
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDocking-based computational analysis of guava (Psidium guajava) leaves derived bioactive compounds as a coagulation factor IXa inhibitor†
Joseph G. De Luna *,
Shanahi Chelledie B. Gonzales,
Jimuel Jan M. Nuqui,
Evalyn S. Capinding and
Corazon D. Sacdalan
*,
Shanahi Chelledie B. Gonzales,
Jimuel Jan M. Nuqui,
Evalyn S. Capinding and
Corazon D. Sacdalan
Department of Chemistry, Technological University of the Philippines, Ayala Boulevard, Ermita, Manila, Philippines
First published on 14th August 2024
Abstract
Thrombotic disorders pose a global health threat, emphasizing the urgent need for effective management strategies. This study explores the potential of bioactive compounds derived from guava leaves in inhibiting coagulation factor IXa (CFIXa) using in silico methods. Using GC-MS, bioactive compounds extracted from guava leaf through ethanol maceration were identified. Pharmacokinetic properties were elucidated using SwissADME. Molecular docking with AutoDock Vina was used to investigate the interactions with CFIXa. CFIXa was modeled with pysimm/LAMMPS and analyzed with CastP for active site identification. The setup with a higher solvent concentration and lower surface area yielded the highest percent yield (78.541 g, 39.27%). Among the 28 identified bioactive compounds, predominantly terpenoids, only seven exhibited suitable pharmacokinetic properties for oral ingestion and drug development. Docking analysis revealed favorable binding of these compounds to CFIXa (−7.6:−5.3). This study shows inhibition of coagulation factor IXa, thus bridging the ambiguity surrounding the effect of guava leaves on hemostasis. These findings also reveal that guava leaf extract harbors bioactive compounds with potential as coagulation pathway inhibitors, promising novel avenues for thrombotic disorder management.
Introduction
The prevalence of thrombotic disorders in society is a common concern across the globe. In 2021, cardiovascular diseases were the leading cause of death globally. It was stated that it represented 32% of all global deaths in 2019, of which 85% were due to illnesses related to thrombin disorder, such as heart attack and stroke,1 implying that these disorders pose a potential threat and health burden for which action needs to be taken.Thrombotic disorders are characterized by the formation of blood clots within blood vessels that can lead to several cardiovascular risks and complications.2 These disorders are commonly caused by a lifestyle that involves minimal physical activity or movement. Moreover, unhealthy lifestyles such as heavy smoking and frequent beer consumption can increase the risk of developing these disorders. Aligned with this, induced bleeding due to certain surgeries can alleviate the possibility of thromboembolic events.3 In terms of managing unwanted blood clot formation, inhibition of specific protein pathways is necessary. Coagulation factor IXa (CFIXa) is the most suitable protein to be inhibited as this factor is directly involved with the coagulation cascade and provides a more balanced and fewer side-effects compared to direct thrombin inhibitors that can increase the risk of excessive bleeding.4 Favorably, bioactive compounds that are derived from plants are considered effective anti-oxidants that possess anti-coagulant properties, which implies their excellence as a potential candidate for treating thrombotic risks.5,6
Guava (Psidium guajava) leaves, in general, have been widely studied for their beneficial health effects and uses;7,8 however, guava is unexplored in terms of the coagulation cascade and is often regarded as a natural coagulant. Thus, in the context of hemostasis, there is still a large gap in the understanding of whether it supports or inhibits blood clotting as past studies have displayed ambiguous results on the particular matter.9
The researchers of this study aimed to evaluate and identify bioactive compounds present in guava leaves using gas chromatography-mass spectrometry (GC-MS) and in silico tools such as python simulation interface for molecular modeling integrated with Large-scale Atomic/Molecular Massively Parallel Simulator (pysimm/LAMMPS),10,11 Py3Dmol for generating molecular structure of the bioactive compound and CFIXa, Computed Atlas of Surface Topography of Proteins (CASTp) for identifying CFIXa active sites,12 SwissADME (absorption, distribution, metabolism, and excretion) for analyzing interactions between bioactive components and various human body enzymes,13 and automated docking with Vina (AutoDock Vina) to perform docking analysis.14–18 This assisted the seclusion of different bioactive compounds present in the guava leaf extract, and understanding its dynamics and binding capabilities with regard to minimizing coagulation disorders, thrombosis prevention, and novelty drug targets exploration.
Results and discussion
Molecular modeling and active sites identification of CFIXa
For CFIXa protein, the PDB ID 6MV4 model was chosen as it was the only available protein structure for CFIXa without an inhibitor attachment.19 The result of CASTp shows that CFIXa (6MV4) constitutes a total of 42 active sites which are selected based on its geometry (Fig. S1 and Table S1†). However, there are only three active site IDs that are significant in the protein function and are common sites of CFIXa inhibition based on literature (Fig. 1), with its residues' different bond lengths ranging as follows: 1.22405–1.55715 (ID II), 1.11949–1.6194 (ID III), and 1.22965–1.56548 (ID V) (Table S2†). The location of the binding for ID II was determined to be at H:LYS224 and H:TYR225, particularly at the S1 specificity pocket, which interacted with the side chain of positively charged arginine and lysine residues.20 This active site, which contained sodium ions also accounts for a vital role in the linkage formation and binding between antithrombin (AT) and CFIXa, in the case of the calcium deficient reaction. It is also surrounded dominantly by a blue region, and slightly gray, which indicates the presence of hydrophobic residues such as H:ILE123, G:ILE227, H:TRP215, H:ALA221, and H:MET221 (Fig. 1B). Specifically, H:TRP215 is reported to form the S4 pocket with PHE174 and TYR99 and has the capacity to accommodate more hydrophobic chains, which resulted in a main chain conformational shift that has become a target for specific IXa inhibitor drug design.21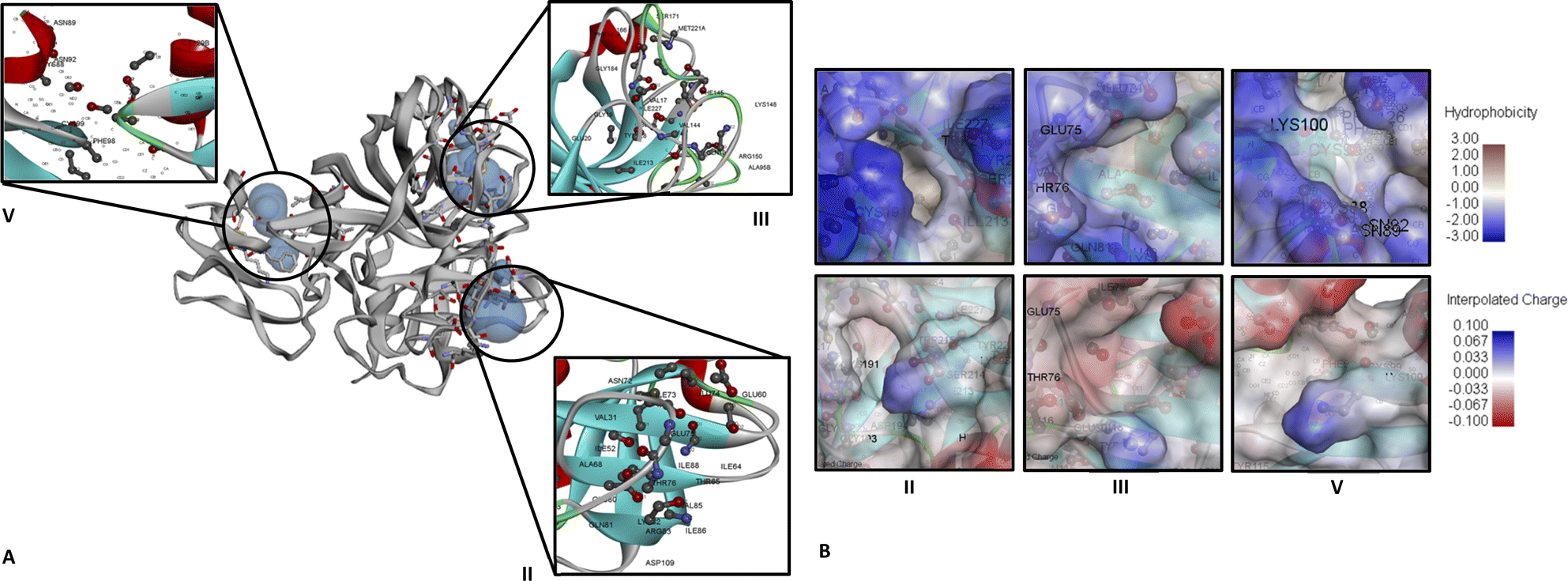 | ||
| Fig. 1 CFIXa (PDB ID 6MV4) active sites identified that have significant biological function. (A) Ribbon diagram of CFIXa and CFIXa active sites ID: II (122.970 Å2:49.494 Å3), ID: III (90.078 Å2:45.053 Å3), and ID: V (55.432 Å2:25.515 Å3). (B) Hydrophobicity and interpolated charge of CFIXa active sites. | ||
Binding occurs in the ID III as well, specifically at H:GLU74, H:GLU75, and H:GLU80, at the 70 s loop of calcium ion binding site, exosite region, wherein the AT and CFIXa interaction occurs. In this active site, the conformational shift in the AT induced by heparin results in optimal binding specificity and affinity of the AT's cleavage in the binding pocket site of CFIXa, hence markedly inhibiting CFIXa.21 It is also surrounded mostly by a blue region, which depicts the presence of hydrophobic residues including H:VAL32, H:VAL67, H:ALA40, H:ALA39, H:ILE73, H:ILE227, and H:THR76 (Fig. 1B). The conformational shift due to calcium-coordinating structure twist of GLU77 residue that was intertwined with the H:THR76 side chain shift. This was necessary for the calcium ion exosite and active site binding which contains the CFIXa inhibitor.22
The binding on ID V occurred at H:ASN72, which interacted intensively with other inert residues, proline and arginine. It is also responsible for the active site cleft formation, efficiently fitted with the S4 pocket, which resulted in a stable structure of AT in the P'wall necessary for CFIXa inhibition.21 The binding on ID 5 also takes place at H:TYR128, which was primarily involved in the interaction of CFIXa protease domain and sulfate ion that leads to the formation of hydrogen bond and salt bridge interaction, particularly at the OH region for about 3.1 Å. This site is necessary to modulate the interaction between the counterpart of heparin upon its binding with CFIXa.19 Also, L:PHE98 is part of the epidermal growth factor (EGF)-2 domain of CFIXa, which was involved in binding with the catalytic domain of the coagulation factor VIIIa and also comprised an area accessible by water, which was determined to be essential for the secretion of CFIXa.23 It was also surrounded by both blue and gray regions, which indicated the presence of both hydrophobic and neutral amino acid residues (Fig. 1B). The hydrophobic residues included H:ILE129B and L:PHE98.
Molecular modeling and pharmacokinetic properties of derived bioactive compounds
The ethanol maceration method was used to extract bioactive compounds from guava (P. guajava) leaves, with set-up using 95% ethanol achieving the highest yield at 78.541 g, 39.27% (Table S3†). The crude extract from this setup was analyzed using GC-MS using electron impact, identifying 28 peaks representing various bioactive compounds. The identification was based on the parent ion peaks observed in the mass spectra, which were compared with reference spectra from the NIST library (Fig. S2†). Major compounds included caryophyllene, eucalyptol, and β-sitosterol acetate. Some synthetic compounds were also detected, which are atypical for guava leaf extracts (Table S4†). Molecular modeling and ADME analysis were conducted using SwissADME to evaluate the pharmacokinetic properties and potential risks of these compounds. Those showing two or more hit markers for potential side effects were ruled out to avoid toxicophores and false-positive biological outputs (Table S5†). The top seven bioactive compounds (Scheme 1) were modelled using their respective SMILES.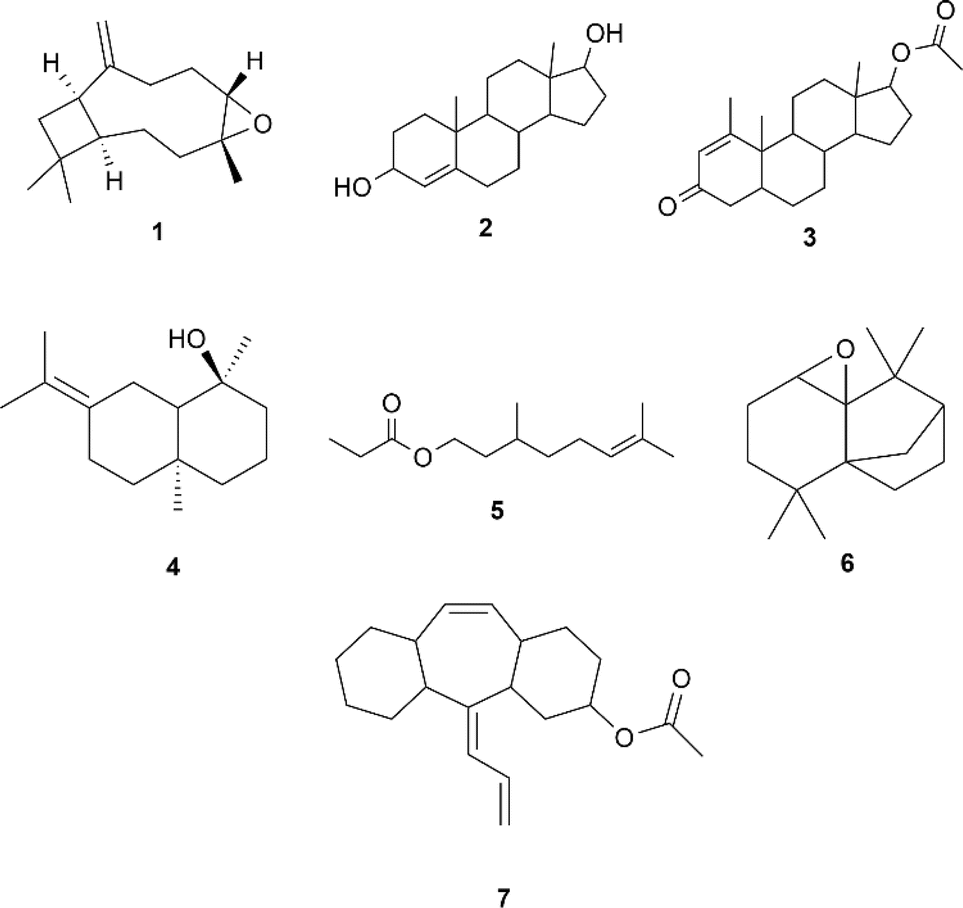 | ||
| Scheme 1 Seven bioactive compounds extracted from guava leaves that contain less than two violations on drug-likeness based on SwissADME: 1 caryophyllene oxide [MS (m/z, %): 177 (M+, 11.2), 79.05 (100)], 2 4-androstene-3α,17β-diol [MS (m/z, %): 220 (M+, 21.9), 159.15 (100)], 3 methenolone acetate [MS (m/z, %): 204 (M+, 4.3), 136.15 (100)], 4 juniper camphor [MS (m/z, %): 204 (M+, 49.4), 81.10 (100)], 5 6-octen-1-ol, 3,7-dimethyl-, propanoate [MS (m/z, %): 137 (M+, 17.6), 81.10 (100)], 6 4a,7-methano-4aH-naphth[1,8a-b]oxirene,octahydro-4,4,8,8-tetramethyl-[MS (m/z, %): 220 (M+, 39.8), 164.10 (100)], and 7 amitriptyline-M–(CH3)2NOH AC [MS (m/z, %): 290 (M+, 43.4), 247.15 (100)]. | ||
These bioactive compounds have garnered a bioavailability score of 0.55. These compounds also typically range from 220 g mol−1 to 288.34 g mol−1, which coincides with the ideal range for a drug's molecular weight of 150 g mol−1 < MW < 500 g mol−1. In comparison with novel drugs, the identified bioactive compounds possess high gastrointestinal (GI) tract absorption, which is a crucial characteristic for orally administered drugs and is all blood–brain-barrier (BBB) permeant, which is essential in reaching targets within the Central Nervous System (CNS). Compounds 1, 3, 4, and 7 inhibit CYP2C9, which is an important enzyme in the liver that processes drugs in specific doses to be safe and effective.24 Too much of these enzymes make the drugs break down too quickly, while too little activity of these enzymes might not break down the drug enough, increasing the risk of side effects. However, although these drugs can potentially inhibit these enzymes, at certain concentrations, they would not exhibit significant effects like apixaban, a coagulation factor Xa inhibitor drug, wherein it did not show cytochrome P450 inhibitions (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4/5) even at concentrations up to 20 μM when tested in primary human liver cells. Implying dosages and concentrations of drugs coincides with achieving the best therapeutic results when minimizing potential risks.25
Compounds 1, 2, 3, 4, 5, and 6 were all within the ADME's radar ideal range for the suitable physicochemical space for bioavailability (Fig. S3†). However, for compound 7 its unsaturation parameter showed that it was more than the ideal Csp3 fraction of 0.25 > Csp3 value > 1. Also, compounds 1, 3, 4, 5, 6, and 7 were P-glycoprotein negative (PGP−). These compounds, which are either PGP inhibitors or non-substrates of PGP, increase the systemic exposure of PGP substrates by inhibiting intestinal PGP. This inhibition enhances bioavailability, allowing the compounds to remain in the system longer and making them more suitable for oral administration, as they are effectively absorbed by the body.26 While compound 2 is the only P-glycoprotein positive (PGP+) compound, meaning, it was predicted to be a substrate of P-glycoprotein that actively pumps from the brain and or gastrointestinal lumen, exhibiting likeliness to be transported out of cells, shortening its duration of action, and decreasing its bioavailability. In contrast, PGP+ compounds can be advantageous as they can help to prevent excess accumulation of the drug in certain tissues and organs, reducing toxicity.
Molecular docking
Prior to docking, qualified bioactive compounds underwent conformational analysis to determine the most kinetically favored conformer and were used for docking analysis (Fig. S4–S10†) using rdkit.27 These kinetically favored conformers were easier to form because the transition state leading to their formation was lower in energy, making the kinetic pathway more accessible. This property allowed the ligand to remain in that kinetically favored conformation, increasing its residence time, therefore providing a longer therapeutic effect, improving ligand selectivity, and potentially acting at lower concentrations.28 Even though flexible docking was employed and these bioactive compound conformers were still adjusted, having kinetically favored conformers as a starting point provided more efficient sampling of conformational space, thereby improving the accuracy of predicting the binding mode of the ligand. The bioactive compounds were docked on each active site of the biomarkers. The more negative docking scores of each bioactive compound, regardless of the active site number, in both Vina and Vinardo scoring functions were analyzed further for their specific interactions. The top three complexes with the highest docking score were recorded in ID II: CFIXa–3 complex (Fig. 3), ID II: CFIXa–7 complex (Fig. 4), and ID V: CFIXa–2 complex (Fig. 2), respectively (Table S6†). Compounds 1, 3, 5, 6, and 7 bind in ID II, and interacts with the catalytic triad H:TYR99, H:PHE174 and H:TRP215 at the S4 pocket of the protein.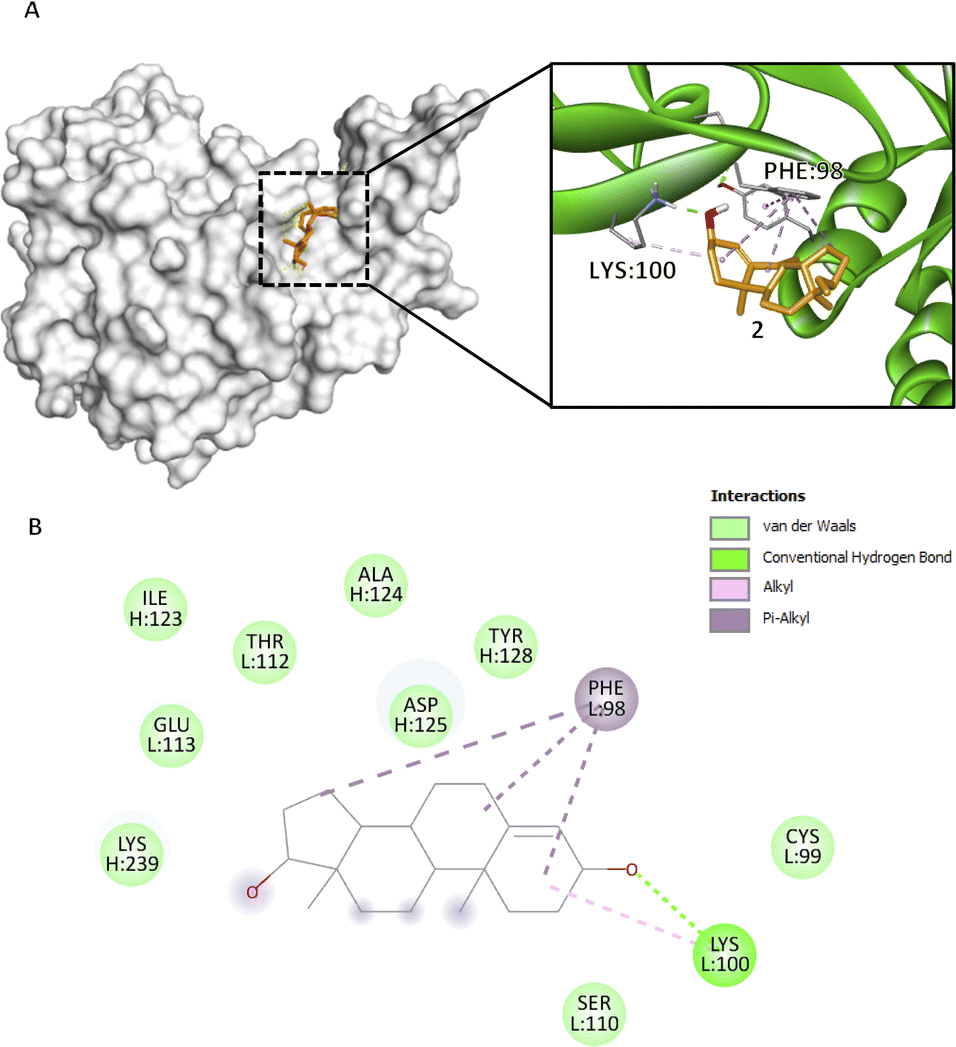 | ||
Fig. 2 Compound 2 binding mode that has the highest Vina and Vinardo docking score (−7.0:−6.1). (A) 3D model of compound 2 ( ) and CFIXa ( ) and CFIXa ( ). (B) 2D model and their respective interactions. ). (B) 2D model and their respective interactions. | ||
Compound 1 methyl group 5 forms an alkyl interaction with H:LYS98 (3.95 Å) and binds in a compact, cyclic shape (Fig. S11†). Particularly, this interaction induced the opening of the CFIXa S4 pocket allowing inhibitors to bind to the cavity. This residue is part of the upper rim of the active site configuration of CFIXa that acts as a hindrance on the CFIXa S4 pocket. When low molecular weight heparin binds to CFIXa, this residue moves and allows an increase in interaction with Kunitz-type inhibitor, BPTI.29 A π–alkyl interaction of methyl group 13 on H:TYR99 (4.77 Å), and methyl group 13, ring B, and ethenyl group 15 on H:TRP215 (4.92 Å, 4.74 Å, 4.14 Å) induces a conformational change on both residue, which implies induced binding affinity, stronger bonds, and formation of more stable protein–ligand complex for compound 1 (Table S7†).
Compounds 2 and 3, bind with active site ID V, particularly at L:PHE98 and L:ASN92. Compound 2 rings A, B, and D form a π–alkyl interaction at L:PHE98 (5.17 Å, 5.05 Å, 4.28 Å) (Fig. 2B). Specifically, this interaction induces the intercalation of the ligand to the binding pocket of the protein by adding hydrophobicity, which allows the association of the non-polar surfaces and the stabilization of the ligand to its binding interface.30 Also, L:PHE98 is part of the EGF-2 domain of CFIXa, which is important in CFIXa–CFVIIIa binding. L:LYS100 forms an alkyl and conventional hydrogen bond on compound 2's ring A (4.91 Å) and carbonyl group 3 (1.97 Å), respectively. Even though this residue does not have a reported significance, the hydrogen bond promotes stability of the ligand within the active site and an allosteric conformational change in CFIXa, specifically at L:PHE98 (Table S7†), that contributes to the disruption of the formation of intrinsic tenase complex.
Compound 3, which obtained the highest docking score, methyl groups 20, 21, and 22 form three π–alkyl with H:TYR99 (4.78 Å, 5.13 Å, 4.54 Å), rings C and D form π–alkyl interactions at H:PHE174 (4.06 Å, 5.17 Å) and ring C at H:TRP215 (5.45 Å) (Fig. 3B), particularly in an elongated, slightly curved shape. The binding conformation is more linear than cyclic, with the compound fitting snugly into the binding pocket (Fig. 3A). These aforementioned residues form the S4 pocket, particularly at the side chain of H:ILE390. The S4 pocket mediates hydrophobic interaction, which resulted in the shifting of the main conformation of the protein, hence considered as a candidate for the novo drug design of CFIXa inhibitor.21 Alkyl interaction of methyl group 20 in H:LYS98 (4.77 Å) allows the opening of the S4 pocket inducing strong interaction with the aforementioned residues. Also, the conventional hydrogen bond of the carbonyl group in C2 in H:ASN97 (2.44 Å) contributes to its high docking score. This interaction aligns with the reported mechanism of aminobenzisoxazole, a CFIXa inhibitor that is extensively researched but not yet established as a standard, against the 5TNT, 5TNO, and 5EGM CFIXa models.4 These interactions produced a significant conformational change to H:TYR99 and H:TRP125, indicating a successful binding (Table S7†). Among the bioactive compounds, it garnered the highest docking score indicating optimal pose for both ligand and CFIXa.
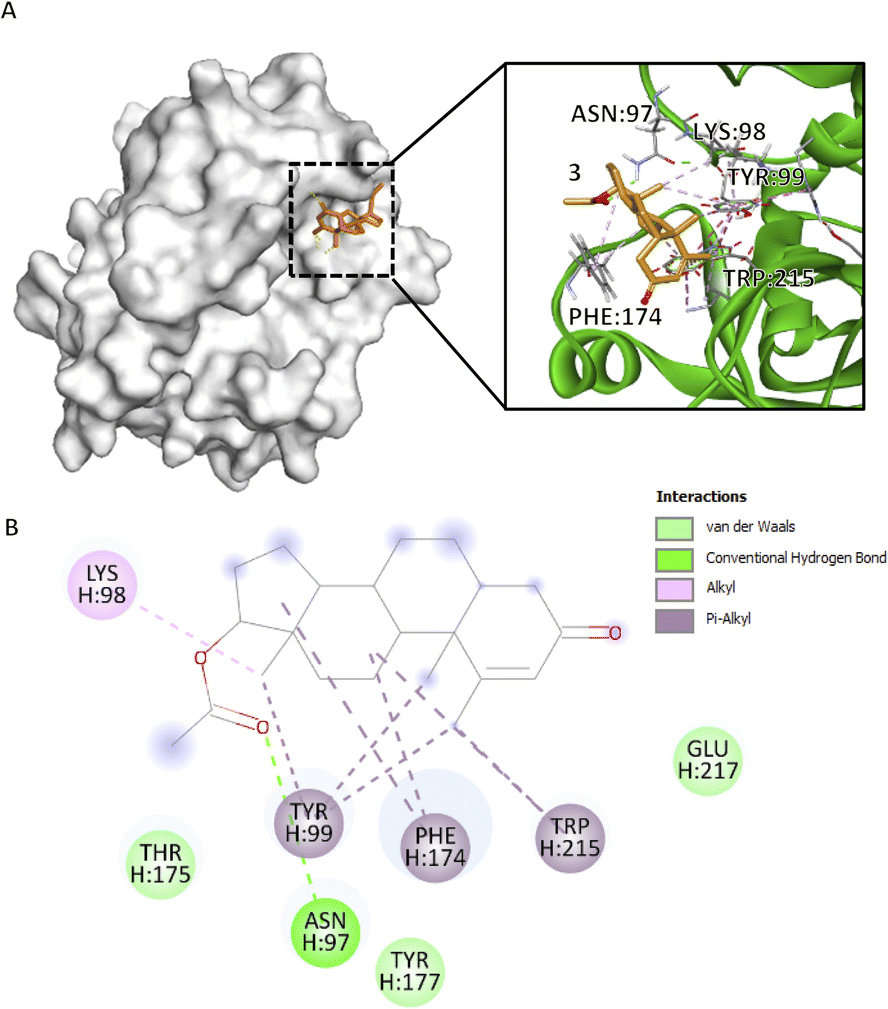 | ||
Fig. 3 Compound 3 binding mode that has the highest Vina and Vinardo docking score (−7.6:−6.8). (A) 3D model of compound 3 ( ), and CFIXa ( ), and CFIXa ( ). (B) 2D model and their specific interaction types. ). (B) 2D model and their specific interaction types. | ||
Compound 4 methyl group 1 forms π–alkyl interaction at H:TYR128 (5.33 Å), H:PHE130 (4.92 Å), and H:PHE133 (4.97 Å) (Fig. S12†). Additionally, methyl groups 3 and 9 formed π–alkyl interactions with H:PHE133 (4.86 Å, 4.62 Å) (Fig. S12†). Primarily, these residues were involved in the formation of aromatic clusters that bury L:PHE98, which was an essential indicator of binding between EGF-2 and the catalytic domain of CFIXa.31 Additionally, these residues are also involved in the loop rearrangement of CFIXa for low-efficiency coagulation. Compound 4 OH group in C1 forms an unfavorable donor–donor interaction with L:ASN92 (2.62 Å), while also forming a conventional hydrogen bond with L:CYS95 (1.83 Å). This suggests a competitive and suboptimal binding environment that could destabilize the protein complex. The lowest bond length was recorded at ID V, particularly at H:TYR128 (CG–CD1) and H:ILE129B (CG1–CD1), which implied the presence of stronger intermolecular bonds (Table S7†).
Compound 5 like compound 3 interacted with the S4 pocket of CFIXa linearly with some angular bends (Fig. S13A†). Its carbonyl group 1a and carbon 1 forms a carbon–hydrogen bond with H:TYR175 (3.42 Å, 3.51 Å) (Fig. S13B†). H:TYR175 along with H:ASN97 AND H:TYR177 was involved in the sulfate mode binding of CFIXa protease domain and heparin.19 This residue induced binding affinity of the heparin to the surface area of CFIXa, resulting in a reactive loop in activation, thus resulting in less stable clot formation. Like in compound 3, compound 5's carbonyl group at C1a forms a conventional hydrogen bond with H:ASN97 (2.41 Å) and together with C1, a carbon–hydrogen bond with H:THR175 (3.42 Å, 3.51 Å). These interactions should contribute to the increase of the complex's docking score. However, CFIXa-5 complexes yielded the least negative docking scores across the compounds ranging from −5.6 to −3.7 (Table S6†). This indicated that the conformer the ligand takes is not its optimal structure, contributing to the increase of its docking score.
Compound 6 is also bound with the S4 pocket of CFIXa interacting with the same residues as in compounds 1, 3, and 5. Compound 6 epoxide 1 constituent forms a carbon–hydrogen bond at residue H:ASN97 (3.53 Å) (Fig. S14†). This interaction has a vital role in the ligand recognition of the binding pocket and protein structure folding, which results in higher binding affinity and the formation of a stable complex.32 Furthermore, H:ASN97 is also involved in the heparin-binding mode of CFIXa, located at the exosite region. This residue interacts with the sulfate ion in the putative heparin binding site, therefore, inducing conformational shift and formation of less stable clot.19 Epoxide 1 constituent also forms a conventional hydrogen bond with H:LYS98 (2.38 Å), which allows the further opening of CFIXa S4 pocket in comparison with the other compounds. The lowest bond length was recorded at the CE1-CZ atom of residue H:TYR99 of ID II, which implies stronger bonds and an indication of the conformational change on the S4 pocket (Table S7†).
Similar to previous compounds, compound 7 binds to the S4 pocket of the CFIXa. Specifically in a S-shape manner (Fig. 4A). However, only three residues form specific interactions (i.e. pi interactions, alkyl interactions, and hydrogen bond), while the rest are involved in van der Waals interactions (Fig. 4B). Specifically, this interaction modulates the core of the protein and also induces the contact of the ligand to the binding pocket.33 Compound 7 mechanism is straightforward. Its C3 forms an alkyl interaction with H:LYS98 (4.31 Å) and that opens the S4 pocket of CFIXa and proceeds to form strong interactions with the catalytic triad. Specifically, compound 7's ring A forms a pi–pi T-shaped interaction and the carbonyl group on C1 forms a conventional hydrogen bond with H:TYR99 (5.53 Å, 2.66 Å), and ring B forms a pi–pi T-shaped and ring A forms a pi–pi stacked interaction with H:PHE174 (5.09 Å, 4.06 Å). These interactions induce conformational changes on H:TYR175 (Table S7†). Specifically, The CG–CD2 atom of this residue at active site ID II constitutes the lowest bond length, which implies stronger bond interaction, compact molecules, and the formation of more stable complexes. It also garnered the second most negative docking score among the bioactive compounds.
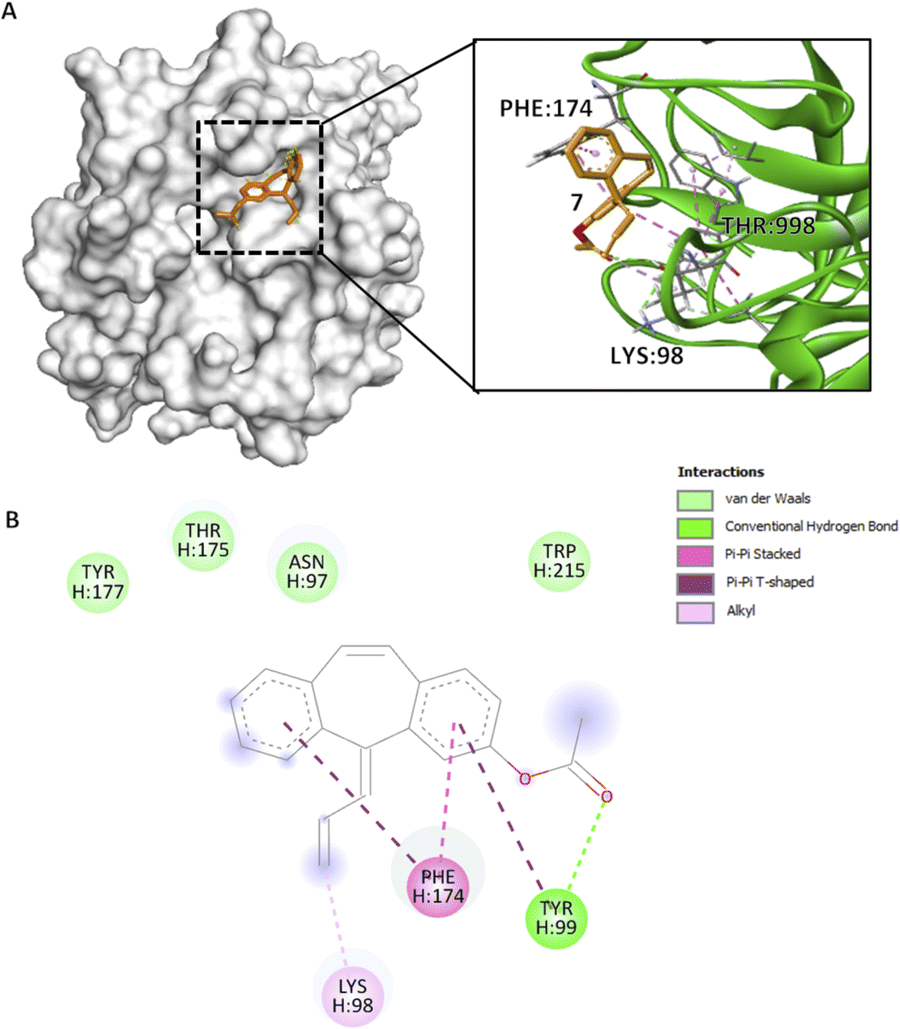 | ||
Fig. 4 Compound 7 binding mode in ID V that has the highest Vina and Vinardo docking score (−7.2:−6.6). (A) 3D model of compound 7 ( ) and CFIXa ( ) and CFIXa ( ). (B) 2D model and their specific interaction types. ). (B) 2D model and their specific interaction types. | ||
Conclusions
The molecular modeling and active site identification of CFIXa revealed three critical binding sites (IDs II, III, and V) essential for the protein's inhibition, as confirmed by structural data and literature. Bioactive compounds were extracted from guava leaves using the ethanol maceration method, which yielded a high concentration of potentially therapeutic agents. Pharmacokinetic analysis indicated high bioavailability and gastrointestinal absorption, with some compounds showing potential to inhibit liver enzymes crucial for drug metabolism. Docking studies demonstrated that these compounds effectively bind to identified active sites, especially within the S4 pocket, which is crucial for CFIXa inhibition. These interactions suggest that the bioactive compounds can induce conformational changes in CFIXa, leading to its inhibition. This behavior was experimentally explored in the cited literature. Implications of these findings highlight the potential of these bioactive compounds as promising candidates for developing novel anticoagulant drugs that target CFIXa with high specificity and efficacy, offering new therapeutic options for conditions requiring anticoagulation. This also gives insights into the ambiguity surrounding the effect of guava leaves on the bleeding time in vivo.Data availability
The data supporting this article have been included as part of the ESI.† The data analysis scripts of this article are available in the interactive notebook Google Collab at https://colab.research.google.com/drive/1LUmEoFpTX9u7z_pjNCofoHN634zZZauu?usp=sharing.Author contributions
De Luna, Joseph G. – conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, software, validation, visualization, writing – original draft, writing – review & editing Gonzales, Shanahi Chelledie B. – investigation, resources, validation, writing – review & editing Nuqui, Jimuel Jan M. – investigation, resources, writing – review & editing Capinding, Evalyn S. – methodology, supervision, writing – review & editing Sacdalan, Corazon D. – supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The researchers extend their gratitude to the Technological University of the Philippines and the Chemistry Department, for their valuable resources and encouragement. Special thanks are also due to Mr Shane Tuballa Mendez for his assistance with research procedures.Notes and references
- Cardiovascular diseases (CVDs), https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds), accessed, 15 May, 2024 Search PubMed.
- A. Dorgalaleh, M. Daneshi, J. Rashidpanah and E. R. Yasaghi, in Congenital Bleeding Disorders, ed. A. Dorgalaleh, Springer International Publishing, Cham, 2018, pp. 3–26 Search PubMed.
- Hemostasis and Thrombosis: Methods and Protocols, ed. E. J. Favaloro and G. Lippi, Springer New York, New York, NY, 2017, vol. 1646 Search PubMed.
- S. Kundu and S. Wu, Molecules, 2021, 26, 5372 CrossRef CAS PubMed.
- W. Ruksiriwanich, C. Khantham, A. Muangsanguan, Y. Phimolsiripol, F. J. Barba, K. Sringarm, P. Rachtanapun, K. Jantanasakulwong, P. Jantrawut, C. Chittasupho, R. Chutoprapat, K. Boonpisuttinant and S. R. Sommano, Plants, 2022, 11, 3514 CrossRef CAS PubMed.
- M. V. B. Sahagun, R. R. Macadangdang Jr, C. C. Abogadie, R. M. C. Asi, A. H. Boncodin, C. F. Julian and K. D. Pedersen, Asian J. Biol. Life Sci., 2021, 10, 238–244 CrossRef CAS.
- M. Kumar, M. Tomar, R. Amarowicz, V. Saurabh, M. S. Nair, C. Maheshwari, M. Sasi, U. Prajapati, M. Hasan, S. Singh, S. Changan, R. K. Prajapat, M. K. Berwal and V. Satankar, Foods, 2021, 10, 752 CrossRef CAS PubMed.
- M. I. Tousif, M. Nazir, M. Saleem, S. Tauseef, N. Shafiq, L. Hassan, H. Hussian, D. Montesano, D. Naviglio, G. Zengin and I. Ahmad, Molecules, 2022, 27, 7016 CrossRef CAS PubMed.
- F. Ebrahimi, M. Torbati, J. Mahmoudi and H. Valizadeh, J. Pharm. Pharm. Sci., 2020, 23, 10–23 Search PubMed.
- M. E. Fortunato and C. M. Colina, SoftwareX, 2017, 6, 7–12 CrossRef.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. In'T Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- W. Tian, C. Chen, X. Lei, J. Zhao and J. Liang, Nucleic Acids Res., 2018, 46, W363–W367 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- M. Brylinski, Chem. Biol. Drug Des., 2018, 91, 380–390 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- M. Murcia and A. R. Ortiz, J. Med. Chem., 2004, 47, 805–820 CrossRef CAS PubMed.
- R. Quiroga and M. A. Villarreal, PLoS One, 2016, 11, e0155183 CrossRef PubMed.
- S. Tang, R. Chen, M. Lin, Q. Lin, Y. Zhu, J. Ding, H. Hu, M. Ling and J. Wu, Molecules, 2022, 27, 3041 CrossRef CAS PubMed.
- K. Vadivel, H. A. Schreuder, A. Liesum, A. E. Schmidt, G. Goldsmith and S. P. Bajaj, J. Thromb. Haemostasis, 2019, 17, 574–584 CrossRef PubMed.
- A. E. Schmidt, J. E. Stewart, A. Mathur, S. Krishnaswamy and S. P. Bajaj, J. Mol. Biol., 2005, 350, 78–91 CrossRef CAS PubMed.
- D. J. D. Johnson, J. Langdown and J. A. Huntington, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 645–650 CrossRef CAS PubMed.
- T. Zögg and H. Brandstetter, Structure, 2009, 17, 1669–1678 CrossRef PubMed.
- Y.-J. Chang, H.-L. Wu, N. Hamaguchi, Y.-C. Hsu and S.-W. Lin, J. Biol. Chem., 2002, 277, 25393–25399 CrossRef CAS PubMed.
- A. Daly, A. Rettie, D. Fowler and J. Miners, J. Pers. Med., 2017, 8, 1 CrossRef PubMed.
- L. Wang, D. Zhang, N. Raghavan, M. Yao, L. Ma, C. A. Frost, B. D. Maxwell, S. Chen, K. He, T. C. Goosen, W. H. Griffith and S. J. Grossman, Drug Metab. Dispos., 2010, 38, 448–458 CrossRef CAS PubMed.
- M. Elmeliegy, M. Vourvahis, C. Guo and D. D. Wang, Clin. Pharmacokinet., 2020, 59, 699–714 CrossRef CAS PubMed.
- G. Landrum, P. Tosco, B. Kelley, R. Rodriguez, D. Cosgrove, R. Vianello, G. Peter, G. Jones, N. Schneider, E. Kawashima, D. Nealschneider, A. Dalke, M. Swain, B. Cole, S. Turk, A. Savelev, A. Vaucher, M. Wójcikowski, I. Take, V. Scalfani, R. Walker, K. Ujihara, D. Probst, G. Godin, A. Pahl, J. Lehtivarjo, F. Berenger, B. Jason, S. Andrew and R. Serina, rdkit/rdkit (version Release_2024_03_2) [object Object], 2024 Search PubMed.
- D. V. Borisov and A. V. Veselovsky, Biochemistry (Moscow) Supplement Series B: Biomedical Chemistry, 2020, 14, 228–240 CrossRef.
- P. F. Neuenschwander, K. J. Deadmond, K. Zepeda and J. Rutland, J. Thromb. Haemostasis, 2012, 10, 382–389 CrossRef CAS PubMed.
- R. Patil, S. Das, A. Stanley, L. Yadav, A. Sudhakar and A. K. Varma, PLoS One, 2010, 5, e12029 CrossRef PubMed.
- K.-P. Hopfner, A. Lang, A. Karcher, K. Sichler, E. Kopetzki, H. Brandstetter, R. Huber, W. Bode and R. A. Engh, Structure, 1999, 7, 989–996 CrossRef CAS PubMed.
- S. Horowitz and R. C. Trievel, J. Biol. Chem., 2012, 287, 41576–41582 CrossRef CAS PubMed.
- G. Bitencourt-Ferreira, M. Veit-Acosta and W. F. De Azevedo, in Docking Screens for Drug Discovery, ed. W. F. De Azevedo, Springer New York, New York, NY, 2019, vol. 2053, pp. 79–91 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04709e |
| This journal is © The Royal Society of Chemistry 2024 |