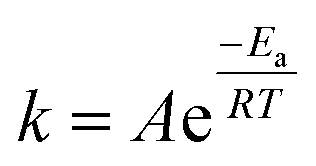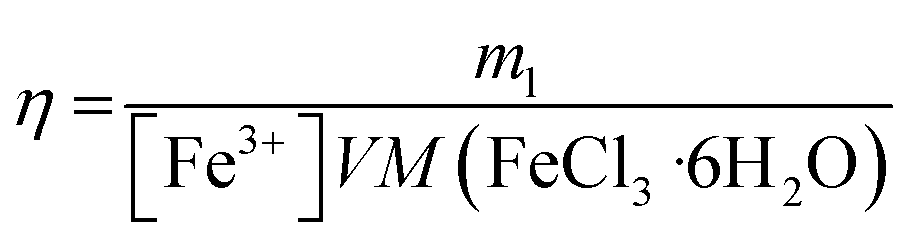DOI:
10.1039/D4RA05135A
(Paper)
RSC Adv., 2024,
14, 29174-29183
Circular recycling concept for silver recovery from photovoltaic cells in Ethaline deep eutectic solvent†
Received
20th August 2024
, Accepted 27th August 2024
First published on 19th September 2024
Abstract
Reducing the number of stages, energy costs and carbon footprint of recycling processes is essential to overcome environmental challenges. The interest in replacing the acids used in traditional hydrometallurgical methods with deep eutectic solvents (DES), which are less toxic and more environmentally friendly, has been growing. The aim of this study is to estimate the potential use of this class of solvents in an ionometallurgical process of leaching and electrodeposition to recover silver as part of the recycling of solar panels, a major challenge of the years to come. In the present work, a circular recycling concept based on an iron redox shuttle was studied to leach and recover silver via electrodeposition. Different DESs were evaluated in combination with a hexahydrated iron(III) chloride oxidizing agent. Ethaline DES has gained significant interest as it can attain a high silver leaching efficiency of 99.9% on crystallized silicon cell scraps at 75 °C. The promising results led to a comprehensive study of limits of this chemical system, focusing on the relation between the concentration of species (iron and water), the interfacial potential of silver (electrochemical measurements), and surface evolution (X-ray photoelectron spectroscopy analysis). Silver leaching was determined as a mixed control mechanism involving chemical and species diffusion dependence. The concentration of iron(III) chloride appeared crucial, determining the kinetic of formation of a poorly soluble AgCl layer. Electrodeposition from leachate highlighted the need to use an oxygen-free atmosphere to produce high-quality silver. Finally, leaching at 75 °C and electrodeposition at 50 °C of silver from crystallized silicon cell scraps were demonstrated using Ethaline (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) + FeCl3·6H2O (0.12 mol L−1) under an argon atmosphere.
:2) + FeCl3·6H2O (0.12 mol L−1) under an argon atmosphere.
Introduction
To face the environmental issues related to the use of carbon-based systems, which generate enormous amounts of CO2, photovoltaic (PV) production of electricity has steadily grown to become an important source of renewable energy around the world. In 2022, installed cumulative capacity overcame 1 TW and is expected to reach 9 TW in 2050.1 The International Renewable Energy Agency estimated that 78 Mt of end-of-life PV modules will have to be managed by 2050, including almost 10 Mt in Europe, which are dominated by PV cells based on crystalline silicon (c-Si).2 Additionally, the global demand for silver has been increasing and will continue to do so, as it is an essential material in the fabrication of photovoltaic cells, while the produced amount of silver remains relatively unchanged.3 Nowadays, most of the PV waste is landfilled due to cost consideration; however, this leads to environmental issues and potential loss of thousands of tons of silver. The dismantling of the first generation of solar panels and the recovery of its materials would be an important source of recycled silver for the manufacturing of new panels in order to fulfil the growing need for green electricity in the future. PV waste is a potential resource, which contains a large number of common materials such as aluminium; glass; and high-value materials such as copper, silver and silicon. Numerous chemical treatments have been considered by various researchers to recover valuable metals via hydrometallurgy. Harsh treatments to remove metallic layers by reactive chemicals were demonstrated in strong mineral acids, caustic solutions that are toxic and require a large amount of wastes and post-treatment before disposal.4–10 Alternative leaching reagents have been proposed as environmentally friendly and non-toxic substitutes for acids, such as halide leaching,11 or methane sulphonic acid (MSA) combined with hydrogen peroxide.12 The deep eutectic solvents (DESs) or assimilated solvents (CaCl2·6H2O:EG, ChCl:2 EG, ChCl:4H2O) were also investigated as green alternatives for leaching.13 The parametric optimization in ChCl:4H2O demonstrated the etching of silver from EoL c-Si PV cells at 65 °C in 0.5 mol L−1 FeCl3.
In these approaches, the silver ions were selectively recovered by AgCl precipitation. The selective AgCl precipitation is a common approach, considering the convenience to add chloride ions in the leachate. However, the precipitation by chloride ions generates large amounts of wastes for reusing the solution in another cycle of treatment. In chemical solutions with sparingly silver chloride solubility (HNO3, H2SO4, MSA, etc.), the contamination of the leachate by chloride ions impedes another leaching step due to the low solubility that would immediately form a passivating layer and prevent further oxidation. In chemical solutions with a higher silver chloride solubility (such as DESs or assimilated), AgCl precipitation requires a strong dilution (10 fold dilution).13 Moreover, silver is not in a higher value-added metal form. Thus, electrochemical metal recovery has been investigated to convert silver chloride under a metallic form via a wet chemical process, which then underwent electrorefining for purification to the 4 N level in MSA.12 These approaches are conceptually limited to promote a circular treatment. These conventional hydrometallurgical flowsheets or unit operations can be described as predominantly “linear,” in the sense that the reagents consumed are not regenerated for subsequent reuse.14 To overcome the disadvantages of AgCl precipitation, electrochemical processes were used for leaching silver by peroxydisulfate generated by sulphuric acid electrolysis (5 mol L−1). The silver dissolved could be electrochemically recovered, but limited at 88% after 24 h and required a large consumption of acid by water electrolysis and H2 formation.15
Thus, it is important to develop innovative, cost-effective, and more eco-friendly methods to extract and recycle materials such as silver and silicon from end-of-life photovoltaic cells. Our team is defining a circular recycling concept based on ionometallurgy to recover metals.16,17 The general principle associates a DES and a redox shuttle as an oxidizing agent for silver leaching. After the leaching, the silver ions are electrochemically deposited to metallic silver, while the redox shuttle is regenerated. The oxidizing agent regenerated during the electrolytic deposition of silver can be reused in the next leaching cycle. This alternative route is included within the PHOTORAMA project (PHOtovoltaic waste management – advanced Technologies of recOvery & recycling of secondary RAw MAterials from end-of-life modules). The concept includes principles of a novel and more sustainable approach promoting the regeneration of reagents, closing solvent loops, preventing waste, using benign chemicals and reducing chemical diversity. A DES is the mixture of chemicals at their eutectic points, and are composed of hydrogen bond acceptors (HBA) and donors (HBD). One category of DESs often consists of a quaternary ammonium salt as the HDA and a carboxylic acid or an amide as the HBD.18–20 The quaternary ammonium salt and the HBD interact with each other, disturbing their crystallographic structure.21,22 The use of DES to extract and refine metals is challenging regarding an industrial approach.23 The circular concept aims to benefit from the DES advantages (low volatility, good chemical stability, etc.), while minimizing the economic disadvantages regarding conventional acidic systems (prices, post-treatment, and established process). A balance must be found regarding the kinetics, the solubility and the diffusion of reactive species to perform the leaching of silver effectively and give acceptable electrochemical properties. As such, the oxidizing agent has to diffuse easily in the medium and silver must be easily complexed to promote the dissolution process. Thus, low viscosity should be preferred to promote the diffusion of the oxidizing redox shuttle.24,25 For eco-compatibility, low toxicity, availability and economic reasons, only choline chloride (ChCl) was tested as the HBA.26–29 Moreover, a high chloride concentration caused by ChCl would insure good solubility of silver as complexes.30 The choice of HBD and its chemical structure is hence essential and governs the DES properties at least as much as the HBA. Its importance is well illustrated by the widely different solubility of metal oxides with the same hydrogen acceptor and should not be neglected in the choice of a well-suited medium for silver recovery.31 The use of DESs as alternatives to the conventional hydrometallurgy in metal processing has previously been mentioned.32 Many studies have highlighted the capability of different DESs based on ChCl, especially when the latter is combined with urea or ethylene glycol (EG), to highly solubilize and electrodeposit metals such as iron, silver, gold, or cobalt.33–40 Recent articles investigated the application of DESs for silver recovery from solar waste,13,41 and none in a complete process allowing to directly recover the metallic form in a circular recycling concept. The leaching in DESs is highlighted and often optimized by a parametric approach, while the efficiency is explained by the speciation (ionic complexes under a chloride form such as [Ag(Cl)x]1−), without further information about the fundamental limitations.13,42
This study aims to understand the silver leaching limitation for defining an efficient recycling concept based on the circularity of reagents through the leaching and electrodeposition of silver. It combined a ChCl-based DES with an iron-based redox shuttle. The suitable DES for silver leaching is evaluated considering different HDBs. The promising results obtained with Ethaline led to a comprehensive study of limits for this chemical system, focusing on the relation between the concentration of species (iron and water), the interfacial potential of silver (electrochemical measurements), and the surface evolution (X-ray photoelectron spectroscopy analysis). The results revealed the importance to control the concentration in reactive species for an efficient silver dissolution. These fundamentals insights were applied successfully to solar cell scraps, demonstrating the efficient leaching and electrodeposition of silver under a metallic form for a circular recycling treatment.
Results and discussion
Choice of the DES
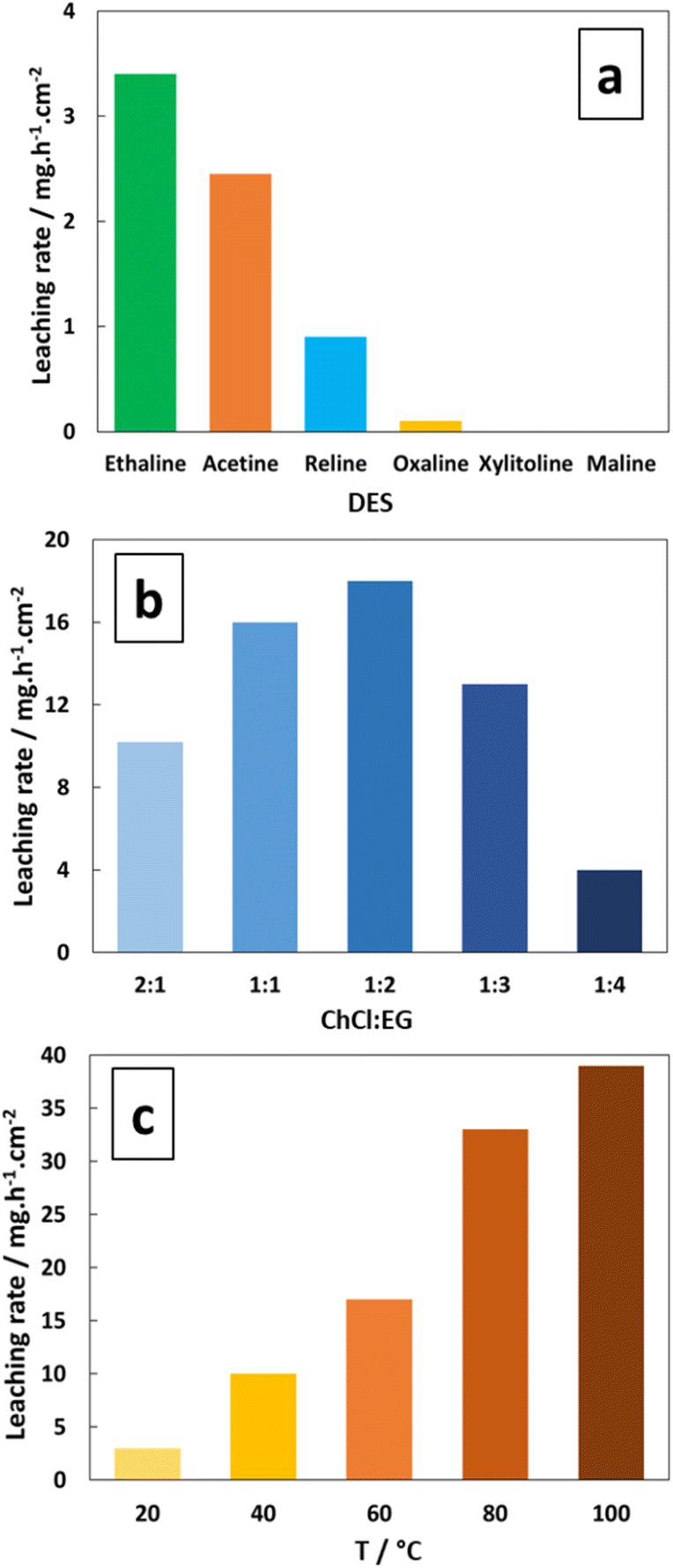
Several ChCl-based DESs detailed in Table 1 were pre-selected as testing media, and the corresponding silver leaching rate is represented in Fig. 1a. The results indicate that the Ethaline solution has the highest leaching rate of 3.4 mg h−1 cm−2 at 20 °C (FeCl3·6H2O at 0.30 mol L−1). The corresponding chemical reactions can be described by the formation of two silver chloride complexes, as expressed using eqn (1) and (2): 13,29| | |
Ag + FeCl3·6H2O ↔ AgCl2− + Fe2+ + Cl− + 6H2O
| (1) |
| | |
Ag + FeCl3·6H2O ↔ AgCl32− + Fe2+ + 6H2O
| (2) |
Table 1 List of tested DESs
| DES |
HBA |
HBD |
Molar ratio HBA:HBD |
| Ethaline |
ChCl |
Ethylene glycol |
1:2 |
| Xylitoline |
Xylitol |
1:1 |
| Acetine |
Acetic acid |
1:2 |
| Oxaline |
Oxalic acid |
1:1 |
| Maline |
Malic acid |
1:1 |
| Reline |
Urea |
1:2 |
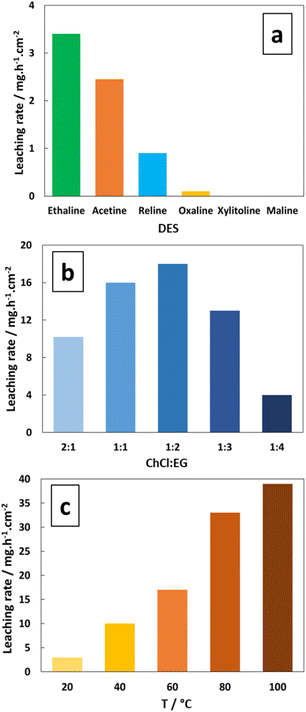 |
| | Fig. 1 Effect of (a) DES, (b) ChCl/EG ratio at 60 °C, and (c) Ethaline 1:2 temperature on the pure Ag leaching rate. [FeCl3·6H2O] = 0.30 mol L−1. | |
In this work, the hexahydrated form of FeCl3 was chosen because Xu et al.43 and Alfurayj et al.44 demonstrated that the iron redox reversibility, charge transfer coefficient and reaction rate constant were higher than those for the dry form, as well as being cheaper. As shown in Fig. 1b, the variation in the molar ratio between HBAs and HBDs in Ethaline indicated an optimum for 1:2 composition, for which the highest leaching rate of 18 mg h−1 cm−2 was obtained at 60 °C. Fig. 1c reports the temperature effect on the leaching rate in Ethaline 1:2 + Fe3+ 0.30 mol L−1. and reveals a strong increase in the leaching rate with temperature from 20 °C to 100 °C, multiplying the kinetic of leaching by 10 (3.5 to 38 mg h−1 cm−2). A recrystallization temperature of 20 °C was observed for Ethaline 1:2, so it is the lower limiting condition of operation. For examining the effect of temperature on the leaching process, the Arrhenius equation can be used to characterise the rate-controlling stage, described using eqn (3):
| |
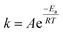 | (3) |
where
k is the leaching rate constant,
A is the frequency factor,
Ea is the so-called activation energy and
R is the universal gas constant. Using the temperature data (
Fig. 1c), the activation energy of leaching was determined at 29.3 kJ mol
−1. The activation energy of the chemically controlled reactions is usually higher than 42 kJ mol
−1; otherwise, the reaction is diffusion controlled (4–13 kJ mol
−1) or reveals a mixed mechanism (20–35 kJ mol
−1).
45 Silver leaching is determined as a mixed control mechanism, involving chemical and species diffusion dependence. Thus, a higher temperature decreases viscosity and increases simultaneously the species diffusion and the rate of the chemical reactions (electronic steps, solubility limit,
etc.) to greatly accelerate the leaching step. However, the temperature was limited to 75 °C, due to the modification of ChCl conformation from α phase to β phase above 78 °C.
46
Effect of FeCl3·6H2O concentration
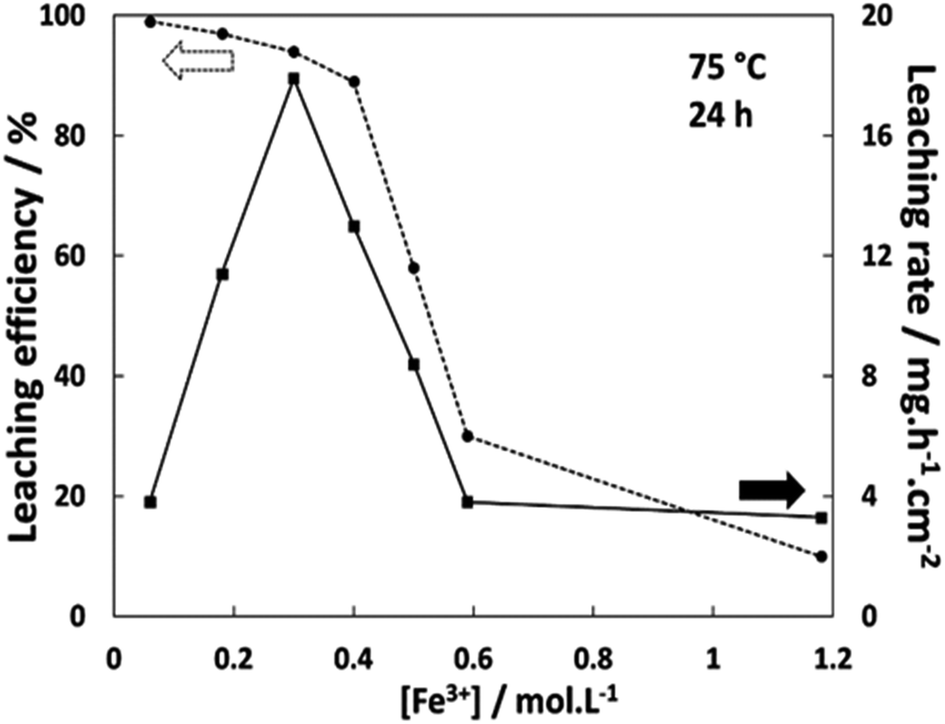
In order to determine the effect of the redox shuttle concentration on the leaching rate, Ag wires were leached at 75 °C with different concentrations of FeCl3·6H2O. In Fig. 2, the leaching efficiency and the average leaching rate over 24 h are reported, as a function of the Fe3+ concentration. The leaching efficiency η was calculated using eqn (4) as the ratio between the silver mass loss ml and the theoretical mass loss corresponding to a complete reaction (100% of Fe3+ reduced) with the volume V:| |
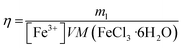 | (4) |
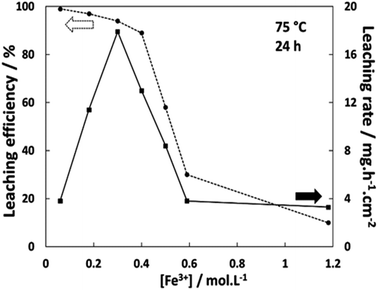 |
| | Fig. 2 Leaching efficiency (circles) at 75 °C after 24 h and average leaching rate (squares) depending on the Fe3+ concentration in Ethaline 1:2. | |
The results indicate a minimum of 94% of leaching efficiency when the concentration of FeCl3·6H2O is lower than 0.30 mol L−1. With 0.40 mol L−1, the leaching efficiency decreases to 89%, dropping to 59% with 0.50 mol L−1 for reaching 30 and 10% using 0.59 and 1.18 mol L−1 of FeCl3·6H2O respectively. In the last two cases, extending the leaching reaction times up to 110 h showed that the percentage of mass loss reached a plateau at 42% and 13% respectively. The simultaneous evolution of the leaching efficiency and the leaching rate reveals a chemical evolution around 0.40 mol L−1 in FeCl3·6H2O. At this critical concentration of FeCl3·6H2O, the rate of leaching are suddenly reduced by a factor of ∼4. The observation of a dark layer at the Ag wire surface suggests the formation of an AgCl layer when using high FeCl3·6H2O concentrations. Considering the solubilisation reaction of AgCl (eqn (5)), the solubility product constant Ks can be expressed as s2 (eqn (6)) with s equal to [Ag+] and being the solubility of AgCl. This solubility s was estimated to be around 0.36 mol L−1, corresponding to 51 g L−1, which is the highest value obtained using eqn (7) for each Fe3+ concentration, as shown in Fig. 2:
| | |
Ks = [Ag+][Cl−] = [Ag+]2 = s2
| (6) |
| | |
s = Ks = [Ag+] = [Fe3+] η
| (7) |
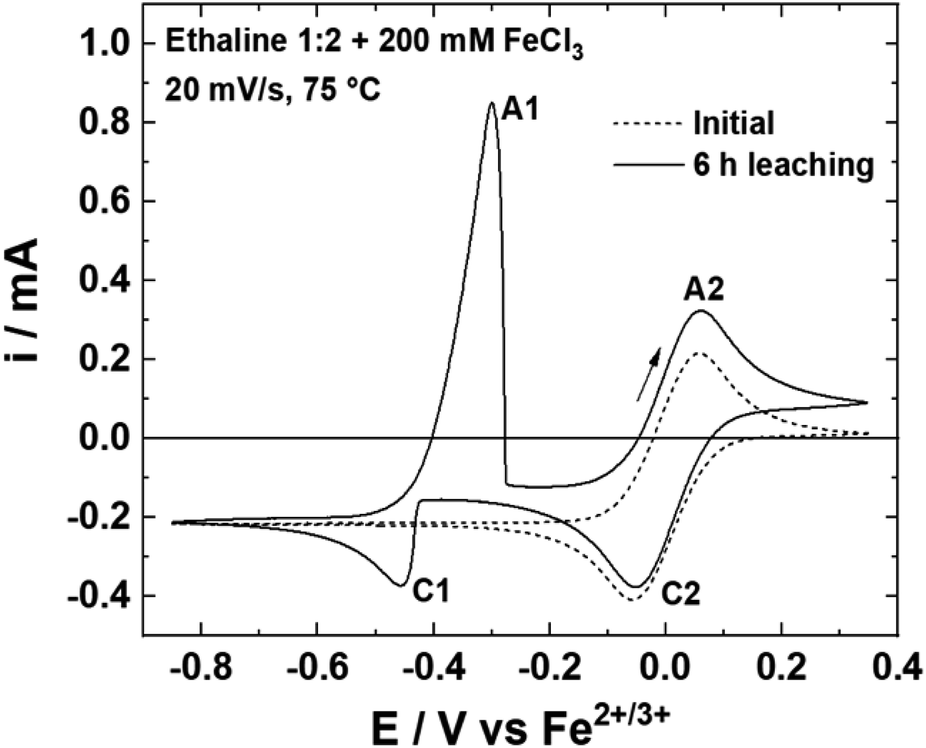
The solubility of silver chloride in aqueous media is around 1 × 10−5 mol L−1 at 20 °C,47 whereas in a DES formed from choline chloride and ethylene glycol, the solubility is 0.2 mol L−1.48 Considering kinetic and solubility limit of AgCl, 0.30 mol L−1 of FeCl3·6H2O appears to be the optimum concentration for reaching more than 90% of theoretical Ag dissolution with the fastest leaching rate (17.9 mg h−1 cm−2 at 75 °C). For a better understanding, the chemical system was studied electrochemically. Fig. 3 displays the cyclic voltammetry (CV) curves of a leaching solution at 75 °C before and after 6 h of silver wire leaching with 0.20 mol L−1 of FeCl3·6H2O. Before leaching, the voltammogram shows only the Fe2+/Fe3+ couple with the oxidation peak (A2) at 0.06 V and the reduction peak at −0.06 V (C2) vs. Fe2+/Fe3+. After 6 h of Ag leaching, the voltammogram indicates an additional reduction peak (C1) of Ag+ to Ag at −0.45 V vs. Fe2+/Fe3+ and the corresponding oxidation peak (A1) of Ag to Ag+ at −0.30 V vs. Fe2+/Fe3+, confirming the silver leaching. In addition, silver wires were leached under the same conditions using different FeCl3·6H2O concentrations: 0.03, 0.23, 0.40 and 0.60 mol L−1 (cf. Fig. S1†). The voltammograms show the increase in Ag oxidation peak intensity in good agreement with silver leached in solutions. The ratio iAg/iFe of reduction peaks increases with the concentration in iron, reflecting the natural consumption in iron with the silver dissolution. This ratio should be proportional with the concentration introduced in oxidative agents, while it drops from 0.40 mol L−1. It reveals a limitation in relation with the FeCl3·6H2O concentration.
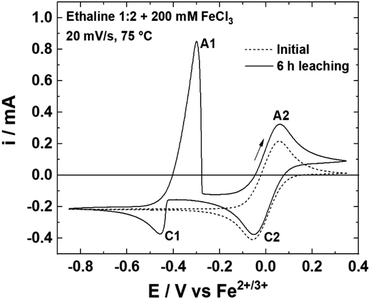 |
| | Fig. 3 Voltammograms of a Pt wire at 20 mV s−1 and 75 °C in an Ethaline 1:2 + Fe3+ 0.20 mol L−1 solution before and after 6 h of leaching. | |
Relation between surface and leaching limitation
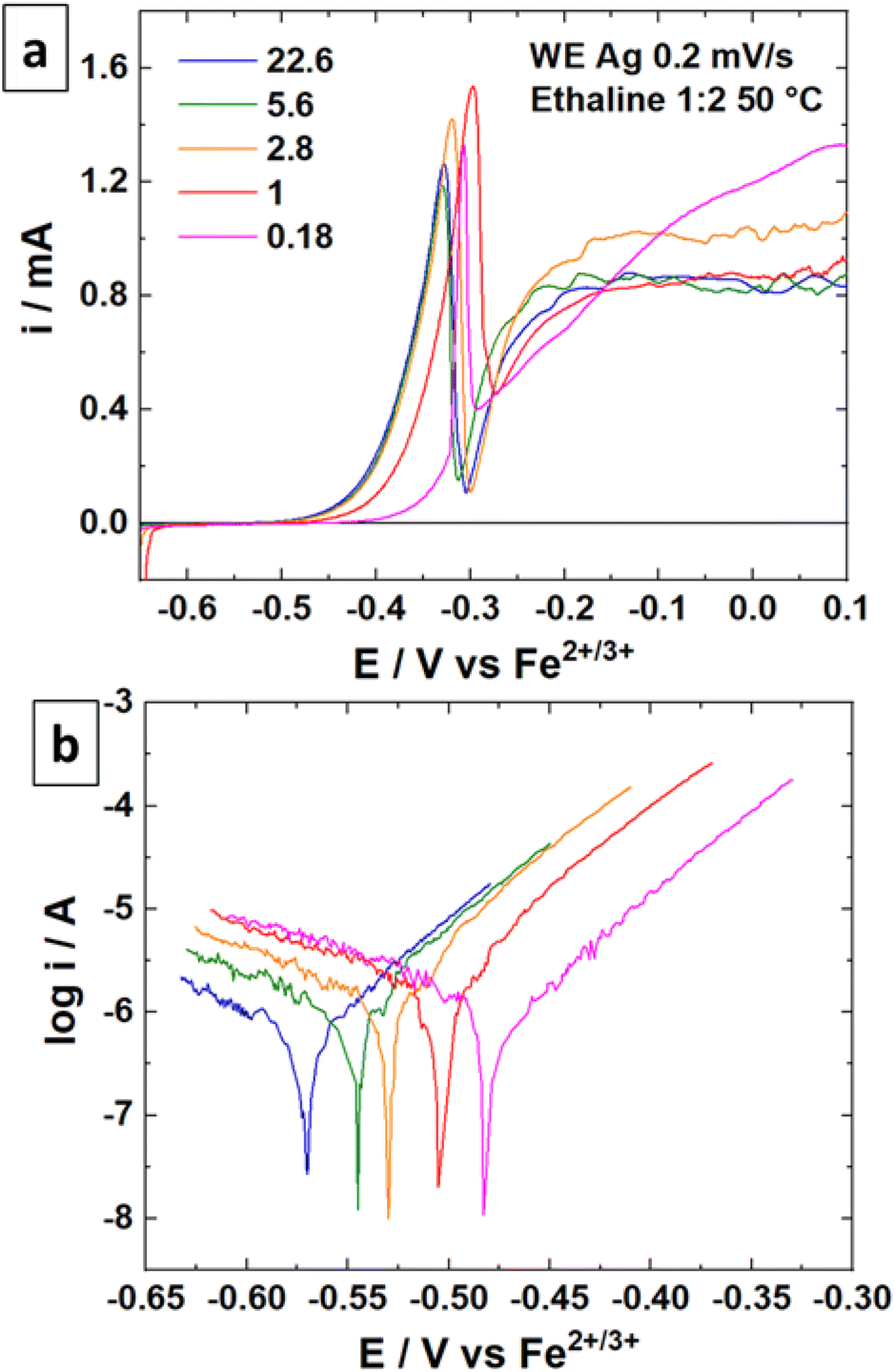
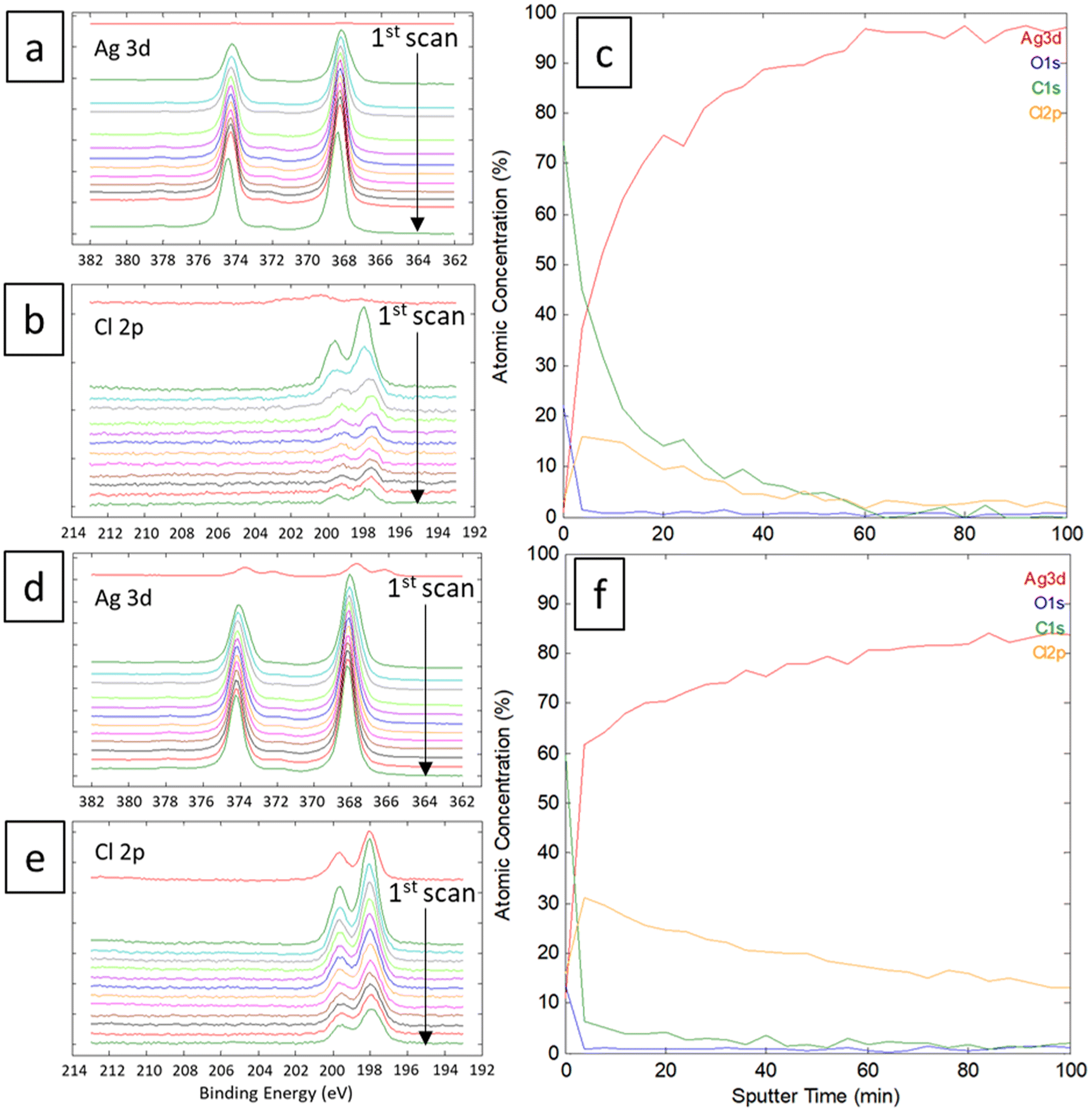
The linear sweep voltammetry (LSV) curves were recorded for the silver material in Ethaline 1:2 at 50 °C with different quantities of water in the solution. The resulting voltammograms and the Tafel plots are displayed in Fig. 4 for different molar ratios of Cl−/water. The higher molar ratio at 22.6 (no water added) indicates an oxidation peak of Ag around −0.50 V vs. Fe2+/Fe3+ followed by a straight drop in current at −0.34 V vs. Fe2+/Fe3+. The trend is confirmed with other water contents. The sudden drop of current can be attributed to the formation of a poorly soluble surface layer, such as AgCl. It is visually confirmed by the blackening of the immerged part of the wire.49 This surface layer is not protective, as demonstrated by a significant current at −0.20 V vs. Fe2+/Fe3+. However, the reaction is limited either by the layer solubility or the mass transport of active species in the layer. Voltammograms with a higher water content indicate a similar profile for Ag oxidation, with a slight positive shift in potential (Fig. 4b). The water content shows a moderate effect if the Cl−/water ratio remains below 1 (corresponding to 6.3 wt% of water). This content corresponds to the proportion of water brought by the oxidative salt FeCl3·6H2O at 0.72 mol L−1. These results are in line with the study of Alfurayj et al.44 who explained that a moderate water content has a low impact up to 10 wt%. Fig. 5 shows the pictures of silver wires after 16 h of leaching for different concentrations in FeCl3·6H2O. For the lowest concentration of 0.03 mol L−1 (Fig. 5a), the wires did not significantly change regarding the size and the visual aspect. At 0.23 mol L−1 (Fig. 5b) and 0.40 mol L−1 (Fig. 5c), the size of the wires diminished, while some silver pieces appeared darkened for the latter. The phenomenon is amplified at 0.60 mol L−1 of FeCl3·6H2O (Fig. 5d), while the silver wire almost retained the same size. This colour change could be attributed to the formation of an AgCl layer, which is favourable at higher FeCl3·6H2O concentrations. These observations are in agreement with the kinetic results of leaching (cf. Fig. 2), and demonstrate a correlation between the surface evolutions with oxidant concentration, which determines the kinetics of leaching. The precipitation and the growth of AgCl on a silver substrate was studied in an aqueous chloride solution, demonstrating a relation between the silver surface and the kinetic of dissolution.49 Thus, the surface evolution was further characterized by X-ray photoelectron spectroscopy (XPS) for Ag wires leached at 0.40 mol L−1 and 0.80 mol L−1 of FeCl3·6H2O. The Ag 3d and Cl 2p core-level spectra depth profile of samples Ag0.4 and Ag0.8 are displayed in Fig. 6a, b, d and e, respectively. Ag 3d and Cl 2p orbital signal pairs are located at 368.2–374.4 eV, and 198.0–199.8 eV, respectively, confirming that the dark layer on the wires is AgCl. The presence of satellite features in Ag 3d peak region at 372 eV and 378 eV is the signature of metallic silver additionally to AgCl.50 In the case of Ag0.4, the Cl signal of the last scan (t = 100 min) is almost invisible, corresponding to a depth of 350 ± 100 nm. However, even if the intensity of the Cl signal also decreases for sample Ag0.8, it is still clearly visible at the same depth. The atomic percentages of Ag and Cl were estimated, and are plotted in Fig. 6c for Ag0.4 and 6f for Ag0.8. At t = 60 min, the Cl atomic percentage of Ag0.4 is already very close to zero, whereas the one of Ag0.8 is still equal to 13% at t = 100 min. The AgCl layer is 210 ± 60 nm thick for Ag0.4, and thicker than 350 ± 100 nm for Ag0.8. It has been previously observed that with a FeCl3·6H2O concentration of 0.40 mol L−1, the leaching efficiency reaches 90% after 24 h, with silver surface partly darkened. On the contrary, at 0.80 mol L−1 of FeCl3·6H2O, the leaching efficiency is around 20% after 24 h, and silver completely turned black. The AgCl layer thickness traduces a lower permeability to the solution at high FeCl3·6H2O concentrations. The thicker AgCl layer reduces the accessibility of the oxidizing species, which is physically limited by the layer's porosity.51 As the maximum leaching rate was found at 0.30 mol L−1 (Fig. 2), it means that the formation of undissolved AgCl layer starts around this concentration. As a result, the FeCl3·6H2O concentration in Ethaline 1:2 will have to be lower to 0.30 mol L−1 to efficiently leach the PV cell scraps without being kinetically limited by the formation of a thick AgCl layer.
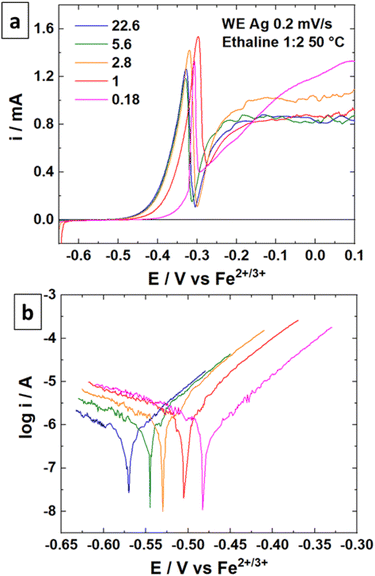 |
| | Fig. 4 (a) LSV curves of a Ag wire at 0.2 mV s−1 and 50 °C in Ethaline 1:2 with different amounts of water. (b) Tafel plots of the LSV curves. | |
 |
| | Fig. 5 Recovered Ag wires after 16 h of leaching with (a) 0.03 mol L−1, (b) 0.23 mol L−1, (c) 0.40 mol L−1 and (d) 0.60 mol L−1 of FeCl3·6H2O. | |
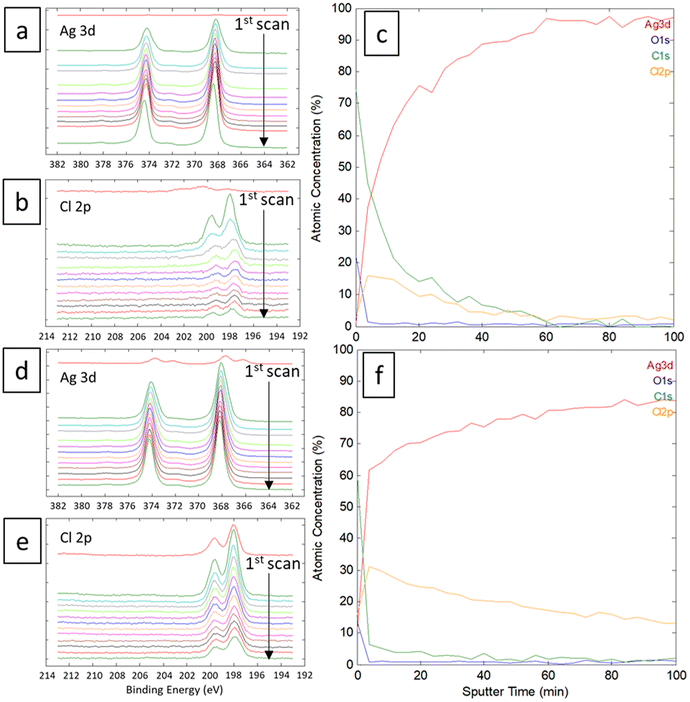 |
| | Fig. 6 XPS spectra focused on Ag and Cl of (a and b) Ag0.4 and (d and e) Ag0.8 wires during abrasion. Calculated atomic concentration in percentage of (c) Ag0.4 and (f) Ag0.8. | |
Leaching of silver from c-Si cell scraps
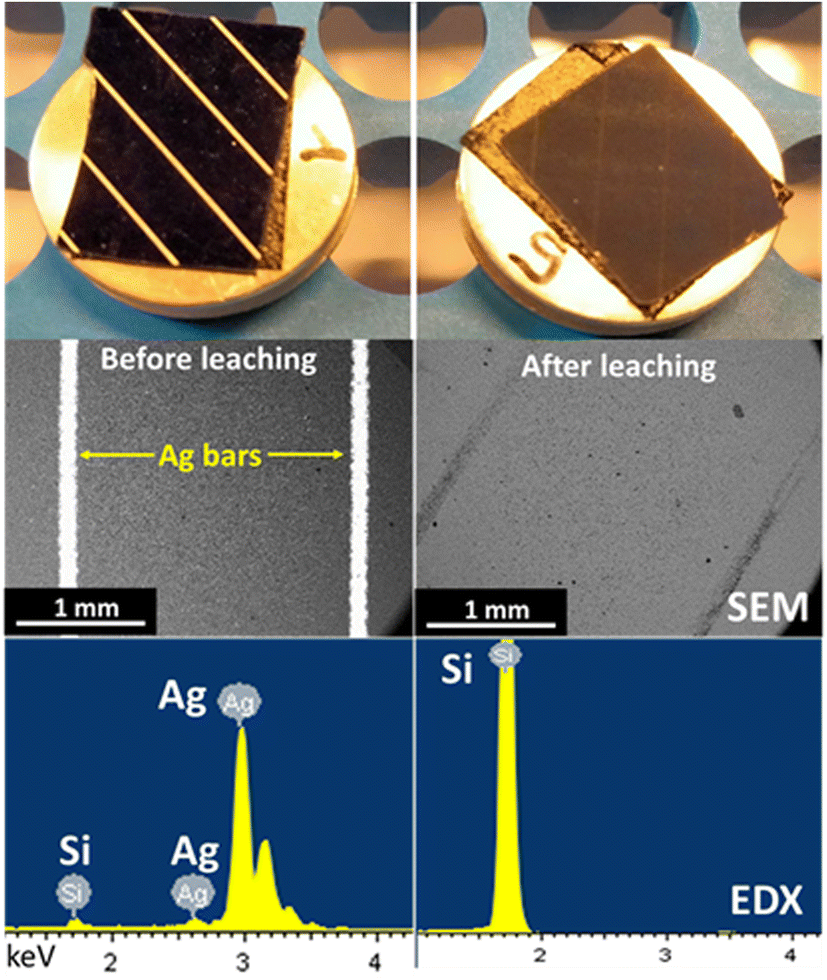
The leaching of silver from crystalline silicon (c-Si) cell scraps was performed in Ethaline 1:2 + 0.10 mol L−1 FeCl3·6H2O, in a glass vessel either opened to air or under an argon atmosphere. For practical operation, the leaching step was performed three times with the same recycled solution to consume all reactants and concentrate the solution in Ag+ ions. After each leaching under an air atmosphere, CVs were recorded and confirmed the enrichment of silver ions (Fig. S2†), characterized by the evolution of the Ag oxidation and reduction peaks between each leaching step. Scanning electron microscopic (SEM) images and energy-dispersive X-ray (EDX) spectra realized before and after leaching confirm the complete dissolution (Fig. 7). After leaching, SEM images indicate that Ag bars disappeared and the EDX analysis spectra reveal only Si on the surface. The ICP-OES analysis of the c-Si residue revealed that the Ag content was reduced from 1.180 wt% to 0.001 wt% during leaching. The chemical association of Ethaline 1:2 + FeCl3·6H2O enabled a silver leaching efficiency of 99.92% from c-Si cells, regardless of the atmosphere. However, a significant colour difference was observed depending on the leaching atmosphere. Before filtration, both leachates were opaque and greenish (Fig. 8a). After filtration, the leachate obtained under argon (Fig. 8b) was yellow-green and very light, whereas the one obtained under air (Fig. 8c) looked orange and darker, indicating a different speciation of iron. To evaluate the complete concept, the Ag electrodeposition was investigated from the silver-enriched solutions.
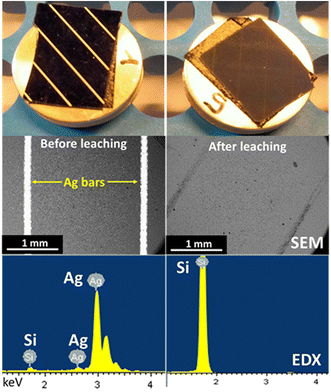 |
| | Fig. 7 Photographs, SEM images and EDX spectra of a c-Si cell piece before (left column) and after (right column) 6 h of leaching with 0.10 mol L−1 of Fe3+. | |
 |
| | Fig. 8 Photographs of PV cell scraps leachate (a) before filtration, and after filtration (b) under an argon atmosphere and (c) opened to air during leaching. | |
Ag electrodeposition
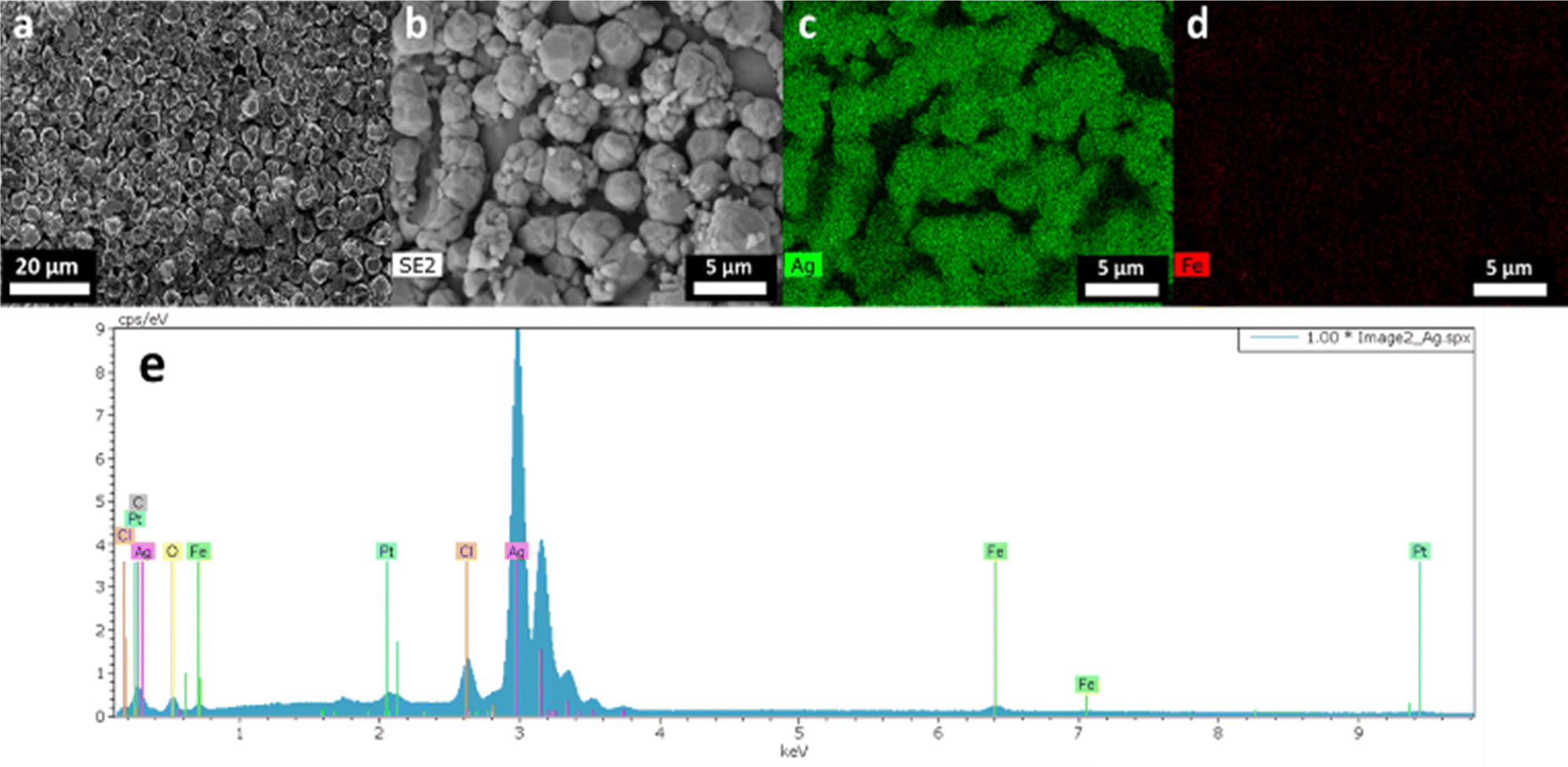
To evaluate the complete concept, the electrodeposition was studied using silver-enriched solutions (leachates obtained under air and argon atmospheres). The electrodeposition was performed by chronoamperometry at −0.47 V vs. Fe2+/Fe3+, 50 °C under constant stirring. For both air and argon leachates, the atmosphere was set to maintain the same conditions. The deposit obtained under air appeared heterogeneous, with an iron-like color and a poor adhesion to the platinum substrate. SEM shows a flake-like morphology of the particles and a poor density (Fig. S3†). On the contrary, the deposit obtained under the argon atmosphere appeared homogeneous, with silver-like color and good adhesion to the substrate. The argon atmosphere deposit was characterized by microscopy. SEM images (Fig. 9a and b) reveal some dense spherical particles of similar sizes. The images (Fig. 9c and d) and the spectrum obtained from energy-dispersive X-ray analysis (Fig. 9e) confirm a deposit composed of Ag containing a few traces of iron and chloride, attributed to incomplete washing. The mass fractions of elements determined by ICP-OES are 98.8 wt% in silver and 1.2 wt% in iron. Thus, the electrodeposition of metallic silver is achievable in a hydrated Ethaline solution, if keeping an air-free atmosphere. The air impact was observed without being elucidated. Further electrochemical investigations would be required to evaluate the secondary reactions and their relations in solutions such as electrochemical reduction of O2 in Ethaline media. Zante et al.13 mentioned the destabilization of the leachate by the oxidation of Fe2+ by oxygen, or the formation of insoluble oxy-hydroxide species.
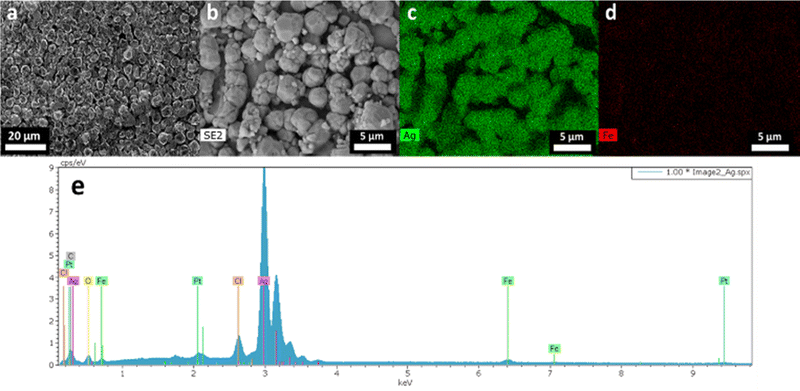 |
| | Fig. 9 (a) SEM image of Ag deposit obtained under an argon atmosphere. (b) SEM image used to do (c) Ag and (d) Fe element cartographies, and (e) EDX spectrum of image (b). | |
Experimental
Materials
Choline chloride (ChCl) (≥98%), ethylene glycol (EG) (≥99%), xylitol (≥99%), oxalic acid (98%), FeCl3·6H2O (≥99%) and FeCl2·4H2O (≥99%) were all purchased from Sigma Aldrich. Acetic acid (99.9%) was purchased from Technic. Nitric acid (69.5%) was purchased from Carlos Erba Reagents. Silver wires of 2 mm diameter were purchased from Alfa Aesar.
DES preparation
ChCl and each HBD were mixed in a closed container at 40 °C respecting desired molar ratio until it became fully liquid. FeCl3·6H2O was then weighed and added under stirring at the same temperature until complete solvation.
Silver wire leaching
The comparison of leaching rates was performed by a gravimetric method to choose the adequate DES. Ag wires of immerged known-area were weighed before leaching in DES containing 0.30 mol L−1 of FeCl3·6H2O, and weighed again after 4 h in the solution under magnetic stirring (Fig. S4†), washed with demineralized water and dried at 90 °C for 4 h. For each experiment, the quantity of silver introduced corresponded to the double of the stoichiometric quantity to not limit the reaction by the metal supply.
XPS analysis
X-ray photoelectron spectroscopy (XPS) scans were recorded on two Ag wires, one leached with 0.40 mol L−1 of FeCl3·6H2O (noted Ag0.4) and the other one leached with 0.80 mol L−1 of FeCl3·6H2O (noted Ag0.8). XPS analysis was performed using Versa-Probe2 from ULVAC-PHI equipped with a localized focused X-ray source consisting of monochromatic Al kα (1486.6 eV) sources and an Ar-ion beam for ionic etching. The wires underwent 2 min steps of ionic abrasion (Ar+, 2 keV) at a constant rate of 7 ± 2 nm min−1, separated by scans of 2 min, corresponding to depth intervals of 14 ± 4 nm. An X-ray beam spot size of 100 μm diameter was used for both sources with a power equal to 25 W. The take-off angle for photoelectron detection was 45°. All experiments were performed with both electron and Ar-ion charge neutralizations enabled under ultrahigh vacuum conditions (p < 10−7 Pa). High-resolution spectra were recorded with a pass energy of 23 eV for both energy sources, corresponding to an energy resolution of about 0.6 eV.
Electrochemical characterizations and electrodeposition
Electrochemical measurements were carried out in a 50 mL laboratory cell with a typical three-electrode arrangement (Fig. S5†). Electrodes consisted of a Pt wire as the working electrode (WE), a glassy carbon (GC) rod as the counter electrode (CE), and a quasi-reference electrode (REF) made of a Pt wire in a tube with a porous glass frit filled with a DES + Fe3+/Fe2+ equimolar solution, from FeCl3·6H2O and FeCl2·4H2O salts. In this way, all potentials reported are expressed in relation to the potential of Fe3+/Fe2+ fixed at 0 V. Cyclic voltammetry (CV) at 20 mV s−1, and open-circuit potential (OCP) measurements were done using this configuration while leaching small pieces of silver wires with an approximate length of 5 mm, in over-stoichiometry compared to iron. Linear sweep voltammetry (LSV) curves were recorded at 0.2 mV s−1 with an Ag wire WE in Ethaline 1:2 containing different amounts of water. Ag deposition using DESs after leaching PV scraps was done at 50 °C in the same cell by chronoamperometry (CA) on a 2 mm diameter platinum disk, previously polished and washed.
PV cell scrap leaching
Commercial monocrystalline silicon photovoltaic cells (c-Si, 15 × 15 cm) were used. The c-Si cells contain an anti-reflective coating, a silicon wafer, a rear passivation layer, silver wire electrical contacts and aluminium backside contact. The cell samples used in this work consisted of crushed pieces of size between 1 and 10 mm, from which aluminium was chemically removed following the process described by Palitzsch and Loser.52 To prepare the leaching solution, FeCl3·6H2O was added to an Ethaline 1:2 solution to obtain a concentration of 0.10 mol L−1 or 0.12 mol L−1. The theoretical amount of PV cells that should be added to correspond to an equimolar ratio between Fe3+ and Ag is too much compared to the volume of the liquid and would make it impossible to stir the mix. The leaching solution was consequently used multiple times to leach different smaller fractions of PV until all Fe3+ ions have reacted. Each leaching step was realized at 75 °C during 6 hours under vigorous stirring (Fig S6 and S7†). At the end of each step, as the solid settles fast, it was easy to recover the solution for another leaching. After the last leaching step, the leachate was filtered under vacuum on a 5–10 μm pore diameter paper filter. The residue was washed three times with water then dried in an oven at 90 °C for 24 h.
ICP-OES characterization of the PV cells and the Ag deposit
Samples of dealuminated crushed PV cell scraps were placed in HNO3 4 mol L−1 at 90 °C under reflux during 4 hours to leach all metals from the silicon. These solutions were diluted with 2% HNO3 to adjust the concentration, and analysed by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES 725 Agilent Technologies) to quantify the mass content of silver in the cell, before and after leaching. The Ag deposit composition was also investigated following the same preparation method.
SEM/EDX characterization
Pieces of c-Si cell scraps and Ag deposit were investigated using a high-resolution LEO 1530 FEG-SEM Scanning Electron Microscope (SEM) at an applied voltage of 3 kV. The surface of the cells and the composition of the deposit were analysed by Energy-Dispersive X-ray Spectroscopy.
Conclusions
A circular concept based on ionometallurgy has been studied to leach and recover silver by electrodeposition. Different deep eutectic solvents were evaluated in combination with a hexahydrated iron(III) chloride oxidizing agent. An Ethaline DES gained great interest to replace the conventional strong acids used in hydrometallurgical processes to treat solar cells. Its high chloride content is favourable for silver solubilisation with a moderate activation energy determined at 29.3 kJ mol−1. In this study, the difference of leaching in the chemical system was not associated to the different speciation, as mentioned for the system formed from a molar ratio of 1 choline chloride:4 water and 0.5 mol L−1 of FeCl3. Silver leaching was determined as a mixed control mechanism, involving chemical and species diffusion dependence. The concentration of iron(III) chloride appeared crucial to ensure proper silver dissolution kinetics. The concentration in the oxidizing agent controlled the formation of a poorly soluble AgCl layer, which required maintaining a concentration below 0.40 mol L−1. It was found that the appropriate condition for the silver leaching was treatment with 0.20–0.30 mol L−1 FeCl3·6H2O at 75 °C. Under appropriate conditions, the silver leaching efficiency from photovoltaic cell scraps reached 99.9 wt%, with a kinetic around 20–30 mg h−1 cm−2. The silver electrodeposition was demonstrated in an air-free atmosphere. The air impact was observed without being elucidated. Further electrochemical investigations would be required to evaluate the secondary reactions and their relations. This study demonstrated a complete chemical/electrochemical recycling process of silver from photovoltaic materials to valorise silver under a metallic form. The optimization of electrodeposition and the estimation of the regeneration rate of the iron(III) chloride will have to be studied in the future, as well as the ageing solution after a large number of processing cycles.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and the ESI.†
Author contributions
C. Lemoine: investigation, methodology, experimental work, formal analysis, writing – original draft. Y. Petit: investigation, methodology, experimental work. T. Karaman: experimental work. G. Jahrsengene: investigation, methodology, experimental work, writing – review & editing. A. M. Martinez: conceptualization, supervision. A. Benayad: XPS studies. E. Billy: conceptualization, supervision, writing – review & editing. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 958223.
References
- International Renewable Energy Agency, https://www.irena.org/-/media/Files/IRENA/Agency/Publication/2019/Nov/IRENA_Future_of_Solar_PV_2019.pdf (accessed November 2019).
- International Renewable Energy Agency, https://www.irena.org/-/media/Files/IRENA/Agency/Publication/2016/IRENA_IEAPVPS_End-of-Life_Solar_PV_Panels_2016.pdf (accessed June 2016).
- The Silver Institute, https://www.silverinstitute.org/wp-content/uploads/2023/04/World-Silver-Survey-2023.pdf (accessed 2023).
- L. S. S. De Oliveira, M. T. W. D. C. Lima, L. H. Yamane and R. R. Siman, Detritus, 2020, 10, 62–74 CrossRef.
- P. Dias, S. Javimczik, M. Benevit, H. Veit and A. M. Bernardes, Waste Manage., 2016, 57, 220–225 CrossRef CAS PubMed.
- Y. Yue, Y. Zhuo, Q. Li and Y. Shen, Resour., Conserv. Recycl., 2022, 186, 106548 CrossRef CAS.
- W.-S. Chen, C. Yen-Jung, K.-C. Yueh, C.-P. Cheng and T.-C. Chang, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 720, 012007 CAS.
- N. Wongnaree, W. Kritsarikun, N. Ma-ud, C. Kansomket, T. Udomphol and S. Khumkoa, Mater. Sci. Forum, 2020, 1009, 137–142 Search PubMed.
- C. E. L. Latunussa, F. Ardente, G. A. Blengini and L. Mancini, Sol. Energy Mater. Sol. Cells, 2016, 156, 101–111 CrossRef CAS.
- S. Preet and S. T. Smith, J. Cleaner Prod., 2024, 448, 141661 CrossRef CAS.
- J. Chung, B. Seo, J. Lee and J. Y. Kim, J. Hazard. Mater., 2021, 404, 123989 CrossRef CAS PubMed.
- E.-H. Yang, J.-K. Lee, J.-S. Lee, Y.-S. Ahn, G.-H. Kang and C.-H. Cho, Hydrometallurgy, 2017, 167, 129–133 CrossRef CAS.
- G. Zante, R. Marin Rivera, J. M. Hartley and A. P. Abbott, J. Cleaner Prod., 2022, 370, 133552 CrossRef CAS.
- K. Binnemans and P. T. Jones, J. Sustain. Metall., 2023, 9, 1–25 CrossRef.
- C. Modrzynski, L. Blaesing, S. Hippmann, M. Bertau, J. Z. Bloh and C. Weidlich, Chem. Ing. Tech., 2021, 93(11), 1851–1858 CrossRef CAS.
- E. Billy, EP Pat., 3388554A1, 2017.
- E. Billy, EP Pat., 4159882A1, 2021.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121(3), 1232–1285 CrossRef CAS PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114(21), 11060–11082 CrossRef CAS PubMed.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126(29), 9142–9147 CrossRef CAS PubMed.
- C. D'Agostino, L. F. Gladden, M. D. Mantle, A. P. Abbott, I. Ahmed Essa, A. Y. M. Al-Murshedi and R. C. Harris, Phys. Chem. Chem. Phys., 2015, 17(23), 15297–15304 RSC.
- V. Migliorati, F. Sessa and P. D'Angelo, Chem. Phys. Lett., 2019, 737, 100001 CrossRef.
- K. Binnemans and P. T. Jones, J. Sustain. Metall., 2023, 9(2), 423–438 CrossRef.
- S. Fryars, E. Limanton, F. Gauffre, L. Paquin, C. Lagrost and P. Hapiot, J. Electroanal. Chem., 2018, 819, 214–219 CrossRef CAS.
- L. Bahadori, M. H. Chakrabarti, N. S. A. Manan, M. A. Hashim, F. S. Mjalli, I. M. AlNashef and N. Brandon, PLoS One, 2015, 10(12), e0144235 CrossRef PubMed.
- A. Yadav, J. R. Kar, M. Verma, S. Naqvi and S. Pandey, Thermochim. Acta, 2015, 600, 95–101 CrossRef CAS.
- A. L. Sazali, N. AlMasoud, S. K. Amran, T. S. Alomar, K. F. Pa’ee, Z. M. El-Bahy, T.-L. K. Yong, D. J. Dailin and L. F. Chuah, Chemosphere, 2023, 338, 139485 CrossRef CAS PubMed.
- K. Radošević, M. Cvjetko Bubalo, V. Gaurina Srček, D. Grgas, T. Landeka Dragičević and I. Radojčić Redovniković, Ecotoxicol. Environ. Saf., 2015, 112, 46–53 CrossRef PubMed.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41(21), 7108–7146 RSC.
- N. Fu, R. Lv, Z. Guo, Y. Guo, X. You, B. Tang, D. Han, H. Yan and K. H. Row, J. Chromatogr. A, 2017, 1492, 1–11 CrossRef CAS PubMed.
- A. P. Abbott, M. Azam, G. Frisch, J. Hartley, K. S. Ryder and S. Saleem, Phys. Chem. Chem. Phys., 2013, 15(40), 17314–17323 RSC.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47(36), 10031–10033 RSC.
- Y. Dai, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, Food Chem., 2015, 187, 14–19 CrossRef CAS PubMed.
- B. Villemejeanne, S. Legeai, E. Meux, S. Dourdain, H. Mendil-Jakani and E. Billy, J. Environ. Chem. Eng., 2022, 10(3), 108004 CrossRef CAS.
- A. P. Abbott, R. C. Harris, F. Holyoak, G. Frisch, J. Hartley and G. R. T. Jenkin, Green Chem., 2015, 17(4), 2172–2179 RSC.
- A. P. Abbott, J. Griffith, S. Nandhra, C. O'Connor, S. Postlethwaite, K. S. Ryder and E. L. Smith, Surf. Coat. Technol., 2008, 202(10), 2033–2039 CrossRef CAS.
- A. P. Abbott, K. E. Ttaib, G. Frisch, K. S. Ryder and D. Weston, Phys. Chem. Chem. Phys., 2012, 14(7), 2443–2449 RSC.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- Y. Fan, Y. Liu, L. Niu, W. Zhang and T. Zhang, Hydrometallurgy, 2020, 197, 105454 CrossRef CAS.
- W. Sánchez-Ortiz, J. Aldana-González, T. Le Manh, M. Romero-Romo, I. Mejía-Caballero, M. T. Ramírez-Silva, E. M. Arce-Estrada, V. Mugica-Álvarez and M. Palomar-Pardavé, J. Electrochem. Soc., 2021, 168(1), 016508 CrossRef.
- C. Zhang, J. Jiang, E. Ma, L. Zhang, J. Bai, J. Wang, Y. Bu, G. Fan and R. Wang, Waste Manage., 2022, 150, 280–289 CrossRef CAS PubMed.
- J. M. Hartley, C.-M. Ip, G. C. H. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53(12), 6280–6288 CrossRef CAS PubMed.
- J. Xu, Q. Ma, H. Su, F. Qiao, P. Leung, A. Shah and Q. Xu, Ionics, 2020, 26, 483–492 CrossRef CAS.
- I. Alfurayj, C. C. Fraenza, Y. Zhang, R. Pandian, S. Spittle, B. Hansen, W. Dean, B. Gurkan, R. Savinell, S. Greenbaum, E. Maginn, J. Sangoro and C. Burda, J. Phys. Chem. B, 2021, 125(31), 8888–8901 CrossRef CAS PubMed.
- T. Havlík, in Hydrometallurgy: Principles and Applications, Woodhead Publishing, Cambridge, 1st edn, 2008, ch. 7, pp. 184–228 Search PubMed.
- R. L. Collin, J. Am. Chem. Soc., 1957, 79(22), 6086 CrossRef CAS.
- O. Dinardo and J. E. Dutrizac, Hydrometallurgy, 1985, 13(3), 345–363 CrossRef CAS.
- A. P. Abbott, G. Frisch, S. H. Garrett and J. Hartley, Chem. Commun., 2011, 47, 11876–11878 RSC.
- X. Jin, J. Lu, P. Liu and H. Tong, J. Electroanal. Chem., 2003, 542, 85–96 CrossRef CAS.
- N. Pauly, F. Yubero and S. Tougaard, Appl. Surf. Sci., 2016, 383, 317–323 CrossRef CAS.
- H. Ha and J. Payer, Electrochim. Acta, 2011, 56, 2781–2791 CrossRef CAS PubMed.
- W. Palitzsch and U. Loser, Presented in Part at 37th IEEE Photovoltaic Specialists Conference, Seattle, WA, USA, 2011 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Ana Maria Martinez
b,
Ana Maria Martinez