
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
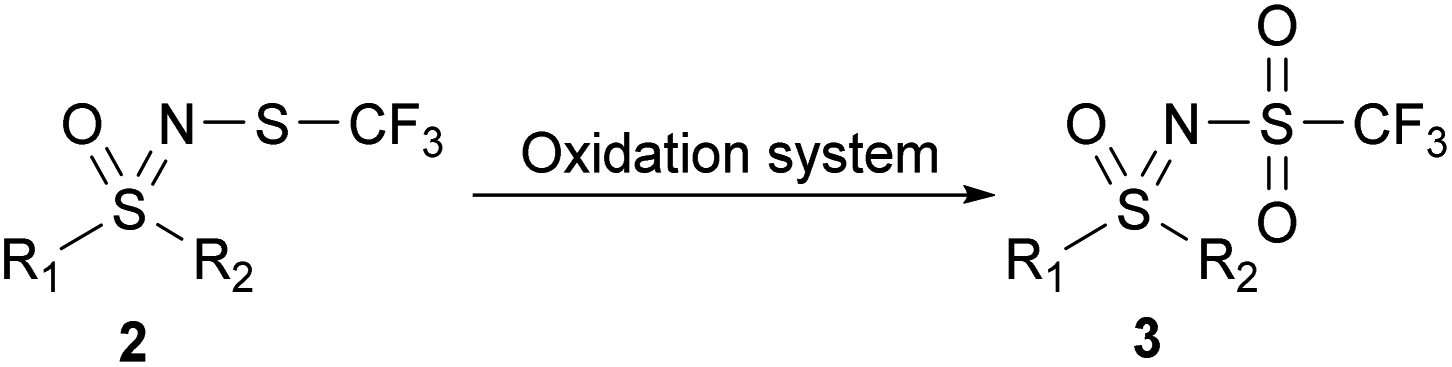
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategies for oxidative synthesis of N-triflyl sulfoximines†
Žan Testen and
Marjan Jereb*
and
Marjan Jereb*
University of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, 1000 Ljubljana, Slovenia. E-mail: mailto:marjan.jereb@fkkt.uni-lj.si
First published on 27th September 2024
Abstract
The oxidation of various structurally different N-trifluoromethylthio sulfoximines was investigated using different oxidizing agents and conditions. Mono- and disubstituted phenyl methyl and phenyl cyclopropyl N-trifluoromethylthio sulfoximines were oxidized with NaOCl·5H2O in water, while sterically hindered substrates bearing bulkier alkyl chains or two phenyl rings required the addition of MeCN to the reaction mixture. Chloro-, bromo-, and cyano-substituted substrates, as well as substrates bearing the benzyl groups, required a completely different approach using m-CPBA in DCM. Each method was tested on a gram-scale, with almost no difference in yield or reaction profile. The methods were also tested on N-p-tolylthio sulfoximine where successful oxidation to the corresponding sulfone derivative was observed. Finally, the N-triflyl sulfoximines acquired in the oxidations were examined in terms of stability and reactivity in Suzuki–Miyaura and Sonogashira coupling reactions, as well as many others. The selective mono- and dinitration of 4-methoxyphenyl N-triflyl sulfoximine was demonstrated by using nitric and sulfuric acid. N-triflyl sulfoximines were found to be stable in concentrated aqueous NaOH and HCl solutions and at elevated temperatures.
Introduction
The synthesis of sulfoximines and their functionalization continues to be an interesting topic, largely due to the properties that these compounds possess. These versatile molecules have found wide-ranging applications, serving as synthons in pharmaceutical and medicinal chemistry,1–6 in agrochemistry,7–9 as chiral auxiliaries,10,11 and more.12,13This article focuses on the functionalization and modification of groups on the nitrogen atom of the sulfoximine group. Recent examples of N–C type functionalizations include the procedures for the synthesis of N-allyl sulfoximines using allylic alcohols in a cocatalytic organoboron/Pd process14 and N-aryl sulfoximines using an inexpensive copper catalyst.15 Alkylation of sulfoximines could be achieved with an operationally simple Mitsunobu-type reaction using alcohols16 or by a copper-catalyzed photochemical reaction with diacyl peroxides.17 NH-sulfoximines can also be functionalized via an epoxide ring opening assisted by a catalytic amount of calcium(II) triflimide to afford 1,2-sulfoximidoyl ethanols.18 Decarboxylative sulfoximination of benzoic acids,19 oxidative decarboxylative sulfoximidation of cinnamic acids,20 C–H sulfoximidation of arenes21 and aerobic acylation with methylarenes led to N-functionalized sulfoximines.22 N-sulfenyl sulfoximines (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–S–R) can be obtained from sulfoximines and various sulfur sources, i.e., thiosulfonates and NaH,23 thiols and I2/H2O2 in PEG 400,24 or from disulfides under mechanochemical25 and electrochemical conditions.26–28 Regioselective chloro-sulfoximidations of allenes under solvent-free mechanochemical29 and photochemical30 conditions also gave S-substituted sulfoximines.
N–S–R) can be obtained from sulfoximines and various sulfur sources, i.e., thiosulfonates and NaH,23 thiols and I2/H2O2 in PEG 400,24 or from disulfides under mechanochemical25 and electrochemical conditions.26–28 Regioselective chloro-sulfoximidations of allenes under solvent-free mechanochemical29 and photochemical30 conditions also gave S-substituted sulfoximines.
N-sulfonylsulfoximines (N–SO2–R), which are similar to the compounds studied in this paper, have already been reported in the literature. The two main approaches to synthesize these compounds are based on the oxidation of N-sulfonyl sulfilimines and the transfer of the –NSO2– functionality to sulfoxides. The oxidation of N-sulfonyl sulfilimines with oxidizing agent/ruthenium tetroxide effectively gave the corresponding N-sulfonyl sulfoximines.31,32 Further oxidations of various sulfilimines were carried out with different oxidizing agents, e.g., m-CPBA (m-chloroperoxybenzoic acid),33 NaOCl/Bu4NBr,34 and electro generated peroxodicarbonate.35 The reaction of sulfoxide and tosyl azide in the presence of copper resulted in the formation of N-benzenesulfonyldimethyl sulfoximine in 97% yield.36 The imination of sulfoxides with various sulfonamides catalyzed by silver(I),37 copper(II),38 copper(I),39 rhodium(II),40 iron(III)41 and uncatalyzed imination of sulfoxides are also known.42 Iminoiodanes can be used as imination agents for sulfoxides.43 Sodium alkyl- and aryl-sulfinates with catalytic amounts of I2 and H2O2 in water undergo oxidative coupling to form N-sulfonyl-protected sulfoximines in good yields.44 Another mild N-sulfonylation of NH-sulfoximines was demonstrated using aryldiazonium tetrafluoroborates, 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) (DABSO) and a cheap copper catalyst.45
The synthetic routes described in the literature for the preparation of N-triflyl sulfoximines are rather limited and are based on the transfer of –NSO2CF3 and –SO2CF3 moieties. By using sulfonylimino-λ3-bromane under mild conditions, the sulfonylimino group was transferred to sulfides and sulfoxides yielding N-triflyl sulfilimines and sulfoximines, respectively (Scheme 1a).46 The other approach is based on the functionalization of sulfoximines using trifluoromethanesulfonic anhydride and pyridine as a base (Scheme 1b).47
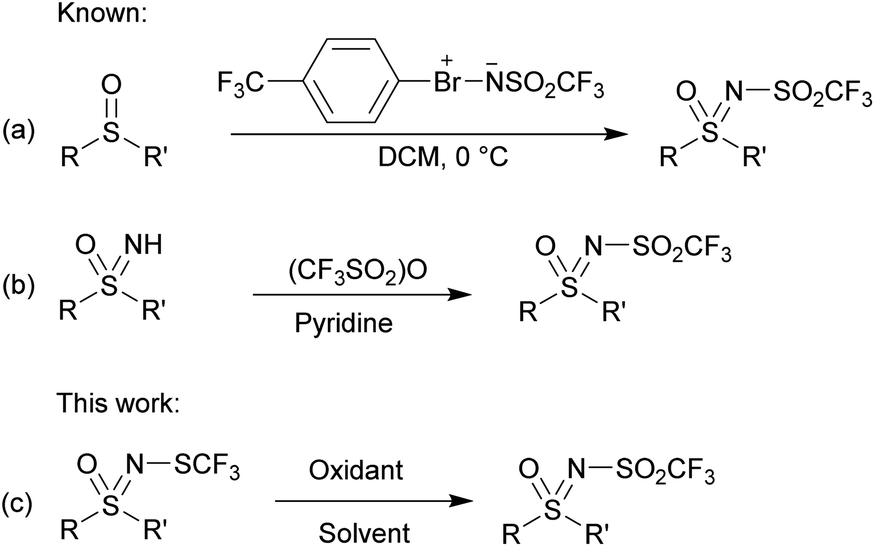 | ||
| Scheme 1 Previous and current work. | ||
N-trifluoromethylthiolated sulfonimidamides and sulfoximines possess various biological activities,48 and we reasoned that their oxidized NSO2CF3 derivatives might also be interesting in this regard. Our aim was threefold: to explore the oxidation of N-trifluoromethylthio sulfoximines to the corresponding N-sulfonyl sulfoximines, to propose an alternative oxidative pathway to obtain structurally different N-triflyl sulfoximines, and, if possible, to keep in mind the sustainability of these reactions. Finally, we wanted to test the stability and reactivity of these compounds with further functionalization reactions. Herein, we report complementary strategies for the synthesis of N-triflyl sulfoximines by oxidation of N-trifluoromethylthio sulfoximines (Scheme 1c),49,50 as well as some further functionalization reactions and stability experiments. For the majority of our products, we used solid sodium hypochlorite pentahydrate (NaOCl·5H2O),51 which we also used in our previous work for the oxidation of N-trifluoromethylthio sulfoximines to their sulfinyl derivatives.52 NaOCl·5H2O in water proved to be a mild oxidation system especially for aryl methyl sulfides, while NaOCl·5H2O in a mixture of water and MeCN worked better for bulkier aryl-, alkyl-, and diaryl-sulfides. Finally, m-CPBA in DCM was used for chloro-, bromo-, and cyano-substituted substrates, as well as for those with benzyl groups which could not be oxidized by the two previous methods.
Results and discussion
NaOCl·5H2O in water was the first oxidation system we tested and it gave good results (Table 1, entry 1). Increasing the excess of NaOCl·5H2O (3.2 equiv.) to convert the leftover sulfoxide 4a produced N–Cl sulfoximine as well as an unknown side-product, having no effect on the conversion of the reaction (Table 1, entry 2). Other solvents were also tested. The reaction does not occur in ethanol (Table 1, entry 3), instead 25% of the N-trifluoromethylthio sulfoximine 2a was converted to the parent NH-sulfoximine. In methanol (Table 1, entry 4) only traces of products 3a and 4a are visible, otherwise no changes to the substrate are observed. We assume that the alcohols are oxidized to their corresponding aldehyde or carboxylic acid instead. MeCN and EtOAc (Table 1, entries 5 and 6) behaved similarly, with the majority of the product being in the form of the sulfoxide 4a. Some N–Cl was also present in EtOAc. DCM, hexane and toluene (Table 1, entries 7, 8 and 9) were all more selective for the sulfoxide 4a, with minimal sulfone 3a being present in the reaction mixture, indicating that more non-polar solvents promote single oxidation to sulfoxide.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Relative ratiob (%) | ||
| 2a | 3a | 4a | ||
| a Reaction conditions: 2a (0.1 mmol), solvent 0.3 mL, NaOCl·5H2O (2.2 equiv.), stirring 16 h.b Determined by 1H and 19F NMR.c Observed the formation of N-chloro sulfoximine.d 25% of SONH present. | ||||
| 1 | H2O | — | 85 | 15 |
| 2 | H2O | —c | 85 | — |
| 3 | EtOH | 75d | — | — |
| 4 | MeOH | 99 | Trace | Trace |
| 5 | MeCN | — | 34 | 66 |
| 6 | EtOAc | —c | 31 | 64 |
| 7 | DCM | 26 | — | 74 |
| 8 | Hexane | 19 | 4 | 77 |
| 9 | Toluene | —c | 3 | 97 |
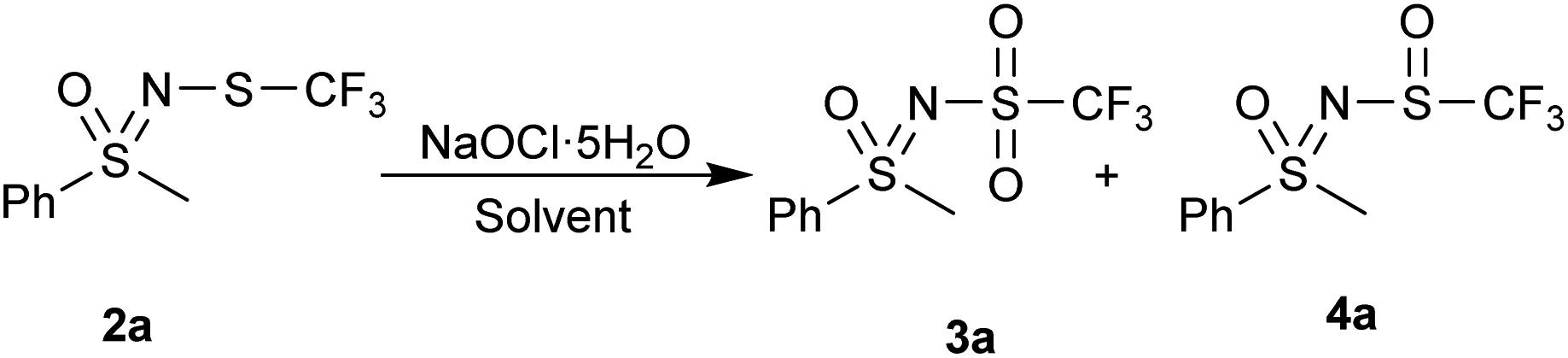
NaOCl·5H2O in water was then chosen as a suitable starting point with a simple water/EtOAc workup extraction to isolate the products (Table 2). If the crude product contained some sulfoxide 4 (less than 20%), the product was purified by flash chromatography. If more sulfoxide 4 was present, the reaction was repeated with more oxidant. If other, less polar impurities were present besides the starting substrate 2 or sulfoxide 4, the product was further purified by column chromatography.
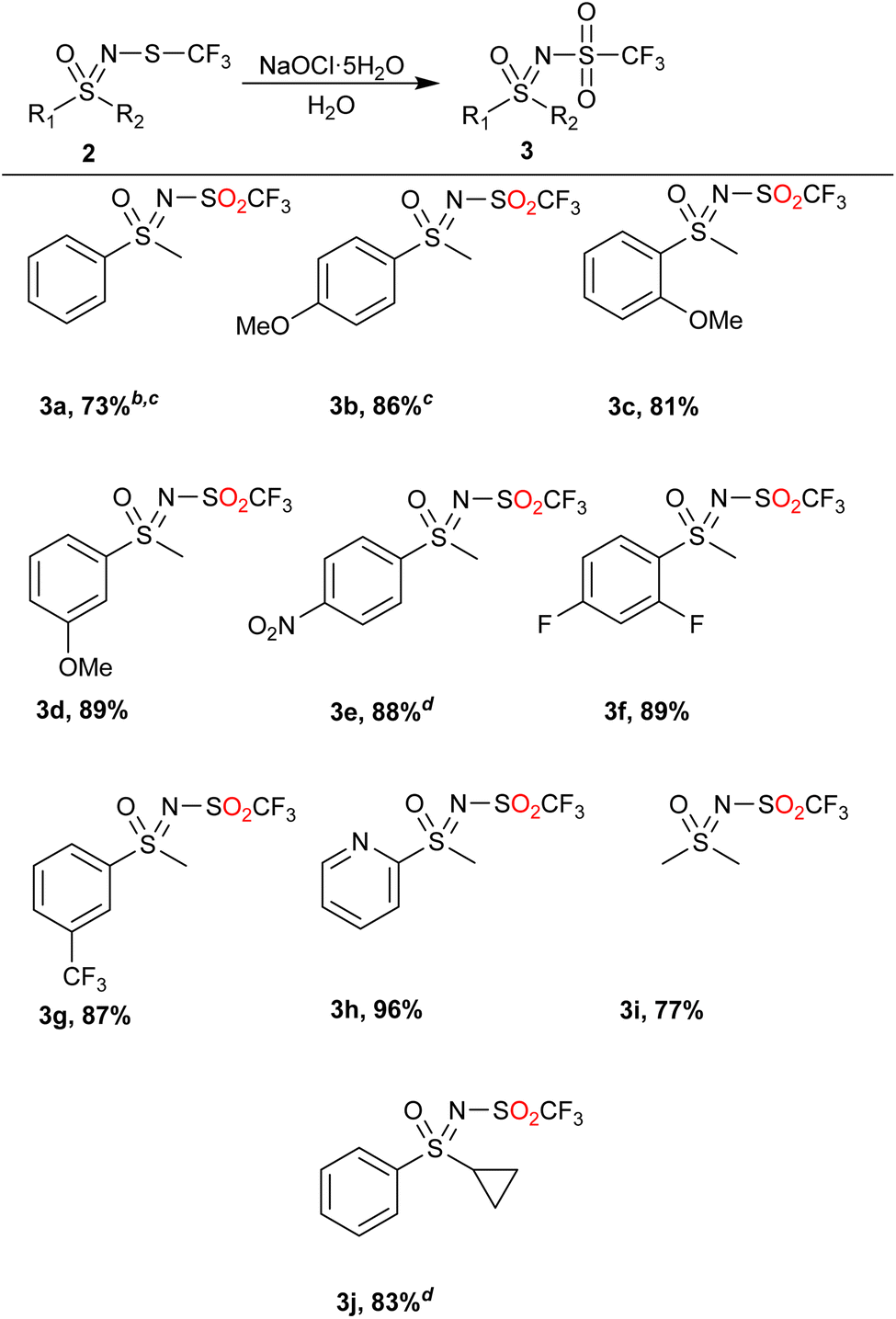
Phenyl methyl sulfide 2a and monosubstituted phenyl rings bearing the methoxy 2b–2d or nitro 2e group as well as the fluoro-disubstituted 2f underwent the reaction in good to excellent yields, with only the products 3a and 3b requiring flash chromatography. Using this method, we also succeeded in obtaining the products 3g and 3h, a trifluoromethyl and heterocyclic product, respectively. Smaller alkyl sulfoximines could also be reacted and furnished the product 3i in 77% yield. Lastly, the substrate 2j, bearing a cyclopropyl group, was investigated. Although more NaOCl·5H2O had to be used, the product was still obtained in good yield. In general, the method exhibited several important green attributes (water as a reaction medium, high selectivity, room temperature and low amount of non-hazardous waste).
The bulkier substrates posed a challenge under the reaction conditions used in Table 2 with sulfoxide 4 as the most isolated product or a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of sulfone 3 and sulfoxide 4. Adding more NaOCl·5H2O did not move the reaction towards sulfones, so an alternative was sought. By adding MeCN to water, more favorable results were obtained and previously difficult to access sulfones were isolated with very good yields and mostly no need for purification (Table 3). We suspect that the increased solubility of the substrates was the decisive factor.
:1 mixture of sulfone 3 and sulfoxide 4. Adding more NaOCl·5H2O did not move the reaction towards sulfones, so an alternative was sought. By adding MeCN to water, more favorable results were obtained and previously difficult to access sulfones were isolated with very good yields and mostly no need for purification (Table 3). We suspect that the increased solubility of the substrates was the decisive factor.
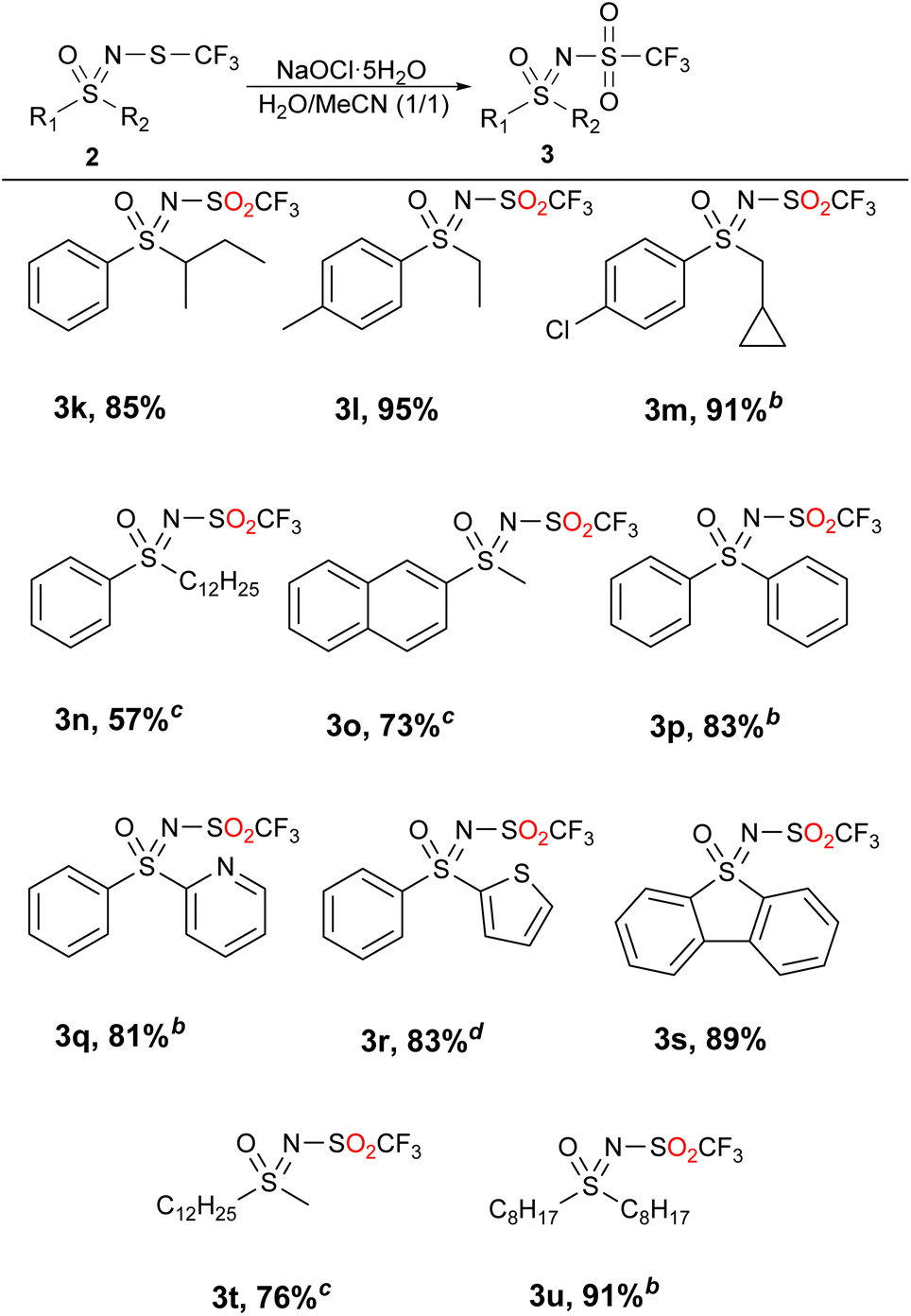
The products with longer and branched alkyl chains 3k, 3l, 3m and 3u were isolated in good to excellent yields, while the substrates with the dodecyl group (2n and 2t) also produced products 3n and 3t in 57% and 76% yield, respectively. The diaryl substrates 2p–2s reacted smoothly, although they produced some traces of sulfoxides, which were easily removed by flash chromatography. The product 3o, containing the bulkier naphthyl group, was also successfully obtained. Although acetonitrile was present in the reaction medium, the transformation is in good accordance with green chemistry principles. A mechanism is probably similar to the previously proposed51 and is comprised of a chlorination of the sulfur atom and subsequent attack by the hydroxide ion (Scheme 2).
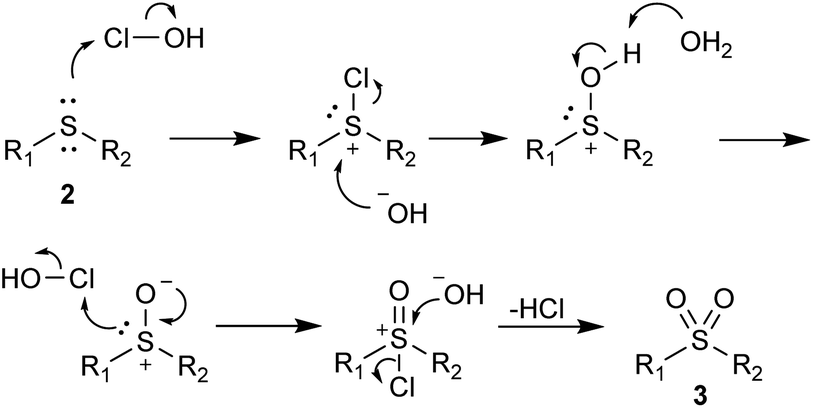 | ||
| Scheme 2 Proposed mechanism for the oxidation of sulfides 2 to their corresponding sulfones 3 with NaOCl·5H2O. | ||
However, the method used in Table 3 was not suitable for 4-chloro 2w, 4-bromo 2x and 4-cyano 2y aryl methyl substrates. 2w could not exceed 60% conversion even when additional NaOCl·5H2O was introduced. 2y produced various chloroform-insoluble compounds, while 2x underwent deprotection of the N-trifluoromethylthio group to NH-sulfoximine. In addition, the methyl group adjacent to the sulfoximine was chlorinated, replacing all the hydrogen atoms (Scheme 3, 5x). The product 5x decomposed rapidly and could not be purified by chromatographical means. The reactivity of 4-bromo derivative 2x is somewhat anomalous, since 4-chloro and 4-methoxy analogues 2w and 2b reacted smoothly, giving 3w and 3b in high yields.
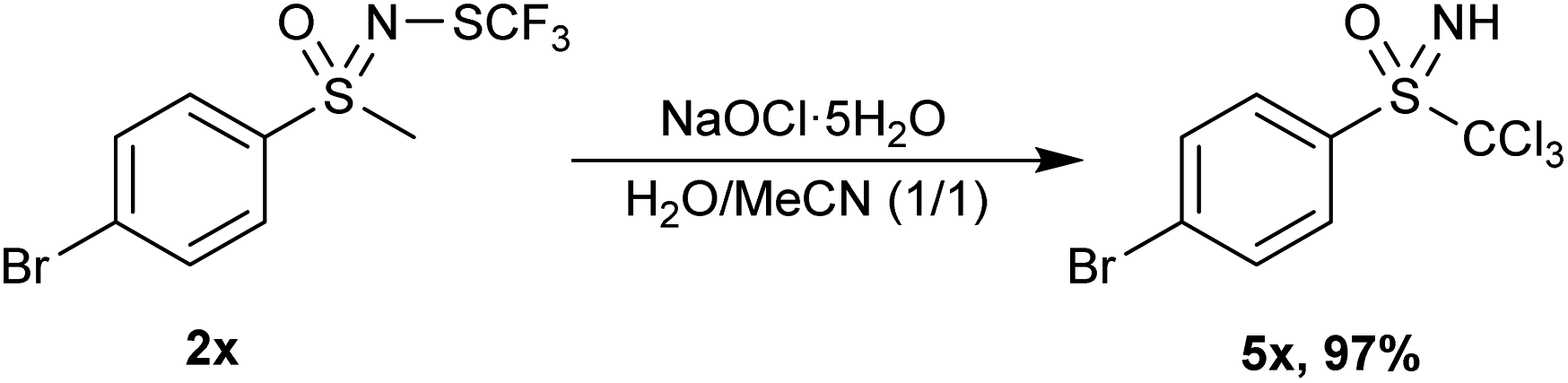 | ||
| Scheme 3 Removal of trifluoromethythio group with concomitant chlorination. | ||
These, and the other substrates that could not be quantitatively oxidized to sulfones by the two previous methods were successfully transformed to N-triflyl sulfoximines 3 using m-CPBA in DCM (Table 4). The latter method can be used with all substrates contained herein, but generates the highest amount of waste (the benzoic acid leftover as well as the Na2CO3 that is needed in the extraction step). With sustainability and waste minimization in mind, we opted to present all three methods instead of using only m-CPBA for all substrates.
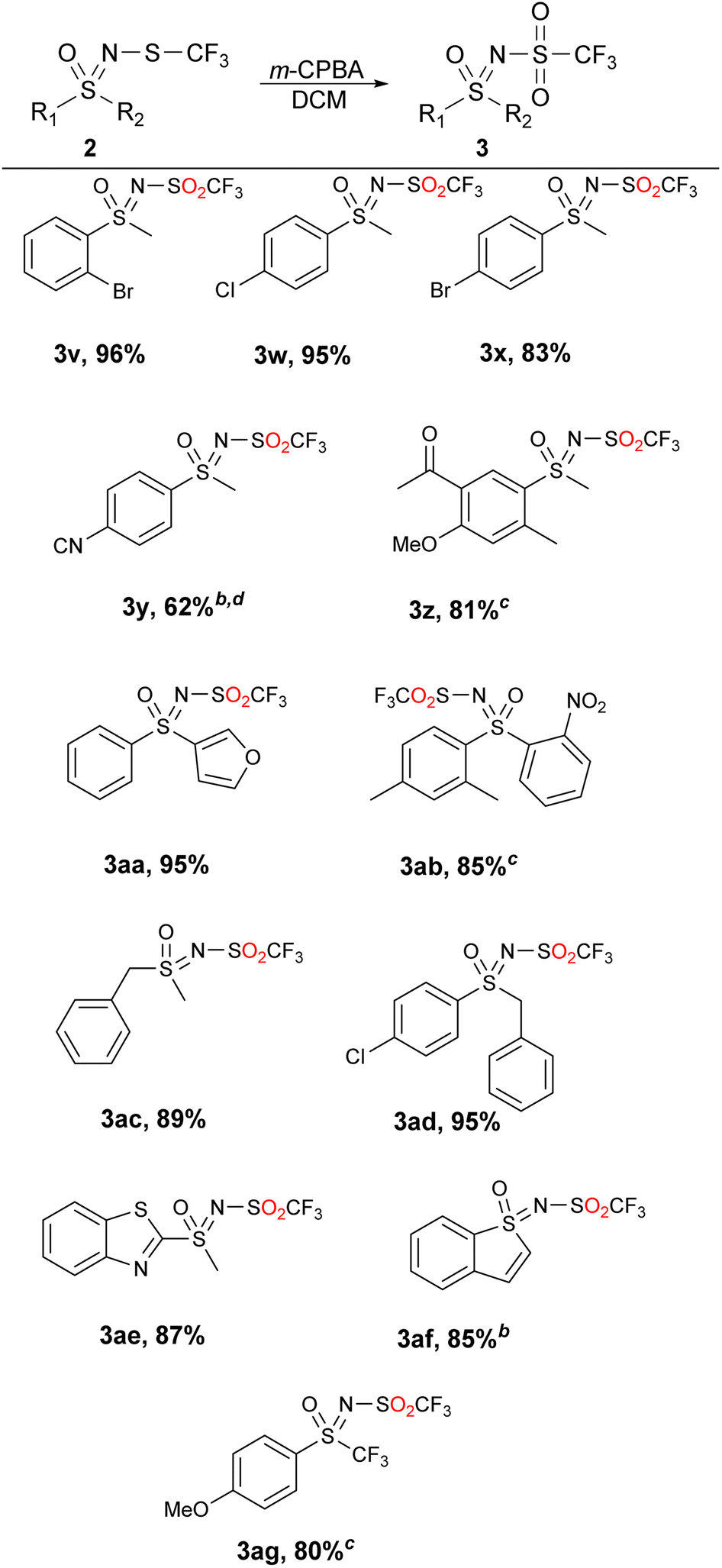
Substrates with halogen-substituted phenyl groups 2v–2x were readily oxidized to the corresponding sulfones 3v–3x. The substrate with the benzonitrile group 2y proved to be challenging and required more oxidant and flash chromatography. 2z, which contains the methyl ketone group, formed a number of impurities in very small amounts and had to be purified by column chromatography. The sterically hindered 2ab produced some minor impurities that required column chromatography, while the functionalization of benzylic substrates 2ac and 2ad proceeded without any major difficulties. Transformation of the heterocyclic substrates 2ae and 2af also proceeded smoothly and with good yields. The product 3ag was also obtained in 80% yield despite the inclusion of the trifluoromethyl group. Mechanistically we hypothesize that the reaction occurs with the nucleophilic attack of the sulfide towards the peroxide, similar as it does with hydrogen peroxide45 and furnishing the m-chlorobenzoic acid (Scheme 4).
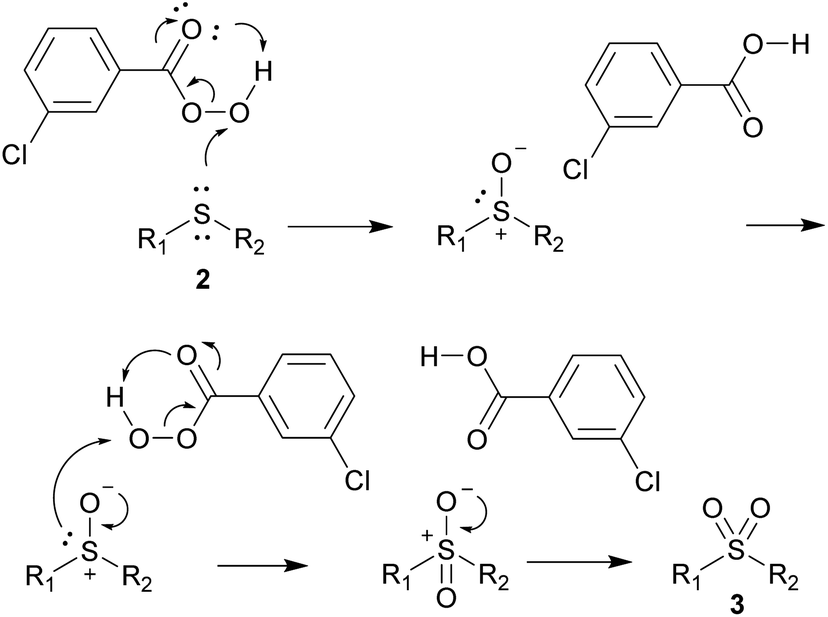 | ||
| Scheme 4 Proposed mechanism for the oxidation of sulfides 2 with m-CPBA. | ||
For further clarification, a table with all substrates and their corresponding oxidation system is provided (Table 5). NaOCl·5H2O in water is suitable for most phenyl methyl substrates and smaller dialkyl substrates. Interestingly, substrates with chloro- and bromo-substituted phenyl rings 2v–2x appear to be outliers from this theory, although both 4-MeO 2b and 4-NO2 2e were successfully converted to their corresponding sulfones 3b and 3e. Reasons might be complex, since they could not be attributed to the stereoelectronic effects and solubility issues. As mentioned above, the addition of MeCN assisted the oxidation of some sterically hindered and more hydrophobic substrates, which most likely suffered from decreased solubility. Products that could not be obtained with these two methods (Tables 2 and 3) were obtained with m-CPBA in DCM (Table 4). It can be concluded that the effect of structure of starting 2 on oxidation is multifaceted, and it is rather challenging to match the method with the structure of the starting material 2.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Substrate | R1 | R2 | Oxidation system | Yield | Substrate | R1 | R2 | Oxidation system | Yield |
| 2a | Ph | Me | NaOCl·5H2O, H2O | 73% | 2r | Ph | 2-Thiophenyl | NaOCl·5H2O, H2O/MeCN | 83% |
| 2b | 4-MeO–C6H4 | Me | NaOCl·5H2O, H2O | 86% | 2s | Dibenzo[b,d]thiophenyl | NaOCl·5H2O, H2O/MeCN | 89% | |
| 2c | 2-MeO–C6H4 | Me | NaOCl·5H2O, H2O | 81% | 2t | Dodecyl | Me | NaOCl·5H2O, H2O/MeCN | 76% |
| 2d | 3-MeO–C6H4 | Me | NaOCl·5H2O, H2O | 89% | 2u | Octyl | Octyl | NaOCl·5H2O, H2O/MeCN | 91% |
| 2e | 4-NO2–C6H4 | Me | NaOCl·5H2O, H2O | 88% | 2v | 2-Br–C6H4 | Me | m-CPBA, DCM | 96% |
| 2f | 2,4-F–C6H3 | Me | NaOCl·5H2O, H2O | 89% | 2w | 4-Cl–C6H4 | Me | m-CPBA, DCM | 95% |
| 2g | 3-CF3–C6H4 | Me | NaOCl·5H2O, H2O | 87% | 2x | 4-Br–C6H4 | Me | m-CPBA, DCM | 83% |
| 2h | 2-Pyridyl | Me | NaOCl·5H2O, H2O | 96% | 2y | 4-CN–C6H4 | Me | m-CPBA, DCM | 62% |
| 2i | Me | Me | NaOCl·5H2O, H2O | 77% | 2z | 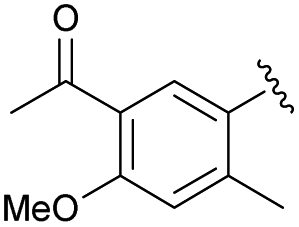 |
Me | m-CPBA, DCM | 81% |
| 2j | Ph | Cyclopropyl | NaOCl·5H2O, H2O | 83% | |||||
| 2k | Ph | sec-Butyl | NaOCl·5H2O, H2O/MeCN | 85% | 2aa | Ph | 3-Furanyl | m-CPBA, DCM | 95% |
| 2l | 4-Tolyl | Et | NaOCl·5H2O, H2O/MeCN | 95% | 2ab | 2,4-Me–C6H3 | 2-NO2–C6H4 | m-CPBA, DCM | 85% |
| 2m | 4-Cl–C6H4 | Cyclopropylmethyl | NaOCl·5H2O, H2O/MeCN | 91% | 2ac | Benzyl | Me | m-CPBA, DCM | 89% |
| 2n | Ph | Dodecyl | NaOCl·5H2O, H2O/MeCN | 57% | 2ad | Benzyl | 4-Cl–C6H4 | m-CPBA, DCM | 95% |
| 2o | Naphthyl | Me | NaOCl·5H2O, H2O/MeCN | 73% | 2ae | Benzo[d]thiazolyl | Me | m-CPBA, DCM | 87% |
| 2p | Ph | Ph | NaOCl·5H2O, H2O/MeCN | 83% | 2af | Benzo[d]thiophenyl | m-CPBA, DCM | 85% | |
| 2q | Ph | 2-Pyridyl | NaOCl·5H2O, H2O/MeCN | 81% | 2ag | 4-MeO–C6H4 | CF3 | m-CPBA, DCM | 80% |
Each method was also tested at the gram-scale, with encouragingly higher yields of 3a, 3p and 3x were obtained compared to the 0.3 mmol scale (Scheme 5).
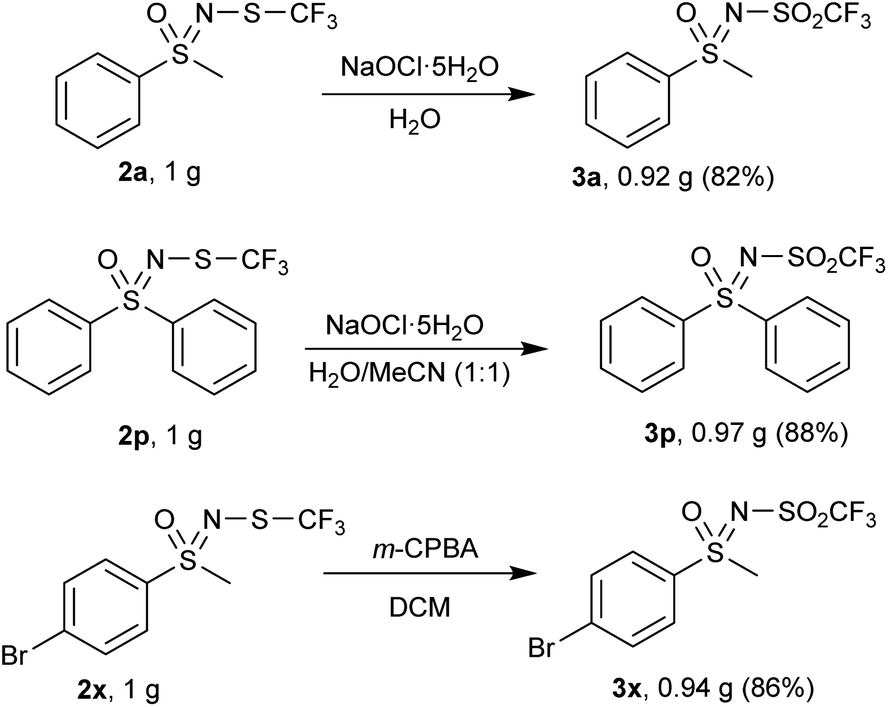 | ||
| Scheme 5 Gram scale reactions. | ||
Although compounds 3 have been synthesized previously, to the best of our knowledge there are few examples of reactions in which they assume the role of reactants.53 We therefore decided to test the stability of N-triflyl sulfoximines 3 under different conditions. 3a was exposed to aqueous solutions of 37% HCl and 50% NaOH, both of which showed no effect even after prolonged exposure at room temperature (24 h). Elevated temperature (120 °C) also had no effect other than melting the compound.
Using standard Suzuki–Miyaura conditions, the coupling of 3x and 4-methylbenzeneboronic acid was achieved and the product 3ah was isolated in 88% yield (Scheme 6a). Similarly, 3x was successfully coupled with phenylacetylene in Et3N with catalytic amounts of palladium and copper furnishing 3ai in 83% yield (Scheme 6b).
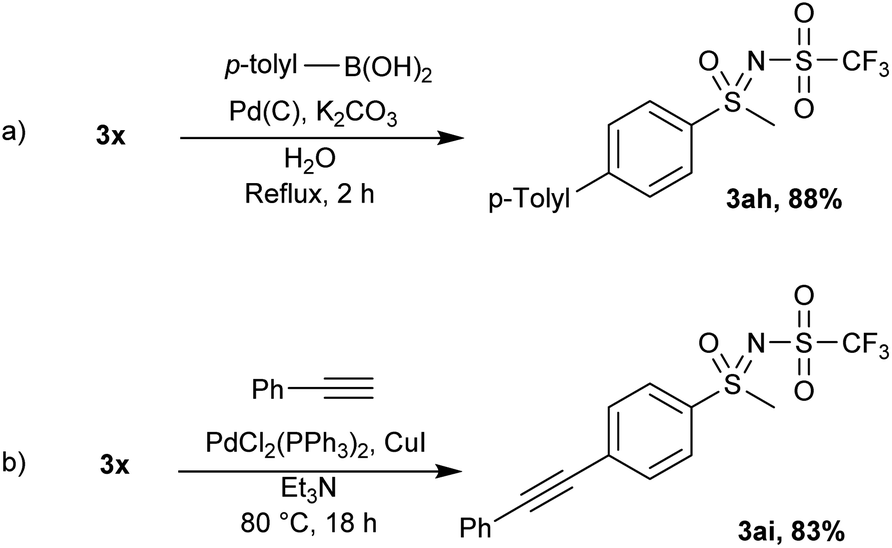 | ||
| Scheme 6 Further functionalizations of 3x. | ||
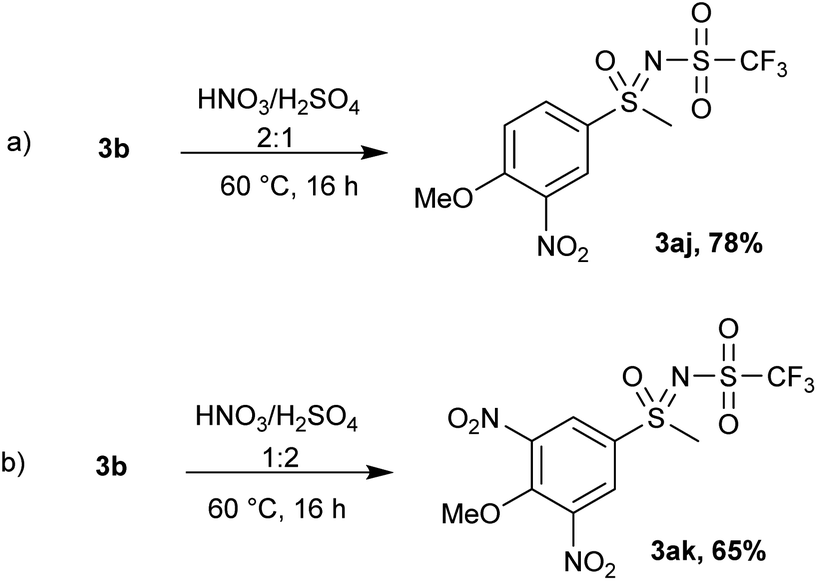
An N-triflyl sulfoximine with an electron-donating group (4-methoxy) 3b was used in an electrophilic aromatic substitution reaction (Scheme 7). By heating 3b in nitric acid (63% solution) overnight, 25% conversion was observed and no further changes in the substrate were detected. Addition of sulfuric acid (ratio of HNO3:H2SO4 = 2:1) led to the mononitrated product 3aj with complete conversion (Scheme 7a), while switching the ratio of acids and using more H2SO4 promoted the formation of a dinitrated product 3ak (Scheme 7b). We reason that the addition of more H2SO4 increases the concentration of the nitronium ion (NO2+), which promotes further nitration. When fuming nitric acid (90% solution) was used, a mixture of both nitration products (3aj and 3ak) was formed.
 | ||
| Scheme 7 Nitration of 3b. | ||
Other reaction conditions were also investigated, which provided further insight into the stability and reactivity of compounds 3. Attempts to reduce the triflyl group to a sulfoxide or sulfide were unsuccessful. The reducing agents used included NaSH·H2O in HCl, NaBH4 in EtOH and LiAlH4 in THF; all of which are used in literature for various reductions of sulfones and sulfoxides. Substrate 3a remained unchanged, only the latter reducing agent led to some decomposition. The product 3x remained unchanged when a Grignard reaction with i-PrMgCl was attempted. Bromination with bromine in acetic acid, the classic Friedel–Crafts reaction with acetyl chloride and AlCl3 in DCM and a variation of this reaction in TFA with acetic anhydride had no effect on substrate 3b. Reactions of 3a with n-butyllithium led to decomposition of the substrate into many unknown compounds. From this, we can infer that compounds 3 have good resistance under a wide range of conditions, implying that the functional group would remain intact in further functionalization reactions of larger compounds.
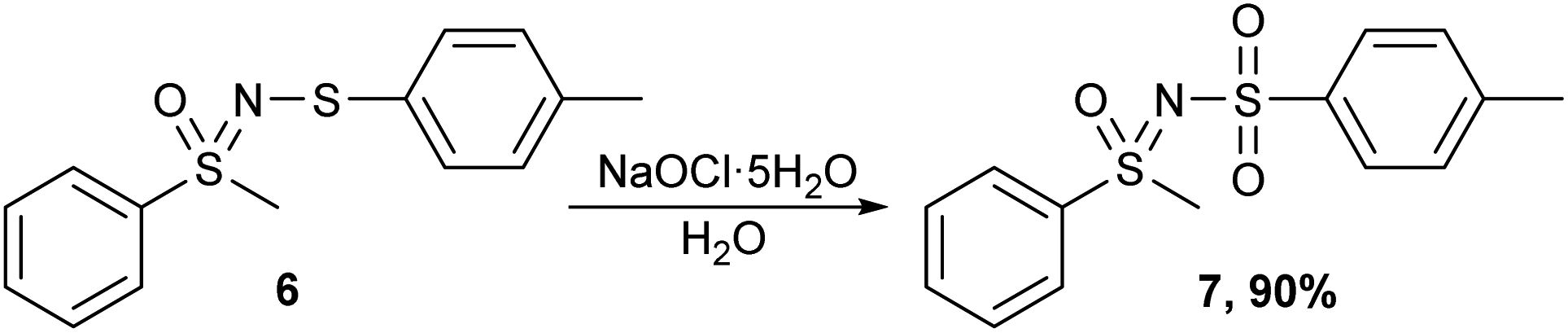
Finally, the methods were tested on a non-N-trifluoromethylthio sulfoximine compound. For this purpose, an N-sulfenylated sulfoximine 6 was prepared and oxidized using all three methods. All three methods successfully afforded the corresponding sulfone 7 (Scheme 8), with m-CPBA being the least desirable oxidizing agent as it also produced some minor impurities that would require purification.
 | ||
| Scheme 8 Oxidation of N-sulfenylated sulfoximine. | ||
Conclusion
In conclusion, new oxidative methods for obtaining N-triflyl sulfoximines as well as N-sulfonyl sulfoximines were reported. By using NaOCl·5H2O in water, this relatively green approach is able to oxidize most phenyl methyl sulfoximines in good yields with little need for additional purification. This method was also used for the oxidation of N-sulfenyl sulfoximines. A mixture of water and MeCN was required for bulky substrates, while m-CPBA in DCM was needed for benzyl, heavily-substituted aryl systems, and bromo-, chloro-, and cyano-substituted rings. The stability of N-triflyl sulfoximines was studied and no degradation was observed in concentrated aqueous NaOH and HCl solutions or at elevated temperatures. Furthermore, mono- and dinitration occurred in concentrated nitric and sulfuric acid. The N-triflyl group also proved to be tolerant to the Suzuki–Miyaura and Sonogashira coupling reaction conditions and gave both products in good yields.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ž. T.: investigation, validation, data curation, writing – original draft and writing – review & editing. M. J.: conceptualization, resources, validation, supervision, writing – original draft and writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the Slovenian Research Agency (Research Core Funding Grant P1-0230 and Young Researcher Grant to Ž. T.). The authors thank Dr Damijana Urankar from the Research Infrastructure Center at the Faculty of Chemistry and Chemical Technology, the University of Ljubljana, for HRMS analyses of small molecules. Dedicated to Dr Slovenko Polanc, Professor Emeritus of University of Ljubljana.References
- U. Lücking, Org. Chem. Front., 2019, 6, 1319–1324 RSC.
- M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225–245 CrossRef CAS PubMed.
- Y. Han, K. Xing, J. Zhang, T. Tong, Y. Shi, H. Cao, H. Yu, Y. Zhang, D. Liu and L. Zhao, Eur. J. Med. Chem., 2021, 209, 112885 CrossRef CAS PubMed.
- P. Mäder and L. Kattner, J. Med. Chem., 2020, 63, 14243–14275 CrossRef PubMed.
- U. Lücking, Chem.–Eur. J., 2022, 28, e202201993 CrossRef PubMed.
- M. L. G. Borst, C. M. J. Ouairy, S. C. Fokkema, A. Cecchi, J. M. C. A. Kerckhoffs, V. L. De Boer, P. J. Van Den Boogaard, R. F. Bus, R. Ebens, R. Van Der Hulst, J. Knol, R. Libbers, Z. M. Lion, B. W. Settels, E. De Wever, K. A. Attia, P. J. Sinnema, J. M. De Gooijer, K. Harkema, M. Hazewinkel, S. Snijder and K. Pouwer, ACS Comb. Sci., 2018, 20, 335–343 CrossRef CAS PubMed.
- P. Devendar and G. F. Yang, Top. Curr. Chem., 2017, 375, 1–44 CrossRef CAS PubMed.
- C. Gnamm, A. Jeanguenat, A. C. Dutton, C. Grimm, D. P. Kloer and A. J. Crossthwaite, Bioorg. Med. Chem. Lett., 2012, 22, 3800–3806 CrossRef CAS PubMed.
- Y. Xie, H. Li, D. Liu, L. Zhao, X. Liu, X. Liu and Y. Li, J. Agric. Food Chem., 2024, 72, 11716–11723 CrossRef CAS PubMed.
- H. Okamura and C. Bolm, Chem. Lett., 2004, 33, 482–487 CrossRef CAS.
- S. Sau, K. Mukherjee, K. Kondalarao, V. Gandon and A. K. Sahoo, Org. Lett., 2023, 25, 7667–7672 CrossRef CAS PubMed.
- P. Ghosh, B. Ganguly and S. Das, Asian J. Org. Chem., 2020, 9, 2035–2082 CrossRef CAS.
- R. Luisi and J. A. Bull, Molecules, 2023, 28, 1120 CrossRef CAS PubMed.
- M. T. Zambri, C. Ho and M. S. Taylor, Org. Lett., 2023, 25, 8274–8278 CrossRef CAS PubMed.
- M. Kumar, A. Rastogi, Raziullah, A. Ahmad, M. K. Gangwar and D. Koley, Org. Lett., 2022, 24, 8729–8734 CrossRef CAS PubMed.
- C. J. Dodd, D. C. Schultz, J. Li, C. W. Lindsley and A. M. Bender, Org. Biomol. Chem., 2023, 21, 5181–5184 RSC.
- P. Shi, Y. Tu, D. Ma and C. Bolm, Adv. Synth. Catal., 2023, 365, 1613–1617 CrossRef CAS.
- N. S. George, M. Kosiuha, A. Moquette and C. Parsy, Eur. J. Org Chem., 2022, 2022, e202201219 CrossRef CAS.
- P. Xu, W. Su and T. Ritter, Chem. Sci., 2022, 13, 13611–13616 RSC.
- N. Chakraborty, K. K. Rajbongshi, A. Dahiya, B. Das, A. Vaishnani and B. K. Patel, Chem. Commun., 2023, 59, 2779–2782 RSC.
- W. Su, P. Xu, R. Petzold, J. Yan and T. Ritter, Org. Lett., 2023, 25, 1025–1029 CrossRef CAS PubMed.
- N. Chakraborty, K. K. Rajbongshi, A. Gondaliya and B. K. Patel, Org. Biomol. Chem., 2024, 22, 2375–2379 RSC.
- X. Kang, H. Wang and Q. Zeng, Eur. J. Org Chem., 2022, 2022, e202201229 CrossRef CAS.
- L. Yang, J. Feng, M. Qiao and Q. Zeng, Org. Chem. Front., 2017, 5, 24–28 RSC.
- D. Kong, D. Ma, P. Wu and C. Bolm, ACS Sustain. Chem. Eng., 2022, 10, 2863–2867 CrossRef CAS.
- S. Zhang, M. Hu, C. Qin, S. Wang, F. Ji and G. Jiang, New J. Chem., 2024, 48, 2576–2583 RSC.
- X. Li, J. Huang, L. Xu, J. Liu and Y. Wei, Adv. Synth. Catal., 2023, 365, 4647–4653 CrossRef CAS.
- W. Zhang, D. Jin, Y. Hu, K. Yin, Q. Zou, L. Tang and P. Qian, J. Org. Chem., 2024, 89, 6106–6116 CrossRef CAS PubMed.
- D. Kong, M. M. Amer and C. Bolm, Green Chem., 2022, 24, 3125–3129 RSC.
- P. Shi, Y. Tu, D. Zhang, C. Wang, K. N. Truong, K. Rissanen and C. Bolm, Adv. Synth. Catal., 2021, 363, 2552–2556 CrossRef CAS.
- H. S. Veale, J. Levin and D. Swern, Tetrahedron Lett., 1978, 19, 503–506 CrossRef.
- D. M. Ketcha and D. Swern, Synth. Commun., 1984, 14, 915–919 CrossRef CAS.
- S. L. Huang and D. Swern, J. Org. Chem., 1979, 44, 2510–2513 CrossRef CAS.
- K. Akutagawa, N. Furukawa and S. Oae, J. Org. Chem., 1984, 49, 2282–2284 CrossRef CAS.
- M. Klein, D. L. Troglauer and S. R. Waldvogel, JACS Au, 2023, 3, 575–583 CrossRef CAS PubMed.
- H. Kwart and A. A. Kahn, J. Am. Chem. Soc., 1967, 89, 1950–1951 CrossRef CAS.
- Y. C. Gae and C. Bolm, Org. Lett., 2005, 7, 4983–4985 CrossRef PubMed.
- Y. Liu, H. Wang and X. Yang, Tetrahedron, 2019, 75, 4697–4702 CrossRef CAS.
- J. F. K. Müller and P. Vogt, Tetrahedron Lett., 1998, 39, 4805–4806 CrossRef.
- H. Okamura and C. Bolm, Org. Lett., 2004, 6, 1305–1307 CrossRef CAS PubMed.
- O. G. Mancheño and C. Bolm, Org. Lett., 2006, 8, 2349–2352 CrossRef PubMed.
- G. Y. Cho and C. Bolm, Tetrahedron Lett., 2005, 46, 8007–8008 CrossRef CAS.
- A. Yoshimura, V. N. Nemykin and V. V. Zhdankin, Chem.–Eur. J., 2011, 17, 10538–10541 CrossRef CAS PubMed.
- W. Zheng, M. Tan, L. Yang, L. Zhou and Q. Zeng, Eur. J. Org Chem., 2020, 2020, 1764–1768 CrossRef CAS.
- H. Nie, Z. Xiong, M. Hu, S. Zhang, C. Qin, S. Wang, F. Ji and G. Jiang, J. Org. Chem., 2023, 88, 2322–2333 CrossRef CAS PubMed.
- M. Ochiai, M. Naito, K. Miyamoto, S. Hayashi and W. Nakanishi, Chem.–Eur. J., 2010, 16, 8713–8718 CrossRef CAS PubMed.
- D. Craig, N. J. Geach, C. J. Pearson, A. M. Z. Slawin, A. J. P. White and D. J. Williams, Tetrahedron, 1995, 51, 6071–6098 CrossRef CAS.
- N. Thota, P. Makam, K. K. Rajbongshi, S. Nagiah, N. S. Abdul, A. A. Chuturgoon, A. Kaushik, G. Lamichhane, A. M. Somboro, H. G. Kruger, T. Govender, T. Naicker and P. I. Arvidsson, ACS Med. Chem. Lett., 2019, 10, 1457–1461 CrossRef CAS PubMed.
- C. Bohnen and C. Bolm, Org. Lett., 2015, 17, 3011–3013 CrossRef CAS PubMed.
- A. Zupanc and M. Jereb, J. Org. Chem., 2021, 86, 5991–6000 CrossRef CAS PubMed.
- M. Kirihara, T. Okada, Y. Sugiyama, M. Akiyoshi, T. Matsunaga and Y. Kimura, Org. Process Res. Dev., 2017, 21, 1925–1937 CrossRef CAS.
- Ž. Testen and M. Jereb, Org. Biomol. Chem., 2024, 22, 2012–2020 RSC.
- D. Craig, F. Grellepois and A. J. P. White, J. Org. Chem., 2005, 70, 6827–6832 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental detail, copies of 1H, 19F and 13C NMR spectra. See DOI: https://doi.org/10.1039/d4ra04992f |
| This journal is © The Royal Society of Chemistry 2024 |