
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrapping of thermally generated ortho- and para-quinone methides by imidazoles and pyrazoles: a simple route to green synthesis of benzopyrone-azole hybrids and their evaluation as α-glucosidase inhibitors†
Andrii S. Myshko ab,
Galyna P. Mruga,
Svitlana P. Bondarenkoc,
Kostyantyn M. Kondratyuka,
Oleksandr L. Kobzara,
Vladyslav M. Buldenkoa,
Andriy V. Kozytskiybd,
Andriy I. Vovka and
Mykhaylo S. Frasinyuk*ab
ab,
Galyna P. Mruga,
Svitlana P. Bondarenkoc,
Kostyantyn M. Kondratyuka,
Oleksandr L. Kobzara,
Vladyslav M. Buldenkoa,
Andriy V. Kozytskiybd,
Andriy I. Vovka and
Mykhaylo S. Frasinyuk*ab
aV. P. Kukhar Institute of Bioorganic Chemistry and Petrochemistry, NAS of Ukraine, 1 Academician Kukhar Str., Kyiv 02094, Ukraine. E-mail: mykhaylo.frasinyuk@ukr.net
bEnamine Ltd., 78 Winston Churchill Str., Kyiv 02094, Ukraine
cNational University of Food Technologies, Kyiv 01601, Ukraine
dChemBioCenter, Taras Shevchenko National University of Kyiv, 64 Volodymyrska Str., Kyiv 01601, Ukraine
First published on 2nd September 2024
Abstract
An efficient green approach for the trapping of in situ generated ortho-and para-quinone methide intermediates by imidazoles and pyrazoles has been developed. A wide range of quinone methide precursors based on simple phenols are compatible with the experimental protocol under mild thermal conditions. This methodology was demonstrated to be suitable for the synthesis of methylene-linked benzopyrone-azole hybrids using naturally occurring coumarin and chromone Mannich bases. In most cases, the products were isolated in good to excellent yields without chromatographic purification. In vitro studies showed that some of the synthesized compounds exhibit inhibitory activity towards α-glucosidase.
Introduction
Chemical properties of five-membered nitrogen-containing heterocycles with two or more heteroatoms commonly named “azoles” depend on the number, nature, and positions of heteroatoms in their skeleton. Among them, imidazoles and pyrazoles with isomeric ring systems containing two nitrogen atoms show the most distinct amphoteric properties, which make it possible to afford a wide range of derivatives using various reactions. The importance of these aromatic ring systems is reflected by their presence in naturally occurring histidine, histamine, purines, and several classes of pharmaceuticals. Recent advances in the synthesis and biological activity of compounds based on these heterocyclic cores are discussed in chapters of a monography1 and a book2 as well as references cited herein.A common and widely used method for the N-alkylation of azoles includes the action of alkyl halides, pseudo halides, or sulfates with azole metal salts or azoles in the presence of a strong base. However, applying these procedures for the synthesis of target compounds bearing phenolic fragments requires the protection of at least the phenolic groups.
On the other hand, the reaction of phenols, azoles, and formaldehyde can be considered as a Mannich reaction whereas azoles play the role of the amino component. Probably due to low nucleophilicity of azoles, the application of only imidazole with simple phenols was reported in the literature,3 and other azoles could be involved in the reaction using 2-naphthol as a very active CH-component.4 However, direct imidazolylmethylation of simple phenols has limitations due to the possible formation of bis-imidazolylmethyl phenols3d or the formation of methylene-bis-phenols.3c
Natural compounds have played a significant role as sources of new drugs over nearly four decades.5 Among them, flavonoids and coumarins are recognized as privileged scaffolds in medicinal chemistry.6 In the framework of benzopyrone-azole hybrids, imidazolylmethylflavones were identified as aromatase inhibitors,7 inhibitors of corticosteroid biosynthesis,8 and antiproliferative agents,9 whereas various 2-azolylmethylchromones were studied as kinase modulators.10 Their synthetic protocols were developed by using the intermediate halomethyl derivatives of benzopyrones and subsequent reaction with azoles. Protection of phenolic groups was required for the obtaining of target hydroxylated derivatives.
Quinone methides with general structures 1 (Fig. 1) are highly polarized and hence reactive under nucleophilic and electrophilic conditions.
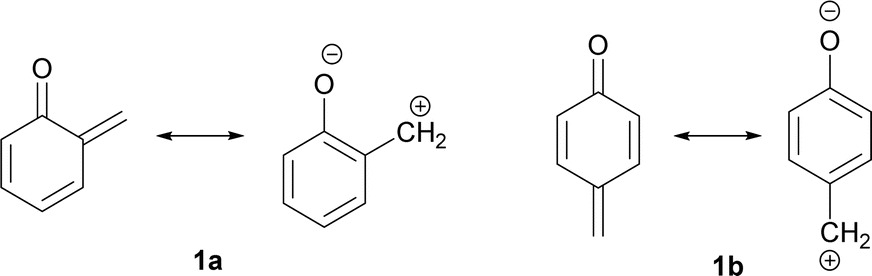 | ||
| Fig. 1 Structural features of ortho- and para-quinone methides. | ||
A few attempts were reported for the synthesis of imidazolylmethylphenols via Michael's addition of imidazole to in situ-generated quinone methides. This route was used for the synthesis of 1H-5-hydroxybenzimidazole,11 6-hydroxyquinoline,12 and 5-hydroxyindole13 derivatives with anti-hepatitis activity, as well as for modification of camptothecin14 for the design of topoisomerase I inhibitors (Fig. 2).
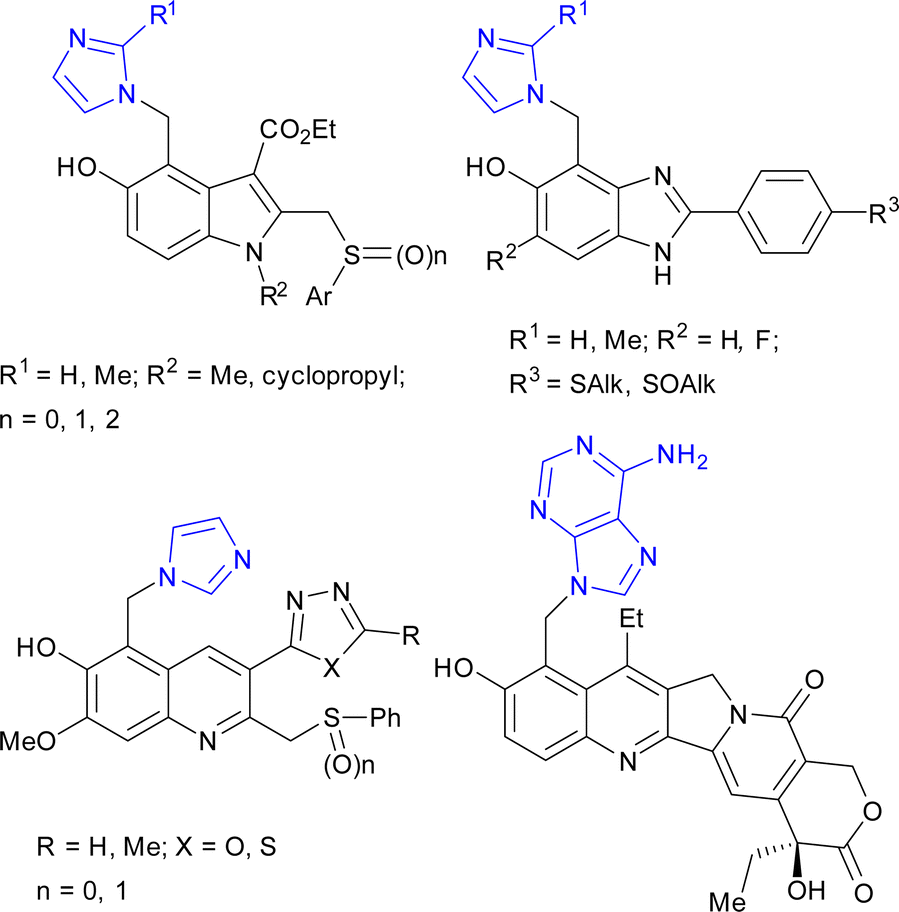 | ||
| Fig. 2 Biologically active compounds synthesized by Michael's addition of imidazoles/adenine to in situ-generated quinone methides. | ||
Considering the obvious multiple advantages of applying quinone methides in the context of the development of synthetic procedures, we embarked on a study of the feasibility of these intermediates for the conjugation with azaheterocycles in environmentally benign conditions. Herein, we report the realization of this goal; specifically, we describe using dimethyl carbonate for the generation of ortho- and para-quinone methides followed by their trapping by azoles. This protocol proved to be effective for using different quinone methide precursors based on simple phenols and naturally occurring benzopyrones.
Results and discussion
Synthetic and commercial availability of quinone methide precursors could facilitate the development of the conjugation protocols of phenols and azoles. Using them has some lacks and advantages and choosing each should be appreciated in a strategy of the synthesis. As ortho-quinone methide precursors, ortho-dimethylaminomethylphenols are easily accessible using Mannich reaction contrary to para-dimethylaminophenols which can be synthesized by reductive amination often with poor yield. Both precursors such as 2-hydroxymethylphenols and 4-hydroxymethylphenols can be synthesized by the reduction of numerous commercially available substituted hydroxylated aldehydes, but their use for the synthesis of azolylmethylphenols requires high temperatures.15 Moreover, using 2- or 4-methoxymethylphenols requires additional steps for their synthesis, but has an advantage in the case of catechols16 or the synthesis derivatives from natural phenolic compounds bearing methoxymethyl group.17As a model reaction for the construction of benzopyrone-azole hybrids, we chose a reaction of ortho- and para-substituted phenols bearing dimethylaminomethyl, hydroxymethyl, or methoxymethyl groups 2a–2c and 3a–3c as possible precursors18 for generation of quinone methides with subsequent trapping them with imidazole, pyrazole, and related benzimidazole or 3,5-dimethylpyrazole (as analogs with low nucleophilicity). However, the reported procedure for the capture by azoles of quinone methides in aqueous conditions19 was not applicable for benzopyrone derivatives due to their poor solubility.
In the first stage, to determine the role of solvents and temperature, we investigated the reaction of ortho-substituted phenol 2a–2c with imidazole at various conditions (Table 1). According to these data, dimethyl carbonate (DMC) was chosen as the most acceptable solvent for further development of phenol–azole conjugate synthesis. However, these data were insufficient for elucidation of the most useful leaving group (LG) for the reaction with other azoles, especially in cases of using naturally occurring compound derivatives. Our next experiments were focused on further optimization of the reaction conditions of compounds 2 and 3 with some azoles.
|
||||
|---|---|---|---|---|
| Compd. (LG) | Solvent | Temp., C | Conversionb, % | Purityc, % |
| a A mixture of quinone methide precursors 2a–2c (2 mmol) and imidazole (4 mmol) in an appropriate solvent (5 mL) was refluxed for 24 h.b Was determinate by LCMS spectra.c Calculated by LCMS spectra excluding intact reagents. | ||||
| 2a (NMe2) | EtOH | 80 | n.r. | — |
| 2a (NMe2) | (MeO)2CO | 90 | 83 | 85 |
| 2a (NMe2) | 1,4-Dioxane | 100 | 53 | 78 |
| 2a (NMe2) | Toluene | 110 | 50 | 85 |
| 2a (NMe2) | (EtO)2CO | 126 | 78 | 82 |
| 2a (NMe2) | DMF | 154 | 72 | 78 |
| 2b (OH) | EtOH | 80 | n.r. | — |
| 2b (OH) | (MeO)2CO | 90 | 43 | 91 |
| 2b (OH) | 1,4-Dioxane | 100 | 39 | 89 |
| 2b (OH) | Toluene | 110 | 35 | 90 |
| 2b (OH) | (EtO)2CO | 126 | 38 | 93 |
| 2b (OH) | DMF | 154 | 53 | 86 |
| 2c (OMe) | EtOH | 80 | n.r. | — |
| 2c (OMe) | (MeO)2CO | 90 | 73 | 92 |
| 2c (OMe) | 1,4-Dioxane | 100 | 45 | 83 |
| 2c (OMe) | Toluene | 110 | 48 | 85 |
| 2c (OMe) | (EtO)2CO | 126 | 69 | 90 |
| 2c (OMe) | DMF | 154 | 67 | 75 |
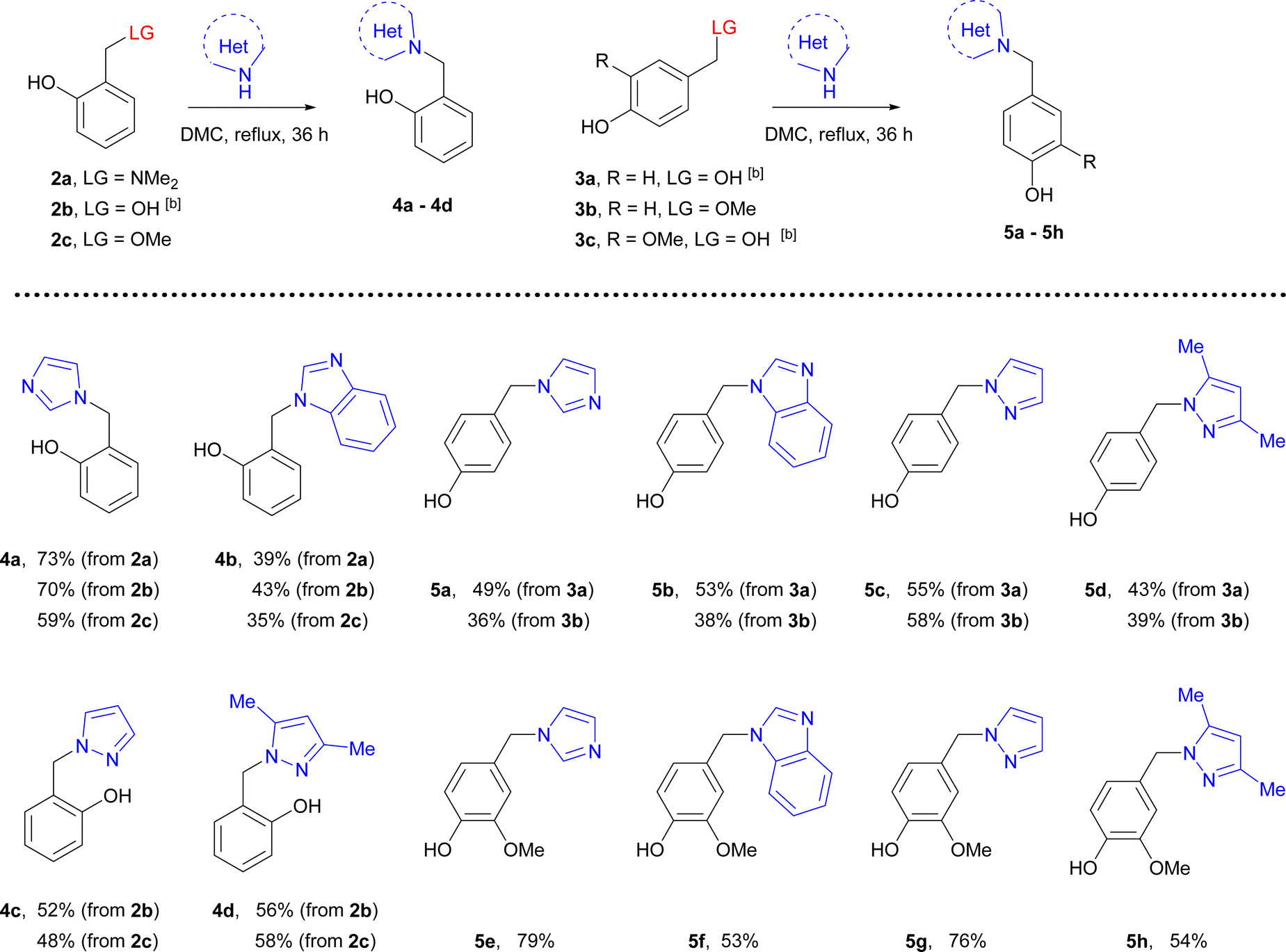
Phenol-methylene azoles 4 and 5 were synthesized by refluxing compounds 2a–2c and 3a–3c with [benz]imidazole and pyrazoles in dimethyl carbonate for 36 h.
Using 2-dimethylaminomethylphenole 2a was successful for the synthesis of imidazoles 4a, 5a and benzimidazoles 4b, 5b. In the cases of 2- and 4-hydroxybenzylphenols 2b, 3a, and 3c, the target hybrids were synthesized with acceptable to good yield with all azoles. However, the addition of acids such as trifluoroacetic or methane sulfonic was needed to complete the reactions. It should be noted that the reaction of 2-hydroxymethylphenol with imidazole requires high temperature.15a The reactions of imidazole or benzimidazole with 2-methoxymethylphenol 2c and 4-methoxymethylphenol 3b were completed in 24 h whereas conversion of these methoxymethyl derivatives with pyrazole or 3,5-dimethylpyrazole was approximately 40% and was not completed after 72 h.
Thus, imidazole derivatives 4a, 5a, and 5e can be synthesized from all precursors with poor to good yield, whereas applying hydroxymethylphenols 2b, 3a, and 3c was more suitable for the synthesis of benzimidazole derivatives 4b, 5b, and 5f. The more effective conditions for the synthesis of related pyrazole and 3,5-dimethylpyrazole derivatives were achieved using hydroxymethyl phenols 2b, 3a, and 3c in the presence of acids (Table 2).
Our findings were extended to the application of [benz]imidazole and pyrazoles for the synthesis of azole-phenol hybrids bearing a coumarin or chromone fragment, as attractive compounds which represented naturally occurring bioactive phenols. Thus, umbelliferone (7-hydroxycoumarin, 6a), 4-methylumbelliferone (6b), 2,3-dimethylchromone (6c), soybean isoflavonoids daidzein (7,4′-dihydroxyisoflavone, 6d), formononetin (7-hydroxy-4′-methoxyisoflavone, 6e), cladrin (7-hydroxy-3′,4′-dimethoxyisoflavone, 6f), pseudobaptigenin (7-hydroxy-3′,4-methylenedioxyisoflavone, 6g), 2-methylformononetin (6h), their fluorinated derivatives 2-trifluoroformononetin (6i) and 7-hydroxy-4′-trifluoromethoxyisoflavone (6j) were used for the synthesis of hybrids with the mentioned azoles.
Taking into account that the efficiency for the generation of ortho-quinone methides 7a–7j with fused pyrone ring from complex compounds can be significantly different from simple phenol derivatives 2 or 3, we carried out the reaction of 8-substituted formononetin derivatives with imidazole. As a result, target formononetin-imidazole hybrid 9e was isolated from reaction imidazole with 8-dimethylaminomethylformononetin 8e, (82% yield, 100% purity), 8-hydroxymetylformononetin20 (78% yield, 95% purity), and 8-methoxymethylformononetin20 (84% yield, 95% purity). According to these data and synthetic availability of 7-hydroxycoumarin or 7-hydroxychromone derivatives for the generation of ortho-quinone methides, we used their 8-dimethylaminomethyl derivatives 8a–8j in reaction with azoles in dimethyl carbonate at reflux. As a result, a series of coumarin- and chromone-azole hybrids were synthesized with good to excellent yield and 95–99% purity at the reflux of Mannich bases 8a–8c and 8e–8j with 2-fold excess of azoles in dimethyl carbonate. However, using of 1,4-dioxane was needed in the case of daidzein Mannich base 8d due to its poor solubility in dimethyl carbonate and the formation of by-pass products (Table 3).
| a Reaction was performed in 1.4-dioxane. |
|---|
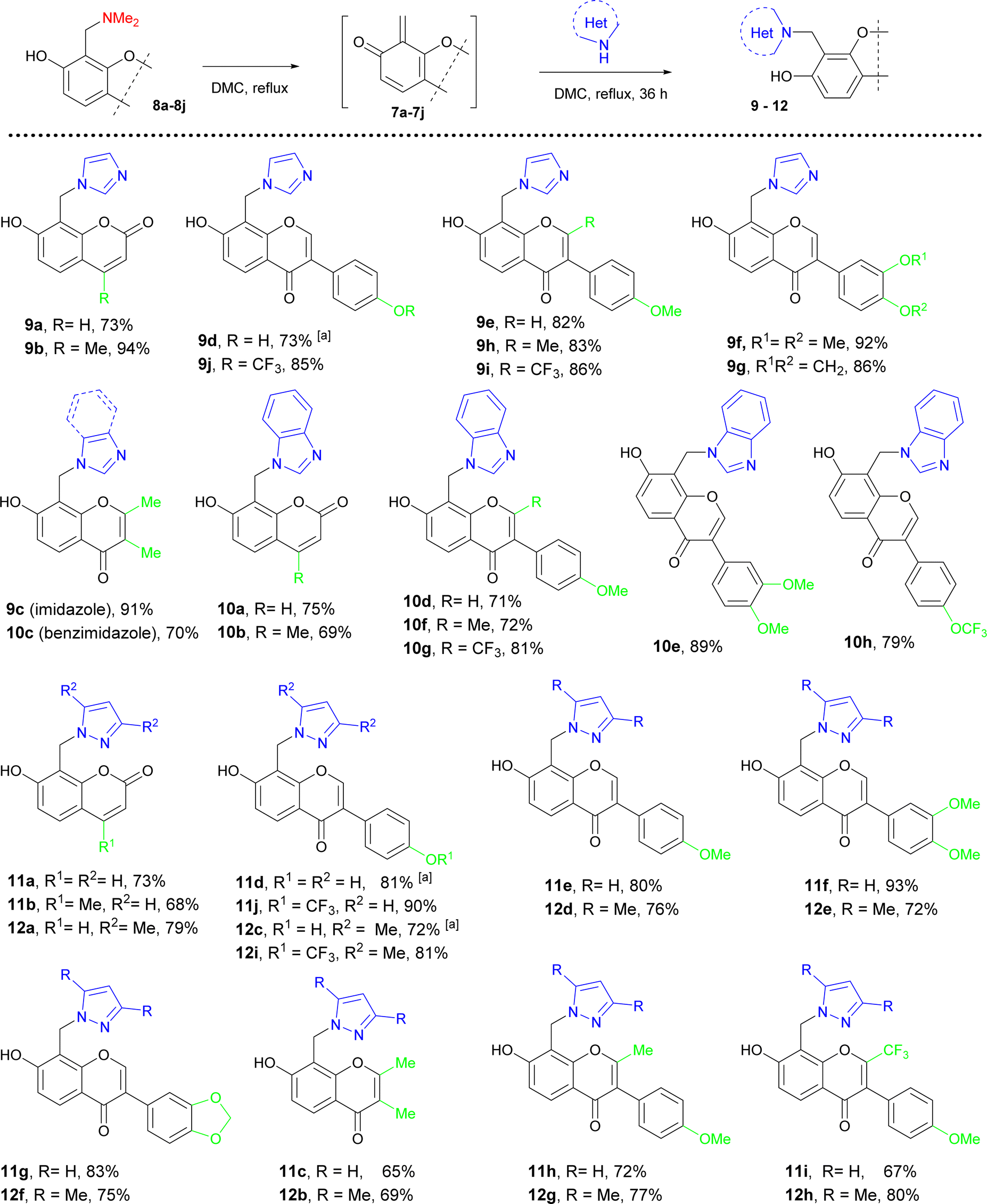 |
The reaction of 7-hydroxycoumarin or formononetin Mannich bases 8a, 8e with indazole led to the formation of both possible isomers, 8-(1-indazolylmethyl)-7-hydroxybenzopyrones 13a, 13b and 8-(2-indazolylmethyl)-7-hydroxybenzopyrones 14a, 14b (Scheme 1) with excess of compounds 14.
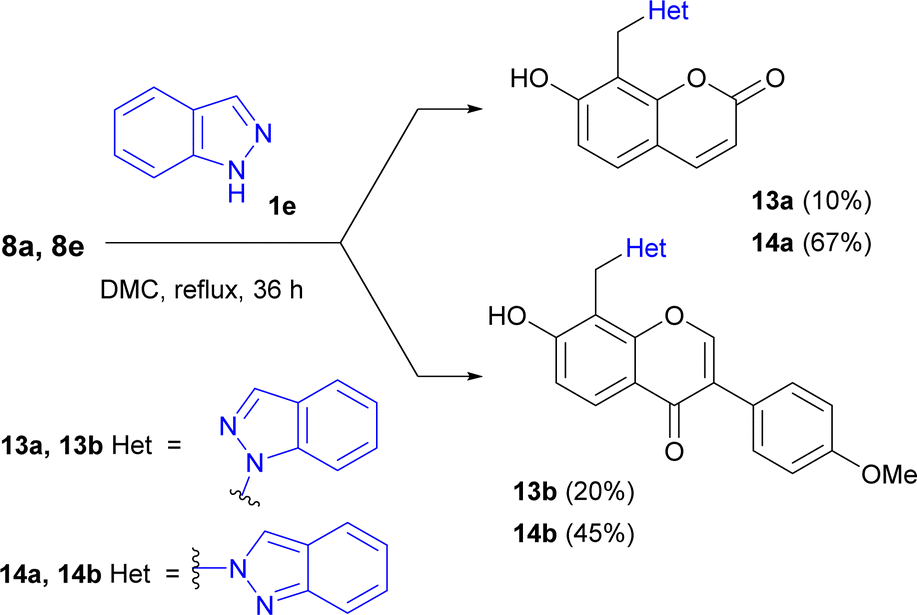 | ||
| Scheme 1 Synthesis of indazole-benzopyrone hybrids 13 and 14. | ||
The structures of synthesized compounds 13 and 14 were elucidated using 1D NOESY, HSQC, and HMBS techniques (Fig. 3).
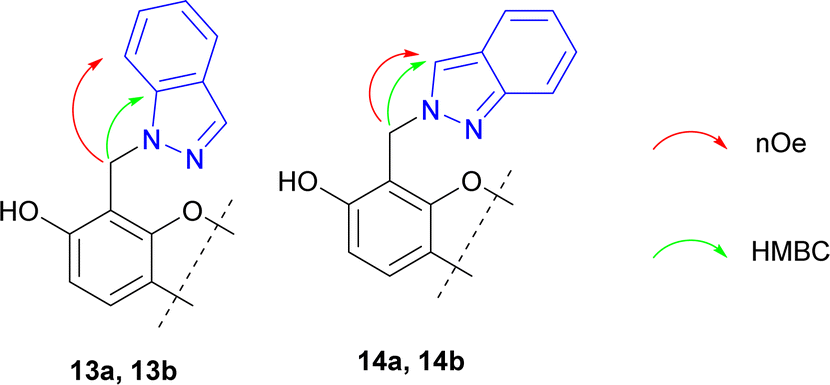 | ||
| Fig. 3 Key correlations for determination of structures 13 and 14. | ||
We presume that using asymmetrical azoles should afford mixture of both possible N-alkylated regioisomers in the ratio, which can depend on various factors, mainly steric hindrance of substituents and reaction time. In our opinion, a steric hindrance determines the key role, which was demonstrated by indazole use.
The naturally occurring flavonoids, their derivatives, and related compounds are widely studied as α-glucosidase inhibitors.21 The various synthetic heterocycle-containing coumarins and chromones were identified as promising inhibitors of this enzyme.22 In the case of isoflavone derivatives, a substituent at position 8 can influence their inhibitory potency.23 Among the compounds synthesized in this study, the sixteen benzopyrone-azaheterocycle hybrids inhibited α-glucosidase activity by 42–99% at the concentration of 25 μM. The determined IC50 values were in the range of 5.8 μM to 27.4 μM (Table 4). The 4′-trifluoromethoxy derivatives 10h and 12i bearing 1-benzimidazolylmethyl or 3,5-dimethyl-1-pyrazolylmethyl groups at position 8 of the isoflavonoid skeleton were found to be the most effective inhibitors with IC50 values of 9.5 μM and 5.8 μM, respectively. At the same time, 8 non-substituted 4′-trifluoromethoxy isoflavonoid 6j exhibited much lower inhibitory activity (39% at 25 μM) against this enzyme.
| Compound | Inhibition at 25 μM (%) | IC50, μM |
|---|---|---|
| a IC50 values are shown as average value ± standard deviation.b Reference compound. | ||
| 9b | 53.4 | 23.6 ± 1.8 |
| 9d | 42.0 | 27.0 ± 0.2 |
| 9i | 73.7 | 20.0 ± 2.0 |
| 10b | 69.3 | 21.1 ± 0.7 |
| 10d | 64.6 | 20.6 ± 2.1 |
| 10f | 48.6 | 25.5 ± 7.9 |
| 10g | 79.6 | 15.9 ± 4.7 |
| 10h | 99.1 | 9.5 ± 1.6 |
| 11d | 62.5 | 16.2 ± 3.4 |
| 11j | 84.1 | 14.3 ± 2.0 |
| 12c | 42.8 | 27.4 ± 1.9 |
| 12d | 67.3 | 20.9 ± 2.5 |
| 12h | 66.9 | 21.2 ± 0.7 |
| 12i | 94.0 | 5.8 ± 0.2 |
| 13b | 73.0 | 17.1 ± 2.8 |
| 14b | 50.3 | 23.9 ± 2.3 |
| Acarboseb | 760.6 ± 120.3 | |
Conclusions
In summary, we have demonstrated that ortho- and para-quinone methides can be efficiently generated under thermal conditions in dimethyl carbonate. In contrast to most known methodologies, this protocol allows using water-insoluble compounds and does not require the use of high temperatures and harmful solvents. Various precursors for the generation of quinone methides showed high efficiency in trapping these intermediates by azoles. Also, coumarin and chromone Mannich bases were used for the synthesis of benzopyrone-azole hybrids in good to excellent yield which makes our protocol well-suited for large-scale syntheses. Our protocol could also be implemented for the generation and trapping of ortho-quinone methide intermediates from various naturally occurring 2-hydroxymethyl- or 2-methoxymethylphenols. Several of the synthesized methylene-linked benzopyrone-azaheterocycle hybrids were found to be α-glucosidase inhibitors with IC50 values of 5.8–27.4 μM.Experimental
Chemistry
1H, 13C, and 19F NMR spectra were recorded on Bruker AVANCE DRX 500 (500/125/470 MHz) or AVANCE III 400 (400/100/376 MHz) spectrometers in CDCl3 [residual CHCl3 (δH = 7.26 ppm) or CDCl3 (δC = 77.16 ppm) as internal standard] or DMSO-d6 [residual SO(CD3)(CD2H) (δH = 2.50 ppm) or SO(CD3)2 (δC = 39.52 ppm) as internal standard]. 2D NMR spectra were recorded on Agilent ProPulse 600 MHz. Melting points were determined in open capillary tubes using the Buchi B-535 apparatus and were uncorrected. IR spectra were recorded on a Bruker Vertex 70. Mass spectra were obtained using an Agilent 1100 spectrometer using APCI (atmospheric-pressure chemical ionization). Elemental analysis was performed on a vario MICRO cube automated CHNS-analyzer. Column chromatography was performed using Macherey-Nagel Silica 60, 0.04–0.063 mm silica gel.Synthesis of Mannich bases 8a,24 8b,25 8e–8i (ref. 26) was described previously. Inhibition of α-glucosidase by compounds 4–5 and 9–14, characteristics of synthesized compounds, and copies of their NMR spectra are provided in ESI†.
7-Hydroxy-3-[4-(trifluoromethoxy)phenyl]-4H-chromen-4-one (7j) was synthesized according to typical procedure.27
Synthesis of indazole derivatives 13 and 14 was carried out similarly to the general procedure. The mixture of isomeric indazole derivatives was separated by column chromatography using 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2–MeOH (for compounds 13a and 14a) or ethyl acetate (for compounds 13b and 14b).
:1 CH2Cl2–MeOH (for compounds 13a and 14a) or ethyl acetate (for compounds 13b and 14b).
Biology
α-Glucosidase from Saccharomyces cerevisiae and p-nitrophenyl α-D-glucoside as a substrate were purchased from Sigma-Aldrich.| % inhibition = 100 − ((Asample × 100)/Acontrol) |
The IC50 values were determined from dose-dependent curves using semi-logarithmic plots (percentage of remaining α-glucosidase activity versus logarithm of inhibitor concentration) and linear trend equations. The dose-dependent curve for each of the compounds was obtained from the results of 2–3 experiments, which included the determination of the remaining activity of α-glucosidase at several inhibitor concentrations.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, A. I. Vovk, M. S. Frasinyuk, O. L. Kobzar; synthesis of compounds, A. S. Myshko, G. P., Mrug, S. P. Bondarenko, K. M. Kondratyuk; elucidation of structures, A. Kozytskiy; biological assays, V. M. Buldenko, O. L. Kobzar; writing – review and editing, M. S. Frasinyuk, O. L. Kobzar, A. I. Vovk.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Academy of Science of Ukraine (project 0124U001926).Notes and references
- (a) L. Yet, in Comprehensive Heterocyclic Chemistry IV, ed. D. S. Black, J. Cossy and C. V. Stevens, Elsevier, Oxford, 2022, pp. 1–112 Search PubMed; (b) G. Varvounis, V. Gkalpinos, P. Theodorakopoulou and E. Tsemperlidou, in Comprehensive Heterocyclic Chemistry IV, ed. D. S. Black, J. Cossy and C. V. Stevens, Elsevier, Oxford, 2022, pp. 113–307 Search PubMed.
- S. Agarwal, Imidazole-Based Drug Discovery, Elsevier, 2022 Search PubMed.
- (a) S. Yu, A. M. Deberardinis, M. Turlington and L. Pu, J. Org. Chem., 2011, 76, 2814–2819 CrossRef CAS PubMed; (b) M. C. de Koning, G. Horn, F. Worek and M. van Grol, Eur. J. Med. Chem., 2018, 157, 151–160 CrossRef CAS PubMed; (c) J.-M. Yan, Z.-J. Zhang, D.-Q. Yuan, R.-G. Xie and H.-M. Zhao, Synth. Commun., 1994, 24, 47–52 CrossRef CAS; (d) L. Yang, L. Luo, S. Zhang, X. Su, J. Lan, C.-T. Chen and J. You, Chem. Commun., 2010, 46, 3938–3940 RSC.
- (a) P. Acharya, M. M. Venkata Ramana, N. Korgavkar, G. Pavale and M. Upadhyay, Lett. Drug Des. Discovery, 2023, 20, 724–737 CrossRef CAS; (b) S. Jone Pradeepa, D. Tamilvendan, M. Susai Boobalan and N. Sundaraganesan, J. Mol. Struct., 2016, 1112, 33–44 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- (a) C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS PubMed; (b) J. Reis, A. Gaspar, N. Milhazes and F. Borges, J. Med. Chem., 2017, 60, 7941–7957 CrossRef CAS PubMed; (c) D. Raffa, B. Maggio, M. V. Raimondi, F. Plescia and G. Daidone, Eur. J. Med. Chem., 2017, 142, 213–228 CrossRef CAS PubMed; (d) M. Costa, T. A. Dias, A. Brito and F. Proença, Eur. J. Med. Chem., 2016, 123, 487–507 CrossRef CAS PubMed; (e) R. Pratap and V. J. Ram, Chem. Rev., 2014, 114, 10476–10526 CrossRef CAS PubMed.
- (a) A. Cavalli, A. Bisi, C. Bertucci, C. Rosini, A. Paluszcak, S. Gobbi, E. Giorgio, A. Rampa, F. Belluti, L. Piazzi, P. Valenti, R. W. Hartmann and M. Recanatini, J. Med. Chem., 2005, 48, 7282–7289 CrossRef CAS PubMed; (b) S. Gobbi, A. Cavalli, A. Rampa, F. Belluti, L. Piazzi, A. Paluszcak, R. W. Hartmann, M. Recanatini and A. Bisi, J. Med. Chem., 2006, 49, 4777–4780 CrossRef CAS PubMed; (c) F. Leonetti, A. Favia, A. Rao, R. Aliano, A. Paluszcak, R. W. Hartmann and A. Carotti, J. Med. Chem., 2004, 47, 6792–6803 CrossRef CAS PubMed; (d) S. Gobbi, S. Martini, R. Rozza, A. Spinello, J. Caciolla, A. Rampa, F. Belluti, N. Zaffaroni, A. Magistrato and A. Bisi, Molecules, 2023, 28, 3047 CrossRef CAS PubMed; (e) M. Recanatini, A. Bisi, A. Cavalli, F. Belluti, S. Gobbi, A. Rampa, P. Valenti, M. Palzer, A. Palusczak and R. W. Hartmann, J. Med. Chem., 2001, 44, 672–680 CrossRef CAS PubMed.
- S. Gobbi, Q. Hu, C. Zimmer, M. Engel, F. Belluti, A. Rampa, R. W. Hartmann and A. Bisi, J. Med. Chem., 2016, 59, 2468–2477 CrossRef CAS PubMed.
- (a) X.-B. Wang, W. Liu, L. Yang, Q.-L. Guo and L.-Y. Kong, Med. Chem. Res., 2012, 21, 1833–1849 CrossRef CAS; (b) L. C. Potey, P. M. Sabale and V. P. Sabale, Lett. Drug Des. Discovery, 2023, 20, 1610–1620 CrossRef CAS.
- (a) M. Muthuppalaniappan, S. Viswanadha, G. Babu and S. K. V. S. Vakkalanka, US Pat., 20110118257, 2011 Search PubMed; (b) D. Nagarathnam, S. K. V. S. Vakkalanka, M. Muthuppalaniappan, S. Viswanadha, G. Babu and P. K. Bhavar, WO Pat., 2012151525, 2012 Search PubMed.
- D. Chen, X. Zhai, Q. H. Yuan, J. Luo, S. C. Xie and P. Gong, Chin. Chem. Lett., 2010, 21, 1326–1329 CrossRef CAS.
- (a) Y. Liu, G. Feng, Z. Ma, C. Xu, Z. Guo, P. Gong and L. Xu, Arch. Pharm., 2015, 348, 776–785 CrossRef CAS PubMed; (b) C. Zhao, Y. Zhao, H. Chai and P. Gong, Bioorg. Med. Chem., 2006, 14, 2552–2558 CrossRef CAS PubMed.
- H. Chai, Y. Zhao, C. Zhao and P. Gong, Bioorg. Med. Chem., 2006, 14, 911–917 CrossRef CAS PubMed.
- B. Naumczuk, W. Bocian, J. Sitkowski, R. Kawęcki and L. Kozerski, New J. Chem., 2019, 43, 18975–18978 RSC.
- (a) D. Ma, Y. Yin, Y.-L. Chen, Y.-T. Yan and J. Wu, RSC Adv., 2021, 11, 15380–15386 RSC; (b) N. E. Sidorina and V. A. Osyanin, Chem. Heterocycl. Compd., 2007, 43, 1065–1071 CrossRef CAS.
- A. I. Poddel'sky, M. V. Arsenyev, T. V. Astaf'eva, S. A. Chesnokov, G. K. Fukin and G. A. Abakumov, J. Organomet. Chem., 2017, 835, 17–24 CrossRef.
- (a) Y. Feng, J. W. Blunt, A. L. J. Cole and M. H. G. Munro, J. Nat. Prod., 2002, 65, 1681–1682 CrossRef CAS PubMed; (b) Y.-P. Liu, Y. Li, X.-H. Cai, X.-Y. Li, L.-M. Kong, G.-G. Cheng and X.-D. Luo, J. Nat. Prod., 2012, 75, 220–224 CrossRef CAS PubMed; (c) R. Mhiri, I. Koubaa, R. Chawech, F. Auberon, N. Allouche and T. Michel, Chem. Biodiversity, 2020, 17, e2000758 CrossRef CAS PubMed.
- H. Sugimoto, S. Nakamura and T. Ohwada, Adv. Synth. Catal., 2007, 349, 669–679 CrossRef CAS.
- S. González-Pelayo and L. A. López, Eur. J. Org Chem., 2017, 2017, 6003–6007 CrossRef.
- M. S. Frasinyuk, G. P. Mrug, S. P. Bondarenko, V. M. Sviripa, W. Zhang, X. Cai, M. Fiandalo, J. L. Mohler, C. Liu and D. Watt, Org. Biomol. Chem., 2015, 13, 11292–11301 RSC.
- D. Sohretoglu and S. Sari, Phytochem. Rev., 2020, 19, 1081–1092 CrossRef CAS.
- (a) R. G. Soengas, V. L. M. Silva, D. Ide, A. Kato, S. M. Cardoso, F. A. Almeida Paz and A. M. S. Silva, Tetrahedron, 2016, 72, 3198–3203 CrossRef CAS; (b) R. Ichale, A. M. Kanhed and A. Vora, Mol. Diversity, 2024, 28, 1239–1247 CrossRef CAS PubMed; (c) M. S. Asgari, M. Mohammadi-Khanaposhtani, M. Kiani, P. R. Ranjbar, E. Zabihi, R. Pourbagher, R. Rahimi, M. A. Faramarzi, M. Biglar, B. Larijani, M. Mahdavi, H. Hamedifar and M. H. Hajimiri, Bioorg. Chem., 2019, 92, 103206 CrossRef CAS PubMed.
- H. Sun, Y. Li, X. Zhang, Y. Lei, W. Ding, X. Zhao, H. Wang, X. Song, Q. Yao, Y. Zhang, Y. Ma, R. Wang, T. Zhu and P. Yu, Bioorg. Med. Chem. Lett., 2015, 25, 4567–4571 CrossRef CAS PubMed.
- G. P. Mrug, N. V. Myshko, S. P. Bondarenko, V. M. Sviripa and M. S. Frasinyuk, J. Org. Chem., 2019, 84, 7138–7147 CrossRef CAS PubMed.
- P. Da Re, G. Bonola and L. Verlicchi, J. Med. Chem., 1964, 7, 162–166 CrossRef CAS PubMed.
- M. S. Frasinyuk, G. P. Mrug, S. P. Bondarenko, V. P. Khilya, V. M. Sviripa, O. A. Syrotchuk, W. Zhang, X. Cai, M. V. Fiandalo, J. L. Mohler, C. Liu and D. S. Watt, ChemMedChem, 2016, 11, 600–611 CrossRef CAS PubMed.
- M. S. Frasinyuk, S. P. Bondarenko, V. P. Khilya, C. Liu, D. S. Watt and V. M. Sviripa, Org. Biomol. Chem., 2015, 13, 1053–1067 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05230g |
| This journal is © The Royal Society of Chemistry 2024 |