
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning and the anticancer activity of chitosan and chitosan oligosaccharide lactate nanobeads loaded with Biginelli hybrid
Nenad Janković *a,
Jovana Ristovski*b,
Željko Žižakc,
Milica Radand,
Sandra Cvijiće,
Katarina Nikolićf and
Nenad L. Ignjatovićg
*a,
Jovana Ristovski*b,
Željko Žižakc,
Milica Radand,
Sandra Cvijiće,
Katarina Nikolićf and
Nenad L. Ignjatovićg
aUniversity of Kragujevac, Department of Science, Institute for Information Technologies Kragujevac, Jovana Cvijića bb, Kragujevac 34000, Serbia. E-mail: nenad.jankovic@uni.kg.ac.rs
bUniversity of Novi Sad, Faculty of Medicine, Hajduk Veljkova 3, Novi Sad 21000, Serbia
cInstitute of Oncology and Radiology of Serbia, Pasterova 14, Belgrade 11000, Serbia
dThe Institute for the Study of Medicinal Herbs “Dr Josif Pančić”, Tadeuša Košćuška 1, Belgrade 11000, Serbia
eUniversity of Belgrade, Faculty of Pharmacy, Department of Pharmaceutical Technology and Cosmetology, Vojvode Stepe 450, Belgrade 11221, Serbia
fUniversity of Belgrade, Faculty of Pharmacy, Department of Pharmaceutical Chemistry, Vojvode Stepe 450, Belgrade 11221, Serbia
gInstitute of Technical Sciences of the Serbian Academy of Sciences and Arts, Knez Mihailova 35/IV, P.O. Box 377, Belgrade, Serbia
First published on 4th October 2024
Abstract
This study focuses on the designing and characterization, and anticancer evaluation of chitosan-based nanoparticles (NPs) loaded (enriched) with a Biginelli hybrid compound (BH). NPs based on chitosan (CH) or chitosan oligosaccharide lactate (CHOL), are carefully designed to encapsulate a tetrahydropyrimidine derivative (BH) with already proven anticancer properties. The formulations were evaluated for their physicochemical properties, including particle size distribution and morphology, using techniques such as infrared spectroscopy, scanning electron microscopy, and X-ray diffraction. The cytotoxicity profiles were assessed on different cancer cell lines, showing a higher selectivity towards HeLa and A549 cells related to BH. BH-CH showed better cytotoxic profile related to BH-CHOL NPs. A cell cycle analysis revealed an accumulation of cells in the G2/M phase after a treatment with these NPs, indicating the ability to induce mitotic arrest in cancer cells. In summary, the results underscore the promising application of CH-based natural nanocarriers for the targeted delivery of Biginelli hybrids, showcasing a significant potential for further in vivo testing.
Introduction
Biginelli reaction is an efficient route in the synthesis of tetrahydrpyrimidines-THPMs [formerly known as 3,4-dihydropyrimidin-2(1H)-ones). Although discovered in the 19th century, the Biginelli reaction is still a notable tool in drug discovery due to the presence of a THPM scaffold in different active pharmaceutical ingredients. Different structures with significant pharmacological properties, including molecules with antiviral, anticancer, anti-inflammatory, antidiabetic, antibacterial, antifungal, anti-epileptic and antimalarial characteristics, are obtained using this multicomponent one-pot reaction. 1–8After the discovery of monastrol, which is a selective inhibitor of kinesin Eg5, Biginelli chemistry has intensively been used in the development and synthesis of novel molecules with anticancer activity. The research focused on creating new variations and combinations of monastrol and assessing their potential to inhibit the growth of cancer cells is an active area of study that has yielded significant outcomes. THPMs in higher concentrations have a disturbing effect on the cell cycle and cause mitotic arrest.9 Given the rapid growth and the survival potential of cancer cells on the one hand, and the numerous side effects of current anticancer drugs on the other, scientists and the pharmaceutical industry are challenged to develop innovative strategies for creating new anticancer medicines.10
Modern technologies have revolutionized cancer diagnosis and treatment. From advanced imaging techniques and AI-driven diagnostics to innovative therapies like immunotherapy and targeted treatment, significant progress has been made. Nanotechnology further enhances these developments by improving drug formulation, delivering targeted therapies more effectively. Each of these advancements speaks to the ongoing commitment to combat cancer and improve patient outcomes.11
The development of innovative drug delivery systems can lead to improved efficacy and selectivity of anticancer agents. The sizes of nanoparticles range from 1 to 100 nm. They find diverse applications in modern medicine, especially as carriers for delivering drugs and genes to tumors. When appropriately prepared, they offer a large surface area and pore volume, as well as a high drug-loading capacity, enabling selective and controlled delivery of therapeutic agents.12 In pharmaceutical nanotechnology, the selection of appropriate coating materials and coating methods are very important steps that can help modify the selectivity of nanoparticles in the delivery process and obtain a final system ensuring improved targeted drug delivery. Present concerns and constraints regarding the utilization of nanotechnology in biomedical settings stem from the insufficient understanding of the toxicological effects of nano systems in living organisms and potentially adverse outcomes.13
For that reason, our research group selected chitosan (CH) as a natural non-toxic material that is widely utilized as a polymer for particle formation and, notably, as a surface coating. Various types of nanoparticles, such as polymeric, lipid-based, and metal or metal oxide-based, serve as carriers for drugs or active substances that have been enhanced with chitosan for diverse applications. The amino group present in chitosan molecule facilitates protonation within acidic to neutral environments. This results in a positive charge on the cationic polysaccharide, which enhances its solubility in water and its adhesive properties, allowing it to effectively promote permeation across negatively charged surfaces, such as mucosal and basement membranes. As outcome, CH plays a significant role in improving the oral bioavailability of polar drugs and their transport through epithelial layers.14,15 Numerous in vitro and in vivo studies have demonstrated that enhancing the surface of these nanocarriers by CH coating offers numerous benefits, such as enhancing physicochemical stability, controlling drug release, promoting mucoadhesiveness and tissue penetration, modulating cell interactions (including cellular uptake and toxicity), boosting antimicrobial effects, and improving bioavailability and drug efficacy. The specific advantages of surface coating can vary in attractiveness depending on the type of nanoparticle and the chemical composition of its components.16
Extensive research on CH has positioned it as an important material in the preparation of new anticancer formulations. Furthermore, chitosan has demonstrated anticancer properties through an antiangiogenic mechanism, which disrupts the balance between proangiogenic and antiangiogenic factors in pathological conditions. Also, research indicates that chitosan's ability to inhibit cancer growth is linked to the enhancement of the immune system, particularly involving tumor-killing immune cells such as cytotoxic lymphocytes and natural killer cells.17 The use of chitosan-based nanoparticles has shown promising anti-tumor effects in laboratory and animal studies, suggesting a wide range of potential clinical applications.18 Traditional chemotherapy drugs like doxorubicin, paclitaxel, cisplatin, 5-fluorouracil, methotrexate, cytarabine and gemcitabine have greatly advanced cancer treatment during the past decades. However, their severe side effects have limited their widespread use in clinical settings. To address this issue, scientists have developed specific Drug Delivery Systems (DDS) to target and release these drugs in a controlled manner, reducing adverse effects. Chitosan-based nanoparticles serve as effective DDS, enabling the encapsulation and targeted delivery of the above-mentioned anti-cancer drugs to specific tumor sites.19
The PI3K-AKT pathway plays a crucial role in cancer development. Abnormal activation of this pathway is linked to various cancers such as endometrial, hepatocellular, breast, colorectal, prostate, and cervical cancer.20 Targeting and blocking key molecules in this pathway could be a promising strategy for cancer therapy. Studies have shown that chitosan and its derivatives can reduce AKT (cellular homolog of murine thymoma virus akt8 oncogene) phosphorylation in different cancer types, potentially inhibiting AKT activities. Additionally, chitosan has been found to induce apoptosis by affecting calcium ion levels, ROS (reactive oxygen species), and mitochondrial membrane potential. Chitosan may also regulate apoptosis-associated proteins, leading to caspase-9 and caspase-3 cleavage and mitochondrial pathway-induced apoptosis. Moreover, chitosan oligosaccharide inhibits the overexpression of PD-L1 in various tumors, enhancing T cell-mediated immune killing through MAPK activation and STAT1 inhibition. This suggests that chitosan oligosaccharide could enhance the effectiveness of current chemotherapy treatments.21–23
In earlier studies, the anti-cancer effects of newly synthesized THPM compounds were examined on human cervical adenocarcinoma (HeLa) cell lines using both 2D and 3D cell culture systems. Flow cytometry was used to monitor alterations in the distribution of cell cycle phases after subjecting spheroids to a 48 hours treatment with five different compounds, revealing that all tested compounds led to an increased percentage of HeLa cells in the subG1 phase compared to the control spheroid sample.24 Also, during previous research, different vanillin-based THPMs with a specific anticancer activity were identified (Fig. 1).25 Fig. 1 displays the structures of selected THPMs. We chose three THPMs from a recent study to emphasize how the number of carbon atoms and the type of functional group attached to the C-4′ position significantly influenced their properties. In the cases presented, the carbon atom counts at the C-4 position were consistently 4, but the functional groups varied: BH (butyl), BH′ (crotyl), and BH′′ (ester). As observed, the anticancer activity varies significantly across cell lines. In this context, tetrahydropyrimidine BH [methyl 4-(4′-butoxy-3′-methoxyphenyl)-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate] exhibited the highest level of activity, demonstrating exceptional selectivity with a selectivity index higher than 10.
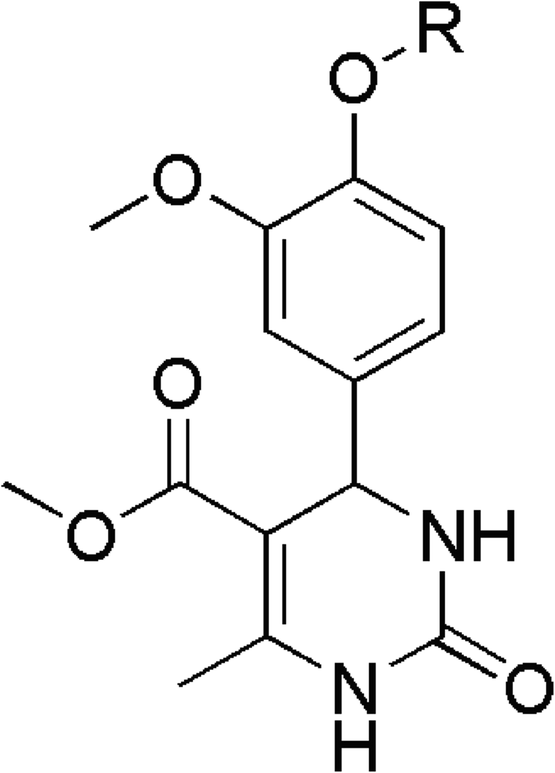 | ||
| Fig. 1 Chemical structures of selected Biginelli hybrids [R = Bu(BH) IC50 = 6 μM (HeLa), 8.3 μM (MCF-7); Crotyl (BH′) IC50 = 50.8 μM (HeLa), 89.5 μM (MCF-7); CH2COOEt (BH′′) inactive].25 | ||
To explore the advantages/limitations of nanoparticle-based drug delivery systems for cancer treatment we developed nanoparticles using CH as a biocompatible and well-established nanocarrier. Additionally, we investigated the feasibility of creating nanosystems enriched with BH by incorporating chitosan oligosaccharide lactate (CHOL) as a coating material in place of CH. The objective of this research is to explore whether the formed nanoparticles (NPs) exhibit better selectivity and anti-tumor efficacy than the pure compounds. The selective viability effect of the BH derivative and the obtained NPs were examined using MTT assays on different cancer cell lines including HeLa, K562, A549, LS174, MDA-MB-453 and MRC-5. Drug release experiments were conducted using high-performance liquid chromatography (HPLC) to quantify the release rates of BH from NPs. Flow cytometry was used to assess the cell cycle and to measure the cellular DNA content. Cell cycle distribution was measured after a 24 hours exposure of the pure BH compound and the prepared nanosystem in the HeLa cell lines.
Results and discussion
Characterization of BH-CH and BH-CHOL
The FTIR spectra (Fig. 3) of the pure Biginelli hybrids (BH) show typical two medium bands near 3300 and 3150 cm−1 assigned to the N–H stretching vibration. These bands correspond to the stretching vibration of the N–H (amide) involved in hydrogen bonding interactions. In the region of ∼1600–1700 cm−1 cyclic amide I bands are found. These strong bands are located around 1720 and 1650 cm−1 and are primarily due to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in amide. The band at 1510 cm−1 originates from amide II. This medium band is formed by a combination of the N–H bending and the C–N stretching vibrations. In the IR spectrum of CH the medium band at ∼3428 cm−1 implies N–H and O–H stretching, along with intramolecular hydrogen bonds. In addition, bands centered around 2920 cm−1 and 2870 cm−1 were assigned to the C–H symmetric and asymmetric stretching motions, respectively. The band at 1653 cm−1 is from amine the N–H bending vibrations.26 In the stacked IR spectra of BH-CH the absence of amide II carbonyl band that was found at 1510 cm−1 in pure BH is observed. After the encapsulation of BH, the N–H and O–H bands are increased and found at ∼3390 and 3250 cm−1. In the IR spectrum of CHOL, the absorption band at 3430 cm−1 corresponds to the N–H and O–H stretching vibrations.27 Additionally, the medium-intensity band observed at 1627 cm−1 arises from the N–H deformation vibrations. The IR of BH-CHOL shows characteristic medium bands from the BH amide function at 1710, 1650, and 1510 cm−1. A broad medium absorption band of the N–H and O–H from CHOL is shifted from 3430 cm−−1 to ∼3280 cm−1 in BH-CHOL. This shift most likely indicates the presence of polymeric hydrogen bonding in the encapsulated BH-CHOL structure. All the data obtained from the FTIR spectra of the synthesized nanopowders (BH-CH and BH-CHOL, Fig. 3) indicate that the pure BH phase was successfully encapsulated in the CH or CHOL.
O bond in amide. The band at 1510 cm−1 originates from amide II. This medium band is formed by a combination of the N–H bending and the C–N stretching vibrations. In the IR spectrum of CH the medium band at ∼3428 cm−1 implies N–H and O–H stretching, along with intramolecular hydrogen bonds. In addition, bands centered around 2920 cm−1 and 2870 cm−1 were assigned to the C–H symmetric and asymmetric stretching motions, respectively. The band at 1653 cm−1 is from amine the N–H bending vibrations.26 In the stacked IR spectra of BH-CH the absence of amide II carbonyl band that was found at 1510 cm−1 in pure BH is observed. After the encapsulation of BH, the N–H and O–H bands are increased and found at ∼3390 and 3250 cm−1. In the IR spectrum of CHOL, the absorption band at 3430 cm−1 corresponds to the N–H and O–H stretching vibrations.27 Additionally, the medium-intensity band observed at 1627 cm−1 arises from the N–H deformation vibrations. The IR of BH-CHOL shows characteristic medium bands from the BH amide function at 1710, 1650, and 1510 cm−1. A broad medium absorption band of the N–H and O–H from CHOL is shifted from 3430 cm−−1 to ∼3280 cm−1 in BH-CHOL. This shift most likely indicates the presence of polymeric hydrogen bonding in the encapsulated BH-CHOL structure. All the data obtained from the FTIR spectra of the synthesized nanopowders (BH-CH and BH-CHOL, Fig. 3) indicate that the pure BH phase was successfully encapsulated in the CH or CHOL.
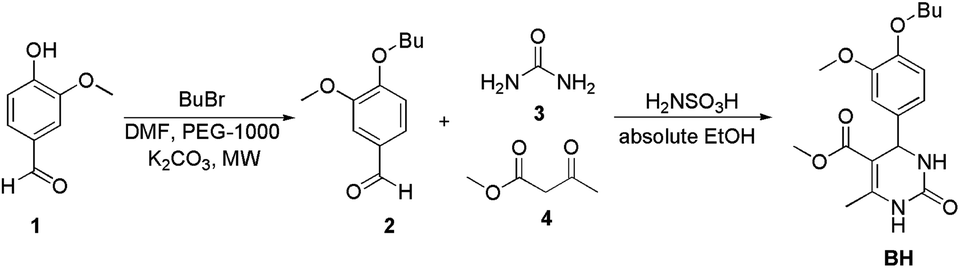 | ||
| Fig. 2 The synthetic path to BH. | ||
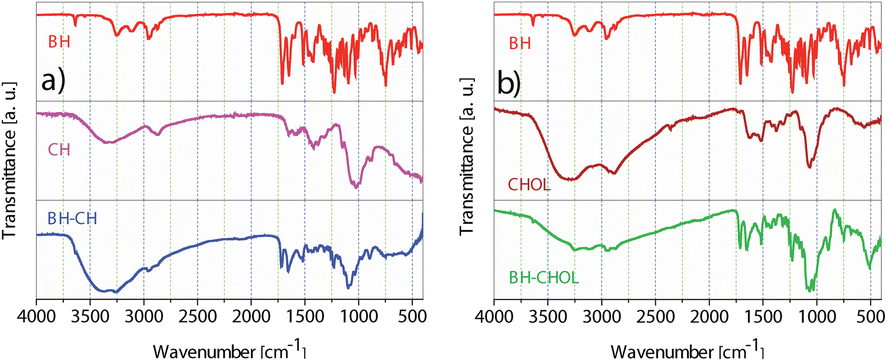 | ||
| Fig. 3 ATR FTIR spectra of (a) BH, CH and BH-CH, (b) BH, CHOL and BH-CHOL. | ||
The particle size distribution (PSD) and the morphology of BH-CH and BH-CHOL powders are shown in Fig. 4. The encapsulation of BH into CH or CHOL leads to the formation of particles with a spherical morphology, for both powders. In general, the particle size for the powder BH-CH is slightly larger (d50 = 115 nm) than the particle size of the powder BH-CHOL (d50 = 90 nm) (d50 is the corresponding particle size when the cumulative percentage reaches 50%).
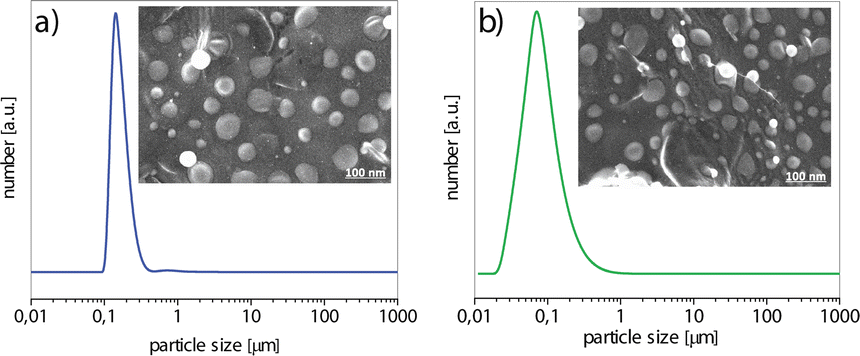 | ||
| Fig. 4 PSD and FE-SEM of (a) BH-CH, (b) BH-CHOL (d50 were 115 nm for BH-CH and 90 nm for BH-CHOL). | ||
The zeta potential is also one of the important parameters that indicate the affinity of the particle towards the cells in the bio-nano interface.28 The zeta potential (ξ) of BH-CH particles (9.30 ± 0.60 mV) is more positive than BH-CHOL (6.80 ± 0.30 mV), and the reason is most likely the higher content of positively charged amine groups in CH than in CHOL. It is most likely that the difference in surface charge affects the affinity and adhesion of particles to cancer cells. Fig. 5 shows the diffractograms of BH, BH-CH and BH-CHOL. The BH (methyl 4-(4′-butoxy-3′-methoxyphenyl)-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate) is characterized by most intensive peaks at the positions 14.4°, 20.6°, 23.5°, 24.6° and 25.6° (marked with * in the Fig. 5). The BH-CH diffractogram confirms the presence of BH and CH. Namely, in addition to the characteristic peaks of BH, characteristic reflection detected at 19.7° originate from CH.29,30 Also, the presence of characteristic reflections of BH and CHOL (at 18.8°) was evident in the diffractogram of powder BH-CHOL. In general, the XRD patterns of CH and CHOL are characterized by typical weak intensity reflections indicating that CH and CHOL are low crystalline polymers.31,32 The presence of characteristic peaks of BH in diffractograms of BH-CH and BH-CHOL powders qualitatively confirms the presence of BH in BH-CH and BH-CHOL. The efficiency of BH encapsulation was also quantitatively determined and was 90% (in the section: Drug release study).
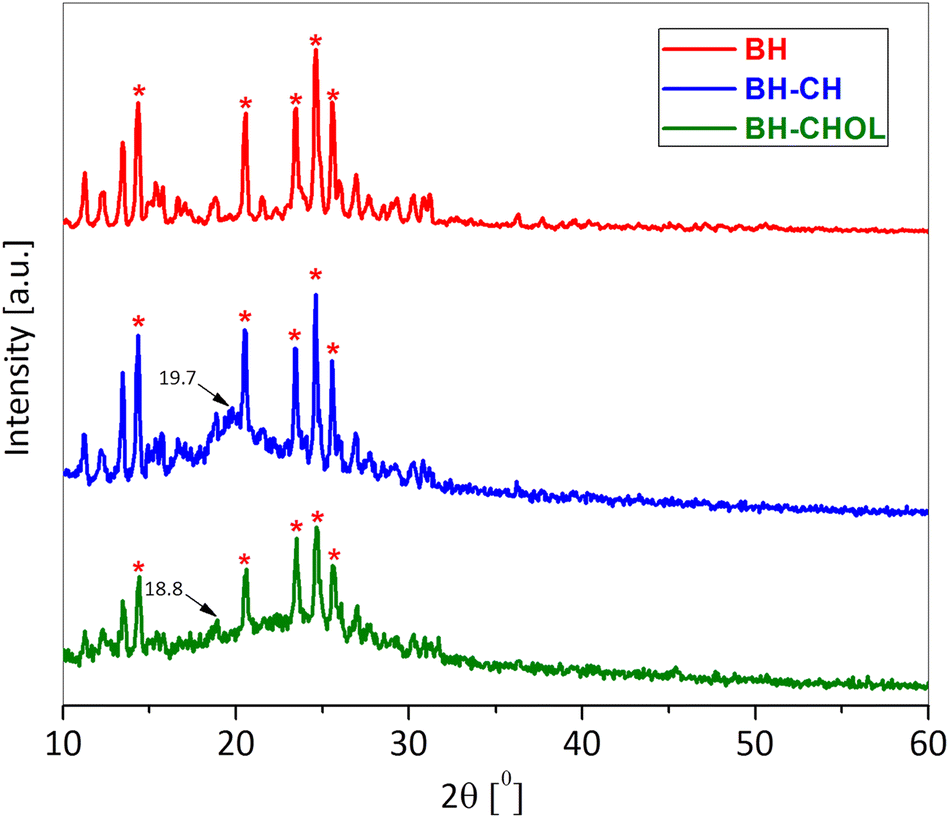 | ||
| Fig. 5 X-ray difraction of BH, BH-CH and BH-CHOL. | ||
Anticancer activity of BH-CH and BH-CHOL
Table 1 shows the IC50 values with standard deviations (SD) in μg mL−1 for the tested nanocomposites (BH-CH and BH-CHOL) against different cell lines. In this study, five cancer (HeLa, K562, A549, LS174, and MDA-MB-453) and one normal cell lines (MRC-5) were employed.| IC50 ± SDa (μg mL−1) | |||||||
|---|---|---|---|---|---|---|---|
| HeLa | K562 | A549 | LS174 | MDA-MB-453 | MRC-5 | SI | |
| a From two independent experiments. | |||||||
| CH | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | — |
| CHOL | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | — |
| BH | 96 ± 7 | 64 ± 12 | 203 ± 4 | 91 ± 3 | 95 ± 4 | 209 ± 6 | 2.2 |
| BH-CH | 77 ± 3 | 100 ± 1 | 113 ± 11 | 106 ± 6 | 91 ± 1 | 201 ± 1 | 2.6 |
| BH-CHOL | 79 ± 7 | 116 ± 7 | 205 ± 8 | 170 ± 7 | 94 ± 1 | 212 ± 4 | 2.7 |
Chitosan, as a natural material, has recently been utilized for tumor-targeting drug delivery systems.33 The investigated biopolymers, CH and CHOL, showed IC50 values greater than 1000 μg mL−1 against all tested cell lines, indicating low or no cytotoxic activity at the concentrations tested. Therefore, BH and BH-CH exhibited varying levels of cytotoxic activity across investigated cancer cell lines. BH-CH shows improved activity against the HeLa and A549 cell lines compared to BH. BH-CHOL shows significant cytotoxic activity against the HeLa cell lines. The selectivity index (SI) values for BH, BH-CH, and BH-CHOL are 2.2, 2.6, and 2.7, respectively. After encapsulation, BH-CH and BH-CHOL exhibit higher cytotoxicity and higher selectivity towards cancer cells compared to normal cells (MRC-5). From a structural perspective, CHOL has a lower molecular weight compared to CH, which makes it more water soluble. This increased solubility is also associated with CHOL's lower degree of acetylation (deacetylation > 90%), suggesting that it has a greater capacity for hydrogen bonding than CH (deacetylation > 80%). In our case, this ability of BH-CHOL to form more hydrogen bonds could negatively affect its cytotoxic activity (Table 1). SI achieved for BH-CH NPs system (SI ≈ 2.6) is improved and slightly higher in comparison to our previous published article (SI ≈ 2.0).26
Cell cycle determination
To examine the mechanisms of action of our nanoparticles in the HeLa cells, the cell cycle distribution was determined. As shown in Fig. 6, 24 hours after exposure to pure BH or BH-loaded nanoparticles, the number of the HeLa cells in the G2/M phase increased for all test compounds. Also, sub-G1 fraction increases moderately for the cells treated with BH or any loaded nanoparticle. The accumulation of cells in the G2/M phase and the increase in the number of cells in the sub-G1 phase is accompanied by a decreased proportion of cells in the G1 and the S phase compared to control the untreated HeLa cells.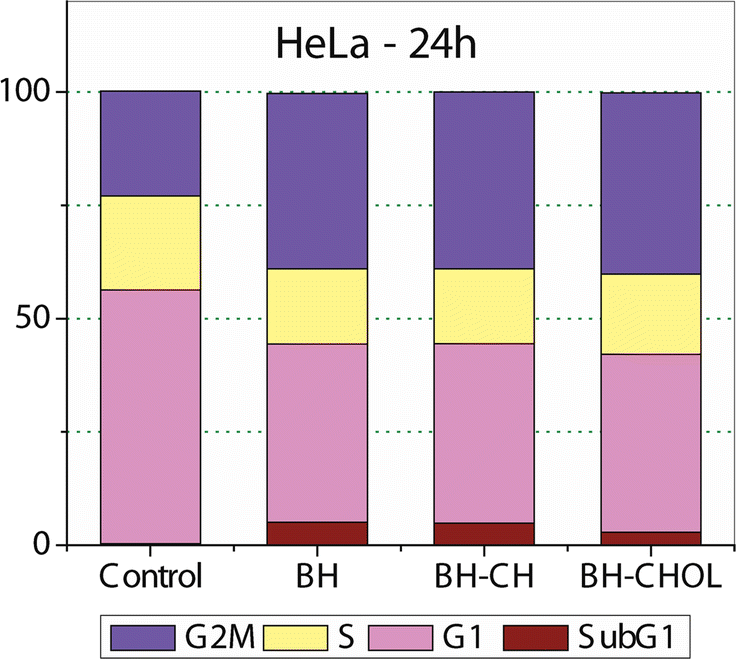 | ||
| Fig. 6 Cell cycle distribution after 24 h, continuous action of investigated BH and nanoparticles loaded with BH. After exposure for 24 h (concentration corresponded to IC50 value obtained in MTT test), cells were harvested, stained with propidium iodide and subjected to flow cytometry. Untreated HeLa cells are labeled as control. | ||
Fig. 7 shows the outcomes of experiments conducted before and after the treatment with specific nanoparticles (NPs). In the control group, no notable apoptosis was detected. However, following the treatment with BH-CH and BH-CHOL systems, in the experimental groups, cells in advanced stages of apoptosis exhibited distinctive crescent-shaped or granular green staining. Notably, staining in the experimental groups was asymmetrically localized within cells, whereas in normal cells, it had a symmetric distribution.
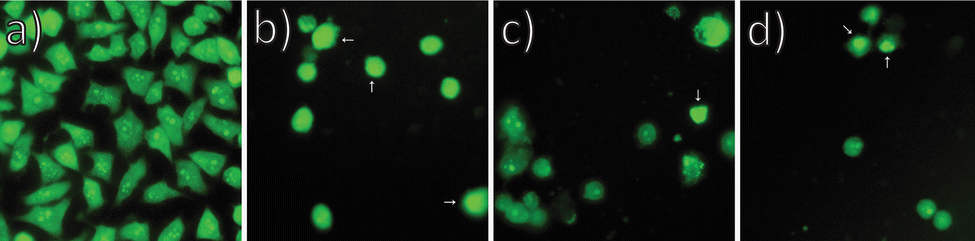 | ||
| Fig. 7 (a) Control (negative control group – normal cells), (b) BH-CH, (c) BH-CHOL and (d) BH show experimental groups (apoptotic cells) after the treatment with the corresponding nanocomposites. Arrows indicate apoptotic cells with fragmented nuclei, while arrowheads mark cells with condensed chromatin. | ||
Drug release study
For a more comprehensive understanding of bioavailability and drug release kinetics, HPLC measurements were performed. The areas corresponding to the released compound from the BH-CH and BH-CHOL formulations in the HPLC chromatograms were compared, and the concentrations of the released BH compound were calculated (BH with a retention time of 15.678 min). The results were illustrated in a cumulative curve in Fig. 8. In the first 1.8 ± 0.2 h hour, over 50% of the pure compound from the BH-CH formulation was released, and by 9 ± 0.2 h hour, approximately 95% was released. In the BH-CHOL formulation, a 50% release of the pure compound was registered after 1.8 ± 0.1 h hour and 95% after 30 ± 0.3 h hour. Keeping in mind that CH and CHOL with medium molecular weights served as the carriers in the formulations, these outcomes were expected. Drug encapsulation efficiency was also determined using HPLC, confirming an efficiency of 90%. With a BH to CH ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 and BH to CHOL ratio of 20:80 the proportion of entrapped BH was calculated to be 18 wt%. The results are expressed as mean values ± standard deviation (SD), and presented as cumulative curves over time (Fig. 7).
:80 and BH to CHOL ratio of 20:80 the proportion of entrapped BH was calculated to be 18 wt%. The results are expressed as mean values ± standard deviation (SD), and presented as cumulative curves over time (Fig. 7).
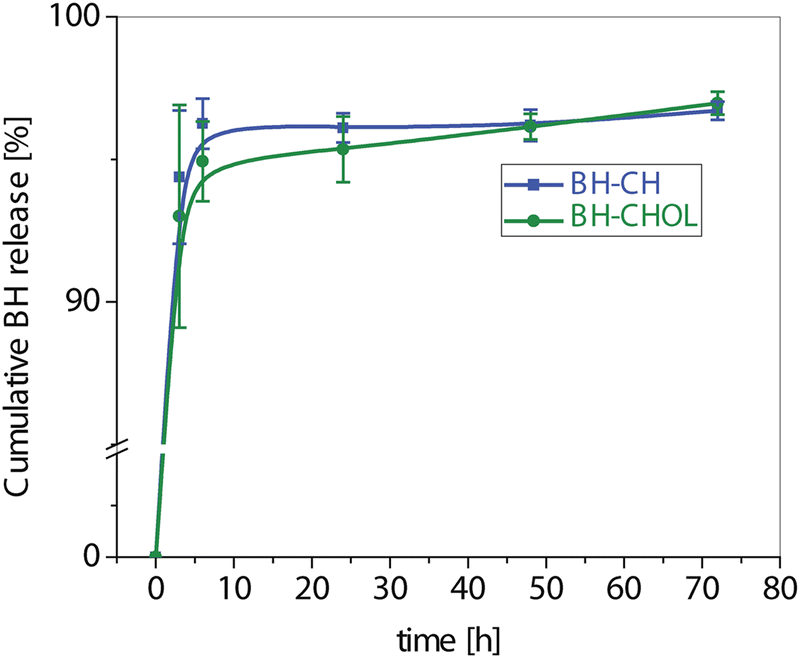 | ||
| Fig. 8 Cumulative BH release profiles (mean ± SD) from the investigated nanocomposites BH-CH and BH-CHOL. | ||
Experimental
Materials
All chemicals and reagents were purchased from Sigma Aldrich without further purification, except for butyl bromide that was distilled, while amidosulfonic acid was dried before use.Synthesis of BH
Designing nanocomposites
Chitosan (CH) with a medium molecular weight (Sigma-Aldrich, deacetylation > 80%), was dissolved in acetic acid (1 wt%), and mixed with an ethanol solution of BH in a weight ratio of CH:BH = 8:2, while stirring with a magnetic stirrer at 400 rpm. The obtained mixture of BH, and CH was slowly poured into a solution of sodium triphosphate pentabasic (STP, Sigma-Aldrich) 0.1 wt% in H2O, while stirring at 21000 rpm for 30 min (IKA, ULTRA- TURRAX T 25). The obtained mixture was then centrifuged at 5000 rpm and 5 °C for 1 h, and the resulting precipitate was subjected to lyophilization (Freeze Dryer, Christ Alpha 1–2/LD Plus) at temperatures ranging from −10 °C to −60 °C and pressures ranging from 0.37 mbar to 0.1 mbar for 1–5 h. The obtained powder was washed with distilled water three times, centrifuged at 4000 rpm and dried again. The final products were dry powders composed of BH-loaded CH (BH-CH). Dry powder composed of BH-loaded CHOL (BH-CHOL) is obtained in the same way, but instead of CH, chitosan oligosaccharide lactate (CHOL), was used, (Sigma-Aldrich, Mn = 4000–6000, deacetylation > 90%).
Characterization
Infrared spectroscopy (ATR FT-IR) was performed on a Nicolet iS10 FT-IR Spectrometer (Thermo Scientific Instruments) in the spectral range from 400 cm−1 to 4000 cm−1. Particle size distribution (PSD) was measured on 10 mg mL−1 of powders dispersed in water using a Mastersizer 2000 (Malvern Instruments Ltd) and a HydroS dispersion unit for liquid dispersants. Field-emission scanning electron microscopy (FE-SEM) was performed on a Carl Zeiss ULTRA Plus microscope at the electron acceleration voltage of 3 kV. Zeta potential of the aqueous suspensions of synthesized powders was analyzed using a Zeta-Sizer Nano (Malvern Instruments Ltd) in deionized water at 25 °C and pH 7.0. The obtained powders were characterized using X–ray powder diffraction (XRPD, Philips PW1050 diffractometer with CuKα1,2 radiation).Cell cultures
Cytotoxicity studies of two nano-particulate systems were performed using six cell lines: human cervix adenocarcinoma (HeLa ATCC CCL-2), human chronic myelogenous leukemia (K-562 ATCC CCL-243), human lung carcinoma (A549 ATCC CCL-185), human colorectal adenocarcinoma, (LS-174 ATCC CL-188), human breast carcinoma (MDA-MB-453 ATCC HTB-131), and normal human lung fibroblasts (MRC-5 ATCC CCL-171). Cells were grown to confluency in nutrient medium (RPMI-1640 without phenol red) supplemented with 3 mM L-glutamine, 100 μg mL−1 streptomycin, 100 IU/mL penicillin, 10% heat inactivated fetal bovine serum (FBS), and 25 mM Hepes, adjusted to pH 7.2 by bicarbonate solution. RPMI-1640, FBS, Hepes, and L-glutamine were products of Sigma Chemical Co., St. Louis, MO. Cell cultures were maintained under standard conditions: at a temperature of 37 °C, in humidified air atmosphere with 5% CO2 and were passaged twice a week. For the experiments, cells between the third and tenth passage were used.MTT assay
HeLa (2500 cells per well), K562 (5000 cells per well), LS-174 (7000 cells per well), A549 (5000 cells per well), MDA-MB-453 (3000 cells per well) and MRC-5 (5000 cells per well) were seeded into 96-well microtiter plates. Twenty hours later, after the cell adherence, five different concentrations of investigated compounds in complete nutrient medium were added to the wells, except for the control cells to which a nutrient medium only was added. Cell survival was determined by a MTT test according to the method of Mosmann, and modified by Ohno and Abe, 72 h after the drug addition.34,35 Shortly later, 20 μL of the MTT solution [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 5 mg mL−1 in phosphate buffered saline] was added to each well. Samples were incubated for four hours more at 37 °C in humidified atmosphere with 5% CO2. Then, 100 μL of 10% SDS was added to the wells. Absorbance was measured 24 h later at 570 nm on the microtiter plate reader (Multiscan Ascent, Thermo Labsystems). To get the cell survival rate, the absorbance at 570 nm of the sample with the cells grown in the presence of various concentrations of investigated agents was divided with the absorbance of the control sample (the absorbance of the cells grown only in the nutrient medium), implying that the absorbance of the blank was always subtracted from absorbance of the corresponding sample with the target cells.Cell cycle
The aliquots of the 106 control HeLa or the cells treated with BH or BH-loaded nanoparticles for 24 h were fixed overnight in ice cold 70% ethanol. The applied concentrations of BH or loaded nanoparticles corresponded to the IC50 values obtained in the MTT test after incubation for 72 h. The cells were pelleted by centrifugation and treated with RNase A (100 μg mL−1) at 37 °C for 30 min and then incubated with 40 μg mL−1 propidium iodide for at least 30 min.Cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences Franklin Lakes, NJ, USA) equipped with a 15 mW, air-cooled 488 nm argon ion laser for excitation of PI. PI fluorescence (FL2) was collected after passing a 585/42 nm band pass filter. The FACSCalibur flow cytometer was equipped with an FL2 upgraded doublet discrimination module (DDM) which allowed for screening and then excluding possible occurrences of cell doublets, clumps and debris by plotting the FL2-area versus the FL2-width signals.36 PI fluorescence data were collected using linear amplification. A minimum of 20000 events were collected for each sample. Finally, data were analyzed using the FlowJo™ Software (for Windows, Version 10.5. Ashland, OR: Becton, Dickinson and Company; 2019).
Morphological analysis of the HeLa cells death
To determine the mode of the HeLa cell death induced by the investigated compounds, a morphological analysis by the microscopic examination of acridine orange and ethidium bromide stained cells was performed. HeLa cells were seeded overnight on coverslips (5 × 104 cells) in 3 mL of the complete medium, and on the next day, they were treated with the investigated compounds for 24 h. The applied concentrations corresponded to double IC50 concentrations of the investigated NPs. Afterwards, the cells were stained with 6 μL of DNA dyes: acridine orange and ethidium bromide (3 mg mL−1 AO and 10 mg mL−1 EB in a 2% solution of ethanol in water) and visualized using a fluorescence microscope with a Fluorescein isothiocyanate (FITC) filter set. In the first phase of apoptosis, only acridine orange entered the cell, ethidium bromide was excluded, and the nucleus was stained green. In the second phase of apoptosis, along with the loss of membrane integrity, both dyes entered the cell, and the nucleus became orange-red.Cell cycle determination
The aliquots of the 106 control HeLa or the cells treated with BH or BH-loaded nanoparticles for 24 h were fixed overnight in ice cold 70% ethanol. The applied concentrations of BH or loaded nanoparticles corresponded to the IC50 values obtained in the MTT test after incubation for 72 h. The cells were pelleted by centrifugation and treated with RNase A (100 μg mL−1) at 37 °C for 30 min and then incubated with 40 μg mL−1 propidium iodide (PI) for at least 30 min.Cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences Franklin Lakes, NJ, USA) equipped with a 15 mW, air-cooled 488 nm argon ion laser for excitation of PI. PI fluorescence (FL2) was collected after passing a 585/42 nm band pass filter. The FACSCalibur flow cytometer was equipped with an FL2 upgraded doublet discrimination module (DDM) which allowed for screening for and then excluding possible occurrence of cell doublets, clumps and debris by plotting the FL2-area versus FL2-width signals.36 PI fluorescence data were collected using linear amplification. A minimum of 20000 events were collected for each sample. Finally, data were analyzed using the FlowJo™ Software (for Windows, Version 10.5. Ashland, OR: Becton, Dickinson and Company; 2019).
Drug release study
The release rate of BH from the tested nanocomposites (BH-CH and BH-CHOL) was assayed in phosphate buffered saline (PBS) with the addition of 0.1% sodium lauryl sulfate (SLS). 10 mg of each sample was placed in a screw-cap vial containing 10 mL of medium, and shaken at 50 rpm in a shaking water bath (LSB Aqua Pro, Grant Instruments, UK) at 37 °C for 72 h. In parallel, the control samples of pure BH were tested under the same experimental conditions. All experiments were performed in triplicate. 1 mL of samples was withdrawn at the predetermined time intervals (3, 6, 24, 48, and 72 h) and replaced with the equal volume of the medium. The samples were filtered and analyzed by high-performance liquid chromatography (HPLC) on an Agilent Technologies 1260 Series device with a DAD detector. Chromatographic separation was performed on a Lichrospher RP-18 (250 × 4.0 mm, particle size 5 μm) HPLC column using a mobile phase consisted of water (A) and acetonitrile (B) under the following gradient elution: 0–5 min, 80–60% A; 5–10 min, 60–40% A; 10–30 min, 40–0% A. The volume of injection was 10 μL, while the solvent flow rate was set to 1 mL min−1. The wavelength detection was carried out at 254 nm. The percent of BH released from the nanocomposites (Qx) at each time point was calculated based on the peak area and the concentration of the released BH from nanocomposites compared to the control sample, using the following equation:| Qx (%) = [(Ax × Vc × Cc)/(Ac × Vx × Cx)] × 100 | (1) |
Conclusions
In summary, the study focuses on the synthesis and characterization of CH and CHOL nanobeads loaded with BH for potential anticancer applications. This compound was encapsulated into chitosan-based NPs (BH-CH) and chitosan oligosaccharide lactate-based NPs (BH-CHOL). The prepared NPs were successfully synthesized and characterized using techniques such as FTIR spectroscopy, particle size distribution analysis, and morphology assessment via SEM. Both types of NPs exhibited spherical shapes with slightly different particle sizes, confirming successful encapsulation of BH into the CH and CHOL matrices. The evaluation of cytotoxicity against multiple cancer cell lines (HeLa, K562, A549, LS174, MDA-MB-453) and normal human lung fibroblasts (MRC-5) shows that BH-CH and BH-CHOL NPs have a better cytotoxic effect and selectivity towards cancer cells than free BH alone. Based on the obtained results, it is observed that the BH-CH nanoparticle system exhibited more selective anticancer activity towards the HeLa and MDA-MB-453 cell lines compared to K562, A549, LS174, and MRC-5. In general, the BH-CH system shows stronger cytotoxicity than the BH-CHOL system for the examined cell lines. Flow cytometry experiments indicate an increase of HeLa cells in the G2/M phase across all tested systems. Additionally, there was a moderate rise in the sub-G1 fraction for the cells treated with the pure compound or any loaded NP. The effect of Monastrol in the HeLa cells resulted in activation of the spindle checkpoint which leads to mitotic arrest and apoptosis. The depletion of the spindle checkpoint proteins BubR1 or Mad2 significantly reduced the duration of drug-induced arrest, leading to a premature mitotic exit without cell division, which could also be the case with investigated THPMs. The release study indicates sustained release of BH from both NP systems, suggesting the potential for controlled drug delivery. Future research could address the improvement of BH-nanoparticle formulations to prolongated their efficacy and thermal stability. In addition, it could involve exploring their therapeutic potential through rigorous preclinical studies and clinical trials to assess their efficacy and safety in living organisms. Further study requires that new BH-based materials exhibit significantly higher selectivity and efficacy for potential applications. These materials need to be designed to decompose safely over time, reducing the likelihood of complications and long-term adverse consequences.Data availability
The datasets produced and/or analyzed in our study will be accessible upon reasonable request. These datasets encompass all raw data and processed data related to our experiments. Researchers interested in obtaining the data can send demand to the corresponding author. We are committed to fostering transparency and reproducibility in our research, and we are delighted to disseminate our data to facilitate additional scientific exploration.Author contributions
Nenad Janković: writing – review & editing, writing – original draft, supervision, methodology, investigation, formal analysis, conceptualization. Jovana Ristovski: writing – review & editing, writing – original draft, investigation, formal analysis. Željko Žižak: writing – review & editing, methodology, formal analysis, data curation. Milica Radan: writing – review & editing, methodology, formal analysis. Sandra Cvijić: writing – review & editing, methodology, formal analysis. Katarina Nikolić: writing – review & editing, formal analysis, methodology. Nenad L. Ignjatović: writing – review & editing, visualization, supervision, methodology, conceptualization.Conflicts of interest
The authors confirm that they do not have any competing financial interests or personal relationships that could have influenced the work reported in this article.Acknowledgements
This research was funded by the Ministry of Science, Technological Development, and Innovation of the Republic of Serbia on the research programs: Grant no. 451-03-66/2024-03/200378 (University of Kragujevac, Institute for Information Technologies) and 451-03-66/2024-03/200175 (Institute of Technical Sciences of SASA). The authors acknowledge the NITRA for financial support to bilateral project between Serbia and Turkey [No (RS) 026-02-06 and No TR (123N941)]. The authors acknowledge the help of Dr Ljiljana Veselinović (ITS SASA, Belgrade, Serbia) for the XRPD analysis.References
- N. Pagano, P. Teriete, M. E. Mattmann, L. Yang, B. A. Snyder, Z. Cai, M. L. Heli and N. D. P. Cosford, Bioorg. Med. Chem., 2017, 25, 6248–6265, DOI:10.1016/j.bmc.2017.03.061.
- M. Matias, G. Campos, A. O. Santos, A. Falcão, S. Silvestre and G. Alves, RSC Adv., 2016, 6, 84943–84958, 10.1039/C6RA14596E.
- G. Lauro, M. Strocchia, S. Terracciano, I. Bruno, K. Fischer, C. Pergola, O. Werz, R. Riccio and G. Bifulco, Eur. J. Med. Chem., 2014, 80, 407–415, DOI:10.1016/j.ejmech.2014.04.061.
- K. L. Dhumaskar, S. N. Meena, S. C. Ghadi and S. G. Tilve, Bioorg. Med. Chem. Lett., 2014, 24, 2897–2899, DOI:10.1016/j.bmcl.2014.04.099.
- T. N. Akhaja and J. P. Raval, Eur. J. Med. Chem., 2011, 46, 5573–5579, DOI:10.1016/j.ejmech.2011.09.023.
- M. Y. Wani, A. Ahmad, S. Kumar and A. J. F. N. Sobral, Microb. Pathog., 2017, 105, 57–62, DOI:10.1016/j.micpath.2017.02.006.
- R. W. Lewis, J. Mabry, J. G. Polisar, K. P. Eagen, B. Ganem and G. P. Hess, Biochemistry, 2010, 49, 4841–4851, DOI:10.1021/bi100119t.
- J. D. Bhatt, C. J. Chudasama and K. D. Pate, Arch. Pharm., 2017, 350, 1700088, DOI:10.1002/ardp.201700088.
- T. M. Kapoor, T. U. Mayer, M. L. Coughlin and T. J. Mitchison, J. Cell Biol., 2000, 150, 975–988, DOI:10.1083/jcb.150.5.975.
- E. . Milović, N. Janković, J. Petronijević, N. Joksimović, M. Kosanić, T. Stanojković, I. Matić, N. Grozdanić, O. Klisurić and S. Stefanović, Pharmaceutics, 2022, 14, 2254, DOI:10.3390/pharmaceutics14102254.
- P. Pandurangan, A. D. Rakshi, M. S. A. Sundar, A. V. Samrat, S. S. Meenambiga, V. Vedanarayanan, R. Meena, S. K. R. Namasivayam and M. Moovendhan, J. Drug Delivery Sci. Technol., 2024, 91, 105197, DOI:10.1016/j.jddst.2023.105197.
- L. A. Frank, G. R. Onzi, A. S. Morawski, A. R. Pohlmann, S. S. Guterres and R. V. Contri, React. Funct. Polym., 2020, 147, 104459, DOI:10.1016/j.reactfunctpolym.2019.104459.
- F. Pinelli, G. Perale and F. Rossi, Gels, 2020, 6, 6, DOI:10.3390/gels6010006.
- A. H. Sharan and Y. P. Nath, Int. J. Biomater., 2018, 2952085 DOI:10.1155/2018/2952085.
- J. Ding and Y. Guo, Front. Pharmacol, 2022, 13, 888740, DOI:10.3389/fphar.2022.88874.
- Y. Luo, Z. Teng, Y. Li and Q. Wang, Carbohydr. Polym., 2015, 122, 221–229, DOI:10.1016/j.carbpol.2014.12.084.
- M. S. Shakil, K. M. Mahmud, M. Sayem, M. S. Niloy, S. K. Halder, M. S. Hossen, M. F. Uddin and M. A. Hasan, Polysaccharides, 2021, 2(4), 795–816, DOI:10.3390/polysaccharides2040048.
- I. Takeuchi, Y. Kamiki and K. Makino, Colloids Surf., B, 2018, 167, 468–473, DOI:10.1016/j.colsurfb.2018.04.047.
- J. Ding and Y. Guo, Front. Pharmacol, 2022, 13, 888740, DOI:10.3389/fphar.2022.888740.
- T. F. Franke, PI3K/Akt: Getting it Right Matters, Oncogene, 2008, 27, 6473–6488, DOI:10.1038/onc.2008.313.
- J. Gao, Y. Zhao, C. Wang, H. Ji, J. Yu, C. Liu and A. Liu, Int. J. Biol. Macromol., 2020, 158, 689–697, DOI:10.1016/j.ijbiomac.2020.05.016.
- D. Wu, Y. Zhao, S. Fu, J. Zhang, W. Wang, Z. Yan, H. Guo and A. Liu, Cell Cycle, 2018, 17, 1579–1590, DOI:10.1080/15384101.2018.1464845.
- C. Wu, Y. Dai, G. Yuan, J. Su and X. Liu, Front. Immunol., 2019, 10, 869, DOI:10.3389/fimmu.2019.00869.
- G. M. Chin and R. Herbst, Mol. Cancer Ther., 2006, 10, 2580–2591, DOI:10.1158/1535-7163.MCT-06-0201.
- J. Ristovski Trifunović, R. Minorics, S. Bartha, N. Janković and I. Zupkó, J. Mol. Struct., 2022, 1254, 132373, DOI:10.1016/j.molstruc.2022.132373.
- J. Ristovski Trifunović, Ž. Žižak, S. Marković, N. Janković and N. Ignjatović, RSC Adv., 2020, 10, 41542–41550, 10.1039/D0RA08085C.
- N. Ignjatović, M. Sakač, I. Kuzminac, V. Kojić, S. Marković, D. Vasiljević Radović, V. Wu, V. Uskoković and D. Uskoković, J. Mater. Chem. B, 2018, 6, 6957–6968, 10.1039/C8TB01995A.
- A. E Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557, DOI:10.1038/nmat2442.
- N. Ignjatović, V. Wu, Z. Ajduković, T. Mihajilov-Krstev, V. Uskoković and D. Uskoković, Mater. Sci. Eng., C, 2016, 60, 357–364, DOI:10.1016/j.msec.2015.11.061.
- L. Qi, Z. Xu, X. Jiang, C. Hu and X. Zou, Carbohydr. Res., 2004, 339, 2693–2700, DOI:10.1016/j.carres.2004.09.007.
- R. S. C. M. De Queiroz Antonino, B. R. P. Lia Fook, V. A. De Oliveira Lima, R. Í. De Farias Rached, E. P. N. Lima, R. J. Da Silva Lima, C. A. Peniche Covas and M. V. Lia Fook, Mar. Drugs, 2017, 15, 141, DOI:10.3390/md15050141.
- T. Si Trung, P. Thi Dan Phuong, N. Cong Minh, N. Thi Nhu Thuong, W. Prinyawiwatkul, H. Nguyen Duy Bao and N. Van Hoa, Int. J. Biol. Macromol., 2023, 244, 125337, DOI:10.1016/j.ijbiomac.2023.125337.
- R. Mariappan, R. A. Praphakar, D. Govindaraj, P. Arulselvan and S. S. Kumar, Mater. Today Chem., 2017, 6, 26–33, DOI:10.1016/j.mtchem.2017.08.002.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- M. Ohno and T. Abe, J. Immunol. Methods, 1991, 145, 199–203 CrossRef CAS PubMed.
- M. Carbonari, T. Tedesco and M. Fiorilli, Cytometry, 2001, 44, 120–125 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |