
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel green and efficient heterogeneous acid catalyst for the one-pot synthesis of benzopyrazine-aminoimidazole hybrids with antiproliferative potential †
Ali Altharawi *a,
Safar M. Alqahtania,
Taibah Aldakhila,
Pawan Sharmabc,
Abhishek Kumarde and
Montather F. Ramadanf
*a,
Safar M. Alqahtania,
Taibah Aldakhila,
Pawan Sharmabc,
Abhishek Kumarde and
Montather F. Ramadanf
aDepartment of Pharmaceutical Chemistry, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia. E-mail: a.altharawi@psau.edu.sa
bDepartment of Chemistry, School of Sciences, Jain (Deemed-to-be) University, Bengaluru, Karnataka 560069, India
cDepartment of Sciences, Vivekananda Global University, Jaipur, Rajasthan 303012, India
dSchool of Pharmacy-Adarsh Vijendra Institute of Pharmaceutical Sciences, Shobhit University, Gangoh, Uttar Pradesh 247341, India
eDepartment of Pharmacy, Arka Jain University, Jamshedpur, Jharkhand 831001, India
fCollege of Dentistry, Al-Ayen University, Thi-Qar, Iraq
First published on 19th August 2024
Abstract
A novel, green, efficient, and stable magnetically heterogeneous nanocatalyst was developed by immobilizing butanesulfonic acid (BuSO3H) onto the surface of MFe2O4 magnetic nanoparticles (MNPs). The resulting core–shell structure of the MFe2O4@PDA@BuSO3H nanocatalyst was thoroughly characterized using various analytical techniques, including Fourier-Transform Infrared Spectroscopy (FT-IR), X-ray Diffraction (XRD), Energy-Dispersive X-ray Spectroscopy (EDS), Field Emission Scanning Electron Microscopy (FESEM), Transmission Electron Microscopy (TEM), Vibrating Sample Magnetometry (VSM), and Brunauer–Emmett–Teller (BET) analysis. A nanocatalyst was used to synthesize 2-benzopyrazine-aminoimidazole hybrid derivatives through a domino multicomponent Knoevenagel–condensation–cyclization reaction (5a–p) in an environmentally friendly manner. The resulting compounds were then tested for their anticancer activity against three types of human cancer cells (MCF-7, A549, and U87) using the MTT assay. The experiment showed that the nanocatalyst had excellent catalytic activity, and the synthesized compounds exhibited promising antiproliferative activity. Notably, compounds 5g and 5h, containing a 2-naphthyl ring, showed the highest antiproliferative effects against MCF-7 cells, with IC50 values of 0.03 and 0.32 μM, respectively. Additionally, the activity of compounds 5g and 5h in tubulin polymerization, apoptosis induction, and cell cycle arrest in MCF-7 cells were investigated. The results demonstrated that these compounds effectively induced apoptosis and cell cycle arrest. The binding of representative compounds to the colchicine binding site of tubulin was confirmed through molecular modeling studies.
1. Introduction
Benzopyrazine (quinoxaline), a heteroaromatic ring containing nitrogen, is a bioisostere of privileged functional scaffolds such as benzimidazole, benzothiophene, and quinoline.1 It has been found to possess diverse biological and therapeutic applications, displaying potential anticancer properties via multiple mechanisms, including inhibition of kinase, tubulin polymerization, and topoisomerase.2–4 Synthetic derivatives of benzopyrazine, particularly those containing 2-amino benzopyrazine, have shown significant medicinal roles as anti-proliferative agents, particularly by inhibiting tubulin polymerization. For instance, Alanazi et al. synthesized a new set of benzopyrazine-based derivatives with anticancer and VEGFR-2 inhibitory properties.5 Compound 1 (Fig. 1) is highly effective against HepG2 and MCF-7 cell lines, with IC50 values of 12.9 μM and 7.5 μM. In another study, a series of novel derivatives based on triazole benzopyrazine were designed and synthesized as inhibitors of VEGFR-2. Among these derivatives, Compound 2 (Fig. 1) has exhibited the most promising cytotoxic effect against HepG2 and MCF-7cell lines, with IC50 values of 4.9 μM and 6.2 μM, in comparison to sorafenib (which shows IC50 values of 3.53 μM and 2.18 μM against these cell lines).6 It is worth noting that these are novel findings and contribute to developing new potential anticancer agents.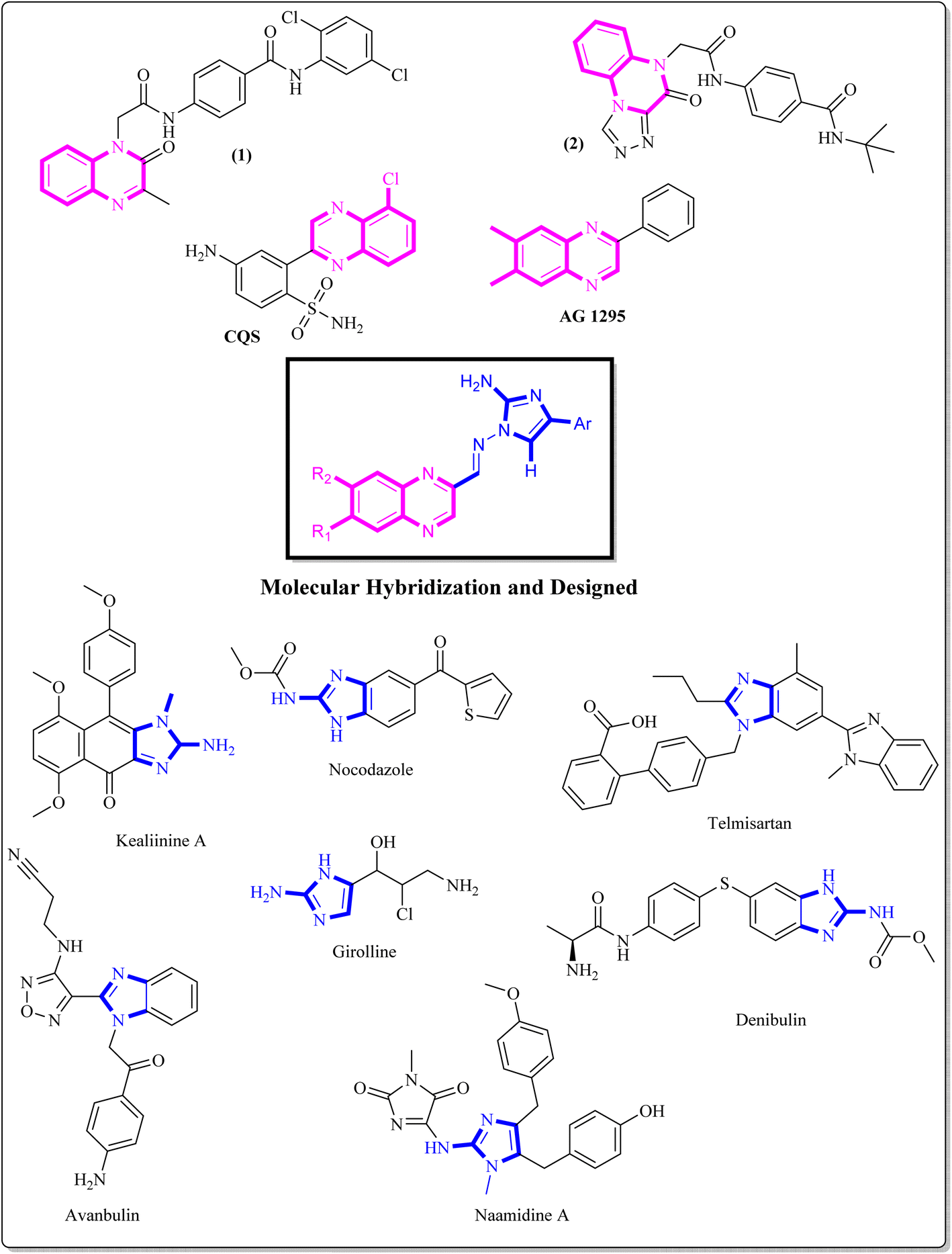 | ||
| Fig. 1 Some benzopyrazine-based drug-like candidates and benzopyrazine-derived synthetic compounds. | ||
The incorporation of an appropriate motif in drug discovery is crucial for generating new medicinal structures that have the potential to act as bioactive agents targeting specific sites. 2-Aminoimidazole, a heterocyclic scaffold, is highly valuable and has wide-ranging applications in various fields, especially as a therapeutic/bioactive agent.7 It exhibits an exciting pattern of hydrogen bond donation and acceptance during target interactions. Numerous studies have highlighted the anticancer properties of benzimidazole derivatives. Denibulin, nocodazole, and avanbulin (Fig. 1) are benzimidazole-based anticancer drugs that exert their effects by inhibiting tubulin polymerization.7–9
Extensive research has been conducted on the anticancer properties of molecules that contain N-benzylated benzimidazoles. Telmisartan G, a representative antihypertensive drug bearing an N-benzylated benzimidazole core, has been reported for its anticancer activity.8 Additionally, the synergistic activity of LLS30 H, a derivative of N-benzylated benzimidazole, which is a hybrid with docetaxel, has been reported by Tsung-Chieh Shih et al. Based on the results, these compounds effectively inhibited the growth and metastasis of prostate cancer cells in vivo.9,10
Despite developing various synthetic methods for highly functionalized 2-aminoimidazole and benzopyrazine core moieties, driven by the increasing demand for natural product synthesis and medicinal chemistry, most of these methods contain lengthy experimental procedures steps.11–13 Furthermore, while numerous protocols have demonstrated advantages, they also possess limitations such as extended reaction durations, toxic and costly reagent requirements, and challenges in separating and recycling catalysts.14–17 Therefore, there is a pressing need to explore sustainable and environmentally friendly protocols and efficient and reusable catalysts to prepare benzopyrazine and 2-aminoimidazole motifs. This is a significant challenge associated with ongoing research.
In recent years, nano heterogeneous acid catalysts have been emerging as effective tools for synthesizing bioactive molecules due to their high surface area and the ability to provide a high density of active sites. These catalysts have not only improved catalytic efficiency but also facilitated greener chemical processes.16,17 Magnetically recoverable nano-catalysts, in particular, offer the dual benefits of easy separation and reusability, making them highly attractive for sustainable chemistry.18,19
In this work, we report a rapid, highly efficient, and environmentally benign synthetic pathway for the preparation of a leak-free, high-performance, and stably attached heterogeneous magnetic nanocatalyst, achieved by grafting sulfonic groups onto the surface of dopamine-coated MNPs.18–22 The polydopamine (PDA) coating on the MNPs endows them with exceptional adhesion properties and outstanding biocompatibility. The surface of these particles can be further modified due to the presence of abundant carboxyl and amino groups. PDA serves as a bridge connecting two functional heads through its amino and quinone groups, which form Schiff base addition adducts via the oxidation of dopamine. This bridging capability results in a strong affinity for the hydroxyl and NH groups on the surface of MFe2O4@PDA MNPs. Consequently, it facilitates the grafting of BuSO3H functionalities, as depicted in Scheme 1. Following the successful synthesis of the organic–inorganic framework MFe2O4@PDA@BuSO3H MNPs, its effectiveness as a core–shell-structured nanocatalyst in the multicomponent one-pot cascade synthesis of a novel series of benzopyrazine derivatives containing 2-aminoimidazole was evaluated. Additionally, in line with the rationale guiding the design of these hybrid ligands, molecular docking simulations focused on the colchicine binding site of tubulin were utilized to investigate the mechanisms underlying their notable anti-proliferative activities.
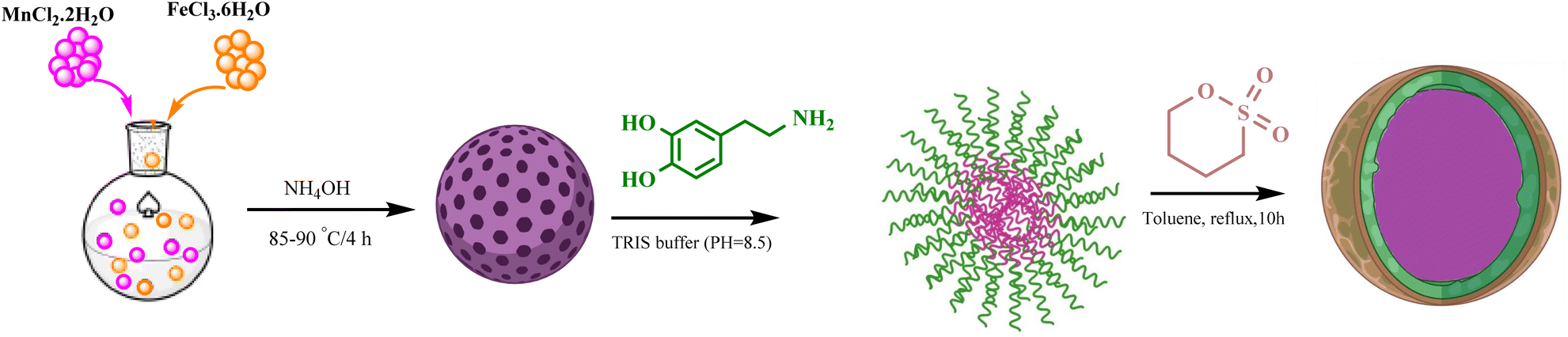 | ||
| Scheme 1 Synthesis of MFe2O4@PDA@ BuSO3H MNPs as a new heterogeneous catalyst. | ||
2. Results and discussion
2.1. Chemistry
 | ||
| Fig. 2 FT-IR spectra (A); XRD spectra (B); BET (C) and VSM (D) diagrams of MFe2O4@PDA@BuSO3H MNPs. | ||
The XRD pattern of the uncoated MNPs is consistent with that of spinel ferrites (Fig. 2B). All peaks' positions and relative intensities match the standard XRD pattern of Fe2O3 MNPs (JCPDS card no. 85-1436), indicating that the MNPs retain their crystalline cubic spinel structure. The XRD patterns of the particles exhibit nine characteristic peaks associated with a cubic iron oxide phase (2θ = 14.80°, 30.10°, 35.50°, 43.10°, 53.00°, 57.00°, 62.80°, 70.50°, 73.90°), which are indexed as (1 1 0), (2 2 0), (3 1 1), (4 0 0), (3 3 1), (4 2 2), (5 1 1), (4 4 0), and (5 3 1), respectively. Based on the XRD pattern data and the Scherrer equation, the crystalline size of MFe2O4@PDA@BuSO3H MNPs is estimated to be 30 nm.
The BET method is essential in determining materials' specific surface area (SSA). To calculate the average particle diameter (nm), the following formula can be utilized: dBET = 6000/(ρs × S), where S represents the specific surface area in m2 g−1, and ρ is the theoretical density in g cm−3.21,23 During the measurement of surface area, when solid particles are not agglomerated, nitrogen gas (N2) permeates the pores and surfaces of the powder, as illustrated in Fig. 2C.
To assess the magnetic characteristics of MFe2O4@PDA@BuSO3H, a VSM was used at room temperature under an applied field of approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe. As shown in Fig. 2D, the saturation magnetization (Ms) values for MFe2O4@ and MFe2O4@PDA@BuSO3H catalysts are about 67 emu g−1 and 55 emu g−1, respectively. Compared to uncoated MFe2O4 MNPs (67.22 emu g−1), the saturation magnetization of the coated catalysts exhibited a significant decrease due to a higher amount of organic compounds on the surface of the nanoparticles.
000 Oe. As shown in Fig. 2D, the saturation magnetization (Ms) values for MFe2O4@ and MFe2O4@PDA@BuSO3H catalysts are about 67 emu g−1 and 55 emu g−1, respectively. Compared to uncoated MFe2O4 MNPs (67.22 emu g−1), the saturation magnetization of the coated catalysts exhibited a significant decrease due to a higher amount of organic compounds on the surface of the nanoparticles.
The EDS spectrum (Fig. 3) clearly shows signals corresponding to Fe, C, O, S, and N, confirming the successful synthesis of MFe2O4@PDA@BuSO3H MNPs, as the iron oxide nanoparticles are now functionalized with sulfonic acid groups. To assess the stability of the MFe2O4@PDA@BuSO3H and the bond formation between MFe2O4 and the organic agent, TGA was performed. The TGA curve (Fig. 4) shows an initial weight loss below 100 °C, which can be attributed to the desorption of solvent and surface hydroxyl groups. The subsequent weight loss is due to the decomposition of sulfonic acid grafted onto the surface of MFe2O4MNPs. The TGA curve indicates that the weight fraction of sulfonic acid is approximately 6.6%.
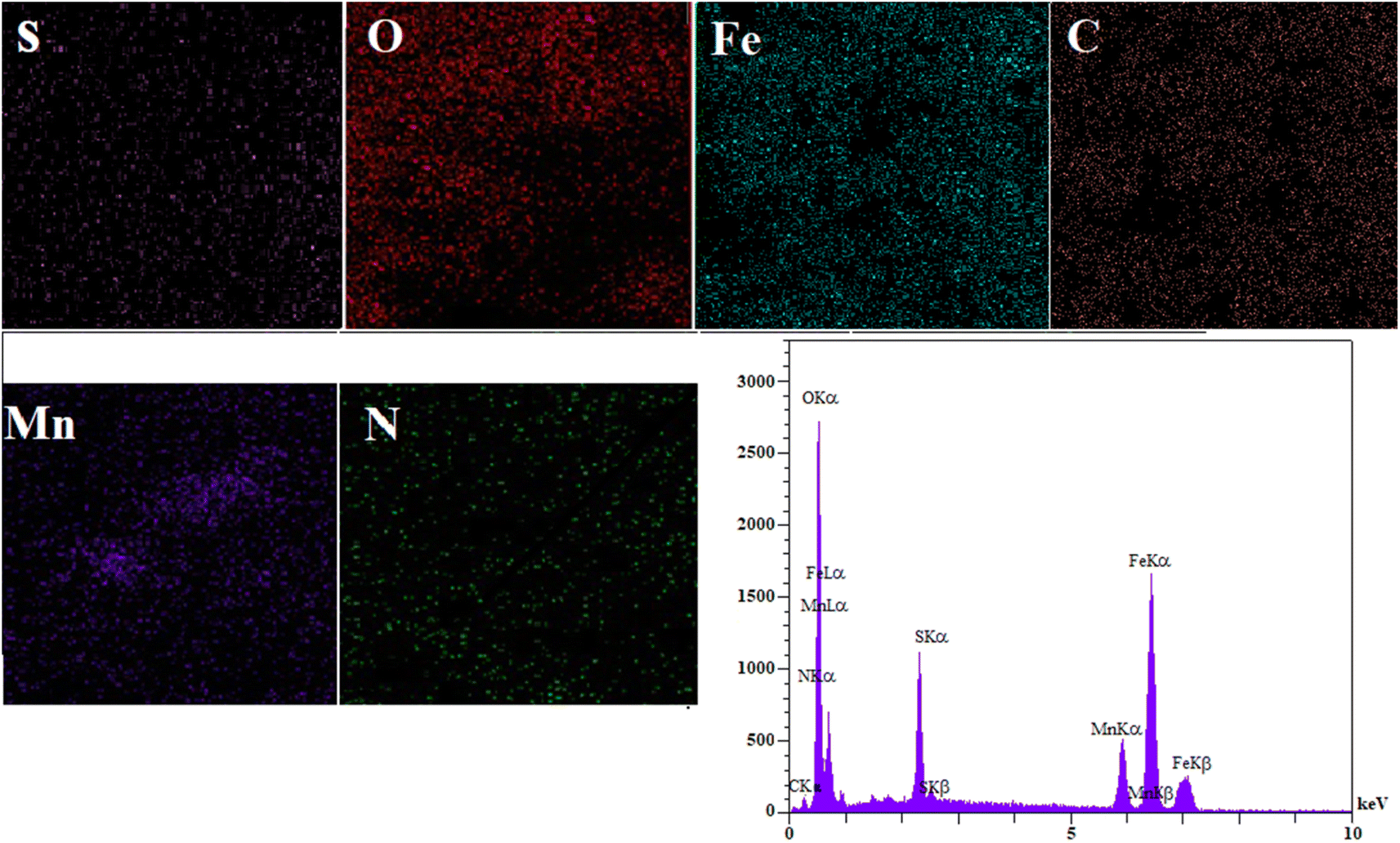 | ||
| Fig. 3 EDS spectra of MFe2O4@PDA@BuSO3H MNPs. | ||
 | ||
| Fig. 4 TGA spectra of MFe2O4@PDA@BuSO3H MNPs. | ||
Surface morphology analysis of the synthesized nanocatalysts was conducted using SEM. The results revealed that the nanocatalysts exhibited a spherical shape, with an average size ranging from 40 to 50 nm (Fig. 5). TEM was employed to observe the particles, revealing their consistent size and shape. The particles exhibited a homogeneous size with an average diameter of 10 nm (Fig. 6).
 | ||
| Fig. 5 SEM image of MFe2O4@DA@BuSO3H. | ||
 | ||
| Fig. 6 TEM image of MFe2O4@PDA@ BuSO3H. | ||
To determine the optimal reaction conditions for synthesizing functionalized derivatives of 2-aminoimidazole-benzopyrazine hybrids, we conducted multicomponent domino reactions using benzopyrazine-2-carbaldehyde 1a (1 mmol), aminoguanidine bicarbonate 2 (1 mmol) and ketones 3a as a model in ethanol (Scheme 2). Initially, we attempted the reaction in ethanol without a catalyst under reflux conditions for 4 hours (Table 1, entry 1). However, the desired product 5a was not obtained, underscoring the necessity of catalytic assistance to drive the reaction and minimize the formation of by-products. When we refluxed 1a and 2 in the presence of BuSO3H and ethanol, a brown solid of 4a formed in less than 10 minutes. Subsequently, ethanol was used to heat the mixture under reflux conditions, and 3a was added. BuSO3H (0.005 g) in ethanol led to the formation of 5a with a yield of 70% (Table 1, entry 2), indicating moderate catalytic activity for the reaction. We further tested the effectiveness of different catalysts in the model reaction, including MFe2O4, MFe2O4@PDA, and MFe2O4@PDA@BuSO3H; all were added in amounts below the stoichiometric ratio (0.05 g), and the reactions were conducted in ethanol under reflux conditions. After a thorough investigation, we established that MFe2O4@PDA@BuSO3H (0.05 g) demonstrated exceptional catalytic activity, resulting in short reaction times and high product yields. Specifically, MFe2O4 alone provided only a 40% yield (Table 1, entry 3), whereas modification with PDA slightly improved the yield to 45% (Table 1, entry 4). Notably, further functionalization with sulfonic acid groups (MFe2O4@PDA@SO3H) significantly enhanced the yield to 87% (Table 1, entry 5), illustrating the crucial role of the acidic groups in catalysis. Additionally, we investigated various solvents for the synthesis of compound 5a. Individual solvents did not yield efficient results. However, when the reaction was carried out in a mixture of ethanol and water (1:1), robust hydrogen bonding ability and a high degree of polarity resulted in increased yields within reduced reaction times. Adhering to the principles of green chemistry, we did not test any other organic solvents in this experiment. After thorough screening, it was determined that the optimal yields and time profiles were achieved by conducting the reaction with 0.05 g of MFe2O4@PDA@BuSO3H MNPs in a mixture of ethanol and water (1:1) under reflux conditions. This resulted in the formation of the corresponding 5a with a yield of 92% in 2 hours (Table 1, entry 7). Further analysis on the effect of temperature variation revealed that lowering the reaction temperature to 50 °C in the H2O–EtOH system reduced the yield to 87%, despite a prolonged reaction time (Table 1, entry 8). Increasing the catalyst amount to 0.07 g did not further improve the yield beyond what was achieved with 0.05 g (Table 1, entry 9). Conversely, reducing the catalyst amount to 0.015 g slightly decreased the yield to 85% (Table 1, entry 10).
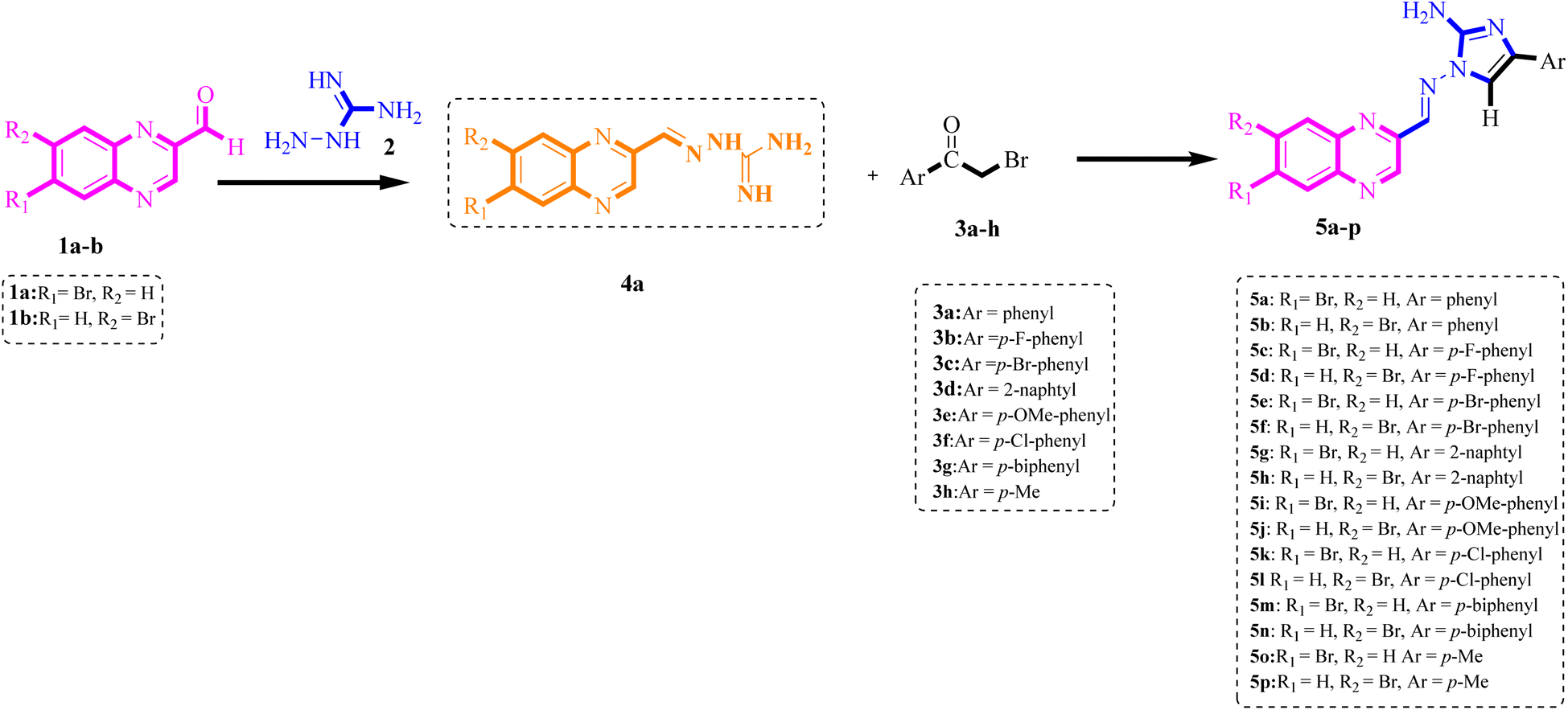 | ||
| Scheme 2 Synthesis of functionalized benzopyrazine-aminoimidazole hybrid derivatives. | ||
| Entry | Catalyst (g) | Reaction conditions | Time (min) | Yielda (%) |
|---|---|---|---|---|
| a Isolated yields. | ||||
| 1 | — | EtOH, reflux | 240 | Nil |
| 2 | BuSO3H (0.005) | EtOH, reflux | 120 | 70 |
| 3 | MFe2O4 (0.03) | EtOH, reflux | 160 | 40 |
| 4 | MFe2O4@PDA (0.03) | EtOH, reflux | 160 | 45 |
| 5 | MFe2O4@PDA@SO3H (0.05) | EtOH, reflux | 120 | 87 |
| 6 | MFe2O4@PDA@SO3H (0.05) | H2O, reflux | 120 | 78 |
| 7 | MFe2O4@PDA@SO3H (0.05) | H2O–EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1), reflux :1), reflux |
120 | 92 |
| 8 | MFe2O4@PDA@SO3H (0.05) | H2O–EtOH (1:1), 50 °C |
180 | 87 |
| 9 | MFe2O4@PDA@SO3H (0.07) | H2O–EtOH (1:1), 70 °C |
120 | 92 |
| 10 | MFe2O4@PDA@SO3H (0.015) | H2O–EtOH (1:1), 70 °C |
120 | 85 |
To generate a limited collection of functionalized derivatives of 2-aminoimidazole benzopyrazine hybrids (5a–p), we employed various substrates to investigate the extent and versatility of the accelerated via a single-step, one-pot Knoevenagel-shift base-annulation reaction under optimized conditions. The outcomes of this exploration are summarized in Table 2. These novel compounds were identified through elemental analysis and spectral characterization, including 1H NMR, 13C NMR, 2D NMR (HHCOSY and HMBC), mass spectrometry (MS) and infrared (IR) spectroscopy. The mass spectra showed peaks at the expected m/z values for the molecular ions. Subsequently, we applied the optimal reaction parameters to examine the reaction's effectiveness for synthesizing 2-aminoimidazole-containing benzopyrazine derivatives. Various 2-bromoacetophenones with different substituents were converted to the desired products with high yields using MFe2O4@PDA@BuSO3H MNPs as a green catalyst. Notably, electron-withdrawing groups on the 2-bromo acetophenone derivatives facilitated the reactions, reducing reaction times and increasing yields compared to electron-releasing groups on the benzene ring.
| Entry | R1 | R2 | Ar | Compound | Time (h) | Mp (°C) | Yield % |
|---|---|---|---|---|---|---|---|
| 1 | Br | H | Phenyl | 5a | 3 | 251–253 | 87 |
| 2 | H | Br | Phenyl | 5b | 3 | 252–254 | 85 |
| 3 | Br | H | p-F-phenyl | 5c | 2 | 250–253 | 90 |
| 4 | H | Br | p-F-phenyl | 5d | 3 | 254–257 | 89 |
| 5 | Br | H | p-Br-phenyl | 5e | 2 | 255–258 | 90 |
| 6 | H | Br | p-Br-phenyl | 5f | 3 | 257–259 | 89 |
| 7 | Br | H | 2-Naphtyl | 5g | 3 | 274–276 | 88 |
| 8 | H | Br | 2-Naphtyl | 5h | 2 | 273–276 | 87 |
| 9 | Br | H | p-OMe-phenyl | 5i | 3 | 243–246 | 89 |
| 10 | H | Br | p-OMe-phenyl | 5j | 2 | 241–243 | 88 |
| 11 | Br | H | p-Cl-phenyl | 5k | 2 | 238–240 | 89 |
| 12 | H | Br | p-Cl-phenyl | 5l | 2 | 238–240 | 87 |
| 13 | Br | H | p-biphenyl | 5m | 2 | 257–260 | 85 |
| 14 | H | Br | p-biphenyl | 5n | 2 | 258–260 | 84 |
| 15 | Br | H | p-Me | 5o | 3 | 247–249 | 84 |
| 16 | H | Br | p-Me | 5p | 3 | 230–232 | 82 |
The recoverability and reusability of catalysts are significant benefits of the green chemistry approach using heterogeneous catalysts, particularly from an industrial standpoint for operations on commercial-scale applications. Accordingly, we investigated the recycling of fresh MNPs using a selected model reaction involving compounds 1a, 2, and 3a in the presence of MFe2O4@PDA@BuSO3H (Table 2, entry 1). After the reaction (monitored by TLC) was completed, a hot ethanol dilution was performed to facilitate the separation of the catalyst from the reaction mixture. The catalyst was then quickly removed using an external magnetic field. The catalyst was then washed with hot ethanol, dried in ambient air, and utilized again for subsequent similar reactions. Notably, the catalyst retained its ability to catalyze reactions for six cycles without appreciable performance diminution (Fig. 7).
 | ||
| Fig. 7 Reusability of nanocatalyst. | ||
Scheme 3 illustrates one of the proposed reaction pathways for the one-pot multicomponent Knoevenagel-shift base reactions using MFe2O4@PDA@BuSO3H MNPs as a heterogeneous Lewis acid catalyst. Although the precise mechanism is not fully understood, it is hypothesized that the surface of the MFe2O4@PDA@BuSO3H MNPs heterogeneous nanocatalyst possesses electron-deficient sites capable of coordinating with the O-donor sites of aldehyde and (substituted) bromoacetophenone. This coordination, especially with the empty orbital of the –SO3H group on the functionalized MFe2O4surface, enhances the electrophilicity of the carbonyl carbon atoms in both formaldehyde and bromoacetophenone. Consequently, the one-pot multicomponent Knoevenagel-shift base reactions efficiently proceed, leading to the formation of the corresponding Knoevenagel-shift base product. In the initial step of this reaction: I. Activation of the nano catalyst's surface through coordination with the –SO3H group; II. Electrophilic attack on the carbonyl carbon atom of formaldehyde by the activated catalyst; III. Formation of a transient intermediate complex between the nano catalyst and the carbonyl compound; IV. Nucleophilic attack by the amine group of the guanidine compound on the carbonyl carbon atom, facilitated by coordination with the nano catalyst; V. Formation of an imine intermediate through the reaction between the carbonyl compound and the guanidine compound; VI. Release of the imine intermediate from the nano catalyst's surface; VII. Formation of an imine intermediate through nucleophilic attack by aminoguanidine on the carbonyl carbon atom in the aldehyde; VIII. Intramolecular proton transfer and rearrangement of bonds occur, resulting in the formation of an imine intermediate; XI. Interaction between the bromoacetophenone derivative and the imine compound leads to the formation of the aminoimidazole ring and the final derivatives; X. In the formation of the final derivative, two moles of imine compounds are required for each mole of alpha-bromo compound; XI. The initial step involves the formation of an imine intermediate between the carbonyl group of the alpha-bromo compound and the imine group; XII. With the entry of the next imine compound, the imidazole ring is formed; XIII. Finally, one extra mole of imine compound is expelled, forming the desired final derivative.
 | ||
| Scheme 3 The proposed mechanism for preparing 2-benzopyrazine-aminoimidazole hybrid derivatives (5a–p) using MFe2O4@PDA@BuSO3H MNPs (0.05 g) as catalyst in H2O–EtOH (1:1), reflux. | ||
This heterogeneous nanocatalyst is functionalized with –SO3H groups, providing Lewis acid sites on its surface.17–19 These acidic centers coordinate with electron-donating groups such as the oxygen in the carbonyl compounds, namely the aldehyde and bromoacetophenone utilized in this reaction. Upon coordination, the catalyst enhances the electrophilicity of the carbonyl carbon atoms, making them more susceptible to nucleophilic attack. The nanocatalyst acts as an electron-withdrawing entity, stabilizing the negative charge on the intermediates formed during the reaction. This stabilization lowers the activation energy required, thus accelerating the reaction process. Moreover, the catalyst facilitates the correct orientation of reactants, which is critical for forming the imine. The amine group of the guanidine compound is positioned in close proximity to the activated carbonyl compound, promoting a more efficient nucleophilic attack to form the imine intermediate. Additionally, the catalyst provides a surface that can adsorb and desorb the reactants and products, optimizing the transformation and release of the desired product. Once the product is formed, it detaches from the catalyst, allowing the catalyst to participate in another catalytic cycle without any permanent structural changes. The entire process benefits from the catalyst's involvement, encompassing sequential steps of activation, intermediate formation, nucleophilic attack, and the eventual creation and release of the final product, all facilitated and accelerated by the catalytic activity of MFe2O4@PDA@BuSO3H MNPs.
The stability of the MFe2O4@PDA@BuSO3H MNPs was demonstrated through multiple catalytic cycles involving the synthesis of benzopyrazine-aminoimidazole hybrids. After each reaction cycle, the nanocatalyst was magnetically separated, washed, and reused. The catalytic activity remained consistent even after six cycles. Structural characterization was conducted using FT-IR and XRD analyses before and after repeated catalytic cycles to assess any structural changes. The spectra and diffraction patterns exhibited no significant alterations, indicating the structural integrity of the nanocatalyst (Fig. 8A and B). TEM observed the morphological stability of the nanocatalyst. The images confirmed that the core–shell structure was retained after repeated catalytic cycles, further substantiating the stability claim (Fig. 8C). In terms of greenness, the synthesis of the nanocatalyst and the catalytic reactions occurred under mild reaction conditions using environmentally friendly solvents such as ethanol or water at ambient or relatively low temperatures. This approach significantly reduces the environmental footprint.
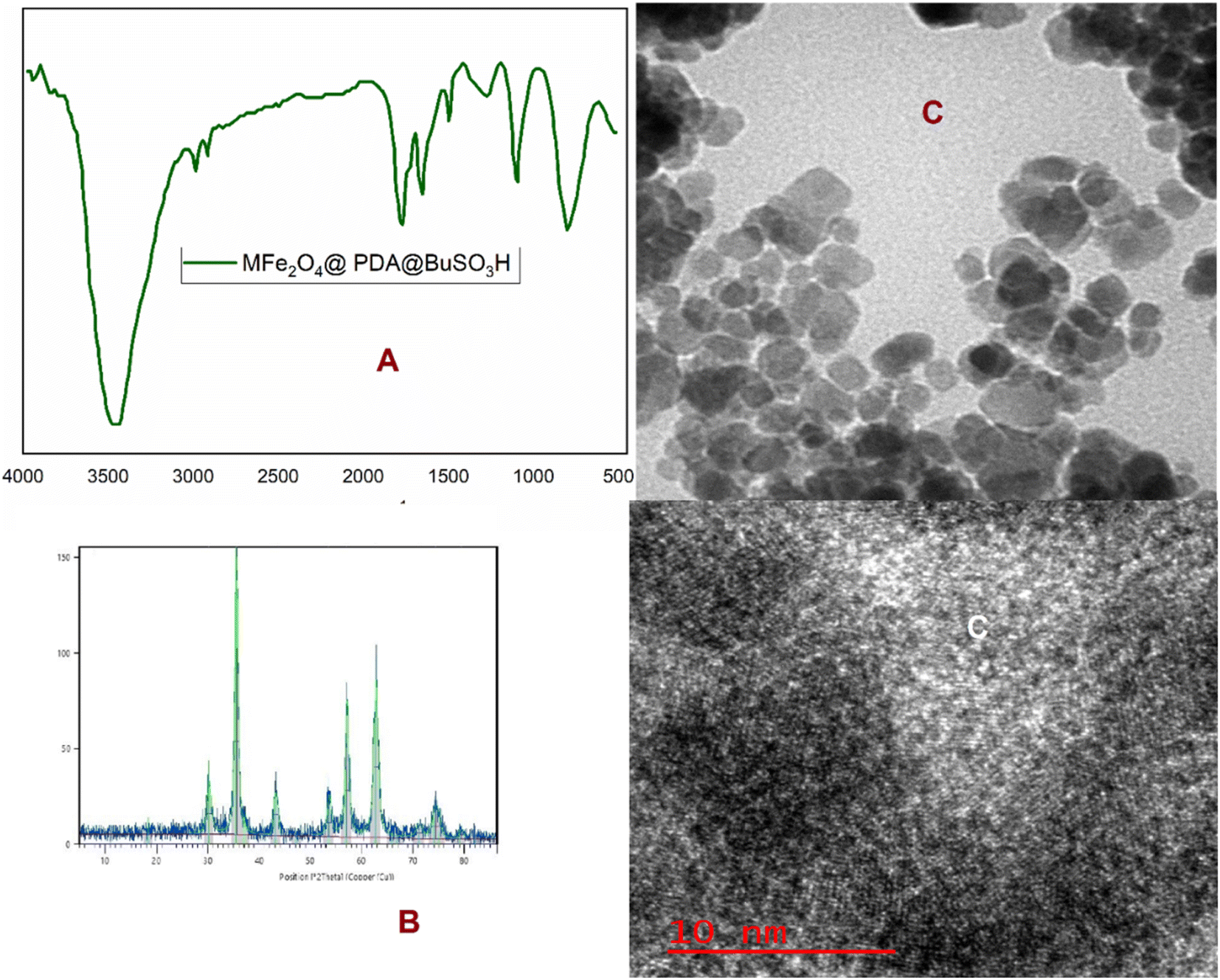 | ||
| Fig. 8 A) FT-IR, (B) XRD, and (C) TEM analysis of the recovered MFe2O4@PDA@BuSO3H MNPs (0.05 g) as a catalyst in H2O–EtOH (1:1), reflux. | ||
2.2. Biological evaluation
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Ar | IC50 (μM) | ||
| MCF7 | SKOV3 | U87 | ||||
| 5a | Br | H | Phenyl | 1.90 | 1.38 | 1.08 |
| 5b | H | Br | Phenyl | 1.11 | 2.99 | 2.00 |
| 5c | Br | H | p-F-phenyl | 2.44 | 2.35 | 2.60 |
| 5d | H | Br | p-F-phenyl | 1.84 | 1.98 | 6.61 |
| 5e | Br | H | p-Br-phenyl | 3.82 | 3.06 | 2.83 |
| 5f | H | Br | p-Br-phenyl | 1.86 | 4.36 | 2.24 |
| 5g | Br | H | 2-Naphtyl | 0.03 | 1.22 | 0.22 |
| 5h | H | Br | 2-Naphtyl | 0.32 | 0.51 | 0.93 |
| 5i | Br | H | p-OMe-phenyl | 2.02 | 1.84 | 4.23 |
| 5j | H | Br | p-OMe-phenyl | 1.56 | 1.78 | 1.28 |
| 5k | Br | H | p-Cl-phenyl | 1.40 | 1.63 | 1.97 |
| 5l | H | Br | p-Cl-phenyl | 3.22 | 1.13 | 1.56 |
| 5m | Br | H | p-Biphenyl | 1.65 | 19.8 | 2.38 |
| 5n | H | Br | p-Biphenyl | 0.11 | 1.93 | 1.97 |
| 5o | H | Br | p-Me | 4.08 | 2.16 | 2.84 |
| 5p | Br | H | p-Me | 2.29 | 2.01 | 2.11 |
| Doxorubicin | 0.50 | 1.76 | 0.31 | |||
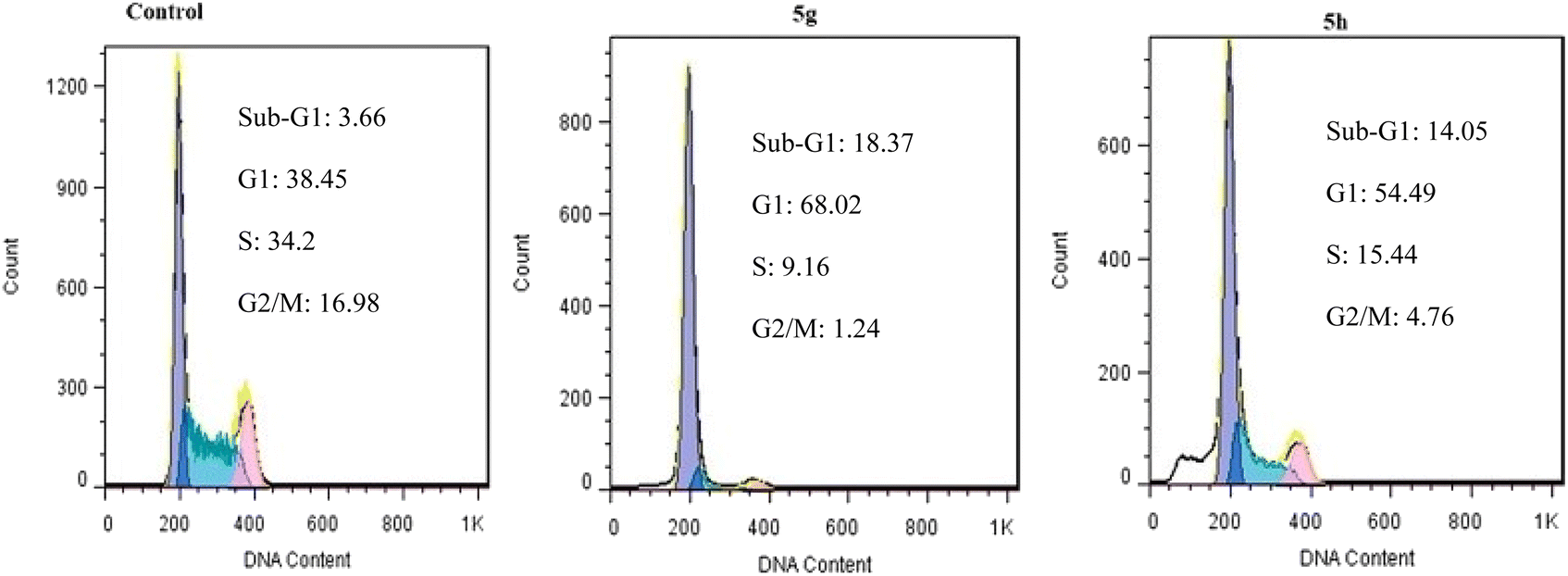 | ||
| Fig. 9 Cell cycle assay. | ||
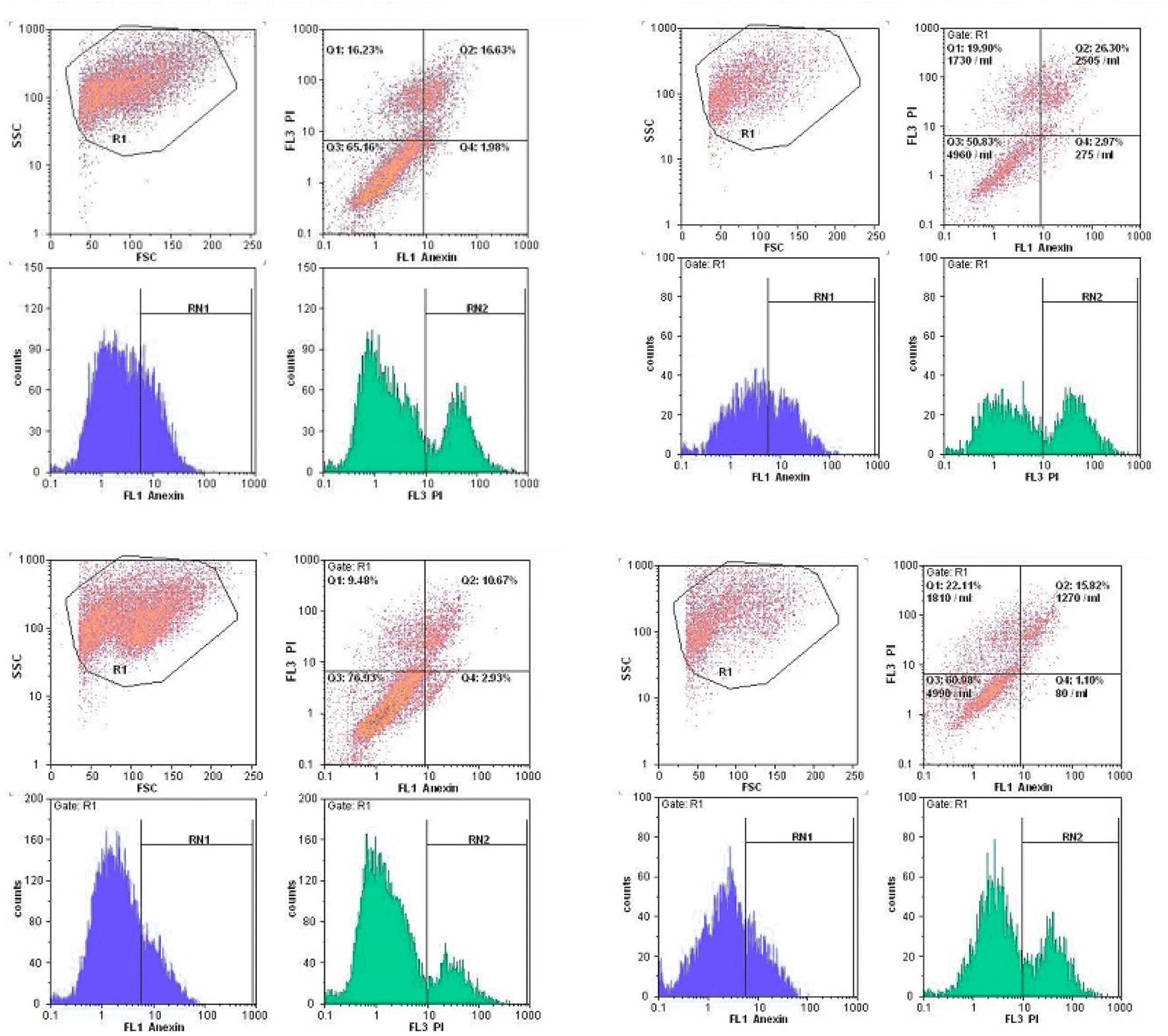 | ||
| Fig. 10 Compounds 5g and 5h induced apoptosis in MCF-7 cells. 1.5 and 3 μM for 48 h. | ||
2.3. Molecular docking study
Molecular docking simulations were conducted on the most potent compounds. The docking investigation demonstrated that promising compounds 5g and 5h colchicine binds with high affinity to the β-subunit of tubulin. Forming essential hydrogen bonds, van der Waals interactions, and polar interactions are responsible for stabilizing the obtained interaction mode. Docking modes shown in Fig. 11A and Fig. 11C demonstrate that the benzopyrazine moiety in compound 5g was interred in the hydrophobic pocket of the colchicine binding site in the β-tubulin and is in contact with amino acids such as βAla250, βLys254, βLeu248, βAsn249. The imidazole ring is buried in the amino acid residues, including βLeu255, βCys241, and βAsp251. Similarly, the 2D representation of compound 5g displayed several secondary interactions like van der Waal interactions with the residues of βAsn258, βAsn349, βAsn249, βThr314, βVal315, βAsp251, βLeu255 and βCys241. Besides this, the formation a pi-alkyl interaction between the 3,4,5-trimethoxyphenyl moiety and βLeu255 and a pi-alkyl interaction between benzopyrazine ring and amino acid residues βAla250, βLys254, βLus248 and between bipheyl ring with the residues of βAla316, βLys352 and, βMet259 were detected, respectively. The molecular docking results for compound 5h (Fig. 11B) showed that the benzopyrazine moiety is encircled by the amino acid residues βLys254, βAla250, βLeu248, and βAsn249. The amino acid residues βAla354, βAsn258, and βLeu255 surrounded the imidazole ring.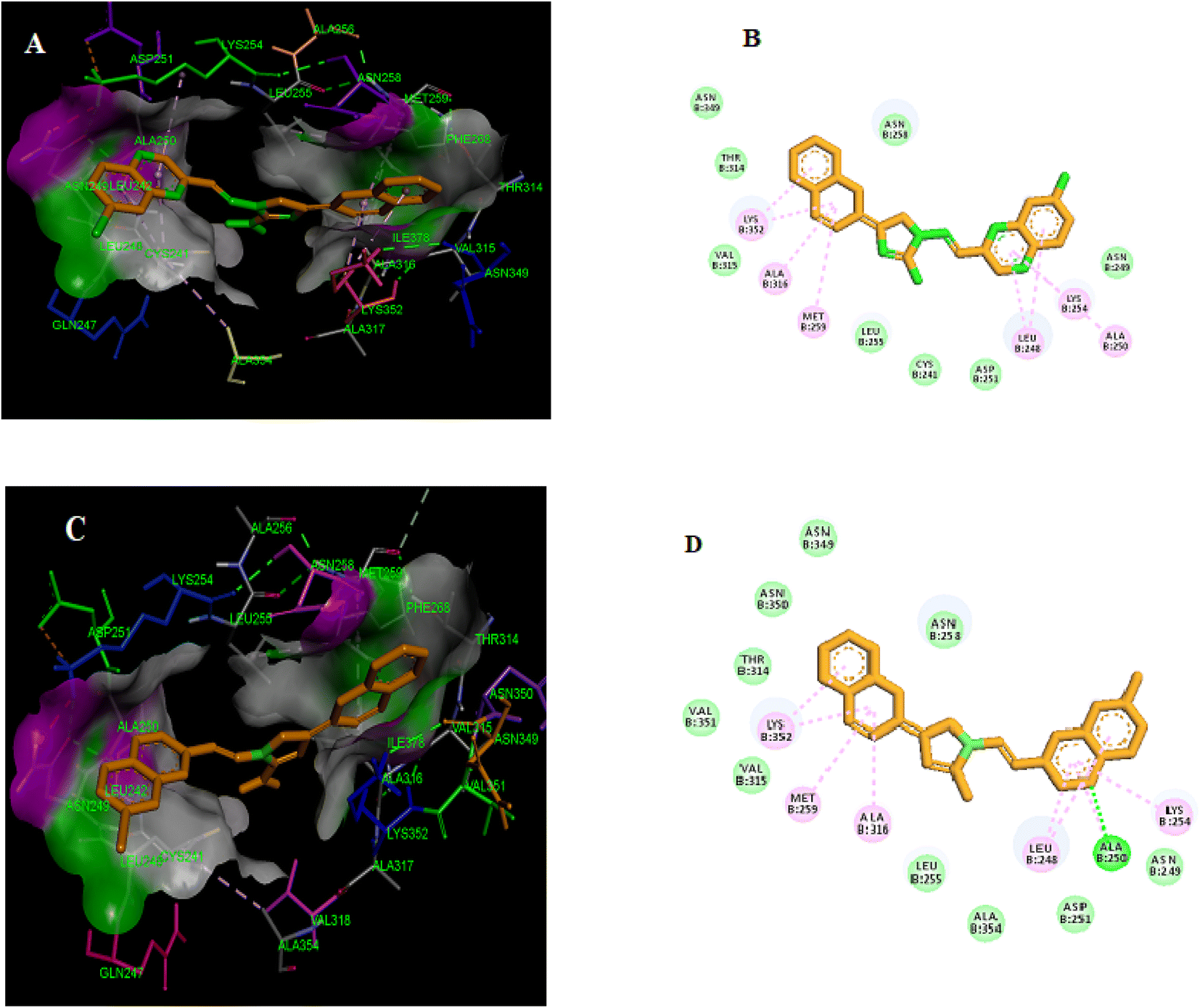 | ||
| Fig. 11 Compounds 5g (A and B) and 5h (C and D) are both potent tubulin polymerization inhibitors that bind to the colchicine binding site. | ||
Furthermore, 2D presentation of the binding interaction mode of compound 5h (Fig. 11D) demonstrates the formation of two pi-alkyl interactions between the benzopyrazine scaffold and βLeu248 and βLys254 and three pi-alkyl interaction between biphenyl ring and βAla316, βLys352, and βMet259. Moreover, several van der Waal interactions between the benzopyrazine ring and biphenyl ring with amino acid residues βAsn249, βAla354, βAsp251, βVal351, βThr314, βAsn350, βAsn349, and βAsn258 were detected. Besides, the formation of a conventional hydrogen bond between benzopyrazine moiety and the amino acid residue of βAla250 was shown.
3. Conclusion
We have reported a novel and efficient acidic nanocatalyst (MFe2O4@PDA@BuSO3H) through single-pot domino Knoevenagel-shift base–annulation reactions, which possess magnetic properties and can overcome the limitations of traditional mineral or homogeneous organic catalysts. MFe2O4@PDA@BuSO3H MNPs is an environmentally friendly, cost-effective, and magnetic nanocatalyst that can be quickly recovered using a simple magnet and reused multiple times without significantly reducing its catalytic performance. We have synthesized aminoimidazole hybrids benzopyrazine derivatives in this nanocatalyst's presence, after which their antiproliferative effects against three human cancer cell lines, namely MCF-7, U87, and A549, were evaluated. All tested derivatives (5a–p) exhibited significant cytotoxic activity. Compound 5g, which possesses a 2-naphthyl ring at the C4 substituent of the C-ring, demonstrated the highest cytotoxicity among other derivatives with IC50 values of 0.03 μM, which were 17-fold more potent than the standard drug doxorubicin. Furthermore, our cell cycle analysis revealed that the disruption caused by compounds 5g and 5h significantly affected microtubule polymerization and resulted in the significant arrest of most MCF7 cells in the G2/M phase.Through our Molecular Docking analysis, we gained insights into the precise binding site of compounds 5g and 5h with tubulin. Moreover, we confirmed that the interaction between these compounds and the colchicine binding site of tubulin is responsible for their anti-proliferative activity.
4. Experimental section
4.1 Chemistry
:1 v/v mixture of H2O and EtOH were combined in a 50 mL flask fitted while stirring with a magnetic stirrer at 70 °C for less than 10 minutes. Then,1 mmol of cyclic ketones 3a–h was added to the flask, and the reaction was heated for the times specified in Table 2. After the reaction was completed, as determined by TLC analysis, hot Ethanol was afforded to the reaction mixture, followed by the catalyst's separation using an external magnet. The catalyst was then washed with hot ethanol, dried, and reused for a consecutive run with the same set of conditions. The reaction mixture was cooled after the completion of the reaction, and the unrefined compound was isolated by filtration. To ensure the highest purity of the product, the raw compound was first subjected to column chromatography using an appropriate solvent system to achieve preliminary purification. Following this step, preparative thin-layer chromatography (TLC) was performed for the final purification of the compound. The purified product was then recrystallized from hot ethanol to obtain the final pure compound.4.2. Biological tests
4.3. Docking simulations
The mode of interaction between promising compounds 5g and 5h with the colchicine binding site of tubulin was examined by employing AutoDock software to investigate the structural basis of the aforementioned biological findings. The structure of compounds was drawn in ChemDraw, and 3D structures were prepared in ChemBioDraw Ultra16.0, following energy minimization using the MM2 force field. The crystal structure of tubulin complexed with colchicine was obtained from the PDB database (PDB code 1SA0). Using the AutoDock protocol,27 final files were prepared. A grid box of 45, 45, and 45 grid points (X, Y, Z dimensions, resp.) and centroid: X = 117.2187, Y = 90.1800, Z = 6.2898 was chosen. The obtained interactions and poses were analyzed, and exhibiting optimal ligand-enzyme interactions were chosen and saved for energy calculations.Data availability
All data generated or analyzed during this study are included in this published article and data will be made available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia, for funding this research work through the project number (PSAU/2023/R/1444). This study is supported via funding from the Prince Sattam Bin Abdulaziz University project number (PSAU/2023/R/1444).References
- J. Lin, P. Wang, Z. Zhang, G. Xue, D. Zha, J. Wang, X. Xu and Z. Li, Synth. Commun., 2020, 50, 823–830 CrossRef CAS.
- E. A. Fayed, Y. A. Ammar, M. A. Saleh, A. H. Bayoumi, A. Belal, A. B. Mehany and A. Ragab, J. Mol. Struct., 2021, 1236, 130317 CrossRef CAS.
- M. Ibrahim, M. Taghour, A. Metwaly, A. Belal, A. Mehany, M. Elhendawy, M. Radwan, A. Yassin, N. El-Deeb and E. Hafez, Eur. J. Med. Chem., 2018, 155, 117–134 CrossRef CAS.
- M. Montana, F. Mathias, T. Terme and P. Vanelle, Eur. J. Med. Chem., 2019, 163, 136–147 CrossRef CAS PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, W. A. Alanazi, A. M. Al-Hossaini, M. A. Alharbi, I. H. Eissa and M. A. Dahab, J. Mol. Struct., 2022, 1253, 132220 CrossRef CAS.
- N. A. Alsaif, A. Elwan, M. M. Alanazi, A. J. Obaidullah, W. A. Alanazi, A. F. Alasmari, H. Albassam, H. A. Mahdy and M. S. Taghour, Mol. Diversity, 2022, 26, 1915–1932 CrossRef CAS.
- D. S. Ermolat'ev, J. B. Bariwal, H. P. Steenackers, S. C. De Keersmaecker and E. V. Van der Eycken, Angew. Chem., 2010, 122, 9655–9658 CrossRef.
- M. MATSUyAMA, K. Funao, K. Kuratsukuri, T. Tanaka, Y. Kawahito, H. Sano, J. Chargui, J.-L. Touraine, N. Yoshimura and R. Yoshimura, Exp. Ther. Med., 2010, 1, 301–306 CrossRef CAS.
- T.-C. Shih, R. Liu, C.-T. Wu, X. Li, W. Xiao, X. Deng, S. Kiss, T. Wang, X.-J. Chen and R. Carney, Clin. Cancer Res., 2018, 24, 4319–4331 CrossRef CAS.
- T.-C. Shih, R. Liu, G. Fung, G. Bhardwaj, P. M. Ghosh and K. S. Lam, Mol. Cancer Ther., 2017, 16, 1212–1223 CrossRef CAS PubMed.
- S. Reyes, R. W. Huigens III, Z. Su, M. L. Simon and C. Melander, Org. Biomol. Chem., 2011, 9, 3041–3049 RSC.
- H. Weinmann, M. Harre, K. Koenig, E. Merten and U. Tilstam, Tetrahedron Lett., 2002, 43, 593–595 CrossRef CAS.
- M. Akhavan and A. Bekhradnia, RSC Adv., 2021, 11, 14755–14768 RSC.
- M. Akhavan, N. Foroughifar, H. Pasdar and A. Bekhradnia, Combinatorial Chemistry & High Throughput Screening, 2019, 22, 716–727 Search PubMed.
- F. Sahin, E. Turan, H. Tumturk and G. Demirel, Analyst, 2012, 137, 5654–5658 RSC.
- Z. Esam, M. Akhavan, A. Bekhradnia, M. Mohammadi and S. Tourani, Catal. Lett., 2020, 150, 3112–3131 CrossRef CAS.
- Z. Esam, M. Akhavan, A. Mirshafa and A. Bekhradnia, RSC Adv., 2023, 13, 25229–25245 RSC.
- M. Akhavan, Z. Esam, A. Mirshafa, M. Lotfi, S. Pourmand, F. Ashori, M. Rabani, G. Ekbatani, S. Tourani and R. Beheshti, RSC Adv., 2024, 14, 22916–22938 RSC.
- Z. Kheilkordi, G. M. Ziarani and A. Badiei, Polyhedron, 2020, 178, 114343 CrossRef CAS.
- P. Diana, A. Martorana, P. Barraja, A. Montalbano, G. Dattolo, G. Cirrincione, F. Dall'Acqua, A. Salvador, D. Vedaldi and G. Basso, J. Med. Chem., 2008, 51, 2387–2399 CrossRef CAS PubMed.
- T. Liang, X. Zhou, L. Lu, H. Dong, Y. Zhang, Y. Xu, J. Qi, Y. Zhang and J. Wang, Bioorg. Chem., 2021, 110, 104793 CrossRef CAS PubMed.
- M. Akhavan, N. Foroughifar, H. Pasdar, A. Khajeh-Amiri and A. Bekhradnia, Transition Met. Chem., 2017, 42, 543–552 CrossRef CAS.
- L. Deferme, J. Briede, S. Claessen, D. Jennen, R. Cavill and J. Kleinjans, Toxicology, 2013, 306, 24–34 CrossRef CAS.
- L. Browne, C. Gude, H. Rodriguez, R. Steele and A. Bhatnager, J. Med. Chem., 1991, 34, 725–736 CrossRef CAS PubMed.
- Y. Koizumi, M. Arai, H. Tomoda and S. Ōmura, Biochim. Biophys. Acta, 2004, 1693, 47–55 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS.
- S. M. D. Rizvi, S. Shakil and M. Haneef, EXCLI J., 2013, 12, 831 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04725g |
| This journal is © The Royal Society of Chemistry 2024 |