
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclodextrin metal–organic framework@SiO2 nanocomposites for poorly soluble drug loading and release†
Shuai Liua,
Yuzhu Xiong b and
Fuping Dong*a
b and
Fuping Dong*a
aDepartment of Polymer Materials and Engineering, Guizhou University, China. E-mail: fpdong@gzu.edu.cn
bCollege of Materials and Metallurgy, Guizhou University, China
First published on 8th October 2024
Abstract
The development of non-toxic drug carrier materials with high loading capacity, sustained release properties, stability, and biocompatibility holds significant medical value and potential for loading and releasing poorly soluble drugs. In this study, we synthesized a biocompatible, non-toxic, environmentally friendly CD-MOF porous material with high specific surface area and tunable structure. By incorporating SiO2 to enhance the stability of MOF materials, the synthesized CD-MOF@SiO2 material shows promising applications in drug delivery. The obtained CD-MOF@SiO2 nanocomposite was utilized as a carrier for the poorly soluble drug, folic acid. Characterization of the drug-loaded composite before and after drug loading was performed using scanning electron microscopy, energy dispersive X-ray spectroscopy, Fourier transform infrared spectroscopy, N2 adsorption–desorption, and X-ray diffraction analyses, showing improved stability as indicated by thermogravimetric analysis and derivative thermogravimetry data. UV spectrophotometry was used to investigate the loading and sustained release of folic acid under different conditions in PBS buffer, demonstrating that the well-structured CD-MOF@SiO2 material exhibits high drug loading and controllable release properties. The CD-MOF@SiO2 achieved a high drug loading efficiency (166.78%) and encapsulation rate (83.39%) for folic acid, leading to a significant increase in apparent solubility from 1.6 μg mL−1 in its free form to 21.74 mg mL−1, a 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 588-fold expansion. This work presents a novel, efficient, and highly valuable approach for the development of carrier materials for loading and releasing poorly soluble drugs.
588-fold expansion. This work presents a novel, efficient, and highly valuable approach for the development of carrier materials for loading and releasing poorly soluble drugs.
1 Introduction
Drug therapy has long been a pivotal component in the treatment of diseases. However, many clinically valuable and highly active drugs are inherently hydrophobic, leading to challenges with poor water solubility. This solubility issue not only hinders the development of drug formulations but also poses challenges when poorly soluble drugs are administered orally. These drugs often require higher doses to compensate for low bioavailability, resulting in poor patient compliance and increased risk of drug toxicity and side effects.1 Therefore, the development of drug delivery systems that can enhance the water solubility of highly potent drugs to improve their bioavailability is essential for advancing modern drug therapy.Research on the efficient encapsulation and delivery of poorly water-soluble drugs with low toxicity has led to the development of various delivery systems, such as solid lipid nanoparticles, liposomes, polymer micelles and microemulsions.2 However, the effectiveness of these delivery systems still needs improvement. For example, in solid lipid nanoparticles, crystalline lipids can reduce drug loading capacity.3 Liposomes face challenges of physical and chemical instability, hindering their clinical utility.4 Polymer micelles raise concerns about clinical safety and drug loading capacity.5 Microemulsions are limited by potential toxicity of surfactants and co-surfactants.6 Metal–organic frameworks (MOFs) are a class of advanced porous materials with a one, two or three-dimensional periodic grid structure formed by coordination bonding between metal ions or clusters and polydentate organic ligands.7 They possess ultra-high surface area, high crystallinity, high porosity, tunable porosity, low density,8,9 diverse composition structures, open metal sites, and ease of chemical modification.10 MOFs have garnered widespread attention in areas such as substance storage and separation, catalysis, sensing, energy, biomedicine, and drug encapsulation and release.11–18 In the field of biomedicine, MOFs have been extensively utilized as carriers for various small molecule drugs such as anti-tumor drugs (Zheng, H. et al. have reported a DOX drug system encapsulated by ZIF-8 nanoparticles),19 anticancer drugs (Lei, B. et al. investigated a responsive MOF nanocarrier for loading a poorly water-soluble anticancer drug: curcumin),20 anti-inflammatory drugs (Sara Rojas et al. have synthesized a carrier loaded with ibuprofen anti-inflammatory drugs using a series of MOF materials),21 as well as large molecule drugs such as proteins, nucleic acids, and polysaccharides.18,22–24 However, due to the toxicity of metal ions or organic ligands and weak coordination bonding, MOFs often exhibit biological toxicity, low biocompatibility, poor mechanical properties, and low stability, leading to rapid degradation in biological systems. This degradation compromises the efficacy of drug encapsulation and release, significantly limiting the widespread application of MOFs in this field.25,26 Therefore, the focus of research in this area has shifted towards developing MOFs with ultra-high surface area, high porosity, good stability and mechanical properties, as well as demonstrating excellent biocompatibility and low toxicity during drug delivery.
Cyclodextrin (CD) is a cyclic oligosaccharide composed of D-(+)-glucose units derived from starch degradation. Different types of cyclodextrin molecules, such as α-CD (6 glucose units), β-CD (7 glucose units), and γ-CD (8 glucose units), have practical significance due to variations in cavity size and glucose unit numbers.27 CD, with its truncated cone structure and unique hydrophobic inner cavity and hydrophilic outer surface, is an eco-friendly and non-toxic drug carrier capable of enhancing the solubility and bioavailability of poorly soluble drugs.28 When cyclodextrin is used directly in biological applications, it suffers from low drug loading capacity and poor stability. Therefore, it is necessary to improve its stability and loading capacity by means of compound modification. While a hybrid system formed with nanoparticles on the basis of cyclodextrin is used for drug delivery, some nanoparticles may have adverse side effects on human tissues due to the problem of toxicity or poor biocompatibility.29 Cyclodextrin-micelle hybrid systems used in biomedical applications may create problems with low drug loading capacity or poor stability.30 In addition to the above examples, modification of cyclodextrin can also be achieved through coordination with various metal ions,31 with low toxicity metals like Ca, Fe, K, and Na being biologically acceptable based on LD50 toxicity data assessment.32 In 2010, a food-grade γ-CD metal–organic framework material was developed using non-toxic γ-CD and alkali metals.33 This three-dimensional porous MOF structure exhibited biocompatibility, low toxicity, and unique MOF advantages, opening up opportunities for drug delivery applications. Researchers have successfully utilized CD-MOFs for delivering poorly soluble drugs, such as Valsartan (VAL)34 and azilsartan (AZL),35 significantly increasing drug loading and apparent solubility compared to traditional CD complexes. Various methods, including ultrasound-assisted crystallization and the “ship in bottle” strategy, have been employed to prepare efficient CD-MOFs for enhancing the solubility of challenging compounds like natural triterpene glycoside and folic acid.36
Although these studies have shown excellent performance in terms of biocompatibility, toxicity, enhancing the loading of poorly soluble drugs, and improving apparent solubility, there has been little focus on the stability of CD-MOF. In addressing the low stability and mechanical properties of MOF materials, the combination of MOFs with functional materials is crucial. Quantum dots, metal nanoparticles/nanorods, graphene, porous silica nanospheres, and magnetic nanoparticles have been used to composite with MOFs to address their poor stability.37–41 Silicon-based nanomaterials possess good mechanical properties, stability, biocompatibility, and degradability, which can support MOF materials and enhance their stability through hydrophobic interactions or covalent bonding.42–44 Trang Thi Thu Nguye et al. effectively modified ZIF-8 nanomaterials by coating them with fluorescent organosilicon to encapsulate curcumin, resulting in composite materials with good bio-interactions that prevent MOF aggregation and enhance stability.45 Our research group also prepared MOF/polysilsesquioxane (PSQ) nanocomposites, where PSQ, known for stability, inertness, and functionality, successfully encapsulated ibuprofen with high efficiency.46
In this study, we synthesized CD-MOF@SiO2 composite by combining CD-MOF with silica material and employing folic acid as a poorly soluble drug model in water. Subsequently, we investigated the drug loading capacity of CD-MOF@SiO2 under different conditions, as well as the factors influencing the drug solubility and the release of folic acid from FA/CD-MOF@SiO2 in PBS buffer. CD-MOF@SiO2 exhibited high drug loading efficiency and encapsulation efficiency of 166.78% and 83.39% for folic acid, respectively, demonstrating effective folic acid loading with a solubility enhancement to 21.74 mg mL−1. The novelty of this study is in the surface composite of SiO2 as armor, which enhanced the stability of CD-MOF and significantly increased the apparent solubility of FA. This research introduces a novel, efficient, and valuable method for designing carriers for poorly soluble drugs. These results indicate the potential applicability of CD-MOF@SiO2 as a nanocarrier for poorly soluble drugs, showcasing promise for the loading and release of folic acid as a model drug with low solubility.
2 Experimental section
2.1 Materials
All reagents and solvents were obtained from commercial suppliers and used as received without additional purification. γ-Cyclodextrin (98%), KOH, ammonia solution (25–28%), CTAB cetyltrimethylammonium bromide (99%), tetraethyl orthosilicate (99%), sodium chloride (99.5%), potassium chloride (99.5%), potassium dihydrogen phosphate (99.5%), and disodium hydrogen phosphate (99%) were purchased from Aladdin Reagents Co. Ltd. Methanol (AR) was obtained from Tianjin Kemiou Chemical Reagent Co., Ltd; isopropanol (AR) and anhydrous ethanol (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. Folic acid (98%) was supplied by Bide Pharmatech Co., Ltd. Deionized water was used in all experimental procedures.2.2 Methods
:CD-MOF@SiO2, loading time and loading temperature. Typically, 0.1 g of FA and 0.2 g of CD-MOF@SiO2 were added to 30 mL of anhydrous ethanol, dispersed thoroughly by ultrasound, and stirred at 60 °C for 6 hours. The mixture was centrifuged at 5000 rpm for 5 minutes, washed five times with anhydrous ethanol, and the precipitate was dried in a 40 °C oven for 12 hours.To determine the apparent solubility of FA in the FA/CD-MOF@SiO2 system, 0.1 g of FA/CD-MOF@SiO2 was thoroughly dispersed in 2 mL of deionized water by ultrasonication. The resulting suspension was then placed on a shaker and incubated at 37 °C for 12 hours. Subsequently, the mixture was centrifuged at 5000 rpm for 5 minutes, and the supernatant was filtered through a 0.22-micron syringe filter. The concentration of FA in the filtrate was determined using UV spectrophotometry, with three measurements taken and averaged.50
For the drug release experiment, 50 mg of FA/CD-MOF@SiO2 composite material was placed in a 5 mL PBS solution and stirred for 30 minutes. Subsequently, the mixture was transferred to a dialysis bag with a molecular weight cut-off of 7000 Da. The dialysis bag was then immersed in 500 mL of PBS buffer solution (pH = 7.4) and maintained at 37 °C with stirring at 300 rpm. 3 mL of the release medium was collected at regular intervals from the 500 mL buffer solution, followed by replacement with 3 mL of fresh PBS solution to maintain a constant release volume in vitro. The absorbance of the extracellular solution at different time points was measured using a UV spectrophotometer to determine the release of FA and construct the drug release profile.
The drug loading capacity of the carriers and the drug sustained release performance of the drug delivery systems are evaluated using drug loading efficiency (DLE), drug encapsulation efficiency (DEE), apparent solubility, and cumulative drug release rate.46
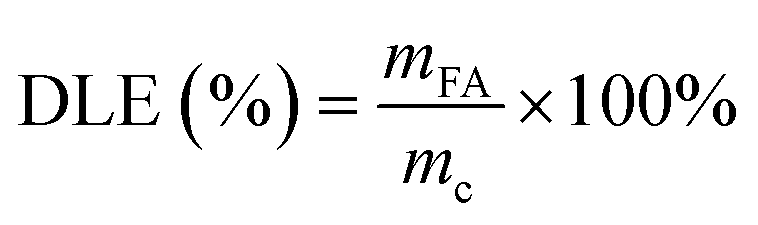 | (1) |
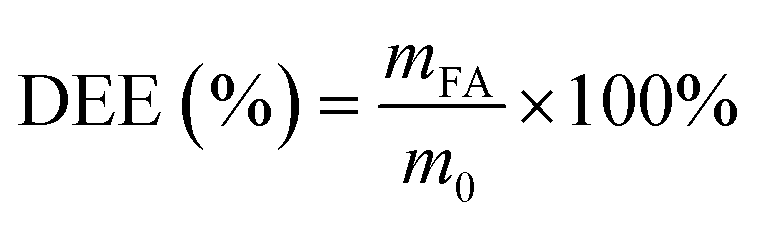 | (2) |
 | (3) |
In eqn (1)–(3), where mc represents the mass of the carrier CD-MOF@SiO2, mg; mFA denotes the mass of FA loaded into CD-MOF@SiO2; m0 is the total mass of FA added; md stands for the mass of the drug FA in the sustained-release system FA/CD-MOF@SiO2; Ve is the volume of the supplemented PBS solution, 3 mL; Ci represents the drug concentration released in the external liquid during the i-th sampling; V0 is the volume of the released external liquid, mL; Cn is the drug concentration released in the external liquid during the (i + 1)-th sampling; and n is the total number of samples taken.
2.3 Characterization
Fourier-transform infrared spectroscopy (FT-IR) analysis was conducted using a Nicolet iS50 FT-IR spectrometer. Powder samples were pressed with KBr and scanned in the wavenumber range of 4000–500 cm−1, with 32 scans. The microstructure of the material was examined using a Zeiss Sigma 300 scanning electron microscope (SEM) after sputter-coating the powder with gold. Elemental analysis was performed using the smart edx spectrometer integrated into the SEM. Thermogravimetric analysis (TG) was carried out using a TG209F1 Libra thermogravimetric analyzer under N2 atmosphere, with a temperature range from room temperature to 800 °C at a heating rate of 10 °C min−1. Prior to analysis, samples were degassed at 50 °C for 5 hours. Adsorption/desorption measurements were conducted using a Micromeritics ASAP2460 instrument under nitrogen conditions. X-ray diffraction (XRD) analysis was performed using a Philips X Pert Powder diffractometer with Cu Kα radiation (λ = 1.5406 Å) and a nickel filter, scanning in the range of 5°–60°(2θ) at a speed of 1° min−1. The absorption of FA in water and PBS buffer was measured using a Shimadzu UV2700 spectrophotometer with a slit width of 0.2, interval of 0.5, and a wavelength range of 200–500 nm.3 Results and discussion
3.1 Morphology and element composition of samples
CD-MOF was formed through the reaction of γ-CD with KOH in an aqueous medium, followed by MeOH vapor diffusion. The methodological difference between the fabrication of um-CD-MOFs and nm-CD-MOFs is the inclusion of a small quantity of methanol as a poor solvent during the fabrication of nm-CD-MOFs. The addition of poor-solvent methanol enhances precursor supersaturation, inducing the burst of nucleation and the generation of numerous nuclei, ultimately leading to particle size reduction.10,51 Fig. 1 depicts scanning electron microscope images of CD-MOF, CD-MOF@SiO2, and FA-loaded CD-MOF@SiO2. In Fig. 1a, nm-CD-MOFs are presented, while Fig. 1b exhibit um-CD-MOFs, and Fig. 1e displays um-CD-MOFs coated with SiO2. Fig. 1c illustrates nm-CD-MOF@SiO2, revealing a smooth surface for CD-MOF without encapsulated SiO2, and a rougher surface after coating with silica. In Fig. 1c, it is evident that the cubic structure of CD-MOF remains intact after SiO2 encapsulation, with a shell formed only on the outer layer. Additionally, energy-dispersive X-ray spectroscopy (EDX) analysis in Fig. 1f confirms the presence of Si elements on CD-MOF, further supporting SiO2 encapsulation. Following the loading of FA, the cubic structure of CD-MOF@SiO2 remained stable, yet displaying a visibly roughened surface. | ||
| Fig. 1 SEM images of (a) nm-CD-MOF, (b) μm-CD-MOF, (c) nm-CD-MOF@ SiO2, (d) FA/nm-CD-MOF@ SiO2, (e) μm-CD-MOF@SiO2, (f) EDX mapping for nm-CD-MOF@ SiO2. | ||
3.2 Chemical composition of samples
Fig. 2 displays the IR spectra of CD-MOF, CD-MOF@SiO2, and the drug-loaded CD-MOF@SiO2 composite. The absorption peaks of nm-γ-CD-MOF and um-γ-CD-MOF are similar to those of γ-CD, indicating that the prepared samples retain the structure of γ-CD. The spectra show a broad peak around 3400 cm−1 corresponding to the –OH stretching vibration of cyclodextrin, peak at around 2926 cm−1 for C–H stretching vibration, and vibrations at 1645 cm−1 and 1420 cm−1 attributed to hydroxyl bending, as well as peaks at 1154 cm−1 and 1022 cm−1 corresponding to C–O stretching vibrations.52 After SiO2 modification, the peak positions based on γ-CD remain unchanged, but the peak intensity decreases, resulting in a smoother curve. The main characteristic peaks of FA include the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1685 cm−1, the CO–NH–CO stretching vibration at 1635 cm−1, methyl bending at 1485 cm−1, and CC skeletal vibrations on the phenyl ring at 1605 cm−1, 1571 cm−1, and 1453 cm−1. The infrared spectrum of FA/CD-MOF@SiO2 after FA loading exhibits both the characteristic peaks of CD-MOF and those of FA, indicating successful loading of FA.
O stretching vibration at 1685 cm−1, the CO–NH–CO stretching vibration at 1635 cm−1, methyl bending at 1485 cm−1, and CC skeletal vibrations on the phenyl ring at 1605 cm−1, 1571 cm−1, and 1453 cm−1. The infrared spectrum of FA/CD-MOF@SiO2 after FA loading exhibits both the characteristic peaks of CD-MOF and those of FA, indicating successful loading of FA.
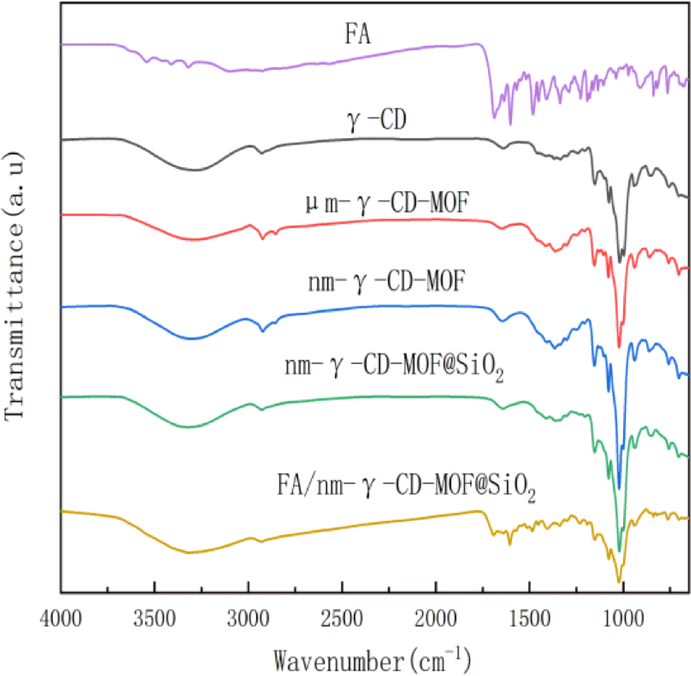 | ||
| Fig. 2 FTIR spectra of FA, γ-CD, um-CD-MOF, nm-CD-MOF, nm-CD-MOF@SiO2, FA/CD-MOF@SiO2. | ||
3.3 Thermal stability of samples
The thermal stability of CD-MOF, CD-MOF@SiO2, and FA/CD-MOF@SiO2 was characterized and analyzed through thermogravimetric analysis from room temperature to 800 °C. As shown in Fig. 3, weight loss below 100 °C in γ-CD, CD-MOF, and CD-MOF@SiO2 is attributed to moisture on the material surface or within pore structures. γ-CD exhibits major weight loss between 310–340 °C, while CD-MOF shows a second stage weight loss between 220–320 °C, consistent with the literature.28 Compared to γ-CD, CD-MOF starts losing weight at relatively lower temperatures, indicating the instability of the MOF crystal structure. Upon SiO2 encapsulation, CD-MOF@SiO2 shows a second stage weight loss starting at 250 °C with a lower rate compared to CD-MOF, resulting in a residue of 30.66%, higher than CD-MOF's 24.35%. The stability of CD-MOF is improved as expected due to successful encapsulation with inert and stable silica. FA exhibits three weight loss intervals below 175 °C, between 230–300 °C, and 381–496 °C, with a final residue of 33.1%. After FA loading, FA/CD-MOF@SiO2 shows a second stage weight loss between 256–350 °C, with the initial temperature increase and final temperature decrease attributed to the interaction between FA and the carrier, confirming successful encapsulation and loading of FA. Weight loss in this range is primarily due to CD-MOF and partial FA loss, resulting in a final residue of 28.86%. The CD-MOF@SiO2 composite displayed notable colloidal and chemical stability, as evidenced by its morphology remaining nearly unchanged even following immersion in a PBS solution for 12 hours (Fig. S1†).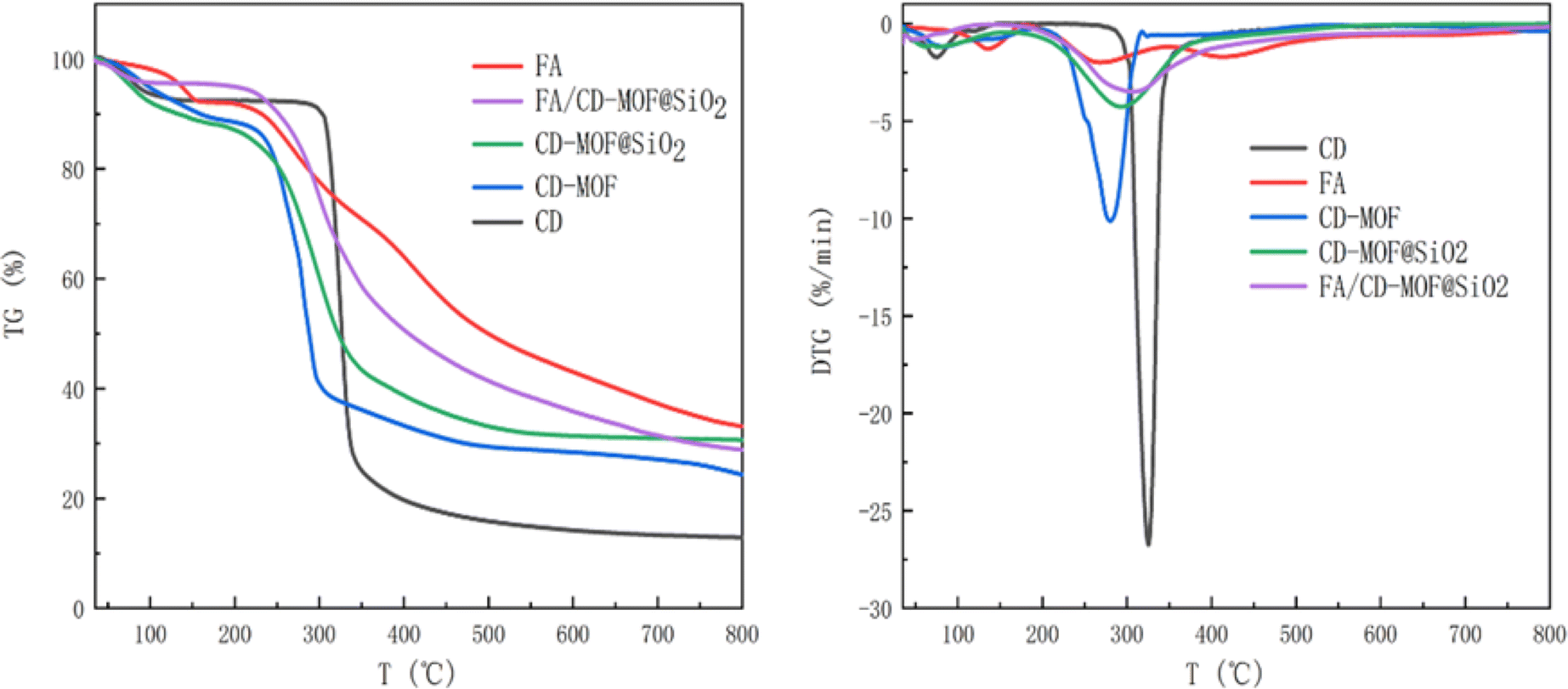 | ||
| Fig. 3 TGA and DTG data for samples. | ||
3.4 Pore structures of samples
The N2 adsorption–desorption isotherms of porous materials can demonstrate the relationship between the specific surface area of the material and the loading substance. Fig. 4 shows the N2 adsorption–desorption isotherms of CD-MOF, CD-MOF@SiO2, and different loading ratios of FA:CD-MOF@SiO2. For CD-MOF and CD-MOF@SiO2 without drug encapsulation, the N2 adsorption amount is higher in the low-pressure stage, showing a type I isotherm. As the relative pressure increases, the adsorption amount quickly reaches a limit without obvious hysteresis, indicating uniform pore size. In contrast, for FA/CD-MOF@SiO2 after drug loading, the adsorption reaching the limit significantly slows down with hysteresis, suggesting the successful loading of FA into the porous structure of the carrier, and affirming the successful synthesis of the porous carrier and its excellent adsorption of poorly soluble drugs. The BET specific surface area of CD-MOF is 299.1 m2 g−1, while that of CD-MOF@SiO2 is 168.2 m2 g−1. The decrease in specific surface area of the material is attributed to the successful composite of CD-MOF with silica. After drug loading, a type H3 hysteresis loop appears around P/P0 of 0.5, and with increasing drug input, the position of the hysteresis loop shifts backwards, indicating the presence of drug nanoclusters on the surface when N2 is adsorbed, and a decrease in nanocluster pore blockage as adsorption deepens into the interior. When the FA:CD-MOF@SiO2 feed ratio is 0.2:1, the BET specific surface area is 75.9 m2 g−1, and at a ratio of 0.5:1, the BET specific surface area is 49.3 m2 g−1, demonstrating successful drug loading as more drug is added, leading to increased drug loading and occupation of more pores.
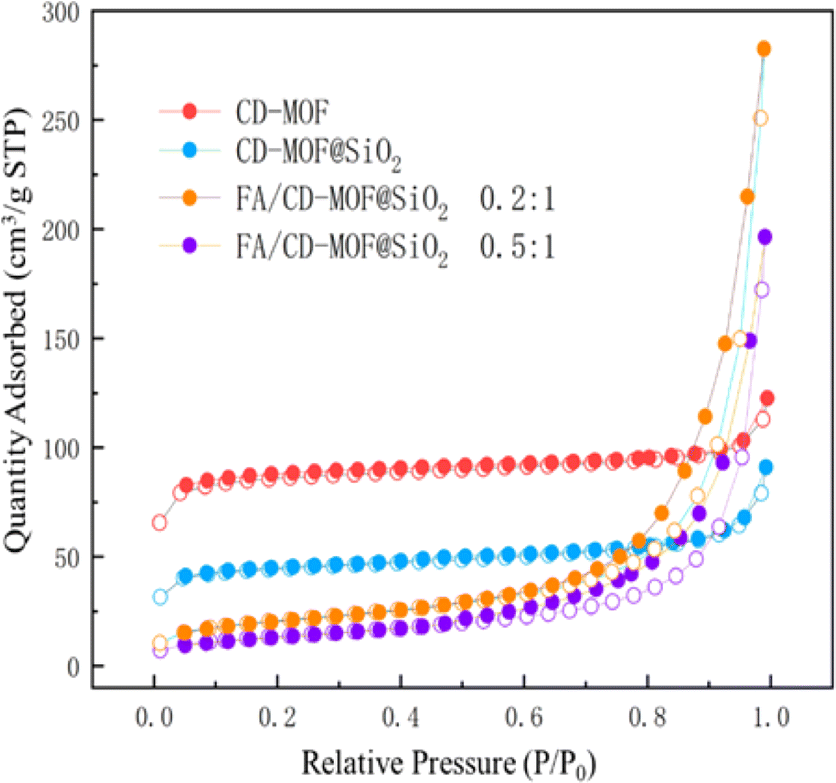 | ||
| Fig. 4 N2 adsorption–desorption curve for CD-MOF, CD-MOF@SiO2 and FA/CD-MOF@SiO2. | ||
3.5 Crystal structure of samples
As shown in Fig. 5, the XRD pattern reveals a strong diffraction peak at 16.6° for CD-MOF, with characteristic peaks at 2θ = 5.6°, 6.9°, 13.4°, and 24°, consistent with a previous report,53 indicating successful synthesis of the MOF with a well-defined crystal structure. CD-MOF@SiO2 retains the characteristic peaks at 6.9°, 13.4°, 16.6°, and 24°, suggesting that the incorporation of SiO2 did not disrupt the original crystal structure of the MOF. Notably, FA exhibits a distinct peak at 26.6°, and with an increasing loading ratio, the intensity of the 26.6° peak in FA/CD-MOF@SiO2 gradually strengthens while the intensity of the 16.6° peak, attributed to CD-MOF, diminishes. These findings suggest that FA initially loads onto the microporous surface of CD-MOF@SiO2, and with further drug loading, the CD-MOF structure becomes obscured.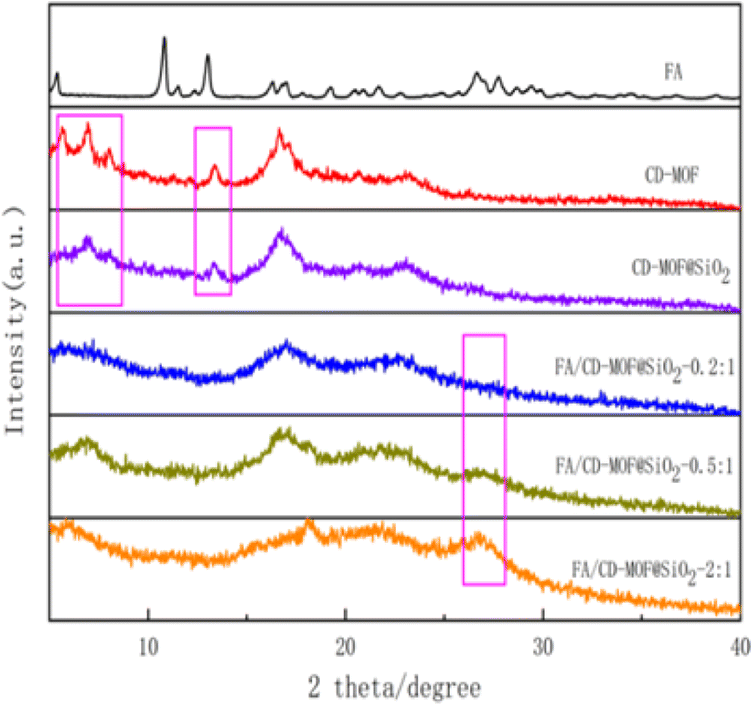 | ||
| Fig. 5 XRD pattern of FA, CD-MOF, CD-MOF@SiO2 and FA/CD-MOF@SiO2. | ||
3.6 Drug loading properties of CD-MOF@SiO2
The drug loading capacity was assessed by investigating the impact of drug feed ratio, temperature, and loading time on drug loading efficiency (DLE), drug encapsulation efficiency (DEE), and apparent solubility, as presented in Table 1 and Fig. 6.| Factor | DLE (%) | DEE (%) | Apparent solubility (mg mL−1) | |
|---|---|---|---|---|
| FA:CD-MOF@SiO2 ratio |
0.1:1 |
9.87 | 98.72 | 2.38 |
| 0.2:1 |
18.22 | 91.09 | 4.57 | |
| 0.5:1 |
45.35 | 90.70 | 9.86 | |
| 1:1 |
87.57 | 87.57 | 13.46 | |
| 2:1 |
166.78 | 83.39 | 21.74 | |
| Load time (h) | 2 | 12.88 | 6.44 | 3.27 |
| 4 | 60.19 | 30.09 | 14.19 | |
| 6 | 166.78 | 83.39 | 21.74 | |
| 12 | 196.31 | 98.16 | 22.56 | |
| Load temperature (°C) | 25 | 192.89 | 96.44 | 5.68 |
| 40 | 168.93 | 84.46 | 14.22 | |
| 60 | 166.78 | 83.39 | 21.74 |
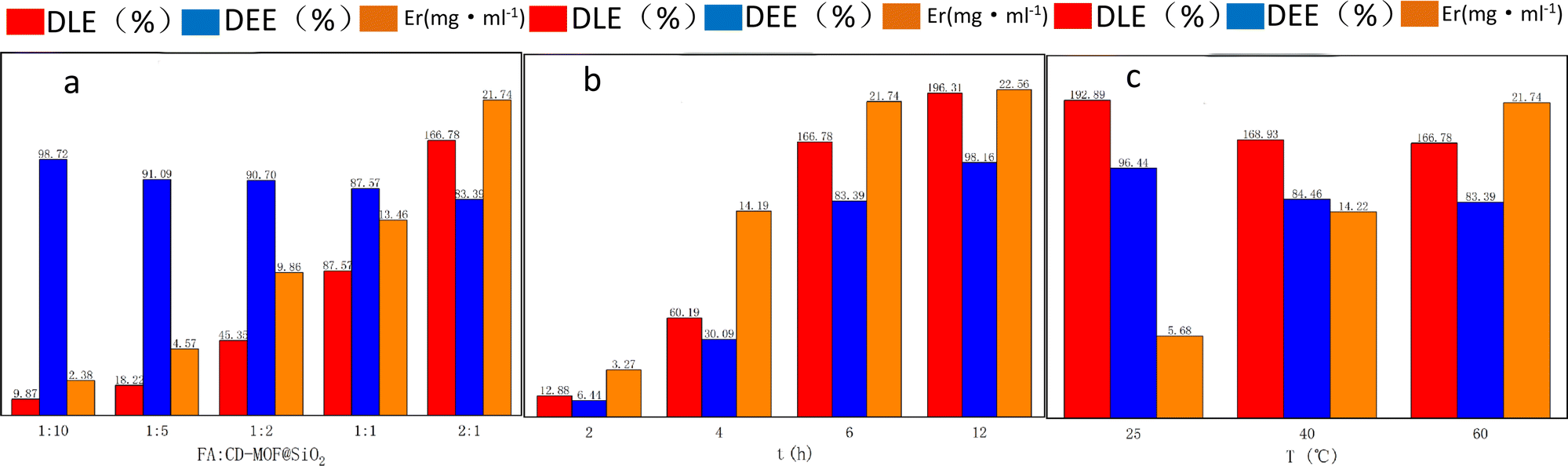 | ||
| Fig. 6 Drug loading efficiency (DLE), drug encapsulation efficiency (DEE), and apparent solubility (Er) at different loading conditions. | ||
The impact of different FA:CD-MOF@SiO2 feeding ratios on drug loading efficiency was investigated, while maintaining a drug loading temperature of 60 °C and a loading time of 6 hours. As shown in Fig. 6, the drug loading efficiency (DLE) increased with higher FA feeding ratios, reaching a maximum of 166.78% at a ratio of 2:1. Conversely, the drug entrapment efficiency (DEE) slightly decreased with increasing FA feeding amounts. This decrease is attributed to the fact that as the feeding amount increases before reaching the entrapment limit of the carrier, the DEE increases. However, once the entrapment limit is reached, the amount that can be entrapped remains constant, leading to a decrease in the calculated entrapment efficiency due to the increase in the input drug mass used for the calculation. The entrapment efficiency reflects the utilization of the added drug, with all ratios in the experiment achieving over 83% DEE, indicating a low wastage rate of the drug during the loading process. The solubility of FA in water is only 1.6 μg mL−1, but after loading FA onto CD-MOF@SiO2, the apparent solubility significantly increased. At a feeding ratio of 2:1, the solubility reached as high as 21.74 mg mL−1, representing a 13588-fold increase, nearly ten times higher than in similar reported works.54
The impact of drug loading time on DLE, DEE, and apparent solubility was also studied under experimental conditions involving a feed ratio of 2:1 and a temperature of 60 °C. The findings revealed that prolonging the duration resulted in an increase in DLE, DEE, and apparent solubility. However, there is little difference in the apparent solubility between the 6 hours and 12 hours loadings.
This study also investigated the impact of loading temperature on DLE, DEE, and apparent solubility, while maintaining a feed ratio of 2:1 and a loading time of 6 hours. As shown in Fig. 6, at 25 °C, DLE and DEE reach their peak, yet the solubility remains at only 5.68 mg mL−1. However, at 40 °C and 60 °C, the apparent solubility increases to 14.22 mg mL−1 and 21.74 mg mL−1, respectively. Elevated loading temperatures favor an increase in the solubility of FA, as higher temperatures facilitate the entry of FA into the pores of the metal–organic framework, thereby enhancing solubility.
3.7 Drug releasing properties and mechanisms of CD-MOF@SiO2
The drug release performance of FA/CD-MOF@SiO2 is illustrated in Fig. 7, showing an initial rapid release followed by a slower rate, with a cumulative release of 90% after 5 hours of sustained release. The drug release kinetics of FA/CD-MOF@SiO2 were fitted using the zero-order release model, first-order release model, Higuchi model, Korsmeyer-Peppas model, and Hixson–Crowell model, as presented in Table 2 and Fig. 7.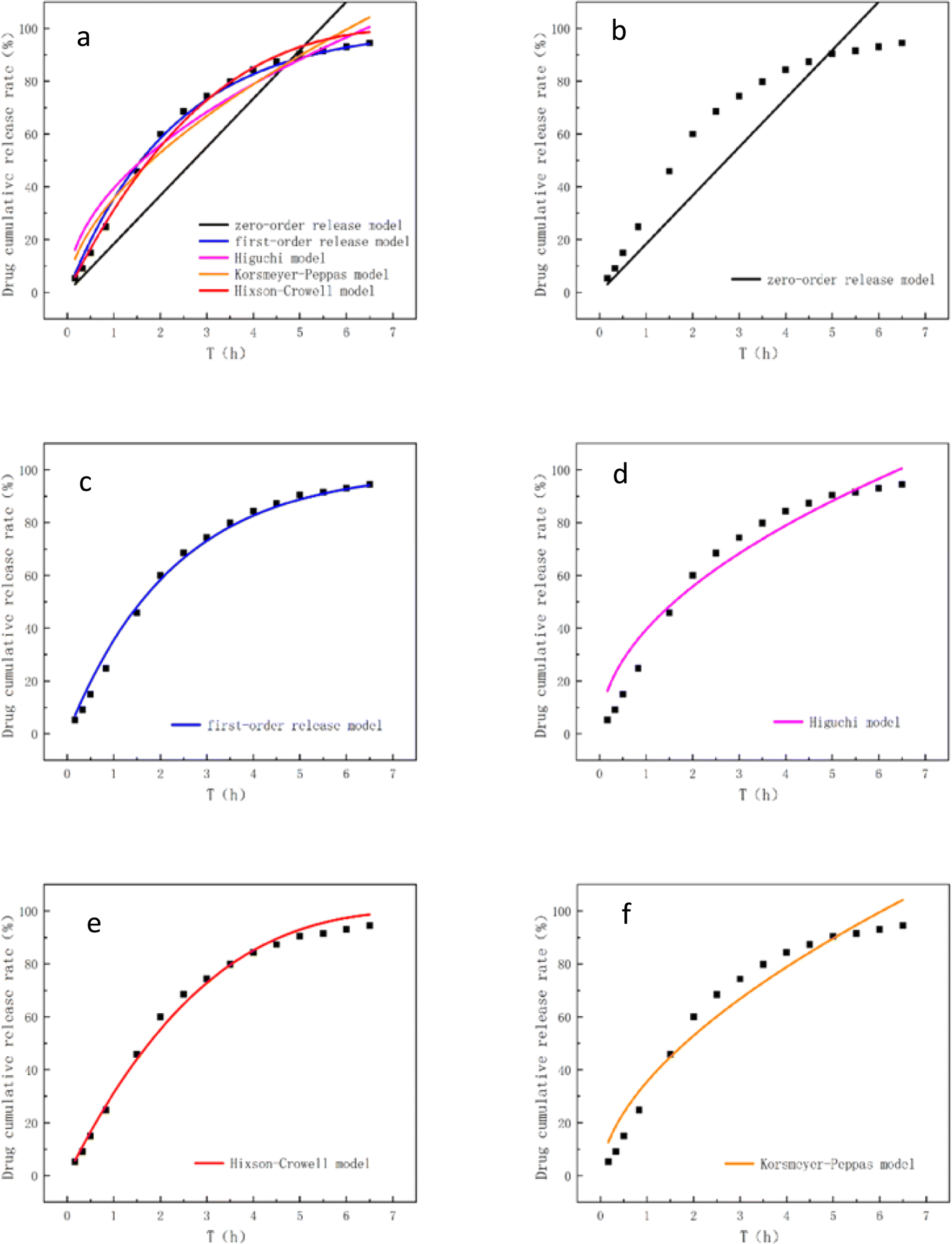 | ||
| Fig. 7 (a) Overview of all models, (b) zero-order release model, (c) the first-order release model, (d) Higuchi model, (e) Hixson–Crowell model, (f) Korsmeyer–Peppas model. | ||
| Model | Formula | Model fitting result | R2 |
|---|---|---|---|
| Zero-order release model | Qt = kt | Qt = 15.274 t | 0.5943 |
| First-order release model | Qt = 100 (1 − e−kt) | Qt = 100(1 − e−0.438t) | 0.9945 |
| Higuchi model | Qt = kt1/2 | Qt = 37.6847t1/2 | 0.9305 |
| Korsmeyer–Peppas model | Qt = ktn | Qt = 38.3373t0.4894 | 0.9264 |
| Hixson–Crowell model | Qt = 100 × [1 − (1 − kt)3] | Qt = 100 × [1 − (1 − 0.1171t)3] | 0.9923 |
The R2 value is commonly used to evaluate the suitability between the release behavior and the release model. According to the data presented in Table 2, the R2 value of 0.5943 obtained from fitting the zero-order release model suggests that the release of FA/CD-MOF@SiO2 in vitro is not characterized by a constant release rate. The variable release rate of FA may be attributed to its dual presence within the CD molecular cavity and on the surface of MOFs micropores.55,56
The fitting of a first-order release model indicates that FA/CD-MOF@SiO2 conforms to the porous matrix drug delivery pattern and the in vitro release is governed by drug diffusion.57 Fitting of the Higuchi model and the Korsmeyer–Peppas model suggests that the in vitro release of FA/CD-MOF@SiO2 involves both diffusion and erosion mechanisms, exhibiting a non-Fickian diffusion pattern. The applicability of the Hixson–Crowell model in simulating water-soluble carrier matrices is confirmed for FA/CD-MOF@SiO2, with an experimental fitting R2 value as high as 0.9923, indicating that FA/CD-MOF@SiO2, characterized by a high surface area, can dissolve and degrade in PBS buffer simulating biological environments. Overall, the sustained release behavior of FA/CD-MOF@SiO2 is better described by the first-order release model (R2 = 0.9945, k = 0.438) and the Hixson–Crowell model (R2 = 0.9923, k = 0.1171). Based on these results, we believe that FA/CD-MOF@SiO2, as a FA carrier holds significant potential for practical applications.
4 Conclusions
The efficient encapsulation and effective release of poorly soluble drugs have long been a focal point in the field of drug delivery. In this study, CD-MOF was prepared using a vapor diffusion method and composite with silica to form CD-MOF@SiO2. Utilizing the synthesized CD-MOF@SiO2 as a carrier matrix, the loading and in vitro release behavior of the poorly soluble drug, folic acid (FA), were investigated. The drug loading and release mechanism of CD-MOF@SiO2 were also investigated. CD-MOF@SiO2 exhibited a high drug loading capacity (166.78%) and encapsulation efficiency (83.39%) for FA, leading to a significant increase in the apparent solubility of FA from 1.6 μg mL−1 in its free state to 21.74 mg mL−1, a 13588-fold expansion. Thus, CD-MOF@SiO2 can serve as a promising drug carrier, offering a new approach and platform for enhancing the solubility and release of poorly soluble drugs.
Data availability
All data generated or analyzed in this study are included in this published article.Author contributions
Conceptualization, S. L. and F. D.; methodology, S. L. and F. D.; software, S. L.; formal analysis, S. L. and F. D.; investigation, S. L.; resources, X. L. and Y. X.; data curation, S.L. and F.D.; writing—original draft preparation, S.L., Y.X. and F. D.; writing—review & editing, S. L. , Y. X. and F. D.; supervision, F. D.; project administration, Y. X. and F. D.; funding acquisition, Y. X. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52063006).References
- K. J. Hartlieb, et al., Mol. Pharm., 2017, 14(5), 1831–1839 CrossRef CAS.
- Z. Li, et al., Drug Delivery Transl. Res., 2022, 12(5), 1096–1104 CrossRef CAS.
- N. Dhiman, et al., Front. Chem., 2021, 9, 19 Search PubMed.
- S. Wang, et al., Int. J. Mol. Sci., 2023, 24(3), 2643 CrossRef CAS.
- Y. Zheng, et al., Nano Today, 2024, 55, 102147 CrossRef CAS.
- Z. Ait-Touchente, et al., Nanomaterials, 2023, 13(10), 1688 CrossRef CAS.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112(2), 673–674 CrossRef CAS.
- H. Furukawa, et al., Science, 2013, 341(6149), 974 CrossRef PubMed.
- M.-X. Wu and Y.-W. Yang, Adv. Mater., 2017, 29(23), 1606134 CrossRef.
- S. Biswas, et al., Chem. Mater., 2022, 34(21), 9760–9774 CrossRef.
- B. Yan, J. Mater. Chem. C, 2019, 7(27), 8155–8175 RSC.
- M. Ding, et al., Chem. Soc. Rev., 2019, 48(10), 2783–2828 RSC.
- A. Kirchon, et al., Chem. Soc. Rev., 2018, 47(23), 8611–8638 RSC.
- Y. Xie and C. Deng, Sci. Rep., 2017, 7, 1162 CrossRef PubMed.
- A. Elsayed, et al., Appl. Energy, 2017, 186, 509–519 CrossRef.
- E. Adatoz, A. K. Avci and S. Keskin, Sep. Purif. Technol., 2015, 152, 207–237 CrossRef.
- L. He, et al., Nanomedicine, 2019, 14(10), 1343–1365 CrossRef PubMed.
- S. Mallakpour, E. Nikkhoo and C. M. Hussain, Coord. Chem. Rev., 2022, 451, 214262 CrossRef.
- H. Zheng, et al., J. Am. Chem. Soc., 2016, 138(3), 962–968 CrossRef.
- B. Lei, et al., ACS Appl. Mater. Interfaces, 2018, 10(19), 16698–16706 CrossRef.
- S. Rojas, et al., ACS Omega, 2018, 3(3), 2994–3003 CrossRef PubMed.
- P.-H. Tong, et al., Chem. Commun., 2021, 57(91), 12098–12110 RSC.
- H. D. Lawson, S. P. Walton and C. Chan, ACS Appl. Mater. Interfaces, 2021, 13(6), 7004–7020 CrossRef.
- M. Kotzabasaki and G. E. Froudakis, Inorg. Chem. Front., 2018, 5(6), 1255–1272 RSC.
- G. Ye, et al., Dalton Trans., 2021, 50(47), 17438–17454 RSC.
- S. He, et al., Acta Pharm. Sin. B, 2021, 11(8), 2362–2395 CrossRef.
- J. Wankar, et al., Adv. Funct. Mater., 2020, 30(44), 1909049 CrossRef.
- S. V. Dummert, et al., Chem. Soc. Rev., 2022, 51(12), 5175–5213 RSC.
- S. Gandhi and P. Shende, J. Controlled Release, 2021, 339, 41–50 CrossRef.
- J.-j. Wang, et al., React. Funct. Polym., 2020, 150, 104542 CrossRef.
- E. Norkus, J. Inclusion Phenom. Macrocyclic Chem., 2009, 65(3–4), 237–248 CrossRef.
- C. Qiu, et al., J. Agric. Food Chem., 2018, 66(16), 4244–4250 CrossRef.
- R. A. Smaldone, et al., Angew. Chem., Int. Ed., 2010, 49(46), 8630–8634 CrossRef PubMed.
- W. Zhang, et al., Pharm. Res., 2019, 36(8), 117 CrossRef CAS.
- Y. He, et al., Acta Pharm. Sin. B, 2019, 9(1), 97–106 CrossRef.
- C. Qiu, et al., J. Colloid Interface Sci., 2019, 553, 549–556 CrossRef PubMed.
- J. Aguilera-Sigalat and D. Bradshaw, Coord. Chem. Rev., 2016, 307, 267–291 CrossRef.
- Z. Long, et al., Microchem. J., 2013, 110, 764–769 CrossRef.
- K. Sugikawa, et al., Chem. Mater., 2013, 25(13), 2565–2570 CrossRef.
- K. Uma, G.-T. Pan and T. C. K. Yang, Materials, 2017, 10(6), 610 CrossRef.
- G. Lu, et al., Nat. Chem., 2012, 4(4), 310–316 CrossRef CAS.
- Q. Qiu, et al., Mater. Lett., 2020, 260, 126907 CrossRef CAS.
- A. M. Hezma, T. A. Elkhooly and G. S. El-Bahy, J. Porous Mater., 2020, 27(2), 555–562 CrossRef CAS.
- M. M. Ferreira and M. A. Bizeto, J. Porous Mater., 2020, 27(4), 1077–1086 CrossRef CAS.
- N. Trang Thi Thu, et al., Microporous Mesoporous Mater., 2022, 338, 111944 CrossRef.
- L. Lu, et al., Pharmaceutics, 2020, 12(2), 98 CrossRef CAS.
- Y. Furukawa, et al., Angew. Chem., Int. Ed., 2012, 51(42), 10566–10569 CrossRef CAS PubMed.
- V. Singh, et al., RSC Adv., 2017, 7(34), 20789–20794 RSC.
- T. Luo, et al., Int. J. Pharm., 2020, 584, 119407 CrossRef CAS.
- X. Zhang, M. Liu and R. Han, J. Environ. Chem. Eng., 2021, 9(6), 106672 CrossRef CAS.
- S. A. Khan and M. Schneider, Macromol. Biosci., 2013, 13(4), 455–463 CrossRef CAS PubMed.
- A. Hamedi, et al., J. Inclusion Phenom. Macrocyclic Chem., 2021, 99(3–4), 245–253 CrossRef CAS.
- T. Ba, et al., RSC Adv., 2022, 12(23), 14639–14643 RSC.
- J. Xu, et al., Int. J. Pharm., 2019, 556, 89–96 CrossRef CAS PubMed.
- M. Ceborska, et al., J. Mol. Struct., 2016, 1109, 114–118 CrossRef CAS.
- M. Ceborska, et al., Carbohydr. Polym., 2018, 184, 47–56 CrossRef CAS PubMed.
- G. Shao, et al., Carbohydr. Polym., 2022, 278, 118915 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04935g |
| This journal is © The Royal Society of Chemistry 2024 |