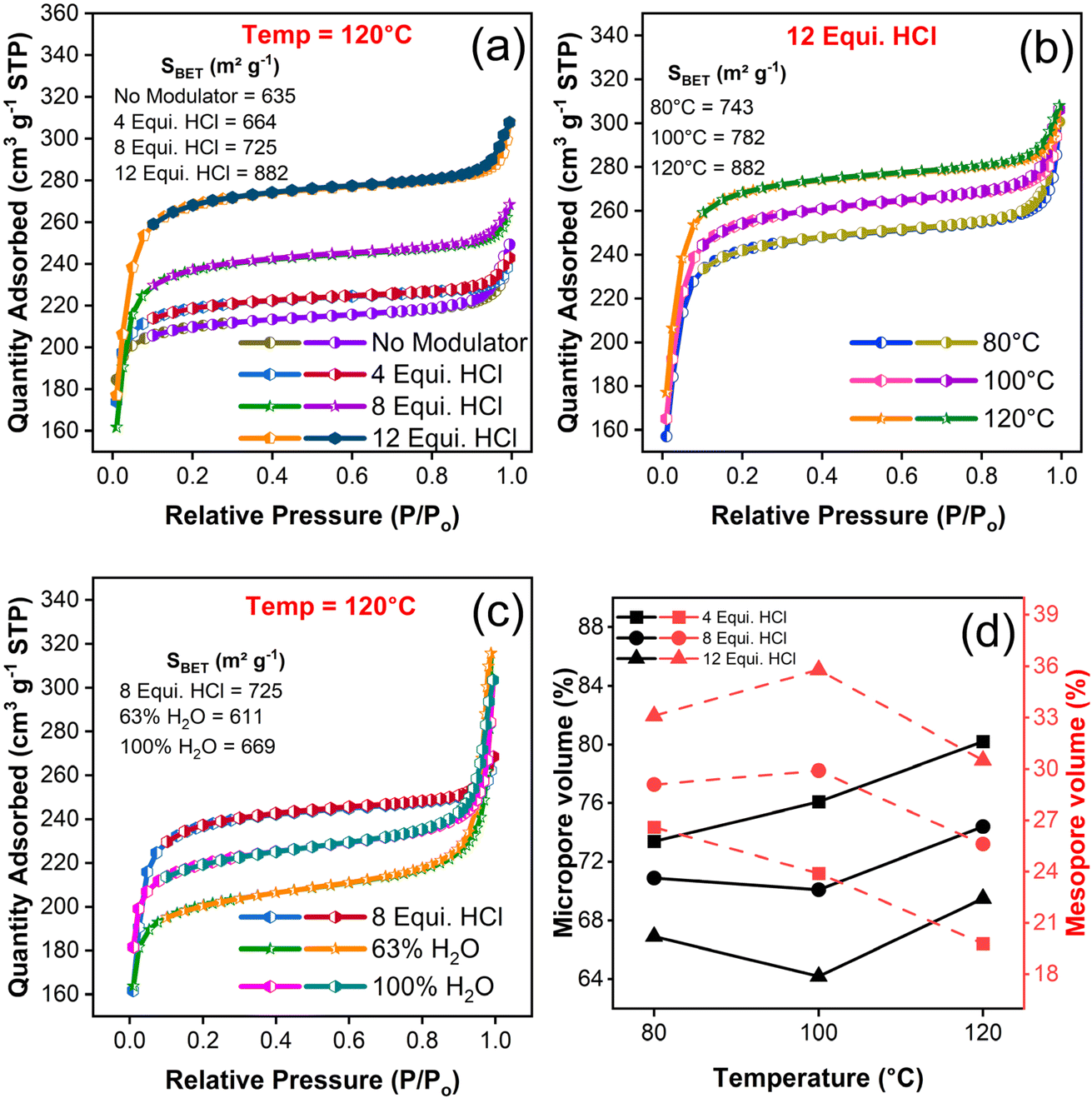 | ||
| Fig. 5 Textural possessions of the as-synthesized UiO-66. N2 adsorption–desorption pattern with respect to their (a) HCl concentration, (b) synthesis temperature, and (c) HCl & water modulation, (d) estimation of micro and mesopore volume across the range of HCl concentration and synthesis temperature. | ||
While analysing the samples in terms of BET surface area estimation, it has been observed that an increase in the amount of HCl concentration have increased the surface area of resultant samples11 (605 to 882 m2 g−1) irrespective of their corresponding synthesis temperatures. It has been inferred that the increase of HCl equivalence augmented the specific surface area in virtue of missing linker defects concentration and the formation of octahedral cavity within a framework structure (Fig. S5(e)†). In specific, 12 equivalence HCl modulated samples synthesized at 120 °C have posed a maximum surface area of 882 m2 g−1 as shown in Fig. 5(a). With respect to the increase of synthesis temperature, the values of surface area followed a sporadic trend (Fig. S5(f)†), whereas the synergetic effect of both HCl modulation and synthesis temperature at elevated levels pose a high surface area of 882.27 m2 g−1. Further, the synthesis modulation with HCl and temperature extended its clout in pores structure wherein, the proportion of pores genre between micro and meso range gets greatly tweaked. This observation may support the hypothesis that change in HCl concentration have influenced the pore structure in virtue of missing cluster or missing linker defects.47 Specifically, the increase of HCl concentration has augmented the magnitude of mesopore volume concomitantly, and vice versa in case of micropore volume and it can be observed in Fig. 5(d). These transition of micro to mesopores supports and confirms the existence of defects due to missing linker in UiO-66 structure.47
For the temperature modulated samples, decrease in the trend of total pore and mesopore volume were observed (Table S4†) to have inversely proportional relation with certain inconsistency. Besides, the surface area continues to increase with an increase in synthesis temperature, which represents the formation of defects in contradiction with the former assertion. On the debate about surface area and variation between defects generation could be claimed based on the hypothesis that the formation of more porous framework is in line with low TGA plateau as reported earlier.48 Aside, the unmodulated and water modulated samples which considered for comparison with HCl modulation, possess trivial contribution to the surface area of resultant samples (Table S4†). The water modulated samples exhibited a large N2 uptake at high relative pressure P/Po indicating the formation of mesopores as depicted in Fig. 5(c). With an increase of water content, the surface area of samples has increased up to 700 m2 g−1, yet less than HCl modulated one. However, the contribution of water molecules towards the formation of pore structures is quite substantial irrespective of elevated pore volume and it can be explained through the estimation of microporous surface area which decreased from 865 to 752 m2 g−1 with the increasing amount of water from 63 to 100% and the trend holds good with an earlier report.49 P. Chammingkwan et al.49 suggested that addition of water molecules during UiO-66 synthesis lead to the formation of missing cluster defects. Even though the mesopores are referred to defect formation, the overwhelming majority of micropores in the structure of all the HCl modulated samples have emphasised the controlled volume of defects. Also, the generation of such defects with missing linkers/open metal centres in a highly microporous environment may enhance the gas uptake at low pressure regime50 and the created mesopore may enable more interaction of guest species especially with bulk molecules.51 From all the above discussions, the dominance role of either HCl modulator or synthesis temperature in the formation of defect concentration has to be further explored, since the surface area and pore relation is not conclusive. Hence, it could conceivably be hypothesised that synergistic trade-off between the HCl concentration and synthesis temperature lead to perceive their relationship with defect concentration.
3.6 Thermal stability
To confirm the thermal stability and emergence of structural defects in UiO-66 samples with respect to the synthesis conditions, TGA was performed. The ideal defect free structural unit Zr6O4(OH)4 (BDC)6 of UiO-66 consisting of 6H2-BDC linker molecules being anchored with each Zr6 cluster. By subjecting the starting material (Zr6O4(OH)4 (BDC)6) to an increase of temperature gradient environment, the end product obtained (ZrO2) is in coherence with the previous studies as described in eqn (1) (ESI, Section S5†). In detail, the removal of various molecular species with the increase of sample temperature (Fig. 6(a)) follows the mentioned steps subsequently: (1) removal of anchored solvent molecules commonly referred to as desolvation occurred at 150 °C, (2) dehydroxylation of [Zr6O4(OH)4]12+ cluster to [Zr6O6]12+ happened at temperature closer to 300 °C, (3) H2-BDC ligand decomposed completely leaving behind dehydroxylated [Zr6O6]12+ cluster at 500 °C, and (4) finally, the formation of ZrO2 end product at 600 °C was in line with the previous report.28 Theoretically, the molecular weight of ideal dehydroxylated Zr6O6 (BDC)6 is approximately 1626.28 g mol−1 followed by reduction to ZrO2 (6 moles) due to complete combustion and it possess ∼739.34 g mol−1.52 The ratio in weight between the dehydroxylated and combusted compounds is 2.20 and thereby the weight of dehydroxylated Zr6O6 (BDC)6 is 220%, when the weight of end product (ZrO2) is normalized to 100%. The detailed calculations are ascribed in ESI (Section S5†).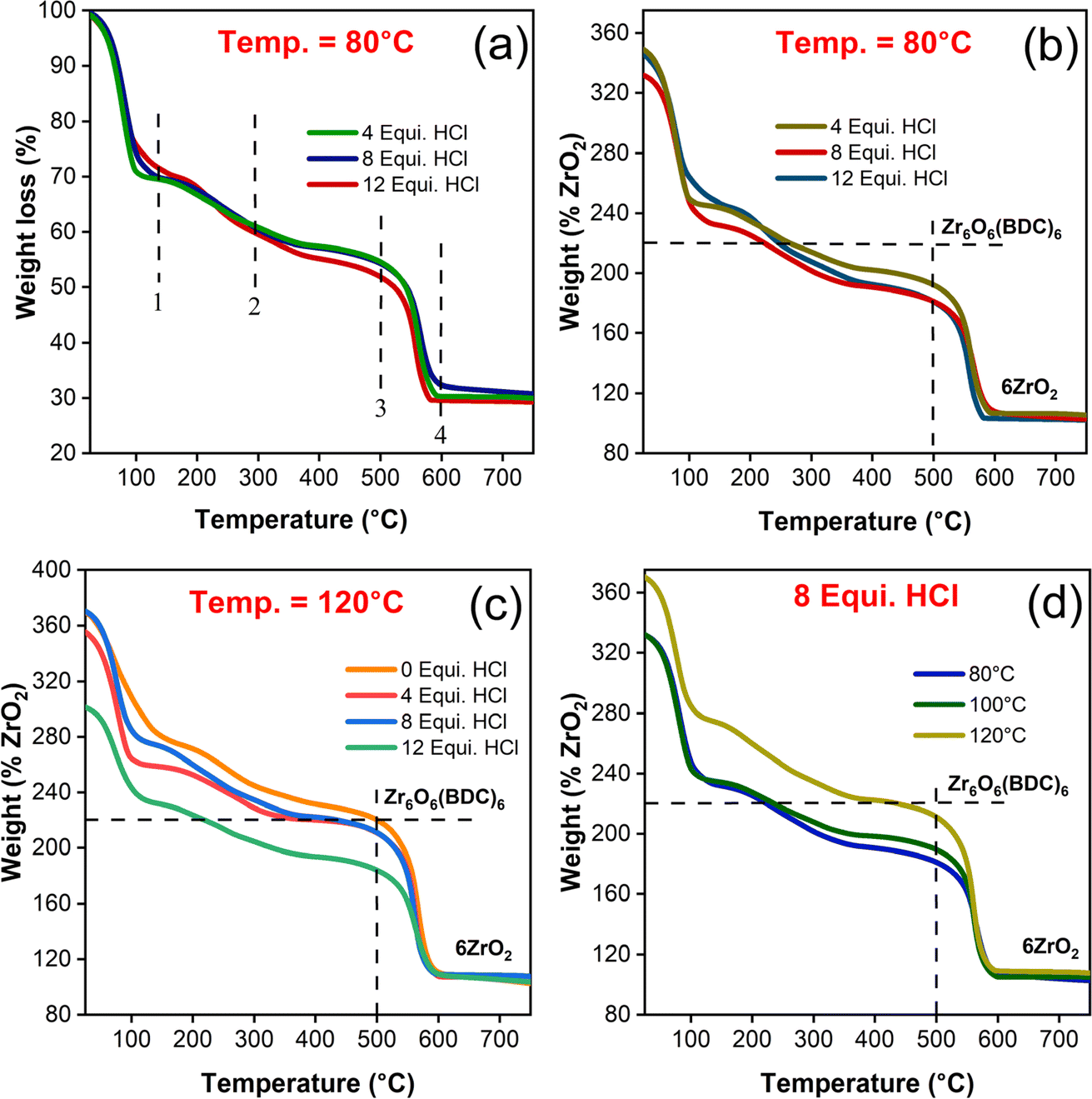 | ||
| Fig. 6 TGA curve of synthesized UiO-66 samples in different batches. (a) Change in weight loss of HCl modulated samples at 80 °C, (b) normalized weight of ZrO2 percentage for HCl modulated samples at 80 °C, (c) comparison of normalized ZrO2 percentage between unmodulated and HCl modulated samples, (d) change of ZrO2 percentage on changing the synthesis temperature. | ||
In view of the fact that weight loss due to combustion during heat treatment is due to the loss of linkers or the severity of linker deficiencies, which might be greater than inorganic cluster defects.53,54 Also, it is impassable to extricate between missing linkers and cluster defects from the TGA residues. It is indeed to be mentioned here that both the missing linkers and cluster defects introduce overall linker deficiencies to the framework. Thus, the material defectivity is fitted with the molecular formula Zr6O6+x(BDC)6−x, where x is the linker deficiencies per Zr6 in the presumed chemical composition. The number of linker deficiencies are estimated as per the calculation elucidated in ESI (eqn (4)†) and their corresponding values are tabulated in Table S5.† From the TGA plateau (Fig. 6(b, c) and S6(c)†), the magnitude of weight loss due to linker combustion is decreased with the increased HCl concentration, depicting the lack of linker coordination to Zr6 node, thus ultimately epitomizes the proportionality of missing linker with defects existence, as anticipated for modulator concentration.55
From Fig. S6(e),† it is elucidated that HCl modulated UiO-66 has more weight loss in which creation of linker deficiency are directly correlated to the modulators with decrease of pKa values. The unmodulated sample has shown the normalized weight loss of 219.87% which exactly matches with the theoretical value of 220% and exhibited the near defect free framework (Fig. 6(c)). With the introduction of HCl, the induced linker deficiency ranged between 0.4 and 1.9 per SBU has been created. For the water modulated samples irrespective of the percentage of added equivalence, the normalized TG curve of %ZrO2 is still less than 220% as shown in Fig. S5(d).† The estimation of defects with respect to the synthesis temperature have posed the inverse proportionality trend, as expected (Fig. 6(d), S6(a & b)†). The phenomenon for such instance is suggested that increase in synthesis temperature has influenced the Zr–O (carboxylate) bonds and tend to form a defect free structure.29 In addition, the complete decomposition of H2-BDC linker molecules in UiO-66 framework towards the linker deficiency estimation has been validated by measuring the FTIR spectrum (Fig. S7†) of resultant TG analyzed samples. From the FTIR spectrum, there is no evidence for the presence of H2-BDC linker molecules and designated that ZrO2 is the final residue. Hence, it could be concluded that, these linker deficiencies are catered by tailoring the synthesis temperature and concentration of HCl added during synthesis.
3.7 Degradation of methyl paraoxon
The catalytic performance of as-synthesized UiO-66 MOFs against the simulant DMNP at room temperature have been evaluated by monitoring the reaction process using UV-vis spectroscopy. The pH and concentration of buffer solution (N-EM) were fixed at ∼10.2 and 0.45 M wherein the catalytic degradation is beneficial with greater reaction rate as reported earlier.56 Upon the addition of UiO-66 as an active catalyst to a reaction medium, the hydrolysis of DMNP leads to the formation of 4-NP, whose absorption peak was found to be centered at 403 nm as shown in Fig. 7(a). UV-vis spectra show an increase in the intensity of peak corresponds to 4-NP with reaction rate and at the same time a decrease in DMNP concentration at 280 nm. The percentage of conversion by the respective UiO-66 catalyst is monitored with regular time interval at constant synthesis temperature, T = 80 °C (Fig. 7(b)) and at constant HCl equivalence of 12 (Fig. 7(c)), and other synthesis conditional samples (Fig. S8(a–d)†). Control experiments have been carried out in the absence of UiO-66, to evaluate the degree of catalytic performance during their presence. In addition, calibration studies for both the 4-NP and DMNP were done for concentrations ranging between 2.5 and 50 μM (Fig. 7(d) & S8(f)†). The obtained linear variation of six different concentrations of 4-NP is represented as inset in Fig. 7(d). Each data point plotted as a function of DMNP conversion with respect to reaction time is the mean value of the triplicated (3N) experimental results. The initial rate of hydrolysis (k) was calculated by plotting the natural log of average concentration of DMNP from triplicate measurements vs. time and the value of slope from the formfitting straight line have been taken as an initial hydrolysis rate. The value of half-life time (t1/2) can be calculated from the relation: t1/2 = ln2/k. The calculated values of k, t1/2 and the maximum percentage of conversion are summarized in Table 1.
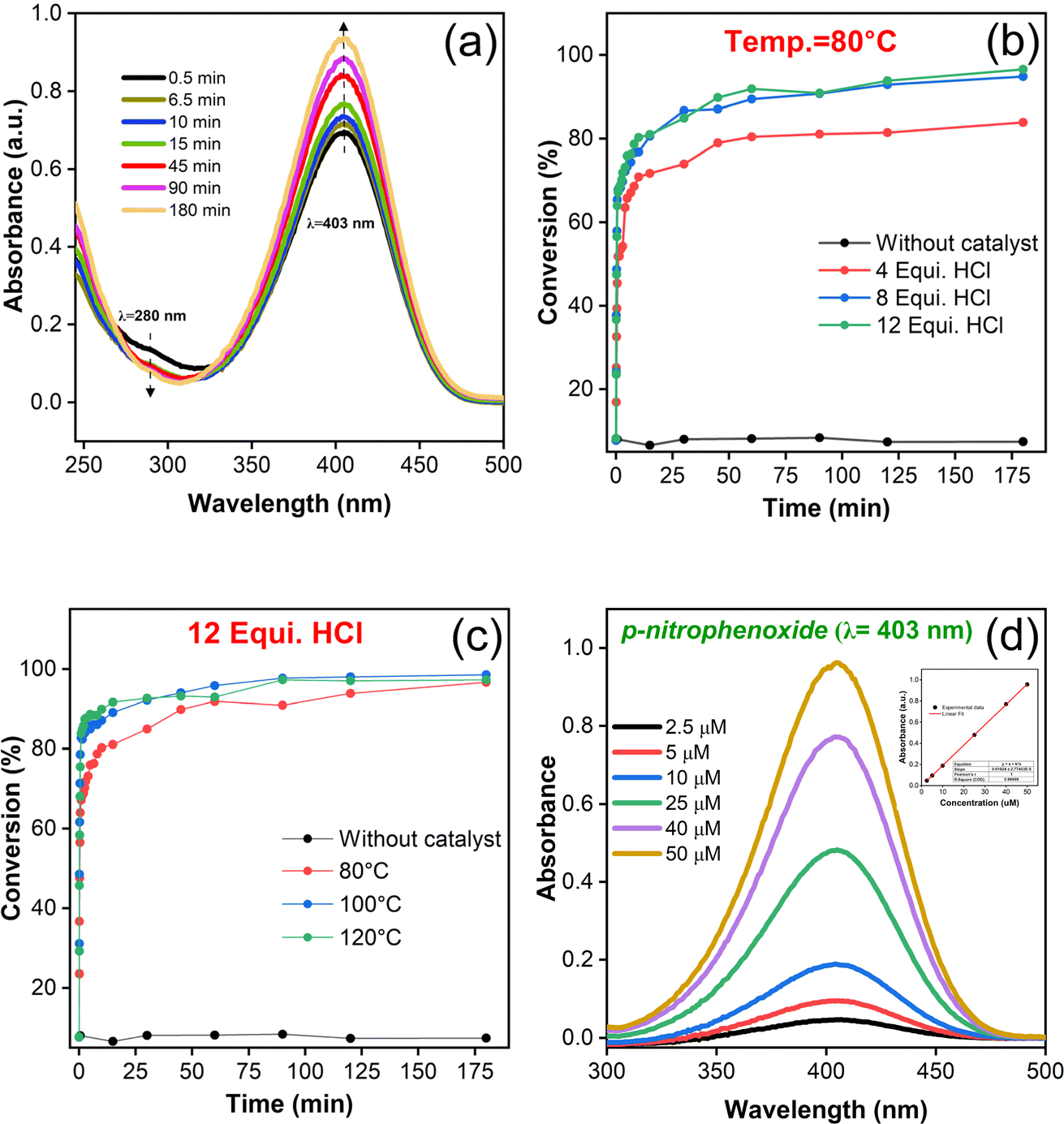 | ||
| Fig. 7 Catalytic hydrolysis of methyl paraoxon. (a) UV-vis spectrum of DMNP degradation catalysed by UiO-66 (8 eq. HCl @ 80 °C), (b) & (c) conversion profile of DMNP to 4-NP with respect to synthesis temperature & HCl equivalence, and (d) calibration of 4-NP for concentration reference. | ||
| Sample description | k1 (min−1) | t1/2 (min) | Conversion% | |
|---|---|---|---|---|
| T = 80 °C | 8 eq. AA | 0.421 | 1.64 | 89.8 |
| 8 eq. FA | 1.260 | 0.55 | 90.0 | |
| 4 eq. HCl | 1.049 | 0.66 | 83.9 | |
| 8 eq. HCl | 1.960 | 0.35 | 94.9 | |
| 12 eq. HCl | 1.881 | 0.36 | 96.6 | |
| T = 100 °C | 4 eq. HCl | 1.440 | 0.48 | 98.0 |
| 8 eq. HCl | 3.104 | 0.22 | 96.9 | |
| 12 eq. HCl | 2.922 | 0.23 | 98.5 | |
| T = 120 °C | 0 eq HCl' | 1.439 | 0.48 | 89.6 |
| 4 eq. HCl | 1.334 | 0.52 | 90.6 | |
| 8 eq. HCl | 1.077 | 0.64 | 90.6 | |
| 12 eq. HCl | 2.651 | 0.26 | 97.2 | |
| 63% H2O | 0.798 | 0.86 | 89.6 | |
| 100% H2O | 0.999 | 0.69 | 89.1 | |
From the estimation, it has been observed that the hydrolysis rate of DMNP to 4-NP aided with UiO-66 catalyst is quite instantaneous with the maximum conversion efficiency of 98.5% and half-life time of less than half a minute. Since, the first time-point value for measurement starts from 30 s, the measurement of such short half-time can be derived from fitting the first two initial data points (i.e., t = 0 & 30 s). The prompt of having such high conversion efficiency might be attributed to the availability of catalytic Zr active sites with more accessibility in the HCl modulated UiO-66 MOFs. The hydrolysis of DMNP molecule is instigated by the substitution of double bonded oxygen (PO) with a labile aqua ligand (Zr–OH2) in UiO-66. Further, negative charges on the leaving group is stabilized by hydroxyl ion (–OH ligand), which then attacks the phosphate ester leading to the cleavage of DMNP to dimethyl phosphonate and 4-NP.57 The possible DMNP hydrolysis pathway proposed in the literature58 is represented in Fig. 8. Hence, it can be elucidated that the mere presence of catalytic sites in the UiO-66 have contributed to the transcendent hydrolysis rate and maximum conversion efficiency.
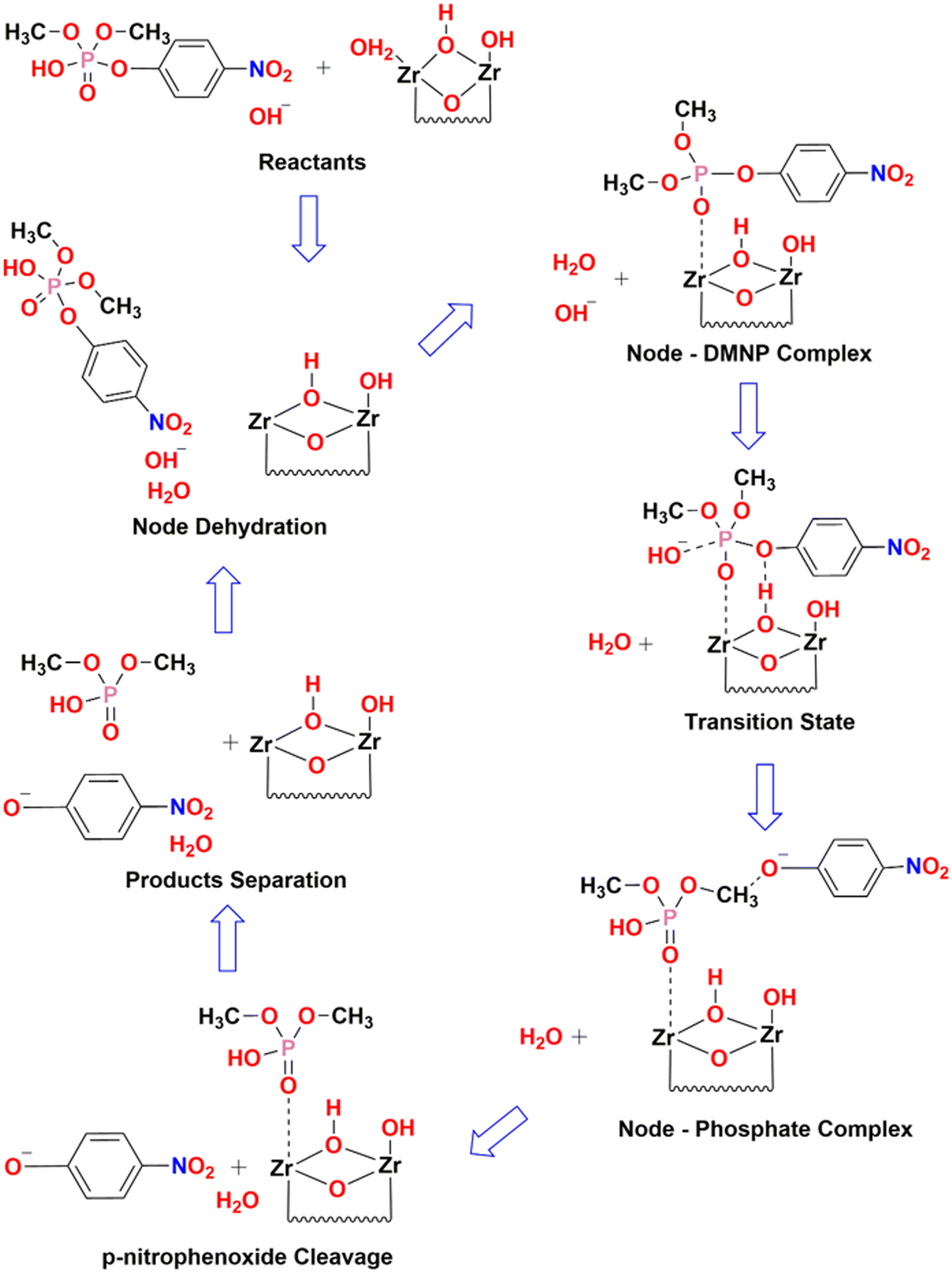 | ||
| Fig. 8 Catalytic hydrolysis pathway cycle of DMNP in N-EM buffer solution. | ||
On the basis of DMNP hydrolysis reaction rate, it is seen that HCl modulated samples have showed greater performance in terms of reaction rate, half-life time and conversion efficiency in comparison to an unmodulated sample (Fig. S8(b)†). For instance, the half-life value is significantly reduced to 0.26 min from 0.48 min (unmodulated UiO-66) after the introduction of HCl (12 eq.) for synthesis. From this observation, it is confirmed that HCl modulated samples uplifted the DMNP degradation in virtue of defects generation. With respect to various modulators pKa synthesized samples (Fig. S8(e)†), it was found that HCl modulated sample have exhibited a greater conversion efficiency of 94.9% in comparison to the AA (88.9%) and FA (90%) modulated samples.
To further analyze the trend, zeta analysis was carried wherein the values of zeta potential (mV) is nearly closer to neutral for HCl modulated sample (13.4 mV) in comparison to AA (31.6 mV) and FA (30.2 mV) modulated samples (Table S6†). The decrease of potential values for samples with decrease of modulator pKa denotes that sample modulated with low pKa value (HCl, −6.3) is slightly acidic and favours to greater degradation of DMNP. Also, an increase of HCl concentration in UiO-66 synthesis have contributed to their conversion efficiency performance (13.4 mV) with mere difference. However, the same constructive interdependence of conversion efficiency is not in terms of synthesis temperature. Also, the correlation between reaction kinetics and conversion efficiency is not litigable with each other; since it followed the tendency that higher reaction kinetics need not to provide the maximum conversion efficiency. The sample with lesser linker deficiency (0.45 from TGA) exhibited the maximum conversion efficiency and half-life of 97.2% and 16 s (0.26 min), respectively. On the other hand, sample with higher linker deficiency (1.95) showed a maximum conversion efficiency of 96.6% with a half-life of 22 s (0.36 min). The subtle differences in conversion performance by all synthesized UiO-66 MOFs with abundant catalytic sites have proven to be the promising candidate towards the catalytic detoxification of CWAs/simulants. It is important to note that the characteristic nature of instantaneous hydrolysis is a propitious one which holds to be UiO-66 as an exemplary contender.
4. Conclusions
Multiple batches of UiO-66 MOFs have been synthesized and tested towards the catalytic hydrolysis of simulant DMNP. The influence of modulators with respect to their pKa constant & concentrations, and synthesis temperature plays a vital role in fine tuning the morphology, crystallinity, surface area and defects of UiO-66. The introduction of HCl had a greater impact on regulating the metal–ligand coordination towards the framework construction at low synthesis temperature. The range of synthesis temperature had a dominance over the crystallization process as well as in the surface area enhancement. From the defects estimation, the value of missing linker defects was more pronounced at low synthesis temperature and this formulation is in line with the values obtained by PXRD and XPS. A series of HCl modulated UiO-66 samples have posed a greater catalytic performance by instantaneous hydrolysis of DMNP to 4-NP with a maximum conversion efficiency of 98.5%. The control experiments proved the detoxification/degradation of targeted molecules in the presence of UiO-66 MOFs. The insights gained from this study will be of high assistance to understand the route of synthesis parameters, which govern the catalytic degradation performance metrics. Moreover, the nearby complete conversion efficiency by as-synthesized UiO-66 through one pot solvothermal approach at relatively low temperatures pruned the need of high-pressure and elevated temperature for the synthesis of highly catalytic MOFs. The present study confirms previous findings and contributes additional evidence that suggests the catalytic performance is greatly influenced by its synthesis practices. Whilst this study partially substantiates the kinetic analyses, the findings suggest that further mechanistic insights from the subsequent studies in UiO-66 will make them as a forerunner in the selection of solid catalyst for CWAs detoxification. The method of UiO-66 MOFs construction in this work paves a way to exercise the same methodology in their isoreticular strategy with different functionalities towards the enhancement of catalytic performance.Data availability
Data will be made available on request.Author contributions
Selva Balasubramanian: methodology, data curation, original draft manuscript preparation; Arockia Jayalatha Kulandaisamy: formal analysis, and reviewing; John Bosco Balaguru Rayappan: conceptualization, supervision, reviewing and fund acquisition. All authors reviewed the results and approved the final version of the manuscript.Conflicts of interest
The authors declare no conflict of financial interests.Acknowledgements
Authors are grateful for the financial support from the Department of Science and Technology (DST), New Delhi under FIST project scheme (SR/FST/ET-I/2018/221 (C)) and CUB-CSR “Clean Energy & Air Project”, India. One of the authors, Mr Selva Balasubramanian wish to acknowledge Council of Scientific and Industrial Research (CSIR), New Delhi for providing him a Senior Research Fellowship (09/1095(0058)/2020 EMR-I). We extend our sincere thanks to SASTRA Deemed University, Thanjavur for infrastructure support to carry out this work.References
- V. Kumar, R. Goel, R. Chawla, M. Silambarasan and R. K. Sharma, J. Pharm. BioAllied Sci., 2010, 2, 220–238 CrossRef CAS PubMed.
- M. Imran, V. V Singh, P. Garg, A. Mazumder, L. K. Pandey, P. K. Sharma, J. Acharya and K. Ganesan, Sci. Rep., 2021, 11, 24421 CrossRef CAS PubMed.
- S. G. Ryu, M.-K. Kim, M. Park, S. O. Jang, S. H. Kim and H. Jung, Microporous Mesoporous Mater., 2019, 274, 9–16 CrossRef CAS.
- Y. Liu, A. J. Howarth, N. A. Vermeulen, S.-Y. Moon, J. T. Hupp and O. K. Farha, Coord. Chem. Rev., 2017, 346, 101–111 CrossRef CAS.
- J. Ru, X. Wang, F. Wang, X. Cui, X. Du and X. Lu, Ecotoxicol. Environ. Saf., 2021, 208, 111577 CrossRef CAS PubMed.
- J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642–5650 RSC.
- R. J. Marshall, C. L. Hobday, C. F. Murphie, S. L. Griffin, C. A. Morrison, S. A. Moggach and R. S. Forgan, J. Mater. Chem. A, 2016, 4, 6955–6963 RSC.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- D. Bůžek, S. Adamec, K. Lang and J. Demel, Inorg. Chem. Front., 2021, 8, 720–734 RSC.
- A. Dhakshinamoorthy, A. Santiago-Portillo, A. M. Asiri and H. Garcia, ChemCatChem, 2019, 11, 899–923 CrossRef CAS.
- M. Katz, Z. Brown, Y. Colón, P. Siu, K. Scheidt, R. Snurr, J. Hupp and O. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- P. Piszczek, A. Radtke, A. Grodzicki, A. Wojtczak and J. Chojnacki, Polyhedron, 2007, 26, 679–685 CrossRef CAS.
- Y. Feng, Q. Chen, M. Jiang and J. Yao, Ind. Eng. Chem. Res., 2019, 58, 17646–17659 CrossRef CAS.
- D. Troya, J. Phys. Chem. C, 2016, 120, 29312–29323 CrossRef CAS.
- L. A. Lozano, C. M. Iglesias, B. M. C. Faroldi, M. A. Ulla and J. M. Zamaro, J. Mater. Sci., 2018, 53, 1862–1873 CrossRef CAS.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC.
- G. González-Rodríguez, I. Taima-Mancera, A. B. Lago, J. H. Ayala, J. Pasán and V. Pino, Molecules, 2019, 24, 3656 CrossRef PubMed.
- G. Jajko, P. Gryta, P. Kozyra, M. Szufla, D. Matoga, D. Majda and W. Makowski, J. Phys. Chem. C, 2022, 126, 9185–9194 CrossRef CAS.
- A. Polyzoidis, S. Reichle, M. Schwarzer, C. G. Piscopo, S. Löbbecke and D. Boskovic, React. Chem. Eng., 2021, 6, 679–684 RSC.
- Y. Chen, X. Zhang, M. R. Mian, F. A. Son, K. Zhang, R. Cao, Z. Chen, S.-J. Lee, K. B. Idrees, T. A. Goetjen, J. Lyu, P. Li, Q. Xia, Z. Li, J. T. Hupp, T. Islamoglu, A. Napolitano, G. W. Peterson and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 21428–21438 CrossRef CAS PubMed.
- B. Shan, J. B. James, M. R. Armstrong, E. C. Close, P. A. Letham, K. Nikkhah, Y. S. Lin and B. Mu, J. Phys. Chem. C, 2018, 122, 2200–2206 CrossRef CAS.
- D. Jiang, C. Huang, J. Zhu, P. Wang, Z. Liu and D. Fang, Coord. Chem. Rev., 2021, 444, 214064 CrossRef CAS.
- K. Y. Cho, J. Y. Seo, H.-J. Kim, S. J. Pai, X. H. Do, H. G. Yoon, S. S. Hwang, S. S. Han and K.-Y. Baek, Appl. Catal., B, 2019, 245, 635–647 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CrossRef CAS PubMed.
- Z. Hu, I. Castano, S. Wang, Y. Wang, Y. Peng, Y. Qian, C. Chi, X. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CrossRef CAS.
- M. Taddei, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS.
- M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1357–1361 CrossRef CAS.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14, 5370–5372 CrossRef.
- D. Yin, C. Li, J. Liu and C. Liang, ChemistrySelect, 2022, 7, e202201106 CrossRef CAS.
- M. G. Goesten, M. F. de Lange, A. I. Olivos-Suarez, A. V Bavykina, P. Serra-Crespo, C. Krywka, F. M. Bickelhaupt, F. Kapteijn and J. Gascon, Nat. Commun., 2016, 7, 11832 CrossRef CAS PubMed.
- F. Ragon, P. Horcajada, H. Chevreau, Y. K. Hwang, U.-H. Lee, S. R. Miller, T. Devic, J.-S. Chang and C. Serre, Inorg. Chem., 2014, 53, 2491–2500 CrossRef CAS PubMed.
- H. Xu, S. Sommer, N. L. N. Broge, J. Gao and B. B. Iversen, Chem.–Eur. J., 2019, 25, 2051–2058 CrossRef CAS PubMed.
- J. Winarta, B. Shan, S. M. Mcintyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2020, 20, 1347–1362 CrossRef CAS.
- L. Chu, J. Guo, L. Wang, H. Liu, J. Yan, L. Wu, M. Yang and G. Wang, Appl. Organomet. Chem., 2022, 36, e6559 CrossRef CAS.
- S. Mukhopadhyay, R. Shimoni, I. Liberman, R. Ifraemov, I. Rozenberg and I. Hod, Angew. Chem., Int. Ed., 2021, 60, 13423–13429 CrossRef CAS PubMed.
- Y. Wang, L. Li, P. Dai, L. Yan, L. Cao, X. Gu and X. Zhao, J. Mater. Chem. A, 2017, 5, 22372–22379 RSC.
- W. Xu, M. Dong, L. Di and X. Zhang, Nanomaterials, 2019, 9, 1432 CrossRef CAS PubMed.
- H. R. Abid, H. Tian, H.-M. Ang, M. O. Tade, C. E. Buckley and S. Wang, Chem. Eng. J., 2012, 187, 415–420 CrossRef CAS.
- C. L. Luu, T. T. Van Nguyen, T. Nguyen and T. C. Hoang, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2015, 6, 25004 Search PubMed.
- M. A. Rodrigues, J. de S. Ribeiro, E. de S. Costa, J. L. de Miranda and H. C. Ferraz, Sep. Purif. Technol., 2018, 192, 491–500 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- J. Qiu, Y. Feng, X. Zhang, M. Jia and J. Yao, J. Colloid Interface Sci., 2017, 499, 151–158 CrossRef CAS PubMed.
- A. Trunschke, Modern Methods in Heterogeneous Catalysis Research: Surface Area and Pore Size Determination, 2017 Search PubMed.
- D. Khan, L. Qiu, C. Liang, K. Mirza, M. Kashif, B. Yang, K. L. Kra, Y. Wang and X. Li, ACS Omega, 2022, 7, 10820–10839 CrossRef CAS PubMed.
- T. K. Vo, V. N. Le, K. S. Yoo, M. Song, D. Kim and J. Kim, Cryst. Growth Des., 2019, 19, 4949–4956 CrossRef CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- P. Chammingkwan, G. Y. Shangkum, L. T. T. Mai, P. Mohan, A. Thakur, T. Wada and T. Taniike, RSC Adv., 2020, 10, 28180–28185 RSC.
- S. E. Bambalaza, H. W. Langmi, R. Mokaya, N. M. Musyoka, J. Ren and L. E. Khotseng, J. Mater. Chem. A, 2018, 6, 23569–23577 RSC.
- P. Yang, F. Mao, Y. Li, Q. Zhuang and J. Gu, Chem. - Eur. J., 2018, 24, 2962–2970 CrossRef CAS PubMed.
- S. Gökpinar, T. Diment and C. Janiak, Dalton Trans., 2017, 46, 9895–9900 RSC.
- W. Xiang, Y. Zhang, Y. Chen, C. Liu and X. Tu, J. Mater. Chem. A, 2020, 8, 21526–21546 RSC.
- S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30, 1704501 CrossRef PubMed.
- X. Ma, L. Wang, Q. Zhang and H.-L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12175–12179 CrossRef CAS PubMed.
- L. Song, T. Zhao, D. Yang, X. Wang, X. Hao, Y. Liu, S. Zhang and Z.-Z. Yu, J. Hazard. Mater., 2020, 393, 122332 CrossRef CAS PubMed.
- Y. Liao, T. Sheridan, J. Liu, O. Farha and J. Hupp, ACS Appl. Mater. Interfaces, 2021, 13, 30565–30575 CrossRef CAS PubMed.
- H. Chen, P. Liao, M. L. Mendonca and R. Q. Snurr, J. Phys. Chem. C, 2018, 122, 12362–12368 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04637d |
| This journal is © The Royal Society of Chemistry 2024 |