DOI:
10.1039/D4RA05191B
(Paper)
RSC Adv., 2024,
14, 30590-30605
Influence of intrinsic spin ordering in La0.6Sr0.4Co0.8Fe0.2O3−δ and Ba0.6Sr0.4Co0.8Fe0.2O3−δ towards electrocatalysis of oxygen redox reaction in solid oxide cell†
Received
17th July 2024
, Accepted 10th September 2024
First published on 25th September 2024
Abstract
The redox reaction of oxygen (OER & ORR) forms the rate determining step of important processes like cellular respiration and water splitting. Being a spin relaxed process governed by quantum spin exchange interaction, QSEI (the ground triplet state in O2 is associated with singlet oxygen in H2O/OH−), its kinetics is sluggish and requires inclusion of selective catalyst. Functionality and sustainability of solid oxide cell involving fuel cell (FC) and electrolyzer cell (EC) are also controlled by ORR (oxygen redox reaction) and OER (oxygen evolution reaction). We suggest that, presence of inherent spin polarization within La0.6Sr0.4Co0.8Fe0.2O3−δ (LSCF6482) (15.86 emu g−1) and Ba0.6Sr0.4Co0.8Fe0.2O3−δ (BSCF6482) (3.64 emu g−1) accounts for the excellent selective electrocatalysis towards ORR and OER. QSEI forms the atomic level basis for OER/ORR which is directly proportional to spin ordering (non-zero magnetization) of the active electrocatalyst. LSCF6482 exhibits (21.5 kJ mol−1@0.8 V for ORR compared to 61 kJ mol−1@0.8 V for OER) improved ORR kinetics whereas BSCF6482 (18.79 kJ mol−1@0.8 V for OER compared to 32.19 kJ mol−1 for ORR@−0.8 V) is best suited for OER under the present stoichiometry. The findings establish the presence of inherent spin polarization of catalyst to be an effective descriptor for OER and ORR kinetics in solid oxide cell (SOC).
Introduction
Spin orbit coupling and subsequent spin polarization enables the mechanistic understanding of spin relaxed reaction (s). The coupling enables the mixing of singlet and triplet states and allows the process to occur at much lower activation barrier.1 Important examples being oxygen evolution (OER) and oxygen reduction (ORR) reaction that are ‘in principle’ forbidden owing to the spin conservation rule.2 However, cellular reaction and solar water splitting are significant examples of spontaneous natural processes which involve ORR and OER. Besides that, the present crisis of energy conversion, storage and utilization based on non-renewables count mainly on the targeted renewables based on hydrogen energy. OER and ORR forms the primary rate determining step for solar water splitting, rechargeable metal air batteries, regenerative fuel cell, water electrolysis etc.3,4a–c,5,6a,b,7 Both OER and ORR involve evolution or reduction of O2 which preferably retains in triplet state involving the associated reactants/products (OH−/H2O) to be in singlet form.8 As a result, the sluggish kinetics during OER/ORR demands for the presence of catalytic surface with inclusion of 3d transition metal oxides (TMO).9a Rehman et al. have also reported the feasibility of transition metal sulphides and selenide monolayers to act as potential catalyst for water splitting reaction.9b It is also reported that incorporation of specific magnetic 3d elements results tunable magnetic structures which could be exploited to achieve magnetically enhanced OER/ORR. Presence of suitable magnetically ordered catalyst thereby tend to lower the binding energies for diffusion and sorption of intermediate species within active electrocatalytic zone and reducing the activation barrier for charge transfer process during ORR and vacancy mediated redox phenomena in OER.10a As already stated, the oxygen species involved during OER/ORR preferably exists in the triplet ground spin state, while the other reactants are poised in singlet spin state. This is further supported by the much higher intrinsic energy of ∼1 eV for singlet oxygen. Many researchers like Yasin et al. and Musa et al. have studied the role of metal free catalyst towards the functionality of water splitting reaction.10b,c However, the utilization of TMO is reported to be more active and efficient towards the redox reaction of oxygen. Prior arts have already reported that electro kinetics of OER/ORR is influenced by the spin polarization of magnetically active TMO's. Gracia et al. have theoretically validated that spin polarized electrons (could be validated through ordered magnetism/ferromagnetism experimentally) in active catalyst favours the generation of triplet spin state for oxygen.11 This phenomenon is termed as the quantum spin-exchange interaction (QSEI) which forms the atomic level basis for OER and ORR and get influenced through polarized spin state within active catalyst site. Even in the absence of active magnetic field, the intrinsic spin ordering/polarization in 3d ferromagnetic catalyst could generate inherent spin sieving for highly spin polarized electrons. Such spin filtering effect has been reported to originate from the exchange splitting of energy levels in the conduction band of ferromagnets.12 The 3d transition metal, Pt based catalyst are essential for oxygen redox reactions.13 However, high temperature operation of solid oxide cell (SOC) in either fuel cell (FC) or electrolyzer cell (EC) mode restricts the usage of Pt compounds as electrocatalyst owing to various incompatibilities reported in prior art.14 SOFC or SOEC are the examples of thermodynamic open system which engages gaseous reactants as fuel and oxygen as oxidant along with active electrode and solid oxygen/proton conducting electrolyte.15,16 High temperature operation necessitates active participation of electrocatalyst at both fuel and air electrode which primarily performs the function of initial dissociation of diffused gasses for effective adsorption on the triple phase boundary surface distributed at the electrode/electrolyte interface.17 For commercialization, the performance of a cell set up in either fuel cell (FC) or electrolyzer cell (EC) mode, redox cyclability along with long term endurance depends on the minimization of nonlinear polarizations like activation (low current density region) and diffusion overpotential (higher external load).18 For such development and commercialization of FC and EC, ORR and OER imposes significant hindrances since they suffer from slow reaction kinetics and higher polarization loses.19 These mentioned overpotential could be nullified by accelerating the reaction kinetics of HOR (hydrogen oxidation reaction)/ORR (FC mode) and HER (hydrogen evolution reaction)/OER (EC mode). The strong dependence of oxygen redox reaction on catalyst structure and functionality is reported to be tailored by certain intrinsic descriptors.20–22 The cataloguing of such descriptors is based on surface structures and morphology involving distortion, rearrangement, tortuosity, electronic environment as temperature driven electrocatalysis, which is a surface driven phenomena.23–27a The modulation of microenvironment and interfacial interactions are also interesting driving factors affecting the electrocatalysis.27b,c Furthermore, in depth analyses of surface sensitive techniques are significant in rendering complexity of local surface, polarity/segregation, space charges, degree of unbalanced surface charge required for adsorption etc.28 The second important descriptor is based on the thermoenergetics of the lattice of the catalyst like charge transfer energetics, Jahn Teller distortion affecting eg/t2g levels (considering octahedral crystal filed), p-band structure, N–V (N and V represents lone pair charge count and average nominal valence charge on B-atoms) etc.29 It has been known form prior arts that, perovskite, double perovskites system with selective doping at A and B-site are efficient electrocatalyst for oxygen redox reaction like La0.7Sr0.3Mn1−xNixO3, AxSr1−xCoyB1−yO3−δ, La1−xSrxMnO3 etc.30,31 In such system, the B-site elements primarily acts as the active catalyst being connected to lattice oxygen through BO6 octahedra. The A-site elements (dopant and active element) influence the spatial orbital overlap among the eg (d-orbital) of the B-site with 2p orbital of oxygen from adsorbates.29,31 In a similar manner, N–V descriptor tend to evaluate the strength and flexibility of B–O bond (B-site atom in catalyst) through spontaneity factor (ΔG, Gibbs free energy).32 Yoo et al.33 have established that, positive free energy accelerates the surface adsorption based evolution mechanism whereas the reverse promotes lattice oxygen route.33 The theoretical predictions have further established oxygen p-band descriptor which limits the highest activity of the catalyst with closest distance from the Fermi level in the density of state plots and charge transfer energy descriptor that visualizes transition and termination of rate limiting steps.34 In this context, the presence of inherent spin polarization of active catalyst is significant as already stated above. Apart from the theoretical study on spin orbital coupling involving energy quantization, the signature of spin polarization (i.e. spin ordering) could also be studied using magnetic study in experimental terms. Goodenough et al. have stated that in spite of being an effective descriptor for QSEI, magnetic studies of catalyst are not yet cultivated.35 The transition metal inclusion are generally capable of exhibiting aliovalency under perturbation owing to d-orbital vacancy and represent resultant non-zero spin state. Magnetic susceptibility is influenced by such bulk/surface electronic structure, coupling of charge spin and orbital, lattice parameter.36 Garcés-Pineda et al. have reported the experimental correlation among magnetism and OER for NiZnFe4Ox system.37
In this connection, doped perovskite oxides (SrFeO3) as La(1−x)SrxCo(1−y)FeyO3−δ and Ba(1−x)SrxCo(1−y)FeyO3−δ are excellent system to evaluate for exhibiting inherent spin polarization towards the catalysis of OER/ORR for solid oxide cells.31 The authors have initially established their work on the novel synthesis route for La0.6Sr0.4Co0.8Fe0.2O3−δ (LSCF6482) and Ba0.6Sr0.4Co0.8Fe0.2O3−δ (BSCF6482) and their electrochemical activity in FC and EC mode.17a,31,38,39 However, literature study and experimentation on the correlation among inherent spin polarization of LSCF6482 and BSCF6482 with the functionality as electrocatalysis in SOC is limited and is not reported till date as depicted in Table 1. The novelty of the present study is to establish the influence of spin ordering of the isovalent doping (of Ba2+ at Sr2+ site) and/or aliovalent doping (of La3+ at Sr2+ site) onto specificity in catalysis during OER and ORR operable in Solid oxide cells. As stated above, the B-site elements like Co and Fe primarily uptake the catalysis which are present in both the compositions. However, A-site element acts the primary charge carrier and controls the charge compensation either through a charge transfer and/or oxygen vacancy mode. The principal quantized energy distribution as a function of either α/β spin state for ABO3 system is independent of B-site element whereas, significantly varied DOS plots are observed by variation of ‘A’ site (La, Sr and Ba) with a constant B-site either Co and/Fe. The detailed observation is described in Section 2 of ESI.† Detailed analysis is reported on the role of intrinsic electronic descriptors (like spin, orbital–spin coupling etc.) towards the spin orientation and hetero-orbital overlap among 2p- orbital of oxygen (gases) with d-orbital of B-site elements.
| S No. |
Catalyst systems reported in literature |
Novelty of research |
| 1 |
SrCo0.9−xNb0.1NixO3−δ (SCNN, 0.1 ≤ x ≤ 0.3). Ref. 9a: Sustain. Energy Fuels, 2020, 4, 1168 |
To establish the mentioned composition as a highly active and stable cathode for oxygen reduction for IT-SOFC |
| 2 |
A set of known perovskite compounds established as effective OER/ORR electrocatalyst. Ref. 10a: Adv. Sci., 2020, 7, 1901614 |
To highlight the intrinsic descriptors which could be engaged as current benchmark activity descriptors for oxygen electrocatalysis as well as their application |
| 3 |
Metal-exsoluted perovskite, La0.52Ca0.28Ni0.06Ti0.94O3 (LCaNT), La0.52Sr0.28Ni0.06Ti0.94O3 (LSrNT) and La0.8Ce0.1Ni0.4Ti0.6O3 (LCeNT), La0.6Sr0.4Ga0.3Fe0.7O3. Ref. 15: Renew. Sustain. Energy Rev., 2021, 149, 111322 |
To review the activity and limitations of reported perovskite, double perovskite family as effective catalyst for OER/ORR |
| 4 |
Perovskites and perovskite/carbon composites. Ref. 19: ACS Catal., 2021, 11, 3094 |
To highlight the intrinsic mechanism governing the ORR/OER and associated intermediates |
| 5 |
(Gd0.5La0.5) BaCo2O5.5+δ and related perovskites. Ref. 25: Nat. Commun., 2019, 10, 3755 |
The prime novelty is to establish the role of perovskites towards hydrogen evolution reaction via unifying ionic electronegativity descriptor |
| 6 |
ORR electrocatalyst compositions. Ref. 26: Nat. Mater., 2018, 17, 827 |
The authors have tried to establish the role of surface distortion factor as the descriptor for analysing the ORR and OER |
| 7 |
Mixed conducting perovskite oxide electrodes. Ref. 28: Materials, 2016, 9, 858 |
The novelty was to highlight the roles of bulk and surface chemistry in the oxygen exchange kinetics and related properties of MIEC perovskite compositions |
| 8 |
LaNiO3 oxide system. Ref. 32: ACS Catal., 2016, 6, 1153 |
An ab initio computation technique has been employed to study the surface structure and OER mechanism for doped LaNiO3 based compounds |
| 9 |
Pr0.5Ba0.5CoO3−δ and SrCoO3−δ based perovskite system. Ref. 33: ACS Catal., 2018, 8, 4628 |
The basic aim of this study is the establish the role of lattice oxygen participation in OER mechanism and its manifestation |
| 10 |
Mixed crystals of La1−xSrxCoO3−λ. Ref. 35: J. Phys. Chem. Solid., 1958, 6, 287 |
Being perovskite system, the magnetic properties are studied and interpreted |
Experimental
Materials and methods
Perovskite oxide powders BSCF6482 and LSCF6482 were produced using solution combustion technique which is self-sustaining by nature as reported in our earlier communications.17a,31,38,39 The detailed mechanistic approach is described in the results and discussion (Fig. S8†). The reaction include the usage of Ba(NO3)2, La(NO3)3, Fe(NO3)2. 9H2O, Sr(NO3)2 and Co(NO3)2. 6H2O as precursors for metal nitrates and citric acid–glycine (for BSCF) and L-alanine (for LSCF) as fuel complexing agents. All the mentioned chemicals were used in pure form from MERCK. ‘δ’ parameter which represents the non-stoichiometry in the oxygen content was quantitatively estimated. The process involves titration of oxide solution of the experimental powders [1 M acidic medium] against 0.02 M sodium thiosulphate (MERCK) solution. Potassium iodide (KI) from (MERCK) is used as the reductant and 0.01% starch solution is utilized as the indicator. The selection of fuels (based on the thermodynamic parameters) serves the dual function of complexing agent as well. The aliovalent substitution of La and isovalent substitution of Ba in SrFeO3 uses L-alanine, glycine and citric acid as the fuel respectively. Irrespective of the substitution at the ‘A’ site, Co2+ is doped at the ‘B’ site. The Ce0.8Gd0.2O2 (GDC) powder required for cell fabrication was also synthesized by solution combustion technique.40 The viscous gels of LSCF6482 and BSCF6482 were studied for thermal analysis through thermogravimetric analysis (TGA) and differential thermal analyses (DTA) using Thermal Analyzer (NETZSCH, STA 449 F3, Jupiter) within the temperature regime of 25–800 °C@5 °C min−1. The synthesized LSCF6482 and BSCF6482 powders were subjected to high temperature calcination at 700–1000 °C calcined in air in the temperature range for 4 h to obtain the phase pure powder of the material. The X-ray diffraction study of the calcined LSCF6482 (at 850 °C) and BSCF6482 (at 1000 °C) powders were investigated by using PANalytical, Philips, Holland with CuKα radiation at 40 kV and 30 mA, in the 2θ range of 20°–80°. Necessary Rietveld refinement of the XRD along with the quantitative phase analysis for the samples were carried out using the MAUD software. Morphology study of the experimental samples was performed using Transmission Electron Microscopy (TEM) [Tecnai G2 30ST (FEI)]. Microstructural analysis of the evaluated cells was studied using Field Emission Scanning Electron Microscopy (FESEM) (Gemini Supra 35-Zeiss). The population density of variable oxidation states of B-site elements and oxygen is analysed through X-ray photoelectron spectroscopy (PHI 5000 Versa probe II spectrophotometer-physical Electronics Inc., USA).
Magnetic behaviour of the experimental samples was studied using a SQUID magnetometer MPMS3 (LOT-Quantum Design inc.). The employed mode during the analyses is vibrating sample magnetometry (VSM).
Details on asymmetric cell fabrication process is already reported in our earlier communication.31 The asymmetric cells fabricated are of Gd-doped CeO2 based electrolyte-supported cell which is fabricated through uniaxial pressing followed by sintering at 1350 °C. The air electrodes (functional LSCF6482 and BSCF-6482) are then screen printed on to the sintered GDC pellets as also reported in our earlier communication.31 The primary novelty undertaken in the present investigation is to reduce the GDC electrolyte thickness considerably (compared to the FC reported by Dey et al.31) so as to minimize the associated polarizations and enhance the self-life and sustainability of the fuel cell. Potential driven EIS of the asymmetric cells [Pt/GDC/GDC: BSCF6482/BSCF6482 and Pt/GDC/GDC: LSCF6482/LSCF6482] were captured using Autolab PGSTAT30 galvanostat-potentiostat. The frequency range employed was within 10−1–106 Hz.
Theoretical study
Theoretical study on the electronic structure of many-body system is significant in understanding the intrinsic mechanism during reaction kinetics. Such theoretical study is undertaken by numerous techniques among which Density functional theory (DFT) is widely used by the researchers. This is the utmost adaptable technique in the arena of computational chemistry.41 It has been employed to get the information about the density of states for the ground level (S0) configurations of the experimental systems. In the present context, generalized-gradient-approximation is coupled with hybrid exchange–correlation functional PBE1PBE. In addition, the valence-only split-valence CEP-121G basis set is utilized.42–44 The mentioned functional was implemented so as to evaluate the accurate energetics of the selected compounds having lanthanides in the present investigation. This is well reported in prior arts.45a Residual charges on the peripheral ions on the truncated crystal has been neutralized by putting charges on the lattice sites. Besides this, the background distribution of charge also provides an appropriate crystal field around each ion in the chosen structure. All the computations reported have been executed with Gaussian 16, Revision B.01 program package. Gauss View 5.0 software was engaged for the visualization of the respective structure.
Results and discussion
Inherent spin polarization of La0.6Sr0.4Co0.8Fe0.2O3−δ (LSCF6482) and Ba0.6Sr0.4Co0.8Fe0.2O3−δ (BSCF6482)
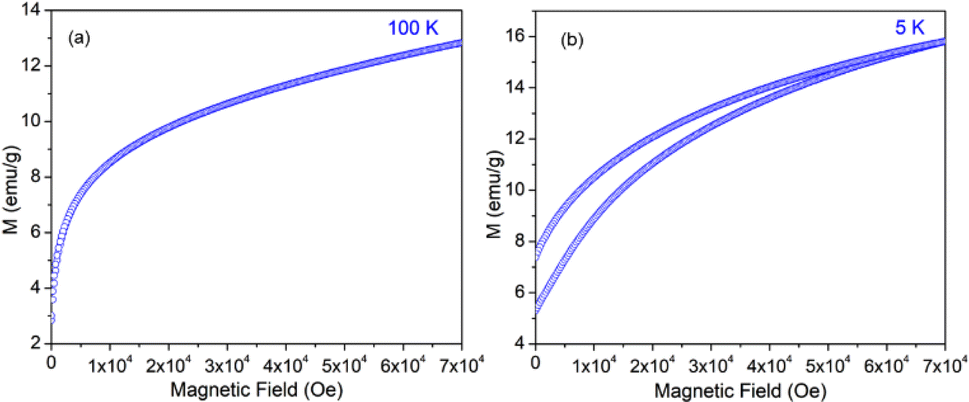
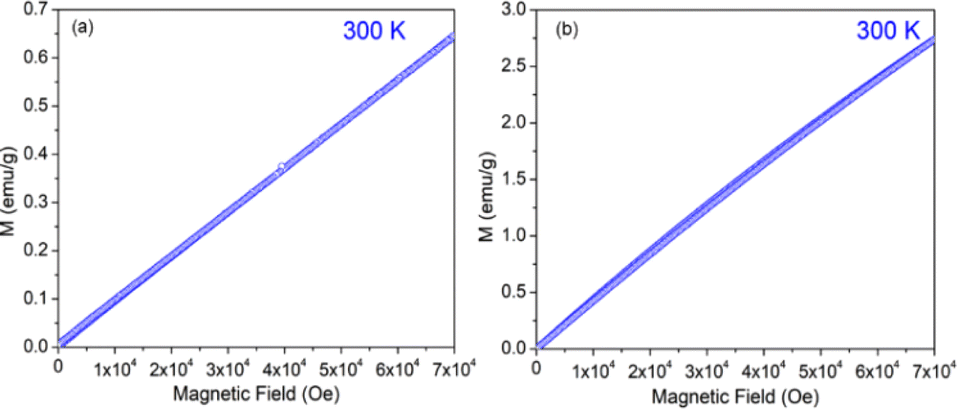
The variation of magnetization (M) of the experimental functional oxides viz. BSCF6482 and LSCF6482 samples have been recorded with magnetic field (H) at different temperatures. Fig. 1a exhibits the variation of magnetization at a temperature of 100 K whereas, the variation at 5 K has been shown in Fig. 1b in the case of BSCF6482 sample. Fig. 2a and b show the variation of magnetization at 100 K and 5 K respectively for LSCF6482 sample. In every case, the maximum applied magnetic field is 70 kOe. The presence of magnetic ordering has been shown at different temperatures. Maximum magnetizations at different temperatures are tabulated in Tables 2 and 3 respectively. Similarly, variation of magnetization at 300 K for BSCF6482 and LSCF6482 is shown in Fig. 3a and b respectively.
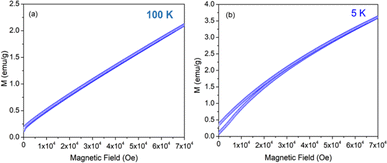 |
| | Fig. 1 MH loop for BSCF6482 at: (a) 100 K and (b) 5 K temperature. | |
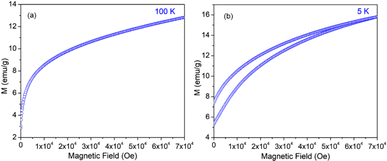 |
| | Fig. 2 MH loop for LSCF6482 at: (a) 100 K and (b) 5 K temperature. | |
Table 2 Influence of temperature on maximum magnetization for LSCF6482 and BSCF6482
| Sample ID |
Temperature (K) |
Maximum magnetization Mmax (emu g−1) |
| LSCF6482 |
5 |
15.86 |
| 100 |
12.89 |
| 300 |
2.75 |
| BSCF6482 |
5 |
3.64 |
| 100 |
2.13 |
| 300 |
0.65 |
Table 3 The value of magnetic susceptibility (χ) at room temperature
| Sample |
Mmax (emu g−1) |
Hmax (Oe) |
χ (M/H) (× 10−5) (emu per gOe) |
| BSCF6482 |
0.65 |
70282 |
0.92 |
| LSCF6482 |
2.77 |
70866 |
3.90 |
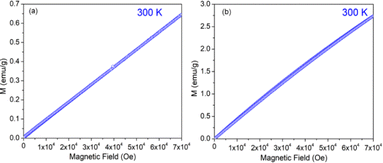 |
| | Fig. 3 MH Loop at 300 K for (a) BSCF6482 and (b) LSCF6482 samples. | |
Fig. 1–3 represents the variation of magnetization with the applied magnetic field at room temperature (300 K) and 5 K for both the sample BSCF6482 and LSCF6482. Nearly linear magnetization shows the field proportionality with a susceptibility value of 0.92 × 10−5 emu per gOe for BSCF6482 and 3.90 × 10−5 emu per gOe for LSCF6482.
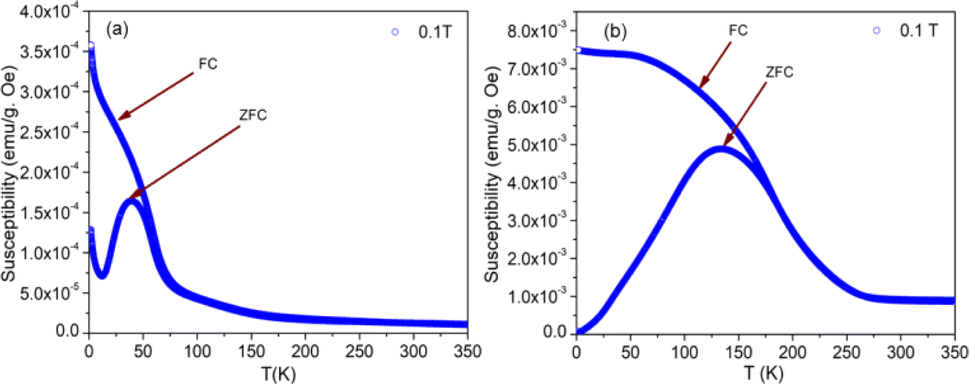
The nature of the plot suggests that the sample is not paramagnetic at room temperature; rather, it should be antiferromagnetic, having the Néel temperature above room temperature. With the lowering of temperature, the magnetization value increases, and at 5 K, a prominent non-linear MH loop is obtained with much higher value of maximum magnetization for both samples where the traceback curve measured during field down does not overlap with the virgin curve. In order to further exemplify the study, Fig. 4 is reported wherein the variation of magnetic susceptibility (χ) is shown with temperature for the samples and Table 3 tabulates the value of magnetic susceptibility at room temperature. Different values of the magnetizations with respect to different temperatures have also been recorded after the cooling of the samples from 350 K under two different conditions. The conditions are (i) in presence of an applied magnetic field i.e., the field-cooled condition (FC) and (ii) in absence of the applied field i.e., zero field-cooled condition (ZFC). To observe this intermediate behavior, a continuous variation of χ with temperature is recorded (Fig. 4) for both the samples with a constant applied field of 1 kOe. Here, a continuous increment of magnetic susceptibility χ(M/H) can be observed by lowering temperature up to a certain range. It is however, observed that, the magnetic susceptibility starts to decrease following a maximum in that variation. The observed maxima are not so sharp as a phase transition peak, and as the sample is already in an ordered state at room temperature, this maximum must not indicate any magnetic phase transition. In that case, this can indicate the presence of superparamagnetism in the sample. To confirm this, we have recorded the variation of magnetic susceptibility χ(M/H) with the temperature at the constant field of 1 kOe during the rise of temperature after cooling the sample from 350 K. The cooling of the sample is performed in the absence of any magnetic field at first, i.e., zero field cooling (ZFC), followed by cooling in the presence of field (1 kOe) (FC). The FC and ZFC magnetization are shown in Fig. 4 for LSCF6482 and BSCF6482 respectively.
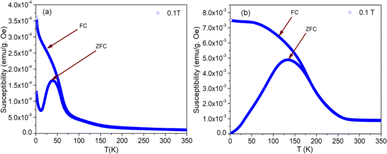 |
| | Fig. 4 Plot of magnetic susceptibility with temperature of: (a) BSCF6482 and (b) LSCF6482 samples at field cooled and zero field cooled conditions. | |
It can be observed from Fig. 4, that both the curve overlaps down to a constant value followed by a bifurcation. The FC curve shows a sharp rise at that temperature where the maximum of the ZFC curve is observed. This temperature at which the maximum of ZFC curve is noted is termed as blocking temperature (TB), the temperature at which the fork (bifurcation) is observed is termed as irreversible temperature (TIrr). Here blocking means arresting of spin fluctuation below that specific temperature. The phenomena of super magnetism are reported to generate if the particle size of the matrix is smaller than the critical particle size of the sample. The resultant magnetic moments can only be exhibited to contribute in ZFC is they are unblocked. The blocked moments however, fail to contribute during ZFC. In contrast, both blocked and unblocked moments are capable of significant contribution during FC measurement producing external measurable moments. Due to distribution of particle size, blocking temperature is assigned to be the temperature at which maximum of blocking occurs in that sample, and the highest temperature where blocking starts to occur is recognized as irreversible temperature (TIrr). It is further noted that the blocking temperature (∼49 K) of BSCF6482 is much lower than that of LSCF6482 (∼145 K). Upon comparing the trend, it can be observed that maximum magnetization is nearly the same at room temperature; however, non-linearity in MH loop is more prominent in LSCF6482 than BSCF6482. In addition, at 5 K, maximum magnetization is much higher (∼4.36 times) in LSCF6482 (15.86 emu g−1) than BSCF6482 (3.64 emu g−1). So, both the sample shows ordered magnetism throughout the temperature range where the magnetic ordering in LSCF6482 is much improved than BSCF6482. It is therefore, stated that both LSCF6482 and BSCF6482 possess polarized spin state which offer affirmative QSEI during OER and ORR phenomena.
Interdependence among spin polarization and electron spin exchange function
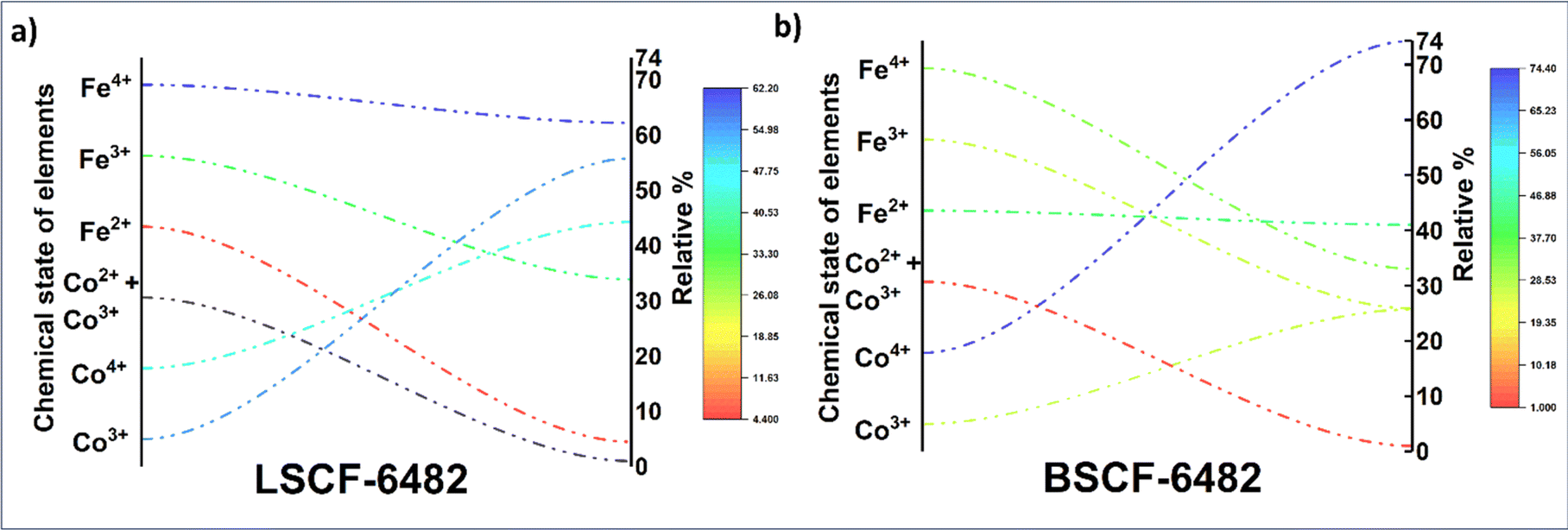
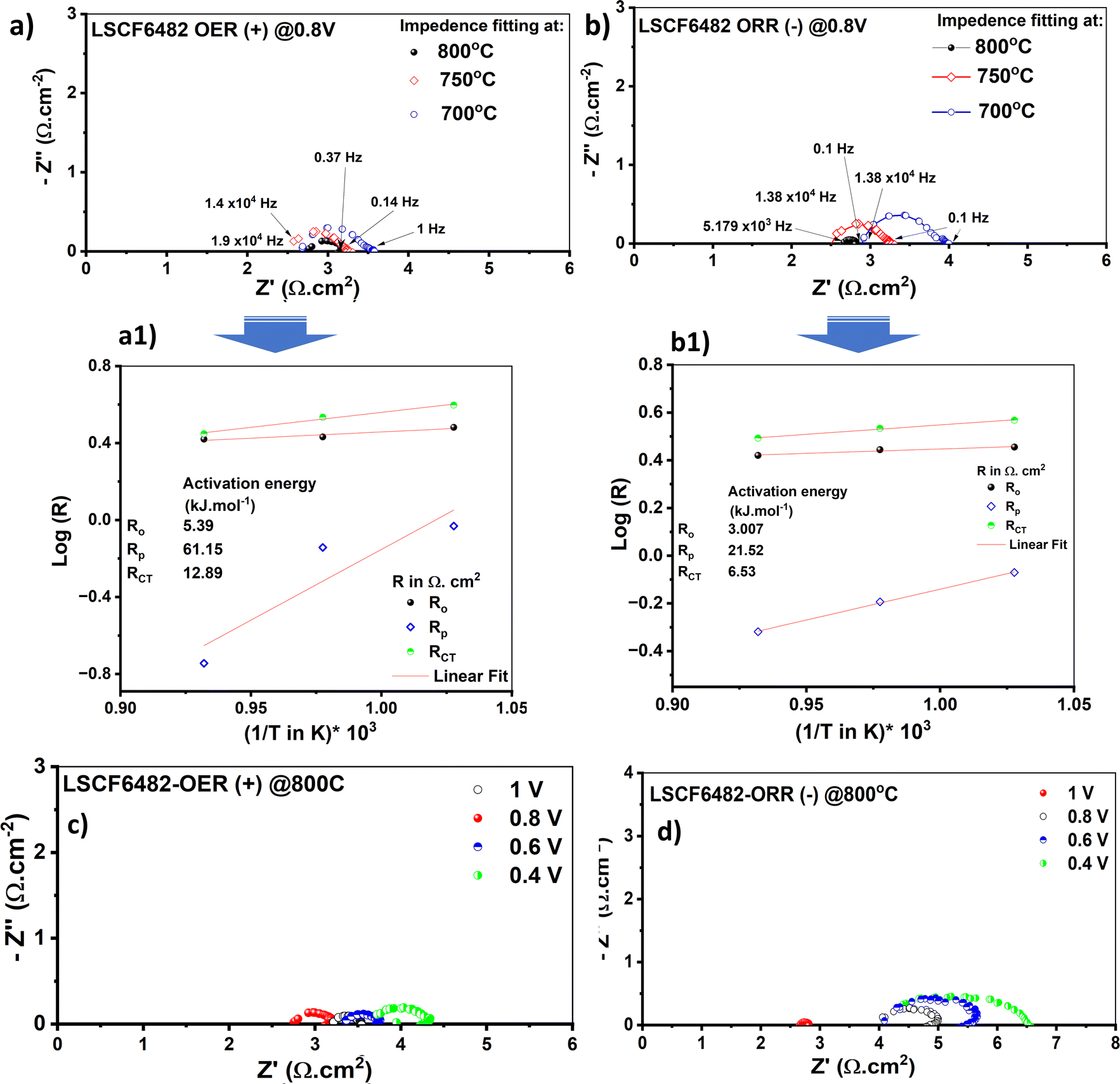
In this section, LSCF6482 and BSCF6482 are evaluated for electrocatalytic property upon applied as air electrode in solid oxide cell. The kinetics of oxygen redox reaction is undertaken through separate charge compensation mechanism in LSCF6482 and BSCF6482. It has been already reported by the authors31 that, the B-site is active for the participation of surface adsorbed OER/ORR. However, the number density of B-site ions with variable oxidation states are influenced by the doping at A-site which controls the principal energy quantization. The details on XPS analyses have been reported in prior arts by the authors. The mode of electrocatalysis is variable for LSCF6482 and BSCF6482 based on the population density of Co3+/4+ as shown in Fig. 5. LSCF6482 favors compensation of charge through aliovalency of metal ions whereas BSCF6482 promotes oxygen vacancy creation and propagation for oxygen exchange reaction, wherein the presence of 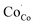 and
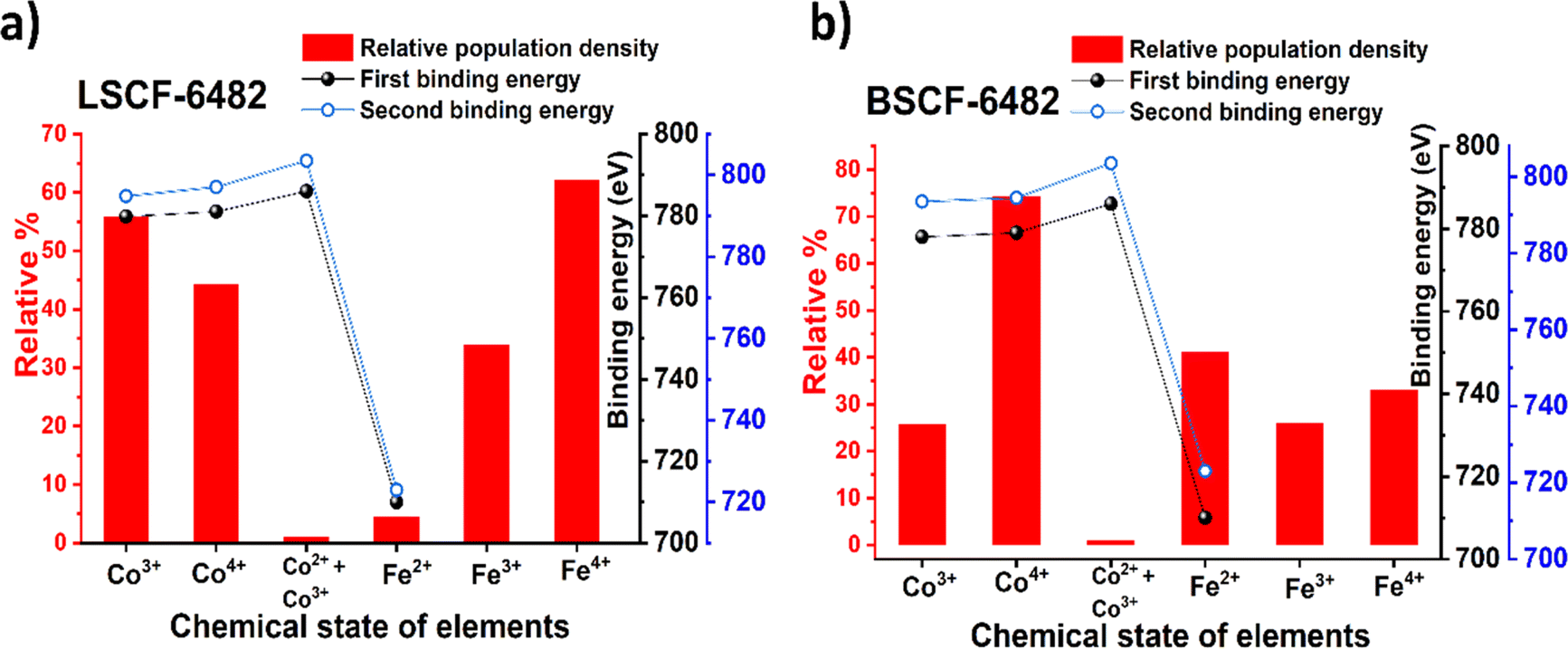
and 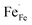 eases the path for the transformation of O2− to O2 during OER. The charge transfer process through aliovalency in LSCF6482 is highly accelerated owing to the presence of higher population density of Co3+ ion which undergoes easy spin state equilibrium shown in Fig. S1.† It could be seen that, the easy spin cross over of Co3+ at even thermal energy results in magnetism owing to the non-zero spin state of ½ and 1 respectively. Similar fact is restored in BSCF, however, due to lesser population density of Co3+ ion, charge compensation route is overshadowed and governed by oxygen vacancy propagation route. This is also responsible for the higher magnetic ordering observed in the matrix of LSCF6482 as shown in Fig. 1–4. Similar population of Co3+ and Co4+ in LSCF 6482 with almost comparable consecutive binding energy (Fig. 6) accelerates ORR through aliovalent charge transfer and restricts the usage of lattice oxygen. Fig. 6 exhibits the distribution of population density for variable oxidation states of B-site ions viz Co and Fe respectively. As already stated, the B-site ions primarily act as the active sites for dissociative adsorption of oxidant followed by catalyzing the redox reaction of oxygen. Doping of isovalent (Ba2+) and/aliovalent (La3+) ion is found to influence the variability in population density of Co (+2 to +4) and Fe (+2 to +4), which alters the specificity in electro kinetics towards OER/ORR. In addition, the quantitative binding energies are plotted as a function of oxidation state. Irrespective of type of ions, the consecutive binding energies are found to follow the similar trend.
eases the path for the transformation of O2− to O2 during OER. The charge transfer process through aliovalency in LSCF6482 is highly accelerated owing to the presence of higher population density of Co3+ ion which undergoes easy spin state equilibrium shown in Fig. S1.† It could be seen that, the easy spin cross over of Co3+ at even thermal energy results in magnetism owing to the non-zero spin state of ½ and 1 respectively. Similar fact is restored in BSCF, however, due to lesser population density of Co3+ ion, charge compensation route is overshadowed and governed by oxygen vacancy propagation route. This is also responsible for the higher magnetic ordering observed in the matrix of LSCF6482 as shown in Fig. 1–4. Similar population of Co3+ and Co4+ in LSCF 6482 with almost comparable consecutive binding energy (Fig. 6) accelerates ORR through aliovalent charge transfer and restricts the usage of lattice oxygen. Fig. 6 exhibits the distribution of population density for variable oxidation states of B-site ions viz Co and Fe respectively. As already stated, the B-site ions primarily act as the active sites for dissociative adsorption of oxidant followed by catalyzing the redox reaction of oxygen. Doping of isovalent (Ba2+) and/aliovalent (La3+) ion is found to influence the variability in population density of Co (+2 to +4) and Fe (+2 to +4), which alters the specificity in electro kinetics towards OER/ORR. In addition, the quantitative binding energies are plotted as a function of oxidation state. Irrespective of type of ions, the consecutive binding energies are found to follow the similar trend.
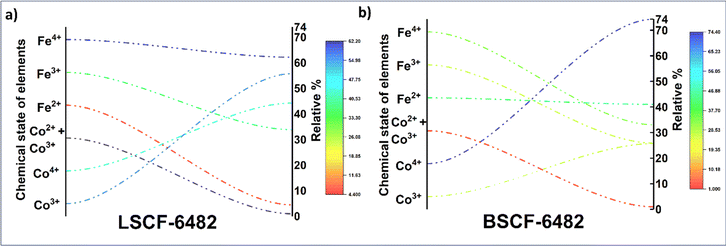 |
| | Fig. 5 Relative population of ions with variable oxidation states calculated from XPS analyses of: (a) LSCF6482 and (b) BSCF6482. | |
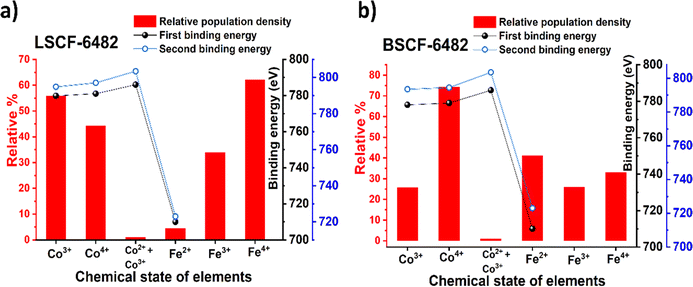 |
| | Fig. 6 Population density of ions with variable oxidation states along with their binding energy (BE1 and BE2) calculated from XPS analyses of: (a) LSCF6482 and (b) BSCF6482. | |
In both compositions, the stoichiometry of Co is higher (80%) which shows spin crossover phenomena with lower activation barrier. The B-site lattice involves the formation of CoO6 octahedra connected through outer hexahedron frame (comprising A-site elements like La/Sr/Ba). The bond length of Co–O is dependent on the oxidation state of Co (+3/+4) and results in distorted octahedra upon increasing Co–O bond length comparison of La/Sr–O dimension. First principle DFT study on LSCF6482 and BSCF6482 exhibits the dependence of orbital overlap (hybridization or electron spin exchange function) bonding of Co–O and its state of bonding as well. In case of shorter bond length as stated above, hybridization among 2p–3d is stronger. The specific role of A-site and B-site dopants in the complex perovskite matrix towards orbital overlap and electronic structure has been undertaken using first principle DFT and is discussed in details in Section 2 of the ESI† (Fig. S2, S3 and Table S1†).45b Based on the literature, Heisenberg's theory correlates orbital overlap integral, A (hybridization) with magnetic parameters (χ)46 by eqn (1):
where,
In eqn (1), z is the coordination number, A is the integral for exchange coefficient, kB is Boltzmann constant, r is the distance among electrons, a2 is the distance of one electron (2) from cation b to the nucleus of cation (a). The orbital overlap among 2p of oxygen and 3d of the active transition metal of the electrocatalyst is the prime factor for electron exchange interaction on which the activation and propagation of OER/ORR is dependent. Since, exchange interaction is proportional to A in the eqn (1), it signifies its linear correlation with the magnetization as well. Since, both LSCF6482 and BSCF6482 possess magnetic ordering throughout the temperature range, these are excellent electrocatalyst for OER/ORR as stated. However, magnetization of LSCF6482 is much higher as shown in Table 3 with improved ordering, the charge transfer process through aliovalency is predominated and results in better activity in ORR compared to BCSCF6482 which shows much reduced non-linear polarization for OER. The mentioned electrocatalysts LSCF6482 and BSCF6482 thereby exhibits specificity towards ORR and OER which bears strong correlation with magnetic properties as depicted form crystal and electronic structure. The influence of intrinsic factor viz spin polarization on extrinsic catalysis (OER/ORR) is described in the subsequent section.
Spin polarized OER and ORR in terms of electrochemical impedance spectroscopy
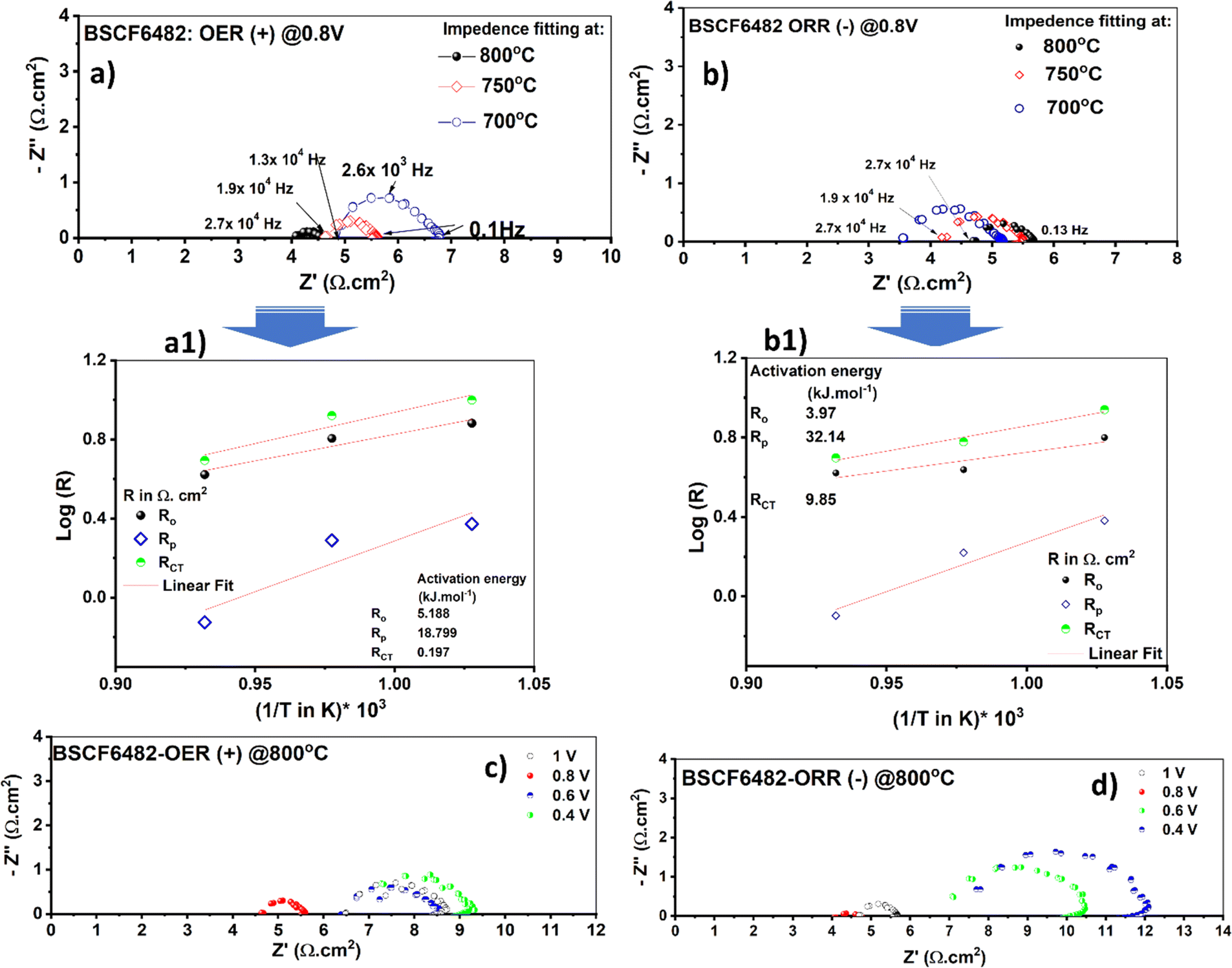
The redox electrocatalysis of LSCF6482 and BSCF6482 is studied by the authors in terms of electrochemical impedance spectroscopy. The respective Nyquist plots are studied for the fabricated asymmetric cells (configurations as mentioned in the Experimental section) in the operating temperature range from 700–800 °C. The cell responses are undertaken within ±0.4 to ±1.0 V at the respective amplitudes of 150 mV and 300 mV respectively. However, the EIS are only reported for the captured amplitudes at 300 mV. The involved electrochemical processes could be well resonated with specific applied frequency domain. The slower process involving surface sorption of active species at air electrode are observed at lower frequency domain (0.1 Hz–10 Hz) which ascertains the diffusion polarization and is operative at even 700 °C. The interfacial mass transfer involving ion transfer process through bulk matrix are operative at mid frequency domain (20 Hz to 5.6 × 104 Hz).47 The cells fabricated either with LSCF6482 and/or BSCF6482 exhibits a steady DC current at either cathodic and anodic potentials of ≥ −0.8V and ≥ +0.8 V. It could be observed from Fig. 7c, d and 8c, d that, irrespective of electrocatalysis mode of either OER and ORR, optimized EIS is obtained at an operating potential of ±0.8 V@800 °C. It has been reported by Dey et al.31 that surface resistance of single cell is minimum at the operating potential of 0.8 V in both OER and ORR mode. Similar fact is also noted to be operative in the single cells reported in the present investigation studied at 700 °C and 750 °C (Fig. S4 and S5†). For LSCF6482 and BSCF6482 systems, both magnetism and electrocatalysis in terms of OER and ORR are engrained in electron exchange interaction. In order to further correlate the specificity of electrocatalysis with magnetic ordering, activation barrier required for non-linear polarization viz. Ro (total overpotential being captured at high operative frequency@104 Hz), Rp (interfacial polarization) and RCT (charge transfer process) are evaluated. The EIS obtained for the single cells at an operating temperature range of 700–800 °C for anodic and cathodic potential (@±0.8 V) is shown in Fig. 7a, b and 8a, b respectively. The respective Arrhenius plots for the single cells are given in Fig. 7a1, b1 and 8a1, b1 and are tabulated in Table 4.
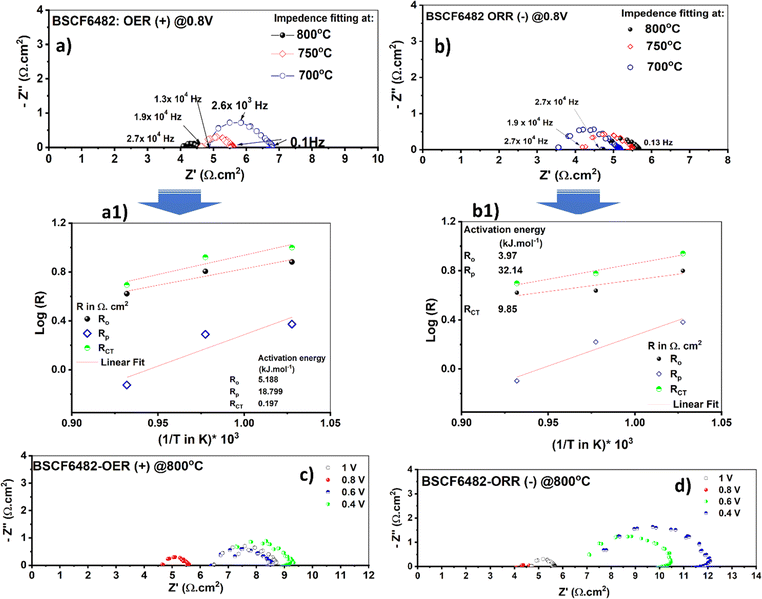 |
| | Fig. 7 Impedance response of single cells fabricated using BSCF6482 in the temperature range of 700–800 °C under: (a) anodic potential of 0.8 V (+) and (b) cathodic potential of 0.8 V (−). The activation energy plot for associated polarizations (Ro, Rp and RCT) for anodic (a1) and cathodic (b1) potential. Nyquist plots for the single cells with BSCF6482 as the active electrocatalyst at an operating temperature of 800 °C as a function of applied potentials (0.4 V–1.0 V) for (c) anodic (OER) and (d) cathodic (ORR) bias. | |
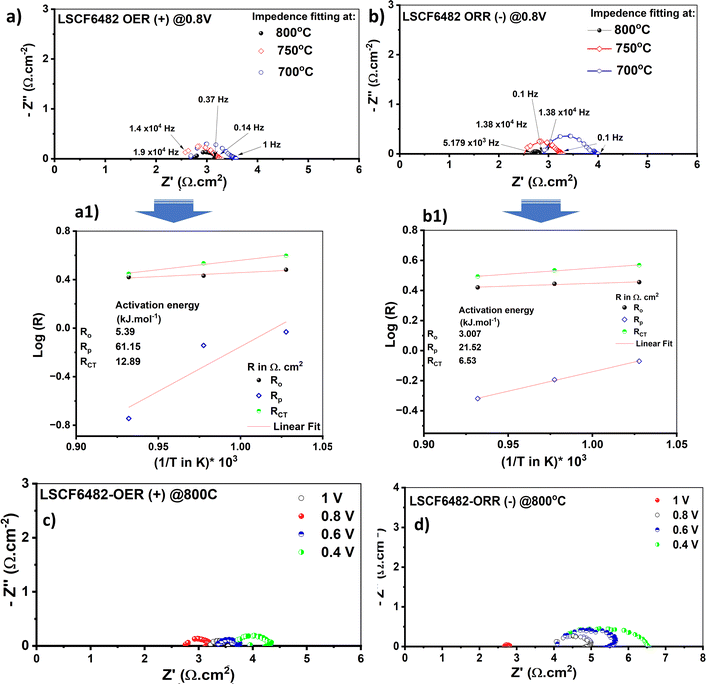 |
| | Fig. 8 Impedance response of single cells fabricated using LSCF6482 in the temperature range of 700–800 °C under: (a) anodic potential of 0.8 V (+) and (b) cathodic potential of 0.8 V (−). The activation energy plot for associated polarizations (Ro, Rp and RCT) for anodic (a1) and cathodic (b1) potential. Nyquist plots for the single cells with LSCF6482 as the active electrocatalyst at an operating temperature of 800 °C as a function of applied potentials (0.4 V–1.0 V) for (c) anodic (OER) and (d) cathodic (ORR) bias. | |
Table 4 Magnitude of non-linear polarization during EIS of single cells fabricated with LSCF6482/BSCF6482 in OER and ORR mode of operation
| Sample and FC mode |
Temperature (°C) |
Activation energy for Ro (EaRo) in kJ mol−1 |
Activation energy for Rp (EaRp) in kJ mol−1 |
Activation energy for RCT (EaRCT) in kJ mol−1 |
Rs in Ω cm2 |
| Operating voltage 1.0 V |
| BSCF@OER (+) |
700 |
2.13 |
9.64 |
4.14 |
0.73 |
| 750 |
0.11 |
| 800 |
0.06 |
| BSCF@ORR (−) |
700 |
15.655 |
41.312 |
21.17 |
0.62 |
| 750 |
0.22 |
| 800 |
0.12 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Operating voltage 1.0 V |
| LSCF@OER (+) |
700 |
30.68 |
51.29 |
36.41 |
0.28 |
| 750 |
0.2 |
| 800 |
0.02 |
| LSCF@ORR (−) |
700 |
3.52 |
30.84 |
2.08 |
0.2 |
| 750 |
0 |
| 800 |
0 |
|
| Operating voltage 0.8 V |
| BSCF@OER (+) |
700 |
5.19 |
18.79 |
0.197 |
0.5 |
| 750 |
0.15 |
| 800 |
0.12 |
| BSCF@ORR (−) |
700 |
3.97 |
32.14 |
9.85 |
0.22 |
| 750 |
0.07 |
| 800 |
0 |
|
| Operating voltage 0.8 V |
| LSCF@OER (+) |
700 |
5.38 |
61.14 |
12.88 |
0.18 |
| 750 |
0.09 |
| 800 |
0 |
| LSCF@ORR (−) |
700 |
3.00 |
21.52 |
6.53 |
0.12 |
| 750 |
0.04 |
| 800 |
0 |
|
| Operating voltage 0.6 V |
| BSCF@OER (+) |
700 |
21.62 |
42.73 |
26.27 |
0.19 |
| 750 |
0.24 |
| 800 |
0 |
| BSCF@ORR (−) |
700 |
22.36 |
45.15 |
28.18 |
0.25 |
| 750 |
0.13 |
| 800 |
0 |
|
| Operating voltage 0.6 V |
| LSCF@OER (+) |
700 |
10.89 |
36.08 |
14.97 |
0.20 |
| 750 |
0.16 |
| 800 |
0.05 |
| LSCF@ORR (−) |
700 |
5.04 |
28.60 |
1.61 |
0.18 |
| 750 |
0.11 |
| 800 |
0 |
|
| Operating voltage 0.4 V |
| BSCF@OER (+) |
700 |
13.47 |
34.84 |
18.62 |
0.24 |
| 750 |
0.17 |
| 800 |
0 |
| BSCF@ORR (−) |
700 |
17.87 |
38.49 |
23.19 |
0.29 |
| 750 |
0.14 |
| 800 |
0 |
|
| Operating voltage 0.4 V |
| LSCF@OER (+) |
700 |
11.06 |
27.43 |
14.89 |
0.25 |
| 750 |
0.16 |
| 800 |
0.08 |
| LSCF@ORR (−) |
700 |
8.97 |
24.28 |
12.30 |
0.15 |
| 750 |
0.10 |
| 800 |
0 |
The cells polarization resistance is found to exhibit Arrhenius dependence which confirms their activation derived out of the temperature dependent impedance values. The thermoenergetics associated with the inherent system resistance is calculated in terms of activation energy (Ea) form the linear fitted slope following eqn (2):
| | |
logR = logA − Ea/RgasT
| (2) |
where,
R,
A,
Rgas and
T corresponds to associated polarization losses, frequency factor, gas constant and operating fuel cell temperature.
Considering 0.8 V to be the optimum applied potential, the associated interfacial polarization of single cell for BSCF6482 is found to be least for OER kinetics (18.79 kJ mol−1@0.8 V for OER compared to 32.19 kJ mol−1 for ORR) and that for LSCF6482 for ORR kinetics (21.5 kJ mol−1@0.8 V for ORR compared to 61 kJ mol−1 for OER) respectively. Similar chronology is observed for the single operation at other applied potentials of ±0.4 V, ±0.6 V and ±0.1 V (Fig. S6 and S7†). The surface adsorption phenomena are accelerated at higher operating temperature of 800 °C as indicated by the tabulated magnitude of Rs (diffusion/surface polarization phenomena). The reported single cell configuration is evaluated electronically through a single equivalent circuit composed of series resistor (representing polarization arc) with Ro connected in parallel with a constant phase element (CPE) to simulate the semicircular axis correlation (Z′ and -Z′′) (Fig. S6g† and S7g). The kinetics of both ORR and OER is controlled by surface sorption of actively diffused species from oxidant and is well correlated with the electron exchange interaction among oxygen species and triple phase boundary (TPB) sites at the interface. Thermoenergetics of fuel cell involves both the involvement of interfacial and bulk exchange interactions. As per the reported content on hybridization among LSCF/BSCF system with active oxidant, 2p orbital of oxygen species (not from electrocatalyst) contributes towards the 2p–3d orbital hybridization during electron exchange interaction in interface. In contrast, during the electron transportation in the bulk crystal matrix, 2p orbital of lattice oxygen in electrocatalyst contributes significantly. The lower Rp for ORR in case of LSCF6482 suggest scavenging of  through the formation of
through the formation of  . In a similar manner, strong hybridization involving lattice oxygen promotes the formation of
. In a similar manner, strong hybridization involving lattice oxygen promotes the formation of 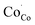 and
and 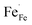 which is responible for oxidation of O2− to O2 in BSCF6482. This lowers the Rp towards OER for BSCF6482 irrespective of operative temperature. It is an established fact that, strongly coupled intrinsic parameters like orbital, spin, charge and lattice acts as descriptors for ORR and/OER processes. In a similar manner, magnetic property like ordering share similar origin from electron exchange interaction and is capable to reveal the mechanistic understanding of electrocatalysis process like OER and ORR. Li et al.46 also proposed the utilization of Curie/Néel temperature as an active oxygen redox process descriptor in the active catalysts with ferro/ferri/antiferromagnetic ground states. Hence, the present functional electrodes (BSCF6482/LSCF6482) which are in function for several years as electrocatalyst could be automatically possess excellent performance as magnetic electrocatalyst as well. In this connection, the future research would share an insight on acceleration and sustenance of OER activity of BSCF6482 and ORR performance of LSCF6482 in presence of tailored magnetic field.
which is responible for oxidation of O2− to O2 in BSCF6482. This lowers the Rp towards OER for BSCF6482 irrespective of operative temperature. It is an established fact that, strongly coupled intrinsic parameters like orbital, spin, charge and lattice acts as descriptors for ORR and/OER processes. In a similar manner, magnetic property like ordering share similar origin from electron exchange interaction and is capable to reveal the mechanistic understanding of electrocatalysis process like OER and ORR. Li et al.46 also proposed the utilization of Curie/Néel temperature as an active oxygen redox process descriptor in the active catalysts with ferro/ferri/antiferromagnetic ground states. Hence, the present functional electrodes (BSCF6482/LSCF6482) which are in function for several years as electrocatalyst could be automatically possess excellent performance as magnetic electrocatalyst as well. In this connection, the future research would share an insight on acceleration and sustenance of OER activity of BSCF6482 and ORR performance of LSCF6482 in presence of tailored magnetic field.
Apart from the dependence on spin polarization of active air electrode catalyst, the electrochemical redox reaction (OER/ORR) is an interfacial phenomenon and is propagated through dissociation, diffusion and consequent adsorption on the active surface termed as triple phase boundary (3D volume element). LSCF6482 and BSCF6482 are established to have oriented spin which accelerates the FC performance with much reduced polarizations. Furthermore, LSCF6482 is more suited for catalyzing ORR and BSCF6482 for OER. Activation and sustenance of OER/ORR also necessitates descriptors which are either molecular surface based viz. distortion, unsaturated metal centers, coordination limit etc or bulk parameters like polarity, space charges, reorganization of atoms, ion paring or segregation etc. As stated earlier, the electrocatalytic process during the operation of FC and/EC are dependent on electron exchange coefficient which is proportional to the extent of hybridization/orbital overlap of the participating electronic cloud having matching i.e. energy and symmetry matrices.46 Section 4.3, intend to evaluate the intrinsic descriptors of SrFeO3 and their variability upon doping at A and B site respectively.
LSCF6482 and BSCF6482 have been established to be effective catalyst for oxygen redox reaction having specificity towards ORR (LSCF6482) and OER (BSCF6482). In this concluding section(s), the influence of A-site doping towards the functional aspect of such compounds are described in details.
Kinetic, mechanistic and thermodynamic analyses on the influence of A-site doping of SrFeO3
In the present context, single fuel like L-alanine and mixed fuel viz. glycine and citric acid (used at a pH of ∼6 using liquor ammonia) to be used for the combustion synthesis of LSCF and BSCF respectively. The total amount of fuel to nitrate ratio is maintained at 1:1. Fig. S8a and b† shows the schematic for the synthesis of doped SrFeO3 by La3+ and Ba2+ at A-site and Co at B-site and the mechanistic approach for the same.
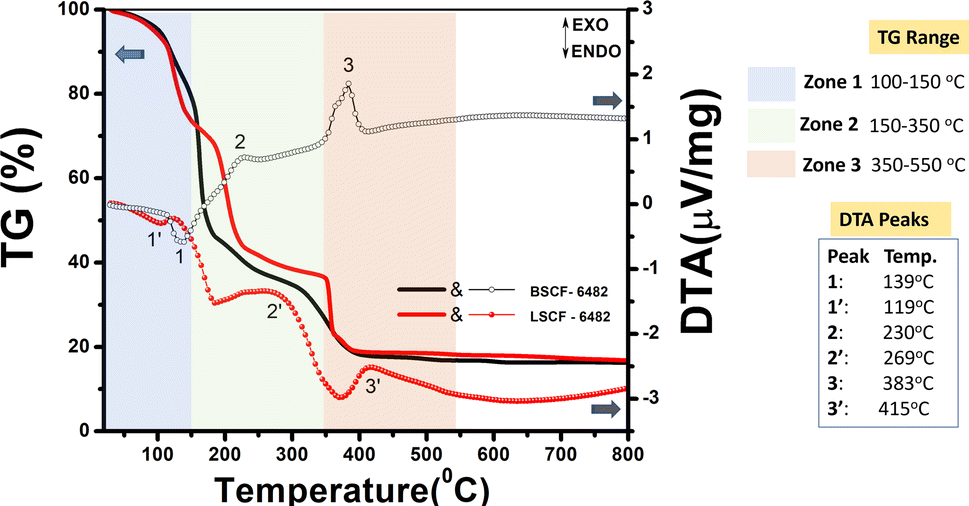
The primary requisite of solution combustion synthesis is the miscibility of reactant salts in a polar solvent like water which tend to solvate the ionized species followed by the formation of a preferable metal–aqua octahedral complex (aquation). The requisite for the formation of an initial symmetric metal–aqua complex is to accelerate the succeeding rate determining step known as anation. As stated in the Experimental section, continuous heating of the metal ion solution at 100–150 °C tend to make it more concentrated and allows easy ligand exchange reaction with the chelating neutral ligands, (or fuel) which is visible during the reaction in the form of gel. Irrespective of the denticity and experimental conditions, L-alanine, glycine and citric acid are all chelating ligands which tend to form stable five/six membered ring (at basic pH due to available O-sites) circumscribing the central metal ion as shown in the Fig. S8a† and hence easily replace monodentate aqua ligand in the first step. La3+ being heavier ion is found to result in higher stability constant with L-alanine as a ligand at a pH ∼7.5. It could be observed from Fig. S8b† that, at a still higher pH ∼8, citric acid results in more stable bi nuclear complex which however, requires a preference of selective metal ion to which La3+ fails to fit. In the present context, BSCF6482 has been optimized using complexation with mixed fuel of glycine and citric acid at a pH of ∼6. The pH criterion of citric acid (availability of all binding sites) is however fulfilled by both ammonia (pH setter in solution) and glycine (co-ligand/co fuel). The relative order of stability constants for metal–ligand (fuel) complex is shown in Fig. S8a.† Relative stability of such formed gel (complex) is significant for obtaining targeted doped perovskite with 100% phase purity prior to calcination. The final stage during the self-sustaining auto combustion route is initiated by external heat treatment (∼300 °C) wherein, the formed chelate complex decomposes and triggers the self-assisted decomposition of the gel. The added fuel plays the dual role of acting as a ligand and also supplies energy towards the breakdown of the gel. As a consequence, the final in situ decomposition of the gel comprises of reorganization or realignment of the metal ions in a stable cubic configuration. This tends to increase the spontaneity of reaction through crystal field stabilization energy thereby propagating and terminating the reaction in an irreversible manner [−ΔGreaction = ΔGproduct (cubic perovskite) – ΔGreactant (octahedral metal–ligand complex)]. The larger metal atoms like La3+, Ba2+ and Sr2+ prefer to occupy the 12-fold cuboctahedral coordination i.e. corners of the cube. However, the net strain developed at the A-site is variable due to the charge difference of the dopants. Relatively smaller ions viz. Co3+/4+ and Fe3+ preferably enter in 6-fold coordination surrounded by an octahedron of anions (O2− here, BO6 octahedra) and finally forms AA′BB′O3−δ (A′ and B′ are doped metal ions). The initial aquation, anation followed by complexation (gelation) and decomposition is observed to result in a multistage decomposition of the resulted doped perovskites as shown in the thermal analyses in Fig. 9.
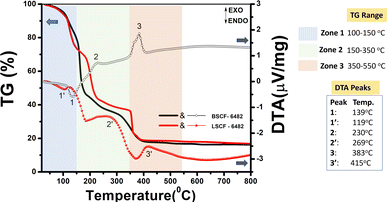 |
| | Fig. 9 Thermogravimetric (TG) and differential thermogravimetric analyses of LSCF6482 and BSCF6482. | |
Decomposition of both LSCF and BSCF shows an initial weight loss between 100–178 °C, which is attributed to the evaporation of the trapped water and are also reflected by the endothermic peaks at 119 °C and 139 °C in the DTA isotherm. The complex gel undergoes double decomposition to yield phase pure doped perovskite powder. However, the nature of decomposition is found to be dependent on the associated ligand/fuel. The decomposition of metal fuel composite is marked at 230 °C for BSCF6482 and at 269 °C for LSCF6482 (exothermic peaks) in the DTA with a mass loss in the regime of 170–250 °C. Double fuel in BSCF synthesis which is capable of forming comparatively stable six membered ring shows relatively sluggish weight loss pattern till the second stage. Loss of residual oxidizers (nitrates) and the fuels in the precursors marked the final stage in thermal analyses and is fast and sudden in BSCF6482 at 383 °C compared to at 415 °C for LSCF6482. The final mass loss continued from 350–600 °C after which the beginning of the crystallization as well as structural transformation initiates to form almost 100% phase pure product.
Influence of A-site substitution on the structural aspect of LSCF6482 and BSCF6482
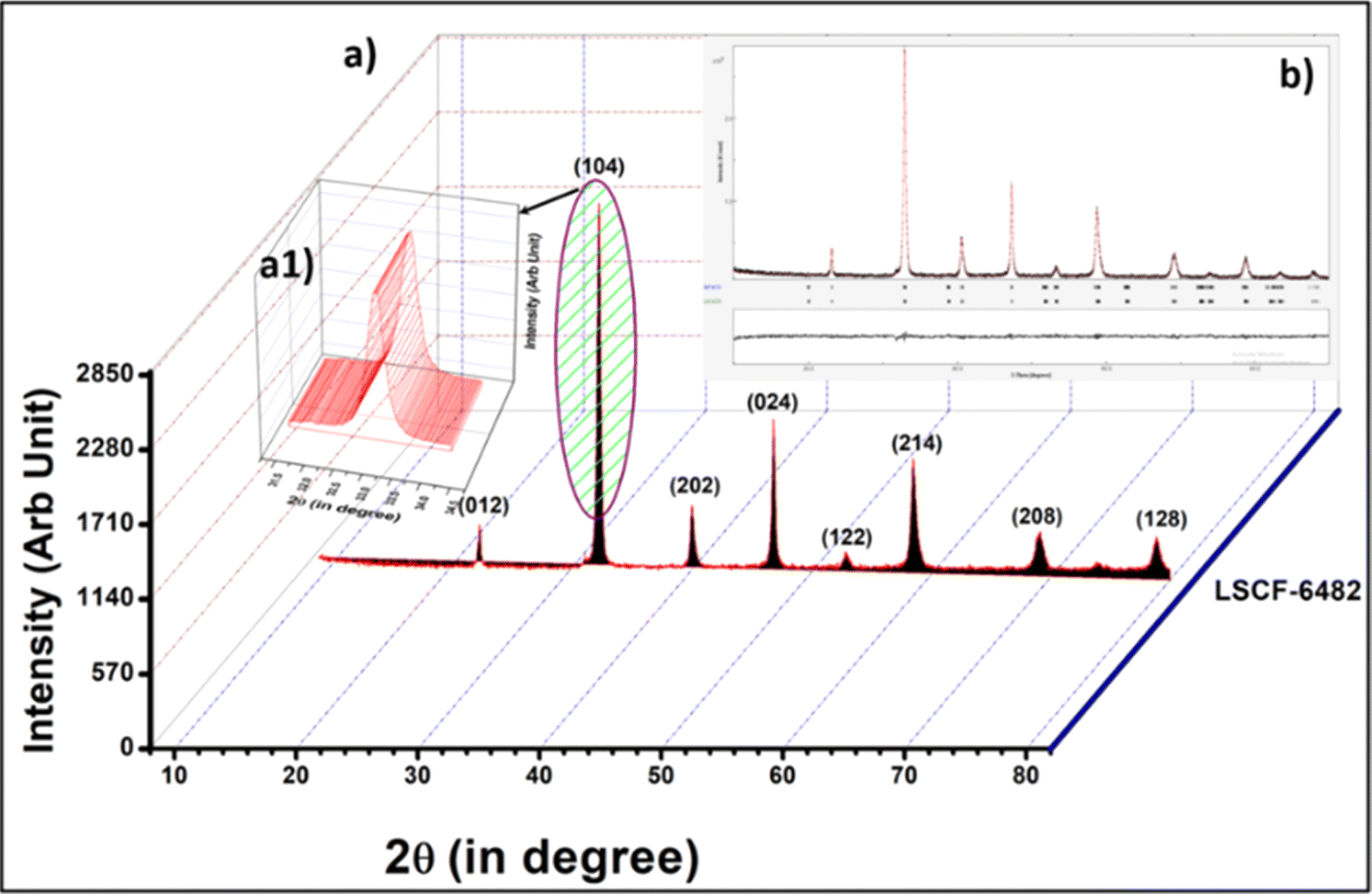
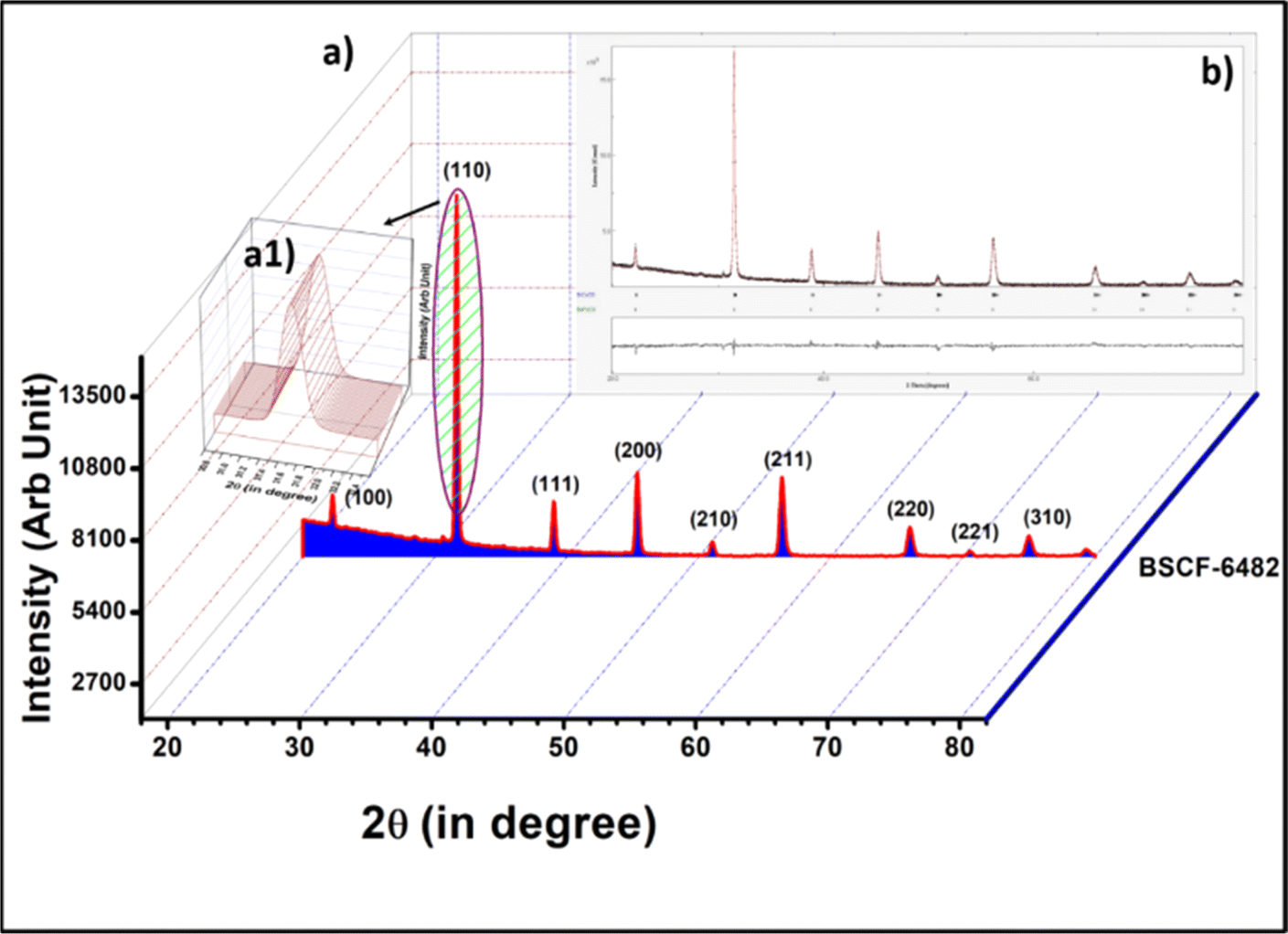
Phase purity of calcined LSCF6482 and BSCF6482 at 850 °C and 1000 °C for 4 h is shown in the X-ray diffraction analyses in Fig. 10a and 11a respectively. The diffraction peaks are indexed using ICDD-JCPDS database which establishes the pure crystalline rhombohedral phase for LSCF and cubic for BSCF sample (JCPDS NO.34-394 and 82-1961) and is also established from the parameters of Rietveld refinement as shown in Tables 5 and 6. The Rietveld refinement study is undertaken using MAUD as shown in Fig. 10b and 11b. A diffraction pattern is first simulated from a series of structural, microstructural (crystallite size and r.m.s (root mean square) macrostrain) constraints, characteristic features of the sample like preferred orientations, residual stress, sample thickness, absorption etc. as well as the background coefficients and profile parameters.
Table 5 Rietveld refinement parameters for LSCF6482a
| Sample |
a |
b |
c |
α |
β |
Γ |
| Volume fraction LaCoO3:SrFeO3 = 0.491:0.509. |
| LaCoO3 |
5.3375 |
5.3375 |
5.3375 |
61.37 |
61.37 |
61.37 |
| LSCF sample |
5.430(6) |
5.427(4) |
5.416(5) |
60.19 (6) |
60.21 (8) |
60.22 (7) |
| SrFeO3 |
5.414(10) |
5.423(11) |
5.448(15) |
60.36 (13) |
60.21 (27) |
60.06 (21) |
Table 6 Rietveld refinement parameters for BSCF6482a
| Sample |
a |
b |
C |
α |
β |
γ |
| Volume fraction BaFeO3:SrCoO3 = 0.293:0.707. |
| BaFeO3 primitive |
4.031 |
4.031 |
4.031 |
90 |
90 |
90 |
| SrCoO3 primitive created |
4.031 |
4.031 |
4.031 |
90 |
90 |
90 |
| BaFeO3 refined |
4.030 |
4.030 |
4.031 |
89.99 |
90 |
90 |
| SrCoO3 |
4.024 |
4.009 |
4.018 |
89.7 |
90.1 |
89.7 |
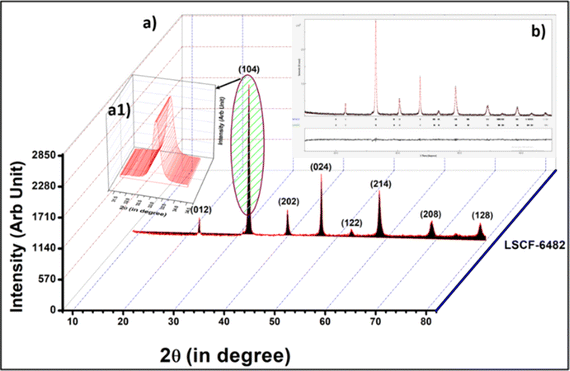 |
| | Fig. 10 (a) X-ray diffractogram of LSCF6482 along with the (a1) inset of peak with 100% peak intensity. (b) Rietveld refinement for XRD of LSCF6482. | |
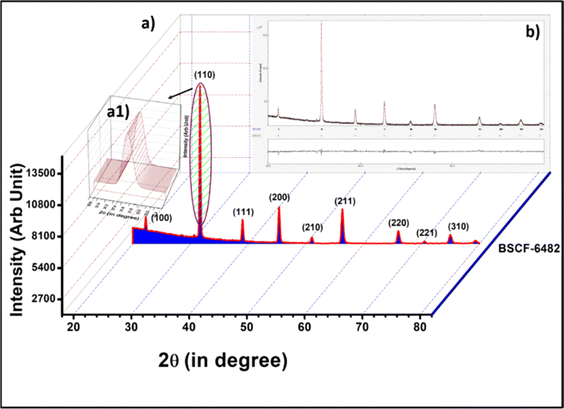 |
| | Fig. 11 (a) X-ray diffractogram of BSCF6482 along with the (a1) inset of peak with 100% peak intensity. (b) Rietveld refinement for XRD of BSCF6482. | |
This is followed by the application of iterative least-squares procedure using which the simulated pattern is compared with the experimentally observed X-ray powder diffraction profile. The technique involves minimization of the residual parameters. The refined unit cell parameters of the samples are listed in the Tables 5 and 6 along with the volume fraction of phases. It could be observed that there are no considerable changes in the output crystalline cell parameters for each sample.
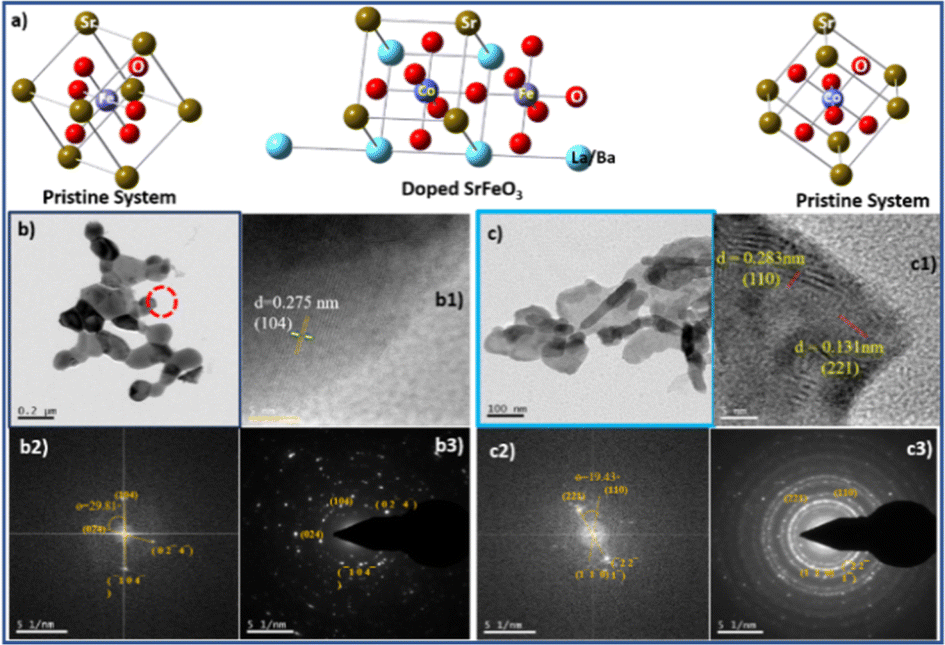
The calculated crystallite size for the 100% peaks for LSCF6482 (32.9° in Fig. 10a1) and BSCF6482(31.6° in Fig. 11a1) are 56 nm and 100 nm respectively using Scherer's equation. Further analyses on the unit cell parameters are reported in details through transmission electron micrography (Fig. 12). Mechanistic analyses for effectivity as oxygen reduction/evolution reaction kinetics by LSCF6482 and BSCF6482 is undertaken presently. Magnetic properties which are dependent on the electron exchange coefficient and are experimentally validated using activation barrier for non-linear polarizations during FC and EC operation. TEM analyses showed the primary nano particulates of size ∼30 nm and ∼50 nm for LSCF6482 and BSCF6482 to be embedded within the soft agglomerates of size in the range of 100–150 nm (Fig. 12b and c). Interplanar spacing (d) of 0.277 nm and 0.283 nm for the crystalline plane (104) and (110) calculated from high resolution TEM (Fig. 12b1 and c1) for LSCF6482 and BSCF6482 are in good agreement with the crystallographic findings. The corresponding SAED patterns (Fig. 12b3 and c3) also confirm the presence of (104) and (024) planes for LSCF6482; (110) and (221) planes for BSCF6482. The interpenetrating angles, calculated from the fast Fourier transformation (FFT) pattern of the two adjacent lattice planes of LSCF6482 and BSCF6482 are found to be 29.81° and 19.43° (Fig. 12b2 and c2). In this present investigation, single stoichiometric composition of LSCF and BSCF are reported to study the correlation among inherent spin polarization of the active electrocatalyst with selective OER and ORR performance. However, the upcoming communication would illustrate stoichiometric as well as non-stoichiometric composition to tailor such selectivity towards SOC performance and long-term sustainability. Hence, understanding of fundamental of quantum spin exchange interaction could enable appropriate selection of active catalyst for oxygen redox reaction in numerous applications. In addition, the spin polarization being established as an effective descriptor for ORR/OER, detailed analyses would be undertaken to correlate the same with model electrode systems reconstructed using FIB-SEM. The numerical simulation comprising linear and non-linear impedance study in terms of reconstructed air electrode would further establish the coherency of multiple atomic level descriptors.
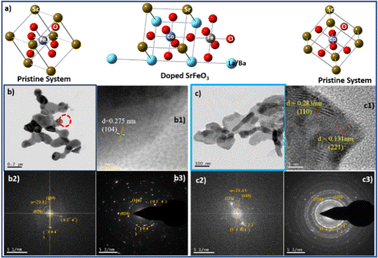 |
| | Fig. 12 (a) Unit cells (of pristine and doped perovskite oxides) generated from single point energy calculation using Gaussian, TEM micrographs [(b) (LSCF6482), (c) (BSCF6482)], high resolution -TEM images (b1 and c1), fast Fourier transform (FFT) pattern (b2 and c2) and SAED pattern (b3 and c3). | |
Conclusions
In this article, we tried to understand the mechanistic overview on redox reaction of oxygen for the functionality in Solid oxide cell (SOC). The prime bottleneck experienced by the researchers is the sluggish reaction rate during oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) which necessities the usage of efficient catalysts. ORR and OER involve spin exchange interaction which enable mixing of triple state of O2 with singlet state of other reactants (H2O, OH− etc.) thereby making it spin allowed process. The present research establishes that, inherent spin ordering within air electrode catalyst during FC and EC mode could act as an intrinsic descriptor (s) for controlling ORR and OER. Magnetic property shares similar origin from electron exchange interaction and is studied for the catalyst (for La0.6Sr0.4Co0.8Fe0.2O3−δ and Ba0.6Sr0.4Co0.8Fe0.2O3−δ) utilized in the present experiment. Magnetization for La0.6Sr0.4Co0.8Fe0.2O3−δ (LSCF6482) (15.86 emu g−1) is found to be ∼4.36 times higher compared to Ba0.6Sr0.4Co0.8Fe0.2O3−δ (BSCF6482) (3.64 emu g−1). Both the compositions show ordered magnetism throughout the operating temperature range wherein magnetic ordering in LSCF6482 is much improved than BSCF6482. Electrocatalysis at air electrode/electrolyte interface needs to be tailored with minimum polarization losses so as to increase the self-life of R-SOC (reversible SOC). In the present context, LSCF6482 with aliovalent substitution at A-site has been evaluated as an effective ORR catalyst with minimum energy barrier for interfacial polarization (21.5 kJ mol−1@0.8 V compared to 61 kJ mol−1 for ORR). In contrast, isovalent substitution at A-site in BSCF6482 shows improved OER kinetics (18.79 kJ mol−1@0.8 V for OER compared to 32.19 kJ mol−1 for ORR) throughout the operating temperature range of 700–800 °C. Heisenberg's theory clarifies the involvement of 2p orbital of active oxygen species from oxidant during interfacial sorption process for hybridization with 3d orbital of electrocatalyst. However, during subsequent dissociation and diffusion process, 2p orbital of oxygen from B-site of electrocatalyst governs the orbital overlap with 3d counterpart. Active catalysts with ferromagnetic ground states like LSCF6482 and BSCF6482 acts as effective electrocatalyst for oxygen redox reaction. The observations of this article strongly correlate spin ordering with OER/ORR mechanism along with selective functionality of LSCF6482 towards ORR and BSCF6482 for OER.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Shoroshi Dey methodology, validation, formal analysis, Investigation, data curation. Rajasekar Saravanan methodology, validation, investigation, data curation. Suprita Hati data curation and validation. Soumyabrata Goswami methodology, software (magnetic study), validation, formal analysis, data curation, writing – review & editing (magnetic study portion). Athira Suresh methodology (magnetic study). Deepshikha Jaiswal-Nagar methodology (magnetic study), investigation. Moupiya Ghosh software (magnetic study), Satadal Paul software (DFT), formal analysis. Abir Bhattacharyya methodology, software (XRD refining), formal analysis, data curation. Madhumita Mukhopadhyay conceptualization, software (impedance spectroscopy and fitting), methodology, validation, formal analysis, data curation, writing – review & editing, project administration, supervision. Jayanta Mukhopadhyay conceptualization, formal analysis, writing – review & editing, supervision, funding acquisition, project administration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
J. M. acknowledges Director CSIR-CGCRI for kind support. CSIR-H2T Mission program is acknowledged for financial and technical support. All the Institutions affiliated in this article are acknowledged.
Notes and references
- S. F. Miller and G. T. Babcock, Heme/Copper Terminal Oxidases, Chem. Rev., 1996, 96, 2889 CrossRef PubMed.
- A. Gupta, Y. Sang, C. Fontanesi and R. Naman, Effect of Anesthesia Gases on the Oxygen Reduction Reaction, J. Phys. Chem. Lett., 2023, 14, 1756 CrossRef CAS PubMed.
- J. L. Young, M. A. Steiner, H. Döscher, R. M. France, J. A. Turner and T. G. Deutsch, Direct solar-to-hydrogen conversion via inverted metamorphic multi-junction semiconductor architectures, Nat. Energy, 2017, 2, 17028 CrossRef CAS.
-
(a) J. Wei, P. Li, J. Shi, M. Huang and H. Wang, Improving the electron transfer in the oxygen reduction reaction by N/S co-doping for high-performance of Zn–air batteries, Sustain. Energy Fuels, 2022, 6, 3383 RSC;
(b) G. Yasin, S. Ali, S. Ibraheem, A. Kumar, M. Tabish, M. A. Mushtaq, S. Ajmal, M. Arif, M. A. Khan, A. Saad, L. Qiao and W. Zhao, Simultaneously Engineering the Synergistic-Effects and Coordination-Environment of Dual-Single-Atomic Iron/Cobalt-sites as a Bifunctional Oxygen Electrocatalyst for Rechargeable Zinc–Air Batteries, ACS Catal., 2023, 2313 CrossRef CAS;
(c) G. Yasin, S. Ibrahim, S. Ibraheem, S. Ali, R. Iqbal, A. Kumar, M. Tabish, Y. Slimani, T. A. Nguyen, H. Xu and W. Zhao, Defective/graphitic synergy in a heteroatom interlinked-triggered metal-free electrocatalyst for high-performance rechargeable zinc–air batteries, J. Mater. Chem. A, 2021, 9, 18222 RSC.
- W. Qi, X. Zang, J. Niu, Z. Zhang, J. Huang, S. Ge and Y. Zhang, Mechanism insight into the oxygen reduction reaction on dual FeN2 embedded graphene for proton exchange membrane fuel cells, Sustain. Energy Fuels, 2022, 6, 4024 RSC.
-
(a) W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Lattice-strained metal–organic-framework arrays for bifunctional oxygen electrocatalysis, Nat. Energy, 2019, 4, 115 CrossRef CAS;
(b) R. Iqbal, S. Ali, A. Saleem, M. K. Majeed, A. Hussain, S. Rauf, A. R. Akbar, H. Xu, L. Qiao and W. Zhao, Electrically conductive Pt-MOFs for acidic oxygen reduction: Optimized performance via altering conjugated ligands, Chem. Eng. J., 2023, 455, 140799 CrossRef CAS.
- H. Koshikawa, H. Murase, T. Hayashi, K. Nakajima, H. Mashiko, S. Shiraishi and Y. o. Tsuji, Single nanometer-sized NiFe-layered double hydroxides as anode catalyst in anion exchange membrane water electrolysis cell with energy conversion efficiency of 74.7% at 1.0 A cm2, ACS Catal., 2020, 10, 1886 CrossRef CAS.
- S. Chretien and H. Metiu, O2 evolution on a clean partially reduced rutile TiO2 (110) surface and on the same surface precovered with Au1 and Au2: the importance of spin conservation, J. Chem. Phys., 2008, 129, 074705 CrossRef.
-
(a) X. Ding, M. Li, X. Zhao, L. Ding, Y. Yan, L. Wang and Z. Wang, A highly active and stable cathode for oxygen reduction in intermediate-temperature solid oxide fuel cells, Sustain. Energy Fuels, 2020, 4, 1168 RSC;
(b) S. U. Rehman, J. Wang, G. Wu, S. Ali, J. Xian and N. Mahmood, Unravelling the photocatalytic potential of transition metal sulfide and selenide monolayers for overall water splitting and photo-corrosion inhibition, J. Mater. Chem. A, 2024, 12, 5693 Search PubMed.
-
(a) J. Liu, H. Liu, H. Chen, X. Du, B. Zhang, Z. Hong, S. Sun and W. Wang, Progress and Challenges Toward the Rational Design of Oxygen Electrocatalysts Based on a Descriptor Approach, Adv.
Sci., 2020, 7, 1901614 CrossRef CAS PubMed;
(b) G. Yasin, S. Ibraheem, S. Ali, M. Arif, S. Ibrahim, R. Iqbal, A. Kumar, M. Tabish, M. A. Mushtaq, A. Saad, H. Xu and W. Zhao, Defects-engineered tailoring of tri-doped interlinked metal-free bifunctional catalyst with lower Gibbs free energy of OER/HER intermediates for overall water splitting, Mater. Today Chem., 2022, 23, 100634 CrossRef CAS;
(c) A. B. Musa, M. Tabish, A. Kumar, M. Selvaraj, M. A. Khan, B. M. Al-Shehri, M. Arif, M. A. Mushtaq, S. Ibraheem, Y. Slimani, S. Ajmal, T. A. Nguyen and G. Yasin, Microenvironment engineering of Fe-single-atomic-site with nitrogen coordination anchored on carbon nanotubes for boosting oxygen electrocatalysis in alkaline and acidic media, Chem. Eng. J., 2023, 451, 138684 CrossRef.
-
(a) J. Gracia, Itinerant spins and bond lengths in oxide electrocatalysts for oxygen evolution and reduction reactions, J. Phys. Chem. C, 2019, 123, 9967 CrossRef CAS;
(b) J. Gracia, R. Sharpe and J. Munarriz, Principles determining the activity of magnetic oxides for electron transfer reactions, J. Catal., 2018, 331 CrossRef CAS;
(c) J. Gracia, Spin dependent interactions catalyse the oxygen electrochemistry, Phys. Chem. Chem. Phys., 2017, 19, 20451 RSC.
- X. Ren, T. Wu, Y. Sun, Y. Li, G. Xian, X. Liu, C. Shen, J. Gracia, H. J. Gao, H. Yang and Z. J. Xu, Spin-polarized oxygen evolution reaction under magnetic field, Nat. Commun., 2021, 12, 2608 CrossRef CAS PubMed.
- Y. Su, Z. Wang, R. Gao, Q. Wu, J. Zhao, G. Zhu, Q. Li, H. Xu, Y. Pan, K. Gu, C. Biz, M. Fianchini and J. Gracia, Ferromagnetic L12-Pt3Co Nanowires with Spin-Polarized Orbitals for Fast and Selective Oxygen Reduction Electrocatalysis, Adv. Funct. Mater., 2024, 34, 2311618 CrossRef CAS.
- Z. Yang, M. Guo, N. Wang, C. Ma, J. Wang and M. Han, A short review of cathode poisoning and corrosion in solid oxide fuel cell, Int. J. Hydrogen Energy, 2017, 42, 24948 CrossRef CAS.
- A. Nechache and S. Hody, Alternative and innovative solid oxide electrolysis cell materials: A short review, Renew. Sustain. Energy Rev., 2021, 149, 111322 CrossRef CAS.
- B. Yang, Y. Li, J. Li, H. Shu, X. Zhao, Y. Ren and Q. Li, Comprehensive summary of solid oxide fuel cell control: a state-of-the-art review, Prot. Control Mod. Power Syst., 2022, 7, 22 CrossRef.
-
(a) S. Dey, S. Chaudhary, D. Parvatalu, M. Mukhopadhyay, A. Das Sharma and J. Mukhopadhyay, Advancing electrode properties through functionalization for Solid Oxide Cells application: A Review, Chem.–Asian J., 2023, e202201222 CrossRef CAS;
(b) M. Mukhopadhyay, J. Mukhopadhyay, A. Das Sharma and R. N. Basu, In-situ patterned intra-anode triple phase boundary in SOFC electroless anode: An enhancement of electrochemical performance, Int. J. Hydrogen Energy, 2011, 36, 7677 CrossRef CAS.
- E. D Wachsman and S. C. Singhal, Solid Oxide Fuel Cell Commercialization, Research, and Challenges, Interface, 2009, 18, 38 Search PubMed.
- C. E. Beall, E. Fabbri and T. J. Schmidt, Perovskite Oxide Based Electrodes for the Oxygen Reduction and Evolution Reactions: The Underlying Mechanism, ACS Catal., 2021, 11, 3094 CrossRef CAS.
- X. Li, Z. Cheng and X. Wang, Understanding the Mechanism of the Oxygen Evolution Reaction with Consideration of Spin, Electrochem. Energy Rev., 2021, 4, 136 CrossRef CAS.
- S. Zhao, C. Tan, C.-T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Structural transformation of highly active metal–organic framework electrocatalysts during the oxygen evolution reaction, Nat. Energy, 2020, 5, 881 CrossRef CAS.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts, Nat. Energy, 2019, 4, 329 CrossRef CAS.
- C. Chen, H. Jin, P. Wang, X. Sun, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Local reaction environment in electrocatalysis, Chem. Soc. Rev., 2024, 53, 2022 RSC.
- J. Liu, H. Liu, H. Chen, X. Du, B. Zhang, Z. Hong, S. Sun and W. Wang, Progress and Challenges Toward the Rational Design of Oxygen Electrocatalysts Based on a Descriptor Approach, Adv. Sci., 2020, 7, 1901614 CrossRef CAS PubMed.
- D. Guan, J. Zhou, Y. C. Huang, C. L. Dong, J. Q. Wang, W. Zhou and Z. Shao, Screening highly active perovskites for hydrogen-evolving reaction via unifying ionic electronegativity descriptor, Nat. Commun., 2019, 10, 3755 CrossRef.
- R. Chattot, O. L. Bacq, V. Beermann, S. Kühl, J. Herranz, S. Henning, L. Kühn, T. Asset, L. Guétaz, G. Renou, J. Drnec, P. Bordet, A. P. A. Eychmüller, T. J. Schmidt, P. Strasser, L. Dubau and F. Maillard, Surface distortion as a unifying concept and descriptor in oxygen reduction reaction electrocatalysis, Nat. Mater., 2018, 17, 827 CrossRef CAS PubMed.
-
(a) Y. Duan, S. Sun, Y. Sun, S. Xi, X. Chi, Q. Zhang, X. Ren, J. Wang, S. J. H. Ong, Y. Du, L. Gu, A. Grimaud and Z. J. Xu, Mastering Surface Reconstruction of Metastable Spinel Oxides for Better Water Oxidation, Adv. Mater., 2019, 31, 1807898 CrossRef;
(b) S. Ajmal, A. Kumar, M. Tabish, M. Selvaraj, M. M. Alam, M. A. Mushtaq, J. Zhao, K. A. Owusu, A. Saad, M. T. Nazir and G. Yasin, Modulating the microenvironment of single atom catalysts with tailored activity to benchmark the CO2 reduction, Mater. Today, 2023, 67, 203 CrossRef CAS;
(c) G. Yasin, S. Ibrahim, S. Ajmal, S. Ibraheem, S. Ali, A. K. Nadda, G. Zhang, J. Kaur, T. Maiyalagan, R. K. Gupta and A. Kumar, Tailoring of electrocatalyst interactions at interfacial level to benchmark the oxygen reduction reaction, Coord. Chem. Rev., 2022, 469, 214669 CrossRef CAS.
- N. H. Perry and T. Ishihara, Roles of Bulk and Surface Chemistry in the Oxygen Exchange Kinetics and Related Properties of Mixed Conducting Perovskite Oxide Electrodes, Materials, 2016, 9, 858 CrossRef.
- Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Spin-State Regulation of Perovskite Cobaltite to Realize Enhanced Oxygen Evolution Activity, Chem, 2017, 3, 812 CAS.
- A. Samreen, M. S. Ali, M. Huzaifa, N. Ali, B. Hassan, F. Ullah, S. Ali and N. A. Arifin, Advancements in Perovskite-Based Cathode Materials for Solid Oxide Fuel Cells: A Comprehensive Review, Chem. Rec., 2023, 24, e202300247 CrossRef.
- S. Dey, S. Das, S. Chaudhary, D. Parvatalu, M. Mukhopadhyay, S. Paul, A. Das Sharma and J. Mukhopadhyay, Advancing Insights towards Electrocatalytic Activity of La/Ba–Sr–Co–Fe–O-based Perovskites for Oxygen Reduction & Evolution Process in Reversible Solid Oxide Cell, Scr. Mater., 2023, 229(1–6), 115380 CrossRef CAS.
- X. Rong, J. Parolin and A. M. Kolpak, A Fundamental Relationship between Reaction Mechanism and Stability in Metal Oxide Catalysts for Oxygen Evolution, ACS Catal., 2016, 6, 1153 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, Role of lattice oxygen participation in understanding trends in the oxygen evolution reaction on perovskites, ACS Catal., 2018, 8, 4628 CrossRef CAS.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides, Energy Environ. Sci., 2017, 10, 2190 RSC.
- J. B. Goodenough, An interpretation of the magnetic properties of the perovskite-type mixed crystals La1−xSrxCoO3−λ, J. Phys. Chem. Solid., 1958, 6, 287 CrossRef CAS.
- W. Araki, Y. Arai and J. Malzbender, Transitions of Ba0.5Sr0.5Co0.8Fe0.2O3−δ and La0.58Sr0.4Co0.2Fe0.8O3−δ, Mater. Lett., 2014, 132, 295 CrossRef CAS.
- F. A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López and J. R. Galán-Mascarós, Direct magnetic enhancement of electrocatalytic water oxidation in alkaline media, Nat. Energy, 2019, 4, 519 CrossRef.
- S. Dey, A. Das Sharma and J. Mukhopadhyay, Effect of oxygen non-stoichiometry and redox phenomena in La/Ba–Sr–Co–Fe–O-based perovskite systems and its heterostructure as applicable in Solid Oxide Cell (SOC) air electrode, Ceram. Int., 2022, 48, 35799 CrossRef CAS.
- S. Dey, J. Mukhopadhyay, R. K. Lenka, P. K. Patro, A. Das Sharma, T. Mahata and R. N. Basu, Synthesis and characterization of Nanocrystalline Ba0·6Sr0·4Co0·8Fe0·2O3 for application as an efficient anode in solid oxide electrolyser cell, Int. J. Hydrogen Energy, 2020, 45, 3995 CrossRef CAS.
-
(a) A. Dutta, A. Kumar and R. N. Basu, Enhanced electrical conductivity in Ce0.79Gd0.20Co0.01O2−δ for low temperature solid oxide fuel cell applications, Electrochem. Commun., 2009, 11, 699–701 CrossRef CAS;
(b) K. B. Ghosh, J. Mukhopadhyay and R. N. Basu, functionally graded doped lanthanum cobalt ferrite and ceria-based composite interlayers for advancing the performance stability in solid oxide fuel cell, J. Power Sources, 2016, 328, 15 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- W. J. Stevens, H. Basch and M. Krauss, Compact effective potentials and efficient shared-exponent basis-sets for the 1st-row and 2nd-row atoms, J. Chem. Phys., 1984, 81, 6026 CrossRef.
- W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Relativistic compact effective potentials and efficient, shared-exponent basis-sets for the 3rd-row, 4th-row, and 5th-row atoms, Can. J. Chem., 1992, 70, 612 CrossRef CAS.
- T. R. Cundari and W. J. Stevens, Effective core potential methods for the lanthanides, J. Chem. Phys., 1993, 98, 5555 CrossRef CAS.
-
(a) Z. J. Wu, J. S. Shi, S. Y. Zhang and H. J. Zhang, Density-functional study of lanthanum, ytterbium, and lutetium dimers, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 064502 CrossRef;
(b) S. Dey, D. Bose, Y. Akinay, M. Mukhopadhyay, A. Das Sharma and J. Mukhopadhyay, Surface Modification and Functionalization of Ceramic Composites, Elsevier Series in Advanced Ceramic Materials, 2023, pp. 255–288, ISBN 9780323858830 Search PubMed.
- X. Li, Y. Bai and Z. Cheng, Revealing the Correlation of OER with Magnetism: A New Descriptor of Curie/Neel Temperature for Magnetic Electrocatalysts, Adv. Sci., 2021, 8, 2101000 CrossRef CAS.
- N. Mushtaq, Y. Lu, C. Xia, W. Dong, B. Wang, X. Wang, M. A. K. Y. Shah, S. Rauf, M. Akbar, E. Hu, R. Raza, M. I. Asghar, P. D. Lund and B. Zhu, Appl. Catal., B, 2021, 298, 120503 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c,
Athira Sureshd,
Deepshikha Jaiswal-Nagar
*c,
Athira Sureshd,
Deepshikha Jaiswal-Nagar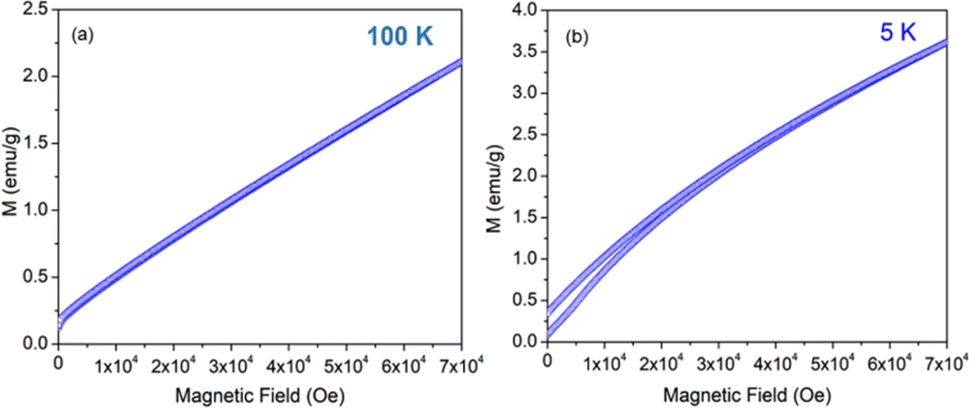
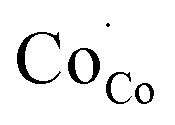 and
and 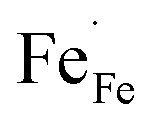 eases the path for the transformation of O2− to O2 during OER. The charge transfer process through aliovalency in LSCF6482 is highly accelerated owing to the presence of higher population density of Co3+ ion which undergoes easy spin state equilibrium shown in Fig. S1.† It could be seen that, the easy spin cross over of Co3+ at even thermal energy results in magnetism owing to the non-zero spin state of ½ and 1 respectively. Similar fact is restored in BSCF, however, due to lesser population density of Co3+ ion, charge compensation route is overshadowed and governed by oxygen vacancy propagation route. This is also responsible for the higher magnetic ordering observed in the matrix of LSCF6482 as shown in Fig. 1–4. Similar population of Co3+ and Co4+ in LSCF 6482 with almost comparable consecutive binding energy (Fig. 6) accelerates ORR through aliovalent charge transfer and restricts the usage of lattice oxygen. Fig. 6 exhibits the distribution of population density for variable oxidation states of B-site ions viz Co and Fe respectively. As already stated, the B-site ions primarily act as the active sites for dissociative adsorption of oxidant followed by catalyzing the redox reaction of oxygen. Doping of isovalent (Ba2+) and/aliovalent (La3+) ion is found to influence the variability in population density of Co (+2 to +4) and Fe (+2 to +4), which alters the specificity in electro kinetics towards OER/ORR. In addition, the quantitative binding energies are plotted as a function of oxidation state. Irrespective of type of ions, the consecutive binding energies are found to follow the similar trend.
eases the path for the transformation of O2− to O2 during OER. The charge transfer process through aliovalency in LSCF6482 is highly accelerated owing to the presence of higher population density of Co3+ ion which undergoes easy spin state equilibrium shown in Fig. S1.† It could be seen that, the easy spin cross over of Co3+ at even thermal energy results in magnetism owing to the non-zero spin state of ½ and 1 respectively. Similar fact is restored in BSCF, however, due to lesser population density of Co3+ ion, charge compensation route is overshadowed and governed by oxygen vacancy propagation route. This is also responsible for the higher magnetic ordering observed in the matrix of LSCF6482 as shown in Fig. 1–4. Similar population of Co3+ and Co4+ in LSCF 6482 with almost comparable consecutive binding energy (Fig. 6) accelerates ORR through aliovalent charge transfer and restricts the usage of lattice oxygen. Fig. 6 exhibits the distribution of population density for variable oxidation states of B-site ions viz Co and Fe respectively. As already stated, the B-site ions primarily act as the active sites for dissociative adsorption of oxidant followed by catalyzing the redox reaction of oxygen. Doping of isovalent (Ba2+) and/aliovalent (La3+) ion is found to influence the variability in population density of Co (+2 to +4) and Fe (+2 to +4), which alters the specificity in electro kinetics towards OER/ORR. In addition, the quantitative binding energies are plotted as a function of oxidation state. Irrespective of type of ions, the consecutive binding energies are found to follow the similar trend.
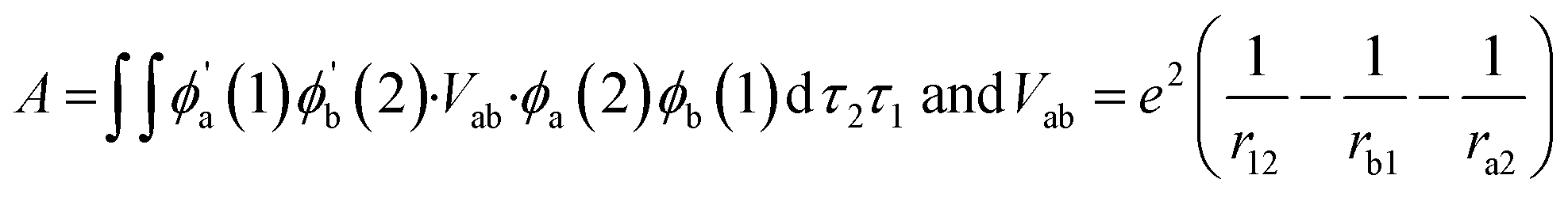
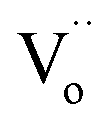 through the formation of
through the formation of 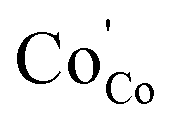 . In a similar manner, strong hybridization involving lattice oxygen promotes the formation of
. In a similar manner, strong hybridization involving lattice oxygen promotes the formation of 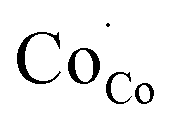 and
and 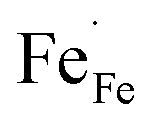 which is responible for oxidation of O2− to O2 in BSCF6482. This lowers the Rp towards OER for BSCF6482 irrespective of operative temperature. It is an established fact that, strongly coupled intrinsic parameters like orbital, spin, charge and lattice acts as descriptors for ORR and/OER processes. In a similar manner, magnetic property like ordering share similar origin from electron exchange interaction and is capable to reveal the mechanistic understanding of electrocatalysis process like OER and ORR. Li et al.46 also proposed the utilization of Curie/Néel temperature as an active oxygen redox process descriptor in the active catalysts with ferro/ferri/antiferromagnetic ground states. Hence, the present functional electrodes (BSCF6482/LSCF6482) which are in function for several years as electrocatalyst could be automatically possess excellent performance as magnetic electrocatalyst as well. In this connection, the future research would share an insight on acceleration and sustenance of OER activity of BSCF6482 and ORR performance of LSCF6482 in presence of tailored magnetic field.
which is responible for oxidation of O2− to O2 in BSCF6482. This lowers the Rp towards OER for BSCF6482 irrespective of operative temperature. It is an established fact that, strongly coupled intrinsic parameters like orbital, spin, charge and lattice acts as descriptors for ORR and/OER processes. In a similar manner, magnetic property like ordering share similar origin from electron exchange interaction and is capable to reveal the mechanistic understanding of electrocatalysis process like OER and ORR. Li et al.46 also proposed the utilization of Curie/Néel temperature as an active oxygen redox process descriptor in the active catalysts with ferro/ferri/antiferromagnetic ground states. Hence, the present functional electrodes (BSCF6482/LSCF6482) which are in function for several years as electrocatalyst could be automatically possess excellent performance as magnetic electrocatalyst as well. In this connection, the future research would share an insight on acceleration and sustenance of OER activity of BSCF6482 and ORR performance of LSCF6482 in presence of tailored magnetic field.