
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of carbon black supports on the hydrogen evolution reaction activity of Pd nanoparticle electrocatalysts synthesized via solution plasma sputtering†
Jidapa Chantaramethakul a,
Chadapat Hussakanab,
Yanisa Yenmankhonga,
Praewpanit Chandeanga,
Ratchatee Techapiesancharoenkijab,
Wisit Hirunpinyopasc,
Kasmuid,
Cepi Kurniawand and
Gasidit Panomsuwan*ab
a,
Chadapat Hussakanab,
Yanisa Yenmankhonga,
Praewpanit Chandeanga,
Ratchatee Techapiesancharoenkijab,
Wisit Hirunpinyopasc,
Kasmuid,
Cepi Kurniawand and
Gasidit Panomsuwan*ab
aDepartment of Materials Engineering, Faculty of Engineering, Kasetsart University, Bangkok 10900, Thailand. E-mail: gasidit.p@ku.ac.th
bInternational Collaborative Education Program for Materials Technology, Education, and Research (ICE-Matter), ASEAN University Network/Southeast Asia Engineering Education Development Network (AUN/SEED-Net), Kasetsart University, Bangkok, 10900, Thailand
cDepartment of Chemistry, Faculty of Science, Kasesart University, Bangkok 10900, Thailand
dChemistry Department, Faculty of Mathematics and Natural Sciences, Universitas Negeri Semarang, Semarang 50229, Indonesia
First published on 7th October 2024
Abstract
The hydrogen evolution reaction (HER) is a pivotal electrochemical process in water electrolysis, essential for hydrogen production. The efficiency and kinetics of HER are significantly influenced by the choice of catalyst and its support material. In this study, we investigated the effect of carbon supports on palladium (Pd) nanoparticle electrocatalysts synthesized via the solution plasma sputtering process for HER. Pd nanoparticles were loaded onto three hierarchically porous carbon black (CB) supports: Vulcan XC-72R, Ketjen Black EC-300J, and Black Pearls 2000. Well-crystalline Pd nanoparticles, ranging in size from approximately 2–6 nm, were distributed on the surface of CB supports with Pd loading contents ranging between 21 and 29 wt%. The catalysts exhibited lower specific surface areas compared to bare CB supports due to a significant decrease in exposed micropores, which were blocked by the Pd nanoparticles at their entrances. Among the CB supports investigated, Pd nanoparticles loaded on Black Pearls 2000 demonstrated the highest HER activity, as evidenced by the lowest overpotential, largest electrochemical surface area, and highest mass activity. This superior activity can be attributed to the unique characteristics of Black Pearls 2000, including its high surface area and abundant micropores. Furthermore, it demonstrated greater HER stability than commercial platinum (Pt)-based catalysts. Our finding suggests that Black Pearls 2000 could serve as a promising CB support for further developing highly efficient and stable HER electrocatalysts.
Introduction
Hydrogen has emerged as a crucial player in the pursuit of cleaner energy solutions. As awareness of environmental issues grows and the need for sustainable energy sources intensifies, hydrogen's potential as a clean energy carrier is gaining significant traction.1 A key pathway to unlocking the full potential of hydrogen is the development of green hydrogen production methods. Green hydrogen refers to hydrogen produced using renewable energy sources, such as solar or wind power, in a process that emits minimal to no greenhouse gases.2 Among the various methods for green hydrogen production, electrochemical water electrolysis stands out as a particularly promising approach.3In electrochemical water electrolysis, water molecules are split into hydrogen and oxygen gases using an electric current.4 The core of this process is the hydrogen evolution reaction (HER), which occurs at the cathode. During HER, protons (H+) from water molecules gain electrons to form hydrogen gas (H2). The efficiency and performance of the HER are crucial in determining the overall efficiency and cost-effectiveness of hydrogen production.5 Currently, platinum (Pt) is the most effective HER electrocatalyst due to its exceptional HER activity.6 However, its high cost and limited availability pose significant barriers to widespread adoption and commercialization. According to the Volcano plot, palladium (Pd) emerges as a promising alternative to Pt due to its lower cost, greater abundance, and comparable exchange current density and hydrogen adsorption energy for HER.7
In the realm of HER electrocatalysis, selecting appropriate support materials for dispersing metal nanoparticles is of paramount importance. Carbon materials are often preferred as support materials due to their outstanding properties, including high surface area, excellent electrical conductivity, affordability, stability in harsh reaction environments, and scalability in production.8 These properties make carbons ideal candidates for hosting metallic nanoparticles and facilitating efficient HER electrocatalysis. Numerous studies have demonstrated that the properties of carbon supports significantly influenced the electrocatalytic activity and stability of HER electrocatalysts.9–11 Exploring the intricate interplay between carbon properties and HER electrocatalysis requires a comprehensive investigation of various factors, such as surface chemistry, porosity, conductivity, and structural characteristics. For instance, surface functional groups and lattice defects of carbon supports can interact with metal nanoparticles, thereby influencing their dispersion and electrocatalytic performance.12–14 Additionally, the surface area and porous structure of carbon supports affect mass transport phenomena, diffusion kinetics, and accessibility of active sites, all of which are critical determinants of HER efficiency.15–18 Despite the recognized importance of carbon supports, there remains a notable gap in understanding how specific properties of carbon supports influence the HER activity of Pd-based electrocatalysts. Unraveling this relationship is crucial for optimizing catalyst design and enhancing overall performance.
Herein, we investigated the effect of different carbon supports on the HER activity of Pd nanoparticles. Three types of carbon blacks (CBs) with varying surface areas and porous structures were selected: Vulcan XC-72R (VC), Ketjen Black EC-300J (KB), and Black Pearls 2000 (BP). Pd nanoparticles were synthesized via the solution plasma sputtering (SPS) and deposited onto the CB supports. The SPS is a simple and chemical-free method for producing high-purity noble metal nanoparticles by directly sputtering metal electrode surfaces (i.e., Pd, Pt, and Au) in a liquid medium at room temperature and atmospheric pressure.19–21 To the best of our knowledge, no prior studies have investigated the effect of carbon supports for the Pd-based electrocatalysts for HER, particularly those prepared using SPS.
Experimental
Chemical and materials
Pd electrodes with a diameter of 1 mm (purity 99.95%) were purchased from Nilaco Co., Ltd. Ethanol (C2H5OH, purity 99.9%), isopropanol (C3H8O, purity 99.8%), and acetic acid (CH3COOH, purity 99.8%) were purchased from RCI Labscan Ltd. A 0.5 M H2SO4 solution was purchased from Thermo Scientific Chemicals. Three types of CB, including Vulcan XC-72R (VC), Ketjen Black EC-300J (KB), and Black Pearls 2000 (BP), were purchased from Fuel Cell Store and Cabot Corporation, respectively. A 20 wt% Pt on Vulcan XC-72R (Pt/C) and perfluorinated resin solution containing Nafion™ 1100W (5 wt% in a mixture of lower aliphatic alcohols and water) were purchased from Sigma-Aldrich. Ultrapure water (18.2 MΩ cm at 25 °C) was obtained from a Direct-Q™ 5 UV Millipore water purification system. All the chemicals were analytical grade and used without further purification.Synthesis of Pd/CB catalysts via SPS
Twenty milligrams of CB supports (i.e., VC, KB, and BP) were dispersed in a mixture comprising 40 mL of ethanol and 40 mL of 0.5 M acetic acid. The CB suspension was stirred for 15 min and then sonicated in an ultrasonic bath for 1 h to obtain a homogeneous dispersion. The experimental setup is illustrated in Fig. 1. A pair of Pd electrodes, each covered with an insulating ceramic tube, was positioned with a gap distance of 0.5 mm at the center of a glass reactor containing the prepared CB suspension. A bipolar-pulse voltage was applied to the Pd electrodes using an MPP-HV04 Pekuris bipolar pulse generator (Kurita Seisakusho Co., Ltd.) with the following parameters: frequency of 20 kHz, pulse duration of 0.8 μs, and discharge time of 15 min. Upon plasma generation, the Pd nanoparticles were produced via sputtering from the electrode surface and deposited onto the CB supports. The catalysts were collected by centrifugal filtration and then dried in an oven. The Pd nanoparticles loaded on VC, KB, and BP were designated as Pd/VC, Pd/KB, and Pd/BP, respectively. | ||
| Fig. 1 A schematic of the experimental setup of SPS for preparation of Pd nanoparticles loaded on CB supports in this work. | ||
Characterization of Pd/CB catalysts
The dispersion of Pd nanoparticles on the carbon supports was investigated using a JEOL JEM-3100F field-emission transmission electron microscope (FE-TEM) operated at an acceleration voltage of 300 kV. The phase structure of the catalysts and bare CB supports was examined with a Panalytical Empyrean X-ray diffractometer using a Cu Kα radiation source (λ = 1.54 Å) in the 2θ range of 5–80° at a scan rate of 4° min−1. The Pd loading content was determined using a PerkinElmer TGA 8000 thermogravimetric analyzer. The catalysts were heated from 30 °C to 800 °C with a heating rate of 10 °C min−1 under an O2 flow. The specific surface area and pore characteristics of bare CB supports and catalysts were determined from N2 adsorption–desorption isotherms recorded at liquid-N2 temperature (−196 °C) using a Micromeritics 3Flex surface area analyzer. The catalysts were degassed using a Smart VacPrep instrument at 150 °C for 6 h under vacuum before measurements.Electrochemical measurements
The catalyst suspension was prepared by dispersing 5 mg of catalyst in a mixture comprising 490 μL of ultrapure water, 490 μL of isopropanol, and 20 μL of Nafion. To ensure a well-dispersed catalyst ink, the suspension was sonicated in an ultrasonic bath for 1 h. A glassy carbon (GC) electrode (3 mm diameter, 0.071 cm2 area) as the working electrode and was successively polished with 1 μm diamond and 0.05 μm alumina slurries on a polishing pad, then cleaned with ultrapure water in an ultrasonic bath for 10 min. Next, 3 μL of catalyst ink was carefully dropped onto the cleaned GC electrode and allowed to dry in ambient air for 1 h, resulting in a catalyst loading of 0.21 mg cm−2. For comparison, a commercial Pt/C modified GC electrode was prepared to serve as a benchmark catalyst.Electrochemical measurements were conducted using a three-electrode system in an N2-saturated 0.5 M H2SO4 solution with a PalmSens4 potentiostat controlled by the PSTrace 5.9 software. The catalyst-modified GC electrode, Pt coil, and Ag/AgCl in a saturated KCl solution were used as the working, counter, and reference electrodes, respectively. Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) measurements were performed at a scan rate of 50 mV s−1 and 5 mV s−1, respectively. The LSV curves for HER activity were recorded with an electrode rotation speed of 1600 rpm after CV measurement for 30 cycles. For the stability test, the LSV curve of the catalyst with the best HER activity was recorded after 2000 cycles at a scan rate of 100 mV s−1.
The measured potentials vs. Ag/AgCl were converted to the reversible hydrogen electrode (RHE) scale using the Nernst equation:22
| ERHE = EAg/AgCl + 0.059 pH + EoAg/AgCl | (1) |
Results and discussion
The morphology and microstructure of both Pd particles and CBs were initially examined using field-emission transmission electron microscopy (FE-TEM) images, as shown in Fig. 2a–f. The FE-TEM images reveal that Pd nanoparticles (dark contrast) were well-dispersed on the surface of KB and BP (bright contrast) with good dispersion. However, Pd nanoparticles on VC exhibited poor dispersion and significant agglomeration. The size of Pd nanoparticles ranged from 2 to 6 nm, as illustrated in Fig. 2g–i. The average size of Pd particles was estimated to be 3.82 ± 0.75, 3.83 ± 0.81, and 3.76 ± 0.71 nm, for Pd/VC, Pd/KB, and Pd/BP, respectively, indicating no significant difference in the size of Pd nanoparticles among the three catalysts. This suggests that the Pd nanoparticles were primarily deposited on the external surface of CBs rather than being confined within the micropores (<2 nm). Additionally, well-defined lattice fringes were observed in the Pd-particle region, indicating a good crystalline structure (see the inset of Fig. 2d–f).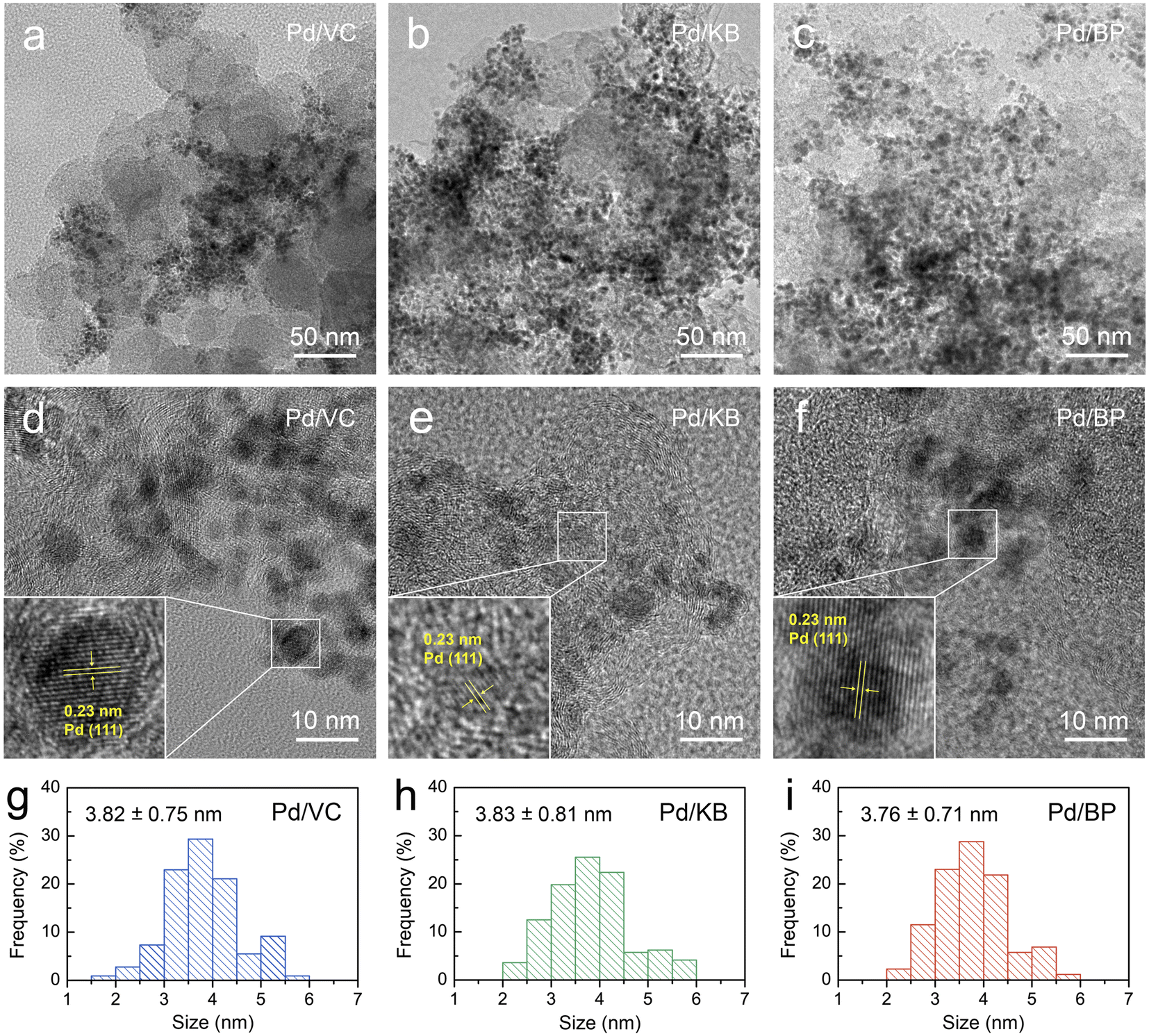 | ||
| Fig. 2 FE-TEM images taken at low and high magnification of (a and d) Pd/VC, (b and e) Pd/KB, and (c and f) Pd/BP. The insets of (d–f) show the enlarged view of Pd nanoparticles with visible lattice fringes. Particle size distribution (counted more than 200 particles) of (g) Pd/VC, (h) Pd/KB, and (i) Pd/BP. | ||
Fig. 3a and S1† show the XRD patterns of the Pd/CB catalysts and their respective bare CB supports, respectively. The XRD patterns of all catalysts exhibited a prominent diffraction peak at around 25°, corresponding to the (002) plane of graphitic carbon. The sharper and more intense (002) peak of VC compared to KB and BP suggests a higher degree of graphitization and larger crystallite size in VC. Moreover, distinct diffraction peaks were observed at 39.4°, 45.9°, and 66.8°, corresponding to the (111), (200), and (220) planes of the face-centered cubic (fcc) structure of crystalline Pd (JCPDS No. 46-1043), respectively, alongside the carbon peaks for all catalysts.
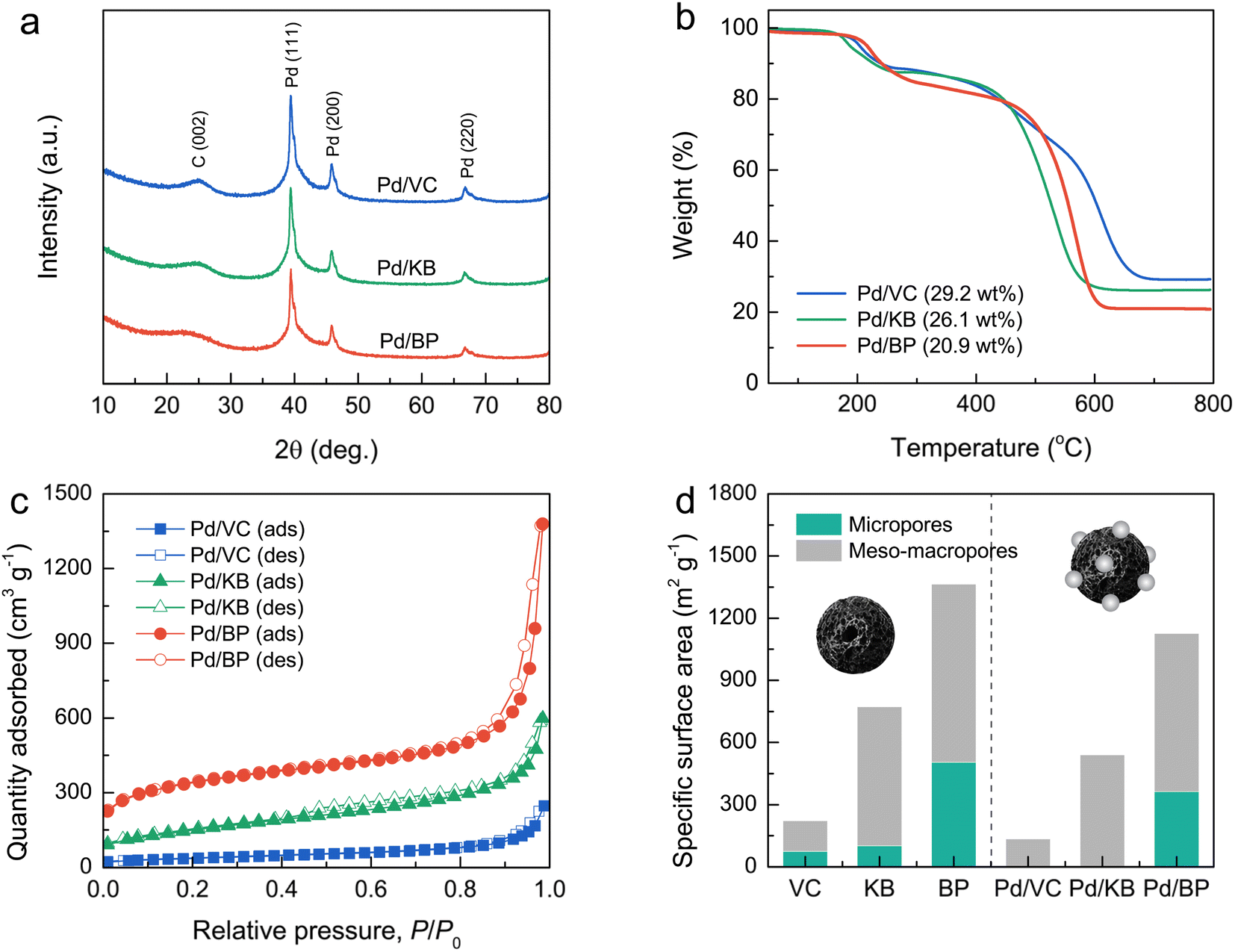 | ||
| Fig. 3 (a) XRD patterns and (b) TGA curves measured at 50–800 °C under O2 atmosphere, (c) N2 adsorption–desorption isotherms (solid and open symbols represent the adsorption and desorption isotherms, respectively), and (d) the specific surface areas contributed by micropores (internal pores) and meso–macropores (external pores) of Pd/VC, Pd/KB, and Pd/BP. | ||
Thermogravimetric analysis (TGA) was used to determine the Pd loading on the CB supports by measuring the residual weight at 800 °C under heating in an O2 atmosphere (Fig. 3b). The Pd loading was found to be 29.2, 26.1, and 20.9 wt% for Pd/VC, Pd/KB, and Pd/BP, respectively. The variation in Pd loading and its dispersion on supports may be attributed to differences in the morphology and surface chemistry of CB, leading to varying Pd–carbon interactions.9
The N2 adsorption–desorption isotherms of bare CB supports and Pd/CB catalysts were recorded to evaluate their specific surface area and porosity (Fig. 3d and S2†). The isotherms for all CB supports exhibited the combination of type I, II, and IV characteristics, indicating a hierarchical porous structure composed of micropores, mesopores, and macropores. The specific surface area, determined using the Brunauer–Emmett–Teller (BET) method, for bare VC, KB, and BP was 222, 773, and 1364 m2 g−1, respectively. According to t-plot analysis, micropores contributed 33%, 13%, and 37% to the specific surface area of VC, KB, and BP, respectively. For the Pd/VC, Pd/KB, and Pd/BP, the isotherms exhibited a significant reduction of quantity adsorbed at low relative pressure; however, their hysteresis loops and isotherm shapes remained unchanged. This finding suggests that the loading of Pd nanoparticles onto the CB supports resulted in the suppression of micropores but did not significantly affect the external pores (i.e., mesopores and macropores). The specific surface area of Pd/VC, Pd/KB, and Pd/BP decreased to 132, 540, and 1128 m2 g s−1, respectively, primarily due to the reduction in micropore area, as illustrated in Fig. 3d. Notably, micropores were absent in Pd/VC and Pd/KB, but 72% of the micropore area was retained in Pd/BP. Conversely, the external surface areas of all three catalysts decreased by less than 20%. These results indicate that the Pd nanoparticles were predominately deposited on the outer surface of CB supports rather than within the internal micropores, as the size of Pd nanoparticles exceeded the diameter of the micropores.23 The deposition of Pd nanoparticles on the CB surface likely blocked the entrance to micropores, preventing the access of N2 molecules into micropores during the adsorption process, thus lowering the micropore area and volume.24 More detailed data from the N2 sorption analysis are summarized in Table 1.
| Sample | SBETa (m2 g−1) | Smicrob (m2 g−1) | Sextc (m2 g−1) | Vtotald (cm3 g−1) | Vmicroe (cm3 g−1) | Vextf (cm3 g−1) |
|---|---|---|---|---|---|---|
| a SBET is the total specific surface area obtained by the BET method.b Smicro is the micropore-specific surface area derived from the t-plot method.c Sext is the external specific surface area calculated by subtracting the micropore-specific surface area from the total specific surface area (Sext = SBET − Smicro).d Vtotal is the total pore volume obtained from the N2 desorption branch isotherm at a relative pressure (P/P0) of 0.95.e Vmicro is the micropore volume determined using the t-plot method.f Vext is the external pore volume calculated by subtracting the micropore volume from the total pore volume (Vext = Vtotal − Vmicro). | ||||||
| VB | 222 | 74 | 148 | 0.299 | 0.039 | 0.260 |
| KB | 773 | 102 | 671 | 0.866 | 0.050 | 0.816 |
| BP | 1364 | 505 | 859 | 1.542 | 0.256 | 1.286 |
| Pd/VB | 132 | 0 | 132 | 0.260 | 0 | 0.260 |
| Pd/KB | 540 | 0 | 540 | 0.695 | 0 | 0.695 |
| Pd/BP | 1128 | 363 | 765 | 1.509 | 0.190 | 1.319 |
The HER activity of all catalysts and a commercial Pt/C was evaluated using a three-electrode system in an N2-saturated 0.5 M H2SO4 solution. Fig. 4a illustrates the LSV curves of all catalysts alongside Pt/C. Notably, the onset potential of Pd/BP commenced at the most positive potential, close to that of Pt/C, indicating the highest HER activity of Pd/BP. The overpotential (η, at a current density of 10 mA cm−2) of Pd/BP was 53 mV, which was significantly lower than Pd/KB (113 mV) and Pd/VC (203 mV). Tafel plots (Fig. 4b) were further employed to explore the underlying HER mechanism of the catalysts. The Pd/BP exhibited the lowest slope of 48.8 mV dec−1 (Volmer–Heyrovsky mechanism) close to that of Pt/C (30.1 mV dec−1, Volmer–Tafel mechanism).25
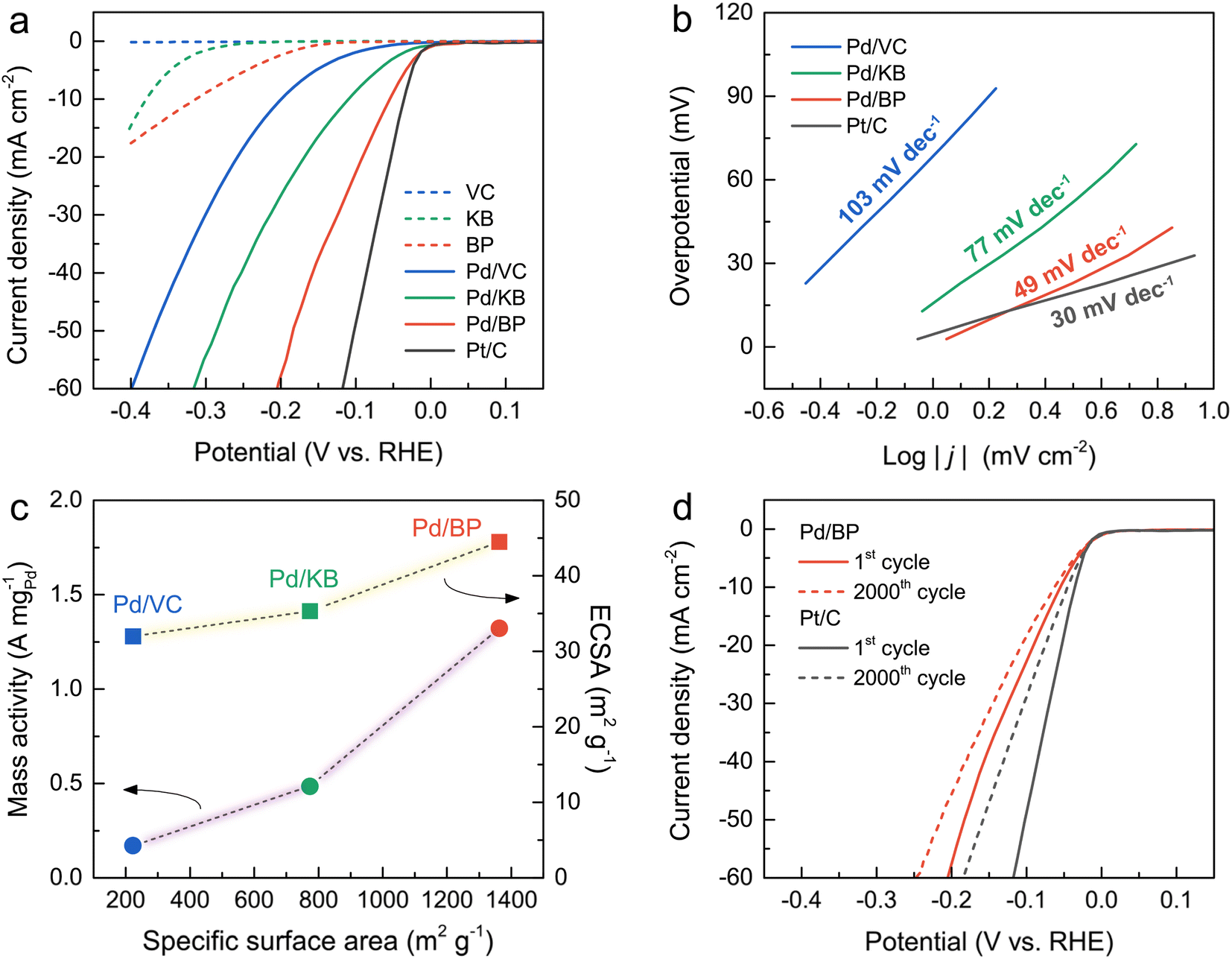 | ||
| Fig. 4 Electrochemical tests in an N2-saturated 0.5 M H2SO4 solution towards HER of the Pd/VC, Pd/KB, Pd/BP, and commercial Pt/C; (a) LSV curves, (b) Tafel plots, (c) mass activity at an overpotential of 200 mV and ECSA versus specific surface area (circle and square symbols represent mass activity and ECSA, respectively), and (d) stability tests of Pd/BP and Pt/C: LSV curves before and after 2000 cycles. | ||
To further quantify the HER activity, the mass activities of the Pd/CB catalysts were calculated, as shown in Fig. 4c. The mass activity of Pd/BP at an overpotential of 200 mV was 1.32 A mgPd−1, which was 2.7 and 7.8 times greater than that of Pd/KB (0.49 A mgPd−1), and Pd/VC (0.17 A mgPd−1), respectively. The electrochemical surface area (ECSA) of the catalysts was determined by the area of hydrogen adsorption (Hads) in the CV curve after subtracting the double-layer charge current (Fig. S3†). The area of Hads region was used for calculation because it provides a more accurate determination than the hydrogen desorption (Hdes) region.26 The ECSA value was calculated using the following eqn (2):27
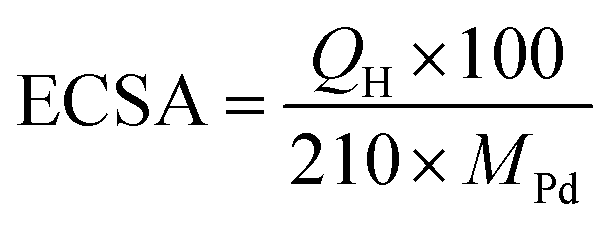 | (2) |
Additionally, electrochemical double-layer capacitance (Cdl) can also represent the ECSA of the Pd-based catalysts for HER.29–31 The CV curves for all catalysts were recorded in the non-faradic potential window (0.4–0.5 V) at various scan rates (10–50 mV s−1), as shown in Fig. S4†. The Cdl values of the catalysts were estimated by the slope of the plots between the ΔJ = (Janodic − Jcathodic)/2 and scan rate. The Cdl value of Pd/BP was 0.65 mF cm−2, which was greater than that of Pd/VC (0.44 mF cm−2) and Pd/KB (0.13 mF cm−2). The ECSA values determined by both Hads and Cdl showed the same trend, corresponding to their surface area. These results highlight that the enhanced HER activity of Pd/BP was attributed to the high surface area of BP support in comparison with Pd/VC and Pd/KB.
Despite Pd/BP having a relatively lower Pd loading compared to Pd/VC and Pd/KB, its HER activity surpassed that of the other catalysts. This discrepancy in HER activity can be attributed to the intrinsic characteristics of the CB supports. Notably, BP exhibited the lowest conductivity due to its lowest degree of graphitization, coupled with the highest surface area. Therefore, it can be inferred that surface area plays a more dominant role than conductivity in promoting HER activity. The superior HER activity of Pd/BP can be attributed to its high surface area and hierarchically porous structures, comprising the combinations of micropores, mesopores, and macropores. The micropores facilitate the diffusion, mass transfer, and assessment of H+ to the active sites during the HER process.32 Importantly, the abundant micropores (<2 nm) on BP promote the dispersion of Pd nanoparticles on the surface, resulting in more exposed active sites (higher ECSA).14,24 Mesopores (2–50 nm) reduce inner-pore ion-transport resistance.33 Additionally, macropores (>50 nm) serve as electrolyte-buffering reservoirs, facilitating electrolyte penetration and minimizing diffusion distances to the internal electrode surfaces.34
To assess stability under prolonged operation, Pd/BP was selected for testing and compared with Pt/C. As depicted in Fig. 4d, the overpotential at 50 mA cm−2 for Pd/BP and Pt/C was negatively shifted by 30 and 56 mV after 2000 cycles, respectively. This indicates that Pd/BP exhibited better stability in HER activity compared to Pt/C, likely due to the strong interaction between Pd and the support. The surface oxygen functional groups on BP, introduced by the plasma process, may serve as anchoring sites for the deposition of Pd nanoparticles with strong interaction.20 The degradation in the HER activity of Pd/BP after 2000 cycles can be attributed to the formation of larger Pd particles (∼20–50 nm), resulting from particle agglomeration and the Ostwald ripening effect during HER stability test (Fig. S5†).35
Conclusions
We synthesized Pd nanoparticles on three types of CB supports using SPS and investigated the effect of the CB support on their HER activity. Characterization results revealed that Pd nanoparticles, with sizes ranging from 2–6 nm, were well-dispersed on the outer surface of CB supports. Upon Pd loading, the micropore surface area (internal pores) was absent in Pd/VC and Pd/KB, but it was still present in Pd/BP. Among the catalysts investigated, Pd/BP exhibited the highest HER activity, close to that of Pt/C. This was evidenced by its lowest overpotential, lowest Tafel slope, and highest mass activity. The enhanced HER activity of Pd/BP is attributed to the unique characteristics of BP, including its high surface area with a hierarchical porous structure, and the abundant micropores. Additionally, Pd/BP demonstrated greater stability than Pt/C. Our findings provide fundamental insights into how the porous structure of CB supports influences the HER activity of Pd-based electrocatalysts synthesized via SPS. This understanding is crucial for the further design and development of HER electrocatalysts, thereby driving progress in sustainable energy technologies.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: J. C., G. P.; data curation: J. C., G. P.; formal analysis: J. C., C. H.; funding acquisition: R. T., G. P.; investigation: J. C., C. H., Y. Y., P. C.; methodology: J. C., C. H.; project administration: G. P.; resource: G. P.; supervision: G. P.; visualization: J. C., G. P.; validation: W. H., R. T., K., C. P., G. P.; writing – original draft: J. C., G. P.; writing – review & editing: G. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the UNNES-KU Matching Grant Research Collaboration 2021. The authors would also like to express their gratitude to the Faculty of Engineering, Kasetsart University, and the ICE-Matter consortium by AUN/SEED-Net, JICA for their partial financial support.Notes and references
- T. T. Le, P. Sharma, B. J. Bora, V. D. Tran, T. H. Truong, H. C. Le and P. Q. P. Nyuyen, Int. J. Hydrogen Energy, 2024, 54, 791–816 CrossRef CAS.
- D. Freire Ordóñez, C. Ganzer, T. Halfdanarson, A. González Garay, P. Patrizio, A. Bardow, G. Guillén-Gosálbez, N. Shaha and N. Mac Dowell, Energy Adv., 2023, 2, 2042–2054 RSC.
- S. S. Kumar and H. Lim, Sustainable Energy Fuels, 2023, 7, 3560–3583 RSC.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- K. Ojha, S. Saha, P. Dagar and A. K. Ganguli, Phys. Chem. Chem. Phys., 2018, 20, 6777–6799 RSC.
- S. Sarker and S. C. Peter, Inorg. Chem. Front., 2018, 5, 2060–2080 RSC.
- A. P. Murthy, J. Maadhavan and K. Marugan, J. Power Sources, 2018, 398, 9–26 CrossRef CAS.
- Y.-X. Xiao, J. Ying, H.-W. Liu and X.-Y. Yang, Front. Chem. Sci. Eng., 2023, 17, 1677–1697 CrossRef CAS.
- F. Liu, P. Wei, J. Zhang, M. Shi, J. Hou, Y. Li and S. Li, Carbon, 2024, 216, 118562 CrossRef CAS.
- J. Li, J. Zhang, J. Zhang, K. Pan, H. Xu, H. Chen, G. Liu, N. Wu, C. Yuan and X. Liu, J. Mater. Chem. A, 2023, 11, 19812–19844 RSC.
- X. Luo, H. Xiao, J. Li, P. Yuan, B. Du, H. Zheng, D. Li and Y. Chen, J. Electroanal. Chem., 2023, 939, 117476 CrossRef CAS.
- K. J. Omann, R. Sharma, P. Morgen, S. Gyergyek, M. J. Larsen and S. M. Andersen, ACS Appl. Energy Mater., 2023, 6, 1294–1307 CrossRef CAS.
- X. Yan, H. Li, J. Sun, P. Liu, H. Zhang, B. Xu and J. Guo, Carbon, 2018, 137, 405–410 CrossRef CAS.
- D. Banham, F. Feng, K. Pei, S. Ye and V. Birss, J. Mater. Chem. A, 2013, 1, 2812–2820 RSC.
- Y. Holade, C. Morais, K. Servat, T. W. Napporn and K. B. Kokoh, Phys. Chem. Chem. Phys., 2014, 16, 25609–25620 RSC.
- Z. Gan, C. Shu, C. Deng, W. Du, B. Huang and W. Tang, Nanoscale, 2021, 13, 18273–18280 RSC.
- Y. Yu, P. Liu, M. Dou, J. Niu, Z. Zhang and F. Wang, Catal. Sci. Technol., 2021, 11, 2997–3001 RSC.
- J. Shi, X. Hu, J. Zhang, W. Tang, H. Li, X. Shen and N. Saito, Prog. Nat. Sci.: Mater. Int., 2014, 24, 593–598 CrossRef CAS.
- C. Mani-Lata, C. Hussakan and G. Panomsuwan, J. Compos. Sci., 2020, 4, 121 CrossRef CAS.
- J. Chantaramethakul, N. Choophun, C. Chokradjaroen, A. Watthanaphanit, N. Saito and G. Panomsuwan, J. Phys. Chem. C, 2023, 127, 3184–3193 CrossRef CAS.
- S. Niu, S. Li, Y. Du, X. Han and P. Xu, ACS Energy Lett., 2020, 5, 1083–1087 CrossRef CAS.
- D. Banham, F. Feng, K. Pei, S. Ye and V. Briss, J. Mater. Chem. A, 2013, 1, 2812 RSC.
- T. Soboleva, K. Malek, Z. Xie, T. Navessin and S. Holdcroft, ACS Appl. Mater. Interfaces, 2011, 3, 1827–1837 CrossRef CAS PubMed.
- O. van der Heijden, S. Park, R. E. Vos, J. J. J. Eggebeen and M. T. M. Koper, ACS Energy Lett., 2024, 9, 1871–1879 CrossRef CAS.
- R. Sharma, S. Gyergyek and S. M. Andersen, Appl. Catal., B, 2022, 311, 121351 CrossRef CAS.
- K. Yoshii, K. Yamaji, T. Tsuda, H. Matsumoto, T. Sato, R. Izumi, T. Torimoto and S. Kuwabata, J. Mater. Chem. A, 2016, 4, 12152–12157 RSC.
- J. Durst, C. Simon, F. Hasché and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, F190 CrossRef CAS.
- P. Chandrasekaran, T. N. J. I. Edison and M. G. Sethuraman, Int. J. Hydrogen Energy, 2020, 45, 28800–28811 CrossRef CAS.
- D. S. Butenko, S. Li, V. O. Kotsyubynsky, V. M. Boychuk, V. I. Dubinko, P. I. Kolkovsky, N. A. Liedienov, N. I. Klyui, W. Han and I. V. Zatovsky, Int. J. Hydrogen Energy, 2021, 46, 21462–21474 CrossRef CAS.
- P. Kaushik, G. Kaur, G. R. Chaudhary and U. Batra, J. Colloid Interface Sci., 2021, 582, 894–905 CrossRef CAS PubMed.
- S. A. Grigoriev, M. S. Mamat, K. A. Dzhus, G. S. Walker and P. Millet, Int. J. Hydrogen Energy, 2011, 36, 4143–4147 CrossRef CAS.
- D. Qiu, C. Kang, M. Li, J. Wei, Z. Hou, F. Wang and R. Yang, Carbon, 2020, 162, 595–603 CrossRef CAS.
- J. Niu, J. Liang, R. Shao, M. Liu, M. Dou, Z. Li, Y. Huang and F. Wang, Nano Energy, 2017, 41, 285–292 CrossRef CAS.
- M. Smiljanic, M. Bele, L. Moriau, F. Ruiiz-Zepeda, M. Šala and N. Hodnik, J. Phys. Chem. C, 2021, 125, 27534–27542 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04809a |
| This journal is © The Royal Society of Chemistry 2024 |