
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deoxyfluorination with superacids – synthesis and characterization of protonated α-fluorohydroxyacetic acid†
Alan Virmani *,
Christoph Jessen,
Alexander Nitzer and
Andreas J. Kornath‡
*,
Christoph Jessen,
Alexander Nitzer and
Andreas J. Kornath‡
Ludwig-Maximilians-Universität München, Butenandtstraße 5-13 (D), D-81377 München, Germany
First published on 4th October 2024
Abstract
α-Fluoroalcohols describe a rare and unstable class of compounds, accessible mainly by fluorination of highly electrophilic carbonyl compounds. In this work, we report the syntheses of α-fluorohydroxyacetic acid (FHA) and its acyl fluoride (FHA-F) by reacting the dihydroxy species glyoxylic acid monohydrate (GAM) with SF4. Surprisingly, only one of the geminal hydroxy groups is substituted when excess SF4 is employed. Implementing GAM with the binary superacid HF/AsF5 also leads to a single yet quantitative deoxyfluorination at the diol group. The reaction pathways are discussed based on NMR experiments, the characterization was carried out using NMR and vibrational spectroscopy as well as single-crystal X-ray diffraction.
Introduction
Organic compounds containing carbon atoms with more than one hydroxy group are known to be labile under regular conditions. According to the rule of Erlenmeyer, they undergo facile dehydration under the formation of the respective carbonyl compound. This also applies to alcohols with a geminal halogen atom, where the hydrogen halide is easily eliminated.1 In the case of fluorinated compounds, only a few examples of α-fluoroalcohols are known. Fluoromethanol CFH2OH, the simplest representative, was synthesized by Olah and Pavláth as early as 1953.2 In 1977, Seppelt was able to generate the perfluorinated alcohol trifluoromethanol CF3OH by reacting CF3OCl with HCl. He operated at low temperatures to prevent the decomposition under the formation of COF2 and HF, which is highly favored.3 30 years later, Christe et al. investigated this equilibrium.4 The addition of HF or F− to a carbonyl group is a convenient way to access (per-)fluorinated alcohols, first shown by Andreades and England in 1961,5 followed by others in recent studies.6,7 The general equation is given below (eqn (1)).
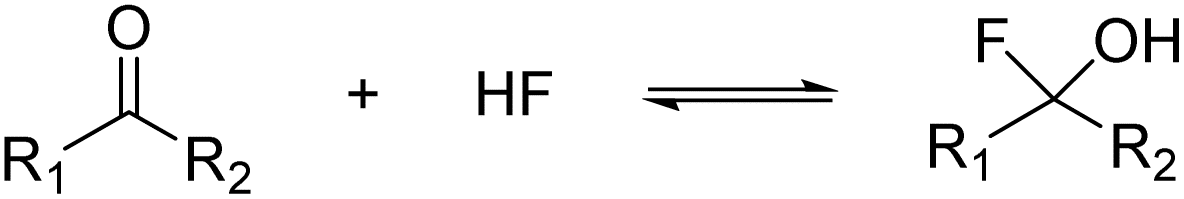 | (1) |
However, the α-fluoroalcohol is only stable when the electrophilicity of the carbonyl group is high enough, similar to the rule of Erlenmeyer.8 The equilibrium of eqn (1) can be shifted to the right by transforming the alcohol into stable derivatives like acetals or oxonium ions.4,9,10 The respective oxonium ions were generated by reacting the carbonyl compounds with the superacidic system HF/SbF5 in anhydrous hydrogen fluoride (aHF). In this way, the perfluorinated oxonium ions of methanol, ethanol, n-propanol,10 and isopropanol11 have been synthesized.
An example of an exception to the rule of Erlenmeyer is glyoxylic acid (GA). The purchasable monohydrate form (GAM) does not imply co-crystallized but chemically bound water and is better described as dihydroxyacetic acid. Its reactivity toward highly acidic systems, in which it can be activated for electrophilic reactions, has been described by Prakash et al.12 The high electrophilicity makes it an interesting target for generating α-fluorohydroxy compounds with an additional functional group in the direct vicinity. To exploit this possibility or to determine if a difluorinated product is formed, we have implemented GAM with the deoxyfluorinating agent SF4 as well as the superacidic medium HF/AsF5. We wish to report the results herein.
Results and discussion
Syntheses and properties
α-Fluorohydroxyacetic acid (FHA, 1) is synthesized by reacting glyoxylic acid monohydrate (GAM) with an equimolar amount of sulfur tetrafluoride (eqn (2)). For the synthesis of α-fluorohydroxyacetyl fluoride (FHA-F, 2), a twofold amount of SF4 is applied (eqn (3)). The formation of difluoroacetyl fluoride was not observed with an excess of SF4.
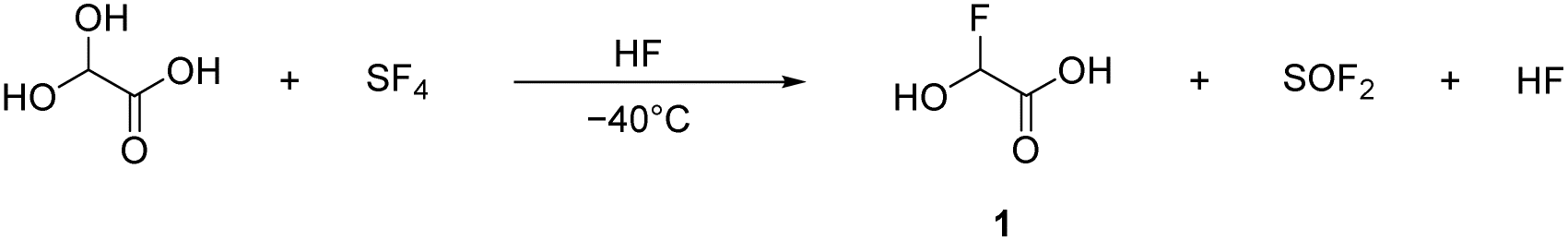 | (2) |
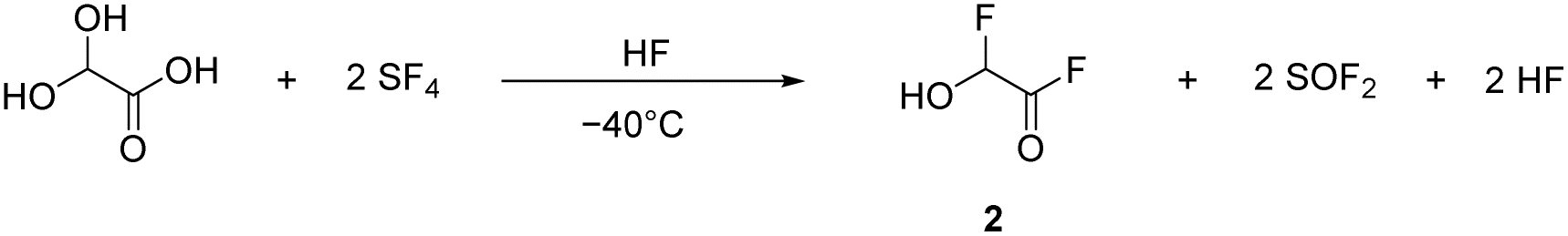 | (3) |
The mechanism of GAM in the system HF/SF4 is proposed based on the literature-reported pathways of similar reactions.13–15 In the first step, SF4 dissociates in aHF according to eqn (4).
 | (4) |
We confirm that the alcohol moiety is more nucleophilic than the carboxy group, which is why the first deoxyfluorination takes place there (Scheme 1). The reactive intermediate is a planar oxonium ion. Since the addition of a nucleophile in this mechanism is not stereoselective, a racemic mixture is expected.
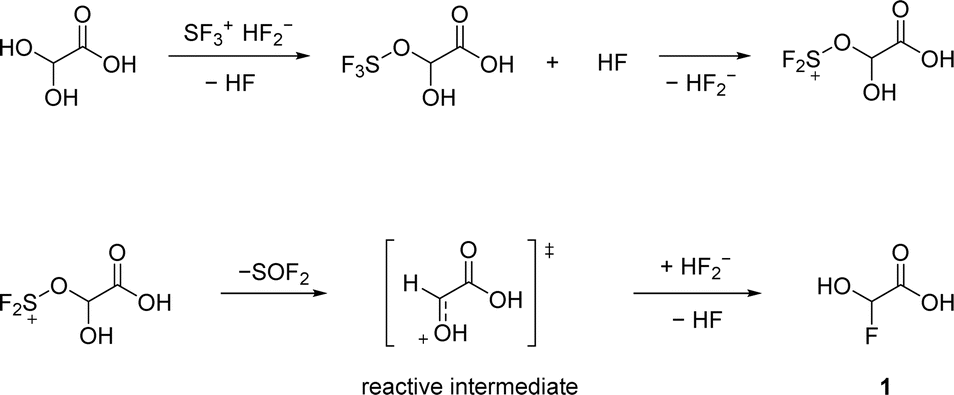 | ||
| Scheme 1 Proposed reaction pathway of GAM with equimolar amounts of SF4. | ||
The second deoxyfluorination of the carboxylic group proceeds in a similar fashion. However, in this case, the formation of a tetrahedral intermediate is likely, as it has been suggested in previous studies about the reactions of carbonyl compounds with HF/SF4.13,15 The proposed mechanism is illustrated in Scheme 2.
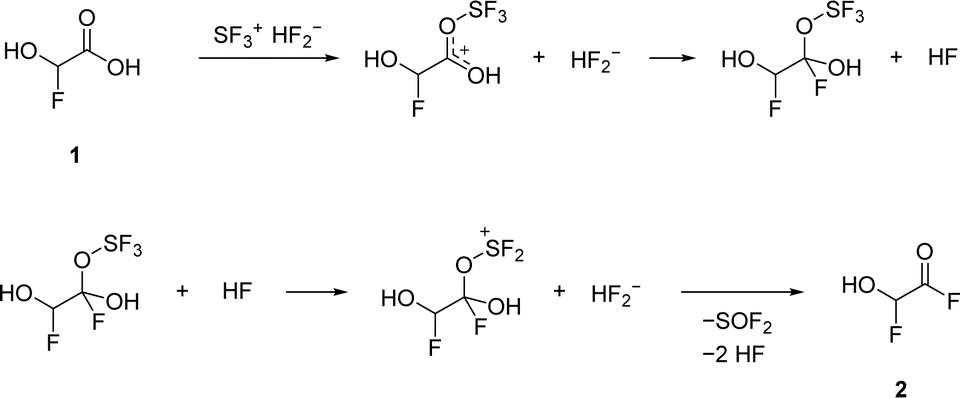 | ||
| Scheme 2 Proposed reaction pathway of the deoxyfluorination reaction at the carboxy group. | ||
Employing three or more equivalents of SF4 did not result in a third deoxyfluorination of the last hydroxy group, hence in 2,2-difluoroacetyl fluoride. Since the cationic intermediate would be a fluoro carbenium ion, it is presumably not sufficiently stabilized.
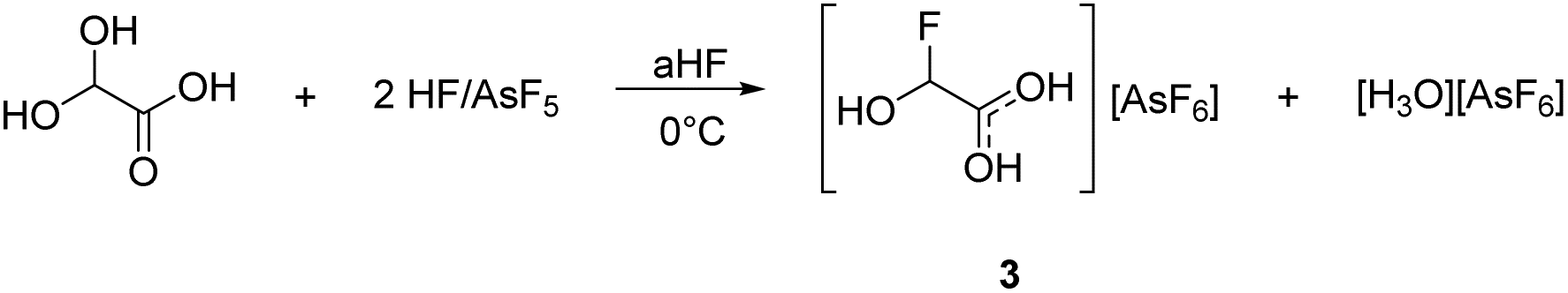
The deoxyfluorination agent SF4 has been investigated in the past.13,15 Interestingly, a similar reaction is observed for GAM reacting with superacids. Protonated α-fluorohydroxyacetic acid [FHA-1H]+ is generated from GAM in the superacidic system HF/AsF5, resulting in [FHA-1H][AsF6] (3). The reaction is visualized in eqn (5).
 | (5) |
According to eqn (5), a two-to-one ratio of AsF5 to GAM would formally suffice to form [FHA-1H][AsF6]. However, full conversion takes place only when three equivalents of Lewis acid are applied. The carboxy group is more basic than the hydroxy groups due to better resonance stabilization, which is why it is likely protonated in the first step. The second protonation occurs at one of the hydroxy groups that subsequently is eliminated as H3O+. The formed substituted ethylene dication [C2(OH)3H]2+ is superelectrophilic enough to add fluoride from the solvent aHF, similar to observations we made in a recent study.16 3 is found as a racemic mixture of the two enantiomers, strongly indicating an SN1 mechanism. The proposed mechanism is displayed in Scheme 3.
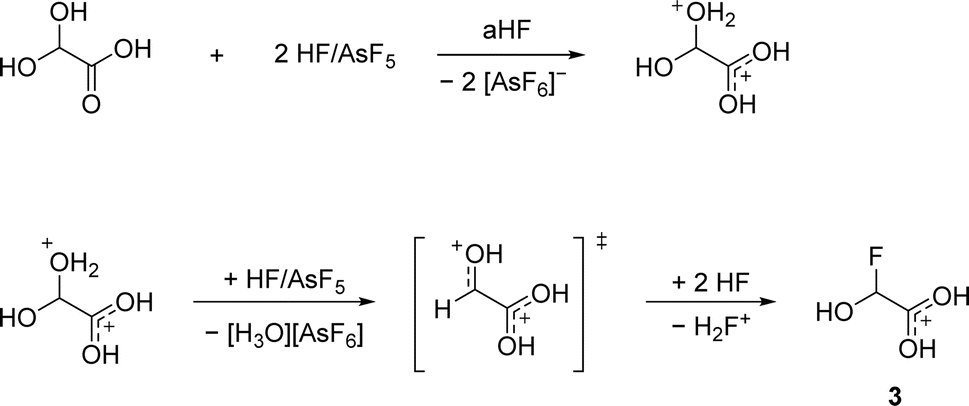 | ||
| Scheme 3 Proposed mechanism of the synthesis of [FHA-1H]+ from GAM. | ||
The necessity of three equivalents of Lewis acid to form 3 leads to the conclusion that a superelectrophilic carbodication is formed as the reactive intermediate.
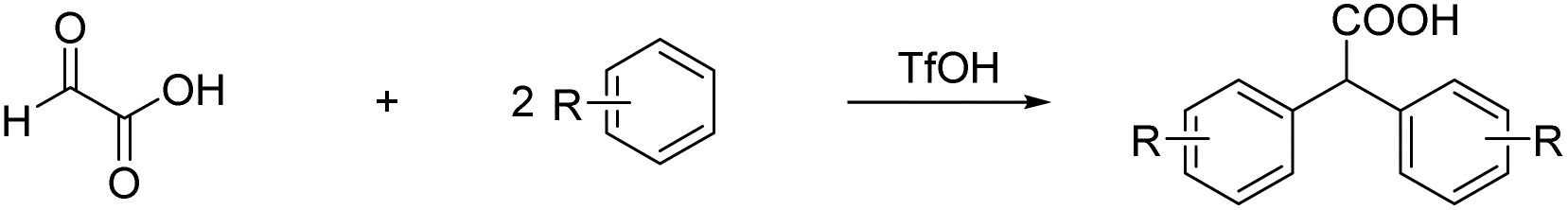
Prakash et al.12 showed that the carbonyl group (or dihydroxy group, respectively) of glyoxylic acid can be activated with strong acids in the presence of aromatic compounds for electrophilic reactions to synthesize diarylacetic acid derivatives (eqn (6)). α-Fluorohydroxyacetic acid may be useful similarly to generate aryl-fluoroacetyl acid derivatives (eqn (7)), while α-fluorohydroxyacetyl fluoride has the potential to be activated with Lewis acids at the C1-atom for Friedel-Crafts-acylations (eqn (8)). However, for this kind of reaction, the protonated species of α-fluorohydroxyacetic acid might be a better fit since the carboxy group is already activated and its [AsF6]− salt is relatively stable (eqn (9)).
 | (6) |
 | (7) |
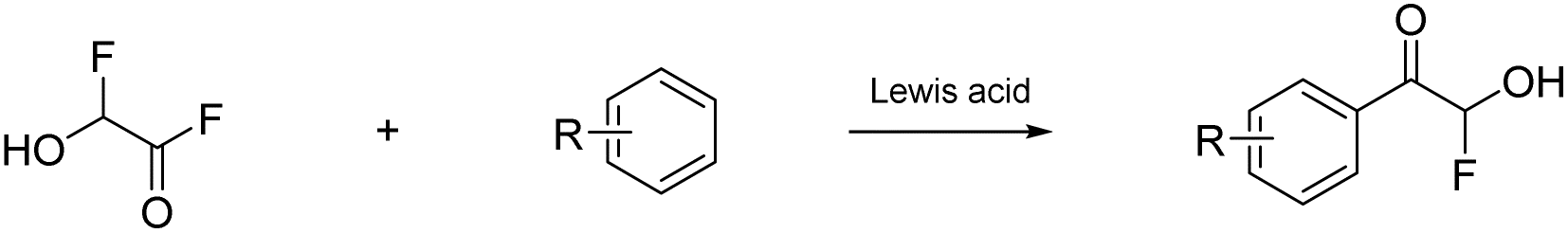 | (8) |
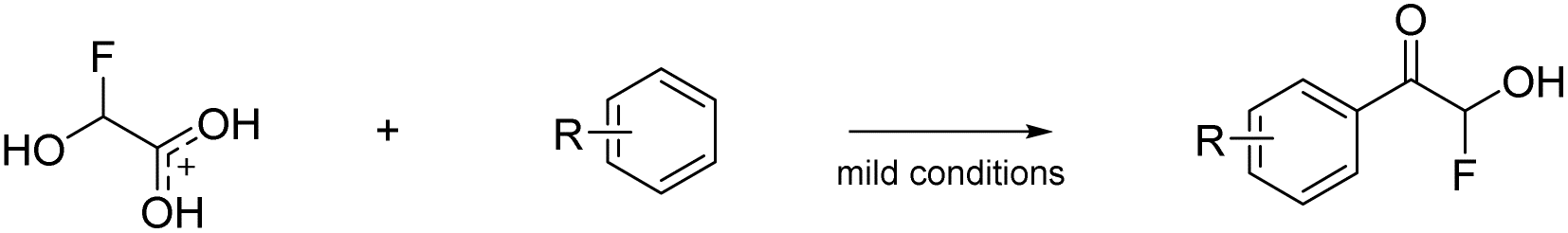 | (9) |
NMR spectroscopy
The reactivity of glyoxylic acid monohydrate (GAM) in the systems HF/SF4, HF/AsF5, or solely aHF can be traced by 1H, 19F, and 13C NMR spectroscopy. The samples were dissolved either in aHF or SO2, and acetone-d6 was employed for external referencing. For more details of the experimental procedure, see the ESI† (Apparatus and Materials). The chemical shifts of 1, 2, and 3 are listed in Table 1. The respective solvent used for the measurements is given in the table footnote. A reference of GAM in D2O is displayed in Fig. S1 and S2.†| FHAa | FHA-Fb | [FHA-1H][AsF6]a | |
|---|---|---|---|
| a aHF as a solvent.b SO2 as a solvent. | |||
| δ [1H] (C–H) | 5.57 (d), J = 54.4 | 7.00 (d) J = 51.2 | 5.61 (d) J = 56.9 |
| δ [1H] (H3O+) | 9.48 (s) | ||
| δ [19F] (C–F) | −130.38 (d) J = 54.4 | −134.67 (d) J = 53.7 | −128.88 (d) J = 53.9 |
| δ [19F] (COF) | 23.63 (d) J = 16.3 | ||
| 22.97 (d) J = 14.1 | |||
| δ [13C] (carboxylic) | 170.98 (d) J = 32.7 | 154.33 (dd) J = 368.7, 34.8 | 184.90 (d) J = 34.0 |
| δ [13C] (tetrahedral) | 96.95 (d) J = 225.1 | 92.59 (dd) J = 241.3, 82.4 | 97.00 (d) J = 227.1 |
GAM has proven to be very reactive to HF. When dissolved in aHF, NMR spectra (Fig. S3–S5, ESI†) show a variety of fluorinated compounds. Doublets in both the 1H (5.65 ppm, J = 61.2 Hz) and the 13C NMR spectra (171.57 ppm, J = 32.6 Hz and 97.99 ppm, J = 225.2 Hz) are very similar to those assigned to 1. This means that deoxyfluorination occurs in aHF, albeit uncontrolled. By first dissolving equimolar amounts of SF4 (compared to GAM) in aHF and secondly adding GAM, 1 becomes the main product with an amount of roughly 74% (NMR spectra displayed in Fig. S6–S8, ESI†), as the doublet at 5.57 ppm is the most intensive one in the 1H NMR spectrum. The 19F signal at −130.38 ppm is assigned to the fluorine atom in FHA since the coupling constant is the same as for the 1H signal. The 13C shift of the carboxy group is observed at 170.98 ppm, and the tetrahedral carbon shift at 96.95 ppm.
By employing two equivalents of SF4, 2 is the most abundant species. The NMR spectra recorded in aHF showed an equilibrium of 2 and its HF-adduct 4 (see eqn (10)). The spectra are displayed in Fig. S9–S11 in the ESI.†
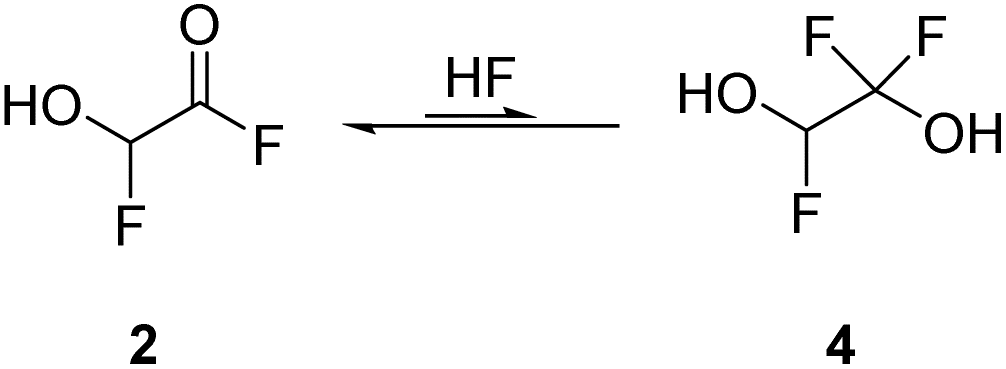 | (10) |
The equilibrium is shifted to 2 by removing all volatile products at −78 °C, successively warming up the residue to 0 °C, and trapping the gas phase into a second vessel at −196 °C. The condensate was dissolved in SO2, and NMR spectra (Fig. S12–S14†) show 2 as the only organic compound, as shown by the intensive doublet at 7.00 ppm in the 1H NMR spectrum. The 13C signal of the acyl fluoride is observed at 154.33 ppm. The dd-splitting pattern shows the coupling to two fluorine atoms, just like the 13C shift of the tetrahedral carbon at 92.59 ppm. In the 19F NMR spectrum, these signals occur at −134.67, 23.63, and 22.97 ppm. Additional signals at 65.17 ppm and 64.48 ppm are assigned to residual SF4 that has not been removed.17
A different approach to generating a derivative of α-fluorohydroxyacetic acid is the deoxyfluorination of GAM with superacids. By dissolving the Lewis acid in aHF in the first step, the superacidic medium is formed. Subsequently adding GAM to the solution led to a quantitative synthesis of [FHA-1H][AsF6] (3) (NMR spectra displayed in Fig. S15–S17, ESI†). The 13C signal of the carboxy group (184.90 ppm) is shifted downfield compared to GAM and 1 due to the protonation in the superacidic system, similar to reported protonated carboxy groups.18 The doublet at 97.00 ppm is assigned to the CHF(OH) group. The 19F signal is observed at −128.88 ppm and the 1H resonance at 5.61 ppm. The 1H signal at 9.48 ppm confirms the formation of H3O+.19
Vibrational spectroscopy
The synthesis of FHA-F and [FHA-1H][AsF6] is confirmed by vibrational spectroscopy. FHA-F (2) was generated by reacting GAM with a twofold amount of SF4 in aHF. The solvent and other volatile products were removed in vacuo overnight at −78 °C. The sample was warmed up to 0 °C and an infrared spectrum of the gas phase was measured at room temperature. In the case of [FHA-1H]+, the [AsF6]− salt (3) was synthesized by reacting GAM with HF/AsF5 in aHF and successively removing the solvent in vacuo at −78 °C. Infrared and Raman spectroscopy of the colorless residue was performed at low temperatures (see ESI† for more details). The spectra are displayed in Fig. 1 and selected observed frequencies are listed in Table 2. Quantum chemical calculations of the respective compounds were performed to support the assignment of the vibrational frequencies. The detailed characterization of the compounds is found in the ESI.† FHA (1) could not be isolated from aHF, so an experimental frequency analysis was not feasible. However, the quantum chemically calculated frequencies and their assignment are listed in Table S3 in the ESI.†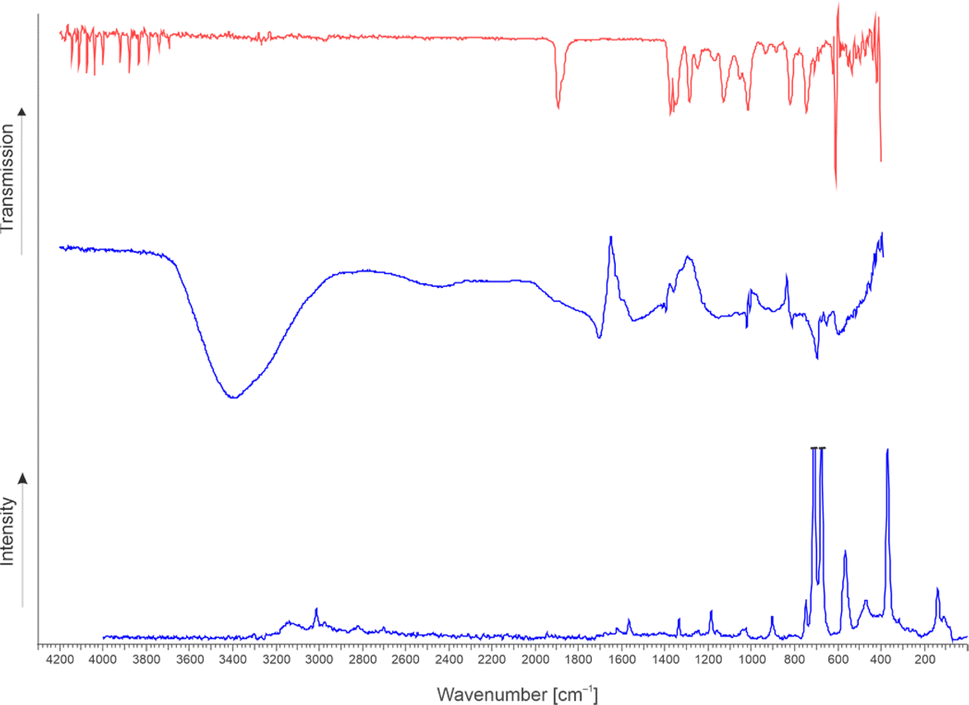 | ||
| Fig. 1 The infrared spectrum of gaseous 2 at room temperature (top, red). Low-temperature infrared (middle) and Raman spectrum (bottom) of 3 (blue). | ||
| FHA-F | [FHA-1H][AsF6] | Assignment | |
|---|---|---|---|
| Exp. IRa | Exp. IRa | Exp. Rab | |
| a Abbreviations: m = medium, a = acid.b Experimental Raman intensities are relative to a scale of 1 to 100. | |||
| 1894 (m) | 1705 (m) | 1567 (7) | ν(COa) |
| 1541 (m) | ν(COa) | ||
| 1173 (m) | ν(C(O)F) | ||
| 1128 (m) | 1151 (m) | 1162 (3) | ν(C–OH) |
| 1016 (m) | 1022 (m) | 1027 (4) | ν(CF) |
| 822 (m) | 897 (m) | 903 (8) | ν(CC) |
The infrared spectrum of 2 shows rotational bands of remaining hydrogen fluoride between 3728 and 4143 cm−1, which was not completely removed after the reaction. The ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibration is observed at 1894 cm−1. This band is distinct for acyl fluorides20–22 and is significantly blue-shifted compared to GAM (1742 cm−1).23 The stretching vibrations of the newly formed CF bonds occur at 1173 (acyl fluoride moiety) and 1016 cm−1 (fluorohydroxy group). The intensive bands down from 708 cm−1 are assigned to residual SF4, which has been observed in the NMR study as well. A reference spectrum of SF4 is illustrated in Fig. S18 in the ESI.†
O) vibration is observed at 1894 cm−1. This band is distinct for acyl fluorides20–22 and is significantly blue-shifted compared to GAM (1742 cm−1).23 The stretching vibrations of the newly formed CF bonds occur at 1173 (acyl fluoride moiety) and 1016 cm−1 (fluorohydroxy group). The intensive bands down from 708 cm−1 are assigned to residual SF4, which has been observed in the NMR study as well. A reference spectrum of SF4 is illustrated in Fig. S18 in the ESI.†
In the IR spectrum of 3, a strong and broad band with a maximum at 3406 cm−1 is found. This might be assigned to H3O+, however, it cannot be excluded that this band is attributed to the measurement method at low temperatures, where water can condense onto the specimen, superposing the OH and CH stretching vibrations. The protonation of the carboxy group can be traced by the νas(CO) band at 1705 cm−1 (IR), which is red-shifted compared to ν(CO) of GAM (1742 cm−1).23 The antisymmetric CO stretching mode of 3 occurs at 1541 (IR) and 1567 cm−1 and is in return blue-shifted concerning ν(C–O) of GAM (1101 cm−1). This convergence of the carboxylic vibrations is a direct result of protonation and has been described in several studies.24,25 The stretching vibration of the newly formed CF bond is observed at 1022 (IR) and 1027 cm−1 (Ra), similar to 2.
Crystal structure of [FHA-1H][AsF6]
Single crystals of 3 were obtained by dissolving the colorless powder in aHF at −55 °C. Colorless needles suitable for single-crystal X-ray diffraction grew as racemic twins within three days. In the following, the S-enantiomer is discussed. 3 crystallizes in the orthorhombic space group P212121 with four formula units per unit cell. The asymmetric unit is displayed in Fig. 2. Table 3 contains selected geometric parameters.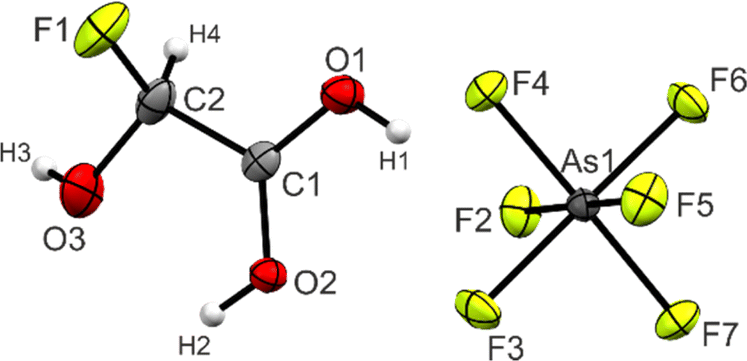 | ||
| Fig. 2 Projection of the asymmetric unit of 3 (50% probability displacement ellipsoids, hydrogen atoms displayed as spheres of arbitrary radius). | ||
| Bond lengths [Å] | Intermolecular interactions D(–H)⋯A [Å] | ||
|---|---|---|---|
| C1–O1 | 1.258(4) | O1(–H1)⋯F2 | 2.579(4) |
| C1–O2 | 1.272(4) | O2(–H2)⋯O3 | 2.587(4) |
| C1–C2 | 1.515(5) | O2(–H2)⋯F6i | 2.669(3) |
| C2–O3 | 1.355(5) | O3(–H3)⋯F3ii | 2.826(4) |
| C2–F1 | 1.376(5) | C1⋯F7 | 2.733(5) |
| Bond angles [deg] | Dihedral angles [deg] | ||
|---|---|---|---|
| O1–C1–O2 | 120.6(3) | O3–C2–C1–O1 | 179.5(4) |
| O1–C1–C2 | 117.4(3) | F1–C2–C1–O1 | −61.2(5) |
| O2–C1–C2 | 122.0(3) | O3–C2–C1–O2 | −1.0(6) |
| O3–C2–C1 | 106.6(3) | F1–C2–C1–O2 | 118.3(4) |
| F1–C2–C1 | 106.1(4) | ||
| O3–C2–F1 | 111.9(4) | ||
The C1–C2 bond of 1.515(5) Å is similar to the starting material glyoxylic acid monohydrate (GAM, 1.522(3) Å),26 yet slightly longer than a regular Csp3–Csp2 single bond (1.502 Å) in carboxylic acids.27 The C1–O1 (1.258(4) Å) and C1–O2 (1.272(4) Å) bond distances are approximately the same, as has been observed in a variety of protonated carboxylic acids.24,25,28 The C2–O3 bond (1.355(5) Å) is significantly shorter than the two C–OH bonds in GAM (1.400(4) and 1.404(3) Å) and even more significant than a regular C–O single bond in primary alcohols (1.426 Å).27 The newly formed C2–F1 of 1.376(5) Å, on the other hand, is longer than a regular C–F bond with an electron-withdrawing group in the geminal position (1.349 Å).27 The nature of this bond relation will be discussed below in the Theoretical Study.
The O1–C1–O2 angle of 120.6(3)° is significantly smaller than in GAM (125.1(2)°)26 as a result of the protonated carboxy group. Subsequently, the O1–C1–C2 is widened from 111.9(2)° in GAM to 117.4(3)° in 3, while the remaining bond angles remain approximately unchanged. Regarding the torsion angles, the O3–C2–C1–O2 dihedral is reduced from 9.9(9)° to −1.0(6)°. This is due to an intramolecular hydrogen bond O2(–H2)⋯O3 with a distance of 2.587(4) Å that is formed upon protonation. The cation exhibits three additional, moderately strong hydrogen bonds29 (Fig. 3) to form layers in the bc-plane (O1(–H1)⋯F2, O2(–H2)⋯F6, and O3(–H3)⋯F3). These layers are connected along the a-axis by nearly perpendicular C1⋯F7 interactions with a distance of 2.733(5) Å (Fig. S21, ESI†), which is about 14% within the sum of the van-der-Waals radii (3.17 Å).30
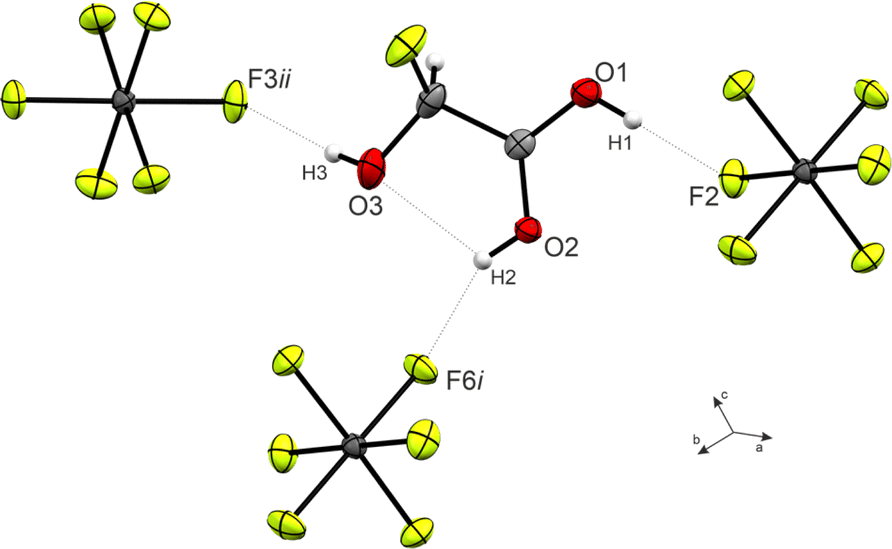 | ||
| Fig. 3 Hydrogen bonds in the crystal packing of 3 (50% probability displacement ellipsoids, hydrogen atoms displayed as spheres of arbitrary radius). | ||
The bond distances of the anion range between 1.698(3) and 1.754(2) Å. As–F bonds involved in donor–acceptor interactions (As1–F2, As1–F2, and As1–F6) are slightly longer than the others, resulting in a distorted octahedral structure. These values have been observed for [AsF6]− anions in literature.28,31,32
Theoretical Study
For FHA (1), FHA-F (2), and the free [FHA-1H]+ cation, quantum chemical calculations were performed. The gas-phase structures were optimized and the vibrational frequencies were computed on the B3LYP/aug-cc-pVTZ level of theory. In the case of the cation, a direct comparison to the experimental X-ray values is possible. The bond lengths are listed in Table 4. The calculated structures are illustrated together with the cation of 3 in Fig. 4. The labeling of the atoms is based on the crystal structure analysis for consistency.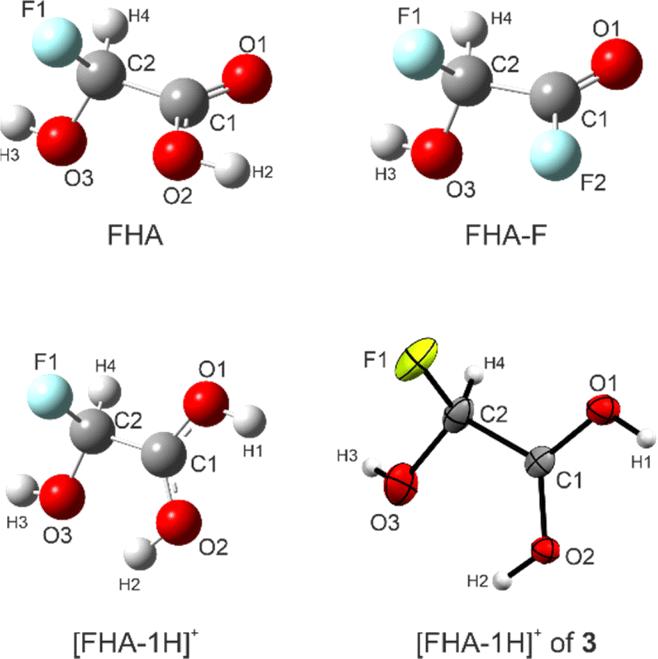 | ||
| Fig. 4 Optimized gas-phase structures of FHA, FHA-F, [FHA-1H]+, and the cation [FHA-1H]+ of 3. Calculated on the B3LYP/aug-cc-pVTZ level of theory. | ||
The C–O bond distances of the protonated carboxy group are in fair agreement with the experimental values. For FHA, the values are consistent with comparable carboxylic acids like GAM or difluoroacetic acid.26,33 The CO bond length of FHA-F is the shortest among the investigated, yet it is in agreement with structural analyses of gaseous acyl fluorides reported in the literature, as well as the C(O)F bond.34,35 The C2–O3 distances of all calculated structures (1.368–1.376 Å) are similar, while the X-ray data of 3 is a little shorter. However, all these values are shorter than a regular C–O single bond in primary alcohols (1.426 Å).27 The calculated C2–F1 bond lengths of FHA and FHA-F are longer than in the case of [FHA-1H]+. The bond distances of α-fluoroalcohols compared to regular C–F and C–OH bonds have been discussed in a study by Krossing et al. Accordingly, the elongation of the C–F bond is suspected to be a result of lone-pair conjugation of the oxygen atom into the antibonding σ*(C–F) orbital, subsequently shortening the C–OH bond.7 This is in agreement with our DFT results. We performed NBO calculations of all three investigated compounds (MP2/aug-cc-pVTZ level of theory) to assess this effect. The stabilization energies according to the second-order perturbation theory analyses of these interactions are summarized in Table 5.
| n(O3) → σ*(C2–F1) | n(F1) → σ*(C1–C2) | |
|---|---|---|
| a Calculated on the MP2/aug-cc-pVTZ level of theory. | ||
| FHA | 109.3 kJ mol−1 | 16.8 kJ mol−1 |
| FHA-F | 111.5 kJ mol−1 | 19.3 kJ mol−1 |
| [FHA-1H]+ | 105.7 kJ mol−1 | 29.0 kJ mol−1 |
The interactions of the oxygen lone-pair with the σ*(C–F) orbital are similar among the investigated compounds, explaining the shortening of the C–OH bond. The C–F distance of the neutral compounds FHA and FHA-F is subsequently elongated. In the case of the protonated species, the calculated C–F bond length rather coincides with a regular distance. Since the protonation has no significant influence on the described interaction, there must be another that strengthens the C–F bond. This is found to be the donation of a fluorine lone-pair into the σ*(C–C) orbital. The stabilization energy of this interaction in [FHA-1H]+ is calculated to be 12.2 kJ mol−1 higher than in FHA. This also explains why the C–C bond of the protonated species is the longest. However, the calculation estimates it longer than the experimental X-ray data shows. Similarly, the C–C bonds of FHA (1.532 Å) and FHA-F (1.529 Å) are longer than expected when compared to the corresponding bonds in difluoroacetic acid and difluoroacetyl fluoride.35 This indicates that solid-state effects might have an additional influence on the bond distances.
Conclusions
For the first time, α-fluorohydroxyacetic acid (FHA), its acyl fluoride (FHA-F), and its protonated species ([FHA-1H]+) are described. The syntheses of FHA and FHA-F are achieved by reacting glyoxylic acid monohydrate (GAM) with HF/SF4. By applying the binary superacid HF/AsF5, [FHA-1H][AsF6] is the only organic compound, allowing a complete characterization by NMR, vibrational spectroscopy, and single-crystal X-ray diffraction. The superacidic deoxyfluorination only occurs when three equivalents of Lewis acid are used, implying that the superelectrophile [C2(OH)3H]2+ is formed intermediately. NBO calculations reveal a complex relation between the C–F and the C–OH bond of the fluorohydroxy group. The use of superacids in aHF could enable convenient access to fluorinated compounds with a high electrophilicity and give deoxyfluorination reagents a new appeal.Data availability
The data supporting this article have been included as part of the ESI.† For full details on vibrational spectroscopy, NMR spectroscopy, X-ray diffraction refinement, and computational details see the ESI.† Crystallographic data for [CH(OH)FC(OH)2][AsF6] has been deposited at the CCDC under the accession number 2173682 and can be obtained from http://www.ccdc.cam.ac.uk.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work by the Ludwig-Maximilian University Munich (LMU), by the Deutsche Forschungsgemeinschaft (DFG), and F-Select GmbH is gratefully acknowledged. Special thanks to Prof. Dr Konstantin Karaghiosoff for his help in the publication process after Prof. Kornath's passing.Notes and references
- W. A. Sheppard and C. M. Sharts, Organic Fluorine Chemistry, Benjamin, New York, 1969 Search PubMed.
- G. A. Olah and A. Pavláth, Synthesis of Organic Fluorine Compounds III. The Preparation of Fluoromethanol, Acta Chim. Acad. Sci. Hung., 1953, 3, 203–207 CAS.
- K. Seppelt, Trifluoromethanol, CF3OH, Angew Chem. Int. Ed. Engl., 1977, 16, 322–323 CrossRef.
- K. O. Christe, J. Hegge, B. Hoge and R. Haiges, Convenient access to trifluoromethanol, Angew Chem. Int. Ed. Engl., 2007, 46, 6155–6158 CrossRef CAS PubMed.
- S. Andreades and D. C. England, α-HALOALCOHOLS, J. Am. Chem. Soc., 1961, 83, 4670–4671 CrossRef CAS.
- A. F. Baxter, J. Schaab, K. O. Christe and R. Haiges, Perfluoroalcohols: The Preparation and Crystal Structures of Heptafluorocyclobutanol and Hexafluorocyclobutane-1,1-diol, Angew Chem. Int. Ed. Engl., 2018, 57, 8174–8177 CrossRef CAS PubMed.
- J. Schaab, M. Schwab, D. Kratzert, J. Schwabedissen, H.-G. Stammler, N. W. Mitzel and I. Krossing, The perfluorinated alcohols c-C6F11OH, c-C6F10-1,1-(OH)2 and c-C6F10-1-(CF3)OH, Chem. Commun., 2018, 54, 9294–9297 RSC.
- L. F. Fieser and M. Fieser, Organische Chemie, Verlag Chemie, Weinheim/Bergstrasse, 1st edn, 1972 Search PubMed.
- G. A. Olah and G. D. Mateescu, Organic fluorine compounds. XXXII. Protonated fluoromethyl alcohol, J. Am. Chem. Soc., 1971, 93, 781–782 CrossRef CAS.
- A. F. Baxter, J. Schaab, J. Hegge, T. Saal, M. Vasiliu, D. A. Dixon, R. Haiges and K. O. Christe, α-Fluoroalcohols: Synthesis and Characterization of Perfluorinated Methanol, Ethanol and n-Propanol, and their Oxonium Salts, Chemistry, 2018, 24, 16737–16742 CrossRef CAS PubMed.
- R. Minkwitz and S. Reinemann, Darstellung, spektroskopische Charakterisierung und Kristallstruktur von (CF3)2C(F)OH2+ Sb2F11−, Z. Anorg. Allg. Chem., 1999, 625, 121–125 CrossRef CAS.
- G. S. Prakash, F. Paknia, A. Kulkarni, A. Narayanan, F. Wang, G. Rasul, T. Mathew and G. A. Olah, Taming of superacids: PVP-triflic acid as an effective solid triflic acid equivalent for Friedel–Crafts hydroxyalkylation and acylation, J. Fluorine Chem., 2015, 171, 102–112 CrossRef CAS.
- W. R. Hasek, W. C. Smith and V. A. Engelhardt, The Chemistry of Sulfur Tetrafluoride. II. The Fluorination of Organic Carbonyl Compounds 1, J. Am. Chem. Soc., 1960, 82, 543–551 CrossRef CAS.
- J. Kollonitsch, S. Marburg and L. Perkins, Selective Fluorination of Hydroxy Amines and Hydroxy Amino Acids with Sulfur Tetrafluoride in Liquid Hydrogen Fluoride, J. Org. Chem., 1975, 40, 3808–3809 CrossRef.
- D. G. Martin and F. Kagan, The Reaction of Sulfur Tetrafluoride with Steroids, J. Org. Chem., 1962, 27, 3164–3168 CrossRef CAS.
- A. Virmani, C. Jessen and A. J. Kornath, Synthesis and Structure of the Small Superelectrophile C2(OH)2Me22+, Chem.–Eur. J., 2024, 30, e202400354 CrossRef CAS PubMed.
- T. V. Oommen, B. Gotthardt and T. R. Hooper, Reactions of lower fluorides of sulfur with hydrogen sulfide, Inorg. Chem., 1971, 10, 1632–1635 CrossRef CAS.
- A. Nitzer, M. Regnat, C. Jessen and A. J. Kornath, Third Time is a Charm – Protonating Tricarboxybenzenes, Eur. J. Org Chem., 2022, 2022(5), e202101488 CrossRef CAS.
- K. O. Christe, C. J. Schack and R. D. Wilson, Novel onium salts. Synthesis and characterization of oxonium hexafluoroantimonate (OH3+SbF6−) and oxonium hexafluoroarsenate (OH3+AsF6−), Inorg. Chem., 1975, 14, 2224–2230 CrossRef CAS.
- J. R. Durig, G. A. Guirgis and T. A. Mohamed, Infrared and raman spectra, conformational stability, normal coordinate analysis, ab initio calculations, and vibrational assignment of difluoroacetyl fluoride, J. Mol. Struct., 1998, 444, 165–182 CrossRef CAS.
- J. R. Durig, W. Zhao, D. Lewis and T. S. Little, Conformational stability, barriers to internal rotation, vibrational assignment, and abinitio calculations of chloroacetyl fluoride, J. Chem. Phys., 1988, 89, 1285–1296 CrossRef CAS.
- J. R. Durig, M. M. Bergana and H. V. Phan, Raman and infrared spectra, conformational stability, barriers to internal rotation, ab initio calculations and vibrational assignment of dichloroacetyl fluoride, J. Raman Spectrosc., 1991, 22, 141–154 CrossRef CAS.
- K. L. Plath, J. L. Axson, G. C. Nelson, K. Takahashi, R. T. Skodje and V. Vaidaa, Gas-phase vibrational spectra of glyoxylic acid and its gem diol monohydrate. Implications for atmospheric chemistry, React. Kinet. Catal. Lett., 2009, 96, 209–224 CrossRef CAS.
- C. Jessen and A. J. Kornath, Syntheses and Structures of Protonated Acetylenedicarboxylic Acid, Eur. J. Inorg. Chem., 2022, 2022 DOI:10.1002/ejic.202100965.
- A. Virmani, M. Pfeiffer, C. Jessen, Y. Morgenstern and A. J. Kornath, Protonation of Pyruvic Acid – Synthesis of a plain Superelectrophile, Z. Anorg. Allg. Chem., 2022, 648(14), e202200005 CrossRef CAS.
- T. Lis, Structure of dihydroxyacetic acid (glyoxylic acid monohydrate), C2H4O4, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 1082–1084 CrossRef.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, in International Tables for Crystallography, ed. E. Prince, H. Fuess, T. Hahn, H. Wondratschek, U. Müller, U. Shmueli, A. Authier, V. Kopský, D. B. Litvin, M. G. Rossmann, E. Arnold, S. Hall and B. McMahon, International Union of Crystallography, Chester, England, 2006, pp. 790–811 Search PubMed.
- M. Schickinger, T. Saal, F. Zischka, J. Axhausen, K. Stierstorfer, Y. Morgenstern and A. J. Kornath, The Tetrahydroxydicarbenium Cation [(HO)2CC(OH)2]2+, ChemistrySelect, 2018, 3, 12396–12404 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford Univ. Press, New York, 1997 Search PubMed.
- A. Bondi, V. van der Waals and J. Radii, Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. Minkwitz, F. Neikes and U. Lohmann, Protonated Trithiocarbonic Acid – Synthesis, Spectroscopic Characterization and the Crystal Structure of C(SH)3+AsF6−, Eur. J. Inorg. Chem., 2002, 2002, 27–30 CrossRef.
- D. Mootz and M. Wiebcke, Fluorides and fluoro acids. 10. Crystal structures of acid hydrates and oxonium salts. 23. Crystal structure of the low-temperature form of oxonium hexafluoroarsenate(V), Inorg. Chem., 1986, 25, 3095–3097 CrossRef CAS.
- J. M. J. M. Bijen and J. L. Derissen, On the molecular structure of difluoroacetic acid as investigated by means of gas electron diffraction, J. Mol. Struct., 1975, 27, 233–240 CrossRef CAS.
- Y. Berrueta Martínez, C. G. Reuter, Y. V. Vishnevskiy, Y. B. Bava, A. L. Picone, R. M. Romano, H.-G. Stammler, B. Neumann, N. W. Mitzel and C. O. Della Védova, Structural Analysis of Perfluoropropanoyl Fluoride in the Gas, Liquid, and Solid Phases, J. Phys. Chem. A, 2016, 120, 2420–2430 CrossRef PubMed.
- B. P. van Eijck, P. Brandts and J. P. M. Maas, Microwave spectra and molecular structures of rotational isomers of fluoroacetic acid and fluoroacetyl fluoride, J. Mol. Struct., 1978, 44, 1–13 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: For full details on vibrational spectroscopy, NMR spectroscopy, X-ray diffraction refinement, and computational details. CCDC 2173682. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05449k |
| ‡ Prof. Kornath passed away in March 2024. |
| This journal is © The Royal Society of Chemistry 2024 |