
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGroup 7 carbonyl complexes of a PNN-heteroscorpionate ligand†
Jorge P. Valdivieso III,
Alexander N. Erickson and
James R. Gardinier *
*
Department of Chemistry, Marquette University, Milwaukee, Wisconsin 53201-1881, USA. E-mail: james.gardinier@marquette.edu
First published on 4th October 2024
Abstract
A series of rhenium and manganese carbonyl complexes of a heteroscorpionate ligand with an atypical N2P-donor set has been prepared to better understand their electronic and CO releasing properties. Thus, the ligand, pz2TTP, with an a,a-bis(pyrazol-1-yl)tolyl group decorated with an ortho-situated di(p-tolyl)phosphanyl reacts with carbonyl group 17 reagents to give [fac-(κ2NP-pz2TTP)Re(CO)3Br], 1, and [fac-(κ3N2P-pz2TTP)M(CO)3](OTf = O3SCF3), 2-M (M = Re, Mn), if care is taken during the preparation of the manganeses derivative. When heated in CH3CN, 2-Mn slowly transforms to [fac,cis-(κ3N2P-pz2TTP)Mn(CO)2(NCCH3)](OTf), 3-Mn. In contrast, the corresponding 3-Re can only be prepared from 2-Re using Me3NO; pure 3-Mn can also be prepared by this method. Experimental and density functional calculations at the M06L/Def2-TZVP/PCM(CH3CN) level show that the replacement of a carbonyl with an acetonitrile solvent decreases the oxidation potential by around 0.8 V per carbonyl released, making decarbonylated species potent reductants. At the same time, the electronic spectrum broadens and undergoes a red-shift, making dicarbonyl complexes more susceptible to photo-initiated decarbonylation reactions than tricarbonyls. When 2-Mn or 3-Mn are irradiated in with 390 nm LED light in aerated solutions, [trans-Mn(pz2TTP = O)2](OTf)2, 4, along with insoluble manganese oxides are rapidly formed.
Introduction
Group 7 carbonyl complexes have found diverse uses in catalysis1–6 and medicinal chemistry.7–14 Their utility is typically predicated on the stabilities of either LM(CO)3+ or LM(CO)2+ cores. Thus, the stability of [Tc(CO)3]+ complexes have been exploited in radiopharmaceutical imaging applications (99mTc, γ emitter, t1/2 = 6.05 h). In contrast, manganese(I) tricarbonyl complexes are visible-light absorbing chromophores that readily lose all carbonyls on photo-excitation, making them useful as photoinduced CO releasing molecules (PhotoCORMs).10,12,13,15,16 Five-coordinate LMn(CO)2 pincer complexes are potent catalysts for a variety of chemical transformations.2,17 Certain Re(CO)3+ complexes can lose one CO with either UV-irradiation (or more commonly by reaction with Me3NO) to give highly reactive Re(CO)2+ species that can facilitate C–H bond activations.18–20 Most recently, rhenium(I) carbonyl complexes have been investigated for their potent anti-cancer properties whose mode of action remains under investigation, but whose activity may be tied to the ability of the Re(CO)x+ (x = 2, 3) to interact with reactive oxygen species after binding to cellular matter.7,8,21,22 Thus, in any new group 7 carbonyl complex, it is desirable to understand their CO releasing capabilities.Facially coordinating tridentate ligands with two pyrazolyl and one non-pyrazolyl Lewis donor are heteroscorpionates23–26 that are related to the more ubiquitous tris(pyrazolyl)methane (Tpm) and tris(pyrazolyl)borate (Tp), C-27 and B-24 scorpionate ligands, respectively.28 For many catalytic and biomedical applications, heteroscorpionates are enticing since the unique donor can be suitably functionalized to elicit a desired reactivity. A variety of group 7 carbonyl complexes of heteroscorpionates with N2N,29–31 N2O,32–39 and N2S40,41 donor sets have been reported. Of these, the CO releasing properties of N2N29 and N2O35,42,43 complexes of Mn(CO)3+ have been studied in detail. It was found that under 365 nm irradiation the N2O complexes with anionic bis(pyrazolyl)acetate, bpza, or bis(pyrazolyl)propionate, bpzp, ligands were more reactive (t1/2 ∼ 8 min−1) than [(Tpm)Mn(CO)3]+ (t1/2 ∼ 11 min−1) while the bulkier N2N heteroscorpionates based on (N-4-R-benzyl)-bis(pyrazolyl)ethanamines, bpeaBzR, were less reactive (t1/2 ∼ 25 min−1) under similar conditions. Moreover, the ultimate manganese products after photoinduced CO dissociation depended on the ligand, with [MnII(Tpm)2]2+ or [(κ3-bpza)MnII(μ-κ2N,κ1O-bpza)]2 and manganese oxides being isolated in the former cases. In bpeaBzR cases, carbonyl-free species were not identified, but the dicarbonyl and, for the first-time, a mono-carbonyl scorpionate species could be detected.29 To our knowledge, the CO releasing properties of softer heteroscorpionates have not yet been reported. In 2014 our group reported on a new heteroscorpionate, pz2TTP (Fig. 1, left),44 with a rare N2P donor set, joining [Fc(PPh2)(Bp*)]− (Fig. 1, right)45,46 as the only other known example with such a donor set. In neither case has the group 7 chemistry been described.
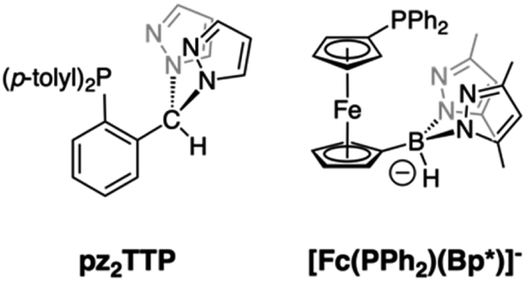 | ||
| Fig. 1 Heteroscorpionate ligands with PNN donor sets. | ||
The incorporation of a phosphorus donor into a heteroscorpionate was envisioned to be advantageous since the bulky and trans-labilizing diarylphosphine group might stabilize unusual low-oxidation state group 7 complexes such as di- or possibly even mono-carbonyls. Additionally, all complexes might be easily characterized by 31P NMR spectroscopy. The findings of our initial investigation into the preparation, electronic properties, and potential CO-releasing reactivity of rhenium(I) and manganese(I) carbonyl complexes of pz2TTP are detailed herein.
Results and discussion
Syntheses
Heating solutions containing pz2TTP and an equimolar quantity of group 7 carbonyl reagents generally produced mixtures of compounds rather than high yields of a single compound. Scheme 1 provides a summary of optimized syntheses of the group 7 carbonyl complexes of pz2TTP. Thus, the reaction between equimolar pz2TTP and Re(CO)5Br in toluene gives the expected (pz2TTP)Re(CO)3Br, 1, in modest yield after crystallization (Scheme 1, left). However, IR, NMR, single crystal and powder X-ray diffraction reveal this analytically pure sample to be a mixture of isomers (vide infra, Fig. S2–S4†). When an equimolar mixture of pz2TTP, Re(CO)5Br, and Tl(O3SCF3 = OTf) are heated in CH3CN, crystals of analytically pure [(pz2TTP)Re(CO)3](OTf), 2-Re, can obtained in modest yield; the crude material before crystallization shows evidence for the formation of minor (but variable) amounts of the dicarbonyl [(pz2TTP)Re(CO)2(NCCH3)](OTf), 3-Re. A similar reaction with Mn(CO)5Br in lieu of the rhenium reactant affords mixtures of tricarbonyl 2-Mn and dicarbonyl 3-Mn, favouring the latter after heating 30 min (Fig. S10†). Instead, pure 2-Mn is best prepared at low temperature by the reaction between pz2TTP and [fac-Mn(CO)3(NCCH3)3](OTf). The pure dicarbonyls 3-M (M = Re, Mn) are best prepared by decarbonylation of 2-M by using equimolar anhydrous Me3NO in CH3CN. Although the reactions appear insensitive to an excess of Me3NO, the separation from this reagent is tedious, so it is easiest to use one equivalent rather than an excess.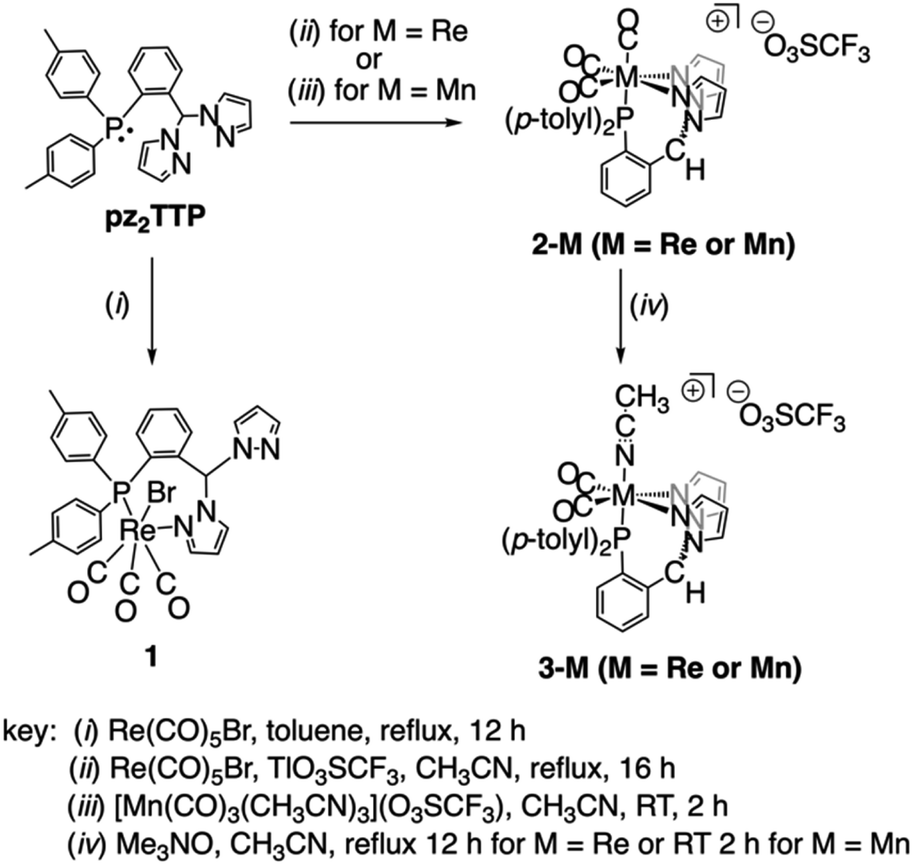 | ||
| Scheme 1 Preparative routes to the various group 7 carbonyl complexes of pz2TTP. | ||
Solid state structures
The solid-state structures of representative rhenium carbonyl complexes of pz2TTP are given in Fig. 2, those of the manganese derivatives are provided in Fig. S1 of the ESI.† Bond distances and angles for each compound are listed in Table 1. It is rare47 for scorpionate complexes of Re(CO)3 to exhibit a κ2-binding mode instead of prototypical κ3-mode. However, the crystals of 1 possess a κ2NP-ligand bound to a fac-Re(CO)3Br moiety; one pyrazolyl ring remains “free”. The resulting seven-membered (Re1–N12–N11–C7–C2–C1–P1) chelate ring folds into a boat conformation with the methine carbon C7 as the bow and the Re1–P1 bond as the stern of the boat. This conformation places the methine hydrogen H7 inside the boat and the “free” pyrazolyl outside (left two structures of Fig. 2). In 1, there is a disorder that places either the bromide (Br1A; 80%) or a carbonyl (C10B–O3B; 20%) inside the boat. The disorder leads to greater uncertainty Re–C9A/B distances than other Re–C distances. For the well-behaved carbonyls, the Re1–C10 bond trans- to P1 (1.977(4) Å in 1) is significantly longer than that trans- to the pyrazolyl nitrogen (Re1–C8: 1.924(4) Å). The Re1–C8 distance compares favourably with the average Re–C distance of 1.92(1) Å in either [fac-(κ3-HCpz3)Re(CO)3]Br48 or [fac-(κ3-CH3CH2Cpz3)Re(CO)3](O3SCF3)·1.5H2O·0.5CH3CN,49 or of 1.91 Å in [fac-(κ3-HCpz3iPr3)Re(CO)3]Br·acetone.50 Thus, the longer Re–C distance in 1 is a manifestation of the significantly greater trans-influence of the ditolylphosphanyl versus a pyrazolyl moiety. It is noted that the Re–N bond distance of 2.209(3) Å in 1 is a little longer than found in other rhenium scorpionates that typically range from 2.14 to 2.19 Å: Re–Navg 2.18 Å for [fac-(κ3-HCpz3)Re(CO)3]Br48 2.16 Å for [fac-(κ3-CH3CH2Cpz3)Re(CO)3]+ (ref. 49) or 2.14 Å for {1,4-C6H4[CH2OCH2Cpz3Re(CO)3]2}2+ for instance.50 The longer Re–N bonds in 1 are due to the charge neutral-nature of the Re(CO)3Br core and to the significantly greater steric profile of the p-tolyl2P group relative to a pyrazolyl. Such long Re–N bonds are also evident in [fac-(κ3-N2O bpza)Re(CO)3] (Re–Navg 2.21 Å)34 and in the complex of a pyridyl based P-scorpionate [fac-(κ2N–P(py)3)ReBr(CO)3] (Re–Navg 2.21 Å).47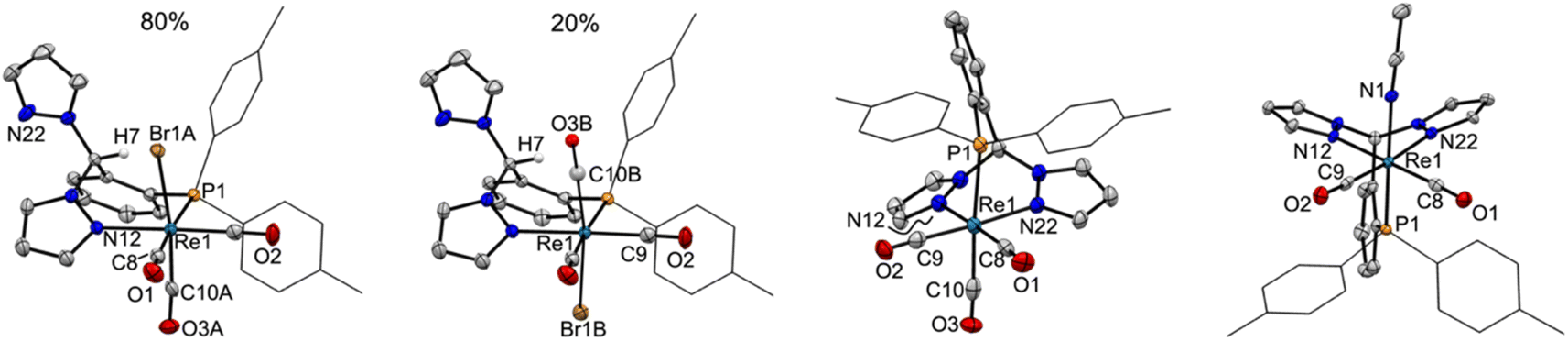 | ||
| Fig. 2 (Left): Views of major and minor disorder components in 1, highlighting isomers; (Centre): view of the cation in 2-Re; (Right): view of the cation in 3-Re. Ellipsoids are drawn at the 50% probability level. | ||
| Compound | 1 | 2-Re | 2-Mn | 3-Re | 3-Mn a |
|---|---|---|---|---|---|
| a Only values for major disorder component of 3-Mn0.922-Mn0.08 are given. | |||||
| Bond | |||||
| M–Br1 | 2.6057(11) | ||||
| M–P1 | 2.4892(10) | 2.4734(10) | 2.3350(5) | 2.3510(10) | 2.2784(3) |
| M–N1 | 2.118(4) | 2.0107(15) | |||
| M–N12 | 2.209(3) | 2.185(3) | 2.0580(14) | 2.180(3) | 2.0550(11) |
| M–N22 | 2.183(4) | 2.0437(14) | 2.158(4) | 2.0359(11) | |
| M–C8 | 1.925(4) | 1.924(5) | 1.8062(17) | 1.914(5) | 1.8005(14) |
| M–C9 | 1.907(9) | 1.936(5) | 1.8139(18) | 1.890(5) | 1.7806(14) |
| M–C10 | 1.976(4) | 1.978(5) | 1.8409(18) | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Angles | |||||
| N12–M–N22 | 84.38(13) | 88.32(5) | 84.82(13) | 88.08(4) | |
| P1–M–N12 | 94.78(9) | 91.27(10) | 89.09(4) | 89.53(9) | 88.74(3) |
| P1–M–N22 | 80.72(9) | 84.90(4) | 90.01(9) | 89.99(3) | |
| C8–M–C9 | 91.9(3) | 86.73(19) | 87.71(7) | 88.79(18) | 86.80(6) |
| C8–M–C10 | 88.50(17) | 89.35(18) | 86.96(7) | ||
| C9–M–C10 | 90.2(3) | 87.97(18) | 91.27(7) | ||
| Br1–M–N12 | 85.60(9) | ||||
| Br1–M–P1 | 93.22(3) | ||||
| N1–M–P1 | 169.78(10) | 171.16(8) | |||
| C10–M–P1 | 175.18(12) | 175.23(13) | 175.48(5) | ||
| N12–M–C8 | 178.37(15) | 176.94(16) | 178.56(6) | 179.53(16) | 176.84(5) |
| N22–M–C9 | 177.11(16) | 177.53(7) | 174.89(15) | 176.27(5) | |
The replacement of the bromide in 1 with a triflate results in a shift in ligand binding mode to give [fac-(κ3N2P-pz2TTP)Re(CO)3](OTf), 2-Re (third structure, Fig. 2) with an unbound anion. The lower electron density at the metal centre in the cation relative to 1 results in shorter metal-scorpionate bonds and, due to lower capacity for pi back-bonding, longer Re–C bonds than in 1. Thus, the Re1–P1 bond in 2-Re of 2.47 Å is 0.02 Å shorter than that in either 1 or its CH2Cl2 solvate. The average Re–N bonds in 2-Re of 2.18 Å are 0.03 Å shorter than in 1 and are comparable to those in [fac-(κ3-HCpz3)Re(CO)3]Br.48 Moreover, the average Re–C bond in 2-Re of 1.95 Å is about 0.01 Å longer than that in 1. As with 1, the Re1–C10 bond in 2-Re 1.978(5) Å is significantly longer than those bonds trans- to pyrazolyl nitrogens (Re1–N12 1.924(5) Å, Re1–N22 1.936(5) Å).
The related complex [fac-(κ3N2P-pz2TTP)Mn(CO)3](OTf), 2-Mn, despite having a formula similar to 2-Re, crystallizes in the monoclinic space group P21/c, rather than P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , like its heavier congener. Many of the structural features observed in 2-Re are preserved in 2-Mn, but the smaller size of Mn relative to Re gives shorter bonds. Thus the average Mn–N bond distance of 2.05 Å is 0.13 Å shorter than in 2-Re but is similar to that found in other C-scorpionate complexes of [Mn(CO)3]+ such as 2.03 Å in [(HCpz3)Mn(CO)3](PF6),51 2.05 Å in [(Hpz(3-CHpz2))Mn(CO)3](O3SCF3),31 or 2.07 Å found in [HC(3-iPrpz)3Mn-(CO)3](O3SCF3).52 The average Mn–C distance of 1.82 Å in 2-Mn is 0.13 Å shorter than that in 2-Re and is comparable to 1.81 Å found for all three of the aforementioned manganese C-scorpionates. Again, in 2-Mn, the Mn1–C10 bond trans- to the P atom is significantly longer than the other two Mn–C bonds (1.841(2) Å vs. 1.806(2) and 1.814(2) Å).
, like its heavier congener. Many of the structural features observed in 2-Re are preserved in 2-Mn, but the smaller size of Mn relative to Re gives shorter bonds. Thus the average Mn–N bond distance of 2.05 Å is 0.13 Å shorter than in 2-Re but is similar to that found in other C-scorpionate complexes of [Mn(CO)3]+ such as 2.03 Å in [(HCpz3)Mn(CO)3](PF6),51 2.05 Å in [(Hpz(3-CHpz2))Mn(CO)3](O3SCF3),31 or 2.07 Å found in [HC(3-iPrpz)3Mn-(CO)3](O3SCF3).52 The average Mn–C distance of 1.82 Å in 2-Mn is 0.13 Å shorter than that in 2-Re and is comparable to 1.81 Å found for all three of the aforementioned manganese C-scorpionates. Again, in 2-Mn, the Mn1–C10 bond trans- to the P atom is significantly longer than the other two Mn–C bonds (1.841(2) Å vs. 1.806(2) and 1.814(2) Å).
Finally, the replacement of the CO trans- to phosphorous in 2-Re, with an acetonitrile gives [fac,cis-(pz2TTP)M(CO)2(NCCH3)](OTf), 3-Re. Interestingly, while the corresponding analytically and spectroscopically pure 3-Mn can be prepared, it has not been possible to obtain single crystals suitable for X-ray diffraction. Instead, suitable X-ray quality single crystals of 3-Mn co-crystallized with small portions of 2-Mn (see experimental) can be obtained. The following structural discussion refers to the metrics found in the major disorder component in 3-Mn0.922-Mn0.08. The lower trans-influence of CH3CN relative to CO affords a shorter M1–P1 bond in 3-M (2.3510(10) Å for M = Re, 2.2784(3) Å for M = Mn) versus that in 2-M (2.4734(10) Å for M = Re and 2.3350(5) Å for M = Mn). A secondary effect of the replacement of CO for CH3CN is a slight shortening of M–C bonds (Re–Cavg = 1.902 Å and Mn–Cavg = 1.795 Å) versus those in 2-M due to an increased electron density at the metal centre that strengthens the pi-back-bonding to the remaining CO groups, as will be detailed more fully below. Surprisingly, while there are multiple structurally characterized dicarbonylrhenium(I) B-scorpionate complexes,53,54 to the best of our knowledge, 3-Re is the first structurally characterized dicarbonylrhenium(I) C-scorpionate complex. C-scorpionates of [Re(CO)2(NO)]+ are known55 but are not structurally characterized. The average Re–C bond in 3-Re is more aligned with that in [(HBpz3 = Tp)Re(CO)2]2(μ-N2) (1.91 Å)54 than those found in either TpRe(CO)2(THF) (1.872 Å) or TpRe(CO)2(PPh3) (1.88 Å).53 The structure of 2-Mn is also noteworthy since there are relatively few structurally characterized C-scorpionate complexes of the [cis-Mn(CO)2]+ moiety, with [(HC(3,5-Me2pz)3 = Tpm*)Mn(CO)2(L)](PF6) (L = PMe3, py)56 being examples. In both of these latter cases, with more electron-donating 3,5-dimethylpyrazolyls, the average Mn–C bond of 1.76 Å is slightly shorter than that in 3-Mn (1.79 Å).
Spectroscopic studies
The identity of each species 1, 2-M, and 3-M can be easily distinguished by 31P and 1H NMR spectroscopy, with the former being easiest to interpret since singlet resonances are observed in most cases (vide infra). That is, while the stable isotopes of group 7 metals used here all have nuclear spin I = 5/2, (abundance: 55Mn (100%), 185Re (37.4%), 187Re (62.6%)), each isotope has a large nuclear quadrupole moment Qo (55Mn: +0.330(10), 185Re: +2.18(2), 187Re: +2.07(2) barn)57 that causes rapid relaxation on the NMR timescale, so that a singlet rather than the expected (1:1:1:1:1:1) sextet resonance is observed in 31P NMR spectrum of each compound. Thus, the 31P NMR spectrum of analytically pure prism crystals of 1 in CD3CN gives two singlets at δP 10.5 (80% rel. int.) and 0.3 (20% rel. int.) ppm for its two co-crystallized isomers (Fig. S3 and S4†). The 1H NMR spectrum of 1 also shows two sets of resonances in a 4:1 ratio (Fig. S3†), that are most easily distinguished by the two unequal pairs of characteristic H4-pyrazolyl resonances near δH ∼6.1 and δH ∼6.3 ppm, assigned to “free” and Re-bound pyrazolyls, respectively, of each isomer in accord with the solid-state structures. These assignments are made by comparison with the spectra of the free ligand (δH = 6.10 ppm) and other Re complexes (vide infra). The 31P NMR spectrum of 2-Re has a singlet resonance at δP 4.5 ppm while that of 3-Re occurs more downfield at 17.9 ppm. Similarly, a singlet is observed for 2-Mn at δP = 33.8 ppm and for 3-Mn at δP 66.3 ppm.‡ The 1H NMR spectrum of each 2-M and 3-M (Fig. S6–S9†) is aligned with expectations that solid state structures are maintained in CD3CN solution with one set of resonances that are significantly shifted with respect to those of the free ligand. Particularly diagnostic are resonances for H4-pyrazolyls. For the rhenium compounds, these occur at δH 6.58 for 2-Re and δH 6.51 for 3-Re, both shifted downfield with respect to that of the free ligand at δH 6.10 ppm. Here, the more electron rich 3-Re is shifted upfield from 2-Re, as expected. The analogous resonances for manganese derivatives are shifted further downfield (δH 6.63 for 2-Mn and δH 6.57 for 3-Mn) than in the rhenium cases, consistent with greater Lewis acidity of the 3rd versus 5th row metal. Other diagnostic resonances occur for tolyl and unique aromatic ring hydrogens. An unusual feature of the 1H NMR spectra of each 2-M and 3-M is that the most downfield resonances for acidic methine Hα, the H5-pyrazolyl ring, and the H3-aryl (positioned ortho- to the CHpz2 group) hydrogens exhibit concentration dependent shifts in the 10−3 to 10−4 M range. The magnitude of concentration-dependent chemical shift changes, |ΔδH|, falls in the order Hα > H5pz > H3Ar consistent with their decreasing relative acidities (Fig. S11†). It is noted that close contacts occur between the triflate ion and these hydrogens in the solid state structures of 2-M. Presumably these concentration dependent shifts are the result of contact ion pairs forming that are favoured at high concentration with triflate oxygens causing the observed shifts to lower fields, as seen in other systems.31,58–65 Finally, although resonances for most carbons could be observed, rapid quadrupolar relaxation in combination with the expected 2JC–P coupling (splitting signal intensity) inhibited observation of resonances for CO groups in the 13C NMR spectrum of tricarbonyl complexes reported here.
The IR spectra of each complex was recorded in both the solid state and in CH3CN solution. A summary of solution data is found in Table 2 while other data are provided in the ESI (Fig. S12 and S13).† In the group 7 tricarbonyl compounds 1 and 2-M three bands were observed for CO stretches consistent with a fac-M(CO)3 arrangement, as observed in the solid state. The low symmetry coordination environment imposed by the scorpionate about the metal in ensures that the A1 and E bands of a nominally C3v symmetric M(CO)3 core are split into two A′ and one A′′ modes in Cs symmetry (2-M) or thee A modes in C1 symmetry (complex 1). The average energy for CO stretches decreases with increasing electron donating ability of the metal centres due to increased metal to ligand π-backbonding. Thus, the average CO stretch in charge neutral 1 (![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/char_e0d5.gif) CO avg = 1953 cm−1) is lower than in cationic 2-Re (CO avg = 1965 cm−1), albeit comparison may be somewhat inappropriate given the different supporting ligands (κ2NP- vs. κ3N2P-). Next, consistent with expectations based on both orbital overlap and spectroscopic electronegativity, the CO bonds in 2-Re are weaker than those in 2-Mn (CO avg = 1976 cm−1). Similar trends are found in TpM(CO)3 (CO avg = 1951 cm−1 for Re20,53,66 and 1964 cm−1 for Mn20,67), [(Tpm)M(CO)3]+ CO avg = 1973 cm−1 for Re50 and 1988 cm−1 for Mn,43,51,52 and in heteroscorpionate cases [(bpza)M(CO)3]34 (CO avg = 1952 cm−1 for Re and 1971 cm−1 for Mn). As noted for TpM(CO)3 versus [(Tpm)M(CO)3]+, charge neutral Tpm derivatives give cationic complexes with higher energy CO stretches than their anionic B-scorpionate counterparts. The electron donating ability of pz2TTP gives complexes with CO stretches intermediate between those of neutral and anionic NNN-based homoscorpionates. The IR spectra of the dicarbonyls, 3-M, consist of two equal-intensity bands for symmetric and asymmetric CO stretches. Again, the heavier group 7 congener has lower energy stretches (CO avg = 1903 cm−1) than the lighter congener (CO avg = 1930 cm−1). The CO stretches in each dicarbonyl 3-M are lower energy than in the tricarbonyl variants 2-M since the replacement of a CO with a less π-acidic CH3CN allows the metals to redistribute their electron density into the remaining CO antibonding orbitals.
CO avg = 1953 cm−1) is lower than in cationic 2-Re (CO avg = 1965 cm−1), albeit comparison may be somewhat inappropriate given the different supporting ligands (κ2NP- vs. κ3N2P-). Next, consistent with expectations based on both orbital overlap and spectroscopic electronegativity, the CO bonds in 2-Re are weaker than those in 2-Mn (CO avg = 1976 cm−1). Similar trends are found in TpM(CO)3 (CO avg = 1951 cm−1 for Re20,53,66 and 1964 cm−1 for Mn20,67), [(Tpm)M(CO)3]+ CO avg = 1973 cm−1 for Re50 and 1988 cm−1 for Mn,43,51,52 and in heteroscorpionate cases [(bpza)M(CO)3]34 (CO avg = 1952 cm−1 for Re and 1971 cm−1 for Mn). As noted for TpM(CO)3 versus [(Tpm)M(CO)3]+, charge neutral Tpm derivatives give cationic complexes with higher energy CO stretches than their anionic B-scorpionate counterparts. The electron donating ability of pz2TTP gives complexes with CO stretches intermediate between those of neutral and anionic NNN-based homoscorpionates. The IR spectra of the dicarbonyls, 3-M, consist of two equal-intensity bands for symmetric and asymmetric CO stretches. Again, the heavier group 7 congener has lower energy stretches (CO avg = 1903 cm−1) than the lighter congener (CO avg = 1930 cm−1). The CO stretches in each dicarbonyl 3-M are lower energy than in the tricarbonyl variants 2-M since the replacement of a CO with a less π-acidic CH3CN allows the metals to redistribute their electron density into the remaining CO antibonding orbitals.
Electronic properties
To facilitate discussion of the electronic properties of the new complexes, it will be useful to briefly examine the frontier orbitals of 2-M and 3-M as elucidated from DFT calculations performed at the M06L/def2-TZVP/PCM (CH3CN) level of theory. As might be expected, the frontier orbitals of the four complexes share many similarities. A view of those orbitals in the cation of 2-Re are given in Fig. 3, others are provided in the ESI, Fig. S18–S20.† The relative energy levels of related orbitals are provided in Fig. 4. First, all four complexes, 2-M and 3-M, possess mirror symmetry (Cs point group) where the mirror is assigned as the yz-plane that bisects the N12–M–N22 bond angle and contains the M-–P bond. As such, the dz2 and dxy orbitals (both A′ representations) are directed along bonds. The dx2–y2 (A′ representation), dyz and dxz (both A′′ representations) orbitals are located between bonds and are involved in pi-bonding interactions. For each complex, the three highest-energy filled orbitals, HOMO(−N) (N = 0, 1, 2), are mainly metal-centred with pi-bonding interaction with the carbonyl carbon, see bottom three orbitals Fig. 3 and those labelled πM–C in the left of Fig. 4. The πM–C orbitals of rhenium complexes are lower energy than the corresponding manganese derivatives. Moreover, those in 2-M are lower energy than those of 3-M. The corresponding antibonding counterparts (labelled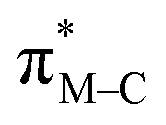 , Fig. 4 left) are found above LUMO(+8) (LUMO+12 to +14 for 2-Re). The LUMO(+N) (N = 0–8) are mainly ligand-centred π-orbitals. For 2-Re, LUMO(+17) and LUMO(+18) exhibit σ*-antibonding interactions between the scorpionate donor atoms and metals' dz2 and dxy orbitals, respectively. Similar interactions are found at higher energy in LUMOs (+19 and +21) in 3-Re. In manganese cases, analogous interactions are found in LUMO(+14) or above, as shown in Fig. 4 and S18.† However, there is extensive mixing of A′ orbitals, so that σ* interactions are also found in LUMOs (+1 and +2) and, for 3-Mn, in LUMOs (+5 and +7).
, Fig. 4 left) are found above LUMO(+8) (LUMO+12 to +14 for 2-Re). The LUMO(+N) (N = 0–8) are mainly ligand-centred π-orbitals. For 2-Re, LUMO(+17) and LUMO(+18) exhibit σ*-antibonding interactions between the scorpionate donor atoms and metals' dz2 and dxy orbitals, respectively. Similar interactions are found at higher energy in LUMOs (+19 and +21) in 3-Re. In manganese cases, analogous interactions are found in LUMO(+14) or above, as shown in Fig. 4 and S18.† However, there is extensive mixing of A′ orbitals, so that σ* interactions are also found in LUMOs (+1 and +2) and, for 3-Mn, in LUMOs (+5 and +7).
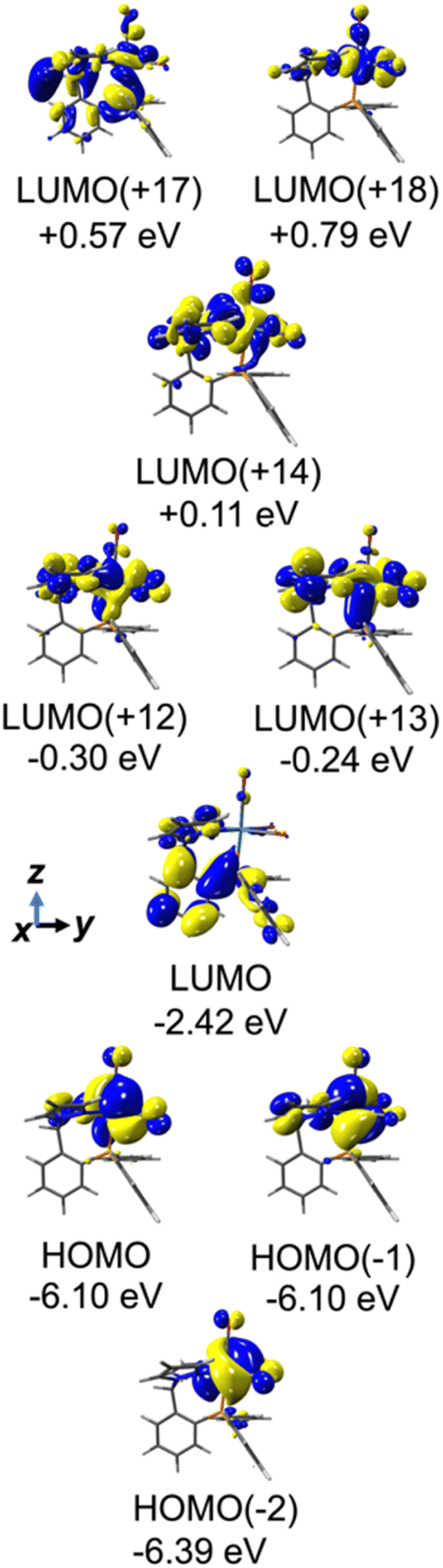 | ||
| Fig. 3 Frontier orbitals of 2-Re. | ||
 | ||
| Fig. 4 Energy levels of frontier orbitals in 2-M and 3-M (M = Re, Mn). | ||
The electronic absorption spectrum for each rhenium complex recorded in CH3CN consists of ligand centred bands below 275 nm (ε > 20000 M−1 cm−1, Fig. S14†) that, at their low energy edge, overlap weak metal ligand charge transfer (MLCT) bands as suggested by TD-DFT calculations at the M06L-TZVP/PCM(CH3CN) level. Electron density difference plots for 2-Re (Fig. S21†) show depletion of charge density at the ReCO3 fragment and accumulation of density at the ligand aryl (for HOMO(−N) to LUMO transitions, N = 0, 1, 2). For 2-Re, the onset of the MLCT band is near 340 nm, and the remaining spectra is featureless in the visible region, consistent with its colourless nature. For 3-Re, the MLCT band onset occurs near 375 nm, giving rise to its faint yellow colour in solution (and the solid). In contrast, 2-Mn and 3-Mn are yellow orange. Their absorption spectrum (Fig. 5) each feature ligand centred bands like those for the rhenium derivatives but also feature medium intensity (ε ∼1000–4000 M−1 cm−1) MLCT bands in the violet (2-Mn) to blue (3-Mn) region of the visible spectrum, assigned based on intensities of bands, comparisons with related compounds, and on TD-DFT calculations. That is, the lowest energy band in the spectrum of 2-Mn occurs at 350 nm (ε = 2800 M−1 cm−1) which is comparable to the similar band in [(Tpm)Mn(CO)3](PF6) (λmax (EtOH) = 349 nm, ε = 2200 M−1 cm−1) or in [(bpza)Mn(CO)3] (λmax (EtOH) = 361 nm, ε = 1800 M−1 cm−1).42 TD-DFT calculations on 2-Mn show that the lowest energy band is comprised of MLCT transitions between filled orbitals HOMO(−N) (N = 0, 1) involving Mn(CO)x (x = 3, 2) chromophores and virtual orbitals on the scorpionate pi-system, LUMO and LUMO(+1). The red shift for similar transitions in 3-Mn compared to 2-Mn is a consequence of the higher energy of the HOMO(−N) (N = 0, 1) in the former versus the latter.
 | ||
| Fig. 5 Overlay of electronic absorption spectra of 2-Mn (solid blue line) and 3-Mn (dashed purple line) in CH3CN. (Inset) Shows a close-up of the lowest energy bands. | ||
The electrochemical properties of the complexes were examined by cyclic voltammetry in CH3CN, as summarized in Table 3. Most voltammograms exhibit irreversible (ipc/ipa ≪ 1) oxidation waves, apart from that of 3-Re whose oxidation appears reversible (Fig. S15†). An irreversible reduction is observed for 2-Mn, but no clear reduction events were observed within the solvent window for other complexes. Given the uncertainties in assigning potentials for electrochemically irreversible waves, the electrochemical processes were also estimated via theoretical calculations (M06L/Def2-TZVP/PCM(CH3CN), Table 3, and ESI†). The computed values are 100 to 400 mV more negative than the observed values, but otherwise mirror observed trends. In each series 2-M or 3-M both the oxidation and reduction of the manganese derivatives are more favourable than for the corresponding rhenium analogues. Thus, oxidation of 2-Re was found to be 320 mV (and calculated to be 300 mV) more positive than 2-Mn. The reduction of 2-Re was calculated to be only 80 mV more negative than 2-Mn, consistent with the mainly ligand-centred LUMO. Moreover, for each metal, the replacement of a carbonyl group with an acetonitrile causes the oxidation to become more negative by about 600 mV experimentally (calculated to be 930 mV and 740 mV more negative for Mn for Re, respectively). Such behaviour mirrors that in other group 7 dicarbonyl scorpionates: [TpRe(CO)2(THF)] E1/2 = +0.42 V vs. NHE54 versus [TpRe(CO)3] E1/2 = +1.41 V vs. NHE,66 [(Tpm)Mn(CO)2(CH3CN)] E1/2 = +0.58 V vs. NHE versus [(Tpm)Mn(CO)3] E1/2 = +1.53 V vs. NHE,43 and [(bpza)Mn(CO)2(CH3CN)] E1/2 = +0.29 V vs. NHE versus [(bpza)Mn(CO)3] E1/2 = +1.09 V vs. NHE43
| Compound |
|
|
λb nm (ε, M−1 cm−1) |
|---|---|---|---|
| a V versus NHE; NBu4PF6 as supporting electrolyte.b Calculated TD-DFT spectra fit as sum of gaussian curves with σ = 0.2 eV. | |||
| 1 | +1.72 | — | 268 (9000), 305 sh (3000), 341 sh (850) |
| 1calc | +1.34 | −2.05 | 279 (3600), 302 sh (2000), 346 (500) |
| 2-Re | +1.95 | — | 243, (34100), 268 (37000) |
| 2-Recalc | +1.82 | −1.92 | 275 (7500), 322 sh (500) |
| 2-Mn | +1.63 | −1.41 | 270 (40800), 350 (2800) |
| 2-Mncalc | +1.52 | −1.84 | 288 (4900), 326 (3150) |
| 3-Re | +1.35 | — | 232 (78400), 270 (30300), 306 (13500) |
| 3-Recalc | +1.08 | −2.12 | 284 (6850), 322 (5200), 389 sh (325) |
| 3-Mn | +0.99 | — | 274 (7500), 300 sh (3900), 405 (900) |
| 3-Mncalc | +0.59 | −2.09 | 314 (4100), 357 (2600), 445 sh (260) |
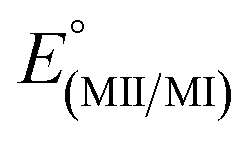
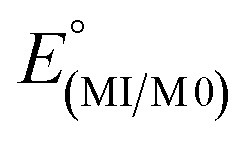
Photo-induced CO release
As expected from the electronic spectra that are devoid of strong absorption bands in the visible region, colourless solutions of 1, 2-Re, or pale-yellow solutions of 3-Re, are stable toward visible light. On the other hand, irradiation of a CH3CN solution 20 mM in 2-Re with a 38 W UV-C 254 nm light causes CO release with t1/2 ∼ 26 min to initially give 3-Re, Fig. 6. This behaviour provides another rare example along with Tp*Re(CO)3, of a rhenium-based CO releasing molecule. Interestingly, complex 3-Re only undergoes slow and partial photodecomposition under these conditions. First, a new species with an asymmetric metal environment is formed, as indicated by two new sets of pyrazolyl resonances in the 1H NMR see the two new H4pz resonances in the top of Fig. 6 at δH 6.62 and 6.44 ppm. The 31P NMR (Fig. S16†) also shows a new resonance at δp = 6.6 ppm. The integrations indicate the new species reaches a maximum concentration of about 20% relative to 3-Re. We tentatively assign this new species as either [cis-(κ2PN-pz2TTP)Re(CO)2(κ2O-OTf)] or [cis-(κ2PN-pz2TTP)Re(CO)2(κ1O-OTf)(CH3CN)]. This assignment is based on the similarity of the 31P NMR resonance and 1H NMR resonances with 1, as well as the shoulders on the low energy side of the two CO stretching bands of 3-Re. It is less likely that the new species is a monocarbonyl, like [cis-(κ3PN2-pz2TTP)Re(CH3CN)2(CO)](OTf), as there is no signal in the expected 1820–1830 cm−1 range. Upon continued irradiation (after 180 min) the NMR and IR signals for all species slowly disappear; the NMR spectra begin to show broad lines associated with paramagnetic species. We have not yet been able to identify the ultimate products of photodecomposition. In contrast, irradiation of 2 mM solutions of 2-Mn in CH3CN under normal atmosphere with a 50 W 390 nm LED light source (a source that matches well with the MLCT band) causes rapid and complete CO release in under 20 minutes with a first order decay half-life of t1/2 = 3 min, indicated by UV-vis and IR spectroscopy, Fig. 7. Here, 3-Mn is an observable intermediate (1880 cm−1 IR band). During irradiation, the original yellow solution becomes a suspension of an insoluble brown precipitate (presumably amorphous manganese oxides) amidst a colourless solution (Fig. S17†). The colourless soluble component was identified as centrosymmetric trans-[MnII(κ2NO-pz2TTPO)2(κ1O-OTf)2], 4, by single crystal X-ray diffraction (Fig. 8). Aside from 3-Mn when starting from 2-Mn, no definitive intermediates have yet been identified which makes it difficult to pinpoint a favoured mechanistic sequence to give 4 at this time. Given the redox properties of 2-Mn and 3-Mn, a photoinduced release of CO's could also produce [(pz2TTP)Mn(CO)(CH3CN)2]+ or [(pz2TTP)Mn(CH3CN)3]+ that are predicted to have Mn2+/Mn+ ground state reduction potentials of +0.39 and −0.35 V vs. NHE, respectively. Neither is sufficiently negative to directly reduce O2 to O2− in CH3CN (−0.6 V vs. NHE)68,69 but with a proton source (adventitious water) or if these act as Lewis acids capable of inner sphere e− transfer, they might be capable or reduction (empirical limit +0.6 V vs. NHE).70,71 If O2 reduction occurs, the resulting manganese(II) species could symmetrize (ligand redistribution) to form [Mn(pz2TTP)2]2+ and [Mn(CH3CN)6]2+ which could react with O2− to form the observed products or the O2− could attack [(pz2TTP)Mn(CH3CN)3]2+ prior to ligand redistribution. Alternatively, and more likely, considering the low energy edge of absorption bands (observed or calculated) and the redox behaviour, the excited state redox potentials of every species in the series [(pz2TTP)Mn(CO)n(CH3CN)3−n]+ (n = 0 to 3) are predicted to be sufficiently negative (<−1.9 V vs. NHE) to reduce oxygen directly. Thus, photooxidation of the manganese(I) complexes might precede CO loss. Our group is continuing to investigate the photoreductive properties of carbonylmanganese complexes and will report our findings in due course.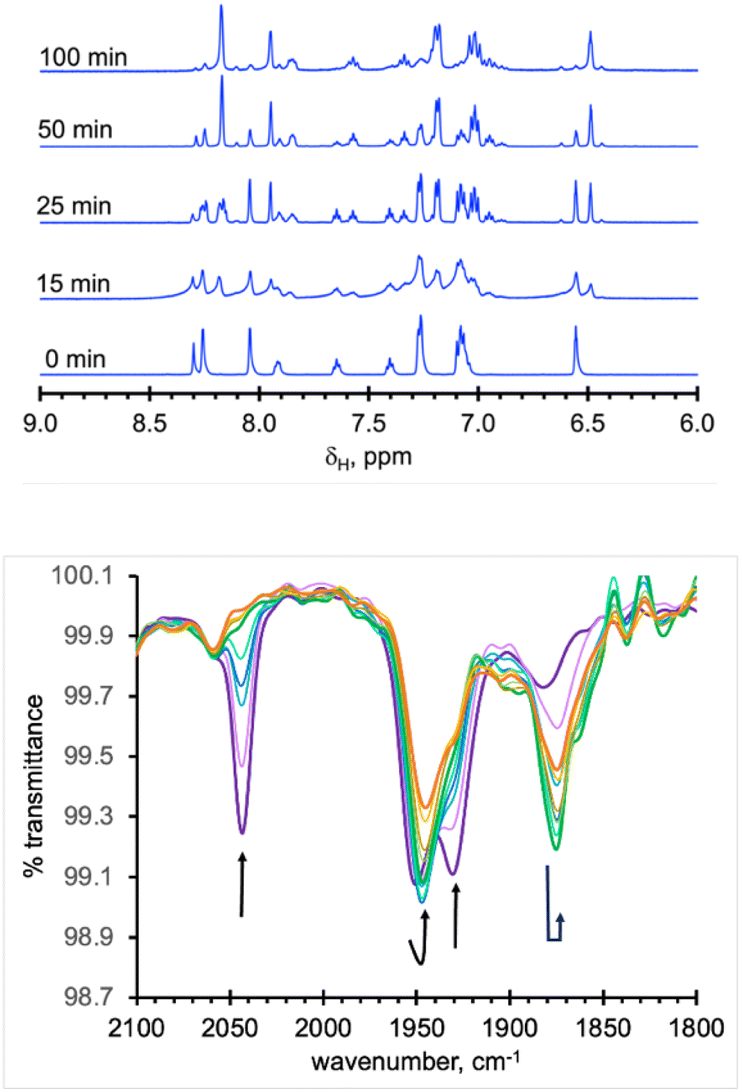 | ||
| Fig. 6 (Top): Overlay of downfield region of 1H NMR spectra of a CD3CN solution initially 20 mM in 2-Re (0 min), then at various stages after irradiation with 254 nm light; (Bottom): overlay of IR spectra of these solutions over similar time period (0 min, thicker purple line) but extended to 360 min (thicker orange line), showing conversion of 2-Re first to 3-Re, then the partial disappearance of the latter after 180 min (thicker green line). | ||
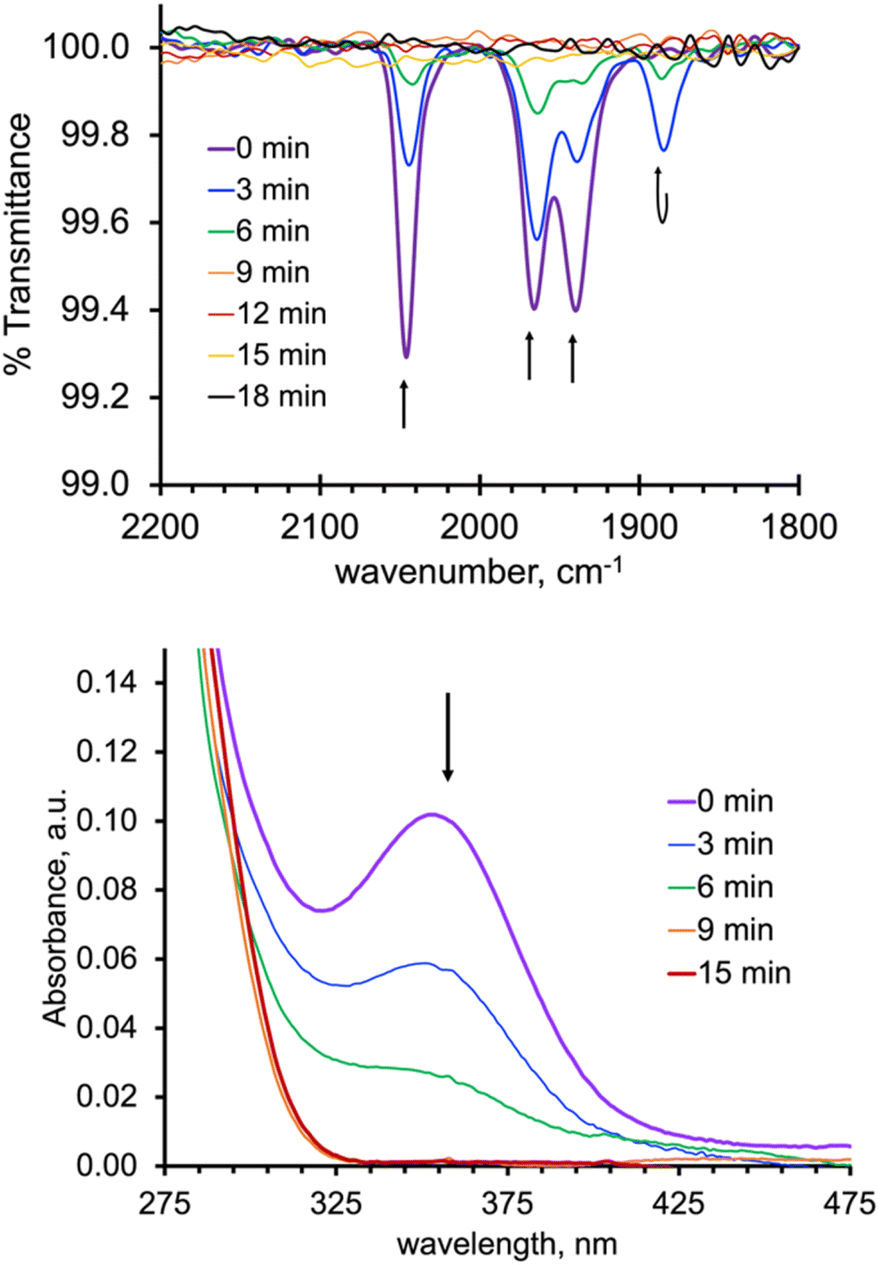 | ||
| Fig. 7 (Top): Overlay of IR spectra of solutions initially 2 mM 2-Mn recorded before and at 3 min intervals while irradiating with 390 nm light; (Bottom): UV-visible spectra of same solutions filtered to remove oxides and diluted to 10−5 M. | ||
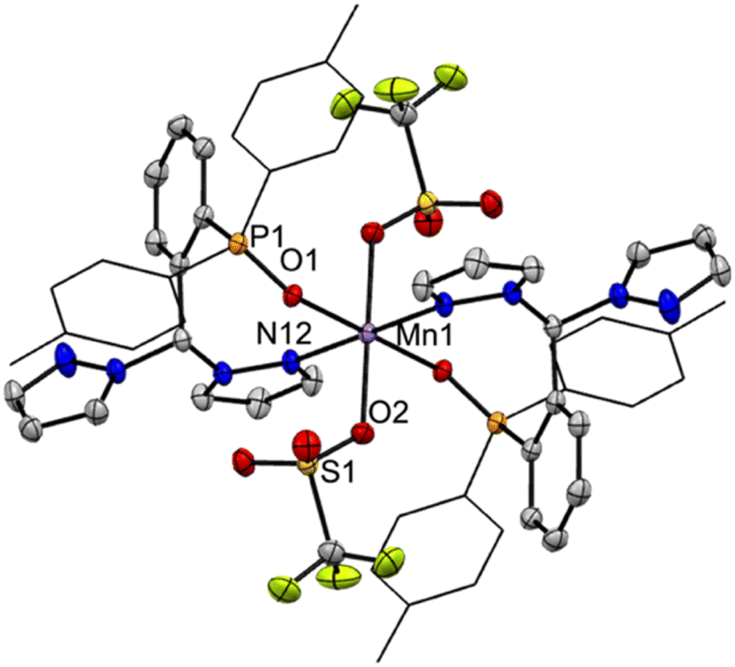 | ||
| Fig. 8 Structure of trans-[Mn(κ2NO-pz2TTPO)2(κ1O-OTf)2], 4, from single crystal X-ray diffraction. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): Mn1–O1 2.1169(10), Mn1–O2 2.2019(11), Mn1–N12 2.3511(14), P1–O1 1.5038(11), S2–O2 1.4571(12); selected bond angles (°): O1–Mn1–O1 180.0, O2–Mn1–O2 180.0, N12–Mn1–N12 180.0, O1–Mn1–O2 88.88(4), O1–Mn1–O2′ 91.12(4), O1–Mn1–N12 94.84(4), O1–Mn1–N12′ 85.16(4), O2–Mn1–N12 86.31(5), O2 Mn1 N12 93.69(5). | ||
Experimental
Materials and methods
The ligand pz2TTP was prepared by the literature route.44 Mn(CO)5Br was used as received from Ambeed whereas Re(CO)5Br was prepared from commercial Re2(CO)10 (Pressure Chemical) by a literature procedure.72 The complex [fac-Mn(CO)3(NCCH3)3](O3SCF3) was prepared by a literature route from Mn(CO)5Br and AgO3SCF3 (Alfa Aesar) in CH3CN.73 Anhydrous Me3NO was prepared from the commercial hydrate (TCI AMERICA) by recrystallization from hot dmf.74 Tl(O3SCF3) was used as received from Sigma Aldrich. It is noted that thallium reagents are highly toxic and extra precautions are required for their safe handling and disposal in accord with local regulations. Midwest MicroLab, LLC, Indianapolis, Indiana 45250, performed all elemental analyses. Melting point determinations were made on samples contained in glass capillaries using an Electrothermal 9100 apparatus and are uncorrected. IR spectra were recorded on solid samples using a Thermo Scientific Nicolet iS5 IR spectrometer equipped with an iD3 Attenuated Total Reflection (ATR) accessory. 1H, 13C, 19F, and 31P NMR spectra were recorded on a Varian 400 MHz spectrometer. Chemical shifts were referenced to solvent resonances at δH 7.26 and δC 77.23 ppm for CDCl3, δH 1.94 and δC 1.32 for CD3CN, or by using external standards δF = 0.00 ppm for CFCl3 or δP = 0.00 ppm for H3PO4 (aq). Abbreviations for NMR and UV-vis: br (broad), sh (shoulder), m (multiplet), ps (pseudo), s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), sept (septet). Electronic absorption (UV-vis/NIR) measurements of solutions were made on an Agilent 8453 Diode array or a Cary 5000 spectrometer. Electrochemical measurements were collected under a nitrogen atmosphere for samples as 0.1 mM solutions in CH3CN with 0.1 M NBu4PF6 as the supporting electrolyte. A three-electrode cell comprised of an Ag/AgCl electrode (separated from the reaction medium with a semipermeable polymer membrane filter), a platinum working electrode, and a glassy carbon counter electrode was used for the voltammetric measurements. Data were collected at scan rates of 50, 100, 200, 300, 400, and 500 mV s−1. With this set up, the ferrocene/ferrocenium couple matched the literature value75,76 with E1/2 = +0.43 V in CH3CN at a scan rate of 200 mV s−1.Crystallography
Single crystal X-ray intensity data from a colourless prism of 1, a light yellow irregular block of 2-Mn, a colourless block of 2-Re, a yellow plate of 3-Mn0.84·2-Mn0.16, a colourless plate of 3-Re, and a colourless block of 4 were each collected at 100.0(1) K with an Oxford Diffraction Ltd. Supernova diffractometer equipped with a 135 mm Atlas CCD detector using Cu(Kα) radiation. Raw data frame integration and Lp corrections were performed with CrysAlisPro (Rigaku Oxford Diffraction, Ltd).77 Final unit cell parameters were determined by least-squares refinement of 12870, 13074, 14655, 12814, 12601, and 11411 reflections of 1, 2-Mn, 2-Re, 3-Mn0.84·2-Mn0.16, 3-Re, and 4 respectively, with I > 2σ(I) for each. Analysis of the data showed negligible crystal decay during collection in each case. Direct methods structure solutions, difference Fourier calculations and full-matrix least-squares refinements against F2 were performed with SHELXTL.78 Numerical absorption corrections based on Gaussian integration over a multifaceted crystal model were applied to the data for each of the complexes as implemented in the SCALE3 ABSPACK scaling algorithm.79 All non-hydrogen atoms were refined with anisotropic displacement parameters with the exception of the minor disorder component CO atoms (20% occupancy) that overlay the Br atom of the major disorder component (80% occupancy) in the structure of 1. Hydrogen atoms were placed in geometrically idealized positions and included as riding atoms. It is also noted that in the structure of 3-Mn0.84·2-Mn0.16 the triflate ion (C14 F1 F2 F3 S3 O3 O4 O5) is most likely disordered across two sites. The careful inspection has revealed the highest Q peak is 0.5 electrons. Modelling such disorder would require heavy restraints due to the low density available. Therefore, we have decided not to model this disorder because no other valuable information would be gained by doing so.
Single crystals of 3-Mn0.92·2-Mn0.08 were grown by vapor diffusion of diethyl ether into acetonitrile at ambient temperature in a closed vial. A small specimen was retrieved, placed on a microscope slide, coated in inert oil, mounted on a MiTeGen Dual Thickness MicroMount and placed on a goniometer head under a cold stream of N2 gas. Data were collected at 100(2) K on a Bruker D8 Venture 4-circle diffractometer coupled to a Photon III C14 CMOS area detector with Cu Kα radiation (λ = 1.54184 Å) provided by a Incoatec IμS 3.0 micro-focus Cu X-ray source running at 50.0 kV and 1.1 mA. All data manipulations for 3-Mn0.92·2-Mn0.08 were carried out using the Bruker APEX-5 Software Suite.80 Data were reduced, correcting for absorption, using the program SADABS.81 Space group assignment was done using XPREP within the SHELXTL suite of programs.82 Using the ShelXle graphical interface, the structure was solved by dual space methods employed by ShelXT and refined by full-matrix least-squares methods against F2 using ShelXL with scattering factors and anomalous dispersion terms taken from the literature.78 All non-hydrogen atoms not apparent from the initial solution were found from difference Fourier maps, and all heavy atoms were refined anisotropically. Special details for 3-Mn0.92·2-Mn0.08. The initial refinement of the structure determined there was additional electron density around the CH3CN molecule (N1 C15 C16), this was initially ignored as it was less than 10% contribution. After further consideration, we have decided to model this additional density as partially occupied CO. The occupancies of both the CO and CH3CN were constrained to a sum of 1 and refined to 0.923(4) for the major component (N1 C15 C16) and 0.077(4) for the minor component (C10 O10). SADI was used for C10 and O10 to keep the bonding appropriate. Additionally, C10 and O10 of the CO molecule and N1 of the CH3CN had their displacement parameters constrained with EADP. Further investigation has determined that the triflate molecule (C14 F1 F2 F3 S3 O3 O4 O5) was disordered across two sites. The occupancies of both components were constrained to a sum of 1 and refined to 0.72(2) for the major component (C14 F1 F2 F3 S3 O3 O4 O5) and 0.28(2) for the minor component (C14A F1A F2A F3A S3A O3A O4A O5A). The minor component was further restrained with SIMU and RIGU. All hydrogen atoms were placed in calculated positions and had their thermal parameters tied to the isotropic thermal parameters of the atoms to which they were bonded (1.5× for methyl, 1.2× for others).
The X-ray crystallographic parameters and further details of data collection and structure refinements are given in Table S1 of the ESI.† CCDC 2371547, 2371549–2371553, and 2383876 contains crystallographic information files. The powder X-ray diffraction (PXRD) patterns were collected at 295 K with a Rigaku MiniFlex II instrument using Cu Ka (1.54178 Å) radiation or at 100 K (for analysis of photolysis products) by using the Oxford Diffraction Ltd instrument.
Synthetic procedures
Photoinduced CO release
Computational methods
Conclusions
The soft C-heteroscorpionate ligand, pz2TTP, binds to rhenium(I) and manganese(I) carbonyl centres in the expected κ3PN2-mode. However, with rhenium(I) the more unusual κ2PN-binding mode has also been found, where the hemilabile pyrazolyl arm dissociates to accommodate metal-ion binding. For [(κ3PN2-pz2TTP)M(CO)3](OTf), 2-M, the CO stretching frequencies indicated a relative donor strength that is intermediate between the traditional C-scorpionate Tpm with an N3-donor set and the anionic hard-heteroscorpionate (bpza) – with an N2O donor set. The electrochemical data of 2-M show that pz2TTP ligand stabilized the univalent state by about 100 mV relative to the Tpm derivative and ca. 500 mV compared to related complexes with anionic scorpionates. The phosphorous donor of pz2TTP labilizes the trans-CO giving cis-[(κ3PN2-pz2TTP)Mn(CO)2(CH3CN)](OTf), 3-Mn simply on heating in CH3CN (t1/2 = 15 min). The rhenium analogue 3-Re is formed exceedingly slowly from 2-Re under these conditions, so is better prepared by decarboxylation with ONMe3. Despite 3-M having stronger M–C bonds than 2-M from IR and single crystal X-ray diffraction data, the 3-M species are more reactive due to two factors. First, 3-M substantially more electron rich than their 2-M counterparts by about 600 mV due to replacement of CO with the poorer π-acceptor CH3CN. Second, the MLCT bands in 3-M are broadened and red-shifted relative to 2-M, increasing their visible light sensitivity. Irradiation of either 2-M or 3-M at their MLCT bands causes total loss of CO over the course of 20 min for Mn derivatives but days for Re compounds. A monocarbonyl was never detected by IR or NMR spectroscopy for either metal. It is likely that, if formed, such species would be even more electron rich and photoreactive than 3-M. The photoreducing properties of 2-Mn were demonstrated by the isolation of [MnII(κ2NO-pz2TTPO)2(κ1O-OTf)2], 4, (and manganese oxides) during photoirradiation under atmospheric conditions, a result of oxygen activation. The potential photoreductive properties of pz2TTP and other scorpionate complexes of M(CO)3+ in organic syntheses warrant further investigation.Data availability
The data supporting this article have been included as part of the ESI.† This data also includes crystallographic data which have been deposited at the CCDC with numbers 2371547, 2371549–2371553 and 2383876, and can be obtained free of charge from https://www.ccdc.cam.ac.uk/structures.Author contributions
JRG was responsible for conceptualizing, supervising, project administration, contributing to writing the original draft, reviewing, and editing subsequent drafts. JPV was responsible for investigation, data curation, formal analysis, and contributed to writing the original draft, reviewing, and editing subsequent drafts. ANE performed and reviewed diffraction experiments and reviewed and edited manuscript drafts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JRG thanks Marquette University for financial support. This research was funded in part by the National Science foundation award CNS-1828649 “MRI: Acquisition of iMARC: High Performance Computing for STEM Research and Education in Southeast Wisconsin”. Crystal data for (CCDC 2383876) was collected at the Marquette University Diffraction Facility with funding for the Bruker D8 Venture diffractometer provided by NSF Award CHE-2320762.Notes and references
- S. Weber and K. Kirchner, Manganese Alkyl Carbonyl Complexes: From Iconic Stoichiometric Textbook Reactions to Catalytic Applications, Acc. Chem. Res., 2022, 55, 2740–2751 CrossRef CAS PubMed.
- K. Das, S. Waiba, A. Jana and B. Maji, Manganese-catalyzed hydrogenation, dehydrogenation, and hydroelementation reactions, Chem. Soc. Rev., 2022, 51, 4386–4464 RSC.
- A. Mukherjee and D. Milstein, Homogeneous Catalysis by Cobalt and Manganese Pincer Complexes, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- S. Hostachy, C. Policar and N. Delsuc, Re(I) carbonyl complexes: Multimodal platforms for inorganic chemical biology, Coord. Chem. Rev., 2017, 351, 172–188 CrossRef CAS.
- Y. Kuninobu and K. Takai, Organic Reactions Catalyzed by Rhenium Carbonyl Complexes, Chem. Rev., 2011, 111, 1938–1953 CrossRef CAS PubMed.
- P. R. Elowe, N. M. West, J. A. Labinger and J. E. Bercaw, Transformations of Group 7 Carbonyl Complexes: Possible Intermediates in a Homogeneous Syngas Conversion Scheme, Organometallics, 2009, 28, 6218–6227 CrossRef CAS.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Anticancer activity of complexes of the third row transition metals, rhenium, osmium, and iridium, Dalton Trans., 2018, 47, 9934–9974 RSC.
- K. Schindler and F. Zobi, Anticancer and Antibiotic Rhenium Tri- and Dicarbonyl Complexes: Current Research and Future Perspectives, Molecules, 2022, 27, 539 CrossRef CAS PubMed.
- R. Alberto, Role of Pure Technetium Chemistry: Are There Still Links to Applications in Imaging?, Inorg. Chem., 2023, 62, 20539–20548 CrossRef CAS PubMed.
- J. Marhenke, K. Trevino and C. Works, The chemistry, biology and design of photochemical CO releasing molecules and the efforts to detect CO for biological applications, Coord. Chem. Rev., 2016, 306, 533–543 CrossRef CAS.
- U. Schatzschneider, PhotoCORMs: Light-triggered release of carbon monoxide from the coordination sphere of transition metal complexes for biological applications, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS.
- E. Kottelat and Z. Fabio, Visible Light-Activated PhotoCORMs, Inorganics, 2017, 5, 24 CrossRef.
- T. T. Tien Vo, Q. C. Vo, V. P. Tuan, Y. Wee, H.-C. Cheng and I.-T. Lee, The potentials of carbon monoxide-releasing molecules in cancer treatment: An outlook from ROS biology and medicine, Redox Biol., 2021, 46, 102124 CrossRef CAS PubMed.
- M. A. Andrade and L. M. D. R. S. Martins, Novel Chemotherapeutic Agents – The Contribution of Scorpionates, Curr. Med. Chem., 2019, 26, 7452–7475 CrossRef CAS PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Visible-to-NIR-Light Activated Release: From Small Molecules to Nanomaterials, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- M. A. Wright and J. A. Wright, PhotoCORMs: CO release moves into the visible, Dalton Trans., 2016, 45, 6801–6811 RSC.
- A. J. Kosanovich, W.-C. Shih and O. V. Ozerov, Synthesis and characterization of unsaturated Manganese(I) and Rhenium(I) dicarbonyl complexes supported by an anionic PNP pincer, J. Organomet. Chem., 2019, 897, 1–6 CrossRef CAS.
- T. B. Gunnoe, M. Sabat and W. D. Harman, Reactions of TpRe(CO)2(THF) with Aromatic Molecules (Tp = Hydridotris(pyrazolyl)borate), J. Am. Chem. Soc., 1998, 120, 8747–8754 CrossRef CAS.
- R. G. Bergman, T. R. Cundari, A. M. Gillespie, T. B. Gunnoe, W. D. Harman, T. R. Klinckman, M. D. Temple and D. P. White, Computational Study of Methane Activation by TpRe(CO)2 and CpRe(CO)2 with a Stereoelectronic Comparison of Cyclopentadienyl and Scorpionate Ligands, Organometallics, 2003, 22, 2331–2337 CrossRef CAS.
- D. M. Tellers, S. J. Skoog, R. G. Bergman, T. B. Gunnoe and W. D. Harman, Comparison of the Relative Electron-Donating Abilities of Hydridotris(pyrazolyl)borate and Cyclopentadienyl Ligands: Different Interactions with Different Transition Metals, Organometallics, 2000, 19, 2428–2432 CrossRef CAS.
- D. Álvarez, M. I. Menéndez and R. López, Computational Design of Rhenium(I) Carbonyl Complexes for Anticancer Photodynamic Therapy, Inorg. Chem., 2022, 61, 439–455 CrossRef PubMed.
- K. Schindler, A. Crochet and F. Zobi, Aerobically stable and substitutionally labile α-diimine rhenium dicarbonyl complexes, RSC Adv., 2021, 11, 7511–7520 RSC.
- A. Otero, J. Fernández-Baeza, A. Lara-Sánchez, J. Tejeda and L. F. Sánchez-Barba, Recent Advances in the Design and Coordination Chemistry of Heteroscorpionate Ligands Bearing Stereogenic Centres, Eur. J. Inorg. Chem., 2008, 2008, 5309–5326 CrossRef.
- S. Trofimenko, Recent advances in poly(pyrazolyl)borate (scorpionate) chemistry, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- A. Otero, J. Fernández-Baeza, A. Lara-Sánchez and L. F. Sánchez-Barba, Metal complexes with heteroscorpionate ligands based on the bis(pyrazol-1-yl)methane moiety: Catalytic chemistry, Coord. Chem. Rev., 2013, 257, 1806–1868 CrossRef CAS.
- C. Pettinari and R. Pettinari, Metal derivatives of poly(pyrazolyl)alkanes: II. Bis(pyrazolyl)alkanes and related systems, Coord. Chem. Rev., 2005, 249, 663–691 CrossRef CAS.
- L. M. D. R. S. Martins, C-scorpionate complexes: Ever young catalytic tools, Coord. Chem. Rev., 2019, 396, 89–102 CrossRef CAS.
- C. Pettinari, Scorpionates II: Chelating Borate Ligands, Imperial College Press, London, 2008 Search PubMed.
- S. Pai, M. Hafftlang, G. Atongo, C. Nagel, J. Niesel, S. Botov, H.-G. Schmalz, B. Yard and U. Schatzschneider, New modular manganese(I) tricarbonyl complexes as PhotoCORMs: in vitro detection of photoinduced carbon monoxide release using COP-1 as a fluorogenic switch-on probe, Dalton Trans., 2014, 43, 8664–8678 RSC.
- D. L. Reger, J. R. Gardinier, T. C. Grattan and M. D. Smith, Tricarbonylmanganese(I) derivatives of [Di(pyrazolyl)(2-pyridyl)methyl]aryl scorpionates, J. Organomet. Chem., 2005, 690, 1901–1912 CrossRef CAS.
- J. R. Gardinier, K. J. Meise, F. Jahan and S. V. Lindeman, Reaction Chemistry of Silver(I) Trifluoromethanesulfonate Complexes of Nitrogen-Confused C-Scorpionates., Inorg. Chem., 2018, 57, 1572–1589 CrossRef CAS PubMed.
- E. Hübner, G. Türkoglu, M. Wolf, U. Zenneck and N. Burzlaff, Novel N, N,O Scorpionate Ligands and Transition Metal Complexes Thereof Suitable for Polymerisation, Eur. J. Inorg. Chem., 2008, 2008, 1226–1235 CrossRef.
- W. Tzegai, M. Reil and N. Burzlaff, Bis(4-carboxylpyrazol-1-yl)acetic acid: a scorpionate ligand for complexes with improved water solubility, Dalton Trans., 2022, 51, 6839–6845 RSC.
- N. Burzlaff, I. Hegelmann and B. Weibert, Bis(pyrazol-1-yl)acetates as tripodal “scorpionate” ligands in transition metal carbonyl chemistry: syntheses, structures and reactivity of manganese and rhenium carbonyl complexes of the type [LM(CO)3] (L = bpza, bdmpza), J. Organomet. Chem., 2001, 626, 16–23 CrossRef CAS.
- F. Strinitz, P. Trautner, H. Pfeiffer, U. Schatzschneider and N. Burzlaff, Synthesis and characterization of heteroscorpionate-based manganese carbonyl complexes as CO-releasing molecules, Tetrahedron, 2015, 71, 2951–2954 CrossRef CAS.
- E. Hübner, N. V. Fischer, F. W. Heinemann, U. Mitra, V. Dremov, P. Müller and N. Burzlaff, N, N,O Ligands Based on Triazoles and Transition Metal Complexes Thereof, Eur. J. Inorg. Chem., 2010, 2010, 4100–4109 CrossRef.
- L. Peters, E. Hübner, T. Haas, F. W. Heinemann and N. Burzlaff, 3,3-Bis(3,5-dimethylpyrazol-1-yl)propionic acid: A tripodal N,N,O ligand for manganese and rhenium complexes – Syntheses and structures, J. Organomet. Chem., 2009, 694, 2319–2327 CrossRef CAS.
- G. Türkoglu, F. W. Heinemann and N. Burzlaff, Transition metal complexes bearing a 2,2-bis(3,5-dimethylpyrazol-1-yl)propionate ligand: one methyl more matters, Dalton Trans., 2011, 40, 4678–4686 RSC.
- L. Peters, E. Hübner, T. Haas, F. W. Heinemann and N. Burzlaff, 3,3-Bis(3,5-dimethylpyrazol-1-yl)propionic acid: A tripodal N,N,O ligand for manganese and rhenium complexes – Syntheses and structures, J. Organomet. Chem., 2009, 694, 2319–2327 CrossRef CAS.
- C. Reus, K. Ruth, S. Tüllmann, M. Bolte, H.-W. Lerner, B. Weber, M. C. Holthausen and M. Wagner, Synthesis, Molecular Structure, and Physical Properties of the Complexes [PhB(pz)2(CH2SMe)2M] (M = MnII, FeII; pz = pyrazol-1-yl) Containing a Novel [N, N,S]-Heteroscorpionate Ligand, Eur. J. Inorg. Chem., 2011, 2011, 1709–1718 CrossRef.
- T. Godau, F. Platzmann, F. W. Heinemann and N. Burzlaff, An enantiopure N, N,S scorpionate ligand derived from (+)-camphor, Dalton Trans., 2008, 254–255 Search PubMed.
- H.-M. Berends and P. Kurz, Investigation of light-triggered carbon monoxide release from two manganese photoCORMs by IR, UV-vis and EPR spectroscopy, Inorg. Chim. Acta, 2012, 380, 141–147 CrossRef CAS.
- U. Sachs, G. Schaper, D. Winkler, D. Kratzert and P. Kurz, Light- or oxidation-triggered CO release from [MnI(CO)3(κ3-L)] complexes: reaction intermediates and a new synthetic route to [MnIII/IV2(μ-O)2(L)2] compounds, Dalton Trans., 2016, 45, 17464–17473 RSC.
- J. R. Gardinier, J. S. Hewage and S. V. Lindeman, Isomer Dependence in the Assembly and Lability of Silver(I) Trifluoromethanesulfonate Complexes of the Heteroditopic Ligands, 2-, 3-, and 4-[Di(1H-pyrazolyl)methyl]phenyl(di-p-tolyl)phosphine, Inorg. Chem., 2014, 53, 12108–12121 CrossRef CAS PubMed.
- M. Abubekerov and P. L. Diaconescu, Synthesis and Characterization of Ferrocene-Chelating Heteroscorpionate Complexes of Nickel(II) and Zinc(II), Inorg. Chem., 2015, 54, 1778–1784 CrossRef CAS PubMed.
- M. Abubekerov, S. M. Shepard and P. L. Diaconescu, Switchable Polymerization of Norbornene Derivatives by a Ferrocene-Palladium(II) Heteroscorpionate Complex, Eur. J. Inorg. Chem., 2016, 2016, 2634–2640 CrossRef CAS.
- M. Y. Petyuk, A. S. Berezin, A. L. Gushchin, I. Y. Bagryanskaya, A. Y. Baranov and A. V. Artem’ev, Luminescent Re(I) scorpionates supported by tris(2-pyridyl)phosphine and its derivatives, Inorg. Chim. Acta, 2021, 516, 120136 CrossRef CAS.
- D. H. Gibson, M. S. Mashuta and H.-Y. He, Tri-carbonyl-[1,1′,1′′-ethylidyne-tris(pyrazole-κN2)]-rhenium(I) bromide and tri-carbonyl-[methylidyne-tris-(pyrazole-κN2)]-rhenium(I) iodide ethanol hemisolvate, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 1135–1137 CrossRef CAS PubMed.
- P. C. Kunz, M. Berghahn, N. E. Brückmann, M. Dickmeis, M. Kettel and B. Spingler, Functionalised Tris(pyrazolyl)methane Ligands and Re(CO)3 Complexes Thereof, Z. Anorg. Allg. Chem., 2009, 635, 471–478 CrossRef CAS.
- D. L. Reger, K. J. Brown and M. D. Smith, Rhenium tricarbonyl complexes of tris(pyrazolyl)methane ligands: first structural characterization of an isomer pair of tris(pyrazolyl)methane derivatives and the supramolecular structure of the homobimetallic complex {1,4-C6H4[CH2OCH2C(pz)3]2[Re(CO)3]2}(Br)2, J. Organomet. Chem., 2002, 658, 50–61 CrossRef CAS.
- J. Niesel, A. Pinto, H. W. P. N'Dongo, K. Merz, I. Ott, R. Gust and U. Schatzschneider, Photoinduced CO release, cellular uptake and cytotoxicity of a tris(pyrazolyl)methane (tpm) manganese tricarbonyl complex, Chem. Commun., 2008, 1798–1800 RSC.
- D. L. Reger, T. C. Grattan, K. J. Brown, C. A. Little, J. J. S. Lamba, A. L. Rheingold and R. D. Sommer, Syntheses of tris(pyrazolyl)methane ligands and {[tris(pyrazolyl)methane]Mn(CO)3}SO3CF3 complexes: comparison of ligand donor properties, J. Organomet. Chem., 2000, 607, 120–128 CrossRef CAS.
- M. Angaroni, G. A. Ardizzoia, G. D'Alfonso, G. L. Monica, N. Masciocchi and M. Moret, Photochemical reactions of tricarbonyl[hydrotris(1-pyrazolyl)borato]rhenium(I) in the presence of neutral donor ligands. X-Ray crystal structures of the substitution derivatives [Re{HB(pz)3}(CO)2L], with L = C4H8O or PPh3, J. Chem. Soc., Dalton Trans., 1990, 1895–1900 RSC.
- T. B. Gunnoe, M. Sabat and W. D. Harman, Reactions of TpRe(CO)2(THF) with Aromatic Molecules (Tp = Hydridotris(pyrazolyl)borate), J. Am. Chem. Soc., 1998, 120, 8747–8754 CrossRef CAS.
- P. Kurz, D. Rattat, D. Angst, H. Schmalle, B. Spingler, R. Alberto, H. Berke and W. Beck, The chemistry of the fac-[Re(CO)2(NO)]2+ fragment in aqueous solution, Dalton Trans., 2005, 804–810 RSC.
- A. J. Hallett, R. A. Baber, A. G. Orpen and B. D. Ward, The co-ordination chemistry of tris(3,5-dimethylpyrazolyl)methane manganese carbonyl complexes: Synthetic, electrochemical and DFT studies, Dalton Trans., 2011, 40, 9276–9283 RSC.
- N. J. Stone, Table of nuclear magnetic dipole and electric quadrupole moments, At. Data Nucl. Data Tables, 2005, 90, 75–176 CrossRef CAS.
- A. M. Camp, M. R. Kita, J. Grajeda, P. S. White, D. A. Dickie and A. J. M. Miller, Mapping the Binding Modes of Hemilabile Pincer–Crown Ether Ligands in Solution Using Diamagnetic Anisotropic Effects on NMR Chemical Shift, Inorg. Chem., 2017, 56, 11141–11150 CrossRef CAS PubMed.
- K. E. Aldrich, B. S. Billow, D. Holmes, R. D. Bemowski and A. L. Odom, Weakly Coordinating yet Ion Paired: Anion Effects on an Internal Rearrangement, Organometallics, 2017, 36, 1227–1237 CrossRef CAS.
- W. M. Ward, B. H. Farnum, M. Siegler and G. J. Meyer, Chloride Ion-Pairing with Ru(II) Polypyridyl Compounds in Dichloromethane, J. Phys. Chem. A, 2013, 117, 8883–8894 CrossRef CAS PubMed.
- E. Kleinpeter, A. Koch, H. S. Sahoo and D. K. Chand, Anisotropic effect of the nitrate anion—manifestation of diamagnetic proton chemical shifts in the 1H NMR spectra of NO3− coordinated complexes, Tetrahedron, 2008, 64, 5044–5050 CrossRef CAS.
- H. S. Sahoo, D. K. Chand, S. Mahalakshmi, M. H. Mir and R. Raghunathan, Manifestation of diamagnetic chemical shifts of proton NMR signals by an anisotropic shielding effect of nitrate anions, Tetrahedron Lett., 2007, 48, 761–765 CrossRef CAS.
- A. Macchioni, Ion Pairing in Transition-Metal Organometallic Chemistry, Chem. Rev., 2005, 105, 2039–2074 CrossRef CAS PubMed.
- G. N. La Mar, Isotropic Shifts of Some Ionic Complexes of Cobalt(II) and Nickel(II) : Evidence for Ion Pairing, J. Chem. Phys., 1964, 41, 2992–2998 CrossRef CAS.
- J. Ammer, C. Nolte, K. Karaghiosoff, S. Thalmair, P. Mayer, R de Vivie-Riedle and H. Mayr, Ion-Pairing of Phosphonium Salts in Solution: C–H⋯Halogen and C–H⋯π Hydrogen Bonds, Chem.–Eur. J., 2013, 14612–14630 CrossRef CAS PubMed.
- K. Wu, D. Mukherjee, A. Ellern, A. D. Sadow and W. E. Geiger, Anodic electrochemistry of Mn and Re tricarbonyl complexes of tris(oxazolinyl)phenyl borate ligands : comparison to tris(pyrazolyl) borate complexes, New J. Chem., 2011, 35, 2169–2178 RSC.
- A. R. Schoenberg and W. P. Anderson, Photochemical reactions of poly(1-pyrazolyl)boratotricarbonylmanganese(I) with phosphorus donor ligands, Inorg. Chem., 1972, 11, 85–87 CrossRef CAS.
- M. E. Peover and B. S. White, Electrolytic reduction of oxygen in aprotic solvents: The superoxide ion, Electrochim. Acta, 1966, 11, 1061–1067 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Superoxide Ion: Generation and Chemical Implications, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- S. O. Kim, C. V. Sastri, M. S. Seo, J. Kim and W. Nam, Dioxygen Activation and Catalytic Aerobic Oxidation by a Mononuclear Nonheme Iron(II) Complex, J. Am. Chem. Soc., 2005, 127, 4178–4179 CrossRef CAS PubMed.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Standard Reduction Potentials for Oxygen and Carbon Dioxide Couples in Acetonitrile and N,N-Dimethylformamide, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS PubMed.
- S. P. Schmidt, W. C. Trogler, F. Basolo, M. A. Urbancic and J. R. Shapley, Pentacarbonylrhenium Halides., Inorg. Synth., 2007, 28, 160–165 CrossRef.
- D. A. Edwards and J. Marshalsea, The synthesis and spectral features of some cationic manganese(I) and rhenium(I) tricarbonyl complexes, J. Organomet. Chem., 1977, 131, 73–91 CrossRef CAS.
- J. A. Soderquist and C. L. Anderson, Crystalline anhydrous trimethylamine N-oxide, Tetrahedron Lett., 1986, 27, 3961–3962 CrossRef CAS.
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple for Studying Solvent Effects on the Thermodynamics of Electron Transfer, J. Phys. Chem. B, 1999, 103, 6713–6722 CrossRef CAS.
- D. Bao, B. Millare, W. Xia, B. G. Steyer, A. A. Gerasimenko, A. Ferreira, A. Contreras and V. I. Vullev, Electrochemical Oxidation of Ferrocene: A Strong Dependence on the Concentration of the Supporting Electrolyte for Nonpolar Solvents, J. Phys. Chem. A, 2009, 113, 1259–1267 CrossRef CAS PubMed.
- CrysAlisPro, Version 1.171.36.28, Agilent Technologies, Santa Clara, CA, 2013 Search PubMed.
- (a) C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: A Qt Graphical User Interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed; (b) G. M. Sheldrick, SHELXT - Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed; (c) G. M. Sheldrick, Crystal Structure Refinement with SHELX, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed; (d) International Tables for Crystallography, Kluwer Academic Publishers, Dordrecht, Netherlands, 3rd edn, 2004, vol. C Search PubMed.
- SCALE3 ABSPACK-An Oxford Diffraction Program (1.0.4.Gui: 1.03)©, 2005 Search PubMed.
- Bruker, Apex-5 (Version 2023.9-4) and SAINT (Version 8.40B), Bruker AXS Inc., Madison, Wisconsin, USA, 2024 Search PubMed.
- (a) SADABS (Version 2016/2), Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed; (b) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of Silver and Molybdenum Microfocus X-ray Sources for Single-Crystal Structure Determination, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- XPREP (Version 2014/2), Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussiañ16 Revision C.01, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- N. Mardirossian and M. Head-Gordon, How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements?, J. Chem. Theory Comput., 2016, 12, 4303–4325 CrossRef CAS PubMed.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model, J. Chem. Phys., 2006, 124, 094107 CrossRef PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- A. D. Laurent and D. Jacquemin, TD-DFT benchmarks: A review, Int. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef CAS.
- L. E. Roy, E. Jakubikova, M. G. Guthrie and E. R. Batista, Calculation of One-Electron Redox Potentials Revisited. Is It Possible to Calculate Accurate Potentials with Density Functional Methods?, J. Phys. Chem. A, 2009, 113, 6745–6750 CrossRef CAS PubMed.
- L. Rulíšek, On the Accuracy of Calculated Reduction Potentials of Selected Group 8 (Fe, Ru, and Os) Octahedral Complexes, J. Phys. Chem. C, 2013, 117, 16871–16877 CrossRef.
- J. H. Letcher and J. R. Van Wazer, Theoretical Interpretation of 31P NMR Chemical Shifts. I, J. Chem. Phys., 1966, 44, 815–829 CrossRef CAS.
- D. Purdela, Theory of 31P NMR chemical shifts. II. Bond-angle dependence, J. Magn. Reson., 1969, 1971(5), 23–36 Search PubMed.
- J. Tong, S. Liu, S. Zhang and S. Z. Li, Prediction of 31P nuclear magnetic resonance chemical shifts for phosphines, Spectrochim. Acta, Part A, 2007, 67, 837–846 CrossRef PubMed.
- D. G. Gorenstein, Phosphorous-31 NMR: Principles and Applications, Academic Press Inc, New York, 1984 Search PubMed.
- J. B. Mann, T. L. Meek, E. T. Knight, J. F. Capitani and L. C. Allen, Configuration Energies of the d-Block Elements, J. Am. Chem. Soc., 2000, 122, 5132–5137 CrossRef CAS.
- L. C. Allen, Electronegativity is the average one-electron energy of the valence-shell electrons in ground-state free atoms, J. Am. Chem. Soc., 1989, 111, 9003–9014 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectra, computational details, xyz files of calculated structures, cif files, checkcif reports. CCDC 2371547, 2371549–2371553 and 2383876. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05287k |
| ‡ As with many other systems (e.g.; see δP's of XPCl3 X = O, S, Se), there are no simple explanations for the observed trends in 31P NMR chemical shifts for the 2-M, and 3-M series. The shift δP depends on many competing factors like bond-distance (diamagnetic contribution) and on paramagnetic terms such as bond angles, relative electronegativities, as well as d- and p- π-bonding contributions.94–97 In the current cases, there is a parabolic relation between the Mulliken charge on phosphorus and chemical shift (see Fig. S22 of the ESI†) that, in turn, may be the result of many of the above factors. The shorter bond distance and greater spectroscopic electronegativity of Mn (χspec (Mn) = 1.75 (Pauling units) vs. χspec (Re) = 1.59)98,99 versus Re may be ultimately responsible for downfield shift in Mn versus Re complexes. It is noted that Pauling electronegativities are reverse order of spectroscopic electronegativities (χPauling (Mn) = 1.55 vs. χPauling (Re) = 1.90). For a given metal, the differences in chemical shifts between 2-M and 3-M counter expectations. The more electron rich 3-M (with shorter M–P bonds due to increased M → P backbonding) is shifted downfield relative to 2-M; the reverse trend might be expected. |
| This journal is © The Royal Society of Chemistry 2024 |