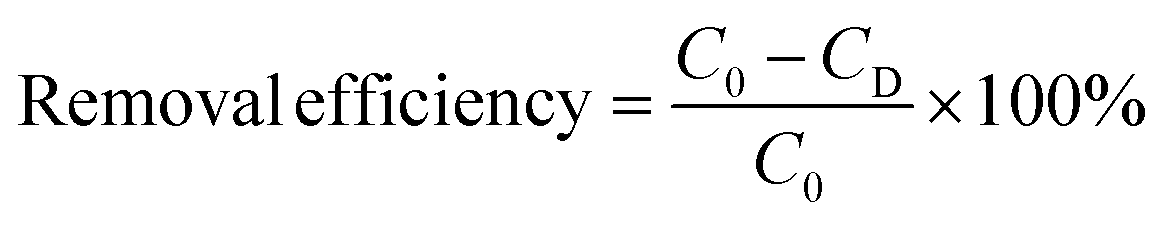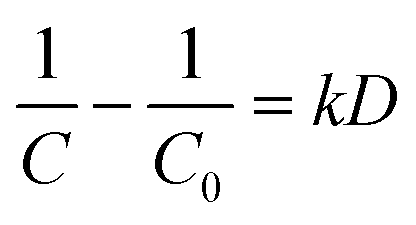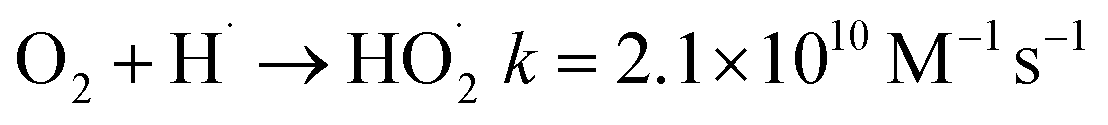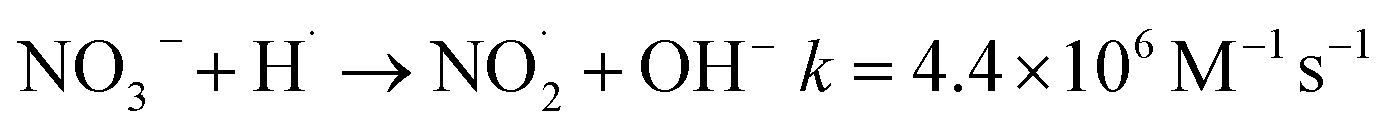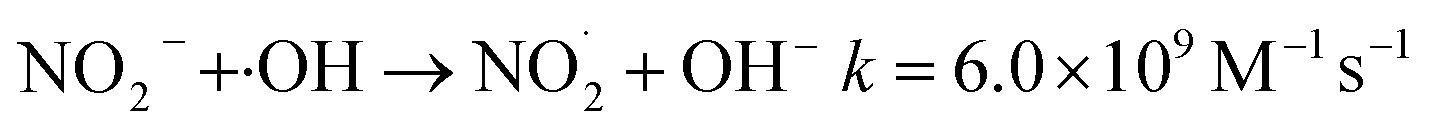DOI:
10.1039/D4RA04993D
(Paper)
RSC Adv., 2024,
14, 38815-38826
Decomplexation of Pb-EDTA by electron beam irradiation technology: efficiency and mechanism†
Received
10th July 2024
, Accepted 7th October 2024
First published on 9th December 2024
Abstract
As a common heavy metal complex in industrial wastewater, Pb-EDTA has garnered much attention due to its detrimental impact on both human health and the ecological environment. The degradation of heavy metal complexes by traditional methods requires subsequent treatment to recover heavy metals. This article attempts to find an effective method to simultaneously degrade both organic matter and heavy metal pollutants. Experimental results indicate that 1 mM Pb-EDTA can be effectively removed at 10 kGy with a degradation efficiency of 91.62%. Most lead ions were still in a stable complex state, with a removal rate of 24.42% (10 kGy). When the absorbed dose increased to 80 kGy, the degradation efficiency of Pb-EDTA was 95.24%. At this time, the removal rate of Pb2+ reached 68.82%. Through radical scavenging experiments and further mechanism analysis, it was demonstrated that electron beam irradiation primarily generates ·OH radicals, disrupting the structure of Pb-EDTA, gradually decarboxylating, and ultimately generating formic acid, acetic acid, and NO3−. The released metal ions were reduced by eaq− and ·H to obtain lead monomers. Residual toxicity analysis indicates that the toxicity of degradation products generated by electron beam irradiation is significantly reduced. Experimental results showed that electron beam irradiation can effectively degrade Pb-EDTA and recover lead ions simultaneously.
1. Introduction
With the continuous development of industrial production, sectors like electroplating and metal smelting generate and discharge a large amount of heavy metal wastewater every year.1 Due to concerns for human health and the ecological environment, heavy metal pollution has grown to be a significant issue today.2 One of the most pervasive heavy metal contaminants is lead (Pb), primarily generated through smelting, mining, coal burning, waste incineration, and leaded gasoline production.3 Lead is a toxic heavy metal that can be inhaled and ingested from a variety of places, including contaminated food, water, soil, and air. Compared with lead alone, synthetic chelating agents like ethylenediamine tetraacetate (EDTA) can create robust complexes with Pb2+, which are more stable and complex.4 As a potent hexadentate chelating ligand, EDTA is mostly utilized in pharmacy, textiles, papermaking, household detergents, and industrial cleaning.5 Even though EDTA is not toxic to mammals at ambient levels, concerns have been raised that it may facilitate the transfer of toxic heavy metals from sewage sludge and sediments since EDTA can reactivate adsorbed or precipitated metal ions.4,5
To degrade these heavy metal complexes with high mobility, a number of treatment techniques have been developed such as adsorption,6 chemical precipitation,7 and ion exchange.8 However, because of the persistent solubility characteristics of metal complexes, these conventional processes often fail to achieve the desired degradation.9,10 Due to the great oxidizing ability, advanced oxidation processes (AOPs) have developed as effective chemical oxidation technologies for the degradation of heavy metal complexes.11 The reaction mechanism of advanced oxidation technology is a process that generates ·OH through different techniques,12 including Fenton oxidation,13,14 electrochemical oxidation,15 photocatalytic oxidation,16,17 and ozone oxidation.18 Once ·OH is formed in the system, the organic matter in the water body is attacked by a series of chain reactions, degrading it to H2O, CO2, or inorganic salts, etc.19 There have been some studies using AOPs to degrade Pb-EDTA. Vohra et al. (2000)20 showed that UV/TiO2 was effective in removing Pb-EDTA complexes from water. Finžgar et al. (2006)21 used ozone/UV to treat heavy metal complexes in soil extractants with actual removal rates of 49.6% for Pb and 19.9% for Zn during the degradation of Pb-EDTA and Zn-EDTA, respectively. However, according to Zhao et al. (2014),22 photocatalytic oxidation can be limited by the rapid compounding of pairs of electron holes and the low use of visible light. Low oxidation efficiency can also be caused by the selectivity and limited solubility of ozone in water.23 In addition, the degradation products of heavy metal complexes treated with AOPs require further alkali treatment and other means to recover the heavy metals. Therefore, with the aim to address the problems that still exist with the present AOPs, it is urgently necessary to further investigate and explore alternative procedures for the degradation of heavy metal–organic complexes.24
Electron Beam (EB) irradiation is a technology that uses high-energy radiation to generate active products such as oxidizing radicals and reducing radicals from water irradiation without changing the radioactivity of the solution, as shown in eqn (1).25,26
| | |
H2O → 2.7·OH + 2.6eaq− + 0.6H· + 2.6H3O+ + 0.45H2 + 0.7H2O2
| (1) |
The numbers in the formula indicate the values of the radiochemical yield (the so-called G-value) in a pure neutral aqueous solution, which was defined as the number of product molecules formed (or initial molecules changed) for every 100 eV of energy absorbed.27 The most important advantage of using EB irradiation is the highly effective in situ generation of the extremely reactive oxidation radical ·OH and unique strong reductive hydrated electrons by water radiolysis, which can achieve advanced oxidation and reduction processes (AO/RP) simultaneously.28 However, there has been limited research on the influencing factors, degradation mechanisms and pathways, and the toxicity of degradation products of EB irradiation.
This study focused on Pb-EDTA as the representative metal complex to study the influencing factors and mechanism of EB irradiation degradation. The experiment explored the impacts of absorbed dose, initial concentration, solution pH, and the co-existing substances in actual environments on degradation efficiency. The main active radicals in the reaction system were determined, the degradation byproducts were analyzed and the possible degradation pathways for Pb-EDTA were proposed. Furthermore, a quantitative structure–activity relationship (QSAR) was used to evaluate the toxicity of Pb-EDTA degradation products.
2. Experimental section
2.1. Chemical reagents
Lead chloride (PbCl2) and ethylenediaminetetraacetic acid disodium salt dihydrate (Na2EDTA) were purchased from Sinopharm Chemical Reagent Co., Ltd (China). Solutions of Pb-EDTA were prepared by mixing a solution of PbCl2 and Na2EDTA to obtain a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of Pb2+/EDTA. Analytical grade hydrogen peroxide, potassium persulfate, and formic acid for use as oxidizers were purchased from Merck and Sigma-Aldrich (China).
:1 molar ratio of Pb2+/EDTA. Analytical grade hydrogen peroxide, potassium persulfate, and formic acid for use as oxidizers were purchased from Merck and Sigma-Aldrich (China).
Analytical grade sodium carbonate, sodium bicarbonate, sodium sulfate, sodium sulfite, nitrite, and sodium nitrite were purchased from Chemical Reagent Shanghai Co., Ltd (China), as anion production reagents. The pH of solutions was adjusted to the desired value by using HCl or NaOH. Since Pb-EDTA was obtained by mixing PbCl2 and Na2EDTA, a high concentration of Cl− was present in the solution itself, so the amount of Cl− introduced during pH adjustment using hydrochloric acid could be almost negligible. Deionized water was used for the preparation and dilution of solutions.
2.2. Irradiation experiment procedure
The Pb-EDTA degradation experiments were conducted with an electron accelerator (GJ-2-II, Xianfeng Electrical Company) supplied by the Institute of Applied Radiation, Shanghai University, China. The beam energy of the device was 1.8 MeV and the variable current was 0–10 mA. Before irradiation, the prepared Pb-EDTA solution was uniformly encapsulated in plastic bags with a volume and thickness of 20 mL and 2 mm, respectively. If required, the samples were purged with high-purity gas (N2 or O2) for half an hour before irradiation. The packaged sample was irradiated by the electron accelerator, with two sets of irradiation doses. One group was set at 4, 6, 8, and 10 kGy, which was a low radiation dose group. The other group was set at 50, 60, 80, 100, and 120 kGy, which was a high radiation dose group. The 10 mL Fricke dosimeter solution samples were stored in 40 mL, 1–2 mm thick polyethylene bags at room temperature (20 ± 2 °C). We placed the Fricke dosimeter and Pb-EDTA solutions under the same conditions for simultaneous irradiation to measure the absorbed dose of the Pb-EDTA solutions. Since the thickness of the irradiation bags was only 1–2 mm, the effect on the penetration of the electron beam was almost non-existent, and therefore the loss of the electron beam irradiation dose was negligible. After irradiation, each sample was centrifuged at 12000 rpm for 15 min and then filtered through a 0.45 μm aqueous polytetrafluoron (PTFE) syringe filter for analysis. All of the experiments were carried out in three sets of parallel experimental measurements.
The remaining metal complexes Pb EDTA, Pb, and total organic carbon content in the irradiated solution were analyzed and detected, and the removal efficiency of each indicator was calculated by eqn (2).
| |
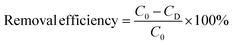 | (2) |
C0: the initial content of metal complexes, metal ions, and TOC.
CD: the residual content of metal complexes, metal ions, and TOC.
2.3. Analytical methods
The concentration of Pb-EDTA was measured by high-performance liquid chromatography (HPLC, Agilent 1260) equipped with a C18 column (250 mm × 4.6 mm × 5 μm). The sample was analyzed with an injection volume of 20 μL. The isocratic elution was comprised of 80% formate buffer (15 mM formic acid, 5 mM sodium formate, and 1 mM TBA-Br) and 20% acetonitrile (v/v) at a temperature of 25 °C. The flow rate was set as 1 mL min−1 and the detection wavelength was 254 nm.9
Atomic Absorption Spectrometry (AA, Agilent 280FS) was utilized to quantify the lead ions concentration. Using acetylene as the fuel gas, the detection wavelength is set at 217.0 nm, with a lamp current of 10 mA. The flame composition is air/acetylene, maintained at an airflow rate of 13.5 L min−1 and an acetylene flow rate of 2.00 L min−1. The concentration measurement range spans from 0 to 20 mg L−1.
The mineralization of Pb-EDTA was evaluated by total organic carbon (TOC) concentration, which was estimated by multi N/C 3100 TOC analyzer (Analytik Jena AG Corporation, Germany).
The distribution of Pb-EDTA species as a function of pH was obtained from the Visual MINTEQ 3.0 software.
The intermediates of Pb-EDTA were identified by HPLC (Agilent 1260) coupled with quadrupole time of flight mass spectrometry (Q-TOF-MS, Agilent 6545) in full-scan mode.
Formic acid, acetic acid, oxalic acid, glycolic acid, and NO3− were detected by ion chromatography (IC, ICS-6000, Thermo Scientific) with a conductivity detector.
After Pb-EDTA complexation, the precipitates were collected by pumping and filtering, and dried in a vacuum freeze drier. The crystalline structure of the precipitate was characterized by X-ray diffraction analysis (XRD, D/max 2550 V, Rigaku) with Cu Kα radiation at a 2θ scanning rate of 8° min−1 (40 kV, 25 mA).
2.4. Toxicity assessment
The software toxicity evaluation software (T.E.S.T.) was applied to calculate the growth inhibition, bioaccumulation factors (BAF), developmental toxicity, and acute toxicity of Pb-EDTA and its degradation intermediates, which was based on quantitative structure–activity relationship (QSAR) models.
3. Results and discussion
3.1 Effect of the initial concentration of Pb-EDTA
To determine the degradation efficiency of Pb-EDTA at varied concentrations, we set up a variety of concentration gradients (0.25, 0.5, 0.75, and 1 mM) in the irradiation experiment with absorbed doses of 4, 6, 8, and 10 kGy. As shown in Fig. 1a, Pb-EDTA was effectively degraded at low absorbed doses, indicating that EB irradiation can degrade heavy metal complexes. The pseudo second order kinetic model can be fitted to describe the decomposition of Pb-EDTA, as indicated in eqn (3):29| |
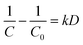 | (3) |
where k represents the pseudo second order rate constant, and D represents the absorbed dose of EB irradiation. As observed in Fig. 1a, the value of k decreased with the increase of Pb-EDTA concentration, indicating that the metal complex degraded more readily when its initial concentration was lower. The primary cause of this phenomenon is that as the absorbed dose increases, more intermediates are generated during the degradation of Pb-EDTA at high concentrations, and these intermediates also react with active radicals to reduce their concentration.30 As shown in Fig. 1c, under the low irradiation dose group, Pb2+ was not effectively removed, and the highest removal rate of Pb2+ was only 24.42% (10 kGy).
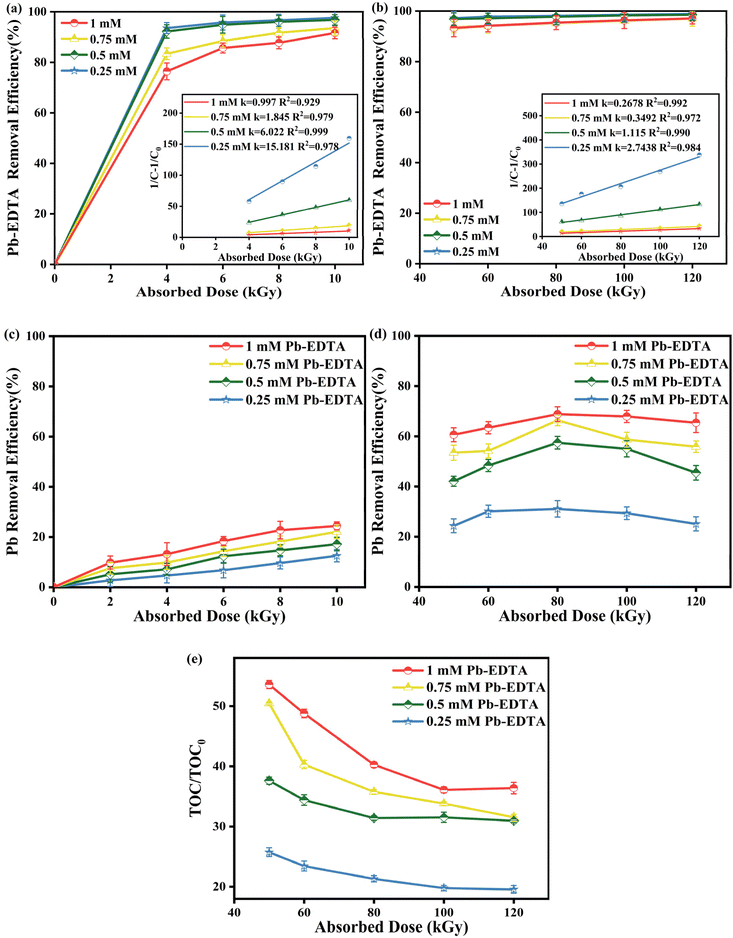 |
| | Fig. 1 (a) The impact of initial concentration on Pb-EDTA degradation under the low dose group; (b) the impact of initial concentration on Pb-EDTA degradation under the high dose group; (c) the impact of initial concentration on Pb2+ removal under the low dose group; (d) the impact of initial concentration on Pb2+ removal under the high dose group; (e) the impact of initial concentration on TOC removal. | |
To achieve synchronous removal of heavy metal ions, we further increased the absorbed dose to 50, 60, 80, 100, and 120 kGy. As shown in Fig. 1b, Pb-EDTA was almost completely removed. Fig. 1d shows that at a specific Pb-EDTA concentration, the removal rate of Pb2+ increased first and then decreased as the EB irradiation absorbed dose increased. With the absorbed dose was 80 kGy and the starting concentration was 1 mM, the removal rate of Pb2+ was 68.82%. By the time the absorbed dose reached 120 kGy, the removal rate of Pb2+ dropped to 65.40%.
This is because when the irradiation dose is low, the concentration of reactive radicals is also low, and the interaction between radicals is negligible, whereas with the gradual increase of irradiation dose, the concentration of reactive radicals increases, and the interactions between radicals are more intense, as shown in eqn (4)–(7),31,32 which reduces the effective concentration of reactive radicals, thus affecting the degradation rate of Pb-EDTA and the removal rate of Pb2+. To clarify the precipitate generated in the solution after irradiation, we characterized it through XRD, and the results proved that the precipitate was the lead elemental precipitate obtained after the reduction of Pb2+. We will discuss this in detail in Section 3.5 identification of degradation intermediates. To investigate the simultaneous removal of the complex and Pb2+, the removal rate of Pb2+ was more suitable for assessing the efficiency of the EB irradiation degradation.
| | |
·OH + ·OH → H2O2k = 5.5 × 109 M−1 s−1
| (4) |
| | |
·H + ·OH → H2Ok = 7.0 × 109 M−1 s−1
| (5) |
| | |
eaq− + ·OH → OH−k = 3.0 × 1010 M−1 s−1
| (6) |
| | |
H2O + ·H + eaq− → H2 + OH−k = 2.5 × 1010 M−1 s−1
| (7) |
Besides the pollutant removal efficiency, the mineralization rate can also be used to assess the degradation effect of EB irradiation.33 To illustrate that the TOC values detected after irradiation did not affected by the degradation of the irradiated bags, we used pure water for comparison with blank irradiation at 50–120 kGy, and the results, shown in Fig. S1,† showed that with increasing irradiation dose, only very low TOC was detected in the pure water, which was negligible. Fig. 1e demonstrates the TOC removal of Pb-EDTA at different concentrations and absorbed doses. It is apparent that the mineralization rate of the complexes continuously rises when the absorbed dose increases. This is mainly because of the rise in absorbed dosage, which causes ·OH to oxidize and break down more intermediate products. The results demonstrate that EB irradiation is a successful technique for pollutant degradation, achieving efficient removal of pollutants and the reduction and precipitation of heavy metals.
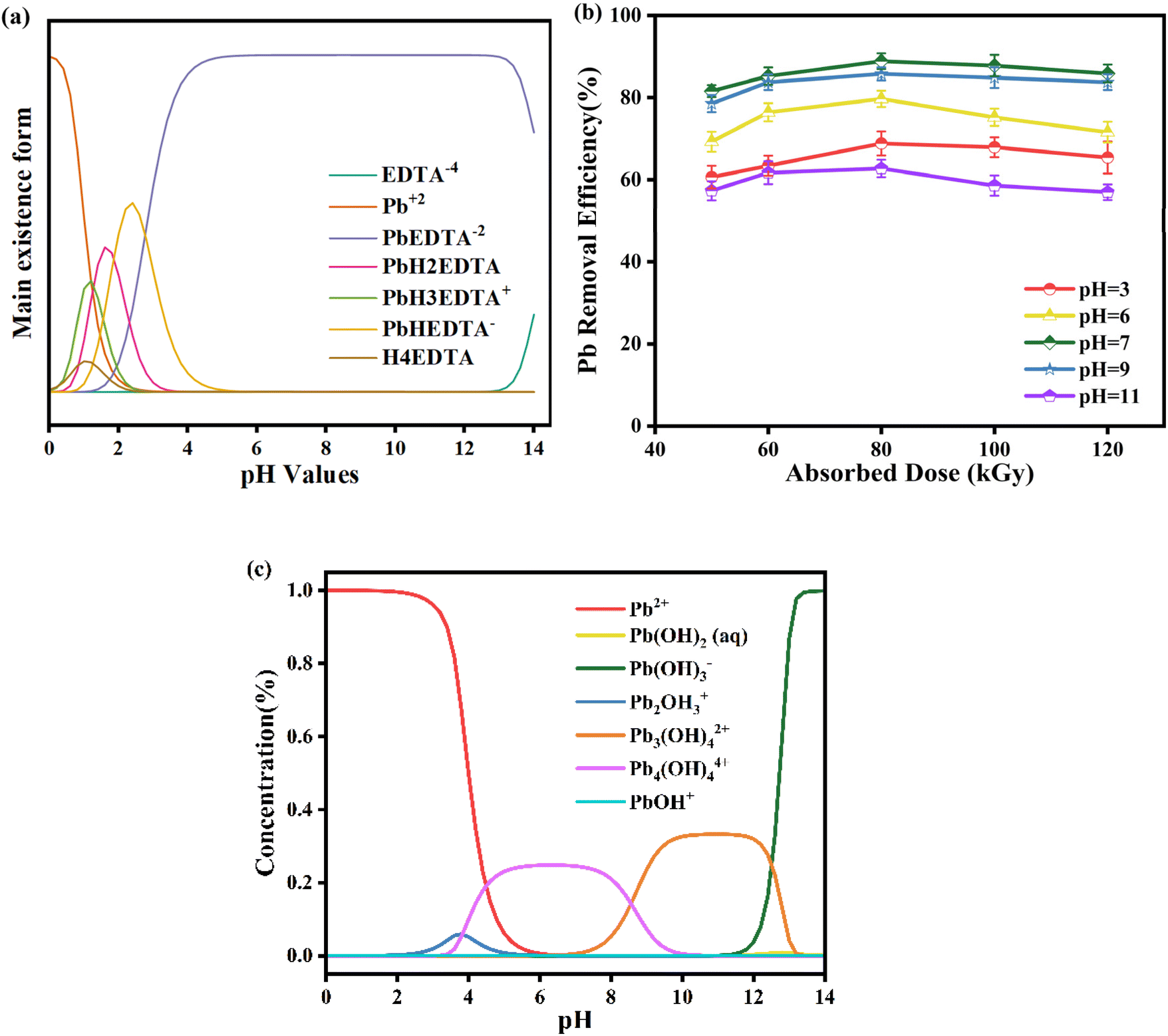
3.2. Effect of the initial pH of Pb-EDTA
The existence form of the heavy metal complexes in the water body changes with the change of pH. The distribution of Pb-EDTA complex morphology and its content change in the pH range of 0–14 were simulated by Visual MINTEQ software, as shown in Fig. 2a. Pb-EDTA is stable over a wide range of pH conditions and mainly exists in the form of PbEDTA−2. Only under acidic conditions at pH 3, Pb-EDTA is protonated into PbHEDTA− and PbH2EDTA−, which are more prone to react with ·OH than PbEDTA−2. Generally speaking, protonated species are more susceptible to the attack of reactive free radicals than deprotonated species, so acidic conditions are favorable for the removal of Pb-EDTA. The removal rate of Pb2+ through the irradiation degradation of Pb-EDTA at pH 3, 6, 7, 9, and 11 was shown in Fig. 2b. We selected a concentration of 1 mM for Pb-EDTA in our experiment, which is similar to the concentration of pollutants in the actual environment, ensuring that the experiment can accurately reflect the removal of Pb-EDTA in water bodies. The results indicated that the efficiency of Pb2+ removal initially increased and then decreased with varying pH levels in the solution. At an absorbed dose of 80 kGy, the Pb2+ removal rate was 68.82% at pH 3. The removal of Pb2+ was more effective when the solution pH was adjusted to neutral, reaching a peak rate of 88.85% at pH 7 (80 kGy). However, as the pH level continued to rise to 11, the Pb2+ removal rate dropped to a minimum of 62.74% (80 kGy). This indicates that the removal of Pb2+ is not only influenced by the initial morphology of Pb-EDTA, but also attributed to two reasons: (1) under the over-acidic condition, eaq− generated by EB irradiation reacts with hydrogen ions, making it fail to reduce Pb2+, as shown in eqn (8).34 The research results are based on earlier studies, as shown in Fig. S2,† where the G value of active radicals generated by EB irradiation changes with pH values. Under alkaline conditions, the reduction of Pb2+ removal rate could be owing to the lower yield of ·OH produced by EB irradiation hydrolysis and the reaction between hydroxyl radicals and hydroxide ions, as shown in eqn (9),35 the degree of reaction between radicals and complexes weakens and thus the removal efficiency is reduced.27 (2) Studies have shown that lead ions exist in different forms at different pH values,36 as seen in Fig. 2c. Under acidic conditions, the main form of lead ions is Pb2+, and when under alkaline conditions, they mainly exist in the form of Pb(OH)3−. In general, the present state of Pb2+ tends to deprotonate as the pH of the solution increases. Compared to deprotonated species, protonated species are more susceptible to attack by reactive radicals, so alkaline conditions are unfavorable for Pb2+ removal compared to acidic conditions.| | |
eaq− + H+ → ·Hk = 2.3 × 1010 M−1 s−1
| (8) |
| | |
OH− + ·OH → ·O−k = 1.2 × 1010 M−1 s−1
| (9) |
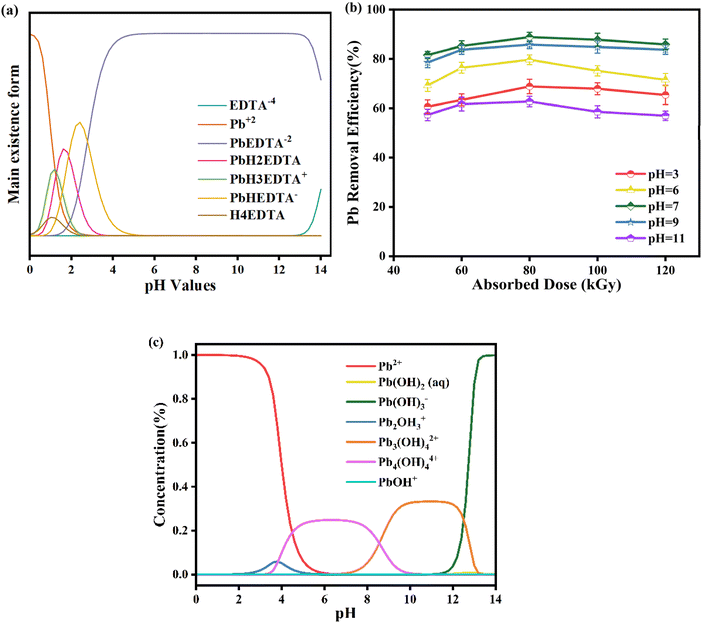 |
| | Fig. 2 (a) The different forms of Pb-EDTA at various pH values; (b) the impact of initial pH on Pb2+ removal; (c) the different forms of lead ions at various pH values. | |
3.3 Effect of reactive species
The effect of reactive radicals generated during irradiation on the removal of Pb-EDTA was determined by adding different types of quenching agents to the solution. O2 can react with eaq− and H· (eqn (10) and (11)).37 Therefore, when the solution is purged with O2, ·OH plays a predominant role in the degradation of Pb-EDTA. When N2 is introduced into the solution, it does not affect the activity of various types of free radicals. This is mainly because N2 is an inert gas used only to degas the solution. The activity of reactive free radicals varies under different pH conditions. When the solution pH is greater than 3, the contribution of H· is minimal. In this case, the primary reactive species here are eaq− and ·OH. However, when the pH of the solution is less than 3, the contribution of eaq− is minimal. In this instance, the primary active ingredients are H· and ·OH.38 Tert-Butanol (TBA) was added separately under the two pH conditions. Since TBA is an effective burster of ·OH (eqn (12)),39 at this time, H· dominates in solutions with a pH above 3, in removing pollutants.| | |
O2 + eaq− → O2˙−k = 9.1 × 1010 M−1 s−1
| (10) |
| |
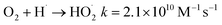 | (11) |
| | |
(CH3)3COH + ·OH → (CH3)2C(·CH2)OH + H2Ok = 1.9 × 1010 M−1 s−1
| (12) |
| | |
Cl− + OH· → ClOH˙−4.3 × 109 M−1 s−1
| (13) |
| | |
ClOH˙− → Cl− + OH·6.1 × 109 M−1 s−1
| (14) |
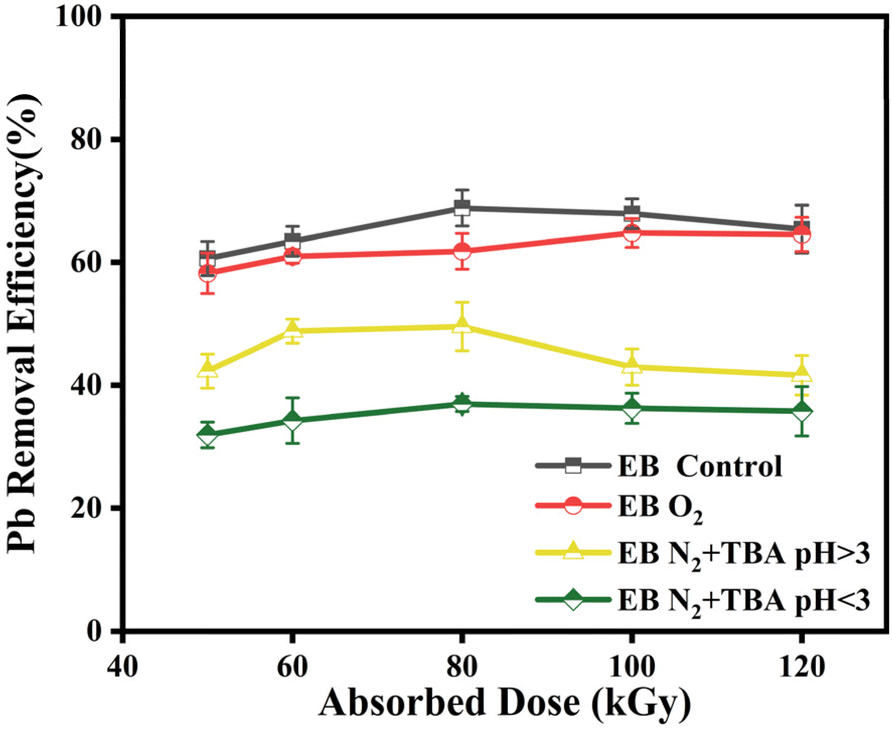
The impact of different reactive radicals on the removal rate of Pb2+ is displayed in Fig. 3. Compared with the initial group, the reduction efficiency of Pb2+ decreased slightly under O2 purification, and ·OH predominated in the degradation of pollutants. After adding TBA, when H· and eaq− were the main free radicals, Pb2+ reduction efficiency significantly decreased. This indicates that the removal of ·OH prevents Pb-EDTA from being oxidatively degraded, and Pb2+ is not effectively released from the metal-complex ligand, thus hindering its reduction and removal. Since the experimental solution was prepared by mixing PbCl2 and Na2EDTA, this result also suggests that the presence of Cl− and the interreaction with hydroxyl radicals (eqn (13) and (14))40 do not scavenge hydroxyl radicals from the system. Therefore, it can be inferred that the electron beam irradiation technique for Pb-EDTA removal is influenced by various active free radicals, primarily the ·OH radicals, which can disrupt the structure of Pb-EDTA, enabling the stepwise decarboxylation and simultaneous generation of low molecular weight compounds. Subsequently, the released metal ions are reduced by eaq− and H· radicals.
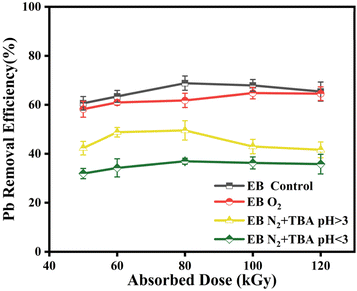 |
| | Fig. 3 The impact of different active radicals on Pb2+ removal. | |
3.4 Identification of degradation intermediates
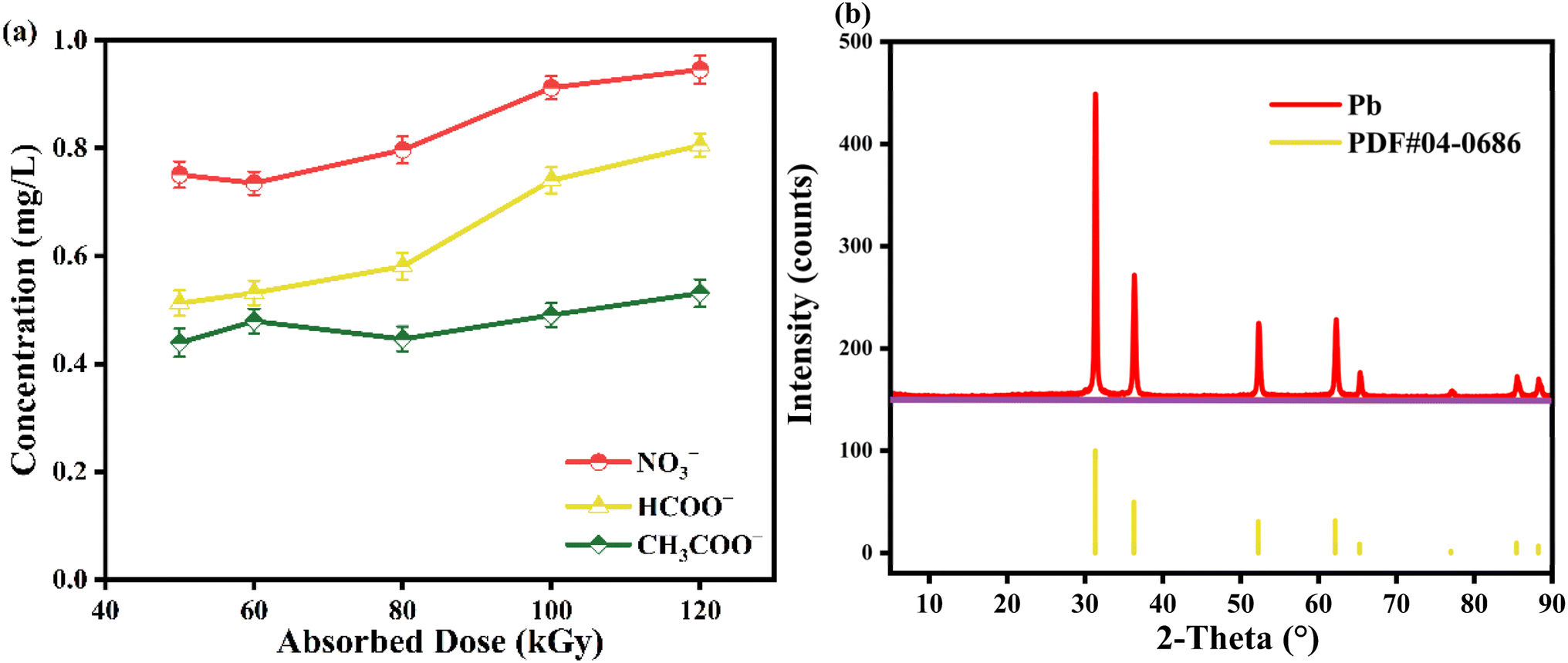
The majority of the pertinent research suggests that metal-EDTA degrades through a similar process in which the complex is progressively decarboxylated by reactive radicals attacking the C–N bond of the ligand.41–43 To investigate the degradation pathway of Pb-EDTA in the EB irradiation process, the degradation by-products were analyzed using a liquid-phase mass spectrometer and an ion chromatography analytical instrument. Table S1† shows the degradation intermediates of Pb-EDTA formed in EB irradiation. As reported in other literature,9,44 Pb-ED3A (m/z = 437.3748), Pb-ED2A (m/z = 381.3546), NTA (m/z = 191.139), and IMDA (m/z = 133.1027) were the major degradation products during the EB irradiation reaction. Analysis of small molecule acids using ion chromatography revealed that formic acid, acetic acid, and NO3− were mainly present in the irradiated solution. Fig. 4a shows that all these small molecule acids gradually increased with increasing absorbed doses. Significant precipitates could be found in the samples after EB irradiation, which were characterized by XRD to further determine the composition and nature of the precipitates. As shown in Fig. 4b, the precipitated phase shows obvious diffraction peaks of Pb monoclinic crystal structure at 2θ values of 31.4°, 36.3°, 52.3°, 62.2°, 65.4°, 85.6°, and 88.2°. Fig. S3† shows the SEM image of the precipitated product.
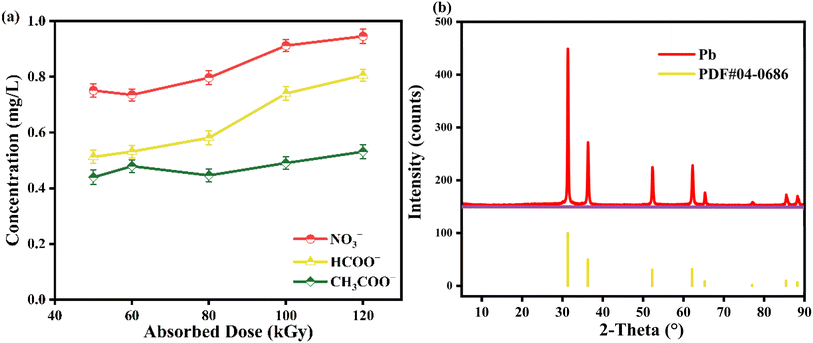 |
| | Fig. 4 (a) The content of formic acid, acetic acid, and nitric acid; (b) XRD of precipitates. | |
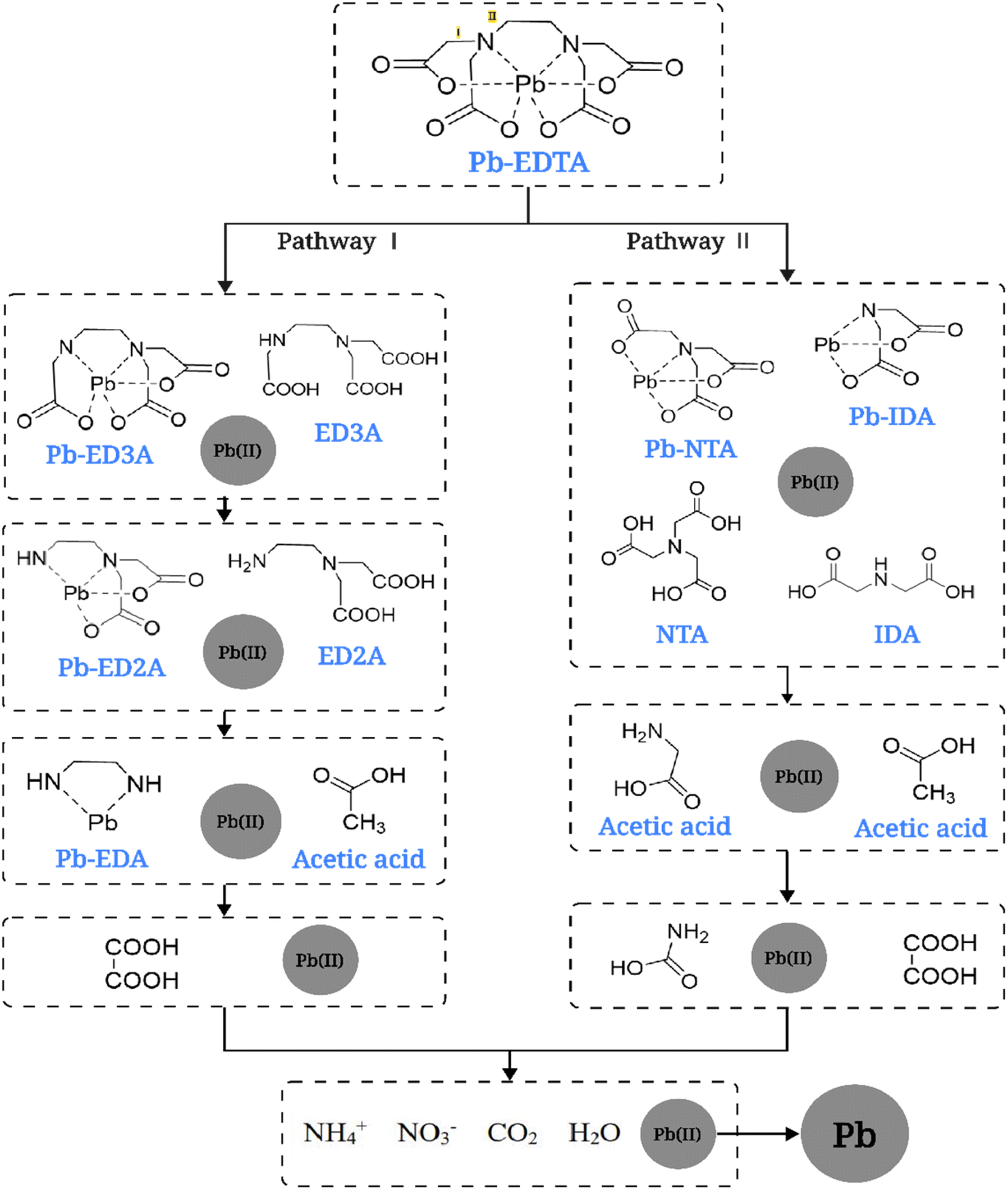
Based on the relevant references and detected degradation intermediates and degradation products,45 the possible degradation pathways of Pb-EDTA during the EB irradiation reaction were proposed, as shown in Fig. 5. Two possible degradation pathways were hypothesized to exist. First, ·OH attacks the C–N bond of Pb-EDTA, causing it to break and split into Pb-ED3A and CH3COOH; then continues to attack the carboxyl site of Pb-ED3A and subsequently forms Pb-ED2A. In the degradation pathway II, the reactive radicals act on the C–N bond to generate NTA and IMDA. Finally, the generated intermediates are further oxidized to small organic acids and inorganic ions including formic acid, acetic acid, and NO3−. The released Pb2+ is reduced to lead monomers by the reducing radicals. In summary, similar to previous studies, the degradation of Pb-EDTA by EB irradiation is mainly achieved by attacking the complex with active radicals and gradually decarboxylating it.
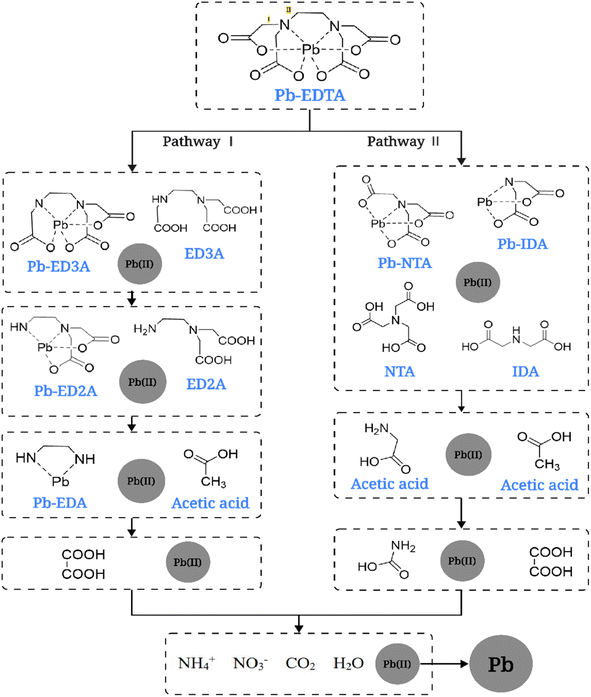 |
| | Fig. 5 Possible degradation pathways of Pb-EDTA. | |
3.5 Comparison of EB, EB/H2O2, EB/K2S2O8, EB/HCOOH process
EB irradiation technology can be combined with different oxidants and reductants to affect the degradation efficiency of Pb-EDTA. In our experiments, we investigated the impact of oxidants hydrogen peroxide (H2O2),46 potassium persulfate (K2S2O8),47 and the reductant formic acid (HCOOH)48 on the removal rate of Pb2+. To have an appropriate comparison of the effect of just the additives, we adjusted the solution pH to a constant value (pH 7) by adding NaOH after adding different oxidizing/reducing agent concentrations.
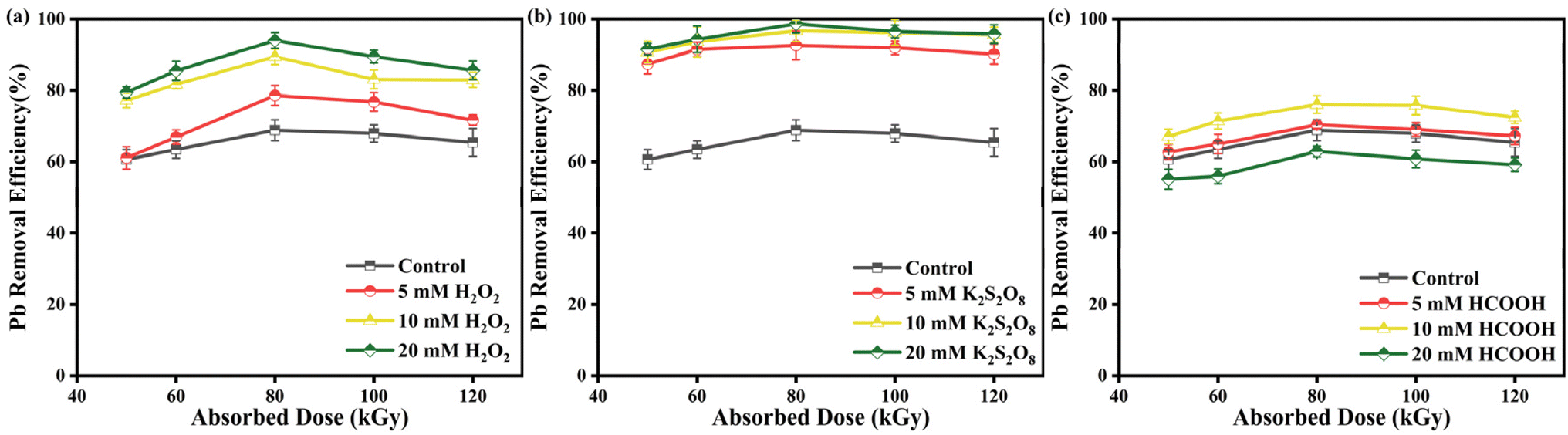
3.5.1 Removal rate of Pb2+ in EB/H2O2 process. Fig. 6a displays the removal rate of Pb2+ through EB irradiation degradation of Pb-EDTA after adding 5, 10, and 20 mM H2O2. At 80 kGy, when the concentration of H2O2 increased to 20 mM, the highest removal rate reached 93.97%. This is mainly because when H2O2 concentration increases, it is conducive to developing oxidizing active free radical ·OH (eqn (15) and (16)),49 which is conducive to the removal of Pb-EDTA.| | |
eaq− + H2O2 → ·OH + OH−k = 1.1 × 109 M−1 s−1
| (15) |
| | |
H· + H2O2 → ·OH + H2Ok = 9.0 × 107 M−1 s−1
| (16) |
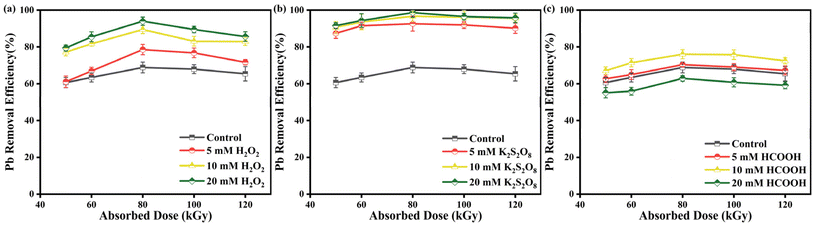 |
| | Fig. 6 (a) The impact of H2O2 dosage on Pb2+ removal; (b) the impact of K2S2O8 dosage on Pb2+ removal; (c) the impact of HCOOH dosage on Pb2+ removal. | |
3.5.2 Removal rate of Pb2+ in EB/K2S2O8 process. Fig. 6b illustrates the removal rate of Pb2+ through EB irradiation degradation of Pb-EDTA after adding 5, 10, and 20 mM K2S2O8, respectively. The highest removal rate of Pb2+ reached 98.60% after adding 20 mM K2S2O8 (80 kGy). It indicates that the addition of K2S2O8 to the system improved the removal rate of Pb2+. This is the result of the rapid reaction of S2O82− with ·H and eaq− converted to reactive ·SO4− (eqn (17) and (18)).37 Because of its high redox potential (E0 = 2.73 V), ·SO4− is regarded as a potent one-electron oxidant akin to ·OH (E0 = 2.60 V).50 Some studies have indicated that ·SO4− could react with S2O82− to form a reductive free radical (eqn (19)).51| | |
S2O82−+ H· → ·SO4− + H+ + SO42−k = 2.5 × 107 M−1 s−1
| (17) |
| | |
S2O82− + eaq− → ·SO4−+ SO42−k = 1.1 × 1010 M−1 s−1
| (18) |
| | |
·SO4−+ S2O82− → S2O8˙− + SO42−k = 1.2 × 106 M−1 s−1
| (19) |
3.5.3 Removal rate of Pb2+ in EB/HCOOH process. Fig. 6c displays the removal rate of Pb2+ through EB irradiation degradation of Pb-EDTA after adding 5, 10, and 20 mM HCOOH. Compared to the previous two oxidants, adding 10 mM HCOOH at 80 kGy resulted in a removal rate of only 76.01% for Pb2+. When the concentration of HCOOH was raised to 20 mM, the removal rate of Pb2+ decreased to 62.87% (80 kGy). This phenomenon is caused by the fact that HCOO− can react with ·OH (eqn (20)).52 Therefore, low concentrations of HCOO− promote the reductive precipitation of Pb2+, whereas when HCOO− is added at too high a dosage, too many reducing species are produced in the system, inhibiting the generation of oxidizing radicals, which negatively affects the generation of reducing species converted from oxidizing species, leading to a significant inhibition of Pb2+ removal.| | |
·OH + HCOO− → ·CO2− + H2Ok = 3.2 × 109 M−1 s−1
| (20) |
Meanwhile, to better illustrate the influence of the dose-effect on the removal efficiency, supplementary experiments were conducted on the removal efficiency of Pb2+ with different additive contents at low doses, and the results are shown in Fig. S4.†
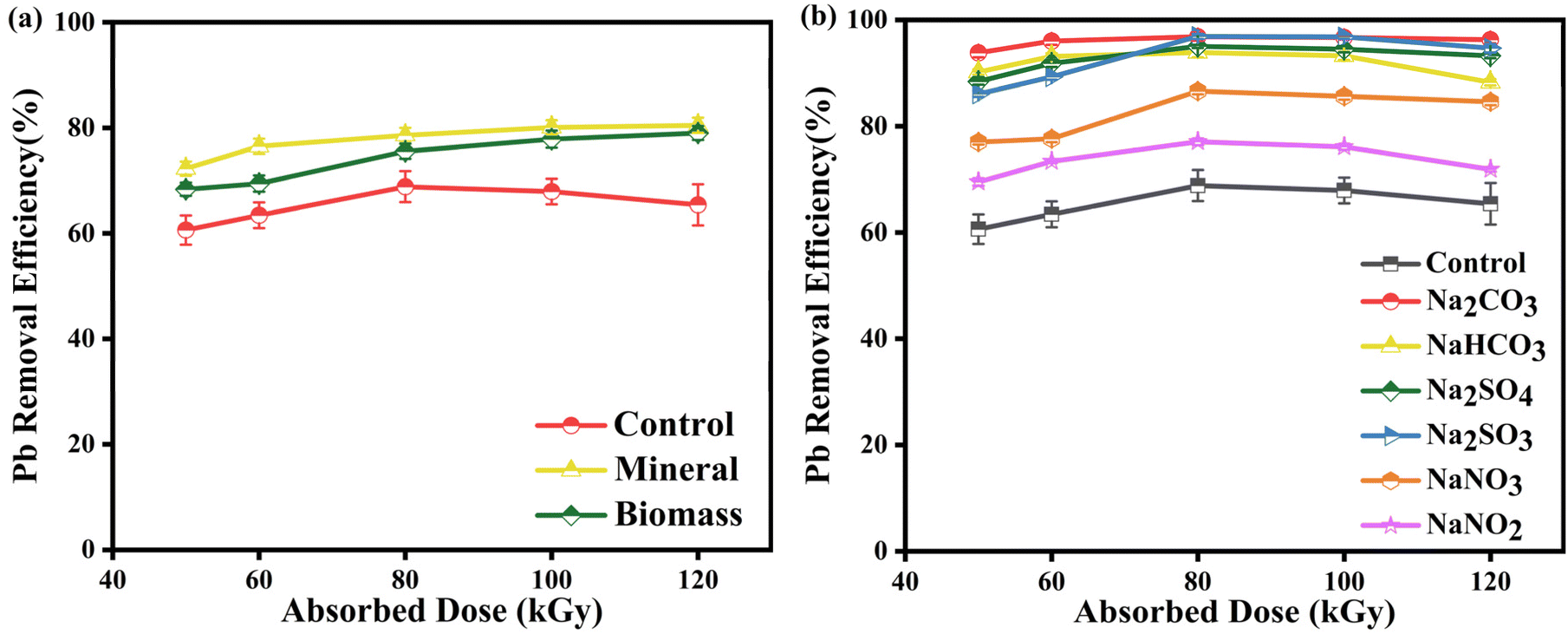
3.6 Effects of common co-existing substances in real water bodies
For the common components in industrial wastewater, we select natural organic matter and inorganic anions to study the impact of coexisting substances on Pb-EDTA degradation in the EB irradiation process.53,54
3.6.1 Effect of humic acid. As shown in Fig. 7a, the addition of a certain amount of humic acid can significantly enhance the precipitation of Pb2+. This is because humic acid itself can act as an electron medium or electron donor, excite the triple excited state of HA (3HA*), and generate a substantial amount of reactive oxidation radicals like ·OH, single molecule oxygen, and H2O2, further contributing to the removal efficiency.54,55 According to studies, humic acid contains a large amount of inorganic ions, which can also promote the precipitation of Pb2+.
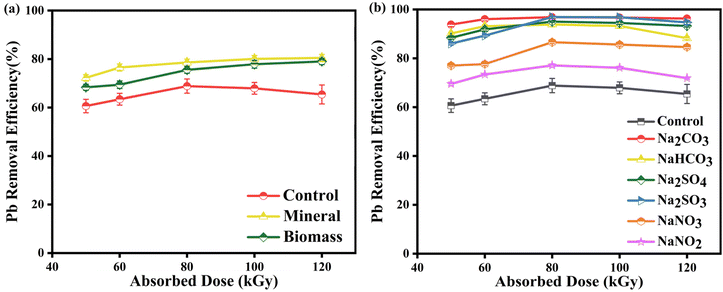 |
| | Fig. 7 (a) The impact of humic acid on Pb2+ removal; (b) the impact of coexisting ions on Pb2+ removal. | |
3.6.2 Effect of coexisting ions. Since common inorganic anions are present in real water, their coexistence in water significantly affects the reaction process, as they may influence the reactions of Pb-EDTA with the reactive radicals generated during irradiation. In this study, these anions (CO32−, HCO3−, NO3−, NO2−, SO42−, and SO32−) were selected (all with a concentration of 10 mM) and added to the Pb-EDTA solution to be irradiated.56 As displayed in Fig. 7b, the addition of various anions enhanced the removal rate of Pb2+ to varying degrees compared to the control group.When CO32− or HCO3− is introduced, they react with ·OH and eaq− to produce CO3˙− (eqn (21)–(25)),57 with CO32− and HCO3− having significantly higher reaction rates with ·OH than with eaq−, resulting in the reduction of ·OH in the EB system,58 which thereby inhibiting the degradation of Pb-EDTA. However, on the other hand, CO32− reacts with ·OH to form CO3˙− also has some ability to oxidatively degrade metal complexes, and compared with ·OH, CO3˙− has a lower redox potential (1.59 V), so the free lead ions released by irradiation degradation will not be re-oxidized due to the strong oxidizing atmosphere in the system, which is more conducive to the removal of lead ions by reduction.
| | |
HCO3− + eaq− → HCO32−k = 6.0 × 105 M−1 s−1
| (21) |
| | |
HCO3− + ·OH → CO3˙− + H2Ok = 8.5 × 106 M−1 s−1
| (22) |
| | |
HCO3− + H· → CO3˙− + H2k = 4.0 × 104 M−1 s−1
| (23) |
| | |
CO32− + eaq− → CO33−k = 3.9 × 105 M−1 s−1
| (24) |
| | |
CO32− + ·OH → CO3˙− + OH−k = 3.9 × 108 M−1 s−1
| (25) |
The addition of SO42− promoted the degradation of Pb-EDTA. This is mainly because SO42− can combine with ·OH to generate highly oxidative compounds ·SO4− (eqn (26)).40 ·SO4−, as an additional oxidizing substance, can cooperate with the oxidizing radicals generated by electron beam irradiation and jointly attack the organic ligand structure, thus enhancing the removal efficiency of the system for Pb-EDTA. When SO32− is added, it interacts with ·OH to generate ·SO3− (eqn (27)), leading to the removal of excess ·OH in the system, thus freeing the de-complexed lead ions from the strong oxidizing atmosphere in the system and making it more conducive to the reductive removal by reducing radicals.
| | |
SO42− + ·OH → ·SO4− + OH−k = 6.5 × 107 M−1 s−1
| (26) |
| | |
SO32− + ·OH → ·SO3− + OH−k = 5.5 × 109 M−1 s−1
| (27) |
The addition of NO3− or NO2− promotes the reduction and removal of Pb2+ to a certain extent. This is because NO3− and NO2− easily react with ·OH, eaq− and ·H to generate ·NO3− and ·NO2− (eqn (28)–(35)),59 with oxidation-reduction potentials of 1.03 V and 0.94 V, respectively. This is much lower than the active potential of free radicals that originally existed in the system, leading to an increase in the reducing atmosphere in the system and further promoting the reduction and removal of Pb2+.
| | |
NO3− + eaq−→ NO3˙2−k = 9.7 × 109 M−1 s−1
| (28) |
| |
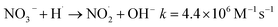 | (29) |
| | |
NO3− + H+ → HNO3k = (4.4–6.0) × 108 M−1 s−1
| (30) |
| | |
HNO3 + ·OH → NO3˙− + H2Ok = (0.88–1.2) × 108 M−1 s−1
| (31) |
| |
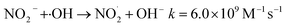 | (32) |
| | |
NO2− + ·OH → HNO3k = 1.0 × 1010 M−1 s−1
| (33) |
| | |
NO2− + H· → NO + OH−k = 7.1 × 108 M−1 s−1
| (34) |
| | |
NO2− + eaq− → NO3˙2−k = 3.5 × 109 M−1 s−1
| (35) |
Furthermore, inorganic ions such as carbonate and sulfate have higher reaction rates with free radicals, but compared to nitrate and nitrite ions, their presence significantly improves the removal efficiency of Pb2+. We speculate that Pb2+ released by the degradation of Pb-EDTA would be precipitated through reactions with inorganic anions, in addition to being reduced directly to Pb monomer. This speculation might be supported by the white precipitate observed in the irradiation bag after irradiation of the solution.
3.7 Residual toxicity analysis
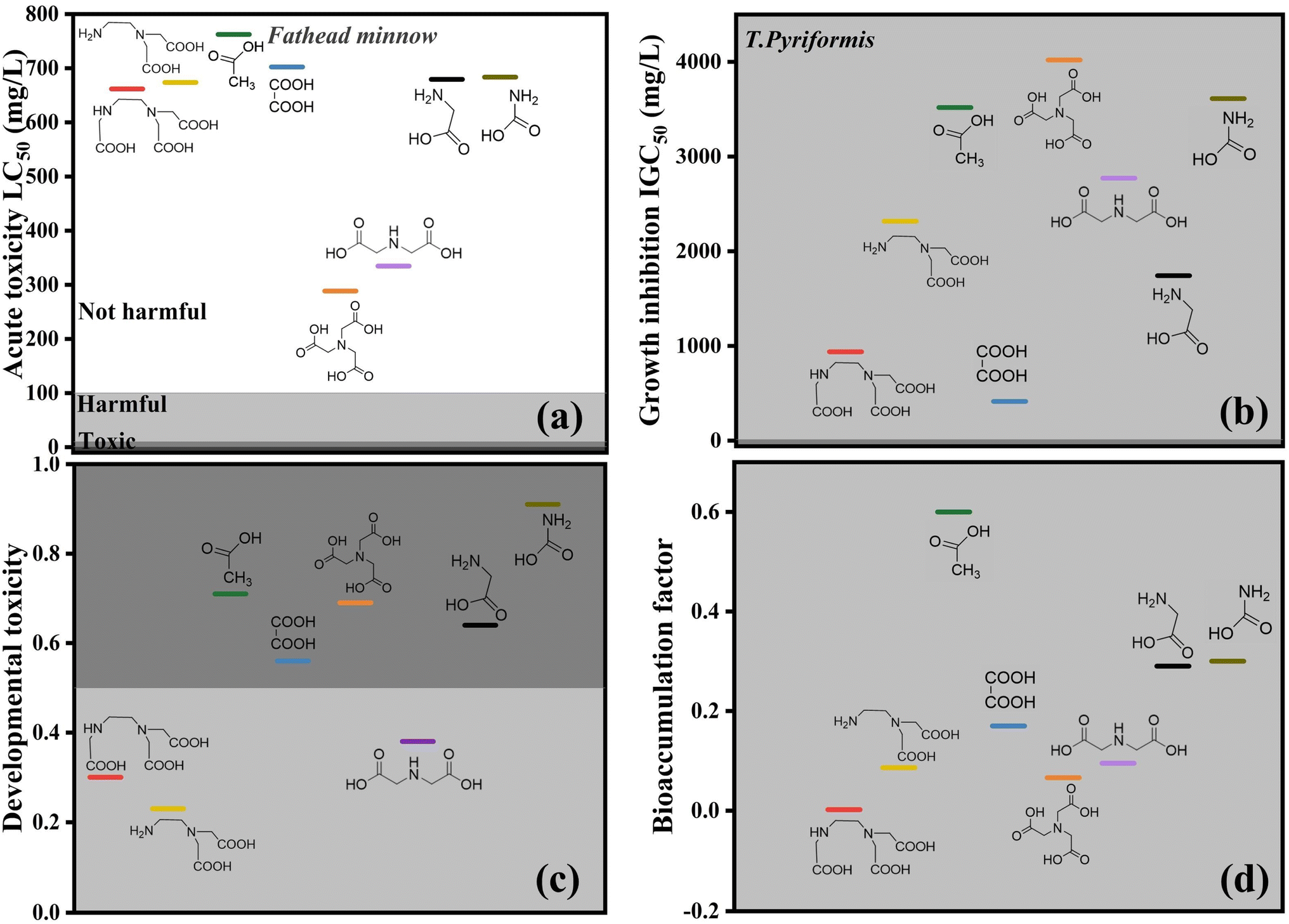
Considering the experimental cost, the toxicity of degradation intermediates was assessed in advance by theoretical calculations to verify the potential toxicity of irradiation-treated solutions. Ecological structure activity relationship (ECOSAR) is a computerized prediction system for toxicity in aquatic systems developed by the US Environmental Protection Agency (US EPA). The system uses mathematical models of example training sets of a range of chemicals to predict chemicals that may cause long-term effects. The model's prediction of toxicity for compounds to be tested presupposes that these compounds to be tested are sufficiently similar to those in the training set. Therefore, in the present study, the toxicity calculation software T.E.S.T (Version 5.1) based on Quantitative Constitutive Effect Relationship (QSAR) modeling was adopted to calculate the toxicity of Pb-EDTA and its degradation intermediates. Acute toxicity LC50, growth inhibition IGC50, developmental toxicity, and bioaccumulation factor (BAF) were selected. By comparing the toxic effects of EDTA before and after irradiation treatment, the hazardous effects of EB irradiation applied in the treatment of degraded Pb-EDTA on environmental organisms were evaluated.
Fig. 8 shows that the degradation intermediates of Pb-EDTA are less toxic, and ED2A and ED3A both show lower toxicity. Table S2† shows the specific toxicity data of the intermediates. The intermediate product was continually oxidized and degraded as the absorbed dose of EB increased. The toxicity of NTA and IDA was higher than that of ED2A and ED3A, but the toxicity of the final degradation products was far less than that of the intermediate products. As a result, the overall toxicity of the solution was trending downward during the degradation process. According to the total toxicity estimations from this investigation, the degradation of Pb-EDTA was accompanied by the generation of toxic intermediates. The toxic effect of the degradation products created by the EB irradiation was, however, significantly reduced, showing that it was an effective means.
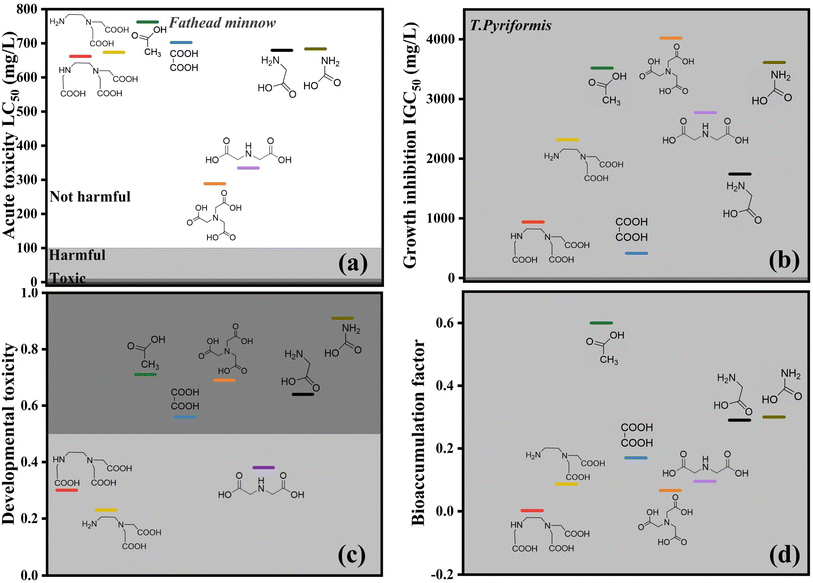 |
| | Fig. 8 Toxicity calculation of Pb-EDTA degradation intermediates generated during EB irradiation: (a) acute oral rat LD50; (b) growth inhibitory concentration of Tetrahymena pyriformis IGC50; (c) developmental toxicity; (d) bioaccumulation factor. | |
4. Conclusions
In this study, electron beam irradiation was used for the oxidative degradation of Pb-EDTA and the reduction of Pb2+ simultaneously. The article explored how various environmental substrates affect electron beam irradiation. The findings show that neutral conditions are favorable to the removal of Pb2+. Compared to other oxidants, K2S2O8 and H2O2 promoted the oxidative degradation of Pb-EDTA. The reactive radicals in the system were impacted by the addition of a certain amount of humic substances and inorganic ions, which altered the efficiency of the degradation. Experiments on mechanism research revealed that ·OH was the main radical for degradation of Pb-EDTA, and eaq− mainly reduced lead ions. The degradation pathway of Pb-EDTA was proposed based on the discovered intermediates and degradation products. Factors such as pH, inorganic ions, and natural organic matter can affect the concentration and types of free radicals. During electron beam irradiation, high concentrations of oxidative and reductive free radicals are generated. Oxidative free radicals attack Pb-EDTA, leading to decomplexation, while reductive free radicals interact with lead ions, causing their transformation into elemental lead.
However, it is important to note that Pb-EDTA removal is a complex process, and changes in the oxidizing and reducing properties of the system can both impact the removal of Pb-EDTA. Inorganic ions and organic molecules in the environment can react with free radicals, altering their concentrations and subsequently affecting the removal efficiency of Pb-EDTA. Experimental results have indicated that most environmental factors can inhibit the removal of Pb-EDTA to some extent, highlighting the need for further research on the transformation of free radicals in different environments.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank National Natural Science Foundation of China (No. 12175133, and 12075148). We would also appreciate the Guizhou Provincial Key Technology R&D Program (No. QKHZC-2024-152).
References
- X. Q. Kan, Y. Q. Dong, L. Feng, M. Zhou and H. B. Hou, Chemosphere, 2021, 267, 128909 CrossRef CAS.
- S. Huang, L. Gu, N. Zhu, K. Feng, H. Yuan, Z. Lou, Y. Li and A. Shan, Green Chem., 2014, 16, 2696–2705 RSC.
- F. Y. AlJaberi, J. Environ. Chem. Eng., 2018, 6, 6069–6078 CrossRef CAS.
- B. Nowack, F. G. Kari and H. G. Krüger, Water, Air, Soil Pollut., 2001, 125, 243–257 CrossRef CAS.
- F. G. Kari and W. Giger, Water Res., 1996, 30, 122–134 CrossRef CAS.
- L. Wu, H. Wang, H. Lan, H. Liu and J. Qu, Sep. Purif. Technol., 2013, 117, 118–123 CrossRef CAS.
- S. Mauchauffée and E. Meux, Chemosphere, 2007, 69, 763–768 CrossRef.
- D. Kołodyńska, H. Hubicka and Z. Hubicki, Desalination, 2008, 227, 150–166 Search PubMed.
- X. Huang, Y. Xu, C. Shan, X. Li, W. Zhang and B. Pan, Chem. Eng. J., 2016, 299, 23–29 CAS.
- J. Rivera-Utrilla, M. Sánchez-Polo, M. Á. Ferro-García, G. Prados-Joya and R. Ocampo-Pérez, Chemosphere, 2013, 93, 1268–1287 CAS.
- C. P. Huang, C. Dong and Z. Tang, Waste Manage., 1993, 13, 361–377 CAS.
- J. L. Wang and L. J. Xu, Crit. Rev. Environ. Sci. Technol., 2012, 42, 251–325 CrossRef CAS.
- P. Bautista, A. F. Mohedano, J. A. Casas, J. A. Zazo and J. J. Rodriguez, J. Chem. Technol. Biotechnol., 2008, 83, 1323–1338 CrossRef CAS.
- L. Szabó, T. Tóth, T. Engelhardt, G. Rácz, C. Mohácsi-Farkas, E. Takács and L. Wojnárovits, Sci. Total Environ., 2016, 551–552, 393–403 CrossRef PubMed.
- C. A. Martinez-Huitle and S. Ferro, Chem. Soc. Rev., 2006, 35, 1324–1340 RSC.
- X. Huang, X. Wang, D.-X. Guan, H. Zhou, K. Bei, X. Zheng, Z. Jin, Y. Zhang, Q. Wang and M. Zhao, Environ. Sci. Pollut. Res., 2019, 26, 8516–8524 CrossRef CAS PubMed.
- F. Rehman, M. Sayed, J. A. Khan, N. S. Shah, H. M. Khan and D. D. Dionysiou, J. Hazard. Mater., 2018, 357, 506–514 CrossRef CAS.
- J. L. Wang and H. Chen, Sci. Total Environ., 2020, 704, 135249 CrossRef CAS PubMed.
- A. O. Oluwole, E. O. Omotola and O. S. Olatunji, BMC Chem., 2020, 14, 62 CrossRef CAS PubMed.
- M. S. Vohra and A. P. Davis, Water Res., 2000, 34, 952–964 CrossRef CAS.
- N. Finžgar and D. Leštan, Chemosphere, 2006, 63, 1736–1743 CrossRef.
- X. Zhao, L. Guo and J. Qu, Chem. Eng. J., 2014, 239, 53–59 CrossRef CAS.
- T. E. Agustina, H. M. Ang and V. K. Vareek, J. Photochem. Photobiol., C, 2005, 6, 264–273 CrossRef CAS.
- M. Trojanowicz, A. Bojanowska-Czajka and A. Capodaglio, Environ.
Sci. Pollut. Res., 2017, 24, 20187–20208 CrossRef CAS.
- W. Jianlong and W. Jiazhuo, J. Hazard. Mater., 2007, 143, 2–7 CrossRef.
- N. K. Vel Leitner, I. Guilbault and B. Legube, Radiat. Phys. Chem., 2003, 67, 41–49 CrossRef.
- J. Wang and L. Chu, Radiat. Phys. Chem., 2016, 125, 56–64 CrossRef CAS.
- M. Trojanowicz, Sci. Total Environ., 2020, 718, 134425 CrossRef CAS PubMed.
- T. Li, S. He, L. Kou, J. Peng, H. Liu, W. Zou, Z. Cao and T. Wang, Sep. Purif. Technol., 2023, 306, 122588 CrossRef CAS.
- A. A. Basfar, H. M. Khan, A. A. Al-Shahrani and W. J. Cooper, Water Res., 2005, 39, 2085–2095 CAS.
- K. Lin, W. J. Cooper, M. G. Nickelsen, C. N. Kurucz and T. D. Waite, Appl. Radiat. Isot., 1995, 46, 1307–1316 CAS.
- B. G. Zheng, Z. Zheng, J. B. Zhang, X. Z. Luo, J. Q. Wang, Q. Liu and L. H. Wang, Desalination, 2011, 276, 379–385 CAS.
- L. J. Xu and J. L. Wang, Appl. Catal., B, 2012, 123, 117–126 Search PubMed.
- Y. Liu, J. Hu and J. Wang, Environ. Technol., 2014, 35, 2028–2034 CrossRef CAS PubMed.
- Z. Guo, F. Zhou, Y. Zhao, C. Zhang, F. Liu, C. Bao and M. Lin, Chem. Eng. J., 2012, 191, 256–262 CrossRef CAS.
- S. He, T. Li, L. Zhang, X. Zhang, Z. Liu, Y. Zhang, J. Wang, H. Jia, T. Wang and L. Zhu, Chem. Eng. J., 2021, 424, 130515 CrossRef CAS.
- Z. Huo, S. Wang, H. Shao, H. Wang and G. Xu, Environ. Sci. Pollut. Res., 2020, 27, 20807–20816 CrossRef CAS.
- S. Wang, J. Hu, S. He and J. Wang, J. Hazard. Mater., 2022, 433, 128727 CrossRef CAS PubMed.
- S.-H. Ma, M.-H. Wu, L. Tang, R. Sun, C. Zang, J.-J. Xiang, X.-X. Yang, X. Li and G. Xu, Nucl. Sci. Tech., 2017, 28, 137 CrossRef.
- J. Wang and S. Wang, Chem. Eng. J., 2021, 411, 128392 CrossRef CAS.
- Y. Cao, X. Qian, Y. Zhang, G. Qu, T. Xia, X. Guo, H. Jia and T. Wang, Chem. Eng. J., 2019, 362, 487–496 CrossRef CAS.
- Z. Xu, C. Shan, B. Xie, Y. Liu and B. Pan, Appl. Catal., B, 2017, 200, 439–447 CrossRef CAS.
- S. Liang, X. Hu, H. Xu, Z. Lei, C. Wei and C. Feng, Appl. Catal., B, 2021, 296, 120375 CrossRef CAS.
- M. Pan, C. Zhang, J. Wang, J. W. Chew, G. Gao and B. Pan, Environ. Sci. Technol., 2019, 53, 8342–8351 CrossRef CAS.
- D. Jiraroj, F. Unob and A. Hagège, Water Res., 2006, 40, 107–112 CrossRef CAS.
- N. S. Shah, J. A. Khan, M. Sayed, Z. U. H. Khan, J. Iqbal, S. Arshad, M. Junaid and H. M. Khan, Sep. Purif. Technol., 2020, 233, 115966 CrossRef CAS.
- X. Chen and J. Wang, J. Cleaner Prod., 2021, 328, 129625 CrossRef CAS.
- Q. Wang, Y. Zhang, Y. Li, J. Ren, G. Qu, T. Wang and H. Jia, Chem. Eng. J., 2022, 427, 131584 CrossRef CAS.
- M. S. Alam, M. Kelm, B. S. M. Rao and E. Janata, Radiat. Phys. Chem., 2004, 71, 1087–1093 CrossRef CAS.
- J. L. Wang and S. Z. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- J. Criquet and N. K. V. Leitner, Chemosphere, 2009, 77, 194–200 CrossRef CAS PubMed.
- H. Ren, Z. Hou, X. Han and R. Zhou, Chem. Eng. J., 2017, 309, 638–645 CrossRef CAS.
- L. Chen, T. Cai, C. Cheng, Z. Xiong and D. Ding, Chem. Eng. J., 2018, 351, 1137–1146 CrossRef CAS.
- J. J. Lopez-Penalver, M. Sanchez-Polo, C. V. Gomez-Pacheco and J. Rivera-Utrilla, J. Chem. Technol. Biotechnol., 2010, 85, 1325–1333 CrossRef CAS.
- S. L. H. Sandvik, P. Bilski, J. D. Pakulski, C. F. Chignell and R. B. Coffin, Mar. Chem., 2000, 69, 139–152 CrossRef CAS.
- N. Liu, W.-Y. Huang, Z.-M. Li, H.-Y. Shao, M.-H. Wu, J.-Q. Lei and L. Tang, Sep. Purif. Technol., 2018, 202, 259–265 CrossRef CAS.
- S. Wang and J. Wang, Chem. Eng. J., 2018, 351, 688–696 CAS.
- N. Liu, T. Wang, M. Zheng, J. Lei, L. Tang, G. Hu, G. Xu and M. Wu, Chem. Eng. J., 2015, 270, 66–72 CAS.
- H.-Y. Shao, M.-H. Wu, F. Deng, G. Xu, N. Liu, X. Li and L. Tang, Chemosphere, 2018, 190, 184–190 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04993d |
| ‡ Mengxin Tu and Lei Chen contributed equally to this article. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ac,
Jianzhong Gu*ac,
Chengkai Maoa,
Yingfei Rena,
Hongyong Wanga and
Gang Xu
ac,
Jianzhong Gu*ac,
Chengkai Maoa,
Yingfei Rena,
Hongyong Wanga and
Gang Xu