
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of non-hydroxamate lipophilic inhibitors of 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR): in silico, in vitro and antibacterial studies†
Sharyu Kesharwania,
Eebab,
Mukesh Tandia,
Nisheeth Agarwalb and
Sandeep Sundriyal *a
*a
aDepartment of Pharmacy, Birla Institute of Technology and Science-Pilani (BITS), Pilani Campus, Vidya Vihar, Pilani, Rajasthan 333 031, India. E-mail: sandeep.sundriyal@pilani.bits-pilani.ac.in
bTranslational Health Science and Technology Institute, NCR Biotech Science Cluster, 3rd Mile Stone, Gurugram-Faridabad Expressway, Faridabad 121001, Haryana, India
First published on 30th August 2024
Abstract
1-Deoxy-D-xylulose 5-phosphate reductoisomerase (DXR) is a key enzyme of the 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway operating in several pathogens, including Mycobacterium and Plasmodium. Since a DXR homologue is not present in humans, it is an important antimicrobial target. Fosmidomycin (FSM) and its analogues inhibit DXR function by chelating the divalent metal (Mn2+ or Mg2+) in its active site via a hydroxamate metal binding group (MBG). The latter, however, enhances the polarity of molecules and is known to display metabolic instability and toxicity issues. While attempts have been made to increase the lipophilicity of FSM by substituting the linker chain and prodrug approach, very few efforts have been made to replace the hydroxamate group with other lipophilic MBGs. We report a systematic in silico and experimental investigation to identify novel MBGs for designing non-hydroxamate lipophilic DXR inhibitors. The SAR studies with selected MBG fragments identified novel inhibitors of E. Coli DXR with IC50 values ranging from 0.29 to 106 μM. The promising inhibitors were also screened against ESKAPE pathogens and M. tuberculosis.
1 Introduction
Antimicrobial resistance (AMR) is a rapidly emerging global threat that has rendered many antibiotics ineffective.1,2 The multidrug-resistant (MDR) strains of several pathogenic fungi, bacteria, viruses, and parasites are rising, increasing mortality, morbidity, and economic burden. The ‘Antibiotic Resistance Threats Report’ published by the Centers for Disease Control and Prevention (CDC) in 2019 estimated 4.95 million deaths attributable to bacterial antibiotic resistance, including 1.27 million deaths directly associated with drug-resistant infections worldwide. The highest number of deaths were reported from sub-Saharan Africa and South Asia. This death toll is estimated to rise to 10 million by 2050 if the issue of AMR is not effectively addressed.3,4 Thus, unique strategies and novel molecular targets must be pursued to combat the challenge of AMR.5The 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway, which is responsible for the synthesis of isoprenoid precursors in several microorganisms, is emerging as the most promising antimicrobial target, especially for the treatment of Tuberculosis (TB) and Malaria.6–9 Since the MEP pathway is present in several clinically relevant bacteria/parasites, it presents an excellent opportunity to combat various infectious diseases.10–13 Additionally, due to the nonexistence of the MEP pathway in mammalian cells, antimicrobials modulating MEP enzymes are anticipated to be less toxic to human hosts.14,15
Among the seven enzymes of the MEP pathway, the DXR enzyme has been widely studied. DXR is the second enzyme of the MEP pathway, operates in many pathogens, and is a well-established antimicrobial target.9,15 The DXR enzyme catalyzes nicotinamide adenine dinucleotide phosphate (NADPH)-dependent intramolecular rearrangement of 1-deoxy-D-xylulose-5-phosphate (DXP) to MEP, utilizing divalent metal ion.16 Knock-out studies of the DXR enzyme have shown the essentiality of the dxr gene in many pathogens, including Escherichia coli and M. tuberculosis. Thus, inhibiting the DXR enzyme could be a promising strategy for developing new bactericidal agents.17–20
Antibiotics fosmidomycin (FSM) and FR900098 (1 and 2, Fig. 1) are the first known natural DXR inhibitors displaying potent activity against several bacteria and Plasmodium falciparum.21–23 A divalent metal ion (Mn2+ or Mg2+) and cofactor β-NADPH are essential for the enzymatic activity. Thus, hydroxamate (an anionic form of hydroxamic acid) of FSM (and its analogues) acts as a metal-binding group (MBG) and chelates the divalent metal ion, thus inhibiting its interaction with the substrate. The phosphonate group interacts with multiple polar amino acid residues via hydrogen bonds (H-bonds).24,25 Detailed SAR studies reported earlier demonstrate the importance of hydroxamate and phosphonic acid fragments and the length (3-carbon) of the linker chain that connects these moieties.9,26,27
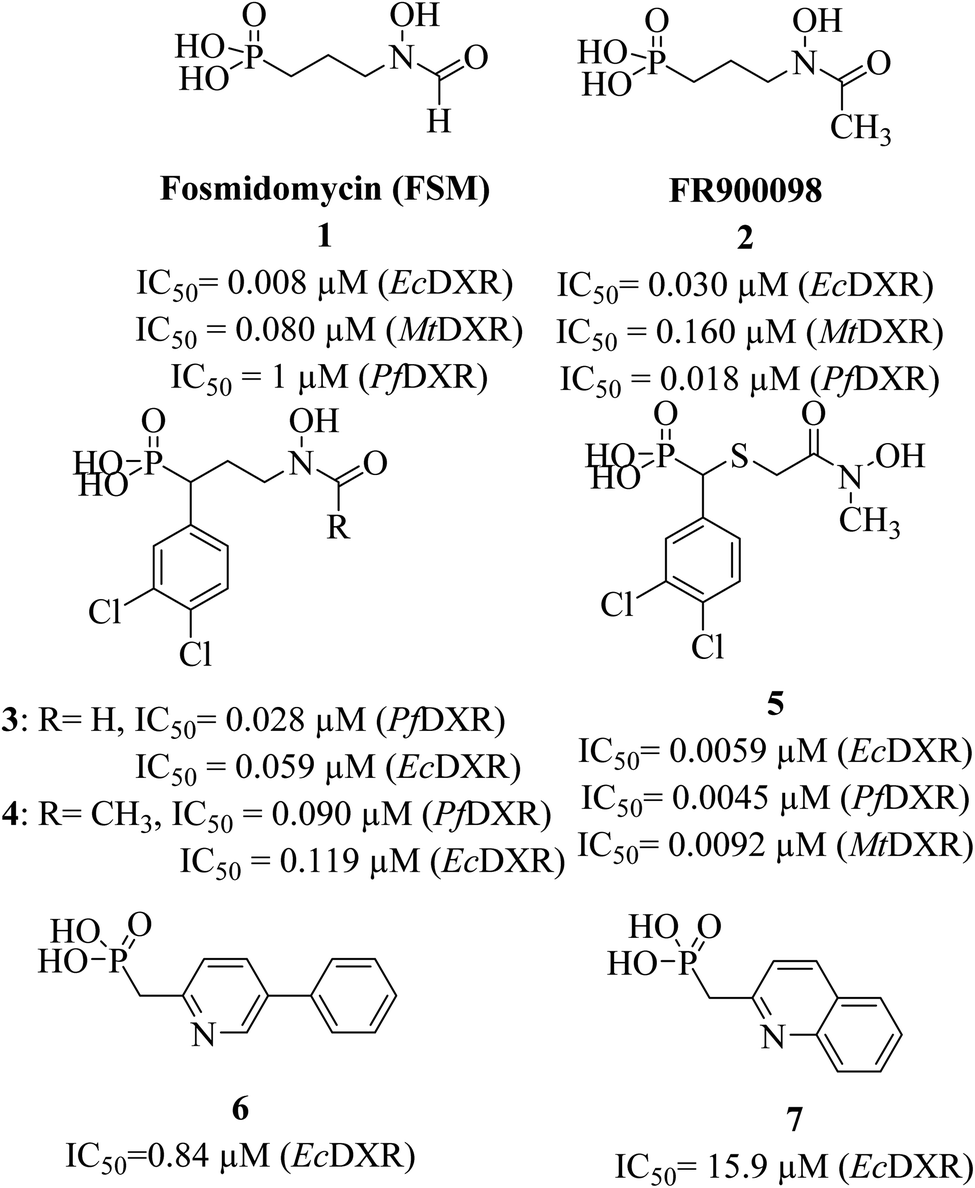 | ||
| Fig. 1 Structures and reported IC50 values of known hydroxamate and non-hydroxamate DXR inhibitors. | ||
Initially discovered DXR inhibitors 1 and 2 are highly polar owing to hydroxamate MBG and phosphonic acid and lack of druglikeness.28–30 These antibiotics exhibit short half-life and poor cellular permeability, limiting their clinical application.15,31–36 Moreover, hydroxamate is a metabolically unstable group that exhibits toxicity through various metabolites.37,38 Several structural modifications were performed around FSM to obtain lipophilic analogues of 1 and 2. For instance, inhibitors with an aromatic ring alpha to the phosphonate groups were designed (3–5) that displayed potent inhibition of E. coli (EcDXR), P. falciparum (PfDXR), and M. tuberculosis (MtDXR).15 Most of these efforts were limited to modifying the linker or using a prodrug strategy (Fig. 2) since the analogues lacking either the hydroxamate (for example, 6 and 7) or phosphonate group are found to be inactive.9
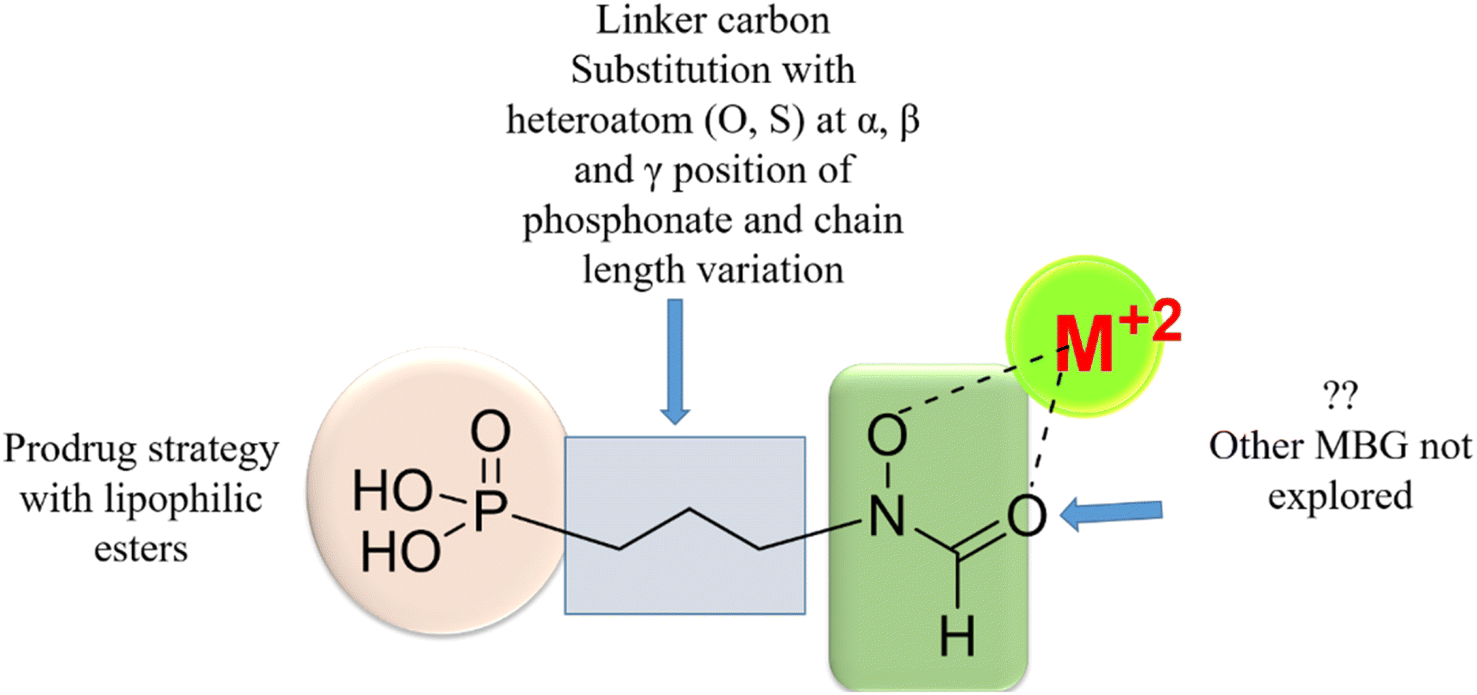 | ||
| Fig. 2 Various approaches reported earlier9,39 to design lipophilic FSM analogues. The systematic replacement of hydroxamate with hydrophobic MBGs has not been explored. | ||
The design of non-hydroxamate ligands has been successfully achieved for many medicinally relevant metalloproteins.40–42 In contrast, no systematic attempts are reported to replace the polar hydroxamate functionality of 1 with other MBGs9 despite computational studies supporting the possibility.43 Thus, most reported DXR inhibitors are close analogues of 1 and lack structural novelty, and only a handful of low-potency non-hydroxamate DXR inhibitors are reported.9
It is reported that the pyridine and quinoline-based lipophilic molecules (6–7) lacking MBG (PDB ID 3ANM and 3ANN) interact with a newly created lipophilic pocket A due to the 180° flipping of Trp211.44 Similarly, a lipophilic pocket B is revealed, which is reportedly occupied by bisphosphonate inhibitors but hidden in the case of the 1-DXR complex.45 Thus, these inducible pockets present an opportunity to design more lipophilic ligands with bulky rings.
In this study, we report a systematic approach to identify potential non-hydroxamate MBGs to design novel DXR inhibitors using molecular modelling studies. Further, we show the effect of the newly synthesized inhibitors on in vitro enzyme inhibition and antibacterial activities.
2 Result and discussion
2.1 MBG library selection based on molecular docking and enzyme inhibition
Intending to design novel non-hydroxamate DXR inhibitors, we started by collating a library of small non-hydroxamate lipophilic MBG fragments. A library reported by Cohen et al. consisting of 96 small metal chelating fragments (Fig. S1, ESI†), referred to as a metal chelating library (MCL), was selected for the study.39 A few other metal chelating fragments not mentioned in the MCL were obtained from the literature, resulting in 103 MBG fragments. With a few exceptions, these fragments fit into the Rule of Three (Ro3) of fragment selection criteria (that is, molecular weight (MW) < 300, clogP ≤ 3, hydrogen bond donor (HBD) ≤ 3, Hydrogen bond acceptor (HBA) ≤ 3, and rotatable bonds (RB) ≤ 3) (Table S1†).46 Since donor moiety is essential for effective metal chelation at the active site, we did not filter the fragments exceeding this criterion. These ring fragments have MBG embedded in the aromatic ring, offering an advantage in increased lipophilicity.9 Since these fragments have MBG locked in the cis configuration, unlike hydroxamate, these are also expected to have an entropic advantage. These ring MBGs also offer an advantage in terms of metabolic resistance to hydrolases, the enzymes which can hydrolyze the hydroxamate group.47 Also, many of these MBGs have other functional groups, providing vectors for further fragment growth.Several reports describe the successful use of molecular docking in designing the inhibitors for various metalloproteins.48 This study employed the Glide docking program49,50 implemented in the Schrodinger Suite.51 The Glide program has consistently shown a high success rate in pose prediction and hit identification in virtual screening campaigns for diverse targets.49,52–56 However, it should be noted that predicting metal–ligand interactions is challenging as subtle changes around the ligand environment can affect the overall binding.57 Also, the estimation of charge on metal atom in the active site is crucial for the accurate prediction pose and binding strength.58–61 A recent study comparing the accuracy of several non-commercial docking programs is reported for metalloproteins.62 To our knowledge, such a study using a specific subset of metalloproteins is not reported for the Glide program. In the Schrodinger Suite, the Epik program63 can be used to generate the ligand's ‘metal binding states’ during ligand preparation. Thus, negatively ionizable functional groups such as phenols and carboxylic acids are deprotonated to access the additional ionization states of ligand-like molecules that are likely to bind to metals in the protein binding pockets. Similarly, interactions between the positively charged metal and negatively charged ligands are recognized and rewarded during docking and scoring. The bidentate interactions of ligand with the metal are also rewarded in which one atom is having a formal negative charge and other belongs to a highly polar functional group.49 This information was kept in mind while selecting MBGs and designing the DXR inhibitors in this study.
To evaluate the suitability of their metal chelating ability, MCL fragments were docked within the EcDXR (PDB 3ANM44 and 3R0I64), PfDXR (PDB 5JAZ),65 and MtbDXR (PDB 2Y1D).66 The cocrystallized poses of all three ligands were successfully reproduced (RMSD < 2 Å) by the Glide (Fig. S2 and Table S2, ESI†). During the course of this study, we used Glide to successfully reproduce the cocrystallized poses of ligands from several other DXR structures (data not shown), suggesting it to be suitable for the design of new DXR inhibitors. The accuracy of docking-based design and MMGBSA modelling studies is further validated experimentally (vide infra). To corroborate experimental data obtained with the recombinant EcDXR, molecular modelling results obtained with the same enzyme (PDB ID 3R0I) are discussed in the following sections.
The inhibitors 1 and its analogues demonstrate favourable interactions with the ‘hard’ Mg2+/Mn2+ ion of DXR active site via a ‘hard’ dioxygen (O,O) donor motif consisting of the deprotonated hydroxyl and the carbonyl oxygen atoms.20 The metal coordination distance between the fragments and dioxygen moiety was found in the 2.10–2.50 Å range. As anticipated, the docked poses of MCL fragments displayed similar binding characteristics (Table 1), where the O,O motif of the MBGs chelated metal ion in a bidentate fashion like the hydroxamate group (Fig. 3A). A few carboxylic acid-containing fragments (F2, F7, F8, F10 and F12) were used as methyl amides for docking studies to mimic the finally designed molecules bearing a phosphonic acid moiety at this position (vide infra).
| Fragment code | Structure of the fragment | % Inhibition at 100 μM | Docking score (kcal mol−1) | Distance from metal (Å) | MMGBSA (kcal mol−1) | |
|---|---|---|---|---|---|---|
| a Docking scores of the corresponding methyl amides are mentioned to mimic the MBG of the final compounds (vide infra). Fosmidomycin is used as a positive control at 100 μM. | ||||||
| F1 |  |
100 | −5.08 | 2.20 | 2.40 | −33.0 |
| F2 (DHBA)a |  |
93.3 | −6.34 | 2.37 | 2.29 | −32.6 |
| F3 |  |
91.4 | −4.52 | 2.19 | 2.46 | −1.02 |
| F4 |  |
97.6 | −4.3 | 2.36 | 2.37 | −15.0 |
| F5 | 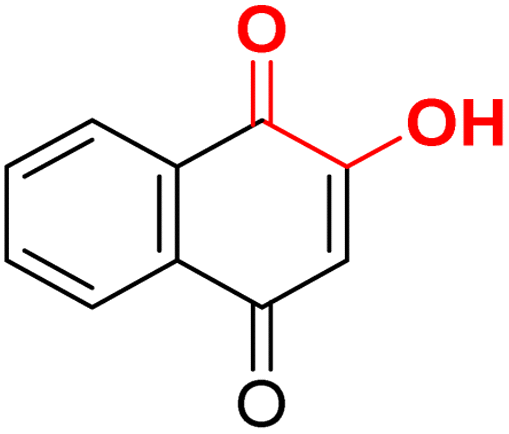 |
100 | −2.9 | 2.36 | 2.38 | −19.7 |
| F6 |  |
98.3 | −4.4 | 2.36 | 2.07 | −15.0 |
| F7 (NA)a |  |
100 | −4.5 | 2.33 | 2.11 | −25.95 |
| F8 (8-HQ)a |  |
100 | −4.76 | 2.37 | 2.21 | −35.4 |
| F9 | 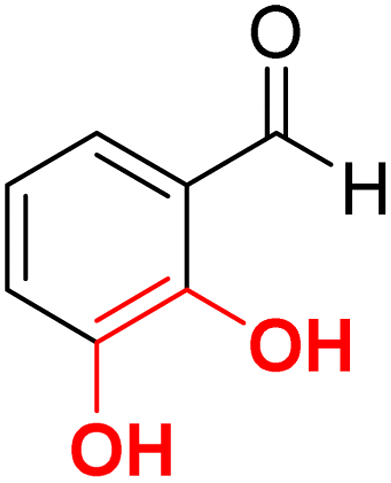 |
86.5 | −5.48 | 2.13 | 2.31 | −15.7 |
| F10 (CCA)a | 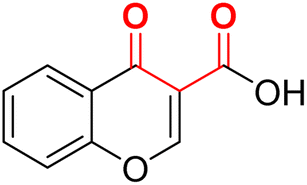 |
100 | −5.03 | 2.20 | 2.32 | −17.4 |
| F11 | 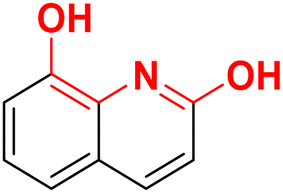 |
100 | −3.58 | 2.15 | 2.45 | −33.9 |
| F12 (SA)a | 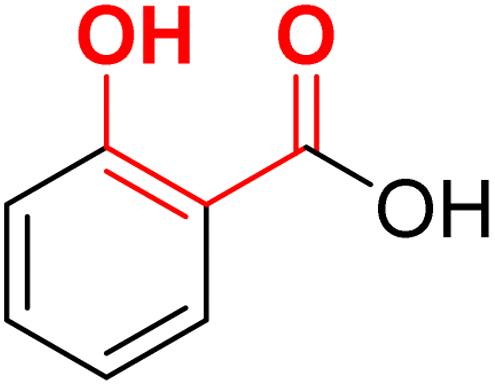 |
87.4 | −4.47 | 2.40 | 2.19 | −22.6 |
| F13 |  |
100 | 3.24 | 2.21 | 2.31 | −33.5 |
| FSM | 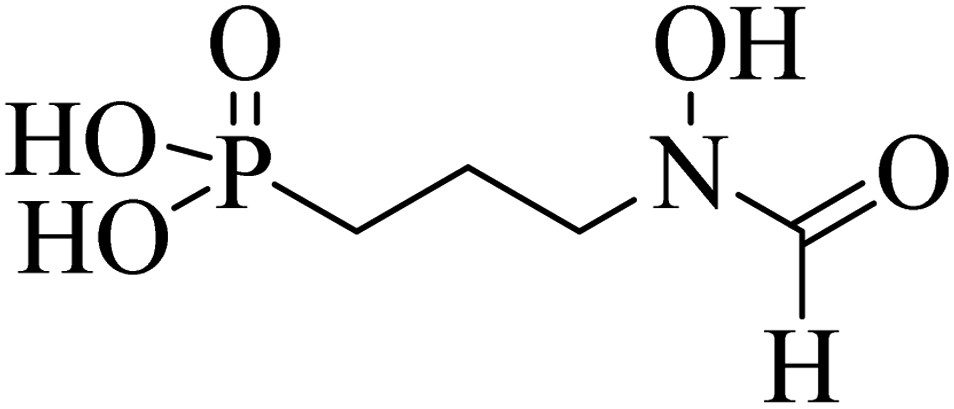 |
100 | — | — | — | — |
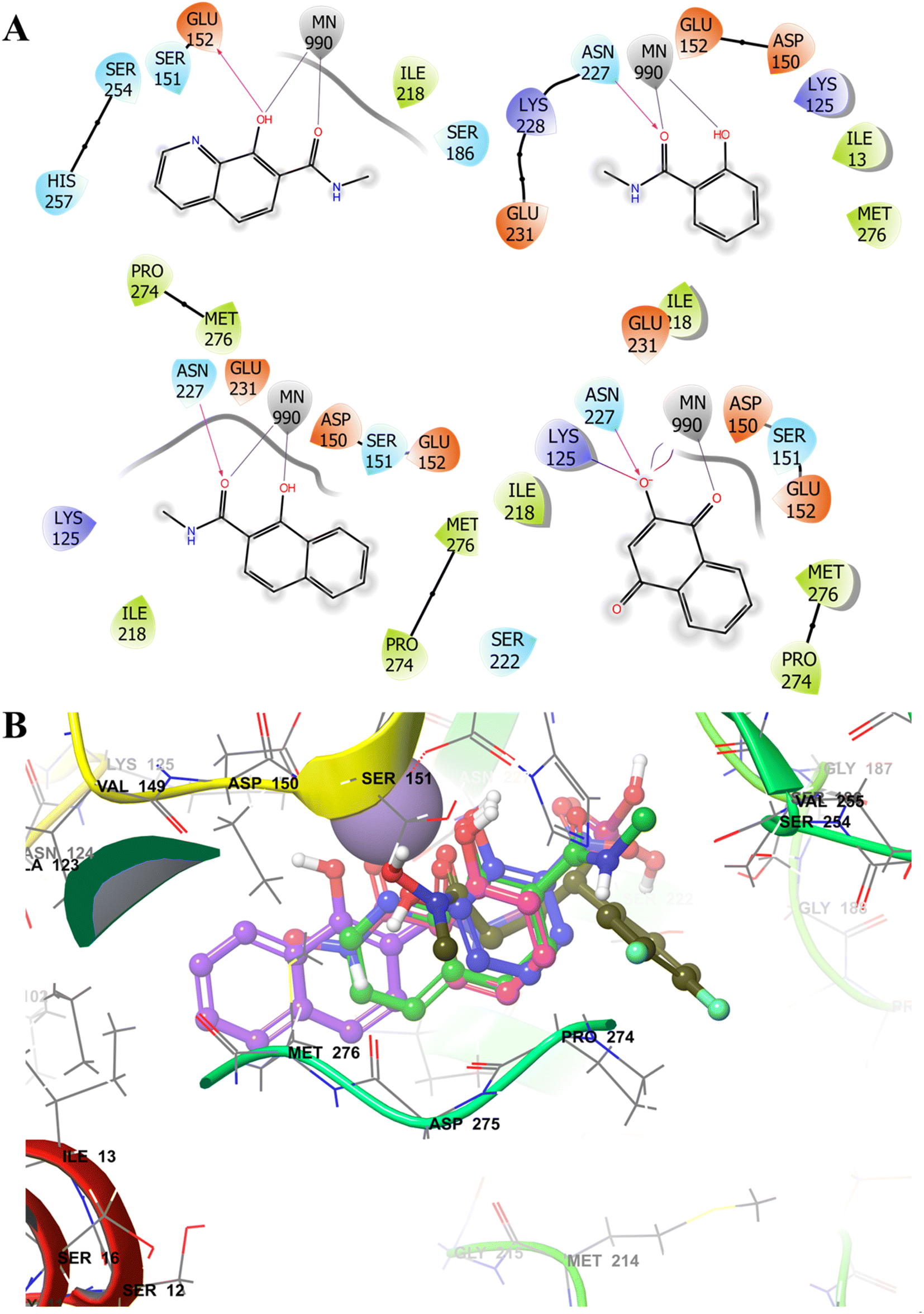 | ||
| Fig. 3 Selective examples of different MBGs (A) showing 2D-interaction map with the Mn2+ ion and other DXR (PDB 3R0I) active site residues. (B) Comparison of the docked poses of MBG fragments F2 (blue), F7 (purple), F8 (green) and F12 (pink) with cocrystallized ligand (moss green ball and sticks). The MBGs show occupation of the hydrophobic Pocket B (lined with Pro274 and Met276). The metal ion is depicted in solid purple ball. | ||
As expected, some MBGs fostered additional interactions with the pocket B residues (Fig. 3B) through their aromatic rings. A few MBGs were also predicted to interact with the polar phosphonic acid binding pocket rather than the active site metal ion, an observation noted in earlier studies.44,67,68 It should be noted that docking score rarely correlates with the experimentally determined inhibition or IC50 values. Thus, in our study docking pose and interactions were considered for the ligand design.
Based on the docking studies and other above-stated advantages, we selected a set of 13 fragments (F1–F13) for the in vitro evaluation. These fragments were purchased and screened against the recombinant E. coli DXR enzyme at 100 μM. FSM was used as the positive control at 100 μM (IC50 = 130 nM under the identical assay conditions). Interestingly, all fragments displayed almost complete DXR inhibition (Table 1) at the tested concentration. Among these, fragments F2, F7, F10, F12 and F9 were selected for additional SAR investigations because they possess a carboxylic acid or aldehyde vector suitable for further functionalization.
2.2 Docking-based design using fragment linking
In addition to MBGs, a phosphonic acid moiety is essential for the potency against the DXR enzyme.26 We adopted a fragment-linking strategy to occupy the phosphonate binding pocket in the DXR active site where the identified MBGs were linked to the α-aminophosphonates. The α-aminophosphonate motifs were deemed suitable due to (i) their ability to supply the necessary phosphonic acid functionality, (ii) straightforward synthesis through Kabachnick–Fields multicomponent reaction,69–71 (iii) the availability of amino group for linking with the selected MBGs, and (iv) the possibility to have a wide range of lipophilic groups (R1) alpha to the phosphonate moiety (mimicking 3–5) derived by selecting an appropriate aldehyde component.Initially, we selected MBGs with carboxylic acid groups (F2, F7, F10, and F12) to employ amide coupling between the acid and the amine functionality of the MBGs and α-aminophosphonates, respectively. Molecular docking studies (with PDB 3R0I) predicted these designed compounds to adopt a comparable conformation, mirroring the ligand cocrystallized with the enzyme (Fig. S3†). In line with our hypothesis, the O′O motif of the MBGs maintained metal chelation, while the aromatic part of the MBGs effectively interacted with pocket B. Moreover, as anticipated, the lipophilic R1 group alpha to the phosphonate moiety occupied pocket A (Fig. S3 and S4†). The occupation of pocket A was particularly prominent with aromatic rings such as phenyl, naphthyl, 3,4-diclorophenyl, and phenylpropyl as R1 attachments (Fig. S4†).
About 50 molecules were designed using molecular docking studies. Overall, the following observations were noted from the modelling studies of the designed molecules (ESI, Tables S2, S3 and Fig. S3–S5†).
(1) MBGs observed metal coordination within the range of 2.10–2.50 Å.
(2) Most ligands, like the cocrystallized ligand, maintained hydrogen bond interactions with the active site residues (Asn227 and Ser186).
(3) The phosphonic acid group interacted with the hydrophilic pocket like the cocrystallized ligand and fostered key H-bond interactions with Ser186, Ser222, Asn227, and Lys228 residues.
(4) Pi–pi stacking was observed between the indole ring of Trp296 and the aromatic ring (R1) of the designed ligands in the P. falciparum DXR protein-ligand complex.
(5) The lipophilic groups (R1) were found to occupy lipophilic pocket A, like the cocrystallized ligands.
(6) All synthesized molecules found to have higher calculated lipophilicity72 (clogP) compared to 1 (Table S3†).
The poses of a few designed molecules also exhibited metal chelation between the phosphonic acid and divalent metal, which aligns with the known metal chelation property of the phosphonic acid functionality.67 Overall, docking studies of the designed molecules supported the optimum metal chelation by MBGs and binding to hydrophobic sub-pockets. Thus, based on the docking poses, chemical synthesis of the derivatives of MBG fragments SA (F12), NA (F7), DBHA (F2), and CCA (F10) was planned (Schemes 2, 3 and Table 2).
| ID | Synthesized molecules | IC50 (μM) | % Inhibition at 50 μM | MMGBSA (kcal mol−1) | Docking score (kcal mol−1) | Distance from metal (Å) | |
|---|---|---|---|---|---|---|---|
| a ND: not determined, IC50 values are based on a single representative experiment performed in duplicates.b Reported by vendor under identical assay conditions. | |||||||
| 20a |  |
33.2 | 94.6 | −48.8 | −6.45 | 2.14 | 2.24 |
| 20b |  |
ND | ND | −43.7 | −5.35 | 2.44 | 2.33 |
| 20c | 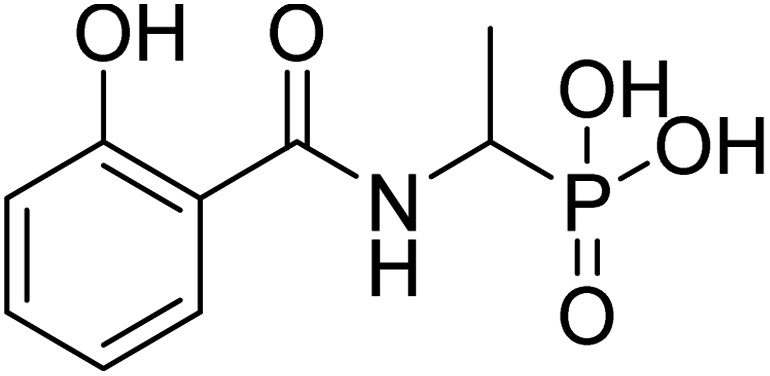 |
ND | ND | −52.9 | −5.77 | 2.20 | 2.14 |
| 20d |  |
32.9 | 81.3 | −41.5 | −6.76 | 2.19 | 2.37 |
| 20e |  |
ND | ND | −58.2 | −6.45 | 2.24 | 2.30 |
| 21a |  |
ND | 66.5 | −53.4 | −6.43 | 2.20 | 2.47 |
| 21b |  |
ND | 68.4 | −29.3 | −5.57 | 2.31 | 2.33 |
| 21c | 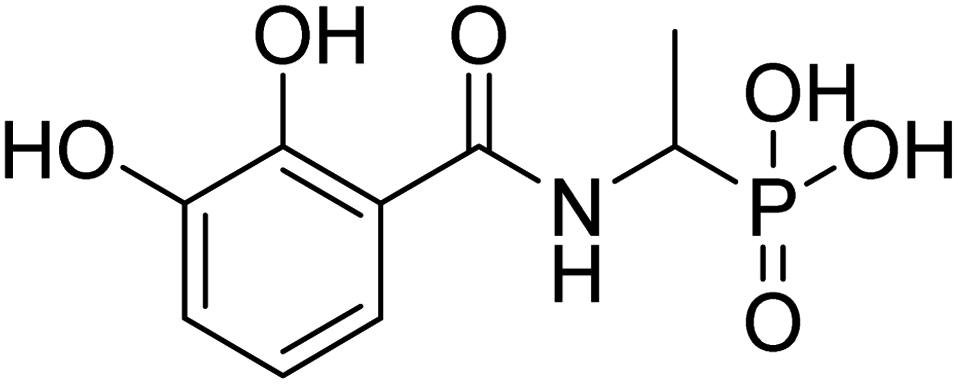 |
ND | 60.13 | −40.0 | −5.59 | 2.35 | 2.17 |
| 21d | 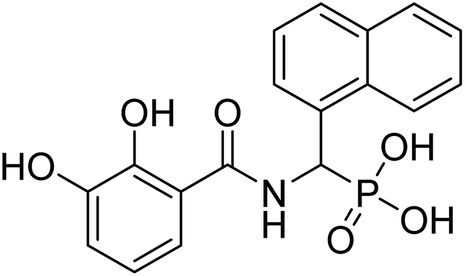 |
ND | 78.48 | −51.1 | −6.71 | 2.29 | 2.21 |
| 22a | 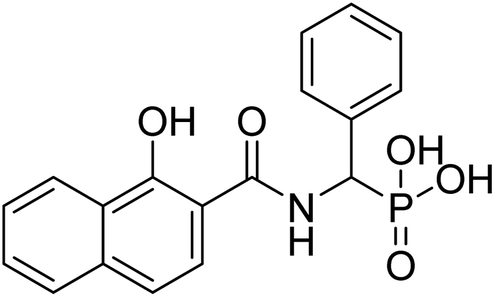 |
16.9 | 70.9 | −72.4 | −3.7 | 2.25 | 2.47 |
| 22b | 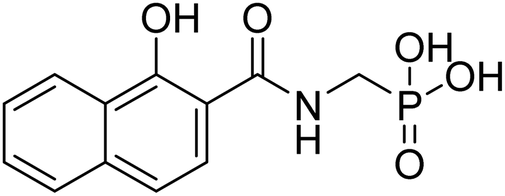 |
0.29 | 76.3 | −38.3 | −5.96 | 2.42 | 2.34 |
| 22c |  |
ND | 84.2 | −78.1 | −6.46 | 2.25 | 2.26 |
| 22d | 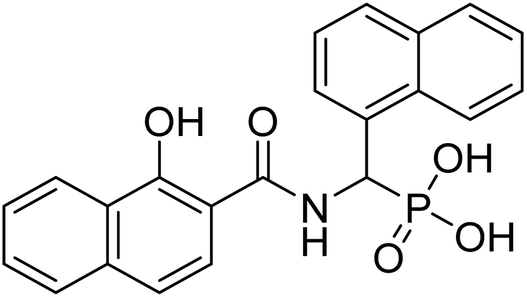 |
4.44 | 73.7 | −47.1 | −6.62 | 2.26 | 2.25 |
| 23a | 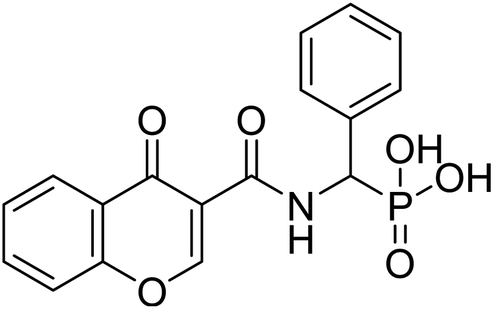 |
6.09 | 79.8 | −15.7 | −6.72 | 2.33 | 2.17 |
| 25a |  |
ND | 30.4 | −44.0 | −2.43 | 2.36 | 2.17 |
| 25b |  |
ND | 4.69 | −21.0 | −4.56 | 2.26 | 2.24 |
| 25c | 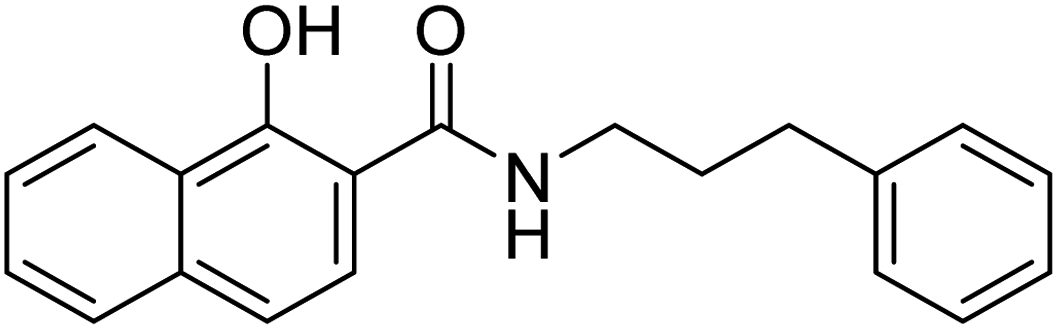 |
ND | 31.3 | −34.4 | 3.95 | 2.19 | 2.33 |
| 28a | 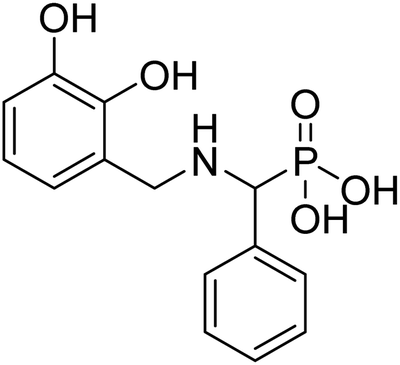 |
106 | 96.8 | −38.7 | −6.6 | 2.22 | 2.24 |
| FSM |  |
0.13b | 100 (at 100 μM) | ||||
2.3 Chemical synthesis
The desired α-aminophosphonates were synthesized employing the well-known Kabachnick–Fields condensation reaction (Scheme 1).69 Thus, a mixture of an aldehyde, ammonium acetate, and diethyl phosphite was heated to obtain phosphonate esters (11a–i) with different R1 substituents with good to moderate isolated yields. | ||
| Scheme 1 Reagents and conditions: (a). reflux, 80 °C, 12 h, (b). diethyl ether, hydrochloric acid, 0 °C, (c) NaOH, pH = 9 (yields, 42–88%). | ||
The carboxylic acid function of SA (F12), NA (F7), DHBA (F2) and CCA (F10) was coupled with the synthesized α-aminophosphonates using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)-mediated coupling under the standard conditions. Subsequently, the phosphonate ester intermediates were hydrolyzed to the desired phosphonic acid compounds (20a–e, 21a–d, 22a–d, and 23a, Scheme 2 and Table 2) using bromotrimethylsilane (TMSBr).20
 | ||
| Scheme 2 Reagents and conditions: (a). EDC, DMAP, various α-aminophosphonates, HOBt, DCM, RT, 12 h (yields, 25–66%); (b). TMSBr, dry DCM, 24 h, RT, THF/H2O, 2 h, RT. | ||
A few NA (F7) derivatives lacking a phosphonic acid moiety were also synthesized (25a–c, Table 2). for the SAR studies. Thus, using the EDC-mediated coupling, NA was coupled with various amines representing the R1 groups (Scheme 3). MBGs 2,3-dihydroxy benzaldehyde (F9) was attached to α-aminophosphonate via reductive amination followed by TMSBr treatment to obtain the desired compound 28a (Scheme 4 and Table 2). In contrast to other amide bond-coupled compounds (Scheme 2), molecule 27a possess a flexible linkage between MBG and the phosphonate moiety.
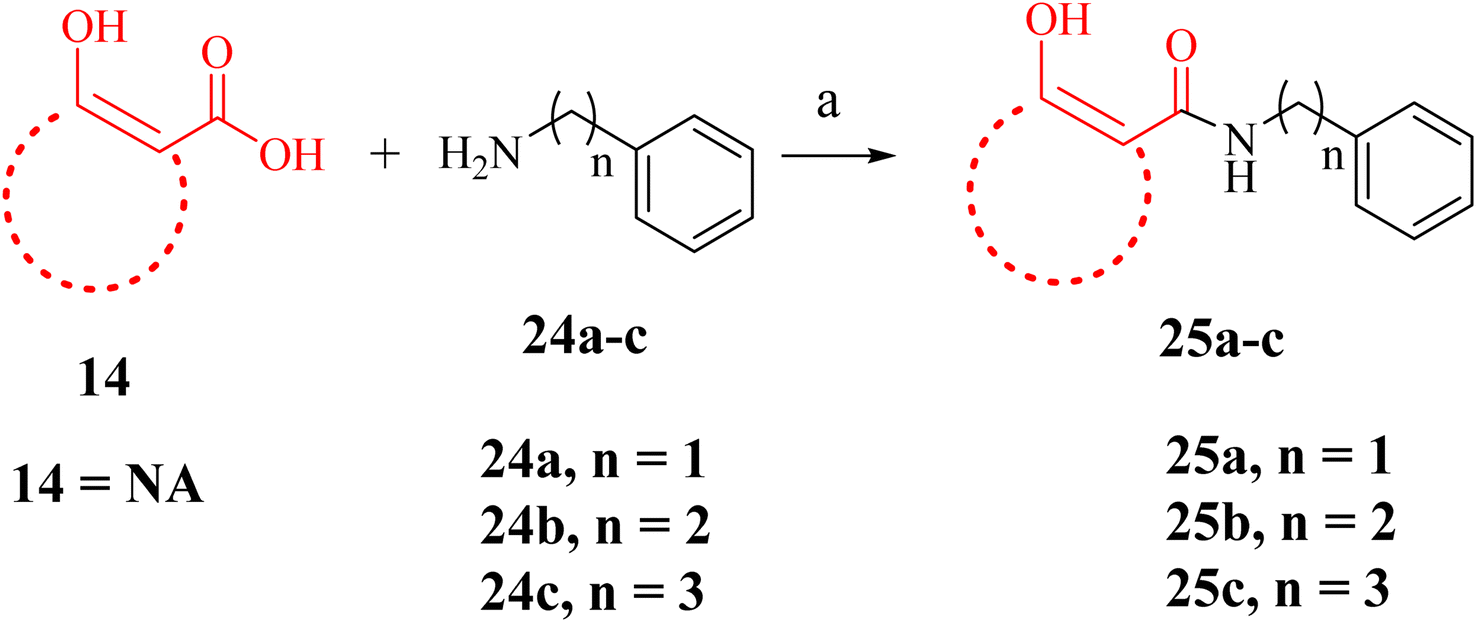 | ||
| Scheme 3 Reagents and conditions: (a). EDC, DMAP, amines, HOBt, DCM, RT, 12 h (yields, 55–62%). | ||
 | ||
| Scheme 4 General synthesis scheme for the derivative of 2,3-dihydroxy benzaldehyde (F9) MBG. Reagents and conditions: (a) acetic acid, sodium cyanoborohydride, DCM, 0 °C to RT, 14 h (yield, 61% for 27a). (b) TMSBr, dry DCM, 24 h, RT, THF/H2O, 2 h, RT. | ||
Thus, a total of 18 molecules based on five MBG fragments (F2, F7, F9, F10, and F12) were synthesized (Schemes 2–4). All the finally tested molecules are novel for which spectral data is reported in the ESI.† The 31P NMR and 1H NMR of a few compounds (e.g. 20a, 22a and 23a) revealed the existence of the syn- and anti-rotamers resulting from restricted rotation around the amide N–C(O) bond.9
2.4 In vitro DXR enzyme inhibition and SAR analysis
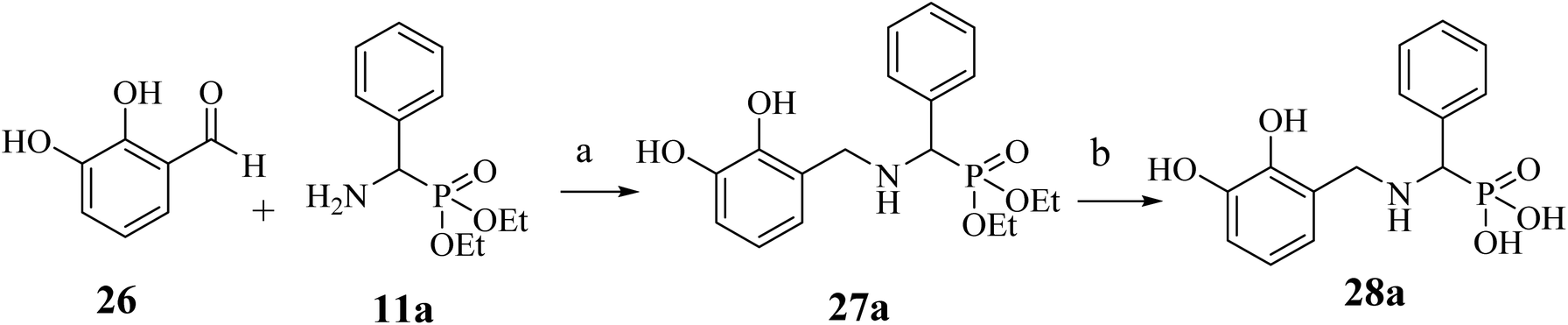
The final derivatives synthesized from Schemes 2–4 (20a–e, 21a–d, 22a–d, 23a, 25a–c and 28a) were evaluated against the recombinant EcDXR using the commercially available assay kit. Out of the 18 molecules, 15 displayed more than 50% inhibition (Table 2) when screened at 50 μM. For 7 molecules that exhibited more than 70% enzyme inhibition, IC50 values were determined. Unfortunately, inhibition at 50 μM could not be determined for a few synthesized molecules as these precipitated during dilution (20b, 20c, and 20e) with the aqueous buffer. However, the synthesis and characterization data for these compounds is reported in the Experimental section.In general, among all the amide-based molecules (Scheme 2), derivatives of NA (F7) (22a–d, Table 2) demonstrated better inhibition compared to other synthesized molecules. Compound 22b with no alpha substituent (R1 = H) showed the highest potency (IC50 = 0.29 μM), which is closest to the potency of 1 (IC50 = 0.13 μM, reported by the vendor) under identical assay conditions (Fig. 4A). The compound 22b closely resembles the binding mode of FSM (1), where the NA's O,O donor motif mimics the hydroxamate group of 1 (Fig. 4). In addition, the bicyclic NA ring of 22b was found to occupy the lipophilic pocket B, similar to its original fragment F7, and expectedly displayed a more negative MMGBSA score (−38.3 kcal mol−1, Table 2) than F7 (−26.0 kcal mol−1, Table 1 and Fig. 4B).
 | ||
| Fig. 4 The binding mode of 22b in the EcDXR (PDB ID 3R0I) active site. (A) The comparison of the predicted pose of 22b (pink ball and stick) with the docked pose of 1 (green-coloured ball and stick). The metal ion (purple-coloured ball) is chelated by the NA ring's O,O donor motif. As hypothesized, NA (F7) occupies the hydrophobic pocket B lined by Pro274 and Met276 (cyan coloured), whereas phosphonic acid of FSM and 22b occupy the hydrophilic region (B) a closer docked view of 22b overlaid with 1 and the cocrystallized ligand (moss green sticks) in the DXR active site (PDB code 3R0I). The phosphonic groups and the 22b and 1 linker atoms are predicted to adopt a similar conformation. | ||
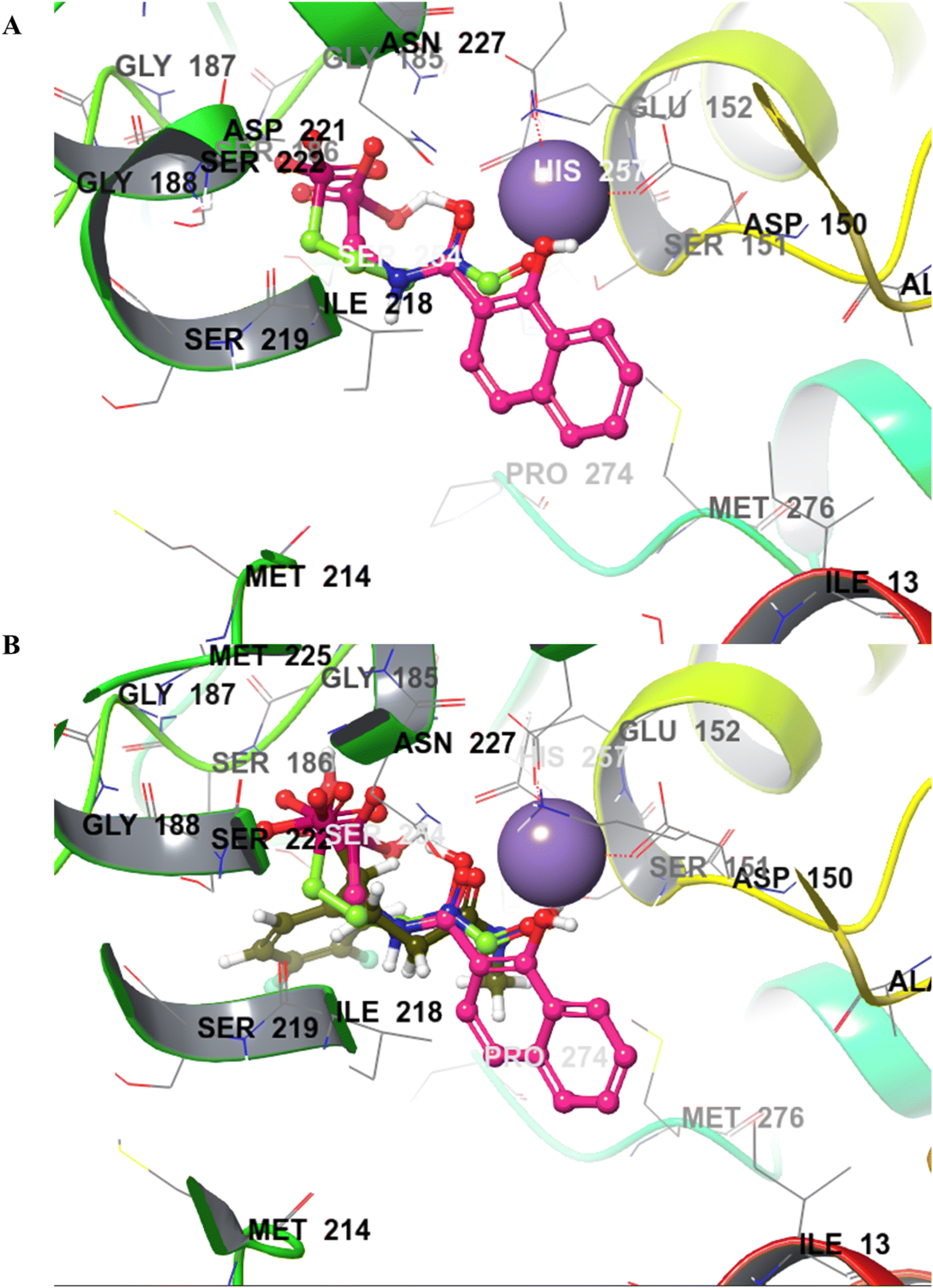
Compound 22a (R1 = phenyl) showed ∼57 folds lower potency (IC50 = 16.9 μM) than 22b despite having higher predicted binding energy (−72.4 kcal mol−1). In docking studies, the NA ring of 22b was found closer to the lipophilic pocket A, while the NA ring of 22a was comparatively flipped in another direction (Fig. 5A). This flipping in 22a resulted in different geometry of the O,O-metal coordination bonds, which might be the reason for its poor potency. The O-metal distances in 22a and 22b are also considerably different (Table 2). When the naphthyl group was attached as a lipophilic substituent in 22d (R1 = naphthyl), a ∼4-fold improvement in IC50 value (4.44 μM) was observed compared to 22a. The docking studies showed a reorientation of the naphthyl ring MBG of 22d as in 22b (Fig. 5A). Also, the bulkier naphthyl substituent alpha to the phosphonate of 22d goes deeper into the pocket “A” as compared to the phenyl ring of 22a (Fig. 5A), which might be a plausible explanation for the better potency of 22d. Thus, the interaction of the NA ring with pocket B seems to improve overall binding as hypothesized. However, MMGBSA energies and docking scores for 22a–22d did not correlate quantitatively with the experimental results, highlighting the known modelling limitations. Also, the Trp211 containing loop is known to be flexible, a vital protein dynamic not considered during the rigid molecular docking.
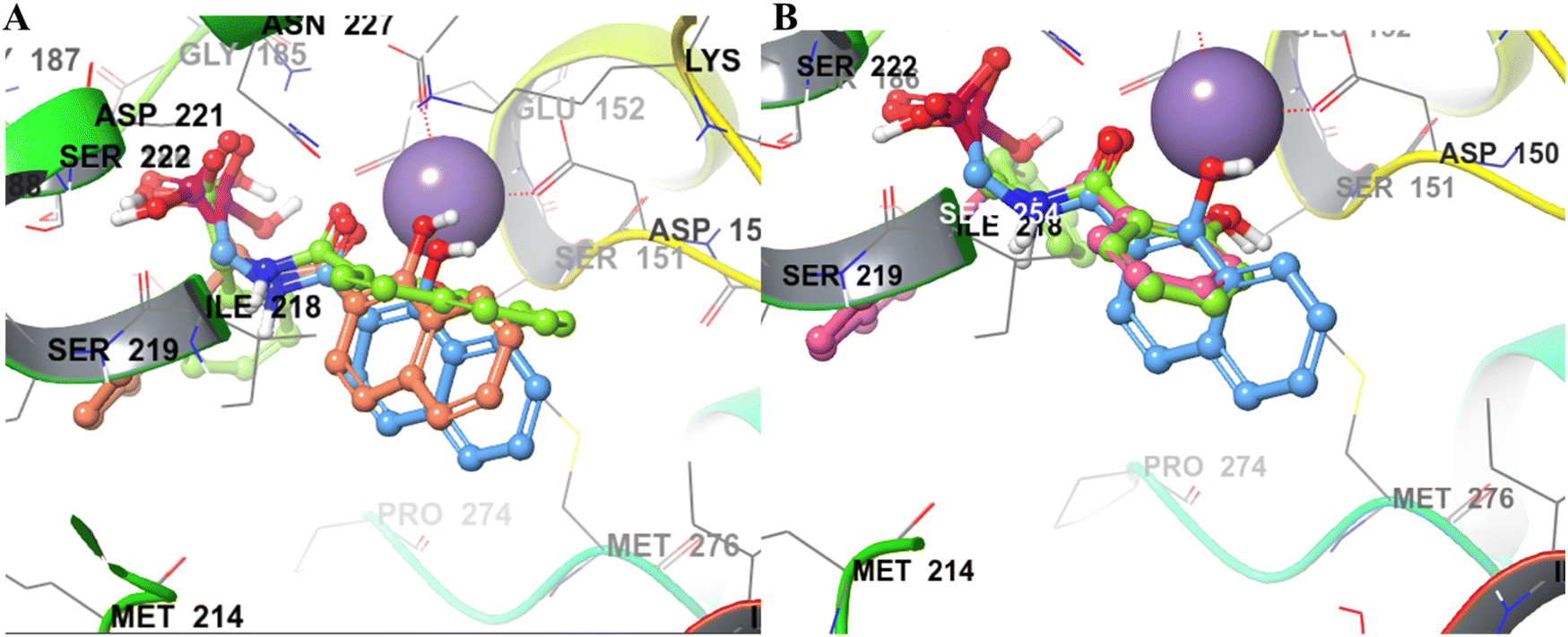 | ||
| Fig. 5 (A) Binding pose comparison of 22a (green), 22b (blue), and 22d (orange). The naphthyl ring of 22a shows considerable flipping compared to 22b and 22d, resulting in poor metal chelation and lower potency than the other two analogues. (B) The binding poses of 20a (green) and 20d (pink) show flipped orientations of the MBGs compared to 22b (blue). | ||
In contrast to NA series, SA (F12) based derivatives showed lower activity. For instance, compound 20a (R1 = phenyl) showed ∼2-fold less potency (IC50 = 33.2 μM) than its counterpart 22a (IC50 = 16.9 μM) in the NA series. Similarly, 20d (IC50 = 32.9 μM) was ∼8 folds less potent than 22d (IC50 = 4.44 μM). In both cases, NA derivatives (22a and 22d) are predicted to have better binding energy (MMGBSA) than the SA analogues (Table 2). The docking poses of both 20a and 20d revealed a perfect overlapping of the SA rings on each other (Fig. 5B). However, the plane of the SA ring in 20a and 20d is found to be perpendicular to the plane of the NA ring of 22b, suggesting different modes of metal chelation (Fig. 5B) and one of the plausible reasons for the observed differences in the potency of the two series. Despite 20d being predicted to have additional interaction with pocket A through its alpha-naphthyl substitution (Fig. 5B), its equipotency to 20a suggests that targeting pocket A with larger phenyl and naphthyl groups may not be appropriate for this series.
The DHBA analogues based on F2 were designed based on the earlier precedence of catechol-based DXR inhibitors.9 The docking studies predicted two different modes of metal chelation by DHBA (F2) derivatives 21a–d. In one mode, phenolic OH groups exhibit metal chelation, while in the second mode, one of the phenolics and the amide carbonyl oxygens participated in chelation (Fig. 6A–C). Experimentally, DHBA analogues 21a–c displayed poor enzyme inhibition at 50 μM (Table 2) compared to the corresponding NA derivatives 22a–c, upholding NA as a better MBG. The predicted binding energies of 21a–c are also significantly lower than their respective NA derivatives. Nevertheless, DHBA compound 21d (R1 = naphthyl) displayed potency similar to the corresponding NA derivative 22d at 50 μM, which agrees with the binding energies of both compounds.
 | ||
| Fig. 6 Two different metal chelation modes observed in the DHBA series. (A) The phenolic groups of 21d participating in metal chelation with Mn2+ (B) predicted pose of phenolic OH and amide oxygen of 21d showing metal chelation with Mn2+ (C) the 3D views of 21d (brown) with different modes of metal chelation. | ||
Comparing docked poses of compounds 20d, 21d, and 22d with a common alpha-substitution (R1 = naphthyl) but different MBGs revealed a remarkable similarity in the positioning of the naphthyl group in pocket A (Fig. 7B). However, compared to 21d and 22d, the metal chelation mode of SA of 20d was predicted to be different, with a slight twisting of the plane of the SA ring (Fig. 7).
 | ||
| Fig. 7 Binding pose comparison for the fragment derivatives. (A) Derivatives of DHBA, 21a (pink), 21b (green), 21c (orange) showing metal chelation by two phenolic OH while 21d (royal blue) showing metal chelation by one phenolic OH and amide O. (B) Derivatives of SA, 20d (green), DHBA, 21d (royal blue), and NA, 22d (orange), showed overlapping of the naphthyl group near pocket A, while DHBA and NA of 21d and 22d showed overlapping near pocket B. However, the NA ring was found closer to pocket B. (C) Derivatives of NA, with phosphonic acid attachment, 22b (cyan) and without phosphonic acid 25a (pink). 22b (cyan) showed a similar binding pose to the cocrystallized ligand (yellow), whereas the NA ring in 25a was oriented towards the phosphonate binding motif. | ||
Compound 23a, a derivative of CCA (F10) with a phenyl α-substituent (R1 = Ph), exhibited better potency (IC50 value = 6.09 μM) compared to its counterparts with other MBGs (20a, 21a, and 22a). However, due to CCA's potential for assay interference73 and significantly weaker calculated binding energy (−15.7 kcal mol−1) discouraged us from studying this MBG further.
We also synthesized a few derivatives (Scheme 3) lacking the phosphonic acid but retaining NA as MBG and an α-substituent. All these compounds (Table 2, 25a–c) were inactive in vitro. For instance, NA derivative 25a displayed only 30% inhibition at 50 μM concentration, much lower than its phosphonic acid-bearing analogue of 22a. The modelling pose predicted that without a phosphonic acid, the NA MBG of 25a could not maintain the metal chelation like 22a. In fact, the NA ring of 25a was oriented towards the phosphonic acid binding region of DXR (Fig. 7C). Increasing the carbon chains to increase the lipophilic interactions in 25b and 25c did not rescue the potency against the enzyme. These observations confirm the significance of the phosphonic acid in determining the potency against DXR and agree with the earlier studies with the hydroxamate-based compounds.9
The compound 28a obtained from Scheme 2 are significantly rigid owing to the usage of the amide group for linking the MBG and phosphonic acid. We used reductive amination to link catechol-based MBG (F9) and phosphonic acid (Scheme 4) to study the effect of increased linker flexibility. Thia amine-based compound, 28a (IC50 = 106), displayed improved DXR inhibition as compared to corresponding amide-based compound 21a (Table 2). Nonetheless, this F9-based compound was found to be weak DXR inhibitor compared to corresponding NA derivative (22a).
Within the NA series, the analogue 22b (R1 = H) showed higher enzyme inhibition than 22a and 22d, suggesting that an α-substitution is not favoured for this MBG. However, comparing 22a and 22d suggests a bicyclic naphthyl group is more favourable than the phenyl ring as an α-substitution. In contrast, 20a and 20d with SA as MBG displayed near identical IC50 against the enzyme, demonstrating no substantial effect of the α-substituent in this series.
Overall, SAR data suggests NA to be the best MBG for developing novel hydrophobic DXR inhibitors compared to other MBGs. However, the effect of α-substituents seems to be MBG dependent, as proposed earlier,9 and cannot be generalized across different MBGs.
2.5 Antibacterial activity
All compounds were evaluated against a panel of Gram-positive and Gram-negative bacteria, including some ESKAPE pathogens. Unfortunately, most of the tested compounds did not inhibit bacterial growth up to the concentration of 500 μM (Table S4, ESI†).Analogues 22b and 22d were also found to be inactive against E. coli despite showing good EcDXR inhibition. Insufficient membrane permeability or metabolic instability of these compounds under the assay conditions might explain these results and remain to be investigated.
With our ongoing interest in anti-TB drug discovery,53,56,74,75 we also screened all compounds against M. tuberculosis, initially at 200 μM (20a, 21a, 22a, 23a and 28a) and 500 μM (20c–e, 21b–d, and 22b–d). At 200 μM concentration, none of the tested compounds affected M. tuberculosis growth. In contrast, four of the nine tested compounds (20e, 20d, 22b, and 22d) inhibited M. tuberculosis growth at 500 μM concentration (Table S4, ESI†). The MIC values for these compounds against M. tuberculosis were subsequently determined using the Alamar Blue Assay (Fig. S6, ESI†). Our results reveal that compound 22d, which is based on NA MBG (IC50 = 4.44 μM against EcDXR), exhibits the most potent antimycobacterial activity (MIC = 125 μM), while other compounds viz., 20d, 20e, and 22b display MIC > 250 μM. It is noteworthy to mention that the most potent inhibitor of E. coli DXR (22b, IC50 = 0.29 μM) shows relatively lower potency (MIC > 250 μM) against M. tuberculosis. Due to the unavailability of recombinant DXR from M. tuberculosis, these molecules could not be tested against MtbDXR.
Overall, the compounds exhibited better activity against Mycobacterium than other bacteria, probably due to the inherent differences in the DXR structures, facilitated uptake mechanisms, or membrane compositions of these pathogens. For instance, 1 requires a GlpT transporter to enter several bacterial cells;32,76 however, GlpT is absent in M. tuberculosis. Compared to 1, molecules in this study are structurally different and probably not recognized by GlpT of these pathogens. Also, the negative charge on these molecules due to phosphonic acid moiety is possibly incompatible with the negatively charged bacterial membranes. This is further supported by the better whole-cell activities of prodrugs of 1 and its analogues with the masked phosphonic acid group.15,43,77–79
Lipophilic prodrugs of DXR inhibitors display better antimycobacterial activity owing to improved membrane diffusion.77–79 In this context, molecules 20e, 20d, 22b, and 22d having polar phosphonic acid may be considered to have fair antimycobacterial activity which can be further improved through masking the phosphonic acid. These derivatives are predicted to have higher lipophilicity than 1 and other analogues (Table S3†) due to the presence of α-substituent or NA-based MBGs. Arguably, the masked prodrugs of these molecules might display improved antibacterial and antimycobacterial activity.
3 Conclusion
In conclusion, we explored a library of 103 non-hydroxamate lipophilic MBGs against the bacterial DXR enzyme. Based on the promising modelling studies, 13 fragments were tested in vitro and showed significant inhibition of the EcDXR activity. Guided by molecular docking studies, the fragments were further grown to link a phosphonic acid moiety, an essential pharmacophoric feature. Seventeen novel DXR inhibitors were synthesized based on 5 non-hydroxamate MBG fragments (F2, F7, F9, F10 and F12). Seven molecules (20a, 20d, 22a–b, 22d, 23a, and 28a) displayed good to moderate enzyme inhibition. One notable observation was that the MMGBSA binding energies qualitatively rationalized the experimental data of analogues with different MBGs but not within a series of the same MBG.Overall, the molecules based on NA (22a–d) displayed the highest potency in enzyme assay than other MBG derivatives. The effect of lipophilic α-substituent, hypothesized to occupy pocket A, seems to vary with varying MBG and highlights the importance of detailed SAR analysis for individual MBGs.9 However, Trp211 containing loop (numbering as in EcDXR PDB 3ANM) of the DXR pocket is flexible; hence, molecular docking studies using rigid protein structures may not be able to rationalize all experimental data.
Unfortunately, most molecules, including 22a–d, did not display potency in the cell-based antibacterial assays. Nonetheless, four compounds (20e, 20d, 22b, and 22d) inhibited M. tuberculosis growth, with 22d being the most potent inhibitor (MIC = 125 μM).
This work positions NA as an important lipophilic MBG for developing novel DXR inhibitors. In future, it would be interesting to evaluate these novel molecules against the DXRs from other species, especially Mycobacterium and Plasmodium. There is a scope to extend SAR-based optimization to improve antibacterial potency. A prodrug approach might also be employed to this end.
4 Methodology
4.1 Fragment screening
The potential metal chelating fragments possessing O,O donor motif were collected from the literature.39 The physicochemical properties of the fragment library were calculated using Datawarrior (v 5.5.0), a versatile open-source program for cheminformatics applications.71,72,80 Molecular docking studies for selected 103 fragments were performed using the Glide program49,50 implemented in the Schrodinger Suite.51 The DXR structures from different species (5JAZ from P. falciparum 3D7, 2Y1D from M. tuberculosis H37Rv and 3R0I and 3ANM from E. coli K-12) were employed for pose prediction following the protocols described below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 63 to generate energetically accessible protonation states and all possible stereoisomers. Metal binding states were generated using Epik.
63 to generate energetically accessible protonation states and all possible stereoisomers. Metal binding states were generated using Epik.4.2 Synthetic methodology
All starting materials were purchased from commercial sources and used as purchased without further purification unless stated otherwise or synthesized via literature procedures. Thin-layer chromatography was used to monitor the progress of the reactions and checked by pre-coated TLC plates (E. Merck Kieselgel 60 F254 with fluorescence indicator UV254). The components were visualized by irradiation with ultraviolet light (254 nm), iodine vapours, or by staining in potassium permanganate solution followed by heating. Compounds were purified over a silica gel (230–400 mesh) column using distilled solvents. All final compounds were characterized by 1H NMR spectroscopy using deuterated solvents, CDCl3 or DMSO-d6. 1H NMR spectra were recorded on a Bruker Advance 400 MHz spectrometer. Chemical shifts are given in parts per million (ppm) (δ relative to residual solvent peak for 1H). Chemical shifts in 31P spectra are measured relative to the standard 70% aqueous H3PO4. High-resolution mass spectrometry (HRMS) analysis was performed using Agilent Technologies 6545 Q-TOF Agilent system. If needed, the LC-MS of compounds were recorded using the Waters TQD system.The purity of all the compounds evaluated against the enzyme or bacteria was determined using a Shimadzu HPLC system (UFLC LC-1020C, Shimadzu Corporation, Japan) with a D2 detector. The Ascentis® C18 (50 mm × 4.6 mm, i.d. 3.0 μm) column was used as a stationary phase for HPLC analysis. The mobile phase consisted of acetonitrile and 10 mM phosphate buffer adjusted to pH 4.7 with orthophosphoric acid. Isocratic mode with a flow rate of 1 mL min−1 was used for the analysis. The purity of all final compounds determined by HPLC was 90% or higher except for two compounds (20a and 21c).
4.2.1.1 Diethyl (amino(phenyl)methyl)phosphonate (11a). Synthesized from benzaldehyde (212.2 mg, 2 mmol), ammonium acetate (77.09 mg, 1 mmol) and diethyl phosphite (129 μL, 1 mmol) according to general procedure 4.2.1. Yellowish semisolid, yield: 57%. 1H NMR (400 MHz, CDCl3) δ 7.42–7.38 (dd, 2H), 7.30 (t, J = 7.6 Hz, 2H), 7.27–7.21 (td, 1H), 4.24–4.17 (d, 1H), 4.05–3.75 (m, 4H), 1.22 (t, J = 7.1 Hz, 3H), 1.12 (t, J = 7.1 Hz, 3H).
4.2.1.2 Diethyl (aminomethyl)phosphonate (11b). Synthesized from paraformaldehyde (600 mg, 20 mmol), ammonium acetate (770.9 mg, 10 mmol), and diethyl phosphite (1.29 mL, 10 mmol) according to general procedure 4.2.1. Yellowish oil, yield: 66%. 1H NMR (400 MHz, CDCl3) δ 4.21–4.08 (m, 4H), 3.36 (d, J = 10.7 Hz, 2H), 1.31 (t, J = 7.1 Hz, 6H).
4.2.1.3 Diethyl (1-aminoethyl)phosphonate (11c). Synthesized from acetaldehyde (3.33 g, 83.32 mmol), ammonium acetate (3.21 g, 41.66 mmol), and triethyl phosphite (6.922 g, 41.66 mmol) according to general procedure 4.2.1. Dark brown liquid, yield: 72%. 1H NMR (400 MHz, CDCl3) δ 5.26 (d, J = 49.3 Hz, 1N–H), 4.14–3.09 (m, 5H), 1.35–1.10 (m, 9H).
4.2.1.4 Diethyl (amino(naphthalen-1-yl)methyl)phosphonate (11d). Synthesized from Naphthaldehyde (4 g, 25.6 mmol), ammonium acetate (0.987 g, 12.8 mmol) and triethyl phosphite (2.127 g, 12.8 mmol) according to general procedure 4.2.1. Creamy white powder, yield: 88%. 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.3 Hz, 1H), 7.89 (dd, J = 7.1, 3.3 Hz, 2H), 7.86 (d, J = 8.5 Hz, 1H), 7.61–7.48 (m, 3H), 4.15–3.74 (m, 4H), 3.65 (d, J = 20.3 Hz, 1H), 1.26–1.22 (m, 3H), 1.07 (t, J = 7.1 Hz, 3H).
4.2.1.5 Diethyl (amino(3,4-dichlorophenyl)methyl)phosphonate (11e). Synthesized from 3,4-dichlorobenzaldehyde (350.02 mg, 2 mmol), ammonium acetate (77.09 mg, 1 mmol) and diethyl phosphite (129 μL, 10 mmol) according to general procedure 4.2.1. Yellowish semisolid, yield: 73.4%. 1H NMR (400 MHz, CDCl3) δ 7.58 (t, J = 2.2 Hz, 1H), 7.43 (t, J = 6.4 Hz, 1H), 7.32 (dt, J = 8.4, 2.1 Hz, 1H), 4.26 (d, J = 17.7 Hz, 1H), 4.13–3.97 (m, 4H), 1.27 (dd, J = 6.5, 2.5 Hz, 6H).
4.2.1.6 Diethyl (amino(furan-2-yl)methyl)phosphonate (11f). Synthesized from 2-furaldehyde (5.33 g, 55.52 mmol), ammonium acetate (2.14 g, 27.76 mmol) and diethyl phosphite (4.622 g, 27.76 mmol) according to general procedure 4.2.1. Dark brown semisolid, yield: 74%. 1H NMR (400 MHz, CDCl3) δ 7.42 (dd, J = 1.7, 0.9 Hz, 1H), 6.51 (t, J = 2.9 Hz, 1H), 6.38–6.36 (m, 1H), 5.01 (d, J = 13.4 Hz, 1H), 4.22–4.09 (m, 4H), 1.31 (t, J = 7.0 Hz, 3H), 1.27–1.22 (m, 3H).
4.2.1.7 Diethyl (1-aminopropyl)phosphonate (11g). Synthesized from Propionaldehyde (2.62 g, 46 mmol), ammonium acetate (1.78 g, 23 mmol), and diethyl phosphite (3.822 g, 23 mmol) according to general procedure 4.2.1. Yellowish oil, yield: 43%. 1H NMR (400 MHz, CDCl3) δ 4.23–4.05 (m, 4H), 4.02–3.83 (m, 2NH), 3.82–3.66 (m, 1H), 1.37–1.32 (m, 6H), 1.18–0.90 (m, 5H).
4.2.1.8 Diethyl (1-amino-3-phenylpropyl)phosphonate (11h). Synthesized from 3-phenylpropanal (6 g, 44.72 mmol), ammonium acetate (1.725 g, 22.36 mmol) and diethyl phosphite (3.715 g, 22.36 mmol) according to general procedure 4.2.1. Orange semisolid, yield: 52%. 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 7.3 Hz, 1H), 7.27–7.17 (m, 5H), 4.31–3.82 (m, 4H), 3.04–2.64 (m, 2H), 2.08–2.02 (m, 2H), 1.42–1.13 (m, 6H).
4.2.1.9 Diethyl ([1,1′-biphenyl]-4-yl(amino)methyl)phosphonate (11i). Synthesized from biphenyl-4-carboxaldehyde (2 g, 11 mmol), ammonium acetate (424 mg, 5.5 mmol) and diethyl phosphite (0.914 g, 5.5 mmol) according to general procedure 4.2.1. Yellow solid, yield: 42%. 1H NMR (400 MHz, CDCl3) δ 7.61–7.58 (dd, 4H), 7.53 (dd, J = 8.3, 2.1 Hz, 2H), 7.46–7.41 (ddd, 2H), 7.35 (ddd, J = 9.4, 5.2, 3.5 Hz, 1H), 4.32 (d, J = 17.3 Hz, 1H), 4.17–3.98 (m, 4H), 1.29 (t, J = 7.1 Hz, 3H), 1.23–1.19 (m, 3H).
4.2.2.1 Diethyl ((2-hydroxybenzamido)(phenyl)methyl)phosphonate (16a). Synthesized using general procedure 4.2.2 by coupling salicylic acid (26 mg, 0.185 mmol) and diethyl (amino(phenyl)methyl)phosphonate (11a, 45 mg, 0.185 mmol). EDCI·HCl (44 mg, 0.28 mmol) and HOBt (13 mg, 0.0925 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (23 mg, 0.185 mmol), according to general procedure 4.2.2. Yield: 47.6%. 1H NMR (400 MHz, CDCl3) δ 8.24 (dd, J = 9.1, 4.0 Hz, 1H), 7.79 (dd, J = 8.0, 1.4 Hz, 1H), 7.60–7.55 (m, 2H), 7.39 (tdd, J = 8.2, 7.7, 1.3 Hz, 3H), 7.05–6.97 (m, 1H), 6.91–6.86 (m, 1H), 5.86–5.75 (m, 1H), 4.28–3.69 (m, 4H), 1.34 (t, J = 6.7 Hz, 3H), 1.14 (t, J = 7.1 Hz, 3H).
4.2.2.2 Diethyl ((2-hydroxybenzamido)methyl)phosphonate (16b). Synthesized using general procedure 4.2.2 by coupling salicylic acid (138.21, 1 mmol) and diethyl (aminomethyl)phosphonate (11b, 243.24 mg, 1 mmol). EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122.17, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 49%; 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.3 Hz, 1H), 7.64–7.59 (m, 1H), 7.50 (dd, J = 11.3, 4.0 Hz, 1H), 6.39 (s, 1H), 4.24–3.75 (m, 4H), 3.44–3.21 (m, 2H), 1.35 (t, J = 7.1 Hz, 3H), 1.28 (t, J = 9.3 Hz, 3H).
4.2.2.3 Diethyl (1-(2-hydroxybenzamido)ethyl)phosphonate (16c). Synthesized using general procedure 4.2.2 by coupling salicylic acid (207 mg, 1.5 mmol) and diethyl (1-aminoethyl)phosphonate (11c, 272 mg, 1.5 mmol), EDCI·HCl (350 mg, 2.25 mmol) and HOBt (101.34, 0.75 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (183.25, 1.5 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 46%; 1H NMR (400 MHz, CDCl3) δ 10.49 (s, 1O–H), 7.85 (d, J = 7.9 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 6.87 (t, J = 7.5 Hz, 1H), 5.51 (p, J = 7.2 Hz, 1H), 4.25–4.12 (m, 4H), 1.58 (dd, J = 16.6, 7.0 Hz, 3H), 1.31 (t, J = 7.0 Hz, 6H).
4.2.2.4 Diethyl ((2-hydroxybenzamido)(naphthalen-1-yl)methyl)phosphonate (16d). Synthesized using general procedure 4.2.2 by coupling salicylic acid (207 mg, 1.5 mmol) and diethyl (amino(naphthalen-1-yl)methyl)phosphonate (11d, 440 mg, 1 mmol). EDCI·HCl (350 mg, 2.25 mmol) and HOBt (101.34 mg, 0.75 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (184 mg, 1.5 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–30% ethyl acetate/hexane). Yield: 61%. 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 8.6 Hz, 1H), 8.17 (dd, J = 8.4, 1.6 Hz, 1H), 7.93 (dd, J = 7.2, 2.9 Hz, 1H), 7.89 (d, J = 8.2 Hz, 2H), 7.68–7.61 (m, 1H), 7.57–7.52 (m, 2H), 7.52–7.48 (m, 1H), 7.23 (d, J = 13.9 Hz, 1H), 6.98 (dd, J = 11.5, 4.4 Hz, 2H), 4.24–3.73 (m, 4H), 1.28 (dd, J = 8.8, 5.4 Hz, 3H), 1.06 (t, J = 7.1 Hz, 3H).
4.2.2.5 Diethyl ((3,4-dichlorophenyl)(2-hydroxybenzamido)methyl)phosphonate (16e). Synthesized using general procedure 4.2.2 by coupling salicylic acid (138.21 mg, 1 mmol) and diethyl (amino(3,4-dichlorophenyl)methyl)phosphonate (11e, 312.13 mg, 1 mmol), EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122.17 mg, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 37%; 1H NMR (400 MHz, CDCl3) δ 10.29 (s, 1O–H), 8.03 (dd, J = 8.0, 1.5 Hz, 1H), 7.66 (t, J = 1.9 Hz, 1H), 7.57–7.46 (m, 2H), 7.45–7.40 (m, 1H), 7.04–6.94 (m, 2H), 6.30 (d, J = 13.8 Hz, 1H), 4.26–4.01 (m, 4H), 1.29 (dd, J = 15.0, 7.2 Hz, 6H).
4.2.2.6 Diethyl (2,3-dihydroxybenzamido)benzylphosphonate (17a). Synthesized using general procedure 4.2.2 by coupling 2,3-dihydroxybenzoic acid (204.2 mg, 1 mmol) and diethyl (amino(phenyl)methyl)phosphonate (11a, 243.24 mg, 1 mmol). EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122.17 mg, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 27%. 1H NMR (400 MHz, CDCl3) δ 12.28 (s, 1H), 7.75 (dd, J = 8.8, 5.2 Hz, 1H), 7.54 (dt, J = 10.7, 5.5 Hz, 2H), 7.43–7.32 (m, 3H), 7.21 (dd, J = 8.2, 1.3 Hz, 1H), 7.07 (dd, J = 7.9, 1.3 Hz, 1H), 6.78 (t, J = 8.0 Hz, 1H), 5.76–5.62 (m, 1H), 4.27–3.68 (m, 5H), 1.37–1.31 (m, 3H), 1.18–1.09 (m, 3H).
4.2.2.7 Diethyl((2,3-dihydroxybenzamido)methyl)phosphonate (17b). Synthesized using general procedure 4.2.2 by coupling 2,3-dihydroxybenzoic acid (204.2 mg, 1 mmol) and diethyl (aminomethyl)phosphonate (11b, 167 mg, 1 mmol). EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122 mg, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 27%. 1H NMR (400 MHz, CDCl3) δ 10.54 (s, 1O–H), 7.43 (dd, J = 8.1, 1.5 Hz, 1H), 7.16 (dd, J = 7.9, 1.5 Hz, 1H), 6.85 (t, J = 8.0 Hz, 1H), 4.67 (d, J = 8.7 Hz, 2H), 4.32–4.21 (m, 4H), 1.41–1.36 (m, 6H).
4.2.2.8 Diethyl(1-(2,3-dihydroxybenzamido)ethyl)phosphonate (17c). Synthesized using general procedure 4.2.2 by coupling 2,3-dihydroxybenzoic acid (204.2 mg, 1 mmol) and diethyl (1-aminoethyl)phosphonate (11c, 181 mg, 1 mmol). EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122 mg, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 37%. 1H NMR (400 MHz, CDCl3) δ 10.61 (s, 1O–H), 7.40 (dd, J = 8.1, 1.5 Hz, 1H), 7.13 (dd, J = 7.9, 1.5 Hz, 1H), 6.79 (dd, J = 10.4, 5.6 Hz, 1H), 5.55 (dq, J = 14.2, 7.1 Hz, 1H), 4.29–4.16 (m, 4H), 1.65–1.58 (m, 3H), 1.35 (dd, J = 9.5, 4.6 Hz, 6H).
4.2.2.9 Diethyl ((2,3-dihydroxybenzamido)(naphthalen-1-yl)methyl)phosphonate (17d). Synthesized using general procedure 4.2.2 by coupling 2,3-dihydroxybenzoic acid (306 mg, 1.5 mmol) and diethyl (amino(naphthalen-1-yl)methyl)phosphonate (11d, 440 mg, 1.5 mmol), EDCI·HCl (350 mg, 2.25 mmol) and HOBt (101 mg, 0.75 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (184 mg, 1.5 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 61%. 1H NMR (400 MHz, CDCl3) δ 10.43 (s, 1O–H), 8.32 (d, J = 8.6 Hz, 1H), 7.89 (dd, J = 13.5, 5.7 Hz, 3H), 7.70–7.62 (m, 2H), 7.54 (dd, J = 18.0, 7.8 Hz, 2H), 7.21 (d, J = 13.9 Hz, 1H), 7.16 (dd, J = 7.9, 1.1 Hz, 1H), 6.90 (t, J = 8.0 Hz, 1H), 4.38–3.70 (m, 4H), 1.28 (t, J = 7.0 Hz, 3H), 1.07 (t, J = 7.1 Hz, 3H).
4.2.2.10 Diethyl ((1-hydroxy-2-naphthamido)(phenyl)methyl)phosphonate (18a). Synthesized using general procedure 4.2.2 by coupling 2-hydroxy naphthoic acid (188.18 mg, 1 mmol) and diethyl (amino(phenyl)methyl)phosphonate (11a, 243.2 mg, 1 mmol). EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122.17 mg, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). White powder. Yield: 58%. 1H NMR (400 MHz, DMSO) δ 8.26 (d, J = 8.2 Hz, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.72–7.60 (m, 3H), 7.55 (dd, J = 14.0, 7.1 Hz, 1H), 7.40 (dd, J = 12.0, 8.2 Hz, 3H), 7.36–7.32 (m, 1H), 5.79 (d, J = 22.1 Hz, 1H), 4.13–3.82 (m, 4H), 1.15 (t, J = 7.0 Hz, 3H), 1.08 (t, J = 7.0 Hz, 3H).
4.2.2.11 Diethyl((1-hydroxy-2-naphthamido)methyl)phosphonate (18b). Synthesized using general procedure 4.2.2 by coupling 2-hydroxy naphthoic acid (188.18 mg, 1 mmol) and diethyl (aminomethyl)phosphonate (11b, 167 mg, 1 mmol). EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122 mg, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–50% ethyl acetate/hexane). Yield: 32.5%; 1H NMR (400 MHz, CDCl3) δ 11.67 (s, 1O–H), 8.44 (d, J = 7.8 Hz, 1H), 7.81 (dd, J = 8.5, 6.2 Hz, 2H), 7.69–7.62 (m, 1H), 7.60–7.54 (m, 1H), 7.33 (d, J = 8.8 Hz, 1H), 5.37 (t, J = 4.7 Hz, 1H), 4.72 (d, J = 8.6 Hz, 2H), 4.40–4.17 (m, 4H), 1.40 (t, J = 7.1 Hz, 6H).
4.2.2.12 Diethyl 1-(1-hydroxy-2-naphthamido)ethylphosphonate (18c). Synthesized using general procedure 4.2.2 by coupling 2-hydroxy naphthoic acid (188.18 mg, 1 mmol) and diethyl (1-aminoethyl)phosphonate (11c, 185 mg, 1 mmol). EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122 mg, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 30%. 1H NMR (400 MHz, CDCl3) δ 11.78 (s, 1O–H), 8.44 (d, J = 8.3 Hz, 1H), 7.81 (dd, J = 11.1, 8.6 Hz, 2H), 7.70–7.61 (m, 1H), 7.60–7.53 (m, 1H), 7.32 (d, J = 8.8 Hz, 1H), 5.62 (dq, J = 14.3, 7.1 Hz, 1H), 4.32–4.16 (m, 4H), 1.67 (dd, J = 16.6, 7.1 Hz, 3H), 1.36 (td, J = 7.1, 1.8 Hz, 6H).
4.2.2.13 Diethyl ((1-hydroxy-2-naphthamido)(naphthalen-1-yl)methyl)phosphonate (18d). Synthesized using general procedure 4.2.2 by coupling 2-hydroxy naphthoic acid (188.18 mg, 1 mmol) and diethyl (amino(naphthalen-1-yl)methyl)phosphonate (11d, 293.3 mg, 1 mmol). EDCI·HCl (233 mg, 1.5 mmol) and HOBt (68 mg, 0.5 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (122 mg, 1 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 66%. 1H NMR (400 MHz, CDCl3) δ 11.63 (s, 1H), 8.42 (t, J = 9.2 Hz, 2H), 8.10 (d, J = 8.8 Hz, 1H), 8.00 (dd, J = 6.9, 2.5 Hz, 1H), 7.95–7.88 (m, 2H), 7.79 (d, J = 8.1 Hz, 1H), 7.72–7.65 (m, 1H), 7.64–7.59 (m, 1H), 7.59–7.49 (m, 3H), 7.38 (d, J = 8.8 Hz, 1H), 7.32 (d, J = 13.9 Hz, 1H), 4.31–3.79 (m, 4H), 1.32–1.30 (m, 3H), 1.09 (t, J = 7.1 Hz, 3H).
4.2.2.14 Diethyl ((4-oxo-4H-chromene-3-carboxamido)(phenyl)methyl)phosphonate (19a). Synthesized using general procedure 4.2.2 by coupling chromone-3-carboxylic acid (95.0 mg, 0.5 mmol) and diethyl (amino(phenyl)methyl)phosphonate (11a, 134 mg, 0.5 mmol), EDCI·HCl (116.5 mg, 0.75 mmol) and HOBt (35 mg, 0.25 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (61 mg, 0.5 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (2–20% ethyl acetate/hexane). Yield: 25%. 1H NMR (400 MHz, CDCl3) δ 11.93 (s, 1H), 7.95 (d, J = 3.5 Hz, 1H), 7.70 (d, J = 7.4 Hz, 1H), 7.57 (d, J = 7.5 Hz, 2H), 7.36 (tt, J = 14.3, 7.0 Hz, 3H), 6.98 (d, J = 8.3 Hz, 1H), 6.85 (t, J = 7.5 Hz, 1H), 5.73 (dd, J = 21.3, 9.2 Hz, 1H), 4.29–3.68 (m, 4H), 1.33 (t, J = 7.1 Hz, 3H), 1.14 (t, J = 7.0 Hz, 3H).
4.2.2.15 N-Benzyl-1-hydroxy-2-naphthamide (25a). Synthesized using the general procedure 4.2.2 by coupling 2-hydroxy naphthoic acid (94.1 mg, 0.5 mmol) and benzylamine (53.6 mg, 0.5 mmol eq.), and EDCI·HCl (116.5 mg, 0.75 mmol) was added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (61 mg, 0.5 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (hexane/ethyl acetate = 95
:5) to yield the desired product. Yield: 56%. 1H NMR (400 MHz, CDCl3) δ 13.81 (s, 1H), 8.46 (ddd, J = 8.3, 4.5, 3.8 Hz, 1H), 7.77 (d, J = 7.5 Hz, 1H), 7.58 (dd, J = 22.2, 8.2, 6.9, 1.3 Hz, 2H), 7.45–7.37 (m, 4H), 7.35–7.24 (m, 3H), 6.62 (s, 1H), 4.72 (d, J = 5.6 Hz, 2H).
4.2.2.16 1-Hydroxy-N-phenethyl-2-naphthamide (25b). Synthesized using the general procedure 4.2.2 by coupling 2-hydroxy naphthoic acid (94.1 mg, 0.5 mmol) and phenylethylamine (61 mg, 0.5 mmol), and EDCI·HCl (116.5 mg, 0.75 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (61 mg, 0.5 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (hexane/ethyl acetate = 95
:5) to yield the desired product. Yield: 62%. 1H NMR (400 MHz, CDCl3) δ 13.84 (s, 1H), 8.47–8.42 (m, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.57 (dd, J = 22.1, 8.1, 6.9, 1.3 Hz, 2H), 7.41–7.36 (m, 2H), 7.33–7.29 (m, 2H), 7.28 (s, 1H), 7.25 (d, J = 8.7 Hz, 1H), 7.16 (d, J = 8.8 Hz, 1H), 6.36 (s, 1H), 3.80 (dd, J = 12.8, 6.9 Hz, 2H), 3.00 (t, J = 6.9 Hz, 2H).
4.2.2.17 1-Hydroxy-N-(3-phenylpropyl)-2-naphthamide (25c). Synthesized using the general procedure 4.2.2 by coupling 2-hydroxy naphthoic acid (94.1 mg, 0.5 mmol) and phenylpropylamine (66.5 mg, 0.5 mmol), and EDCI·HCl (116.5 mg, 0.75 mmol) were added under a nitrogen environment at 0 °C. The reaction mixture was charged with DMAP (61 mg, 0.5 mmol), according to general procedure 4.2.2. After work-up, the crude residue was purified using column chromatography (hexane/ethyl acetate = 95
:5) to yield the desired product. Yield: 55%. 1H NMR (400 MHz, CDCl3) δ 13.86 (s, 1H), 8.45 (d, J = 8.2 Hz, 1H), 7.76 (d, J = 7.9 Hz, 1H), 7.57 (dd, J = 23.6, 8.2, 6.9, 1.3 Hz, 2H), 7.34 (ddd, J = 17.1, 10.7, 6.1 Hz, 2H), 7.29–7.21 (m, 4H), 7.06 (dd, J = 8.5, 4.1 Hz, 1H), 6.25 (s, 1H), 3.58 (dd, J = 12.7, 6.9 Hz, 2H), 2.80 (t, J = 7.4 Hz, 2H), 2.12–2.00 (m, 2H).
:1). After 30 min, the solvent was removed in vacuo, and the solid residue was given washings with cold dichloromethane. The resulting residue was dried in vacuo overnight and further characterized by NMR and mass spectroscopy.
4.2.3.1 ((2-Hydroxybenzamido)(phenyl)methyl)phosphonic acid (20a). Ester diethyl ((2-hydroxybenzamido)(phenyl)methyl)phosphonate (16a) was hydrolyzed according to the given general procedure 4.2.3. Yield = 55.6%. 1H NMR (400 MHz, DMSO) δ 7.90 (d, J = 7.5 Hz, 1H), 7.39 (d, J = 5.1 Hz, 3H), 7.32 (t, J = 7.3 Hz, 2H), 7.29–7.22 (m, 1H), 7.01–6.90 (m, 2H), 5.34 (d, J = 21.1 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 134.32–133.69 (m), 130.66–130.56 (m), 128.60–128.50 (m), 128.21–128.03 (m), 127.69–127.57 (m), 120.09–119.92 (m), 120.04–119.92 (m), 117.96–117.81 (m), 117.32–117.17 (m). 31P NMR (162 MHz, DMSO) δ 18.26, 18.13. HRMS (ESI) for C14H14NO5P ([M+H]+): calculated 307.061, found 307.0625. Purity 89.3% [mobile phase, ACN
:buffer (70:30); RT: 3.087 min].
4.2.3.2 ((2-hydroxybenzamido)methyl)phosphonic acid (20b). Ester diethyl ((2-hydroxybenzamido)methyl)phosphonate (16b) was hydrolyzed according to the given general procedure 4.2.3. Yield = 38.6%. 1H NMR (400 MHz, DMSO) δ 7.93 (t, J = 12.9 Hz, 1H), 7.64 (dd, J = 14.1, 6.0 Hz, 1H), 7.07 (dd, J = 12.1, 7.9 Hz, 2H), 4.51 (d, J = 6.9 Hz, 2H). HRMS (ESI) for C8H10NO5P ([M+H]+): calculated 231.0297, found 231.0542.
4.2.3.3 (1-(2-Hydroxybenzamido)ethyl)phosphonic acid (20c). Ester diethyl (1-(2-hydroxybenzamido)ethyl)phosphonate (16c) was hydrolyzed according to the given general procedure 4.2.3. Yield = 86.6%. 1H NMR (400 MHz, DMSO) δ 7.84 (dd, J = 7.9, 1.6 Hz, 1H), 7.54 (ddd, J = 8.8, 5.7, 1.7 Hz, 1H), 7.05–6.94 (m, 2H), 5.22 (d, J = 1.2 Hz, 1H), 1.45 (dd, J = 15.4, 7.0 Hz, 3H). 13C NMR (400 MHz, DMSO) δ = 168.23 (s), 160.65 (s), 136.32 (s), 130.78 (s), 119.81 (s), 117.82 (s), 113.51 (s), 67.82 (s), 66.19 (s), 15.73 (s). 31P NMR (162 MHz, DMSO) δ 17.01. MS (-ESI) for C9H12NO5P ([2M+HCOOH–H]−): calculated 536.10, found 535.14. Purity 94.2% [mobile phase, ACN
:buffer (70:30); RT: 2.982 min].
4.2.3.4 ((2-Hydroxybenzamido)(naphthalen-1-yl)methyl)phosphonic acid (20d). Ester diethyl ((2-hydroxybenzamido)(naphthalen-1-yl)methyl)phosphonate (16d) derivative was hydrolyzed according to the given general procedure 4.2.3. Yield = 49.7%. 1H NMR (400 MHz, MeOD) δ 8.38 (d, J = 8.5 Hz, 1H), 8.24 (dd, J = 8.0, 1.5 Hz, 1H), 7.90 (dt, J = 10.4, 4.2 Hz, 3H), 7.63 (t, J = 7.2 Hz, 1H), 7.58–7.51 (m, 3H), 7.19 (d, J = 13.7 Hz, 1H), 7.05–7.00 (m, 1H), 6.95 (s, 1H). 13C NMR (100 MHz, DMSO) δ = 167.69 (s), 160.61 (s), 136.52 (s), 133.63 (s), 132.30 (s), 131.09 (s), 128.93 (s), 126.70 (s), 126.22 (s), 125.83 (s), 124.61 (s), 119.99 (s), 117.97 (s), 113.42 (s), 70.44 (s), 68.76 (s). 31P NMR (162 MHz, DMSO) δ 13.76. HRMS (ESI) for C18H16NO5P ([M+H]+): calculated 357.0766, found 357.075. Purity 99.7% [mobile phase, ACN: buffer (70
:30); RT: 3.43 min].
4.2.3.5 ((3,4-Dichlorophenyl)(2-hydroxybenzamido)methyl)phosphonic acid (20e). Ester diethyl ((3,4-dichlorophenyl)(2-hydroxybenzamido)methyl)phosphonate (16e) derivative was hydrolyzed according to the given general procedure 4.2.3. Yield = 47.2%. 1H NMR (400 MHz, DMSO) δ 7.99 (dd, J = 7.9, 1.6 Hz, 1H), 7.73 (t, J = 1.8 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.60–7.53 (m, 1H), 7.47 (dt, J = 8.4, 1.8 Hz, 1H), 7.05–6.99 (m, 2H), 6.12 (d, J = 13.6 Hz, 1H). 13C NMR (100 MHz, DMSO) δ = 167.25 (s), 160.33 (s), 137.15 (s), 136.40 (s), 131.29 (s), 131.00 (d, J = 26.2), 129.72 (d, J = 4.7), 128.14 (d, J = 4.8), 119.93 (s), 117.94 (s), 113.61 (s), 72.35 (s), 70.77 (s). 31P NMR (162 MHz, DMSO) δ 13.10. (s). HRMS (ESI) for C14H12Cl2NO5P ([M+H]+): calculated 374.983, found 374.9815. Purity 91.9% [mobile phase, ACN
:buffer (55:45); RT: 4.873 min].
4.2.3.6 ((2,3-Dihydroxybenzamido)(phenyl)methyl)phosphonic acid (21a). Ester diethyl ((2,3-dihydroxybenzamido)(phenyl)methyl)phosphonate (17a) was hydrolyzed according to the given general procedure 4.2.3. Yield = 80.3%. 1H NMR (400 MHz, MeOD) δ 7.88 (s, 1H), 7.43 (m, 5H), 6.84 (m, 2H), 5.40 (m, 2H). 13C NMR (100 MHz, DMSO) δ 147.26 (s), 146.32 (s), 138.15 (s), 128.48 (s), 127.91 (s), 120.00 (s), 118.99 (s), 117.89 (s), 49.10 (s). 31P NMR (162 MHz, DMSO) δ = 16.16. LCMS (-ESI) ([M–H]−): m/z calculated for C14H14NO6P: 322.0480, found: 321.96. Purity 94.8% [mobile phase, ACN
:buffer (60:40); RT: 3.02 min].
4.2.3.7 ((2,3-Dihydroxybenzamido)methyl)phosphonic acid (21b). Ester diethyl ((2,3-dihydroxybenzamido)methyl)phosphonate (17b) was hydrolyzed according to the given general procedure 4.2.3. Yield = 52.6%. 1H NMR (400 MHz, MeOD) δ 7.95 (s, 1H), 7.69–7.39 (m, 1H), 7.21–7.00 (m, 1H), 6.82 (m, 1H), 4.62 (s, 2H). 13C NMR (400 MHz, DMSO) δ 168.93 (s), 149.80 (s), 146.55 (s), 121.38 (s), 120.34 (s), 119.43 (s), 113.35 (s), 59.02 (s). 31P NMR (162 MHz, DMSO) δ = 14.15.5. MS (-ESI) for C8H10NO6P ([M–H]−): calculated 247.02, found 245.75.
4.2.3.8 (1-(2,3-Dihydroxybenzamido)ethyl)phosphonic acid (21c). Ester diethyl (1-(2,3-dihydroxybenzamido)ethyl)phosphonate (17c) was hydrolyzed according to given general procedure 4.2.3. Yield = 71.2%. 1H NMR (400 MHz, DMSO) δ 7.28 (d, J = 7.4 Hz, 1H), 7.04 (d, J = 7.0 Hz, 1H), 6.77 (t, J = 7.9 Hz, 1H), 5.25–5.16 (m, 1H), 1.43 (dd, J = 15.4, 6.9 Hz, 3H). 13C NMR (100 MHz, DMSO) δ 173.12–172.44 (m), 169.03–168.51 (m), 150.40–150.14 (m), 149.85 (s), 146.44 (s), 121.53–121.12 (m), 120.47–120.40 (m), 119.39 (s), 113.72–113.32 (m), 67.88–67.49 (m), 15.66 (s). 31P NMR (162 MHz, DMSO) δ 14.15. MS (-ESI) for C9H12NO6P ([2M+HCOOH–H]−): calculated 568.09, found 567.55. Purity 85.7% [mobile phase, ACN
:buffer (60:40); RT: 2.729 min].
4.2.3.9 ((2,3-Dihydroxybenzamido)(naphthalen-1-yl)methyl)phosphonic acid (21d). Ester diethyl ((2,3-dihydroxybenzamido)(naphthalen-1-yl)methyl)phosphonate (17d) was hydrolyzed according to the given general procedure 4.2.3. Yield = 52.1%. 1H NMR (400 MHz, MeOD) δ 8.38 (d, J = 8.5 Hz, 1H), 7.87 (dd, J = 15.3, 7.1 Hz, 4H), 7.78–7.73 (m, 1H), 7.59 (t, J = 7.5 Hz, 1H), 7.49 (dd, J = 15.8, 8.4 Hz, 3H), 7.23 (t, J = 12.2 Hz, 1H), 7.10–7.05 (m, 1H), 6.85 (t, J = 8.0 Hz, 1H). 31P NMR (162 MHz, DMSO) δ 19.38, 13.34. 13C NMR (400 MHz, DMSO) δ 168.58 (s), 149.83 (s), 146.50 (s), 133.59 (s), 132.40 (s), 131.03 (s), 129.63–129.21 (m), 128.95 (s), 128.94–127.69 (m), 126.68 (s), 126.22 (s), 125.80 (s), 124.59 (s), 121.55 (s), 120.69 (s), 119.62 (s), 113.42 (s), 68.96 (s). HRMS (ESI) for C18H16NO5P ([M+H]+): calculated 357.0766, found 357.0766. Purity 96.4% [mobile phase, ACN
:buffer (60:40); RT: 3.189 min].
4.2.3.10 ((1-Hydroxy-2-naphthamido)(phenyl)methyl)phosphonic acid (22a). Ester diethyl ((1-hydroxy-2-naphthamido)(phenyl)methyl)phosphonate (18a) was hydrolyzed according to the given general procedure 4.2.3. Yield = 64.3%. 1H NMR (400 MHz, DMSO) δ 8.24 (d, J = 8.1 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.95–7.81 (m, 1H), 7.70–7.61 (m, 1H), 7.55 (s, 4H), 7.49–7.24 (m, 4H), 5.52 (d, J = 21.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 159.61 (s), 137.53 (s), 136.39 (s), 129.39 (s), 128.87 (s), 128.45 (s), 127.92 (s), 127.66–127.57 (m), 126.24 (s), 125.01 (s), 123.99 (s), 123.50 (s), 118.12 (s), 107.98–107.87 (m). 31P NMR (162 MHz, DMSO) δ 17.19. HRMS (ESI) for C18H16NO5P ([M+H]+): calculated 357.0766, found 357.0756. Purity 97.2% [mobile phase, ACN
:buffer (70:30); RT: 4.300 min].
4.2.3.11 ((1-Hydroxy-2-naphthamido)methyl)phosphonic acid (22b). Ester diethyl ((1-hydroxy-2-naphthamido)methyl)phosphonate (18b) was hydrolyzed according to the given general procedure 4.2.3. Yield = 53.8%. 1H NMR (400 MHz, DMSO) δ 8.31 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.72 (t, J = 7.2 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 4.50 (d, J = 8.6 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 159.90 (s), 137.26 (s), 130.28 (s), 128.17 (s), 126.75 (s), 124.85 (s), 124.44–124.38 (m), 123.66 (s), 119.32 (s), 105.92–105.71 (m). 31P NMR (162 MHz, DMSO) δ 13.37. (s). HRMS (ESI) for C12H12NO5P ([M+H]+): calculated 281.0453, found 281.0462, purity 98.2% [mobile phase, ACN
:buffer (70:30); RT: 3.973 min].
4.2.3.12 (1-(1-Hydroxy-2-naphthamido)ethyl)phosphonic acid (22c). Ester diethyl (1-(1-hydroxy-2-naphthamido)ethyl)phosphonate (18c) was hydrolyzed according to the given general procedure 4.2.3. Yield = 55.9%. 1H NMR (400 MHz, DMSO) δ 11.76 (s, 1H), 8.32 (d, J = 8.2 Hz, 1H), 7.94 (d, J = 8.1 Hz, 1H), 7.82 (d, J = 8.8 Hz, 1H), 7.73 (dd, J = 11.0, 4.0 Hz, 1H), 7.63 (t, J = 7.6 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 5.35–5.26 (m, 1H), 1.50 (dd, J = 15.4, 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 159.99 (s), 137.28–137.09 (m), 130.32 (s), 128.20 (s), 126.79 (s), 124.88 (s), 124.44–124.35 (m), 123.66 (s), 106.04 (s), 68.22–68.09 (m), 66.60–66.48 (m), 15.80 (s). 31P NMR (162 MHz, CDCl3) δ 17.43. HRMS (-ESI) for C13H14NO5P ([M–H]−): calculated 295.06, found 293.70, purity 99.3% [mobile phase, ACN
:buffer (70:30); RT: 2.729 min]. Calculated 295.06, found 293.70.
4.2.3.13 ((1-Hydroxy-2-naphthamido)(naphthalen-1-yl)methyl)phosphonic acid (22d). Ester diethyl ((1-hydroxy-2-naphthamido)(naphthalen-1-yl)methyl)phosphonate (18d) was hydrolyzed according to the given general procedure 4.2.3. Yield = 61%. 1H NMR (400 MHz, DMSO) δ 11.47 (s, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.28 (d, J = 8.3 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 8.01–7.88 (m, 3H), 7.80 (dd, J = 7.1, 1.7 Hz, 1H), 7.76–7.69 (m, 1H), 7.64 (dd, J = 13.5, 7.0 Hz, 2H), 7.60–7.55 (m, 3H), 6.99 (d, J = 13.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 169.30 (d, J = 9.5 Hz), 160.12 (s), 137.38 (s), 133.65 (s), 132.05 (s), 130.47 (s), 129.00 (d, J = 10.2 Hz), 126.81 (d, J = 9.6 Hz), 126.27 (s), 125.84 (s), 124.90 (s), 124.55 (s), 124.38 (s), 123.71 (s), 119.58 (s), 105.81 (s), 70.61 (s), 69.01 (s). 31P NMR (162 MHz, DMSO) δ 14.06. HRMS (ESI) for C18H16NO5P ([M+H]+): calculated 357.0766, found 357.0766. Purity 96.4% [mobile phase, ACN
:buffer (60:40); RT: 3.189 min].
4.2.3.14 ((4-Oxo-4H-chromene-3-carboxamido)(phenyl)methyl)phosphonic acid (23a). Ester diethyl ((4-oxo-4H-chromene-3-carboxamido)(phenyl)methyl)phosphonate (19a) was hydrolyzed according to the given general procedure 4.2.3. Yield = 90.7%. 1H NMR (400 MHz, MeOD) δ 7.98 (d, J = 7.5 Hz, 1H), 7.54 (d, J = 7.4 Hz, 2H), 7.38 (dd, J = 15.6, 7.9 Hz, 3H), 7.30 (t, J = 7.2 Hz, 1H), 6.96 (dd, J = 16.1, 7.9 Hz, 2H), 5.66 (t, J = 17.3 Hz, 1H). 13C NMR (100 MHz, DMSO) δ 165.85 (s), 157.59 (s), 138.27 (s), 133.75 (s), 130.73 (s), 128.65–128.06 (m), 127.39 (s), 119.74 (s), 118.11 (s), 117.33 (s), 52.88 (s), 51.42 (s). 31P NMR (162 MHz, DMSO) δ 17.87. MS (-ESI) for C17H14NO6P ([M+HCOOH–H]−): calculated 405.06, found 403.89. Purity 97.17% [mobile phase, ACN
:buffer (70:30); RT: 4.3 min].
4.2.4.1 (((2,3-Dihydroxybenzyl)amino)(phenyl)methyl)phosphonate (27a). Synthesized by using 2,3-dihydroxybenzaldehyde (207.18 mg, 1.5 mmol), diethyl (amino(phenyl)methyl)phosphonate (11a, 365 mg, 1.5 mmol), acetic acid (90 mg, 1.5 eq.) and sodium cyanoborohydride (141 mg, 2.25 mmol) according to general procedure 4.2.4. After work-up, the crude residue was purified using column chromatography (2–50% ethyl acetate/hexane). Yield: 61%. 1H NMR (400 MHz, CDCl3) δ 7.45–7.34 (m, 5H), 6.87 (dd, J = 8.0, 1.4 Hz, 1H), 6.68 (td, J = 7.8, 4.1 Hz, 1H), 6.39 (dd, J = 7.6, 1.2 Hz, 1H), 4.21–3.87 (m, 5H), 3.76–3.62 (m, 2H), 1.38–1.32 (m, 3H), 1.09 (t, J = 7.1 Hz, 3H).
4.2.4.2 (2,3-Dihydroxybenzylamino)benzylphosphonic acid (28a). Ester diethyl (((2,3-dihydroxybenzyl)amino)(phenyl)methyl)phosphonate (27a) was hydrolyzed according to the given general procedure 4.2.3. Yield = 34%. 1H NMR (400 MHz, CD3OD) δ 7.52 (d, J = 19.8 Hz, 5H), 6.89 (d, J = 7.7 Hz, 1H), 6.72 (t, J = 7.8 Hz, 1H), 6.60 (d, J = 7.4 Hz, 1H), 4.49 (d, J = 17.1 Hz, 1H), 4.20 (dd, J = 52.2, 13.1 Hz, 2H). 13C NMR (100 MHz, DMSO) δ = 145.77 (s), 144.78 (s), 131.74 (d, J = 1.6), 130.06 (s), 130.01 (s), 130.05–128.64 (m), 121.73 (s), 119.54 (s), 118.60 (s), 116.63 (s), 49.07 (s), 46.02 (s). 31P NMR (162 MHz, DMSO) δ = 11.92. HRMS (ESI) for C14H16NO5P ([M+H]+): calculated 309.0766, found 309.0747.
4.3 Biological evaluation
ESKAPE pathogens (obtained from Dr Bhabatosh Das, THSTI) were cultured in the Luria Bertani (LB) medium at 37 °C with shaking at 200 rpm to turbidity.
Abbreviations
| AMR | Antimicrobial resistance |
| CCA | Chromone-3-carboxylic acid |
| CDC | Centers for Disease Control and Prevention |
| CDCl3 | Chloroform |
| DCM | Dichloromethane |
| DHBA | 2,3-Dihydroxy benzoic acid |
| DMAP | 4-Dimethylaminopyridine |
| DMF | Dimethyl formamide |
| DMSO | Dimethyl sulfoxide |
| DXP | 1-Deoxy-D-xylulose-5-phosphate |
| DXR | 1-Deoxy-D-xylulose 5-phosphate reductoisomerase |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| FSM | Fosmidomycin |
| H-bonds | Hydrogen bonds |
| HBA | Hydrogen bond acceptor |
| HBD | Hydrogen bond donor |
| HOBt | Hydroxybenzotriazole |
| HPLC | High-pressure liquid chromatography |
| HRMS | High-resolution mass spectrometry |
| IC50 | Half maximal inhibitory concentration |
| MBG | Metal-binding group |
| MCL | Metal chelating library |
| MDR | Multidrug resistant |
| MeOH | Methanol |
| MEP | 2-C-Methyl-D-erythritol 4-phosphate |
| MIC | Minimal inhibitory concentration |
| MMGBSA | Molecular mechanics with generalized born and surface area solvation |
| Mtb | Mycobacterium tuberculosis |
| MW | Molecular weight |
| NA | 1-Hydroxy-2-naphthoic acid |
| NaCl | Sodium chloride |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NaOH | Sodium hydroxide |
| NMR | Nuclear magnetic resonance |
| PDB | Protein data bank |
| RMSD | Root mean square deviation |
| Ro3 | Rule of Three |
| RPM | Rate per minute |
| RT | Room temperature |
| SA | Salicylic acid |
| SAR | Structure activity relationship |
| SP | Standard precision |
| TB | Tuberculosis |
| TLC | Thin layer chromatography |
| TMSBr | Bromotrimethylsilane |
| XP | Extra precision |
Data availability
The data supporting this article have been included in the Methodology section and as part of the ESI.†Author contributions
Conceptualization: S. S.; data curation: S. K., Eeba, and M. T.; investigation: S. K., Eeba, and M. T; methodology: S. K., Eeba, and M. T; validation: S. S., and N. A.; supervision: S. S., and N. A.; writing – review & editing: S. K., S. S., and N. A.; funding acquisition: S. S, and N. A. Finally, all authors revised and approved the final submitted manuscript.Conflicts of interest
The authors declared no conflict of interest.Acknowledgements
The authors acknowledge the Department of Science and Technology, Science & Engineering Research Board (DST-SERB), New Delhi, for financial assistance through the Core Research Grant (CRG/2018/001527). We also acknowledge DST, New Delhi, for sponsoring the HRMS and LCMS facility at BITS Pilani under the FIST program. We thank Dr William Jacobs at the Albert Einstein College of Medicine, USA, for providing M. tuberculosis H37Rv mc2 6206 strain and Dr Bhabatosh Das at THSTI for providing us with the ESKAPE pathogens. Funding support from the Department of Biotechnology, Govt. of India to THSTI core is acknowledged.References
- Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2022 1, 2022 Search PubMed.
- F. Prestinaci, P. Pezzotti and A. Pantosti, Antimicrobial resistance: A global multifaceted phenomenon, Pathog. Global Health, 2015, 109(7), 309–318, DOI:10.1179/2047773215Y.0000000030.
- CDC. Antibiotic Resistance Threats in the United States 2019, CDC, 2019;10(1), DOI:10.1186/s13756-020-00872-w.
- Collaborators AR articles, Global Burden of Bacterial Antimicrobial Resistance in 2019: a Systematic Analysis, 2022, vol. 399, DOI:10.1016/S0140-6736(21)02724-0.
- A. Coates, Y. Hu, R. Bax and C. Page, The future challenges facing the development of new antimicrobial drugs, Nat. Rev. Drug Discovery, 2002, 1(11), 895–910, DOI:10.1038/nrd940.
- A. Frank and M. Groll, The Methylerythritol Phosphate Pathway to Isoprenoids, Chem. Rev., 2017, 117(8), 5675–5703, DOI:10.1021/acs.chemrev.6b00537.
- L. Zhao, C. W. chen, Y. Xiao, H. W. Liu and P. Liu, Methylerythritol Phosphate Pathway of Isoprenoid Biosynthesis, Annu. Rev. Biochem., 2013, 82(1), 497–530, DOI:10.1146/annurev-biochem-052010-100934.
- X. Wang and C. S. Dowd, The Methylerythritol Phosphate Pathway: Promising Drug Targets in the Fight against Tuberculosis, ACS Infect. Dis., 2018, 4(3), 278–290, DOI:10.1021/acsinfecdis.7b00176.
- S. Kesharwani and S. Sundriyal, Non-hydroxamate inhibitors of 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR): A critical review and future perspective, Eur. J. Med. Chem., 2021, 213, 113055, DOI:10.1016/j.ejmech.2020.113055.
- R. L. Edwards, I. Heueck and S. G. Lee, et al., Potent, specific MEPicides for treatment of zoonotic staphylococci, PLoS Pathog., 2020, 16(6), e1007806, DOI:10.1371/journal.ppat.1007806.
- S. Wang, M. Li and X. Luo, et al., Inhibitory Effects of Fosmidomycin Against Babesia microti in vitro, Front. Cell Dev. Biol., 2020, 8, 247, DOI:10.3389/fcell.2020.00247.
- S. Heuston, M. Begley, C. G. M. Gahan and C. Hill, Isoprenoid biosynthesis in bacterial pathogens, Microbiology, 2012, 158(6), 1389–1401, DOI:10.1099/mic.0.051599-0.
- L. Imlay and A. R. Odom, Isoprenoid Metabolism in Apicomplexan Parasites, Curr. Clin. Microbiol. Rep., 2014, 1(3–4), 37–50, DOI:10.1007/s40588-014-0006-7.
- N. Singh, G. Chevé, M. A. Avery and C. R. McCurdy, Targeting the methyl erythritol phosphate (MEP) pathway for novel antimalarial, antibacterial and herbicidal drug discovery: inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) enzyme, Curr. Pharm. Des., 2007, 13(11), 1161–1177, DOI:10.2174/138161207780618939.
- E. R. Jackson and C. S. Dowd, Inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr): a review of the synthesis and biological evaluation of recent inhibitors, Curr. Top. Med. Chem., 2012, 12(7), 706–728 CrossRef CAS PubMed.
- P. K. Ajikumar, K. Tyo, S. Carlsen, O. Mucha, T. H. Phon and G. Stephanopoulos, Terpenoids: Opportunities for biosynthesis of natural product drugs using engineered microorganisms, Mol. Pharm., 2008, 5(2), 167–190, DOI:10.1021/mp700151b.
- H. Seto, H. Watanabe and K. Furihata, Simultaneous operation of the mevalonate and non-mevalonate pathways in the biosynthesis of isopentenyl diphosphate in Streptomyces aeriouvifer, Tetrahedron Lett., 1996, 37(44), 7979–7982, DOI:10.1016/0040-4039(96)01787-X.
- A. Argyrou and J. S. Blanchard, Kinetic and Chemical Mechanism of Mycobacterium tuberculosis 1-Deoxy-D-xylulose-5-phosphate Isomeroreductase, Biochemistry, 2004, 43(14), 4375–4384, DOI:10.1021/bi049974k.
- A. C. Brown and T. Parish, Dxr is essential in Mycobacterium tuberculosis and fosmidomycin resistance is due to a lack of uptake, BMC Microbiol., 2008, 8, 78, DOI:10.1186/1471-2180-8-78.
- L. Deng, S. Sundriyal, V. Rubio, Z. Z. Shi and Y. Song, Coordination chemistry based approach to lipophilic inhibitors of 1-deoxy-D-xylulose-5-phosphate reductoisomerase, J. Med. Chem., 2009, 52(21), 6539–6542, DOI:10.1021/jm9012592.
- M. Okuhara, Y. Kuroda and T. Goto, et al., Studies on new phosphonic acid antibiotics. I. FR-900098, isolation and characterization, J. Antibiot., 1980, 33(1), 13–17, DOI:10.7164/antibiotics.33.13.
- E. Iguchi, M. Okuhara, M. Kohsaka, H. Aoki and H. Imanaka, Studies on New Phosphonic Acid Antibiotics. II. Taxonomic Studies on Producing Organisms of the Phosphonic Acid and Related Compounds, J. Antibiot., 1980, vol. 33, pp. 19–23, accessed June 24, 2020, https://pubmed.ncbi.nlm.nih.gov/7372546/ Search PubMed.
- T. Kuzuyama, T. Shimizu, S. Takahashi and H. Seto, Fosmidomycin, a specific inhibitor of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis, Tetrahedron Lett., 1998, 39(43), 7913–7916, DOI:10.1016/S0040-4039(98)01755-9.
- L. M. Henriksson, T. Unge, J. Carlsson, J. Åqvist, S. L. Mowbray and T. A. Jones, Structures of Mycobacterium tuberculosis 1-deoxy-D-xylulose-5-phosphate reductoisomerase provide new insights into catalysis, J. Biol. Chem., 2007, 282(27), 19905–19916, DOI:10.1074/jbc.M701935200.
- A. M. Sweeney, R. Lange and R. P. M. Fernandes, et al., The crystal structure of E. coli 1-deoxy-D-xylulose-5-phosphate reductoisomerase in a ternary complex with the antimalarial compound fosmidomycin and NADPH reveals a tight-binding closed enzyme conformation, J. Mol. Biol., 2005, 345(1), 115–127, DOI:10.1016/j.jmb.2004.10.030.
- C. Zinglé, L. Kuntz, D. Tritsch, C. Grosdemange-Billiard and M. Rohmer, Isoprenoid biosynthesis via the methylerythritol phosphate pathway: Structural variations around phosphonate anchor and spacer of fosmidomycin, a potent inhibitor of deoxyxylulose phosphate reductoisomerase, J. Org. Chem., 2010, 75(10), 3203–3207, DOI:10.1021/jo9024732.
- E. R. Jackson, G. S. Jose and R. C. Brothers, et al., The effect of chain length and unsaturation on Mtb Dxr inhibition and antitubercular killing activity of FR900098 analogs, Bioorg. Med. Chem. Lett., 2014, 24(2), 649–653, DOI:10.1016/j.bmcl.2013.11.067.
- C. A. Lipinski, Drug-like properties and the causes of poor solubility and poor permeability, J. Pharmacol. Toxicol. Methods, 2000, 44(1), 235–249, DOI:10.1016/S1056-8719(00)00107-6.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25, DOI:10.1016/s0169-409x(96)00423-1.
- P. D. Leeson and R. J. Young, Molecular Property Design: Does Everyone Get It?, ACS Med. Chem. Lett., 2015, 6(7), 722–725, DOI:10.1021/acsmedchemlett.5b00157.
- H. P. Kuemmerle, T. Murakawa and F. De Santis, Pharmacokinetic evaluation of fosmidomycin, a new phosphonic acid antibiotic, Chemioterapia, 1987, 6(2), 113–119 CAS.
- S. C. Nair, C. F. Brooks and C. D. Goodman, et al., Apicoplast isoprenoid precursor synthesis and the molecular basis of fosmidomycin resistance in Toxoplasma gondii, J. Exp. Med., 2011, 208(7), 1547–1559, DOI:10.1084/jem.20110039.
- R. K. Dhiman, M. L. Schaeffer, A. M. Bailey, C. A. Testa, H. Scherman and D. C. Crick, 1-Deoxy-D-xylulose 5-phosphate reductoisomerase (IspC) from Mycobacterium tuberculosis: Towards understanding mycobacterial resistance to fosmidomycin, J. Bacteriol., 2005, 187(24), 8395–8402, DOI:10.1128/JB.187.24.8395-8402.2005.
- M. Nishida, Pharmacokinetics of Fosmidomycin, a New Phosphonic Acid Antibiotic, Antimicrob. Agents Chemother., 1982, 21(2), 224–230 CrossRef PubMed.
- K. Na-Bangchang, R. Ruengweerayut, J. Karbwang, A. Chauemung and D. Hutchinson, Pharmacokinetics and pharmacodynamics of fosmidomycin monotherapy and combination therapy with clindamycin in the treatment of multidrug resistant falciparum malaria, Malar. J., 2007, 6, 1–10, DOI:10.1186/1475-2875-6-70.
- E. Uh, E. R. Jackson and J. G. San, et al., Antibacterial and antitubercular activity of fosmidomycin, FR900098, and their lipophilic analogs, Bioorg. Med. Chem. Lett., 2011, 21(23), 6973–6976, DOI:10.1016/j.bmcl.2011.09.123.
- S. Shen and A. P. Kozikowski, Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors-What Some May Have Forgotten or Would Rather Forget?, ChemMedChem, 2016, 11(1), 15–21, DOI:10.1002/cmdc.201500486.
- J. J. McClure, X. Li and C. J. Chou, Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics, Adv. Cancer Res., 2018, 138, 183–211, DOI:10.1016/bs.acr.2018.02.006.
- R. L. Edwards, R. C. Brothers and X. Wang, et al., MEPicides: Potent antimalarial prodrugs targeting isoprenoid biosynthesis, Sci. Rep., 2017, 7(1), 1–11, DOI:10.1038/s41598-017-07159-y.
- S. M. Cohen, A Bioinorganic Approach to Fragment-Based Drug Discovery Targeting Metalloenzymes, Acc. Chem. Res., 2017, 50(8), 2007–2016, DOI:10.1021/acs.accounts.7b00242.
- J. A. Day and S. M. Cohen, Investigating the selectivity of metalloenzyme inhibitors, J. Med. Chem., 2013, 56(20), 7997–8007, DOI:10.1021/jm401053m.
- P. Chen, L. B. Horton and R. L. Mikulski, et al., 2-Substituted 4,5-dihydrothiazole-4-carboxylic acids are novel inhibitors of metallo-β-lactamases, Bioorg. Med. Chem. Lett., 2012, 22(19), 6229–6232, DOI:10.1016/j.bmcl.2012.08.012.
- S. L. Williams, C. A. F. de Oliveira, H. Vazquez and J. A. McCammon, From Zn to Mn: The Study of Novel Manganese-binding Groups in the Search for New Drugs against Tuberculosis, Chem. Biol. Drug Des., 2011, 77(2), 117–123, DOI:10.1111/j.1747-0285.2010.01060.x.
- L. Deng, K. Endo, M. Kato, G. Cheng, S. Yajima and Y. Song, Structures of 1-deoxy-D-xylulose-5-phosphate reductoisomerase/lipophilic phosphonate complexes, ACS Med. Chem. Lett., 2011, 2(2), 165–170, DOI:10.1021/ml100243r.
- S. Yajima, K. Hara and J. M. Sanders, et al., Crystallographic structures of two bisphosphonate:1-deoxyxylulose-5- phosphate reductoisomerase complexes, J. Am. Chem. Soc., 2004, 126(35), 10824–10825, DOI:10.1021/ja040126m.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, A “Rule of Three” for fragment-based lead discovery?, Drug Discovery Today, 2003, 8(19), 876–877, DOI:10.1016/S1359-6446(03)02831-9.
- P. Hermant, D. Bosc and C. Piveteau, et al., Controlling Plasma Stability of Hydroxamic Acids: A MedChem Toolbox, J. Med. Chem., 2017, 60(21), 9067–9089, DOI:10.1021/acs.jmedchem.7b01444.
- L. Riccardi, V. Genna and M. De Vivo, Metal–ligand interactions in drug design, Nat. Rev. Chem, 2018, 2(7), 100–112, DOI:10.1038/s41570-018-0018-6.
- R. A. Friesner, J. L. Banks and R. B. Murphy, et al., Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, J. Med. Chem., 2004, 47(7), 1739–1749, DOI:10.1021/jm0306430.
- T. A. Halgren, R. B. Murphy and R. A. Friesner, et al., Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening, J. Med. Chem., 2004, 47(7), 1750–1759, DOI:10.1021/jm030644s.
- Schrödinger Release 2022-2: Maestro, Version 13.2, Schrödinger, LLC, New York, NY, 2016 Search PubMed.
- J. B. Cross, D. C. Thompson and B. K. Rai, et al., Comparison of several molecular docking programs: Pose prediction and virtual screening accuracy, J. Chem. Inf. Model., 2009, 49(6), 1455–1474, DOI:10.1021/ci900056c.
- P. Raj, K. Selvam and K. Roy, et al., Identification of a new and diverse set of Mycobacterium tuberculosis uracil-DNA glycosylase (MtUng) inhibitors using structure-based virtual screening: Experimental validation and molecular dynamics studies, Bioorg. Med. Chem. Lett., 2022, 76, 129008, DOI:10.1016/j.bmcl.2022.129008.
- M. Sándor, R. Kiss and G. M. Keseru, Virtual fragment docking by glide: A validation study on 190 protein-fragment complexes, J. Chem. Inf. Model., 2010, 50(6), 1165–1172, DOI:10.1021/ci1000407.
- M. Agostillo, C. Jene, T. Boyle, P. A. Ramsland and E. Yuriev, Molecular docking of carbohydrate ligands to antibodies: Structural validation against crystal structures, J. Chem. Inf. Model., 2009, 49(12), 2749–2760, DOI:10.1021/ci900388a.
- S. Kesharwani, P. Raj and A. Paul, et al., Crystal structures of non-uracil ring fragments in complex with Mycobacterium tuberculosis uracil DNA glycosylase (MtUng) as a starting point for novel inhibitor design: A case study with the barbituric acid fragment, Eur. J. Med. Chem., 2023, 258, 115604, DOI:10.1016/j.ejmech.2023.115604.
- D. P. Martin, P. G. Blachly, J. A. McCammon and S. M. Cohen, Exploring the Influence of the Protein Environment on Metal-Binding Pharmacophores, J. Med. Chem., 2014, 57(16), 7126–7135, DOI:10.1021/jm500984b.
- L. Mercklé, A. De Andrés-Gómez, B. Dick, R. J. Cox and C. R. A. Godfrey, A fragment-based approach to understanding inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase, ChemBioChem, 2005, 6(10), 1866–1874, DOI:10.1002/cbic.200500061.
- S. C. Hoops, K. W. Anderson and K. M. Merz, Force Field Design for Metalloproteins, J. Am. Chem. Soc., 1991, 113(22), 8262–8270, DOI:10.1021/ja00022a010.
- L. David, P. Amara, M. J. Field and F. Major, Parametrization of a force field for metals complexed to biomacromolecuales: Applications to Fe(II), Cu(II) and Pb(II), J. Comput.-Aided Mol. Des., 2002, 16(8–9), 635–651, DOI:10.1023/A:1021962616650.
- D. Santos-Martins, S. Forli, M. J. Ramos and A. J. Olson, AutoDock4Zn: An improved AutoDock force field for small-molecule docking to zinc metalloproteins, J. Chem. Inf. Model., 2014, 54(8), 2371–2379, DOI:10.1021/ci500209e.
- S. S. Çlnaroǧlu and E. Timuçin, Comparative Assessment of Seven Docking Programs on a Nonredundant Metalloprotein Subset of the PDBbind Refined, J. Chem. Inf. Model., 2019, 59(9), 3846–3859, DOI:10.1021/acs.jcim.9b00346.
- J. Shelley, A. Cholleti, L. Frye, J. Greenwood, M. Timlin and M. Uchimaya, Epik: a software program for pK a prediction and protonation state generation for drug-like molecules, J. Comput.-Aided Mol. Des., 2007, 21(12), 681–691, DOI:10.1007/s10822-007-9133-z.
- C. T. Behrendt, A. Kunfermann and V. Illarionova, et al., Reverse fosmidomycin derivatives against the antimalarial drug target IspC (Dxr), J. Med. Chem., 2011, 54(19), 6796–6802, DOI:10.1021/jm200694q.
- S. Sooriyaarachchi, R. Chofor and M. D. P. Risseeuw, et al., Targeting an Aromatic Hotspot in Plasmodium falciparum 1-Deoxy-d-xylulose-5-phosphate Reductoisomerase with β-Arylpropyl Analogues of Fosmidomycin, ChemMedChem, 2016, 11, 2024–2036, DOI:10.1002/cmdc.201600249.
- M. Andaloussi, L. M. Henriksson and A. Wieckowska, et al., Design, synthesis, and X-ray crystallographic studies of α-aryl substituted fosmidomycin analogues as inhibitors of mycobacterium tuberculosis 1-deoxy-d-xylulose 5-phosphate reductoisomerase, J. Med. Chem., 2011, 54(14), 4964–4976, DOI:10.1021/jm2000085.
- T. Bodill, A. C. Conibear, G. L. Blatch, K. A. Lobb and P. T. Kaye, Synthesis and evaluation of phosphonated N-heteroarylcarboxamides as DOXP-reductoisomerase (DXR) inhibitors, Bioorg. Med. Chem., 2011, 19(3), 1321–1327, DOI:10.1016/j.bmc.2010.11.062.
- F. Cheng and E. Oldfield, Inhibition of isoprene biosynthesis pathway enzymes by phosphonates, bisphosphonates, and diphosphates, J. Med. Chem., 2004, 47(21), 5149–5158, DOI:10.1021/jm040036s.
- E. K. Fields, The Synthesis of Esters of Substituted Amino Phosphonic Acids, J. Am. Chem. Soc., 1952, 74(6), 1528–1531, DOI:10.1021/JA01126A054/ASSET/JA01126A054.FP.PNG_V03.
- M. Tandi and S. Sundriyal, Recent trends in the design of antimicrobial agents using Ugi-multicomponent reaction, J. Indian Chem. Soc., 2021, 98(8), 100106, DOI:10.1016/j.jics.2021.100106.
- M. Tandi, N. Tripathi, A. Gaur, B. Gopal and S. Sundriyal, Curation and cheminformatics analysis of a Ugi-reaction derived library (URDL) of synthetically tractable small molecules for virtual screening application, Mol. Diversity, 2024, 28, 37–50, DOI:10.1007/s11030-022-10588-1.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, DataWarrior: An open-source program for chemistry aware data visualization and analysis, J. Chem. Inf. Model., 2015, 55(2), 460–473, DOI:10.1021/ci500588j.
- J. B. Baell and G. A. Holloway, New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays, J. Med. Chem., 2010, 53(7), 2719–2740, DOI:10.1021/jm901137j.
- A. Mehta, P. Raj, S. Sundriyal, B. Gopal and U. Varshney, Use of a molecular beacon based fluorescent method for assaying uracil DNA glycosylase (Ung) activity and inhibitor screening, Biochem. Biophys. Rep., 2021, 26, 100954, DOI:10.1016/j.bbrep.2021.100954.
- A. Bhanot, A. Lunge and N. Kumar, et al., Discovery of small molecule inhibitors of Mycobacterium tuberculosis ClpC1: SAR studies and antimycobacterial evaluation, Results Chem., 2023, 5, 100904, DOI:10.1016/j.rechem.2023.100904.
- Y. Sakamoto, S. Furukawa, H. Ogihara and M. Yamasaki, Fosmidomycin resistance in adenylate cyclase deficient (cya) mutants of Escherichia coli, Biosci., Biotechnol., Biochem., 2003, 67(9), 2030–2033, DOI:10.1271/bbb.67.2030.
- E. Uh, E. R. Jackson and J. G. San, et al., Antibacterial and antitubercular activity of fosmidomycin, FR900098, and their lipophilic analogs, Bioorg. Med. Chem. Lett., 2011, 21(23), 6973–6976, DOI:10.1016/j.bmcl.2011.09.123.
- S. Ponaire, C. Zinglé, D. Tritsch, C. Grosdemange-Billiard and M. Rohmer, Growth inhibition of Mycobacterium smegmatis by prodrugs of deoxyxylulose phosphate reducto-isomerase inhibitors, promising anti-mycobacterial agents, Eur. J. Med. Chem., 2012, 51, 277–285, DOI:10.1016/j.ejmech.2012.02.031.
- G. S. Jose, E. R. Jackson and A. Haymond, et al., Structure-Activity Relationships of the MEPicides: N-Acyl and O-Linked Analogs of FR900098 as Inhibitors of Dxr from Mycobacterium tuberculosis and Yersinia pestis, ACS Infect. Dis., 2016, 2(12), 923–935, DOI:10.1021/acsinfecdis.6b00125.
- H. Valluri, A. Bhanot, S. Shah, N. Bhandaru and S. Sundriyal, Basic Nitrogen (BaN) Is a Key Property of Antimalarial Chemical Space, J. Med. Chem., 2023, 66(13), 8382–8406, DOI:10.1021/acs.jmedchem.3c00206.
- Schrödinger Release 2022-2: Protein Preparation Wizard, Epik, Schrödinger, LLC, New York, NY, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05083e |
| This journal is © The Royal Society of Chemistry 2024 |