
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEthylene oxide graft copolymers reduce the immunogenicity of lipid nanoparticles†
Yalin Qi‡
 ab,
Hesong Han‡ab,
Albert Liuab,
Sheng Zhaoab,
Atip Lawanprasertab,
Josefine Eilsø Nielsencd,
Hema Choudharyab,
Dengpan Liangab,
Annelise E. Barronc and
Niren Murthy*ab
ab,
Hesong Han‡ab,
Albert Liuab,
Sheng Zhaoab,
Atip Lawanprasertab,
Josefine Eilsø Nielsencd,
Hema Choudharyab,
Dengpan Liangab,
Annelise E. Barronc and
Niren Murthy*ab
aDepartment of Bioengineering, University of California, Berkeley, Berkeley, California 94720, USA. E-mail: nmurthy@berkeley.edu
bInnovative Genomics Institute (IGI), Berkeley, California 94704, USA
cDepartment of Bioengineering, School of Medicine, Stanford University, Stanford, California 94305, USA
dDepartment of Science and Environment, Roskilde University, Roskilde 4000, Denmark
First published on 20th September 2024
Abstract
Lipid nanoparticle (LNP)/mRNA complexes have great therapeutic potential but their PEG chains can induce the production of anti-PEG antibodies. New LNPs that do not contain PEG are greatly needed. We demonstrate here that poly-glutamic acid-ethylene oxide graft copolymers can replace the PEG on LNPs and outperform PEG-LNPs after chronic administration.
Introduction
Lipid nanoparticle (LNP)/mRNA complexes have the potential to transform medicine and are being intensely investigated for treating diseases ranging from cancer to obesity.1–8 However, LNPs contain PEG and patients that receive LNPs generate antibodies against PEG.9 Anti-PEG antibodies have the potential to become a serious problem for the development of LNP products.10–14 Over one billion people have received LNP-based vaccines, and now have high levels of anti-PEG antibodies in their blood. The presence of anti-PEG antibodies in patients will cause several problems for LNP based products.15,16 For example, patients with high levels of anti-PEG antibodies have high levels of reactogenicity to LNP based vaccines, and this can lead to life threatening anaphylaxis. In addition, anti-PEG antibodies are likely to cause even more significant problems for intravenously administered LNPs, and could potentially cause their inactivation and high levels of inflammation through complement mediated activation.17Although the immunogenicity of PEG is well understood, developing new LNP formulations without PEG has been challenging. LNPs based on polysarcosine (PS) or poly(2-ethyl-2-oxazoline) (PEOZ) have been developed, which can transfect mRNA in vivo as efficiently as PEG based LNPs.18–20 However, PS and PEOZ induce the generation of anti-polymer antibodies and lose efficacy after chronic administration, and it is unclear if they are suitable replacements for PEG.20,21 Poly(2-methyacryloyloxyethyl phosphorylcholine) (PMPC) has also been investigated as a PEG replacement polymer for LNPs but has never been investigated in vivo, and it is unclear if PMPC based LNPs can function in vivo.22 There is consequently a great need for new classes of LNPs that do not contain PEG and can be chronically injected without losing efficacy.
Polyamides that contain short PEG grafts have great potential for replacing the PEG on LNPs. Polyamides with short PEG grafts do not cause the generation of anti-polymer antibodies after chronic administration, even after conjugation to immunogenic proteins or nanoparticles.23–26 This unique property is due to the specific binding mechanism of anti-PEG antibodies to PEG. The crystal structure of anti-PEG antibodies complexed with PEG shows that anti-PEG antibodies bind to an ethylene oxide fragment 7 units in length.26 The affinity of anti-PEG antibodies for oligoethylene oxides dramatically declines for oligoethylene oxides <7 units in length, and is probably too low to stimulate antibody production from B cells in vivo. In addition, polymers with short PEG grafts can stabilize LNPs effectively and generate LNPs that do not bind anti-PEG antibodies.27 Despite their potential, polyamides with short PEG grafts have never been investigated as PEG replacement polymers for LNPs.
In this report, we demonstrate that polyamides composed of a poly(L-glutamic acid) backbone esterified (PGE) with oligoethylene-oxide grafts 2–5 units in length can replace the PEG on LNPs and generate LNPs that outperform PEG-LNPs after chronic administration (see Fig. 1 for structures). In particular, PGE-LNPs with 3 unit ethylene oxide grafts transfected the liver with luciferase and human erythropoietin (hEPO) mRNA as efficiently as PEG-LNPs and outperformed PEG-LNPs after chronic administration due to their lower immunogenicity. In addition, PGE-LNPs with ethylene oxide grafts 2, 4 and 5 units in length also transfected cells efficiently. Collectively, these experiments demonstrate that PGEs have great potential as polymeric stabilizers for LNPs.
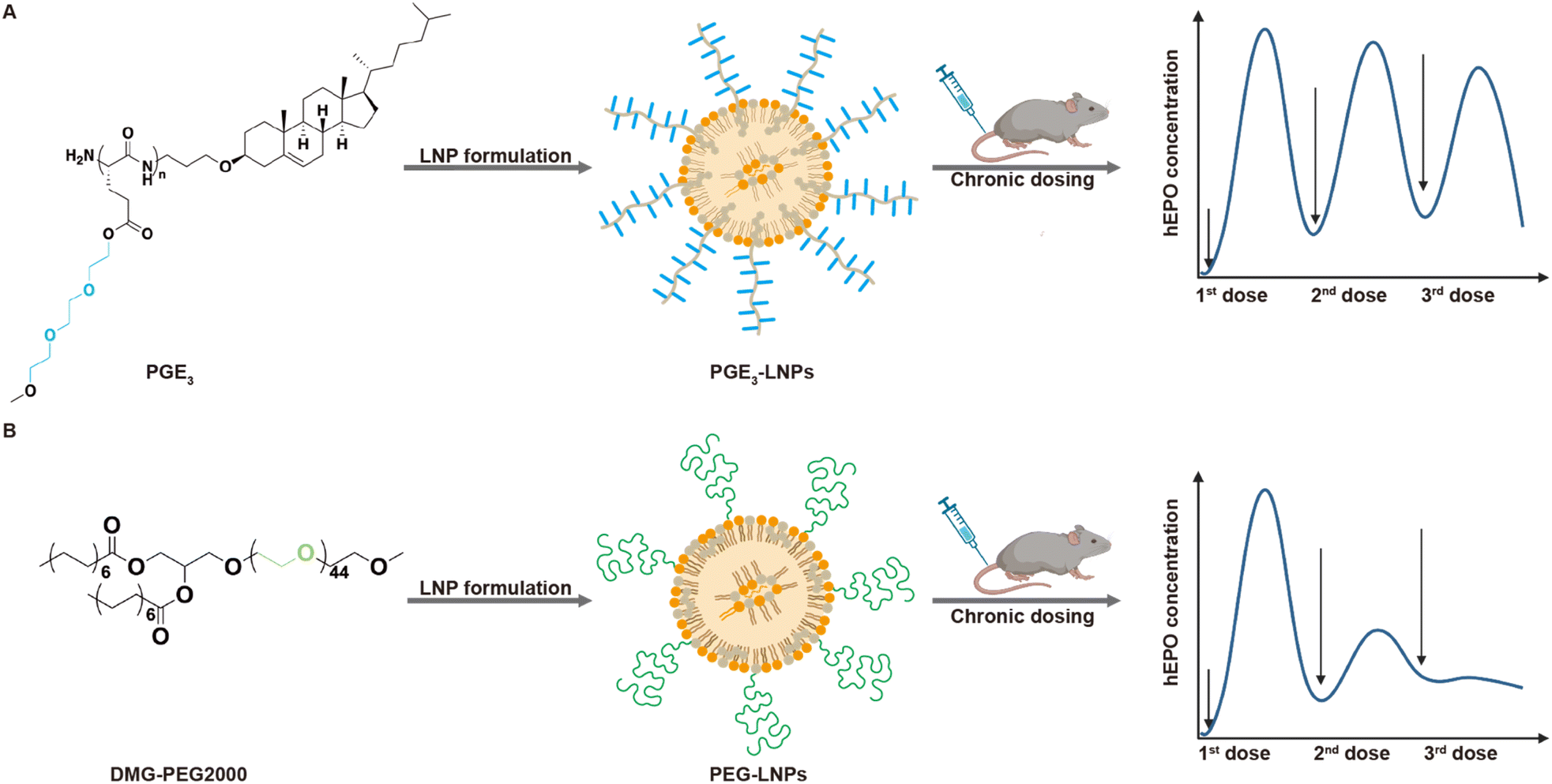 | ||
| Fig. 1 LNPs made with the polyamide PGE have low immunogenicity and outperform PEG-LNPs after chronic administration. Lipid nanoparticle (LNP)/mRNA complexes have great therapeutic potential but their PEG coating can induce the production of anti-PEG antibodies, and there is consequently great interest in developing new LNPs that do not contain PEG. In this report we demonstrate that LNPs made with PEG replacement polymer polyglutamic acid-ethylene oxide (PGE) outperform PEG-LNPs after chronic administration due to their lower immunogenicity. PGE is composed of a polyamide with short PEG grafts conjugated to cholesterol. The chemical structure of PGE3, one of the most promising PGEs identified is shown above. Mice administered multiple intravenous injections of PGE3-LNPs made with human erythropoietin (hEPO) mRNA generated higher hEPO levels in serum (A) compared to those administered PEG-LNPs made with hEPO mRNA (B). | ||
Results and discussion
Synthesis of PGE2-5
The chemical structures of the PGEs are shown in Scheme 1, they are composed of PGEs with ethylene-oxide side chains 2–5 units (PGE2-5) in length conjugated to cholesterol. Cholesterol was selected as the LNP anchoring domain in the PGEs because of its high affinity for phospholipid membrane,28 and its simple synthesis in comparison to traditional phospholipids. PGEs were synthesized by the route shown in Scheme 1. An amine functionalized cholesterol was used as an initiator for the ring opening polymerization of N-carboxyanhydride (NCA) monomers of L-glutamic acid esterified with oligoethylene-oxides 2–5 units in length. The resulting polymers were purified via precipitation in cold ether and had molecular weights between 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–37000 Daltons (see Scheme 1 for details). Poly(L-glutamic acid) was selected as the polymer backbone for the PGEs instead of D or DL poly(glutamic acid) because poly(L-glutamic acid) has lower immunogenicity than the D or DL isoforms.23,24
000–37000 Daltons (see Scheme 1 for details). Poly(L-glutamic acid) was selected as the polymer backbone for the PGEs instead of D or DL poly(glutamic acid) because poly(L-glutamic acid) has lower immunogenicity than the D or DL isoforms.23,24
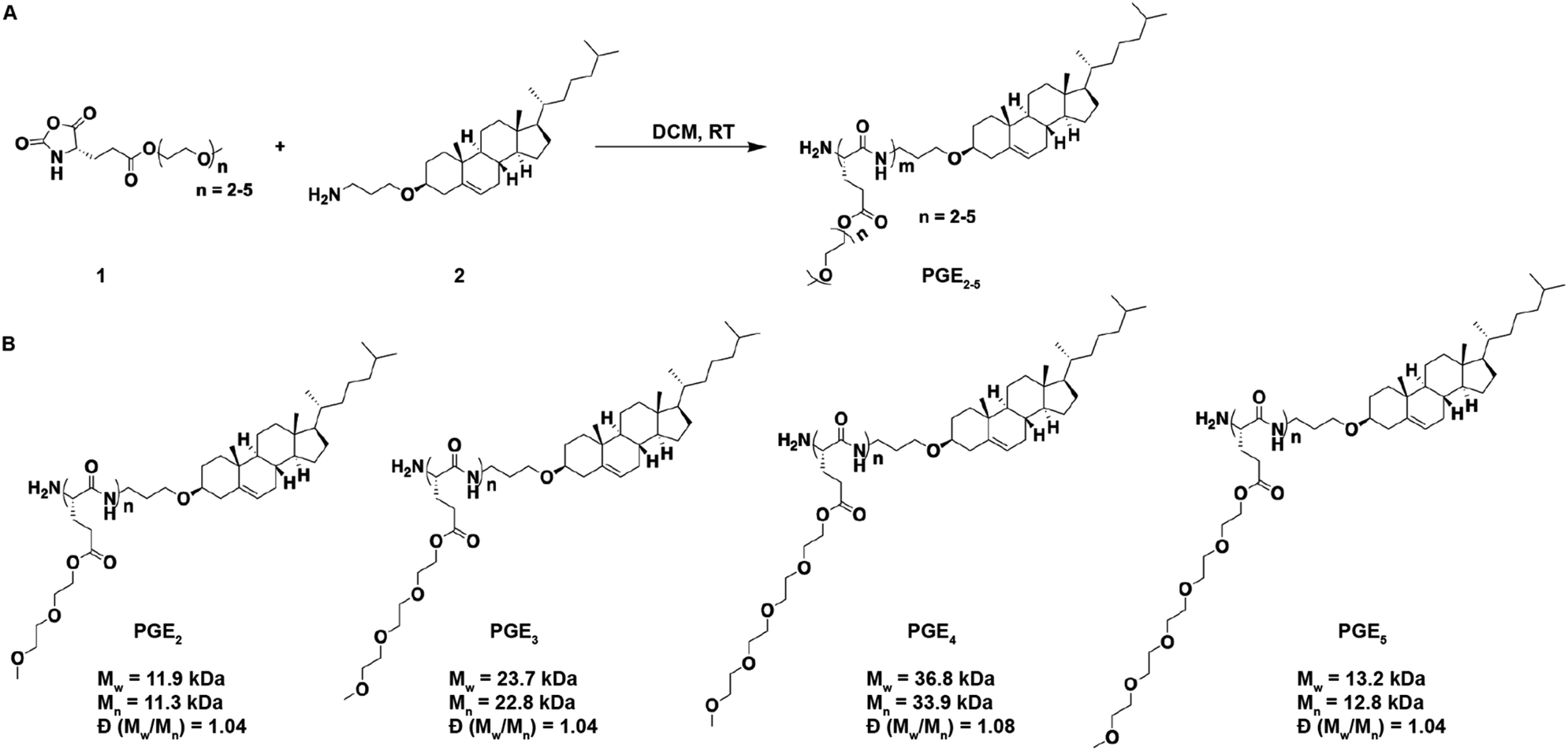 | ||
| Scheme 1 Synthesis of the PEG replacement polymers PGE2-5, and their chemical structures and molecular weights. (A) PGE2-5 were synthesized via ring opening polymerization of the N-carboxyanhydride monomer 1. The polymerization was initiated with the amine containing cholesterol derivative 2. (B) Mw, Mn and Đ (Mw/Mn) values were determined by gel permeation chromatography (GPC). | ||
PGE-LNPs have similar sizes, mRNA encapsulation efficiencies, and internal structures as PEG-LNPs
LNPs formulated with PGE2-5 were characterized for their hydrodynamic diameter, polydispersity index (PDI), encapsulation efficiency, and Zeta-potentials, and compared against PEG based LNPs. LNPs made with PGE2-5 demonstrated comparable encapsulation efficiencies, sizes and Zeta-potentials to PEG-LNPs, despite their very different molecular architectures and molecular weights (see Fig. 2A, B and S1†). The particle size of PGE2-LNPs and PGE3-LNPs were 195 nm, which were larger than those of PEG based LNPs (145 nm), whereas PGE4-LNPs and PGE5-LNPs had diameters of approximately 110 nm, which were smaller than PEG based LNPs. The smaller size of the PGE4-LNPs and PGE5-LNPs are anticipated based upon their longer PEG grafts, which should prevent LNPs made with them from fusing during the self-assembly process. Furthermore, small-angle X-ray scattering (SAXS) analysis indicates that PGE3-LNPs made with 0.84 mole% of PGE3 exhibit a peak at q = 0.118 Å−1, very close to that of PEG-LNPs (q = 0.117 Å−1), which demonstrates that PGE3-LNPs containing 0.84 mole% of PGE3 has similar internal structures to PEG-LNPs (Fig. S2†).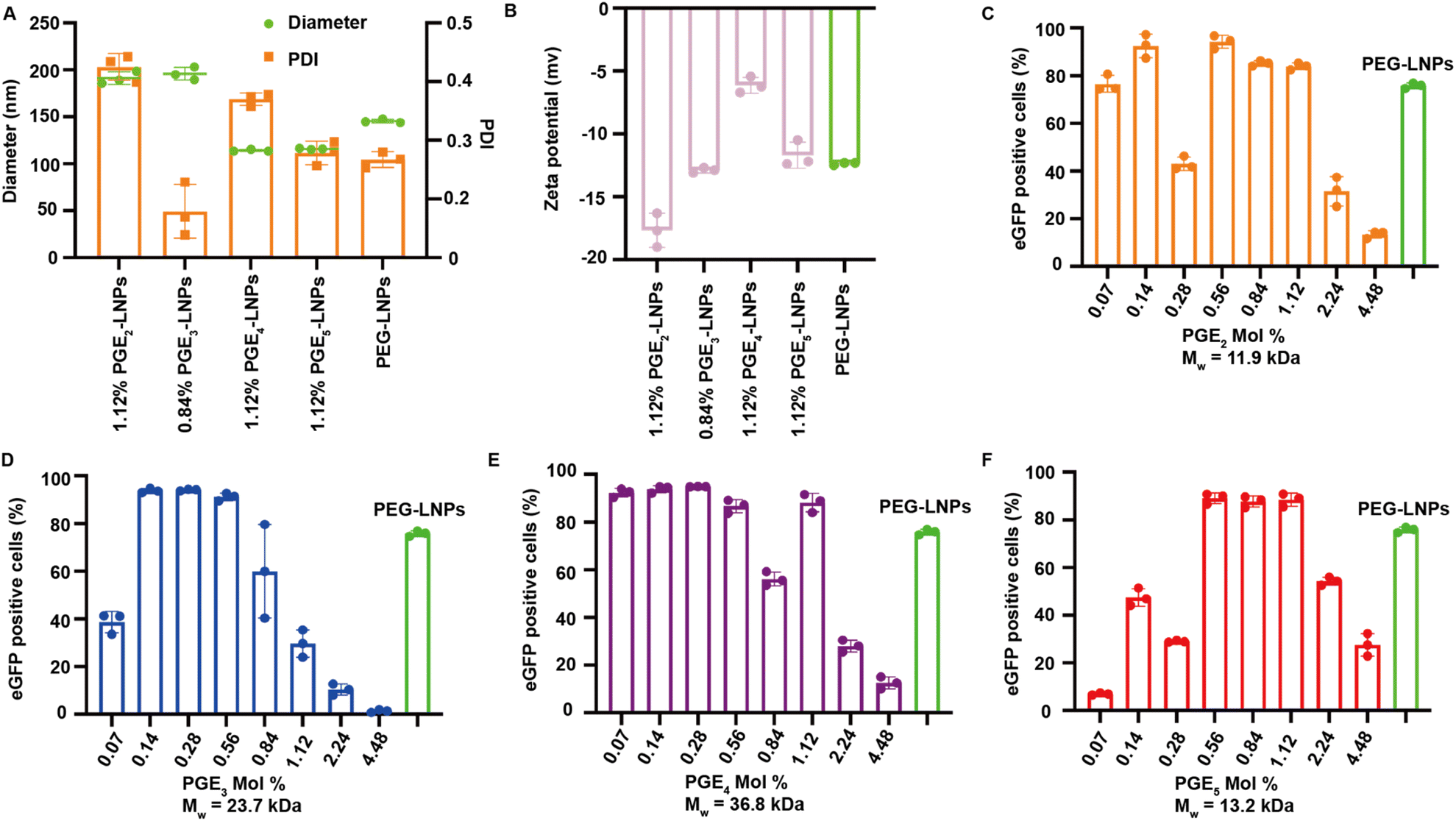 | ||
| Fig. 2 PGE2-5 can replace the PEG on LNPs and can transfect HEK 293T cells in vitro as efficiently as PEG-LNPs. (A) Diameter and PDI of PGE2-5-LNPs. (B) Zeta potential of PGE2-5-LNPs. (C–F) Transfection of HEK 293T cells with eGFP mRNA encapsulated in either PGE2-5-LNPs or PEG-LNPs. LNPs were formulated with PGE2-5 at mole percentages ranging between 0.07%–4.48% and compared against LNPs formulated with DMG-PEG2000 (1.18 mole%). PGE2-5-LNPs transfected cells efficiently at multiple different mole ratios and were comparable to PEG-LNPs, despite their high molecular weights. (C) Comparison of PGE2-LNPs and PEG-LNPs. (D) Comparison of PGE3-LNPs and PEG-LNPs. (E) Comparison of PGE4-LNPs and PEG-LNPs. (F) Comparison of PGE5-LNPs and PEG-LNPs. Data are represented as mean ± standard deviation, n = 3. | ||
PGE2-5 can replace the PEG on LNPs and can transfect HEK 293T cells in vitro as efficiently as PEG-LNPs
We performed experiments to investigate if PGE-LNPs could deliver mRNA to cells and investigated their ability to transfect human embryonic kidney 293T cells (HEK 293T), using eGFP as the reporter mRNA. LNPs were formulated with PGE2-5 at mole percents ranging from 0.07–4.48%, incubated with HEK 293T cells for 12 hours, and analyzed via flow cytometry. All the PGE-LNPs investigated were able to transfect HEK 293T cells efficiently. For example, all four PGE-LNPs made with 0.56 mole% of PGE2-5 transfected >80% of HEK 293T cells, which was comparable to PEG based LNPs (Fig. 2C–F). The molecular weights of the PGEs are between 11000–37000 Daltons and are much larger than DMG-PEG, which has a PEG chain of 2000. The PGE-LNPs with 0.56–1.12 mole% of PGE contain 5–10 times the ethylene oxide mass ratio as PEG-LNPs yet still transfected cells efficiently. This efficacy could be attributed to the higher persistence length of PGE2-5, their propensity to form α-helices and their graft co-polymer architecture. In addition, the off-rate of PGE2-5 from the LNP surface could also be much higher than DMG-PEG2000, due to the large molecular weight of their hydrophilic domains.
PGE2-5 can replace the PEG on LNPs and can transfect mice in vivo as efficiently as PEG-LNPs
We investigated if LNPs made with PGEs could deliver mRNA after an i.v. injection and compared their efficacy against PEG based LNPs (Fig. 3A and B). We selected PGE3 as the initial polymer to investigate because it robustly transfected cells in vitro. In addition, poly(L-glutamic acid) esterified with ethylene oxides 3 units in length do not generate PEG antibodies after chronic administration, even after conjugation to proteins or nanoparticles. PGE3-LNPs made with luciferase mRNA containing 0.84 mole% and 1.12 mole% of PGE3 were investigated in vivo after i.v. administration, at an N/P ratio of 4:1. Fig. 3E–H demonstrate that PGE3-LNPs containing 0.84 mole% of PGE3 have a transfection efficiency equivalent to PEG-LNPs in vivo, after either an i.v. or i.m. injection. PGE3-LNPs made with luciferase mRNA containing 1.12 mole% of PGE3 were unable to deliver mRNA in vivo efficiently (data not shown).
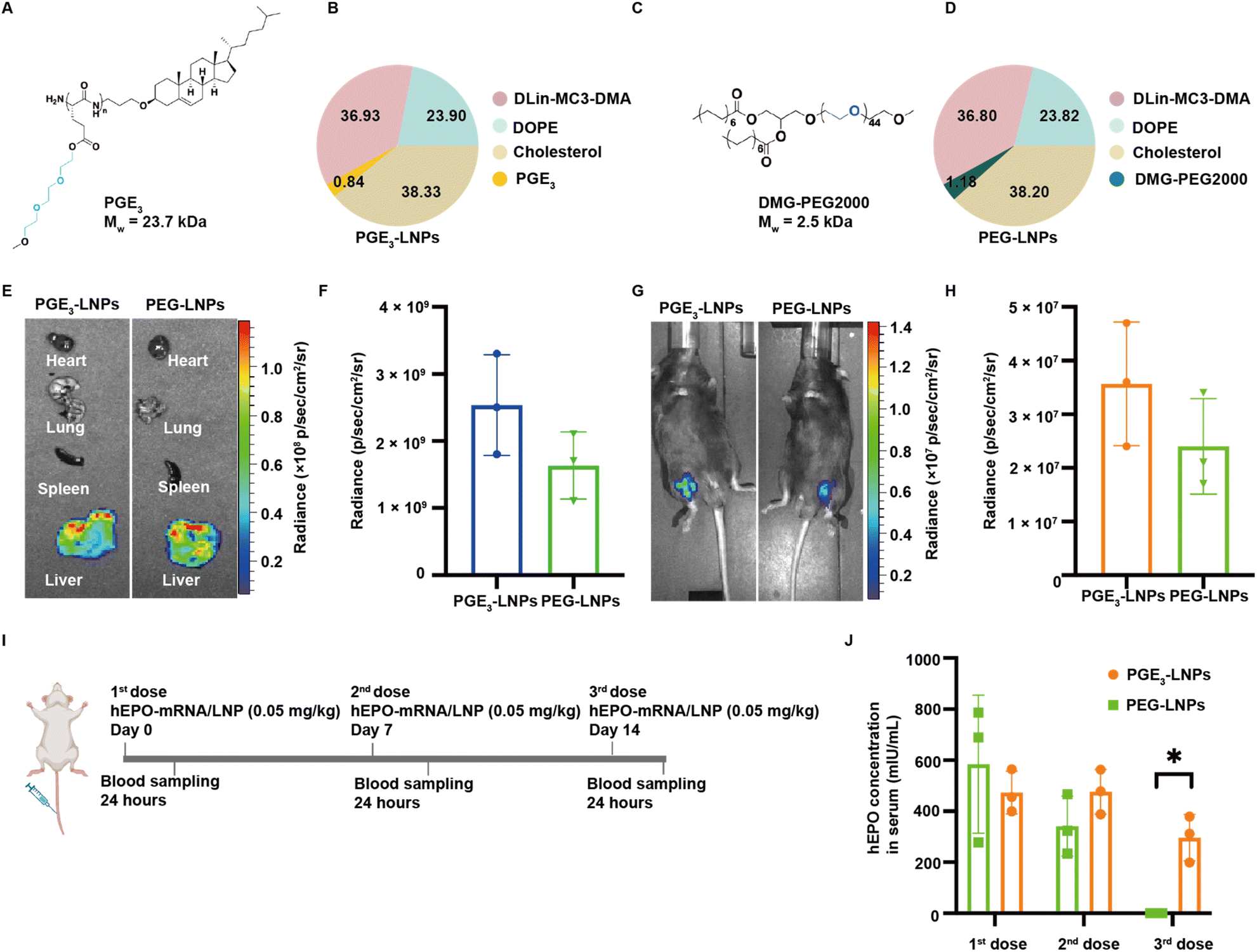 | ||
| Fig. 3 PGE3-LNPs outperform PEG-LNPs after chronic administration. (A) Structure of PGE3. (B) Molar ratios of the different lipid components in PGE3-LNPs. (C) Structure of DMG-PEG2000. (D) Molar ratios of the different lipid components in PEG-LNPs. (E) PGE3-LNPs or PEG-LNPs containing luciferase mRNA were injected intravenously into mice (0.25 mg per kg mRNA dose) and the liver, spleen and other organs were removed and imaged in an IVIS machine. PGE3-LNPs transfect the liver after an intravenous injection. (F) Quantification of the luciferase signal of organs in E from PGE3-LNPs and PEG-LNPs. PGE3-LNPs and PEG-LNPs transfect the liver with similar efficacy (n = 3 mice). (G) PGE3-LNPs or PEG-LNPs containing luciferase mRNA were injected intramuscularly into mice (0.25 mg per kg mRNA dose) and imaged in an IVIS machine. (H) Quantification of the luciferase signal in G from PGE3-LNPs and PEG-LNPs. PGE3-LNPs and PEG-LNPs transfect muscle tissue with similar efficacy (n = 3 mice). (I) Experiment schedule for chronic LNP dosing experiment with hEPO mRNA. Mice were intravenously injected with PGE3-LNPs containing hEPO mRNA (0.05 mg kg−1) or PEG-LNPs containing hEPO mRNA (0.05 mg kg−1) on days 0, 7 and 14. Serum samples were collected 24 hours post-injection (n = 3 mice). (J) Quantitation of hEPO protein levels in the blood of mice treated with PGE3-LNPs or PEG-LNPs. PGE3-LNPs generate high levels of hEPO protein in the blood after three consecutive injections, whereas PEG-LNPs lose >99% of their efficacy. Data are represented as mean ± standard deviation, n = 3. Statistical analyses were performed with a Student's t-test with *P < 0.05. | ||
PGE3-LNPs outperform PEG-LNPs after chronic administration
Finally, we investigated if PGE3-LNPs could outperform PEG-LNPs after chronic administration due to the lower immunogenicity of the PGE3 polymer in comparison to PEG. We used hEPO mRNA as the therapeutic cargo for these experiments. Mice were given a 0.05 mg per kg hEPO mRNA once a week for three weeks, and 24 hours after each dose the hEPO protein levels in the blood were measured (Fig. 3I). Fig. 3J demonstrates that PGE3-LNPs are significantly better than PEG-LNPs after chronic administration. PEG-LNPs lost over 99% of their activity on their third injection. In contrast, PGE3-LNPs retained >60% of their activity on their third dose. Furthermore, we assessed the immunogenicity of the PGE3 polymer by conducting an ELISA to detect anti-PEG antibodies in mice treated with PGE3 formulated LNPs. Mice were first administered PBS intravenously, followed by three intravenous doses of PGE3 formulated LNPs containing 0.05 mg per kg hEPO mRNA. Fig. S4† demonstrates PGE3 formulated LNPs did not induce the production of anti-PEG antibodies after repeated administration.Conclusions
In summary, in this report we demonstrate that PGE3-LNPs can replace the PEG on LNPs and can generate LNPs that are as effective as PEG-LNPs at delivering mRNA in vivo. In addition, PGE3-LNPs outperformed PEG-LNPs after chronic administration due to their lower tendency to generate anti-PEG antibodies. Collectively, these experiments demonstrate that PGE-LNPs have great potential as PEG replacement polymers for LNPs.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. M. thanks the California Institute for Regenerative Medicine (CIRM) DISC2-14045 and DISC2-14097, and the NIH for R01MH125979-01. N. M. also thanks the Innovative Genomics Institute and the CRISPR Cures for Cancer Initiative. A. E. B would like to thank the NIH for funding this work with a Pioneer Award, grant # 1DP1OD029517-01 and Dr James J. Truchard and the Truchard Foundation. J. E. N was funded by grant NNF21OC0068675 from the Novo Nordisk Foundation and the Stanford Bio-X Program. The authors would like to thank SLAC for SAXS beamtime, and Dr Thomas Weiss for support during the SAXS experiment. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894).Notes and references
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS.
- N. Chaudhary, D. Weissman and K. A. Whitehead, Nat. Rev. Drug Discovery, 2021, 20, 817–838 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS.
- Y. Eygeris, M. Gupta, J. Kim and G. Sahay, Acc. Chem. Res., 2022, 55, 2–12 CrossRef CAS PubMed.
- C. Liu, Q. Shi, X. Huang, S. Koo, N. Kong and W. Tao, Nat. Rev. Cancer, 2023, 23, 526–543 CrossRef CAS.
- K. Paunovska, D. Loughrey and J. E. Dahlman, Nat. Rev. Genet., 2022, 23, 265–280 CrossRef CAS PubMed.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef CAS PubMed.
- P. S. Kowalski, A. Rudra, L. Miao and D. G. Anderson, Mol. Ther., 2019, 27, 710–728 CrossRef CAS.
- Y. Ju, J. M. Carreño, V. Simon, K. Dawson, F. Krammer and S. J. Kent, Nat. Rev. Immunol., 2023, 23, 135–136 CrossRef CAS PubMed.
- M. F. S. Deuker, V. Mailänder, S. Morsbach and K. Landfester, Nanoscale Horiz., 2023, 8, 1377–1385 RSC.
- P. Zhang, F. Sun, S. Liu and S. Jiang, J. Controlled Release, 2016, 244, 184–193 CrossRef CAS PubMed.
- M. Estapé Senti, C. A. de Jongh, K. Dijkxhoorn, J. J. F. Verhoef, J. Szebeni, G. Storm, C. E. Hack, R. M. Schiffelers, M. H. Fens and P. Boross, J. Controlled Release, 2022, 341, 475–486 CrossRef.
- S. Zalba, T. L. M. ten Hagen, C. Burgui and M. J. Garrido, J. Controlled Release, 2022, 351, 22–36 CrossRef CAS PubMed.
- G. Besin, J. Milton, S. Sabnis, R. Howell, C. Mihai, K. Burke, K. E. Benenato, M. Stanton, P. Smith, J. Senn and S. Hoge, Immunohorizons, 2019, 3, 282–293 CrossRef CAS PubMed.
- R. Tenchov, J. M. Sasso and Q. A. Zhou, Bioconjugate Chem., 2023, 34, 941–960 CrossRef CAS.
- Y. Ju, W. S. Lee, E. H. Pilkington, H. G. Kelly, S. Li, K. J. Selva, K. M. Wragg, K. Subbarao, T. H. O. Nguyen, L. C. Rowntree, L. F. Allen, K. Bond, D. A. Williamson, N. P. Truong, M. Plebanski, K. Kedzierska, S. Mahanty, A. W. Chung, F. Caruso, A. K. Wheatley, J. A. Juno and S. J. Kent, ACS Nano, 2022, 16, 11769–11780 CrossRef CAS.
- R. Münter, E. Christensen, T. L. Andresen and J. B. Larsen, Mol. Ther.–Methods Clin. Dev., 2023, 29, 450–459 CrossRef.
- S. S. Nogueira, A. Schlegel, K. Maxeiner, B. Weber, M. Barz, M. A. Schroer, C. E. Blanchet, D. I. Svergun, S. Ramishetti, D. Peer, P. Langguth, U. Sahin and H. Haas, ACS Appl. Nano Mater., 2020, 3, 10634–10645 CrossRef CAS.
- D. D. Kang, X. Hou, L. Wang, Y. Xue, H. Li, Y. Zhong, S. Wang, B. Deng, D. W. McComb and Y. Dong, Bioact. Mater., 2024, 37, 86–93 CAS.
- A. J. D. S. Sanchez, D. Loughrey, E. S. Echeverri, S. G. Huayamares, A. Radmand, K. Paunovska, M. Hatit, K. E. Tiegreen, P. J. Santangelo and J. E. Dahlman, Adv. Healthcare Mater., 2024, 2304033, 1–8 Search PubMed.
- C. J. Kim, E. Hara, N. Watabe, I. Hara and S. Kimura, J. Pept. Sci., 2017, 23, 889–898 CrossRef CAS.
- P. Khunsuk, C. Pongma, T. Palaga and V. P. Hoven, Biomacromolecules, 2023, 24, 5654–5665 CrossRef CAS PubMed.
- Y. Hou, Y. Zhou, H. Wang, J. Sun, R. Wang, K. Sheng, J. Yuan, Y. Hu, Y. Chao, Z. Liu and H. Lu, ACS Cent. Sci., 2019, 5, 229–236 CrossRef CAS.
- J. Sun, J. Chen, Y. Sun, Y. Hou, Z. Liu and H. Lu, Bioact. Mater., 2024, 32, 333–343 CAS.
- I. Ozer, G. A. Pitoc, J. M. Layzer, A. Moreno, L. B. Olson, K. D. Layzer, A. M. Hucknall, B. A. Sullenger and A. Chilkoti, Adv. Mater., 2022, 34, 1–12 CrossRef PubMed.
- Y. Qi, A. Simakova, N. J. Ganson, X. Li, K. M. Luginbuhl, I. Ozer, W. Liu, M. S. Hershfield, K. Matyjaszewski and A. Chilkoti, Nat. Biomed. Eng., 2016, 1, 0002 CrossRef PubMed.
- J. Chen, A. Rizvi, J. P. Patterson and C. J. Hawker, J. Am. Chem. Soc., 2022, 144, 19466–19474 CrossRef CAS PubMed.
- D. Hu and Z. Qian, Cholesterol in Drug Delivery Systems, in Cholesterol, Elsevier, 2022, pp. 797–824 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05007j |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |