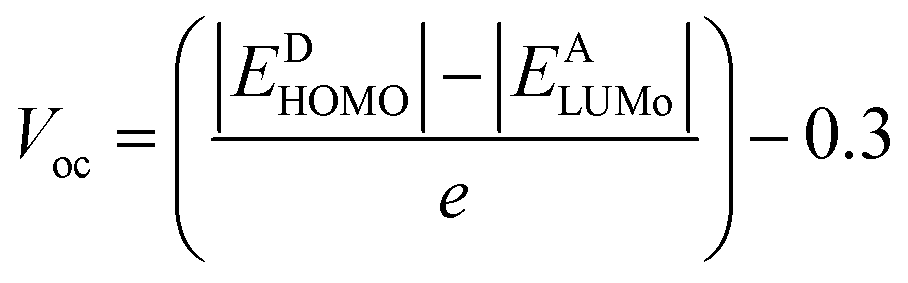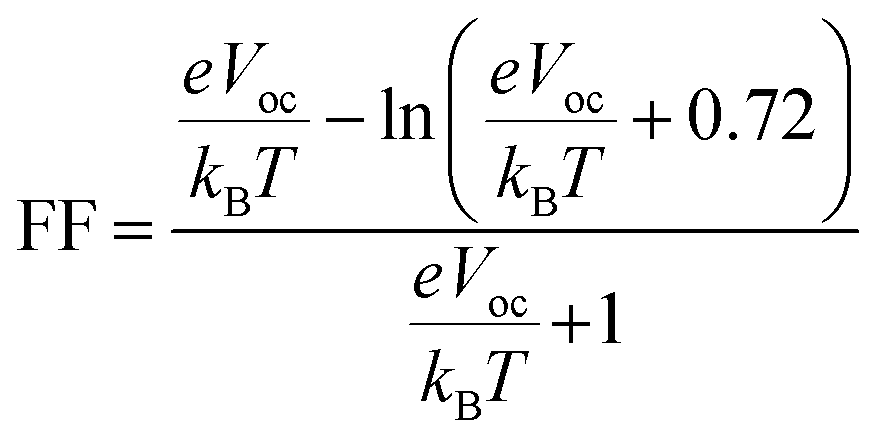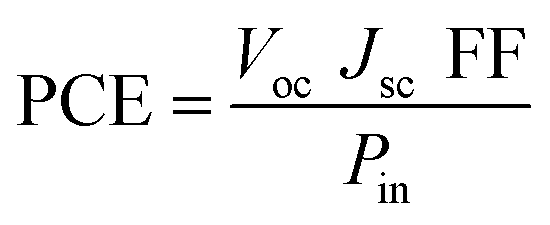DOI:
10.1039/D4RA04818K
(Paper)
RSC Adv., 2024,
14, 29942-29954
Theoretical investigation of benzodithiophene-based donor molecules in organic solar cells: from structural optimization to performance metrics†
Received
3rd July 2024
, Accepted 12th September 2024
First published on 20th September 2024
Abstract
Achieving high power conversion efficiency (PCE) remains a significant challenge in the advancement of organic solar cells (OSCs). In the field of organic photovoltaics (OPVs), considerable progress has been made in optimizing molecular structures to improve the PCE. However, innovative material design strategies specifically aimed at enhancing PCE are still needed. Here, we have designed BDTS-2DPP-based molecules and propose a molecular design approach to develop donor materials that can significantly improve the PCE of OSCs. Density functional theory (DFT) and time-dependent DFT (TD-DFT) methods have been adopted in both gas and solvent phases. Our newly designed molecule M1 shows the highest absorption value (λmax = 846 nm), highest electron reorganization energy (λe = 0.18 eV), and the lowest energy gap (Eg = 1.81 eV) among all the designed molecules. M1 molecule also exhibits the highest dipole moment in both gas (10.62 D) and solvent phase (13.62 D), and their ground and excited state dipole moment difference is also higher (μe − μg = 2.99 D), which enhances its separation to make it a suitable candidate for charge transfer between HOMO–LUMO (97%). Newly designed molecule M3 is observed to have the highest voltage when the current is zero (Voc = 1.15 V) highest PCE value (21.90%) and highest fill factor (FF) value (89.42%). The lowest excitation binding energy is estimated by newly designed molecule M2 (Eb = 0.30 eV), which indicates a higher rate of dissociation during the excitation as observed in transition density matrix (TDM) plots. Utilizing electron density difference maps, the newly designed molecules in dichloromethane solvent exhibited consistent intramolecular charge transfer (ICT). The designed molecules were evaluated against reference molecule R to determine if they exhibit superior optoelectronic capabilities. It is found that all designed molecules (M1–M5) exhibit reduced band gaps, are red-shifted in wavelength in comparison to a reference molecule R, and have remarkable charge motilities in terms of reorganisation energies.
1. Introduction
A scarcity of energy resources has been a direct result of the continuous increase in the global population over the last few decades.1,2 The world's fossil fuel reserves and other conventional energy sources are rapidly depleting. In addition to having a negative effect on the climate, burning fossil fuels is a major cause of environmental damage leading to a threatening level of global warming by the greenhouse effect. Renewable, greener, safer, and more eco-friendly options must be promptly pursued in order to solve the problem of limited energy supply and to control environmental pollution.3 Replacing carbon based fuels with nuclear power, biomass, solar power, hydropower, and wind power ensures little or no carbon emissions to the environment. Solar power offers the most promising potential among all these alternatives in terms of sustainable, long-term, and inexpensive energy sources for the globe. Modern society is tremendously encouraging solar cells because of their many useful properties, such as their low cost, great efficiency, and lack of carbon emissions.4 Inorganic solar cells based on silicon are extensively used due to two primary factors; they decrease the costs of producing solar cells and enhance the efficiency of power conversion. Additionally, they possess exceptional attributes like high thermal stability, lower toxicity, and thermal resilience. The advantages of inorganic solar cells are limited due to some disadvantages, like being frail and fixed levels of energy that cannot be adjusted.5,6 Organic solar cells (OSCs) are drawing increasing attention with their power conversion efficiency (PCE) raised above 19%.7,8 There are primarily two categories of OSCs: those designed from polymers and those derived from small molecules.9 The synthesis method and the size of the individual moieties are the foundations of this categorisation. The first perovskite solar cell (PSC) was created by Yu and colleagues using a combination of C60 fullerene and poly[2-methoxy-5-(2′-ethylhexyloxy)-p-phenylenevinylene] (MEH-PPV), with a reported PCE of 3%.10 Mineral solar cells have been superseded by bulk heterojunction solar cells because of their superior PCE. A fullerene derivative that accepts electrons and a π-conjugated polymer that donates electrons are combined in the same phase.11 Utilising derivatives of fullerene as electron acceptors and a combination of multiple donor moieties, organic solar cells based on fullerene achieve remarkable properties like wide absorption range, tailored HOMO and LUMO levels for efficient charge separation, high absorbance, and significantly enhanced hole mobility which boosts up the PCE tremendously in bulk heterojunctions. As electron acceptors, fullerene derivatives have several limitations, thus researchers are looking for alternatives with better properties.12 Non-fullerene derived organic solar cells have high potential and superior performance, aligning with the features seen in fullerene-based OSCs.13 Non-fullerenes acceptors have enhanced light absorption in the near-infrared (NIR) and visible regions, resulting in increased photocurrent generation in photovoltaic systems. Fused ring acceptors of electrons (FREAs) are very significant in non-fullerene based on OSCs due to their convenient synthesis, purification, and ability to tune energy levels and exhibit considerable absorption in the visible range.14 Devices based on FREAs have been claimed to have an efficiency of around 20%.15 Small molecules and polymer groups are common components of both the donor and acceptor groups. A new class of molecules with characteristics shared by both small molecules and polymers, known as oligomer molecules (OMs), has recently evolved.16,17 Polymers have molecular weights that are more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, while small molecules typically have a molecular weight lower than 1000. The molecular weight range of oligomers is between that of small molecules and polymers, with values between 1000 and 10000. For organic photovoltaic systems, the benzo[1,2-b:4,5-b′] dithiophene unit (BDT) is an excellent donor material.18,19 The planarity and π-conjugation of its structure, which is both rigid and asymmetrical, allows for efficient charge transfer. To make oligomer, BDT has been linked to diketopyrrolopyrrole (DPP) units in the past. PCE of around 6.03% was achieved in earlier research by switching out BDT units with alkyl-thio units.20 The rate of hole transmission is hugely boosted as a result of this replacement. Due to the elongation of π–π stacking on sides of chains that include thiophene groups, the BDT unit with linear alkyl-thio groups experiences an enhanced rate of hole transfer. Charge transfer was further improved by adding strong electron-withdrawing groups like DPP, which is planar like BDT.21
000, while small molecules typically have a molecular weight lower than 1000. The molecular weight range of oligomers is between that of small molecules and polymers, with values between 1000 and 10000. For organic photovoltaic systems, the benzo[1,2-b:4,5-b′] dithiophene unit (BDT) is an excellent donor material.18,19 The planarity and π-conjugation of its structure, which is both rigid and asymmetrical, allows for efficient charge transfer. To make oligomer, BDT has been linked to diketopyrrolopyrrole (DPP) units in the past. PCE of around 6.03% was achieved in earlier research by switching out BDT units with alkyl-thio units.20 The rate of hole transmission is hugely boosted as a result of this replacement. Due to the elongation of π–π stacking on sides of chains that include thiophene groups, the BDT unit with linear alkyl-thio groups experiences an enhanced rate of hole transfer. Charge transfer was further improved by adding strong electron-withdrawing groups like DPP, which is planar like BDT.21
In this study, we have designed five new BDTS-2DPP22 (reference molecule (R)) based donor molecules named as M1, M2, M3, M4 and M5. The central unit in molecule R is a donor and the acceptor groups are attached at its terminal end to make it A–π–D–π–A. All the new molecules contain spacers in between to connect donor and acceptor moieties for efficient charger transfer. All these molecules are linked with five different acceptor groups on the end caps as represented in Fig. S1.† Molecule R serves as the reference point with a well-balanced donor–acceptor structure, providing a baseline for understanding the effects of structural modifications introduced in the other molecules. Molecule M1 features an extension of the conjugated system by adding a thiophene unit, which is intended to enhance electron delocalization. This modification aims to improve light absorption and charge transport properties by increasing the effective conjugation length. Molecule M2 introduces electron-withdrawing groups on the donor part of the molecule and extends the conjugated system through the incorporation of a benzothiadiazole unit. These changes are designed to lower the HOMO level, thereby increasing the Voc and potentially improving the PCE. The additional conjugated units in M2 also contribute to better light-harvesting capabilities. Molecule M3 incorporates additional conjugated units, specifically a thieno[3,2-b]thiophene group, along with structural modifications that increase the planarity of the molecule. The enhanced planarity is expected to improve π–π stacking interactions, which are crucial for efficient charge transport, leading to better charge mobility and higher overall efficiency. Molecule M4 features a more complex structure with both electron-donating groups and an extension of the conjugated system by incorporating a fluorinated benzotriazole unit. These modifications further extend the π-system and are expected to enhance the charge separation process by optimizing the balance between electron donation and withdrawal. The structural changes in M4 are aimed at increasing the FF and overall device performance. Finally, molecule M5 represents the most advanced design, combining the beneficial aspects of M1 through M4. It includes strategic modifications to both the donor and acceptor units, along with additional conjugated systems such as the integration of a fused aromatic ring system (e.g., dithieno[3,2-b:2′,3′-d]thiophene). These are designed to maximize light absorption, charge separation, and transport, leading to the highest theoretical PCE among the five molecules.
The significance of studying these molecules lies in their potential to push the boundaries of organic photovoltaic performance. By systematically varying the molecular architecture, including the addition of specific conjugated units like thiophene, benzothiadiazole, thieno[3,2-b]thiophene, fluorinated benzotriazole, and fused aromatic rings, we aim to identify the key features that contribute to higher efficiency, stability, and scalability in organic solar cells. The insights gained from this study are crucial for guiding the design of next-generation materials that could significantly advance renewable energy technologies. Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) serve as powerful tools for conducting computational analysis and exploring the intricate optoelectronic properties of designed molecules. The designed molecules were evaluated against reference molecule (R) to determine if they exhibit superior optoelectronic properties. It was observed that all designed molecules have reduced band gaps, hence red-shifted in assessment to reference molecule R and have remarkable charge motilities.
2. Computational methodology
The calculations were conducted utilising the Gaussian09 simulation package,23,24 and the results were visualised using GaussView 6.0. To confirm which DFT method would be the best for the designed molecules, reference molecule R was initially optimised using four different functionals: B3LYP,25 CAM-B3LYP,26 MPW1PW91,27 WB97XD.28 Once the optimisation was completed using DFT method,29 the λmax of reference molecule (622 nm)30 was found to be the best correlated with the MPW1PW91 functional (683 nm). We found that for further photovoltaic study, MPW1PW91 is the best functional because correlates with the experimental value of λmax of reference molecule. So, all designed molecules were optimised using the MPW1PW91/6-31G(d,p) functional method. PyMOlyze-1.1 (ref. 31) was used for plotting and visualising the graphs of the density of states (DOS). We also computed other geometric factors like dipole moment (μ),32 binding energies (Eb)33 and open circuit voltage (Voc).34 In order to predict the nature of transitions for the reference and all designed molecules, the transition density matrix (TDM)35 was analysed using Multiwfn software.36 For analyzing the charge transfer in the molecules, a critical factor known as reorganisation energy has been used.37,38 The reorganization energy has a very strong relation with the electron and hole motilities, and it is computed by using equations from Marcus theory.39 The equations for calculating the reorganisation energy of electrons and holes are as follows:| | |
λe = [E−° − E−] + [E°− − E°]
| (1) |
| | |
λh = [E+° − E+] + [E°+ − E°]
| (2) |
E− is the anionic energy that has been optimised, and E°− is the anionic energy from the neutral molecule after optimisation. E+ is the cationic energy that has been optimised, and E°+ is the cationic energy from neutral molecules of optimised structures. E° are single-point energy calculations for both the positively charged cation and the negatively charged anion whereas, E+°, E−°, represents the cationic and anionic neutral energies from their optimised structures.
3. Optoelectronic characteristics
3.1 Structural optimization
In this work, we optimised BDTS-2DPP (reference molecule R) using four distinct functional such as; B3LYP, CAM-B3LYP, WB97XD, and MPW1PW91 with basis set 6-31G(d,p). MPW1PW91 is the best chosen functional as the value of λmax for this functional turns out to be 683 nm which matches well with the experimental value i.e. 622 nm as depicted in Fig. S2.† The reference molecule is altered by slight modifications on the terminal side with different acceptor groups connecting with spacers. These modifications are performed through the spacer groups. Therefore, all the molecules followed the acceptor–spacer–donor–spacer–acceptor combination. The reference BDTS-2DPP and all designed molecules (M1–M5) are optimised with the chosen functional MPW1PW91 to explore the optoelectronic properties.
3.2 UV-visible spectral characteristics
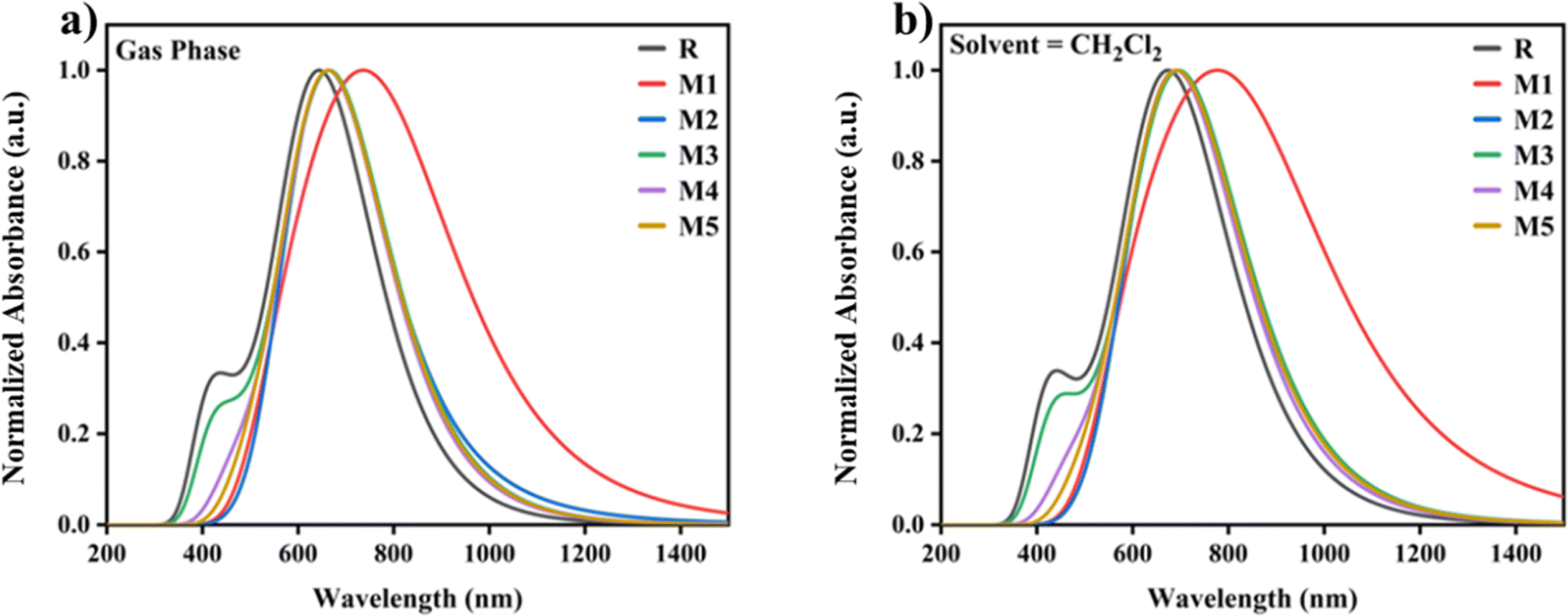
To explore more useful insight into DFT functions, the geometry of molecule R is optimised using a variety of approaches. In the solvent phase, the molecule R is optimised using different functionals such as B3LYP, MPW1PW91, CAM-B3LYP, and WB97XD. The values of different parameters like absorption profile (λmax), light harvesting efficiency (LHE), percentage assignments (S0–S1), dipole moments (μ), excitation energy (EX), oscillator strength (f) for gas phase and solvent phase are shown in Tables S1 and S2.† The change in λmax values of all the designed molecules (M1–M5) in contrast with reference molecule R is due to the introduction of acceptor groups to the donor core of the material. All the designed molecules (M1–M5) have greater λmax and are red-shifted in absorption spectra in contrast to the reference molecule R. The highest λmax is shown by molecule M1 (846 nm) in dichloromethane (CH2Cl2). The λmax values of all the designed molecules using dichloromethane came out to be M1 (846 nm), M2 (748 nm), M3 (710 nm), M4 (699 nm), and M5 (707 nm) respectively, and are shown in Fig. 1(a) and (b). As molecule M1 shows the maximum absorption strength of 846 nm and minimum band gap of 1.81 eV as depicted in Fig. S3† and tabulated in Table 1, hence it is the proficient donor material among all designed molecules.
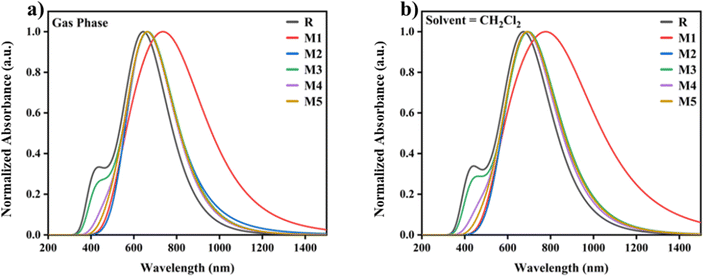 |
| | Fig. 1 Graphically illustration of UV-vis absorption of reference molecule R and designed molecules (M1–M5), in gas phase as well as in solvent (CH2Cl2). | |
Table 1 The theoretically calculated HOMO–LUMO and energy gap (Eg) values of reference molecule R and designed molecules (M1–M5) using the MPW1PW91/6-31G(d,p) level of theory
| Molecules |
EHOMO (eV) |
ELUMO (eV) |
Eg (eV) |
| R |
−5.05 |
−2.81 |
2.24 |
| M1 |
−5.14 |
−3.33 |
1.81 |
| M2 |
−5.07 |
−3.12 |
1.95 |
| M3 |
−5.15 |
−2.98 |
2.17 |
| M4 |
−5.07 |
−2.86 |
2.21 |
| M5 |
−5.12 |
−3.01 |
2.11 |
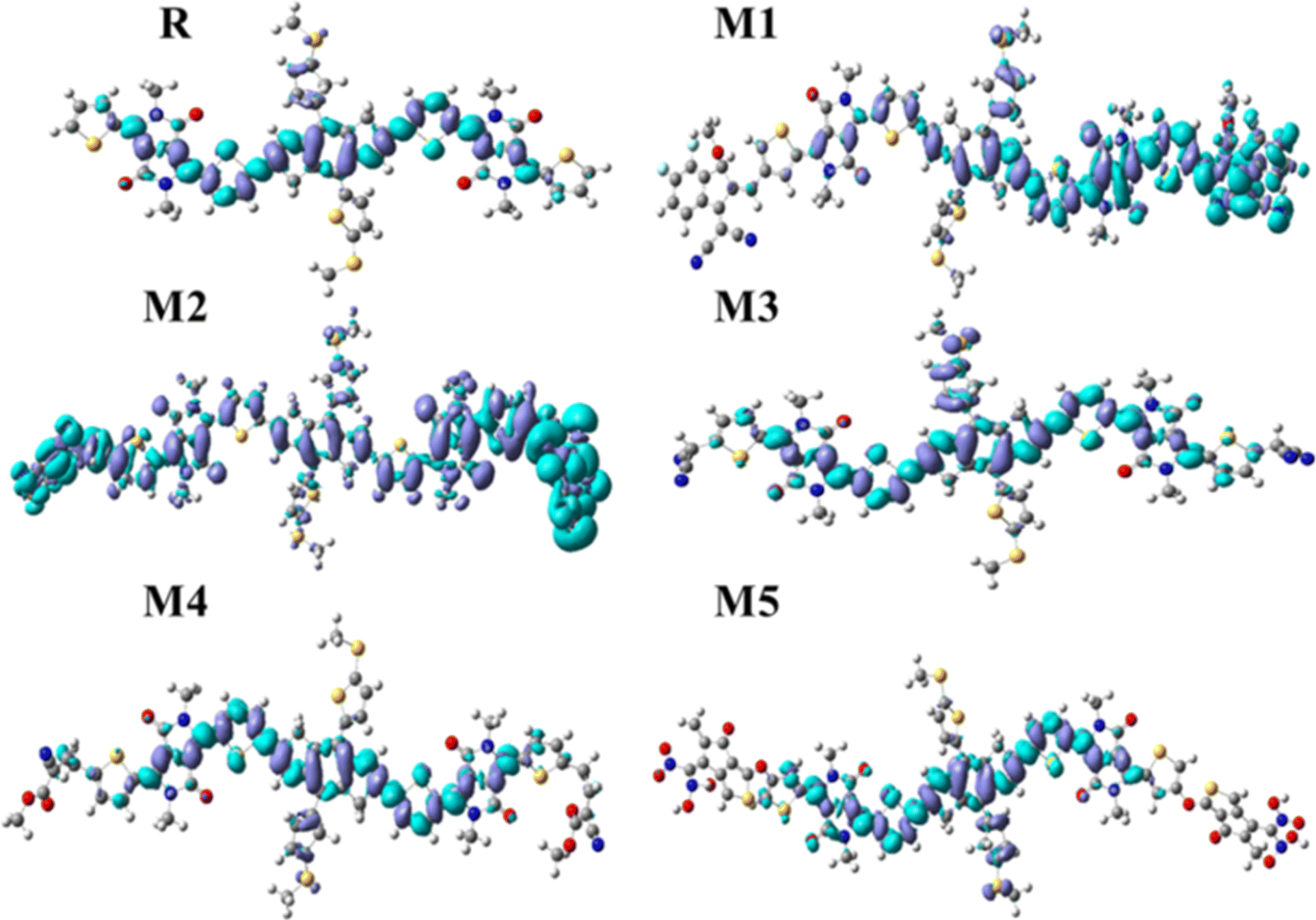
3.3 The frontier molecular orbitals (FMOs)
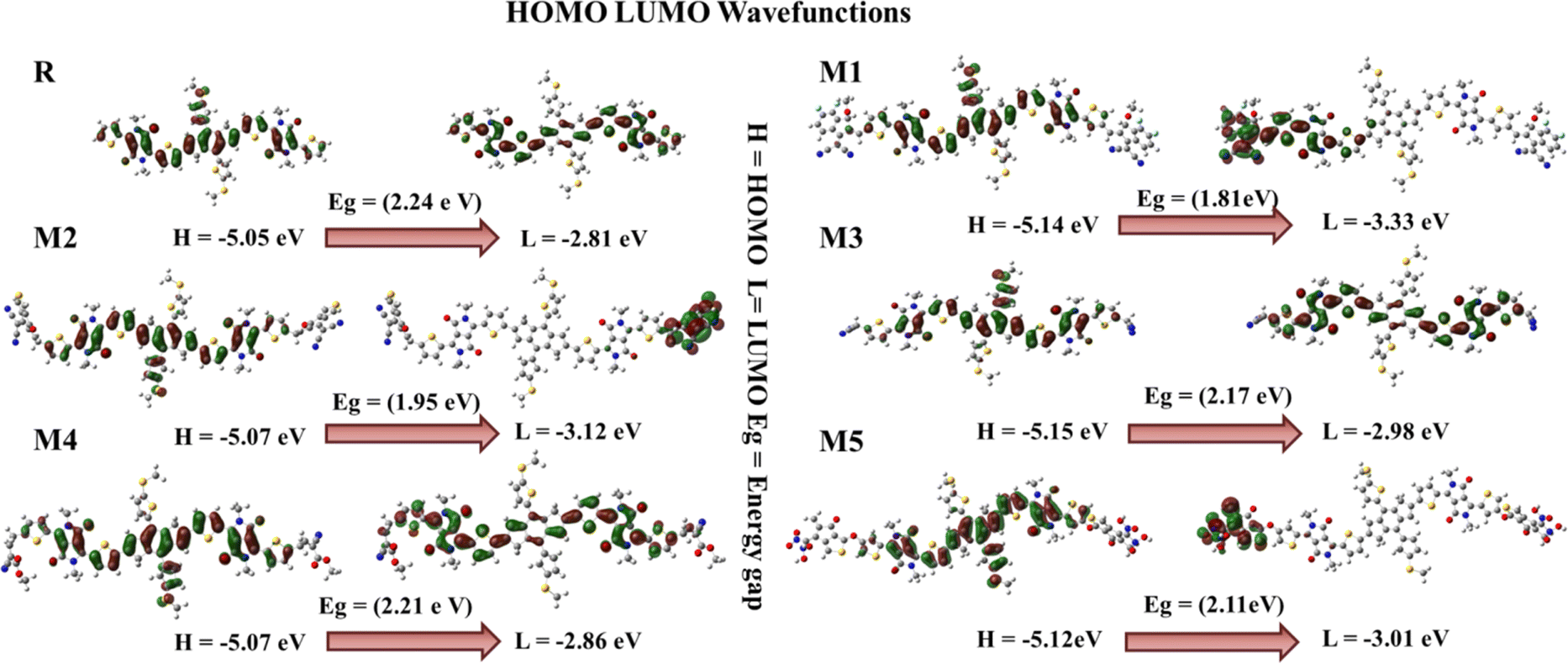
When a bridge unit connects a donor and acceptor region, there is a less energy band gap that facilitates efficient charge transfer. The donor segment is positioned in the centre of the molecule, with end-capped acceptor segments located at the terminals representing two states of energy: one in which electrons are located and another in which they can be promoted. An important factor in a material's electrical and optical properties is its band gap. For efficient charge movement and light absorption, an ideal band gap is required. The linkage between the donor and acceptor units is established using thiophene bridges.40 The band gap for reference molecule R is 2.24 eV, as reported by the FMOs, with HOMO energy of −5.05 eV and LUMO energy of −2.81 eV. When an electron density is excited, it moves from an electron donor to an electron acceptor region. The electron density of the HOMO and LUMO in reference molecule R is mostly concentrated on the whole molecule, including the donor, acceptor, and bridge fractions. The HOMO of the molecule M1 is formed by contributions from the donor, acceptor, and bridge. Conversely, the LUMO signal within the molecule M1 exclusively illuminates the acceptor bridge electron density. The electron density in the HOMO of molecules M2, M3, M4, and M5 is mostly generated by the donor and spacer units, with no contribution from the acceptor moieties in these molecules. Absorption of longer wavelengths of light is possible when the energy gap is reduced. Having the capacity to absorb light in the red and near-infrared parts of the spectrum is a practical property of materials with a low energy gap. The energy levels in these zones are lower than those of blue or UV light. The LUMO of molecules M2 and M5 is determined by the electron density located on the acceptor moieties, whereas the LUMO of molecules M3 and M4 is influenced by the contribution of both the acceptor and the bridge unit. Fig. 2 illustrates that all the designed molecules have the capability to efficiently transmit charges. The Eg values of designed molecules M1–M5 and the reference molecule R are listed in Table 1, along with the HOMO and LUMO energies. M1, with its lowest bandgap (1.81 eV), has been selected as the most effective molecule among all molecules. The lowest bandgap is seen in molecule M1, suggesting that it may easily shift between the HOMO to the LUMO (Fig. S3†). Therefore, it may be inferred that M1 has the lowest band gap, as seen in Fig. S3.† It exhibits the smoothest transitions among all newly engineered molecules.
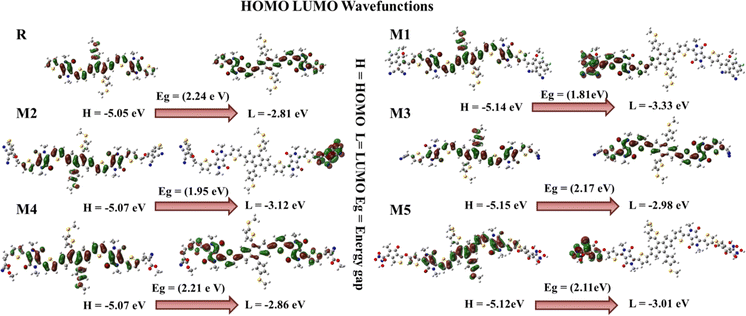 |
| | Fig. 2 FMOs diagram of reference molecule R and all newly designed molecules (M1–M5). | |
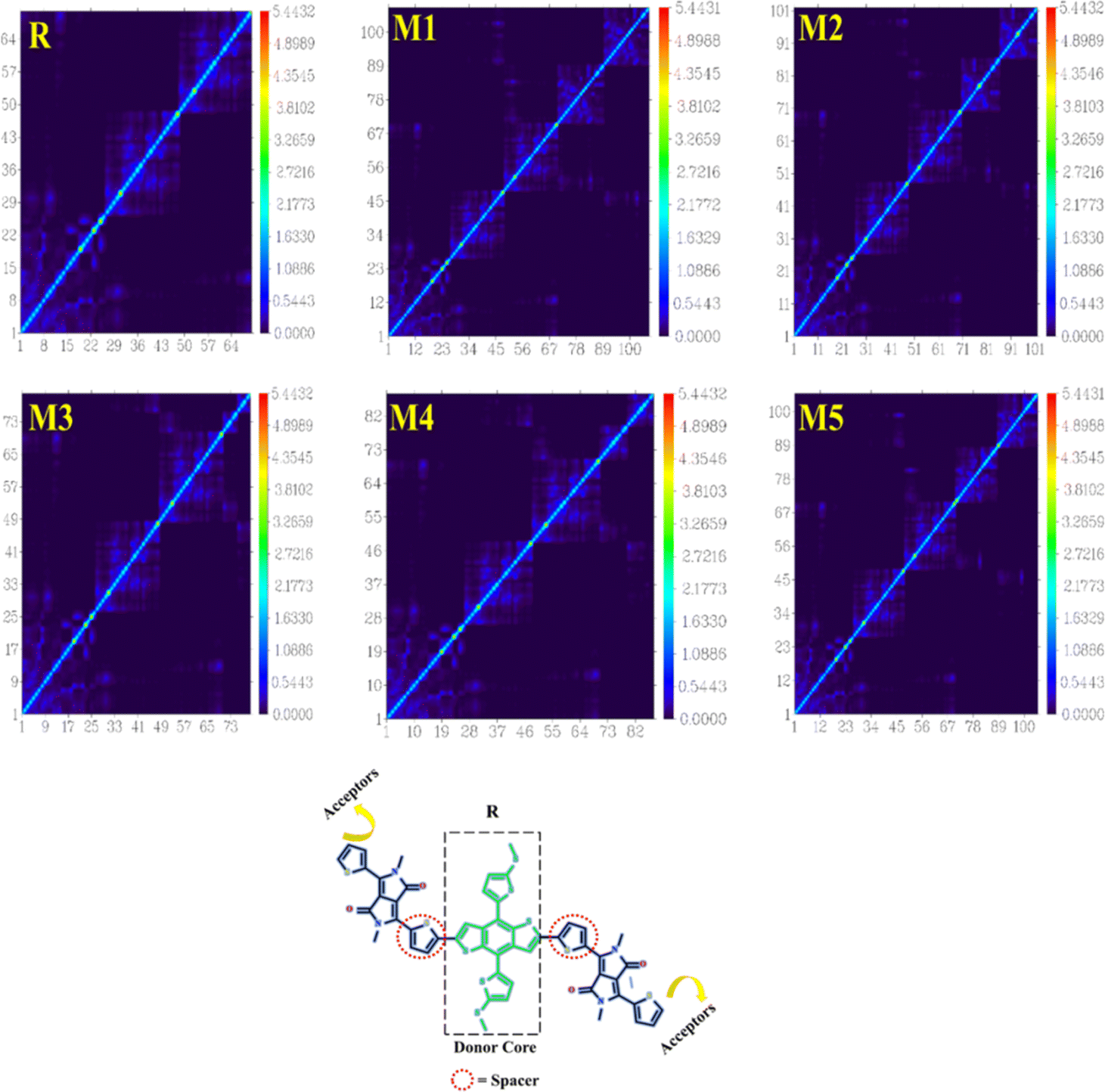
3.4 Density of state analysis (DOS)
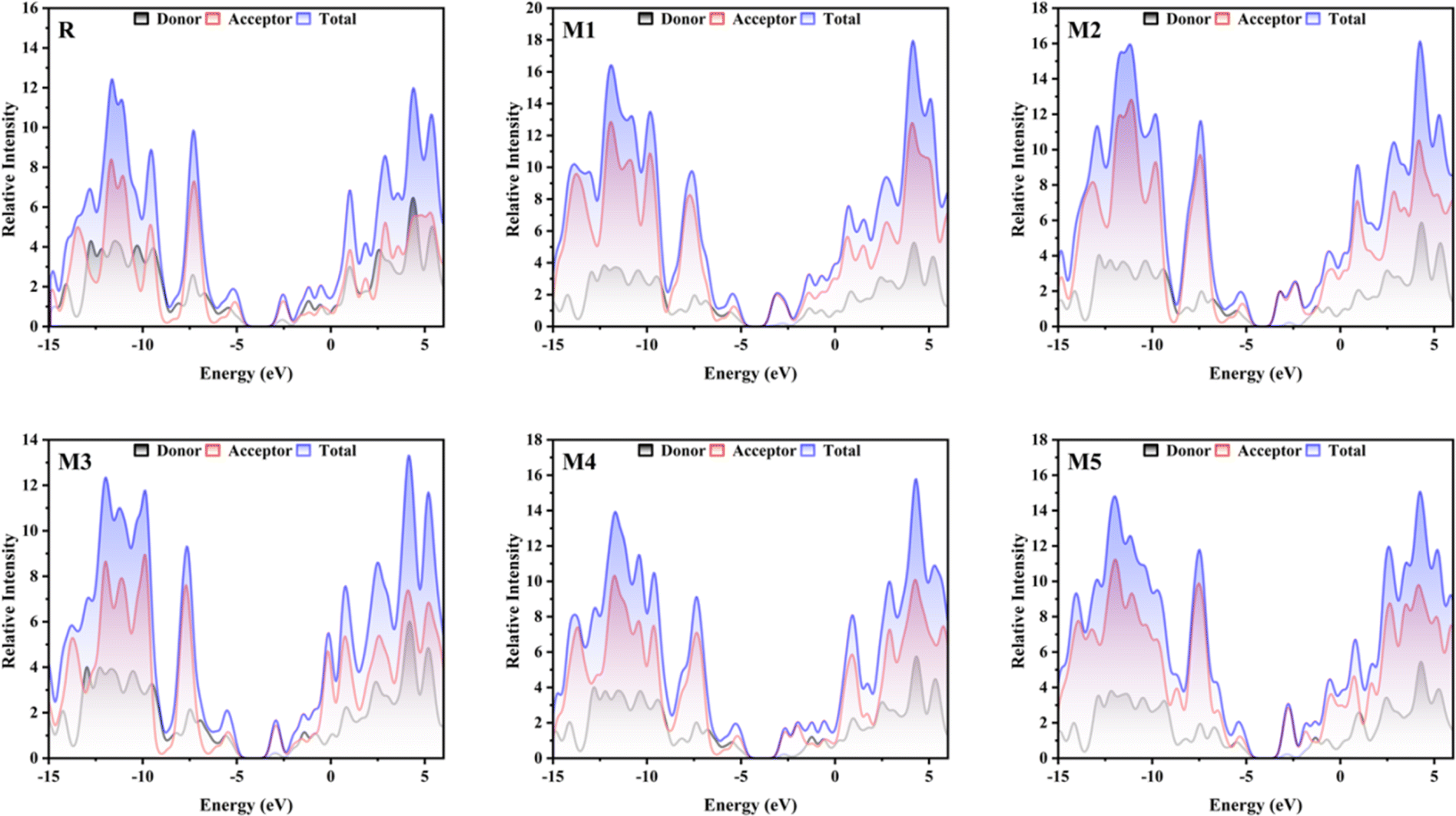
The DOS gives information regarding the quantity of electronic states that are accessible at each energy level. The significance of this factor in OSCs lies in its direct influence on the creation of electron–hole pairs (also known as exciton) when light is absorbed, as well as their subsequent separation and the collection of charges at electrodes. In order to examine the electronic distribution and analyse the electron density contribution in both the conduction and valence bands, DOS calculations were conducted using the MPW1PW91/6-31G(d,p). The different colours in Fig. 3, are used to represent distinct donor and acceptor groups. Additionally, they provide the individual and collective contributions of each segment in the DOS spectrum. The elevated peaks in the DOS graphs, resulting from increased electron density, indicate a greater degree of attraction between the connected groups. The PyMOlyze-1.1 program was used to analyse DOS plots. Fig. 3 provides a clear picture about how the donor core transmits electron density to the acceptor units. The bridging units are what make this density transfer possible. The donor peaks exhibit strong intensity in the HOMO, whereas the acceptor intensity is notably high in the LUMO. This observation is most precisely seen in molecules M1, M2, and M5. The DOS spectra of the M1 molecule demonstrate the segregation of red peaks (representing the acceptor unit) from black peaks (representing the donor unit) in the LUMO region. This provides compelling evidence for the existence of a very potent acceptor unit in the M1 molecule. The examination of the spectra indicates that the primary factors that affect the transfer of charge involve the acceptor and donor components via the spacer group. The findings obtained from DOS spectra are consistent with the FMOs.
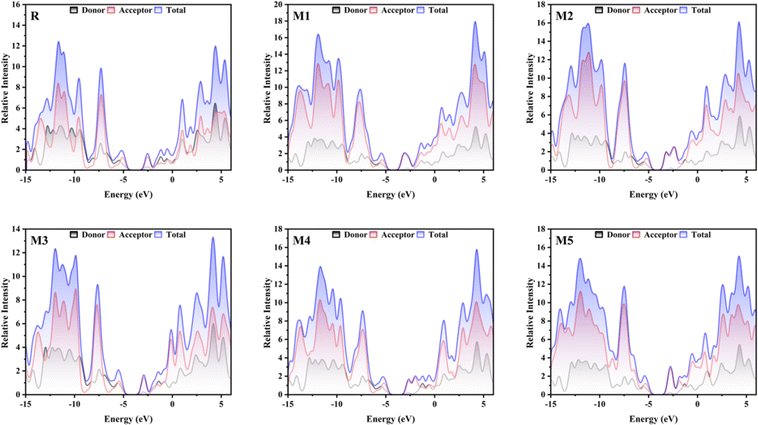 |
| | Fig. 3 DOS graphs for reference molecule R and designed molecules. | |
3.5 Reorganization energy (RE)
RE (λ) plays a vital role in defining the improved photovoltaic properties of materials designed for future applications in organic solar cells. It facilitates the investigation of the energies of electrons (λe) and holes (λh) during molecular excitations and has an inverse relationship with charge transfer. Determining the effect of molecular reorganization on the transport of charges, namely electrons and holes, inside organic solar cell materials is the main reason for calculating RE. The symbol λ can be separated into two components: λint, which specifies the interior geometries, and λext, which defines the exterior polarisation environment. The value of λint can be determined using equations from Marcus' theory since the energies of anions, cations, and neutral molecules influence it. Marcus theory describes the rate at which an electron can transfer between two molecules, considering factors such as the reorganization energy (λ) and the driving force of the reaction (ΔG). The theory provides a relationship between the rate of electron transfer and these key parameters, predicting that the rate increases with decreasing reorganization energy and reaches a maximum at a specific point where the driving force equals the reorganization energy. The significance of Marcus theory in our study lies in its ability to explain the impact of molecular structure on electron transfer efficiency. By analyzing the reorganization energies of the molecules, we can predict their electron transfer rates and, consequently, their performance in photovoltaic applications. Lower reorganization energies, as suggested by Marcus theory, are indicative of more efficient charge transport, which is crucial for achieving higher power conversion efficiencies in organic solar cells.41 The reorganisation energies are shown in Table 2. RE for electron transfer of all the designed molecules is lower than reference molecule R. Therefore, it is evident that all designed molecules exhibit excellent electron mobility. The molecule M3 has the lowest RE value for electron transfer, λext indicating that it may effectively transfer charges. RE for electron transfer exhibits an ascending trend as: M3 < M2 = M4 < M5 < M1 < R.
Table 2 The reorganization energies of holes (λh) and electrons (λe) for the reference molecule R and designed molecules (M1–M5) using the MPW1PW91/6-31G(d,p) level of theory
| Molecules |
λh |
λe |
| R |
0.143 |
0.146 |
| M1 |
0.185 |
0.113 |
| M2 |
0.157 |
0.099 |
| M3 |
0.164 |
0.078 |
| M4 |
0.139 |
0.099 |
| M5 |
0.119 |
0.108 |
3.6 Dipole moment (μ)
The dipole moment serves as a promising factor in assessing the material's solubility in organic solvents and plays a crucial role in enhancing the effectiveness of OSCs.42 The energy levels of materials can be changed by molecular dipole moments, particularly the HOMO and LUMO values. This may improve energy balance at donor–acceptor interfaces in OSCs. If a material's polarisation is strengthened by a strong dipole moment, it is feasible to achieve a rise in the dielectric constant. This improved polarisation may lead to an increase in charge mobility and a lower recombination rate of separate charges (electrons and holes), hence decreasing the coulombic affinity between them. For the charges to be transferred between the materials and collected at the electrodes efficiently, this alignment is crucial. A direct relation between the dipole moment and solubility means that the higher the dipole moment of any material, the higher will be its solubility. Higher mobility improves molecular symmetry, which increase charge mobility and facilitates charge movement. The dipole moment of all designed molecules is determined by DFT-MPW1PW91/6-31G(d,p). Table 3 shows the μ values in the ground and excited states of the solvent. Molecule M1 shows the highest μ in both states i.e. ground state as well as in excited state, hence it will be the best molecule for its use in film formation because higher μ leads to enhanced crystallinity.43 All the designed molecules show greater μ values than the reference molecule R. All of the designed molecules and the reference molecule R have μ values in the order of: M1 > M3 > M5 > M2 > M4 > R. Hence, M1 will show better photovoltaic stability as well as solubility in dichloromethane solvent.
Table 3 The dipole moments of reference molecule R and designed molecules (M1–M5) at the ground state (μg) and the excited state (μe) using the MPW1PW91/6-31G(d,p) level of theory
| Molecules |
μg |
μe |
μe − μg |
| R |
3.143 |
3.942 |
0.799 |
| M1 |
10.625 |
13.624 |
2.999 |
| M2 |
4.211 |
5.688 |
1.477 |
| M3 |
7.723 |
9.786 |
2.063 |
| M4 |
4.041 |
4.228 |
0.187 |
| M5 |
6.360 |
6.360 |
0.000 |
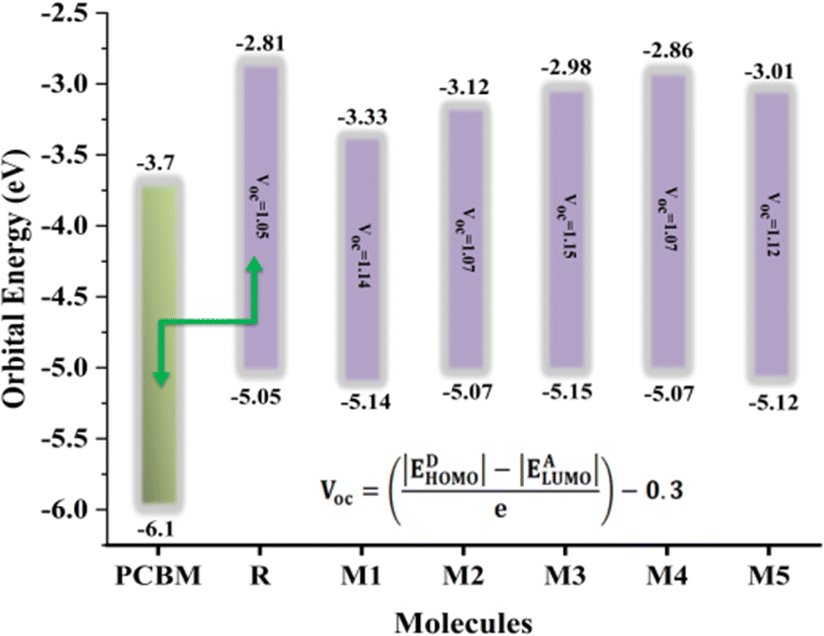
3.7 Open circuit voltage (Voc)
PCE of OSCs is defined by a critical factor named open-circuit voltage. It is the highest voltage by a device when the device is present at zero current. A high Voc requires an acceptor molecule with a high LUMO energy level and a donor molecule with a low HOMO energy level.44 The energy disparity between an acceptor material's LUMO and HOMO, as compared to the substance of the material being applied, is the primary determinant of a material's Voc. To enhance charge separation and extraction, the position of these energy levels can be tuned to achieve a greater Voc. All the designed molecules show properties of electron donation hence their HOMO values are correlated with some acceptor LUMO values. The acceptor used in the current work is PCBM. The equation used to find the values of open circuit voltage is as follows:| |
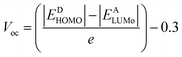 | (3) |
In the above equation, energies are represented by E, the charge on the discrete molecule is represented by e, and 0.3 is the empirical factor. The Voc values of reference molecule R and designed molecules are shown in Fig. 4. The Voc values of reference molecule R and designed molecules (M1–M5) are 1.05 V, 1.14 V, 1.07 V, 1.15 V, 1.07 V and 1.12 V respectively. The molecular structures have variations in conjugation and the presence of electron withdrawing or electron donating groups, which directly affect these energy levels. For instance, molecules M1 and M3, which incorporate additional conjugated units or electron withdrawing groups, exhibit higher Voc values. These structural modifications result in a larger energy gap between the HOMO and LUMO, thereby increasing the Voc compared to the reference molecule R. All designed molecules have more Voc in comparison to reference molecule R, hence they are better donor materials than reference molecule R. M3 exhibits the highest Voc (1.15 V) value, hence it is the most efficient molecule among all designed molecules and it has the potential to serve as donor molecules for solar cells applications.
 |
| | Fig. 4 Voc values of reference molecule R and all designed molecules (M1–M5) calculated with regard to the PCBM. | |
3.8 Transition density matrix (TDM)
The electron–hole and excitation motilities are effectively elucidated through a comprehensive analysis of TDM graphs.45 The charge density distribution in fragments as well as in associations between donor and acceptor subunits can be investigated using TDM graphs.46 The hydrogen atoms have been excluded because of their minor contribution. In order to analyse the TDM graphs properly, the fragments of our designed molecules have been generated i.e. A (end-capped acceptor groups) and D (donor core unit) as shown in Fig. 5. TDM graphs of reference molecule R and designed molecules are depicted in Fig. 5. In reference molecule R and designed molecules, the electron density resides on the donor unit and is distributed efficiently towards the acceptor units through the bridge unit. The charge transfers and exciton dissociation vary inversely with the interaction coefficient. The designed molecule M2 exhibits better charge distribution hence, gives better results by the reference molecule R.
 |
| | Fig. 5 TDM plots of reference molecule R and designed donor molecules (M1–M5) at the same level of theory. | |
3.9 Exciton binding energy (Eb)
The exciton binding energy, which is the energy required to separate an electron hole pair into free charge carriers, plays a crucial role in determining the performance of photovoltaic materials. When the exciton binding energy is reduced, the probability of exciton dissociation increases, leading to a higher generation of free charge carriers. This can improve the Jsc and overall PCE of the material. Conversely, if the exciton binding energy is high, excitons are less likely to dissociate, which can result in significant recombination losses, thereby reducing the device's efficiency. The dissociation of exciton potential can be assessed by exciton binding energy denoted by Eb.47 Both electron and hole pairs have a direct relation with Eb. For that reason, it is the superior method to study the photophysical and optoelectronic characteristics of designed molecules.32,48 The output of a device will be good if the Eb is lower. The lower Eb will consequently make the coulombic interactions lower as it has a direct relation and will give higher charge dissociation because charge dissociation is inversely related to the binding energy. Eb is calculated using the equation below49where, Eg is the energy bandgap, and Eopt is the minimum energy required for the first transition. Table S3† displays the theoretically computed Eb of reference molecule R and each of the proposed molecules.
We observed that molecule M1 has a slightly lower binding energy compared to the reference molecule R, suggesting that the exciton in M1 is less tightly bound. This could facilitate better charge separation upon excitation, potentially enhancing device efficiency by increasing the generation of free charge carriers. Molecule M2 exhibits the lowest binding energy among the molecules studied, indicating the weakest exciton binding. This characteristic is generally favorable for photovoltaic applications, as it promotes the efficient dissociation of excitons into free carriers, which could significantly boost device performance. On the other hand, molecule M4 has the highest binding energy (0.44 eV), indicating very strong exciton binding. While this could lead to lower photovoltaic efficiency due to reduced free carrier generation, it might be advantageous in systems where controlled exciton diffusion is required before charge separation. Overall, the variation in exciton binding energy across the molecules demonstrates its significant impact on their photovoltaic performance, with lower binding energies generally promoting better charge separation and higher efficiency. As molecule M2 has the smallest Eb value out of the five molecules that were designed; hence, it improves charge separation more than reference molecule R and the other molecules. The increasing Eb order is as M2 < M1 < M5 < M3 = R < M4.
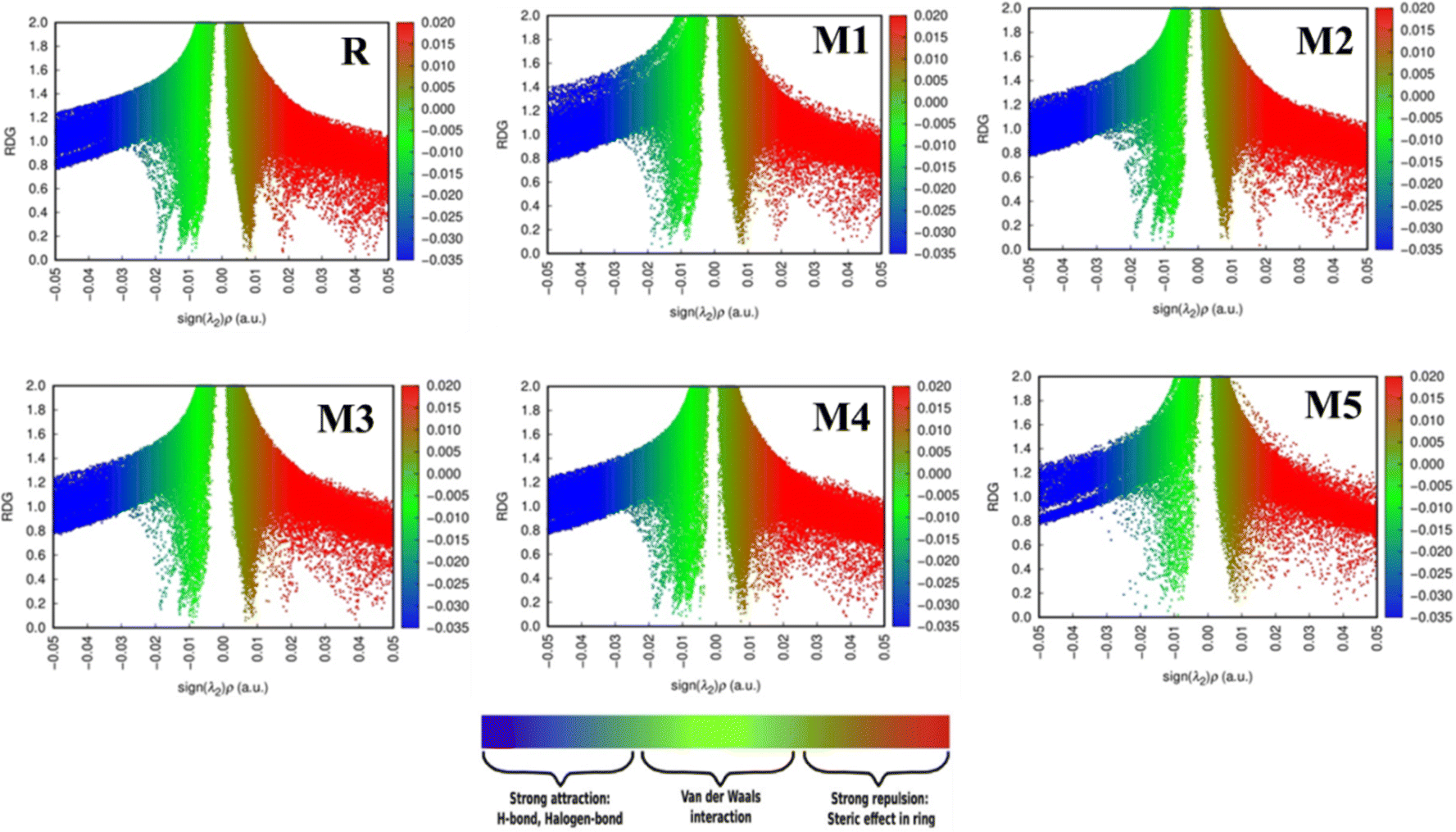
3.10 Non-covalent interaction (NCI)
Covalent bonds are used to examine the disposition of all bonded atoms inside the primary structure of a molecule. On the other hand, NCI like hydrogen bonds, aerogen bonds, chalcogen bonds, π–π and n–π* stacking, hydrophobic interactions, and π anion and cation are used to regulate aggregation, conformation, stabilisation, tertiary structure, and quaternary structures.50,51 Non-covalent interactions are affected by the local environment created by coordinated atoms. These interactions help in maintaining the structure and functioning of molecules.52,53 Steric hindrance, hydrogen bonds (HBs), and van der Waals interactions (vdW) are examined in the NCI plot.54 Kinetic energy and electronic densities are used for analysing NCI. NCI plots rely on electron density (ρ) and reduced density gradient (s).55,56 ρ, and s combine to give an estimation of bonding regions. Low ρ and low s give NCI. Interaction can be repulsive or attractive according to the sign of Hessian eigenvalue (λ2). This distinguishes steric hindrance or non-bonding interactions (λ2 > 0) from bonding interactions. Cut-off values are necessary to obtain a 3D isosurfaces plot. Reduced-gradient Iso-value is referred to as the first cut-off.57 Mapping ρ against s-isosurfaces results in two cut-off values referred to as ρ− cut and ρ+ cut. These cut-offs are required for setting the scale of colours. The blue scheme represents ρ− cut (attractive) while the red-green-blue scale combined with red denotes ρ+ cut (repulsive) as shown in Fig. 6. Electron density itself can differentiate hydrogen bonds from other intermolecular interactions. The peak of the hydrogen bond is observed at approximately ρ ≈ −0.05.57 It appears as a blue localised NCI domain. Peaks appearing within the ρ− region indicate weaker interactions and are depicted in green colour. These interactions lie in the centre of the NCI plot. Highly dense localised blue region of hydrogen bond shows strong intermolecular interaction as compared with wide regions of other weaker interactions.58 By default, RDG isosurfaces are kept at 0.5. A high-quality grid (0.016 Bohr) is used because the number of atoms in designed molecules is large and is necessary to obtain satisfactory graphical quality. In molecule R, a dense blue colour region indicates a hydrogen bond while a dense green region at −0.01 au indicates vdW interactions as depicted in Fig. 6. These interactions predominate over the scattered steric clashes which appear between 0.02 and 0.05 au. In M1, the less dense blue region shows slightly weak hydrogen bonds. Red-coloured steric clashes are much lesser when compared with the reference molecule R. Also, less dense green region indicates scattered vdW interactions. In M2, both steric clashes and vdW interactions are less dense where hydrogen bond predominates. M3 shows a similar trend to R in all three regions. There is a slight difference in the density of green region in M3. M4 shows a solid dense blue region indicating stronger hydrogen bonds. Regions of steric hindrance and vdW interactions are also scattered more. M5 shows a less dense blue region of hydrogen bonds. A less dense green region shows scattered vdW interactions. These interactions do not show a clear peak of vdW interactions. Steric clashes are also scattered and do not show clear peaks. If we compare these regions among reference molecules and all other designed molecules, M1 and M5 do not show dense blue regions of hydrogen bonds. There are many scattered areas of green region showing scattered vdW interactions. Reference molecule R and M4 show a more similar region of steric clashes lying between 0.01 and 0.05 au. Reference molecule R and M3 show a similar region of vdW interactions. Hydrogen bonds in blue region dominate over all other interactions but this approach is not well defined in molecule M5 which shows much scattered regions. Reference molecule R, M1, M2, M3 and M4 show vdW interactions in the range of −0.01 to 0.02 au. There is no clear spike of these interactions in M5. In M1 and M5 more scattered vdW interactions are present. Fig. 6 shows all these interactions in reference molecule R and all designed molecules.
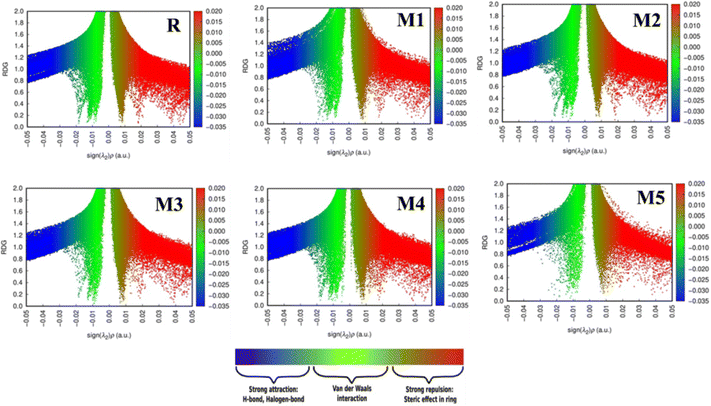 |
| | Fig. 6 Non-covalent interactions in reference molecule R and all designed molecules (M1–M5). | |
3.11 Electron density differences maps (EDDMs)
Essential for understanding the stability of molecules, the EDDM quantifies the electronic charge transfer. Disparities in the accumulation and decline in charge density in different regions may explain why the studied molecules exhibit different levels of intermolecular charge transfer (ICT). Reference molecule R and proposed molecules 2D EDD maps are highlighted in Fig. 7. The molecules under study have holes only and electrons at their acceptor and donor sites, respectively. Consequently, we used dichloromethane as a solvent to simulate EDD maps of the investigated compounds at the MPW1PW91/6-31G(d,p) at a theoretical level. The cubegen and cubeman tools from the G09 package were used for this analysis. Whereas different colours (like cyan-blue and purple) appear on EDD maps, which represent different electron densities. The purple area represents a loss in electron density resulting from charge transfer, whereas the cyan-blue area represents an increase in electron density. The purple area acquires its density from the cyan-blue zone. All of the EDD map plots showed that there was constant ICT. All of these points to the same ICT via bridges between the donor core in the middle and the acceptor fragment at the end as the likely source of the red shift. So, the studied compounds might be used as efficient acceptor materials in organic photovoltaic cells, leading to better efficiency.
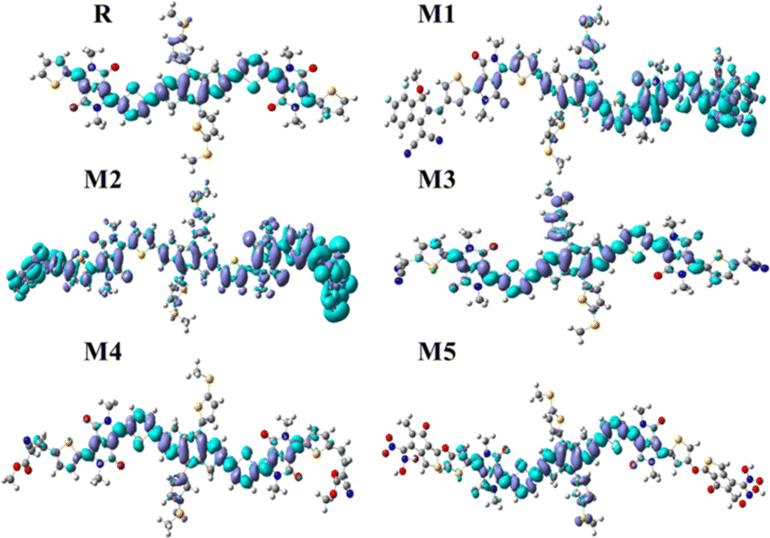 |
| | Fig. 7 Electron density differences maps of reference molecule R and designed donor molecules (M1–M5). All maps are constructed using the MPW1PW91/6-31G(d,p) level of theory at A–π–D–π–A combination in dichloromethane solvent (purple = loose electronic cloud, cyan-blue = gain electronic cloud). | |
3.12 Fill factor (FF)
The FF plays a significant role in influencing the power conversion efficiency of an organic solar cell. FF is calculated by the following formula:59| |
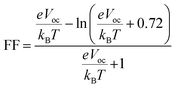 | (5) |
where, Voc, kB, T, and e represent the open-circuit voltage, Boltzmann constant, the temperature at 300 K, and the elementary charge of a molecule respectively. Table 4 shows the calculated FF of all molecules. The calculated FF value of the reference molecule R is 88.66%. Furthermore, the estimated FF value of designed molecules ranges from 88.82% to 89.42%. It describes that the designed molecules exhibit a higher FF value than the reference molecule R. The planarity and rigidity of the molecular structures significantly influence π–π stacking interactions, which in turn affect charge mobility. Molecules M1 and M3, which show the highest FF values benefit from enhanced π–π stacking due to their more planar and rigid structures. These characteristics reduce charge recombination and allow for more efficient charge transport, leading to higher FF values. In contrast, molecules with less optimized stacking, like R, display slightly lower FF due to less efficient charge mobility. Higher FF values indicate the efficacy of newly designed molecules assembled as future solar devices. Hence, the FF increasing pattern is as follows an increasing order of R < M2 = M4 < M5 < M1 < M3. Among the designed molecules, M1 exhibits the second highest FF and PCE values which make it a suitable candidate to be used as a potential future solar cell device.
Table 4 Theoretically calculated value of open circuit voltage (Voc), fill factor (FF), and power conversion efficiency (PCE) of reference molecule R and designed molecules (M1–M5)
| Molecules |
Voc (V) |
FF (%) |
PCE (%) |
| R |
1.05 |
88.66 |
19.82 |
| M1 |
1.14 |
89.33 |
21.69 |
| M2 |
1.07 |
88.82 |
20.24 |
| M3 |
1.15 |
89.42 |
21.90 |
| M4 |
1.07 |
88.82 |
20.24 |
| M5 |
1.12 |
89.18 |
21.27 |
3.13 Power conversion efficiency (PCE)
The PCE of a solar cell is measured by adding up all of its output results to see how well the material is made for practical purposes. The equation is provided below:| |
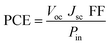 | (6) |
where Pin stands for the power of the incident rays. The JSC is an experimental value of 21.3 mA cm−2 obtained from previous research.60 Fig. S4† shows the graphical representation of the PCE value of all investigated molecules concerning the reference molecule. The increase in PCE value results from alterations at the terminal side, where various electron withdrawing groups are attached. These groups help disperse the electronic cloud between the donor core and the acceptor part of the molecule. The molecular structures that enhance Voc and FF also contribute to higher PCE. Molecules like M1 and M3, with their extended conjugation and optimized electronic properties, show higher PCEs due to their ability to better harness light, efficiently separate charges, and transport them with minimal losses. The structural modifications in these molecules enhance their ability to absorb a broader spectrum of light and facilitate charge transport, leading to improved overall efficiency.
The M3 molecule has the highest estimated PCE value of 21.90%, followed by M1 with a value of 21.69%. Therefore, by making appropriate alterations to the reference molecule at the end, the new molecules exhibit the highest PCE, FF, and Voc values compared to the reference molecule. These new molecules can be utilised in future solar devices to achieve the highest PCE value through practical application, as determined by experimentalists.
4. Conclusions
In summary, our study presents a comprehensive study into the molecular design and characterisation of A–π–D–π–A end-capped structured molecules for improving the photovoltaic efficiency of organic solar cells (OSCs). We have successfully designed five BDTS-2DPP-based donor molecules and employed theoretical approaches, including DFT and TD-DFT methods, to evaluate their performance in OSCs. Our study demonstrates that molecule M1 exhibits exceptional photovoltaic characteristics, including maximum absorption at 846 nm, and highest λe of 0.18 eV, with a narrow energy band gap of 1.81 eV. Furthermore, our findings reveal that M1 displays a high dipole moment in both the gas phase (10.62 D) and solvent phase (13.62 D) and efficient charge transfer capability between the HOMO and LUMO states with efficiency of up to 97%, making it a promising candidate for enhancing charge separation and transport in OSCs. Molecule M3 also shows potential with its high Voc of 1.15 V, highest PCE value (21.90%) and highest fill factor (FF) value (89.42%), indicating its potential for efficient charge collection. The minimum excitation binding energy determined by molecule M2 (Eb = 0.30 eV) suggests a higher dissociation occurring during excitation. The high electron density seen in the newly formed M2 molecule when dissolved in solvent is likely a result of its strong dissociation in a solvation environment. The comparison of the designed compounds with the reference molecule R reveals that all newly designed molecules (M1–M5) exhibit a reduced band gap, are red-shifted towards longer wavelengths relative to reference molecule R, and demonstrate remarkable charge motilities. Overall, our findings contribute to the advancement of organic photovoltaic and offer potential avenues of device engineering for the development of next-generation OSCs with improved efficiency and stability.
Data availability
Data for this article obtained from density functional theory (DFT) and time-dependent density functional theory (TD-DFT) methods, including geometry optimisation, density of states (DOS) analysis, natural transition orbitals (NTO), transition density matrix (TDM) calculations, and total energy computations, are available at Mendeley Data at https://doi.org/10.17632/64tf9v78n3.2.
Author contributions
S. Chang and F. Abbas conceived and directed the project; S. M. K. A. Naqvi, M. K. Shehzad, S. Bibi, and Y. Zhu prepared the models, carried out the simulations and prepared the figures; S. M. K. A. Naqvi, N. Alhokbany, Zahid Nazir, H. Long, R. B. Vasiliev and S. Chang analysed the data, interpreted the results and wrote the manuscript.
Conflicts of interest
No potential conflicts of interest have been disclosed by the authors.
Acknowledgements
This work is supported by the 2022 Shenzhen Fundamental Research Program (20220818150511001), the National Natural Science Foundation of China (22173009, 22211530439), and the National Key Research and Development Program for Young Scientists (2021YFB3601700). The authors extend their sincere appreciation to Researchers Supporting Project number (RSP2024R253), King Saud University, Riyadh, Saudi Arabia.
References
- J. Wang and W. Azam, Natural resource scarcity, fossil fuel energy consumption, and total greenhouse gas emissions in top emitting countries, Geosci. Front., 2024, 15(2), 101757 CrossRef CAS.
- S. Mouassa, A. Alateeq, A. Alassaf, R. Bayindir, I. Alsaleh and F. Jurado, Optimal power flow analysis with renewable energy resource uncertainty using dwarf mongoose optimizer: case of ADRAR Isolated electrical network, IEEE Access, 2024, 12, 10202–10218 Search PubMed.
- L. Chen, Y. Zhang, Z. Chen, Y. Dong, Y. Jiang, J. Hua, Y. Liu, A. I. Osman, M. Farghali and L. Huang, et al., Biomaterials technology and policies in the building sector: a review, Environ. Chem. Lett., 2024, 22(2), 1–36 Search PubMed.
- M. U. Khan, F. Shafiq, M. R. S. A. Janjua, M. Khalid, J. Yaqoob, M. Arshad, S. M. Alshehri and R. A. Khan, Predicting benzodithiophene based donor materials with enhanced 19.09% PCE, Open-circuit voltage and optoelectronic attributes for solar cell applications: photochemical insights from DFT, J. Photochem. Photobiol., A, 2024, 446, 115115 CrossRef.
- B. Zhang, G. Lei, S. You, W. Zhao and H. Liu, DFT Investigation of structural stability, optical properties, and PCE for all-inorganic Csx(Pb/Sn)yXz halide perovskites, Inorg. Chem., 2024, 63(7), 3303–3316 CrossRef CAS PubMed.
- M. Kandasamy, S. Suresh, M. D. Kumar, S. Murugesan, M. Selvaraj, K. P. Ananth, N. Kumar, A. Husain, N. Pugazhenthiran and P. Sathishkumar, Combined experimental and DFT investigation on photovoltaic performance of low-cost metal-free organic dye-sensitized solar cells, Inorg. Chem. Commun., 2024, 164, 112406 CrossRef CAS.
- K. Liu, Y. Jiang, G. Ran, F. Liu, W. Zhang and X. Zhu, 19.7% efficiency binary organic solar cells achieved by selective core fluorination of nonfullerene electron acceptors, Joule, 2024, 8(3), 835–851 CrossRef CAS.
- Z. Chen, W. Song, K. Yu, J. Ge, J. Zhang, L. Xie, R. Peng and Z. Ge, Small-molecular donor guest achieves rigid 18.5% and flexible 15.9% efficiency organic photovoltaic via fine-tuning microstructure morphology, Joule, 2021, 5(9), 2395–2407 CrossRef CAS.
- H. Yao and J. Hou, Recent advances in single-junction organic solar cells, Angew. Chem., Int. Ed., 2022, 61(37), e202209021 CrossRef CAS PubMed.
- F. G. Brunetti, R. Kumar and F. Wudl, Organic electronics from perylene to organic photovoltaics: painting a brief history with a broad brush, J. Mater. Chem., 2010, 20(15), 2934–2948 RSC.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Bulk heterojunction solar cells: morphology and performance relationships, Chem. Rev., 2014, 114(14), 7006–7043 CrossRef CAS PubMed.
- J. Yi, G. Zhang, H. Yu and H. Yan, Advantages, challenges and molecular design of different material types used in organic solar cells, Nat. Rev. Mater., 2024, 9(1), 46–62 CrossRef CAS.
- J. Xie, W. Lin, D. Wang, Z. H. Lu, K. Zheng and Z. Liang, Sequentially N-doped acceptor primer layer facilitates electron collection of inverted non-fullerene organic solar cells, Adv. Funct. Mater., 2024, 34(9), 2309511 CrossRef CAS.
- X. Zhang, X. Gu and H. Huang, Low-cost nonfused-ring electron acceptors enabled by noncovalent conformational locks, Acc. Chem. Res., 2024, 57(6), 981–991 CrossRef CAS PubMed.
- A. Karki, A. J. Gillett, R. H. Friend and T. Nguyen, The path to 20% power conversion efficiencies in nonfullerene acceptor organic solar cells, Adv. Energy Mater., 2021, 11(15), 2003441 CrossRef CAS.
- H. Wang, C. Cao, H. Chen, H. Lai, C. Ke, Y. Zhu, H. Li and F. He, Oligomeric acceptor: a “two-in-one” strategy to bridge small molecules and polymers for stable solar devices, Angew. Chem., Int Ed., 2022, 134(23), e202201844 CrossRef.
- A. F. González, A. P. Mariman, R. A. Hauyon, D. Pavez-Lizana, C. Saldías, E. Schott, X. Zarate, L. Garcia, C. M. González-Henríquez and I. A. Jessop, et al., Thiophene- and bithiophene-based π-conjugated schiff base oligomers containing binaphthalene moieties in the backbone. properties and computational simulations, Polym. Chem., 2024, 15(7), 639–651 RSC.
- C. An and J. Hou, Benzo[1,2-b:4,5-B′] dithiophene-based conjugated polymers for highly efficient organic photovoltaics, Acc. Mater. Res., 2022, 3(5), 540–551 CrossRef CAS.
- Z. Chen, Z. Liu, X. Yang, Z. Sun, C. Zhou, L. Chen, Z. Wang, W. Li, S. Zhang and J. Chen, et al., The dopant-free hole transport material based on benzo[1,2-b:4,5-b′]dithiophene with high efficiency in inverted perovskite solar cells, Mater Today Chem., 2024, 37, 102026 CrossRef CAS.
- D. Zhou, Y. Wang, S. Yang, J. Quan, J. Deng, J. Wang, Y. Li, Y. Tong, Q. Wang and L. Chen, Recent advances of benzodithiophene-based donor materials for organic solar cells, Small, 2024, 20(8), 2306854 CrossRef CAS PubMed.
- J. Li, Z. Zhang, S. Jiang, M. Kang, D. Wang, X. Li, P. Huang, X. Gao, D. Wang and B. Z. Tang, NIR-II excitable semiconducting polymers with AIE characteristics for fluorescence-photoacoustic imaging-guided synergistic phototherapy, Adv. Funct. Mater., 2024, 2401627 CrossRef CAS.
- A. Naveed, R. A. Khera, U. Azeem, I. Zubair, A. Farhat, A. R. Ayub and J. Iqbal, Tuning the optoelectronic properties of benzodithiophene based donor materials and their photovoltaic applications, Mater. Sci. Semicond. Process., 2022, 137, 106150 CrossRef CAS.
- S. M. K. A. Naqvi, Y. Zhu and S. Chang, Manipulating interfacial performance of TPBi–FAPbI3 perovskite interfaces based on tpbi orientations: a theoretical perspective, J. Phys. Chem. C, 2023, 127(46), 22752–22759 CrossRef CAS.
- F. Abbas, U. Ali, A. Tallat, H. M. R. Ahmad, S. A. Siddique, Z. Zeb and M. B. A. Siddique, An optoelectronic study to design better benzodithiophene (BDT) donor unit based non-fullerene organic solar cells (OSCs): the DFT approaches, Chem. Pap., 2022, 76(8), 4977–4987 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41(2), 157–167 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393(1–3), 51–57 CrossRef CAS.
- Y. Zhao, J. Pu, B. J. Lynch and D. G. Truhlar, Tests of second-generation and third-generation density functionals for thermochemical kinetics, Phys. Chem. Chem. Phys., 2004, 6(4), 673 RSC.
- H. Fang and Y. Kim, Excited-state tautomerization in the 7-azaindole-(H2O)n (n = 1 and 2) complexes in the gas phase and in solution: a theoretical study, J. Chem. Theory Comput., 2011, 7(3), 642–657 CrossRef CAS PubMed.
- J. Antony and S. Grimme, Density functional theory including dispersion corrections for intermolecular interactions in a large benchmark set of biologically relevant molecules, Phys. Chem. Chem. Phys., 2006, 8(45), 5287 RSC.
- J. Wang, K. Shi, Y. Suo, Y. Lin, G. Yu and X. Zhan, Monodisperse macromolecules based on benzodithiophene and diketopyrrolopyrrole with strong NIR absorption and high mobility, J. Mater. Chem. C, 2016, 4(17), 3781–3791 RSC.
- N. M. O’boyle, A. L. Tenderholt and K. M. Cclib, A library for package-independent computational chemistry algorithms, J. Comput. Chem., 2008, 29(5), 839–845 CrossRef PubMed.
- J. J. Intemann, K. Yao, F. Ding, Y. Xu, X. Xin, X. Li and A. K. Jen, Enhanced performance of organic solar cells with increased end group dipole moment in indacenodithieno[3,2-b]thiophene-based molecules, Adv. Funct. Mater., 2015, 25(30), 4889–4897 CrossRef CAS.
- S. Y. Leblebici, T. L. Chen, P. Olalde-Velasco, W. Yang and B. Ma, Reducing exciton binding energy by increasing thin film permittivity: an effective approach to enhance exciton separation efficiency in organic solar cells, ACS Appl. Mater. Interfaces, 2013, 5(20), 10105–10110 CrossRef CAS PubMed.
- Y. Zhong, A. Tada, S. Izawa, K. Hashimoto and K. Tajima, Enhancement of VOC without loss of JSC in organic solar cells by modification of donor/acceptor interfaces, Adv. Energy Mater., 2014, 4(5), 1301332 CrossRef.
- M. Ans, M. Paramasivam, K. Ayub, R. Ludwig, M. Zahid, X. Xiao and J. Iqbal, Designing alkoxy-induced based high performance near infrared sensitive small molecule acceptors for organic solar cells, J. Mol. Liq., 2020, 305, 112829 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- C.-P. Hsu, Reorganization energies and spectral densities for electron transfer problems in charge transport materials, Phys. Chem. Chem. Phys., 2020, 22(38), 21630–21641 RSC.
- V. Vehmanen, N. V. Tkachenko, H. Imahori, S. Fukuzumi and H. Lemmetyinen, Charge-transfer emission of compact porphyrin–fullerene dyad analyzed by marcus theory of electron-transfer, Spectrochim. Acta Part A, 2001, 57(11), 2229–2244 CrossRef CAS PubMed.
- J. M. Mayer, Understanding hydrogen atom transfer: from bond strengths to marcus theory, Acc. Chem. Res., 2011, 44(1), 36–46 CrossRef CAS PubMed.
- F. Abbas, U. Ali, H. M. R. Ahmad, A. Tallat, A. Shehzad, Z. Zeb, I. Hussain, A. Saeed and M. Tariq, Role of iodo-substituted subphthalocyanine (subpcs) π-conjugated aromatic N-fused Di-iminoisonidole units on the performance of non-fullerene small organic solar cells, Comput. Theor. Chem., 2022, 1207, 113508 CrossRef CAS.
- L. Eberson, Electron-transfer reactions in organic chemistry, Adv. Phys. Org. Chem., 1982, 18, 79–185 CrossRef CAS.
- R. Lin, H. Zhou, X. Xu and X. Ouyang, Improved charge transport based on donor-acceptor type solid additive with large dipole moment for efficient organic solar cells, Dye Pig., 2024, 224, 111980 CrossRef CAS.
- H. Wang, Y. Wang, X. Cao, L. Zhang, M. Feng and G. Lan, Simulation of electronic density of states and optical properties of PbB4O7 by first-principles DFT method, Phys. Status Solidi, 2009, 246(2), 437–443 CrossRef CAS.
- F. B. Kooistra, J. Knol, F. Kastenberg, L. M. Popescu, W. J. H. Verhees, J. M. Kroon and J. C. Hummelen, Increasing the open circuit voltage of bulk-heterojunction solar cells by raising the LUMO level of the acceptor, Org. Lett., 2007, 9(4), 551–554 CrossRef CAS PubMed.
- S. Zahid, A. Rasool, M. Ans, M. Salim Akhter, J. Iqbal, M. S. Al-Buriahi, S. Alomairy and Z. A. Alrowaili, Environmentally compatible and highly improved hole transport materials (HTMs) based on benzotrithiophene (BTT) skeleton for perovskite as well as narrow bandgap donors for organic solar cells, Sol. Energy, 2022, 231, 793–808 CrossRef CAS.
- J. Rukhsar, M. Waqas, M. S. Akhter, M. Shaban, S. I. Al-Saeedi, M. S. Mahr, T. H. A. Hasanin, M. A. A. Ibrahim, N. S. Alatawi and R. A. Khera, A DFT approach toward designing selenophene-based unfused small molecule acceptors by end-capped modification for improving the photovoltaic performance of organic solar cells, J. Phys. Org. Chem., 2024, 37(3), e4587 CrossRef CAS.
- S. Kashani, J. J. Rech, T. Liu, K. Baustert, A. Ghaffari, I. Angunawela, Y. Xiong, A. Dinku, W. You and K. Graham, Exciton binding energy in organic polymers: experimental considerations and tuning prospects, Adv. Energy Mater., 2024, 14(6), 2302837 CrossRef CAS.
- M. Knupfer, Exciton binding energies in organic semiconductors, Appl. Phys. A, 2003, 77(5), 623–626 CrossRef CAS.
- N. Qiu, H. Zhang, X. Wan, C. Li, X. Ke, H. Feng, B. Kan, H. Zhang, Q. Zhang and Y. Lu, et al., A new nonfullerene electron acceptor with a ladder type backbone for high-performance organic solar cells, Adv. Mater., 2017, 29(6), 1604964 CrossRef PubMed.
- M. Giese, M. Albrecht and K. Rissanen, Anion–π interactions with fluoroarenes, Chem. Rev., 2015, 115(16), 8867–8895 CrossRef CAS PubMed.
- S. K. Singh and A. Das, The N→ Π* interaction: a rapidly emerging non-covalent interaction, Phys. Chem. Chem. Phys., 2015, 17(15), 9596–9612 RSC.
- S. A. Cook, E. A. Hill and A. S. Borovik, Lessons from nature: a bio-inspired approach to molecular design, Biochemistry, 2015, 54(27), 4167–4180 CrossRef CAS PubMed.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Insights into metalloenzyme microenvironments: biomimetic metal complexes with a functional second coordination sphere, Chem. Soc. Rev., 2013, 42(21), 8360 RSC.
- R. A. Klein, Modified van der waals atomic radii for hydrogen bonding based on electron density topology, Chem. Phys. Lett., 2006, 425(1–3), 128–133 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132(18), 6498–6506 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: a program for plotting noncovalent interaction regions, J. Chem. Theory Comput., 2011, 7(3), 625–632 CrossRef PubMed.
- J. Contreras-García, W. Yang and E. R. Johnson, Analysis of hydrogen-bond interaction potentials from the electron density: integration of noncovalent interaction regions, J. Phys. Chem. A, 2011, 115(45), 12983–12990 CrossRef PubMed.
- A. M. Pendás, E. Francisco, M. A. Blanco and C. Gatti, Bond paths as privileged exchange channels, Chem.–Eur. J., 2007, 13(33), 9362–9371 CrossRef PubMed.
- E. U. Rashid, J. Iqbal, M. I. Khan, Y. A. El-Badry, K. Ayub and R. A. Khera, Synergistic end-capped engineering on non-fused thiophene ring-based acceptors to enhance the photovoltaic properties of organic solar cells, RSC Adv., 2022, 12(20), 12321–12334 RSC.
- M. Zhang, J. Wang, L. Li, G. Zheng, K. Liu, M. Qin, H. Zhou and X. Zhan, High-mobility P-type organic semiconducting interlayer enhancing efficiency and stability of perovskite solar cells, Adv. Sci., 2017, 4(9), 1700025 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Faheem Abbas
abc,
Faheem Abbas