
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic stabilization energies in excited states at the multiconfigurational level: assessment in archetypal organic rings†
Ricardo Pino-Rios *ab
*ab
aQuímica y Farmacia, Facultad de Ciencias de la Salud, Universidad Arturo Prat, Casilla 121, Iquique, 1100000, Chile. E-mail: rpinorios@unap.cl
bInstituto de Ciencias Exactas y Naturales (ICEN), Universidad Arturo Prat, Playa Brava 3256, 1111346, Iquique, Chile
First published on 23rd October 2024
Abstract
In this study, the excited state (anti)aromaticity of archetypal rings: benzene, cyclobutadiene, and cyclooctatetraene, was investigated using the energetic criterion by calculating aromatic stabilization energies. Calculations were performed at the multiconfigurational level, including dynamic correlation effect corrections using the N-electron valence state perturbation theory (NEVPT2) method. Results were compared with previously published data based on the magnetic and delocalization criteria. Aromaticity was assessed for the ground state, singlet excited states (S1, S2, and S3), and triplet excited states (T1, T2, T3, and T4). (Anti)aromaticity assignments using the energetic criterion demonstrate both agreement and discrepancies with the other criteria, particularly for higher energy electronic states demonstrating the complexity of aromaticity assignment beyond the ground state. Finally, an approximate equation is proposed for the calculation of aromatic stabilization energies in excited states using experimental data such as formation enthalpies and well-resolved absorption spectra.
Introduction
The modulation of (anti)aromaticity in the ground and excited states has gained much attention in recent years as it opens the door to the intelligent design of new chemical compounds. The modification of ground-state aromaticity has an impact on excited-state antiaromaticity and vice versa. Moreover, the assembly between aromatic and antiaromatic units in the ground state has a clear effect on the electronic structure, which allows to obtain antiaromatic–aromatic species in excited states that can exhibit interesting photochemical properties.1–6For the assignment of aromaticity, Hückel7 and Baird8 rules are the cornerstone. These rules via a simple electron (π for organic species) count determine whether a compound will be aromatic or antiaromatic. For more complex species such as polycyclic systems, we have other rules such as the Platt's perimeter9 model, Clar's rules10,11 for benzenoids and the Glidewell–Lloyd rule for systems containing non-benzenoid rings.12–14
More recently, several criteria have been developed to assign (anti)aromatic behavior and to quantify this property. These include magnetic,15 delocalization,16,17 energetic,18 and other criteria.19–23 The first two are the ones that have shown the greatest development and popularity owing to their effectiveness in quantification and because of the simplicity of their computation due to the development of different programs.24–28
(Anti)aromaticity assignment in excited states is often complicated beyond the lowest energy excited states (beyond T1 or S1) mostly owing to the multiconfigurational character of electronic states, which requires the use of methods to account for these features, such as the complete active space self-consistent field (CASSCF) method.29 Magnetic and delocalization criteria have had success since some indicators can be obtained using these multiconfigurational methods;30–32 however, they have some disagreements concerning (anti)aromaticity assignments.30,33
The energetic criterion is probably the oldest quantitative criterion for the study of aromaticity, dating back to 1933 when Pauling and Wheland calculated the aromatic stabilization energy for benzene.34 However, in many cases, the quantification of (anti)aromaticity using this criterion can be a non-trivial exercise, and in some cases, it only allows the study of the low-lying excited state.35,36 This is primarily due to the need to use reference reactions that avoid the occurrence of effects unrelated to aromaticity and due to the complexity of calculations that need to be performed.18,23,35,37–40
Recently, the fulvenization approach41 was proposed as a simple and reliable method for the calculation of isomerization stabilization energies (ISEs) originally proposed by Schleyer in 2002.37 This approach uses as reference the fulvenic isomer of the N-membered ring to be studied. It is just as simple to obtain as the original but has certain advantages. For example, it allows the calculation of systems that the original method could not calculate due to problems with the reference isomer. Additionally, the non-aromatic reference in the fulvenization (Fulv-ISE) approach is a planar conjugated system, which avoids the appearance of possible effects not related to (anti)aromaticity.
In this work, it is shown that this approach also allows assigning and quantifying the aromaticity in excited states through the use of multiconfigurational tools. For this purpose, the author quantified the (anti)aromaticity using the isomerization stabilization energies of the low-lying excited states of organic archetypes, namely, benzene, cyclobutadiene and cyclooctatetraene. The choice of multi-configurational methods over less computationally expensive techniques, such as time-dependent density functional theory (TD-DFT), stems from the limitations of the latter in accurately describing excited states. Different functionals can yield highly variable results, requiring comparison with experimental data. Moreover, the presence of ghost states in TD-DFT can lead to erroneous energy predictions. Although these ghost states can be mitigated using functionals with a high Hartree–Fock exchange component, there are instances where the predicted excitation energies still do not align closely with the experimental values.42–44 Additionally, the main objective of this study is to quantify for the first time the (anti)aromaticity in electronic states beyond the first excited state as well as to compare the assignments delivered by this method with other (anti)aromaticity criteria in order to evaluate its performance. Therefore, it is advisable to use more robust tools when available.
Computational methods
Geometrical optimizations have been carried out using Gaussian16 (ref. 45) at the PBE0 (ref. 46)/def2-TZVP47 level. The cases of cyclobutadiene (C4H4) and cyclooctatetraene (C8H8) were optimized at two different symmetries. D4h and D2h symmetries were used for the former, while geometries in D8h and D4h symmetries were used for the latter. For the case of benzene, the D6h symmetry was preserved. The CASSCF calculations were then performed with the PBE0 geometries (Franck–Condon approximation) using the ORCA program.48 The active space used corresponds to all the π-electrons and orbitals possessed by each of the systems: (6,6) for C6H6, (4,4) for C4H4, and (8,8) for C8H8 and for their respective fulvene isomers (for this, the rotate command was used so that the π-orbitals correspond to active space, see Fig. S1–S16 in the ESI†). State-average approach was employed (all electronic states contribute equally) and the resolution of identity approximation in conjunction with the def2-TZVP basis, which includes auxiliary functions for a reduction in the computational cost. The calculated states correspond to the fundamental S0 and the vertical S1, S2, S3, T1, T2, T3, and T4 states, which have been made separately according to their multiplicities, and the weights were assigned equally.Energy corrections due to the inclusion of dynamic correlation effects have been made following the N-electron valence state perturbation theory (NEVPT2).49 The calculation of the aromatic stabilization energies under the fulvenization approach are obtained from the isomerization energy when an N-atom ring converts into its fulvenic isomer. The equation for obtaining the energies is shown below
| ΔEFulv-ISE = EFulv-Isomer − EN-Ring | (1) |
Results and discussion
The results obtained through the application of the fulvenization approach have been compared with those obtained with the magnetic and delocalization criteria reported in the literature. The reference reactions used for the quantification of aromaticity using isomerization stabilization energies through the fulvenization approach of the ground state and the vertical excited states correspond to those presented in Scheme 1. Excited state energies including corrections for dynamic correlation effects (NEVPT2) offer better agreement with the available experimental/computational data31,32,50–52 with respect to CASSCF (see Tables S1 and S3 in the ESI†). For this reason, the quantification of (anti)aromatic (de)stabilization was calculated at this level.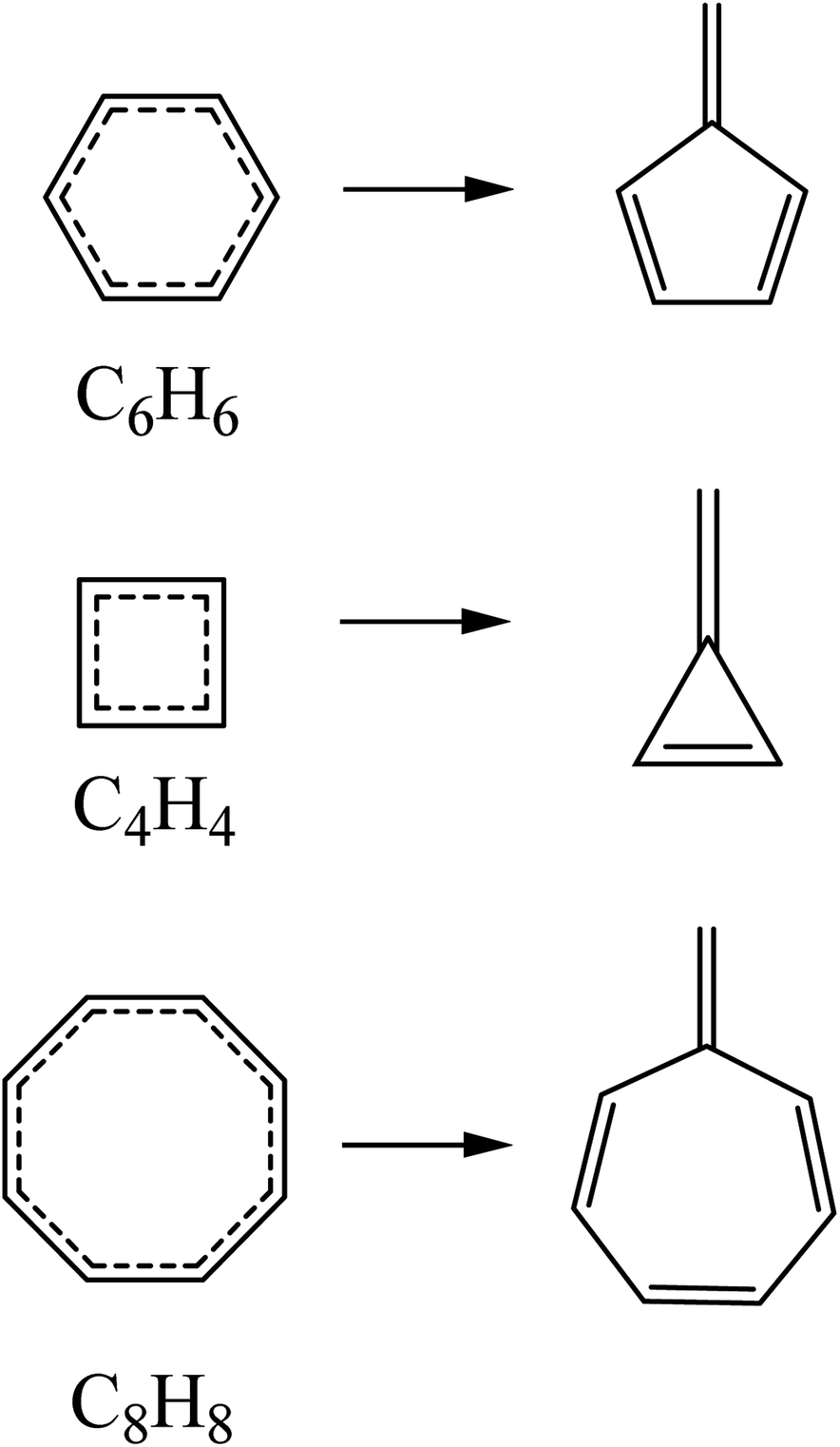 | ||
| Scheme 1 Reference reactions used for the calculation of aromatic stabilization energies using the fulvenization approach. | ||
Initially, the values of Fulv-ISE for benzene and its respective electronic states configurations can be found in Table 1. For the case of S0, the value is similar to that reported previously, indicating an aromatic behavior, while for the case of T1, the value is different from that reported at the DFT level;41 however, since it is a negative value, it is consistent with the antiaromatic character. For the case of S1, the values reported by Karadakov et al.33 and Feixas et al.30 using the magnetic and delocalization criteria respectively indicate antiaromaticity; however, the value obtained under the fulvenization approach is −5.2 kcal mol−1, suggesting a slightly antiaromatic or even non-aromatic behaviour. In the original article where the fulvenization approach was proposed, calculations of non-aromatic systems were not performed; however, in this work, it is shown that the aromatic stabilization energies obtained under this approach present values of −3.4 and −0.4 kcal mol−1 for cyclohexene and cyclohexadiene, respectively (see Fig. S17 in ESI†), thus providing a small range (−4.7 to 0.0 kcal mol−1) for the recognition of non-aromatic compounds at the DFT level.
| C6H6 (D6h) | ||
|---|---|---|
| State | Configuration | Fulv-ISE |
| S0 | π21π22π23π04π05π06 | 34.5 |
| S1 | π21π12π23π14π05π06 | −5.2 |
| π21π22π13π04π15π06 | ||
| S2 | π21π22π13π14π05π06 | 35.5 |
| π21π12π23π04π15π06 | ||
| S3 | π11π22π23π14π05π06 | −22.4 |
| π21π22π03π24π05π06 | ||
| π21π22π13π04π05π16 | ||
| π21π02π23π24π05π06 | ||
| T1 | π21π22π13π14π05π06 | −8.5 |
| π21π12π23π04π15π06 | ||
| π11π22π23π04π05π16 | ||
| T2 | π21π12π23π04π15π06 | −5.5 |
| π21π22π13π14π05π06 | ||
| π11π12π23π14π15π06 | ||
| T3 | π21π22π13π04π15π06 | 51.2 |
| π21π12π23π14π05π06 | ||
| T4 | π21π12π23π14π05π06 | 62.5 |
| π21π22π13π04π15π06 | ||
Regarding S2, the magnetic criteria indicates that in this state, benzene is aromatic, while the delocalization criterion indicates antiaromaticity. Using Fulv-ISE, a value of 35.5 kcal mol−1 is obtained; thus, this electronic state would present an aromatic behavior in agreement with the magnetic criterion. It is necessary to mention that the delocalization criterion indicates that S2 and S3 are antiaromatic because the CASSCF calculations reported by Feixas et al.30 indicate that both states are degenerate; however, the calculations obtained in this work at the CASSCF level show a difference of 0.2 eV between S2 and S3, which becomes even more noticeable when the dynamic correlation effects are included (1.7 eV). In the case of S3, a value of −22.4 kcal mol−1 is obtained; thus, it would be an antiaromatic state. This assignment is in agreement with Feixas et al. under the delocalization criteria while for the magnetic criterion, no values have been reported. The electronic delocalization indices also indicate that the T2, T3 and T4 states are antiaromatic.
Special attention must be paid to the case of T2 and T3, which are degenerate states with excitation energies of 5.1 eV at the NEVPT2 level. The fulvenization approach indicates that despite this degeneracy, the assignments are antiaromatic/non-aromatic for the case of S2 and aromatic for the case of S3 (see Table 1). These differences can be attributed to the non-degenerate electronic states of the reference fulvene; thus, it is possible that under this criterion, one has different (anti)aromaticity assignments, which could result in a limitation of the method in highly symmetric systems. The same pattern will be observed later for the case of cyclooctatetraene. For the case of T4, the value obtained is 62.4 kcal mol−1, which indicates an aromatic behavior; however, due to the results and the possible degeneracies between the electronic states, it is recommended that for the assignment of (anti)aromaticity of N electronic states, at least N + 1 states should be calculated.
The Fulv-ISE values for the S0 and T1 states of C4H4 reported in Table 2 are in agreement with the Hückel and Baird rules; on the other hand, the singlet excited states in all cases indicate aromaticity for both D4h and D2h symmetry. Some discrepancies are observed when compared with the magnetic and delocalization criteria. The states S1, S2 and S3 present aromatic behavior according to Fulv-ISE; however, Karadakov et al.33 indicated aromaticity for S1 and antiaromaticity for S2 (they do not report values for S3). On the other hand, the study by Feixas et al.30 indicates that there is antiaromaticity for S1 and S3 and aromaticity for S2.
| C4H4 | |||||
|---|---|---|---|---|---|
| D4h | D2h | ||||
| State | Fulv-ISE | Configuration | State | Fulv-ISE | Configuration |
| S0 | −30.7 | π21π12π13π04 | S0 | −14.1 | π21π22π03π04 |
| S1 | 49.8 | π21π22π03π04 | S1 | 29.1 | π21π12π13π04 |
| π21π02π23π04 | |||||
| S2 | 98.8 | π21π02π23π04 | S2 | 47.7 | π21π02π23π04 |
| π21π22π03π04 | |||||
| S3 | 38.2 | π21π12π03π14 | S3 | 54.3 | π21π12π03π14 |
| π11π12π23π04 | π11π22π13π04 | ||||
| T1 | 50.7 | π21π12π13π04 | T1 | 31.7 | π21π12π13π04 |
| T2 | −22.8 | π21π12π03π14 | T2 | −17.3 | π21π12π03π14 |
| π11π12π23π04 | π11π12π23π04 | ||||
| T3 | 8.0 | π11π22π13π04 | T3 | −17.3 | π21π02π13π14 |
| π21π02π13π14 | π11π12π23π04 | ||||
| T4 | 18.6 | π21π02π13π14 | T4 | 16.1 | π11π22π13π04 |
| π11π22π13π04 | π21π12π03π14 | ||||
For the case of triplet excited states, the Fulv-ISE values and delocalization indices coincide in an antiaromatic assignment for T2. For the case of T3 and T4, the delocalization criterion indicates antiaromaticity, while the energy criterion applied in this work indicates aromaticity. However, when switching from D4h to D2h symmetry, curiously, the T3 state becomes antiaromatic, coinciding with the delocalization criterion whose values were obtained at this symmetry. This switch from aromaticity to antiaromaticity in T3 is due to different electronic transitions. However, due to the inclusion of dynamic correlation effects, the occupancies of the states obtained in this work with respect to the one performed by Feixas et al. are different, which may explain the differences with respect to the aromaticity assignment.
The case of C8H8 is less controversial since the D8h symmetry coincides perfectly with the magnetic criterion. The S0 state is antiaromatic while the T1, S1 and S2 states are aromatic; indeed, Karadakov et al.53 report that S2 is much more aromatic than T1 and S1 and the Fulv-ISE value for S2 is much higher when compared to the other two electronic states with D8h symmetry. The delocalization criterion indicates that S1 as well as T2 and T3 are antiaromatic (no values are reported for T4 in Karadakov et al.'s work). The results with Fulv-ISE agree with the assignment given for T2 and T3; however, for the case of T3, the value for D8h symmetry is close to zero; therefore, in this symmetry, it could be considered non-aromatic, but if we reduce the symmetry to D4h, the value becomes more negative. The opposite case is presented in T1 wherein on reducing the symmetry to D4h, the value obtained indicates a non-aromatic behavior. T4 is an electronic state that presents antiaromaticity, becoming even more antiaromatic when going from D8h to D4h symmetry (Table 3).
| C8H8 | |||||
|---|---|---|---|---|---|
| D8h | D4h | ||||
| State | Fulv-ISE | Configuration | State | Fulv-ISE | Configuration |
| S0 | −31.5 | π21π22π23π14π15π06π07π08 | S0 | −23.3 | π21π22π23π24π05π06π07π08 |
| S1 | 24.5 | π21π22π23π24π05π06π07π08 | S1 | 11.4 | π21π22π23π04π25π06π07π08 |
| π21π22π23π04π25π06π07π08 | π21π22π23π24π05π06π07π08 | ||||
| S2 | 66.5 | π21π22π23π04π25π06π07π08 | S2 | 30.7 | π21π22π23π14π15π06π07π08 |
| π21π22π23π24π05π06π07π08 | |||||
| S3 | −3.8 | π21π22π13π24π15π06π07π08 | S3 | −15.8 | π21π22π13π24π15π06π07π08 |
| π21π12π23π14π25π06π07π08 | π21π12π23π14π25π06π07π08 | ||||
| π21π22π23π04π15π06π17π08 | π21π22π23π14π05π16π07π08 | ||||
| π21π22π23π14π05π16π07π08 | π21π22π23π04π15π06π17π08 | ||||
| T1 | 13.6 | π21π22π23π14π15π06π07π08 | T1 | −0.3 | π21π22π23π14π15π06π07π08 |
| T2 | −43.6 | π21π12π23π24π15π06π07π08 | T2 | −45.8 | π21π22π13π24π15π06π07π08 |
| π21π22π13π14π25π06π07π08 | |||||
| π21π22π23π04π15π06π17π08 | π21π22π23π14π05π16π07π08 | ||||
| π21π22π23π14π05π16π07π08 | |||||
| T3 | −0.9 | π21π22π13π24π15π06π07π08 | T3 | −3.1 | π21π12π23π24π15π06π07π08 |
| π21π12π23π14π25π06π07π08 | |||||
| π21π22π23π04π15π16π07π08 | π21π22π23π14π05π06π17π08 | ||||
| π21π22π23π14π05π06π17π08 | |||||
| T4 | −2.6 | π21π22π13π14π25π06π07π08 | T4 | −28.4 | π21π22π13π14π25π06π07π08 |
| π21π12π23π24π15π06π07π08 | |||||
| π21π22π23π14π05π16π07π08 | π21π22π23π04π15π16π07π08 | ||||
| π21π22π23π04π15π06π17π08 | |||||
A relationship between the aromatic stabilization energies derived from the fulvenization approach and experimental data is proposed. Initially, it was suggested that the aromatic stabilization energy in the ground state could be estimated using experimental enthalpies of formation. For benzene in its ground state, this approach yields a value of 33.7 kcal mol−1, which is in excellent agreement with the energy calculated at the PBE0/def2-TZVP level (33.2 kcal mol−1). Consequently, eqn (1) can be approximated as follows.
| ΔEFulv-ISE ≈ ΔHFulv-isomerf − ΔHN-ringf | (2) |
Additionally, the energies of the excited electronic states can be obtained from
| Estate = E0 + ΔEexc | (3) |
| ΔEFulv-ISEexc ≈ (ΔHFulv-isomerf − ΔHN-ringf) + (ΔEFulv-isomerexc − ΔEN-isomerexc) | (4) |
| Compound | ΔHf | S1 | S2 | T1 | T2 |
|---|---|---|---|---|---|
| Benzene | 19.8 | 119.9 | 143.0 | 92.2 | 110.7 |
| Fulvene | 53.5 | 79.3 | 121.8 | 54.2 | 71.5 |
| Eqn (4) | 33.7 | −6.9 | 12.5 | −4.3 | −5.5 |
Conclusions
The capacity of the recently proposed fulvenization approach for calculating the aromatic stabilization energies (Fulv-ISE) and assigning (anti)aromaticity in vertical excited states has been tested using three archetypal rings, namely, benzene, cyclobutadiene and cyclooctatetraene. The calculated energies were obtained through multiconfigurational calculations including dynamic correlation effect corrections (NEVPT2). In all cases, there are agreements and differences with the magnetic and delocalization states, the latter especially in the case of higher energy electronic states due to the inclusion of dynamic correction effects and the occupancies of the electronic states being different. Additionally, this work once again shows the complexity of aromaticity assignment when going beyond the ground state.Results indicate that benzene shows aromaticity in the S0, S2, T3 and T4 vertical states, while the other computed states show antiaromatic behavior. Regarding the degenerate states T2 and T3, different assignments are given since fulvene does not show degeneracy in these states, resulting in a limitation of the method due to the use of reference isomers. Additionally, it is recommended that if the N electronic states are to be analyzed, at least N + 1 states should be calculated to confirm that the states do not exhibit degeneracy.
In the case of cyclobutadiene, all the calculated states present aromaticity with the exception of S0 and T2; however, the T3 state, which presents aromaticity in D4h symmetry, becomes antiaromatic in D2h symmetry. Cyclooctatetraene, on the other hand, presents an aromatic character only in the S1 and S2 states, in agreement with the magnetic criterion, while the rest of the results at D8h symmetry present antiaromaticity. In addition, it is shown that the aromatic stabilization energies under this approach can be obtained from the experimental data such as formation enthalpies and absorption peaks in well-resolved absorption spectra.
These results extend the capability of the energy criterion for aromaticity assignment beyond the first excited states. In addition, the aromatic stabilization energies for higher energy electronic states can be calculated in a straightforward manner.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks the financial support of the National Agency for Research and Development (ANID) through FONDECYT Project 1230571.References
- H. Ottosson, Nat. Chem., 2012, 4(12), 969–971 CrossRef CAS PubMed.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114(10), 5379–5425 CrossRef CAS PubMed.
- W. Zeng, D. W. Szczepanik and H. Bronstein, J. Phys. Org. Chem., 2023, 36(1), e4441 CrossRef CAS.
- J. Kim, J. Oh, A. Osuka and D. Kim, Chem. Soc. Rev., 2022, 51(1), 268–292 RSC.
- J. Yan, T. Slanina, J. Bergman and H. Ottosson, Chem.–Eur. J., 2023, 29(19), e202203748 CrossRef CAS PubMed.
- D. Dunlop, L. Ludvíková, A. Banerjee, H. Ottosson and T. Slanina, J. Am. Chem. Soc., 2023, 145(39), 21569–21575 CrossRef CAS PubMed.
- E. Hückel, Z. Phys., 1931, 70(3), 204–286 CrossRef.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94(14), 4941–4948 CrossRef CAS.
- J. R. Platt, J. Chem. Phys., 1949, 17(5), 484–495 CrossRef CAS.
- E. Clar, The Aromatic Sextet, J. Wiley, 1972 Search PubMed.
- M. Solà, Front. Chem., 2013, 1, 1–8 Search PubMed.
- C. Glidewell and D. Lloyd, Tetrahedron, 1984, 40(21), 4455–4472 CrossRef CAS.
- R. Pino-Rios and M. Solà, J. Phys. Chem. A, 2021, 125(1), 230–234 CrossRef CAS PubMed.
- R. Báez-Grez and R. Pino-Rios, Phys. Chem. Chem. Phys., 2024, 12162–12167 RSC.
- R. Gershoni-Poranne and A. Stanger, Chem. Soc. Rev., 2015, 44(18), 6597–6615 RSC.
- F. Feixas, E. Matito, J. Poater and M. Solà, Chem. Soc. Rev., 2015, 44(18), 6434–6451 RSC.
- I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, in Aromaticity, ed. I. Fernandez, Elsevier, 2021, pp. 235–259 Search PubMed.
- M. Alonso and I. Fernández, in Aromaticity, ed. I. Fernandez, Elsevier, 2021, pp. 195–235 Search PubMed.
- H. Szatylowicz, P. A. Wieczorkiewicz and T. M. Krygowski, in Aromaticity, ed. I. Fernandez, Elsevier, 2021, pp. 71–99 Search PubMed.
- D. R. Roy and P. K. Chattaraj, in Aromaticity, ed. I. Fernandez, Elsevier, 2021, pp. 285–319 Search PubMed.
- D. Yu, T. Stuyver, C. Rong, M. Alonso, T. Lu, F. De Proft, P. Geerlings and S. Liu, Phys. Chem. Chem. Phys., 2019, 21(33), 18195–18210 RSC.
- R. Báez-Grez and R. Pino-Rios, ACS Omega, 2022, 7(25), 21939–21945 CrossRef PubMed.
- H. Szatylowicz, P. A. Wieczorkiewicz and T. M. Krygowski, Sci, 2022, 1–22 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- T. A. Keith, AIMAll (Version 190213), Overland Park KS, USA, 2019, https://aim.tkgristmill.com Search PubMed.
- D. Inostroza, V. García, O. Yañez, J. J. Torres-Vega, A. Vásquez-Espinal, R. Pino-Rios, R. Báez-Grez and W. Tiznado, New J. Chem., 2021, 45(18), 8345–8351 RSC.
- M. Orozco-Ic, J. L. Cabellos and G. Merino, Aromagnetic, Cinvestav-Mérida, Mérida, Mexico, 2016 Search PubMed.
- E. Matito, ESI-3D: Electron Sharing Indices Program for 3D Molecular Space Partitioning, Institute of Computational Chemistry and Catalysis (IQCC), University of Girona, 2006 Search PubMed.
- J. W. Park, R. Al-Saadon, M. K. MacLeod, T. Shiozaki and B. Vlaisavljevich, Chem. Rev., 2020, 120(13), 5878–5909 CrossRef CAS PubMed.
- F. Feixas, J. Vandenbussche, P. Bultinck, E. Matito and M. Solà, Phys. Chem. Chem. Phys., 2011, 13(46), 20690–20703 RSC.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112(31), 7303–7309 CrossRef CAS PubMed.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112(49), 12707–12713 CrossRef CAS PubMed.
- P. B. Karadakov, P. Hearnshaw and K. E. Horner, J. Org. Chem., 2016, 81(22), 11346–11352 CrossRef CAS PubMed.
- L. Pauling and G. W. Wheland, J. Chem. Phys., 1933, 1(6), 362–374 CrossRef CAS.
- Y. Mo and P. v. R. Schleyer, Chem.–Eur. J., 2006, 12(7), 2009–2020 CrossRef CAS PubMed.
- I. Fernández and G. Frenking, Faraday Discuss., 2007, 135(0), 403–421 RSC.
- P. v. R. Schleyer and F. Pühlhofer, Org. Lett., 2002, 4(17), 2873–2876 CrossRef CAS PubMed.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101(5), 1385–1420 CrossRef CAS PubMed.
- Y. García-Rodeja and M. Solà, in Exploring Chemical Concepts through Theory and Computation, 2024, pp. 223–250 Search PubMed.
- A. R. Katritzky, K. Jug and D. C. Oniciu, Chem. Rev., 2001, 101(5), 1421–1450 CrossRef CAS PubMed.
- L. Leyva-Parra and R. Pino-Rios, ACS Omega, 2024, 9(1), 1436–1442 CrossRef CAS PubMed.
- F. Furche and D. Rappoport, in Theoretical and Computational Chemistry, ed. M. Olivucci, Elsevier, 2005, pp. 93–128 Search PubMed.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42(3), 845–856 RSC.
- M. Campetella, F. Maschietto, M. J. Frisch, G. Scalmani, I. Ciofini and C. Adamo, J. Comput. Chem., 2017, 38(25), 2151–2156 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.02, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110(13), 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152(22), 224108 CrossRef CAS PubMed.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350(3), 297–305 CrossRef CAS.
- E. N. Lassettre, A. Skerbele, M. A. Dillon and K. J. Ross, J. Chem. Phys., 1968, 48(11), 5066–5096 CrossRef CAS.
- J. P. Doering, J. Chem. Phys., 1969, 51(7), 2866–2870 CrossRef CAS.
- M. H. Palmer, S. V. Hoffmann, N. C. Jones, M. Coreno, M. de Simone and C. Grazioli, J. Chem. Phys., 2019, 151(8), 084304 CrossRef PubMed.
- P. B. Karadakov and N. Preston, Phys. Chem. Chem. Phys., 2021, 23(43), 24750–24756 RSC.
- R. D. Brown, P. J. Domaille and J. E. Kent, Aust. J. Chem., 1970, 23(9), 1707–1720 CrossRef CAS.
- F. Negri and M. Z. Zgierski, J. Chem. Phys., 1995, 102(13), 5165–5173 CrossRef CAS.
- K. R. Asmis, M. Allan, O. Schafer and M. Fülscher, J. Phys. Chem. A, 1997, 101(11), 2089–2095 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05147e |
| This journal is © The Royal Society of Chemistry 2024 |