DOI:
10.1039/D4RA05352D
(Paper)
RSC Adv., 2024,
14, 28160-28167
Hydrothermal synthesis, structures, and catalytic performance of five coordination compounds driven by 5-aminoisophthalic acid†
Received
24th July 2024
, Accepted 21st August 2024
First published on 3rd September 2024
Abstract
An amino-functionalized-dicarboxylic acid, 5-aminoisophthalic acid (H2aipa), was used as a versatile building block to synthesize a series of five novel coordination compounds under hydrothermal conditions and formulated as [Co(μ3-aipa)(2,2′-H2biim)]n (1), [Ni2(μ-aipa)2(2,2′-H2biim)2(H2O)4]·4H2O (2), {[Cd(μ3-aipa)(2,2′-H2biim)]·H2O}n (3), {[Ni(μ-aipa)(μ-bpb)]·0.5bpb·H2O}n (4), and {[Ni2(μ-aipa)(μ3-aipa)(μ-dpea)2(H2O)][Ni(μ-aipa)(μ-dpea)(H2O)]·8H2O}n (5). Three supporting ligands (2,2′-biimidazole (H2biim),1,4-bis(pyrid-4-yl)benzene (bpb), and 1,2-di(4-pyridyl)ethane (dpea)) were used in the synthesis. The structures of the studied products 1–5 vary significantly, ranging from a 0D dimer (2), 2D sheets (1, 3 and 4) to 3D + 2D interpenetrated frameworks (5). Furthermore, these compounds were evaluated as heterogeneous catalysts for the Knoevenagel reaction, achieving high product yields under optimized conditions. In addition, we also investigated various reaction parameters, substrate scope, and assessed the feasibility of catalyst recycling. This thorough investigation highlights the versatility of H2aipa as a dicarboxylate building block in the formation of functional coordination polymers.
Introduction
Coordination polymers (CPs) are an important class of crystal materials that are constructed using self-assembly of metal ions with organic bridging ligands.1–3 Their versatile topologies and chemical properties4,5 make them ideal candidates for diverse applications such as gas adsorption and separation,6–9 heterogeneous catalysis,10–12 sensing,8,13,14 and biomaterials.15 To study the process of mineral formation under supercritical conditions in the laboratory, researchers initially employed hydrothermal and solvothermal synthesis methods. These days, hydrothermal synthesis is extensively utilized in the construction of CPs, especially for the preparation of CPs crystals.16–19 The self-assembly in these reactions is highly sensitive to several variables including the types of metal ion, the concentration of reactants as well as the pH value of the solution.20–23 Polycarboxylate ligands are commonly used as versatile organic linkers in the synthesis of metal clusters and polymers.24,25 This preference stems from their partially or fully deprotonated sites, which enable the development of different structural topologies and exhibit a variety of coordination modalities.26 These ligands, in particular, can function as terminal unidentate, chelating, bridging bidentate and bridging tridentate ligands.27
During the last few years, our research has focused on designing new functional coordination polymers using polycarboxylic ligands.10,18,28 In this study, we chose 5-aminoisophthalic acid (H2aipa, Scheme 1) as a amino-functionalized dicarboxylate building block.
 |
| | Scheme 1 Structures of H2aipa block and auxiliary ligands. | |
H2aipa exhibits several intriguing properties that make it an attractive linker: (a) H2aipa has five coordination sites, including one amino nitrogen and four carboxyl oxygen atoms. (b) The free amino nitrogen atom can act as a base site for catalysis, facilitating reactions such as the Henry reaction, cyanosilylation and Knoevenagel condensation.28–30
Here, we report the synthesis and characterization of five novel coordination compounds derived from metal(II) salts, H2aipa, and three auxiliary ligands. These compounds exhibit diverse structures, ranging from a 0D dimer (2), 2D sheets (1, 3 and 4) to 3D + 2D interpenetrated frameworks (5), specifically [Co(μ3-aipa)(2,2′-H2biim)]n (1), [Ni2(μ-aipa)2(2,2′-H2biim)2(H2O)4]·4H2O (2), {[Cd(μ3-aipa)(2,2′-H2biim)]·H2O}n (3), {[Ni(μ-aipa)(μ-bpb)]·0.5bpb·H2O}n (4), and {[Ni2(μ-aipa)(μ3-aipa)(μ-dpea)2(H2O)][Ni(μ-aipa)(μ-dpea)(H2O)]·8H2O}n (5).
In addition, we have also evaluated the catalytic activities of these compounds in the Knoevenagel reaction between pyridine-3-aldehyde with propanedinitrile.
Experimental section
General methods
All chemicals were purchased commercially utilized exactly as supplied. To record the FTIR spectra (KBr discs), a Bruker EQUINOX 55 spectrometer was utilized. Elemental analyses (EA) for carbon, hydrogen, and nitrogen in compounds 1–5 were conducted using an ElementarVario EL elemental analyzer. Thermogravimetric analyses (TGA) were conducted using a LINSEIS STA PT1600 thermal analyzer under a nitrogen flow and heating at a rate of 10 °C min−1. The powder X-ray diffraction patterns (PXRD) of the compounds were collected using a Rigaku-Dmax 2400 diffractometer (CuKα radiation; λ = 1.54060 Å, Rigaku Corporation, Tokyo, Japan). For 1H NMR experiments, a JNM ECS 400 M spectrometer was used with CDCl3 as the solvent.
Synthesis of [Co(μ3-aipa)(2,2′-H2biim)]n (1)
A 20 mL Teflon cup was sealed with the following contents: H2aipa (36.2 mg, 0.2 mmol), H2biim (26.8 mg, 0.2 mmol), CoCl2·6H2O (47.6 mg, 0.2 mmol), NaOH (16.0 mg, 0.4 mmol), and H2O (10 mL). After three days of heating the mixture at 160 °C, it was allowed to cool to ambient temperature. Purple block-shaped crystals were extracted and rinsed with water. Yield: 43% based on H2aipa. Computed for C14H11CoN5O4: C 45.18, N 18.82, H 2.98%. Found: C 45.43, N 18.49, H 3.00%. FTIR (KBr, cm−1): 3578 w, 3309 w, 1620 w, 1595 m, 1517 m, 1412 s, 1327 s, 1244 w, 1100 w, 1058 w, 995 w, 961 w, 920 w, 845 w, 778 m, 730 w, 666 w, 621 w 580 w, 547 w.
Synthesis of [Ni2(μ-aipa)2(2,2′-H2biim)2(H2O)4]·4H2O (2)
Compound 2 was synthesized using a method analogous to CP 1 by substituting NiCl2·6H2O (47.6 mg, 0.2 mmol) for CoCl2·6H2O. The synthesis yielded green block-shaped crystals of compound 2, with a 42% yield based on H2aipa. Computed for C28H38Ni2N10O16: C 37.87, N 15.77, H 4.31%. Found: C 38.03, N 15.85, H 4.28%. FTIR (KBr, cm−1): 3273 m, 3168 w, 3120 w, 1617 w, 1554 s, 1487 w, 1371 s, 1270 w, 1192 w, 1128 w, 1054 w, 991 w, 964 w, 924 w, 864 w, 818 w, 785 m, 748 w, 715 m, 610 w, 570 w.
Synthesis of {[Cd(μ3-aipa)(2,2′-H2biim)]·H2O}n (3)
CP 3 was synthesized using a method analogous to CP 1 by substituting CdCl2·H2O (40.2 mg, 0.2 mmol) for CoCl2·6H2O. The synthesis yielded yellow block-shaped crystals of CP 3, with a 45% yield based on H2aipa. Computed for C14H13CdN5O5: C 37.90, N 15.78, H 2.95%. Found: C 38.15, N 16.01, H 2.96%. FTIR (KBr, cm−1): 3304 m, 3266 m, 3180 w, 1613 w, 1580 m, 1546 s, 1397 s, 1349 s, 1323 s, 1252 w, 1114 m, 1021 w, 964 w, 920 w, 853 w, 782 m, 734 w, 673 w, 595 w, 540 w.
Synthesis of {[Ni(μ-aipa)(μ-bpb)]·0.5bpb·H2O}n (4)
A 20 mL Teflon cup was sealed with the following contents: H2aipa (36.2 mg, 0.2 mmol), bpb (46.4 mg, 0.2 mmol), NiCl2·6H2O (47.6 mg, 0.2 mmol), NaOH (16.0 mg, 0.4 mmol), and H2O (10 mL). After three days of heating the mixture at 160 °C, it was allowed to cool to ambient temperature. Green block-shaped crystals were extracted and rinsed with water. Yield: 46% based on H2aipa. Computed for C32H25N4NiO5: C 63.61, N 9.27, H 4.17%. Found: C 63.90, N 9.23, H 4.20%. FTIR (KBr, cm−1): 3359 m, 3309 m, 3083 w, 1614 s, 1546 s, 1480 w, 1386 s, 1226 w, 1110 w, 1073 w, 1013 w, 905 w, 804 m, 726 w, 678 w, 618 w, 573 w.
Synthesis of {[Ni2(μ-aipa)(μ3-aipa)(μ-dpea)2(H2O)][Ni(μ-aipa)(μ-dpea)(H2O)]·8H2O}n (5)
A 20 mL Teflon cup was sealed with the following contents: H2aipa (36.2 mg, 0.2 mmol), dpea (36.8 mg, 0.2 mmol), NiCl2·6H2O (47.6 mg, 0.2 mmol), NaOH (16.0 mg, 0.4 mmol), and H2O (10 mL). After three days of heating the mixture at 160 °C, it was allowed to cool to ambient temperature. Green block-shaped crystals were extracted and rinsed with water. Yield: 43% based on H2aipa. Computed for C60H71N9Ni3O22: C 49.83, N 8.72, H 4.95%. Found: C 50.17, N 8.75, H 4.91%. FTIR (KBr, cm−1): 3445 w, 3363 m, 3236 w, 1621 m, 1546 s, 1468 w, 1431 m, 1382 s, 1222 w, 1099 w, 1069 w, 1024 w, 957 w, 894 w, 831 w, 789 m, 726 m, 673 w, 606 w, 547 w.
Single crystal X-ray diffraction & topological analysis
A Bruker APEX-II CCD diffractometer was used to collect the crystal data of compounds 1–5 using graphite-monochromated Mo/CuKα radiation; λ = 0.71073/1.54178 Å). SHELXS-97 and SHELXL-97 software were used for the determination of the structures.31 Detailed crystal parameters and structural refinements can be found in Table 1, with selected bond parameters are listed in Tables S1 and S2 (ESI).† The supplementary crystallographic data for compounds 1–5 are available in CCDC 2367606–2367610.
Table 1 Crystal data for 1–5
| Compound |
1 |
2 |
3 |
4 |
5 |
| R1 = Σ||Fo| − |Fc||/|Fo|. wR2 = {Σ[w(Fo2 − Fc2)]2/Σ[w(Fo2)]2}1/2. |
| Chemical formula |
C14H11CoN5O |
C28H38Ni2N10O16 |
C14H13CdN5O5 |
C32H25N4NiO5 |
C60H71N9Ni3O22 |
| Formula weight |
372.21 |
888.10 |
443.68 |
604.27 |
1446.24 |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
P21/n |
P1 |
| a/Å |
7.6951(3) |
11.9238(3) |
7.89650(10) |
11.0968(4) |
10.1262(3) |
| b/Å |
8.0250(2) |
8.0959(2) |
8.16800(10) |
22.3594(7) |
13.2389(3) |
| c/Å |
11.2161(6) |
18.3412(5) |
11.4250(2) |
11.6156(4) |
13.3575(3) |
| α/° |
83.335(3) |
90 |
83.7430(10) |
90 |
88.8838(17) |
| β/° |
76.981(4) |
95.998(2) |
77.408(2) |
107.065(4) |
75.6606(19) |
| γ/° |
81.112(3) |
90 |
81.2660(10) |
90 |
74.270(2) |
| V Å−3 |
664.34(5) |
1760.85(8) |
708.630(19) |
2755.14(18) |
1667.75(7) |
| T/K |
303(2) |
303(2) |
285(2) |
293(2) |
303(2) |
| Z |
2 |
2 |
2 |
4 |
1 |
| Dc/g cm−3 |
1.861 |
1.675 |
1.995 |
1.457 |
1.297 |
| μ/mm−1 |
1.326 |
1.159 |
12.657 |
0.754 |
1.530 |
| F(000) |
378 |
920 |
420 |
1252 |
674 |
| Refl. measured |
2470 |
3277 |
2577 |
5127 |
8599 |
| Unique refl. (Rint) |
2264 (0.0319) |
2914 (0.0332) |
2463 (0.0291) |
4389 (0.0261) |
8204 (0.0380) |
| GOF on F2 |
1.136 |
1.031 |
1.037 |
1.051 |
1.066 |
| R1[I > 2σ(I)]a |
0.0369 |
0.0324 |
0.0226 |
0.0300 |
0.0420 |
| wR2[I > 2σ(I)]b |
0.0759 |
0.0864 |
0.0579 |
0.0788 |
0.1184 |
Topological analysis of the obtained coordination polymers was conducted using ToposPro software. This involved generating a simplified underlying net, wherein bridging ligands were reduced to the centroids.32,33
Catalytic activity in Knoevenagel reaction
The following ingredients were mixed in a suspension and stirred at 25 °C for a required reaction time: catalyst (2.0 mol%), aromatic aldehyde (0.50 mmol), propanedinitrile (1.0 mmol), and solvent (1.0 mL, usually CH3OH). Centrifugation was subsequently used to extract the catalyst. A crude solid product was obtained by evaporating the filtrate using a rotary evaporator. The amount of this solid product was ascertained by 1H NMR spectroscopy (JNM ECS 400M spectrometer) after it was dissolved in CDCl3 (Fig. S7, ESI†). The catalyst was recovered by centrifugation, washed with methanol, allowed to dry at room temperature, and subsequently reused in further reactions, following the same procedure for recycling experiments.
Results and discussion
Hydrothermal synthesis of compounds 1–5
A series of reactions were conducted under hydrothermal conditions to assess the potential of 5-aminoisophthalic acid (H2aipa) as a building block for synthesizing coordination polymers. These reactions involved a mixture of metal(II) salts, H2aipa, sodium hydroxide, and an auxiliary ligand selected from the following H2biim, bpb, and dpea. The hydrothermal synthesis was carried out at 160 °C for three days, followed by a gradual reduction in temperature to promote the crystallization of CPs. The resulting products were collected in substantial yields and analyzed using standard techniques. The synthesis repeatability of the five complexes is good and the yields are stable. The experimental PXRD patterns of products 1–5 agree with the computed diffractograms (Fig. S3†), suggesting their homogeneous phase purity. Structural discrepancies among compounds 1–5 could be attributed to the varying auxiliary ligands and the coordination properties of the metal(II) centers.
Description of structures
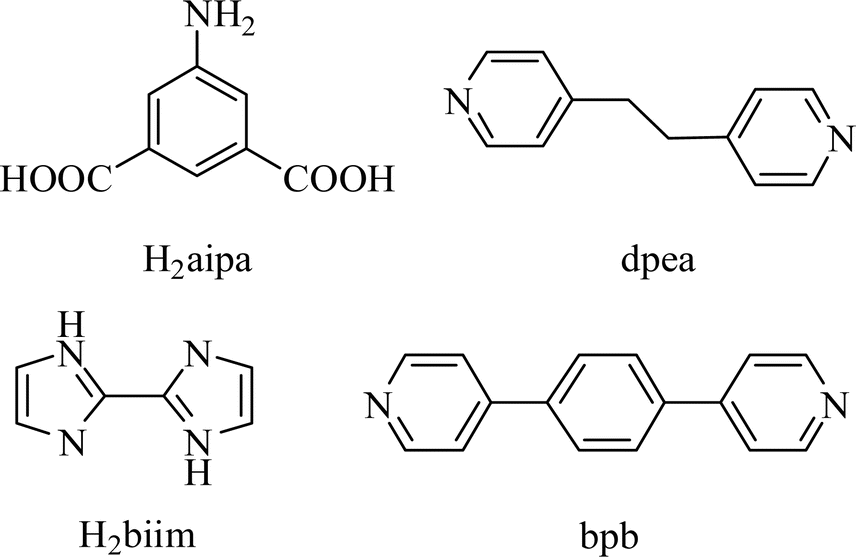
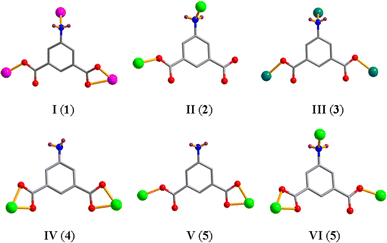
[Co(μ3-aipa)(2,2′-H2biim)]n (1). The asymmetric unit of CP 1 contains a μ3-aipa2− block, a Co(II) center, and a H2biim auxiliary ligand (Fig. 1a). The Co1 center is six-coordinate with the Co1 center being coordinated by three carboxyl oxygen and one N atoms from three μ3-aipa2− linkers and two N donors from the H2biim moiety and forms a distorted octahedral {CoN3O3} geometry. The Co–O and Co–N bond lengths, which measure 2.044(2)–2.399(2) and 2.087(2)–2.230(3) Å, are within the typical ranges for Co(II) compounds.18,28 In CP 1, the aipa2− ligand exhibits the coordination mode I (Scheme 2) with two monodentate or bidentate COO− groups. Furthermore, the nitrogen atom of the NH2 is involved in coordination. The μ3-aipa2− blocks connect the Co(II) centers, forming a 2D sheet (Fig. 1b) with hcb topology (Fig. 1c).
 |
| | Fig. 1 Structure of CP 1. (a) Coordination environment at cobalt(II) atom. (b) The two-dimensional layer viewed along the c axis. (c) The sheet exhibiting a hcb topology, observed along the c axis. | |
 |
| | Scheme 2 The coordination modes of aipa2−. | |
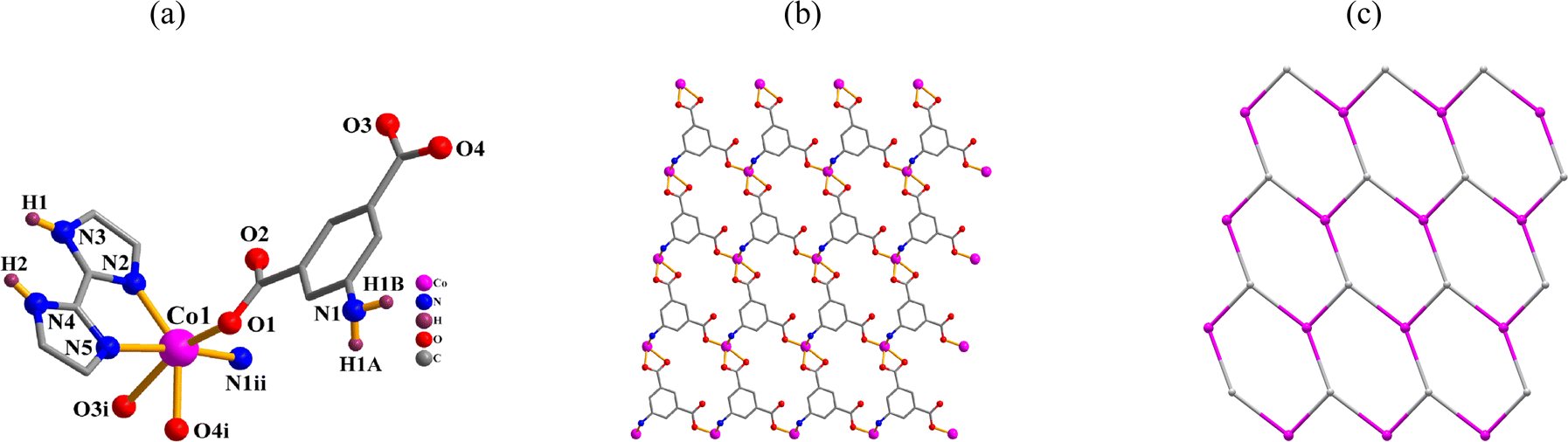
[Ni2(μ-aipa)2(2,2′-H2biim)2(H2O)4]·4H2O (2). One Ni(II) atom, one μ-aipa2− block, one H2biim, two H2O ligands and two lattice water molecules form the asymmetric unit of compound 2 (Fig. 2a). The six-coordinate Ni1 atom shows a distorted octahedral {NiN3O3} environment. It comprises one carboxyl oxygen and one N atoms from two μ-aipa2− blocks, two O donors from two H2O ligands, and a pair of N atoms of a H2biim moiety. The Ni–O [2.021(2)–2.120(2) Å] and Ni–N [2.072(2)–2.164(2) Å] bond lengths fall within the expected ranges.18,28 The cpic2− block functions as a μ-linker (mode II, Scheme 2). Fig. 2b shows that a Ni2 molecule is formed when two μ-aipa2− blocks link two Ni1 atoms. These Ni2 molecules are further assembled into a 3D H-bonded supramolecule framework (Fig. 2c).
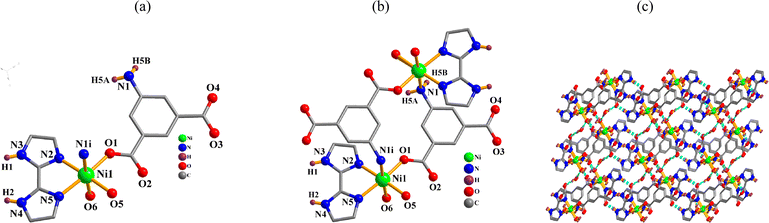 |
| | Fig. 2 Structure of dimer 2. (a) Coordination environment at nickel(II) atom. (b) The Ni2 molecule. (c) A 3D H-bonded supramolecule framework; view along the b axis. | |
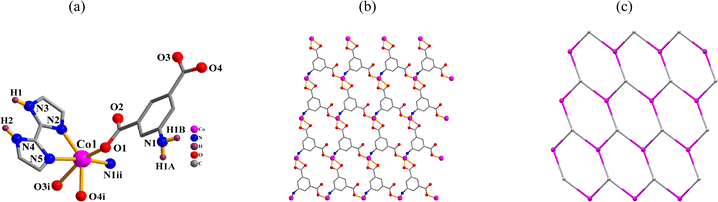
{[Cd(μ3-aipa)(2,2′-H2biim)]·H2O}n (3). CP 3 is an asymmetric unit with one μ3-aipa2− block, one cadmium(II) center, one H2biim supporting ligand, and one lattice water molecule. The five-coordinate Cd1 atom has a distorted trigonal bipyramidal {CdO2N3} geometry (Fig. 3a), made up of one nitrogen and two carboxyl oxygen atoms from three distinct μ3-aipa2− blocks and two Ndonor from the H2biim ligand. The Cd–O [2.248(2)–2.285(2) Å] and Cd–N [2.257(2)–2.447(3) Å] distances are comparable to those in Cd(II) derivatives.28,29 In 3, the aipa2− block behaves as a μ3-spacer (mode III, Scheme 2). The adjacent cadmium(II) atoms are linked by μ3-aipa2− blocks, generating a 2D sheet (Fig. 3b) with a hcb topology (Fig. 3c).
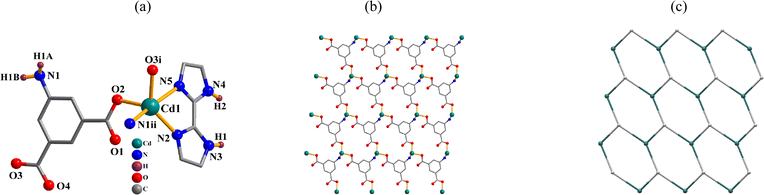 |
| | Fig. 3 Structure of CP 3. (a) Coordination environment at cadmium(II) center. (b) The two-dimensional layer viewed along the c axis. (c) Sheet exhibiting a hcb topology, observed along the c axis. | |
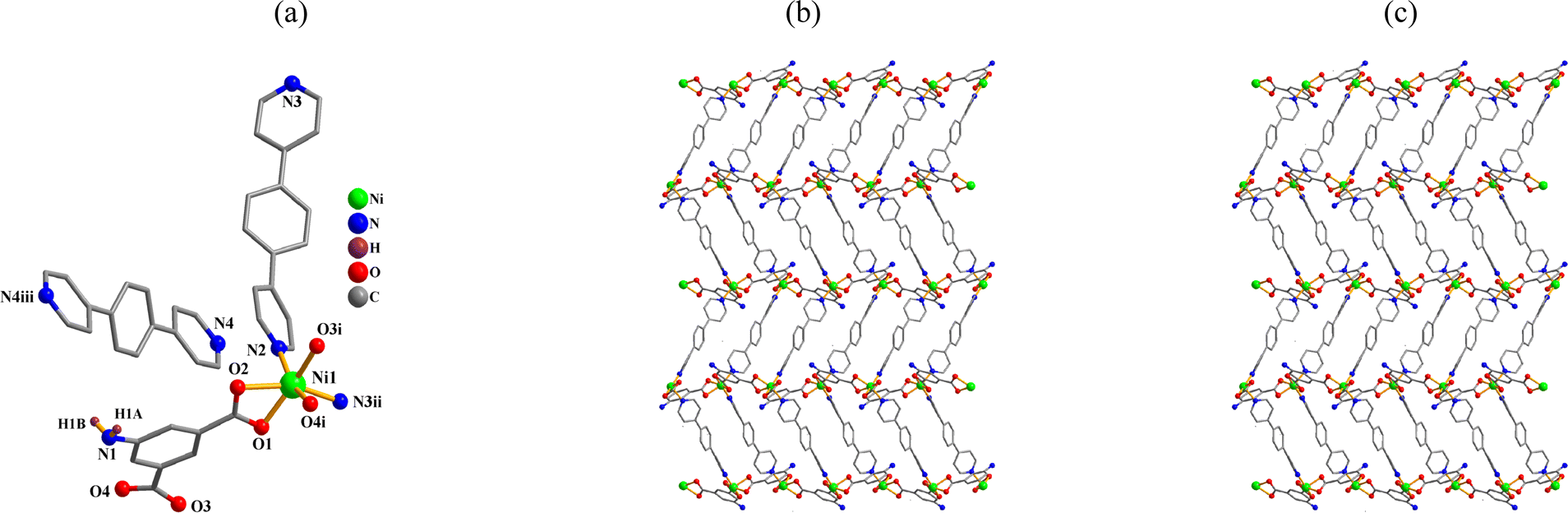
{[Ni(μ-aipa)(μ-bpb)]·0.5bpb·H2O}n (4). CP 4 is an asymmetric unit with one Ni(II) center, one μ-aipa2− block, a half of μ-bpb, moiety and one free water molecule. The compound forms a two-dimensional coordination polymer (Fig. 4a). As shown in Fig. 4a, the nickel center is six-coordinate and form a distorted octahedral {NiO4N2} geometry. The geometry is completed by two Nbpb atoms from two μ-bpb moieties and four carboxyl oxygen atoms from two μ-aipa2−. The Ni–O [2.052(2)–2.192(2) Å] and Ni–N [2.050(2)–2.062(2) Å] bond lengths fall within expected ranges.18,28 The aipa2− moiety adopts μ-coordination fashion (mode IV, Scheme 2). The aipa2− and bpb ligands linked neighbouring Ni(II) centers, resulting a 2D layer (Fig. 4b) with a new topology and point symbol of (84.122)(8)2 (Fig. 4c).
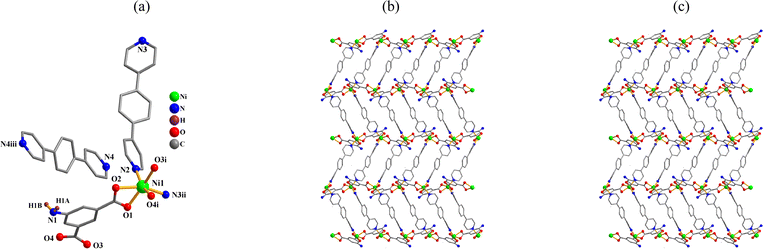 |
| | Fig. 4 Structure of 4. (a) Coordination environment at the nickel(II) atom. (b) The two-dimensional layer viewed along the a axis. (c) Sheet exhibiting a new topology, viewed along the a axis. | |
{[Ni2(μ-aipa)(μ3-aipa)(μ-dpea)2(H2O)][Ni(μ-aipa)(μ-dpea)(H2O)]·8H2O}n (5). The structure consists of a [Ni(μ-aipa)(μ-dpea)(H2O)]n 2D sheet and a [Ni2(μ-aipa)(μ3-aipa)(μ-dpea)2(H2O)]n 3D framework. CP 5 is an asymmetric unit with three distinct nickel(II) centers, three aipa2− blocks, three dpea, two coordinated water molecules, and eight lattice water molecules. The Ni1 and Ni3 atoms exhibit distorted octahedral {NiO4N2} coordination geometries, which include three carboxyl oxygen atoms from two distinct aipa2− blocks, one O atom from the H2O ligand, and two N donors from two dpea ligands (Fig. 5a). The six-coordinate nickel 2 center forms a distorted octahedral {NiO3N3} configuration. It is surrounded by one nitrogen and three oxygen atoms from three distinct aipa2− blocks, as well as two N atoms of two dpea ligands. The Ni–O [2.002(3)–2.158(4) Å] and Ni–N [2.070(4)–2.145(4) Å] bond lengths are similar to those observed in analogous Ni(II) derivatives.18,28 The aipa2− blocks act as μ- or μ3-linkers (modes IV and V, Scheme 2). The dpea shows a bridging coordination fashion. The μ-aipa, μ3-aipa and μ-dpea blocks linked Ni1 and Ni2 centers to form a 3D framework (Fig. 5b). Meanwhile, the Ni3 atoms were connected by the μ3-aipa and μ-dpea ligands to generate a 2D layer (Fig. 5c). Finally, 3D + 2D interpenetrated frameworks were formed (Fig. 5d) with a new topology and point symbol of (84·122)(8)2 (Fig. 5e and f).
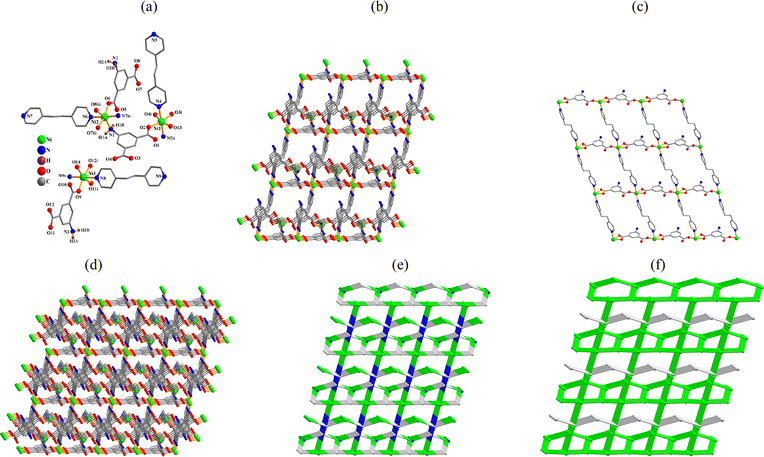 |
| | Fig. 5 Structure of 5. (a) Coordination environment at nickel(II) atoms. (b) Three-dimensional framework along the c axis. (c) Two-dimensional metal–organic network along the b axis. (d) 3D + 2D interpenetrated frameworks along the c axis. (e) Topological representation of the framework along the c axis. (f) Two 3D + 2D interpenetrated frameworks shown by different colours (green and gray). | |
TGA & PXRD data
The assessment of thermal stability for compounds 1–5 was carried out using thermogravimetric analysis (TGA) under a N2 atmosphere within a temperature range of 23–800 °C (Fig. 6). The CP 1 does not contain coordinated or crystallization water, so its network is stable up to 395 °C. At temperatures 72 to 292 °C, dimer 2 lost four lattice and four coordinated water molecules (exptl 15.9%; calculated 16.2%), whereas the dehydrated sample held steady until 316 °C. For CP 3, a weight loss of 4.3% (calcd 4.1%) occurred at temperatures from 28 and 193 °C, associated with the release of one lattice water molecule. In CP 4, one lattice water and a half of bpb were lost at 28–193 °C (exptl, 22.0%; calcd, 22.2%), with the dehydrated sample is stable up to 343 °C. For CP 5, a mass loss between 29–190 °C indicated the release of eight lattice water molecules and two coordinated water molecules (exptl 12.1%, calculated 12.4%), whereas the sample held steady until 335 °C.
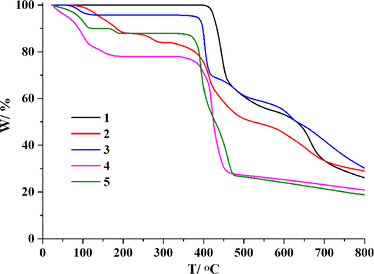 |
| | Fig. 6 TGA plots of 1–5. | |
Power X-ray diffractograms were acquired at 25 °C for compounds 1–5 (Fig. S3†). Phase purity of all samples was established by analyzing the experimental patterns and comparing them with the simulated ones (based on CIF data).
Catalytic Knoevenagel reaction
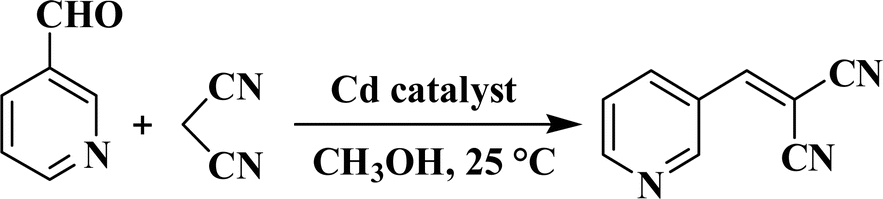
We investigated compounds 1–5 for their potential as heterogeneous catalysts in the Knoevenagel reaction, which involves a combination of propanedinitrile and a variety of aldehydes, considering the possibility of different coordination complexes acting as catalysts in this reaction.28–30,34,35 Using pyridine-3-aldehyde as a model substrate, we reacted it with malononitrile at 25 °C in methanol to synthesize the related product. As a model substrate, pyridine-3-aldehyde was reacted with malononitrile at 25 °C in methanol (Scheme 3 and Table 2). Furthermore, we conducted a thorough investigation of various reaction parameters, which included the duration of the reaction, the type of solvent used, catalyst loading, potential for catalyst reuse, and the scope of substrates involved. CP 3 exhibited the highest activity, achieving a >99 conversion rate of pyridine-3-aldehyde (Table 2). It was then used to study the effects of different reaction parameters. The yield increased from 55 to >99% when the reaction time was extended from 10 to 60 min (Table 2, entries 1–6). The impact of catalyst amount was also examined, and the results showed that increasing the loading of catalyst from 1 to 2 mol% increased product yield from 95 to >99% (entries 6 and 11). It is worth mentioning that various solvents, including methanol, were tested in these reactions. Chloroform, acetonitrile, water, and ethanol all have lower product yields (66–98% product yields). Compounds 1, 2, 4 and 5 exhibit reduced activity when compared to CP 3, achieving product yields in the 82–90% range (entries 12–15, Table 2). It is worth mentioning that the Knoevenagel reaction of pyridine-3-aldehyde is much less efficient without a catalyst (only 20% product yield) or when H2aipa or CdCl2 are used as catalysts (28% yield or 24% yield), respectively under comparable reaction conditions (entries 16–18, Table 2). The improved performance of CP 3 is likely due to the presence of unsaturated coordination sites in the Cd(II) centers, which makes it easier to access the substrates.28,30,32
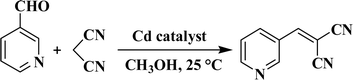 |
| | Scheme 3 Knoevenagel reaction of pyridine-3-aldehyde with propanedinitrile. | |
Table 2 Knoevenagel reaction of pyridine-3-aldehyde with propanedinitrilea
| Entry |
Catalyst |
Reaction time (min) |
Catalyst loading (mol%) |
Solvent |
Product yieldb (%) |
| Conditions: pyridine-3-aldehyde (0.5 mmol), propanedinitrile (1.0 mmol), catalyst (1–2 mol%), solvent (1.0 mL), 25 °C. Yield based on 1H NMR analysis: [moles of product per mol of aldehyde substrate] × 100%. |
| 1 |
3 |
10 |
2.0 |
CH3OH |
55 |
| 2 |
3 |
20 |
2.0 |
CH3OH |
75 |
| 3 |
3 |
30 |
2.0 |
CH3OH |
84 |
| 4 |
3 |
40 |
2.0 |
CH3OH |
91 |
| 5 |
3 |
50 |
2.0 |
CH3OH |
96 |
| 6 |
3 |
60 |
2.0 |
CH3OH |
>99 |
| 7 |
3 |
60 |
2.0 |
H2O |
98 |
| 8 |
3 |
60 |
2.0 |
C2H5OH |
96 |
| 9 |
3 |
60 |
2.0 |
CH3CN |
86 |
| 10 |
3 |
60 |
2.0 |
CHCl3 |
66 |
| 11 |
3 |
60 |
1.0 |
CH3OH |
95 |
| 12 |
1 |
60 |
2.0 |
CH3OH |
82 |
| 13 |
2 |
60 |
2.0 |
CH3OH |
87 |
| 14 |
4 |
60 |
2.0 |
CH3OH |
85 |
| 15 |
5 |
60 |
2.0 |
CH3OH |
90 |
| 16 |
Blank |
60 |
— |
CH3OH |
20 |
| 17 |
CdCl2·2H2O |
60 |
2.0 |
CH3OH |
32 |
| 18 |
H2aipa |
60 |
2.0 |
CH3OH |
26 |
A range of substituted benzaldehyde substrates was evaluated to determine the substrate scope in the Knoevenagel reaction with propanedinitrile. These reactions were carried out under optimized conditions (2.0 mol% 3, methanol, 25 °C, 1 h). Table 3 shows that the yields of the respective products ranged from 33 to >99%. Benzaldehydes with strong electron-withdrawing groups, such as nitro, chloro, bromo and fluoro substituents, demonstrated the highest efficiency (entries 2–7, Table 3). This increased efficiency is likely due to the higher electrophilicity of these substrates. However, benzaldehydes with electron-donating groups, such as methyl or methoxy groups, resulted in lower product yields (entries 9 and 10, Table 3).
Table 3 Substrate scope for Cd-catalyzed Knoevenagel reaction of substituted benzaldehydes with propanedinitrilea
| Entry |
Substituted benzaldehyde |
Product yieldb (%) |
| Conditions: aldehyde (0.5 mmol), propanedinitrile (1.0 mmol), catalyst 3 (2.0 mol%), CH3OH (1.0 mL), 25 °C. Yield on the basis of 1H NMR analysis: [moles of product per mol of aldehyde substrate] × 100%. |
| 1 |
Benzaldehyde |
>99 |
| 2 |
2-Nitrobenzaldehyde |
>99 |
| 3 |
3-Nitrobenzaldehyde |
>99 |
| 4 |
4-Nitrobenzaldehyde |
>99 |
| 5 |
4-Chlorobenzaldehyde |
>99 |
| 6 |
4-Bromobenzaldehyde |
>99 |
| 7 |
4-Fluorobenzaldehyde |
>99 |
| 8 |
Pyridine-4-aldehyde |
>99 |
| 9 |
4-Methylbenzaldehyde |
97 |
| 10 |
4-Methoxybenzaldehyde |
66 |
| 11 |
4-Hydroxybenzaldehyde |
33 |
The recyclability of catalyst 3 was evaluated. After every reaction cycle, the catalyst was separated by centrifugation, rinsed inmethanol, air-dried at approximately 25 °C, and reused in the subsequent cycle. The data show that CP 3 retains its activity for at least five more reaction cycles (Fig. S5†). Additionally, PXRD patterns show that the structure of CP 3 remains intact (Fig. S6†), despite the occurrence of multiple new signals or widened peaks. These changes are likely due to the existence of certain or a decrease in crystallinity after multiple catalytic cycles.
Referring to previous studies in this area,36–40 we propose a possible reaction mechanism for the Knoevenagel condensation catalyzed by 3 (Scheme S1, ESI†). The unsaturated Cd(II) metal centers of the catalyst (5-coordinate cadmium centers) eventually act as the Lewis acid sites interacting with the H–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionality of pyridine-3-aldehyde, leading to its polarization and an enhanced electrophilicity of the corresponding carbon atom. Such a polarization can facilitate a nucleophilic attack of this site by propanedinitrile acting as a nucleophile precursor. On the other hand, an interaction between the Lewis acid site and the –CN group of malononitrile augments an acidic character of the methylene functionality and enhances its deprotonation. The basic sites present in 3 (O-carboxylate sites) can easily abstract H+ from the –CH2– group to give rise to a nucleophile that would attack the H–CO moiety of pyridine-3-aldehyde and result in the C–C bond formation, followed by the dehydration to give the 2-(pyridin-3-ylmethylene)malononitrile product.
O functionality of pyridine-3-aldehyde, leading to its polarization and an enhanced electrophilicity of the corresponding carbon atom. Such a polarization can facilitate a nucleophilic attack of this site by propanedinitrile acting as a nucleophile precursor. On the other hand, an interaction between the Lewis acid site and the –CN group of malononitrile augments an acidic character of the methylene functionality and enhances its deprotonation. The basic sites present in 3 (O-carboxylate sites) can easily abstract H+ from the –CH2– group to give rise to a nucleophile that would attack the H–CO moiety of pyridine-3-aldehyde and result in the C–C bond formation, followed by the dehydration to give the 2-(pyridin-3-ylmethylene)malononitrile product.
Conclusions
This study showed the use of H2aipa as a amino-functionalized dicarboxylate precursor to synthesize five novel coordination compounds using a hydrothermal method. These compounds 1–5 exhibit diverse structural features, ranging from 0D dimer (compound 2), 2D sheets (CPs 1, 3 and 4) to 3D + 2D interpenetrated frameworks (CP 5). The catalytic potential of compounds 1–5 was evaluated in Knoevenagel reaction, which involved pyridine-3-aldehyde and propanedinitrile. CP 3 showed significant catalytic efficiency in this reaction.
Data availability
The data supporting this article have been included as part of the ESI.† CCDC-2367606–2367610 (compounds 1–5) contain the supplementary crystallographic data for this paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Science and Technology Projects by State Nickel Cobalt New Material Engineering Technology Research Center, Lanzhou, China. The grant number is GCZX2023JSKF0005.
Notes and references
- G. Chakraborty, I. H. Park, R. Medishetty and J. J. Vittal, Chem. Rev., 2021, 121, 3751–3891 CrossRef CAS.
- G. Maurin, C. Serre, A. Cooper and G. Ferey, Chem. Soc. Rev., 2017, 46, 3104–3107 RSC.
- Z. J. Chen, K. O. Kirlikovali, P. Li and O. K. Farha, Acc. Chem. Res., 2022, 55, 579–591 CrossRef CAS PubMed.
- L. K. Macreadie, R. Babarao, C. J. Setter, S. J. Lee, O. T. Qazvini, A. J. Seeber, J. Tsanaktsidis, S. G. Telfer, S. R. Batten and M. R. Hill, Angew. Chem., Int. Ed., 2020, 59, 6090–6098 CrossRef CAS PubMed.
- X. Y. Dong, Y. Si, J. S. Yang, C. Zhang, Z. Han, P. Luo, Z. Y. Wang, S. Q. Zang and T. C. W. Mak, Nat. Commun., 2020, 11, 3678 CrossRef CAS PubMed.
- S. F. Gu, X. H. Xiong, L. L. Gong, H. P. Zhang, Y. Xu, X. F. Feng and F. Luo, Inorg. Chem., 2021, 60, 8211–8217 CrossRef CAS PubMed.
- L. Fan, Z. Liu, Y. Zhang, D. Zhao, J. Yang and X. Zhang, Inorg. Chem. Commun., 2019, 107, 107463 CrossRef CAS.
- D. Wu, J. Liu, J. Jin, J. Cheng, M. Wang, G. Yang and Y. Y. Wang, Cryst. Growth Des., 2019, 19, 6774–6783 CrossRef CAS.
- Y. Zhao, L. Wang, N. N. Fan, M. L. Han, G. P. Yang and L. F. Ma, Cryst. Growth Des., 2018, 18, 7114–7121 CrossRef CAS.
- X. Q. Kang, C. Ren, Z. Z. Mei, X. X. Fan, J. J. Xue, Y. L. Shao and J. Z. Gu, Molecules, 2023, 28, 7474 CrossRef CAS.
- Y. Zheng, Q. Shen, Z. Li, X. Jing and C. Duan, Inorg. Chem., 2022, 61, 11156–11164 CrossRef CAS PubMed.
- D. Markad and S. K. Mandal, Dalton Trans., 2018, 47, 5928–5932 RSC.
- M. Mörtel, J. Oschwald, A. Scheurer, T. Drewello and M. M. Khusniyarov, Inorg. Chem., 2021, 60, 14230–14237 CrossRef PubMed.
- A. Rashid, S. Mondal and P. Ghosh, Molecules, 2023, 28, 1231 CrossRef CAS PubMed.
- P. Q. Hung, P. Y. Lin, X. H. Wang and J. A. Ho, J. Chin. Chem. Soc., 2023, 70, 1284–1296 CrossRef CAS.
- Y. Qin, P. She, X. Huang, W. Huang and Q. Zhao, Coord. Chem. Rev., 2020, 416, 213331 CrossRef CAS.
- L. C. Hong, L. Y. Lin, L. Wei and K. Y. Fei, Chin. J. Struct. Chem., 2021, 40, 363–368 Search PubMed.
- X. Q. Kang, J. H. Wang and J. Z. Gu, Chin. J. Inorg. Chem., 2023, 39, 2385–2392 CAS.
- R. S. Singh, R. P. Paitandi, R. K. Gupta and D. S. Pandey, Coord. Chem. Rev., 2020, 414, 213269 CrossRef CAS.
- S. Islam, S. Tripathi, A. Hossain, S. K. Seth and S. Mukhopadhyay, J. Mol. Struct., 2022, 1265, 133373 CrossRef CAS.
- D. Fonseca, A. F. Pérez-Torres, J. Cobo, J. Zapata-Rivera, J. J. Hurtado and M. A. Macías, CrystEngComm, 2022, 24, 2982–2991 RSC.
- M. Xu, G. Liang, S. Wang, X. Ma, G. Liang and Q. Ni, J. Mol. Struct., 2020, 1217, 128411 CrossRef CAS.
- J. J. Kong, D. Shao, J. C. Zhang, Y. X. Jiang, C. L. Ji and X. C. Huang, CrystEngComm, 2019, 21, 749–757 RSC.
- Q. P. Li and J. J. Qian, RSC Adv., 2014, 4, 32391–32397 RSC.
- J. J. Qian, F. L. Jiang, K. Z. Su, J. Pan, L. J. Zhang, X. J. Li, D. Q. Yuan and M. C. Hong, J. Mater. Chem. A, 2013, 1, 10631–10634 RSC.
- R.-W. Huang, B. Li, Y.-Q. Zhang, Y. Zhao, S.-Q. Zang and H. Xu, Inorg. Chem. Commun., 2014, 39, 106–109 CrossRef CAS.
- X. Chai, H. Zhang, S. Zhang, Y. Cao and Y. Chen, J. Solid State Chem., 2009, 182, 1889–1898 CrossRef CAS.
- X. X. Fan, H. Y. Wang, J. Z. Gu, D. Y. Lv, B. Zhang, J. J. Xue, M. V. Kirillova and A. M. Kirillov, Inorg. Chem., 2023, 62, 17612–17624 CrossRef CAS PubMed.
- Z. W. Zhai, S. H. Yang, Y. R. Lv, C. X. Du, L. K. Li and S. Q. Zang, Dalton Trans., 2019, 48, 4007–4014 RSC.
- Y. Q. Cai, Y. Q. Peng and G. H. Song, Catal. Lett., 2006, 109, 61–64 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97; Program for X-Ray Crystal Structure Determination, University of Gottingen, Göttingen, Germany, 1997 Search PubMed.
- V. A. Blatov, IUCrCompComm Newsletter, 2006, vol. 7, pp. 4–38 Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- H. Chen, L. Fan, T. Hu and X. Zhang, Inorg. Chem., 2021, 60, 3384–3392 CrossRef CAS PubMed.
- M. Almasi, V. Zelenak, M. Opanasenko and J. Cejka, Dalton Trans., 2014, 43, 3730–3738 RSC.
- N. Seal, A. Karmakar, P. P. Mondal, S. Kundu and S. Neogi, ACS Appl. Mater. Interfaces, 2024, 16, 41721–41733 CrossRef CAS PubMed.
- B. Parmar, P. Patel, V. Murali, Y. Rachuri, R. I. Kureshy, N.-u. H. Khan and E. Suresh, Inorg. Chem. Front., 2018, 5, 2630–2640 RSC.
- R. Pandey, D. Singh, N. Thakur and K. K. Raj, ACS Omega, 2021, 6, 13240–13259 CrossRef CAS PubMed.
- M. Saghian, S. Dehghanpour and Z. bayatani, Sci. Rep., 2023, 13, 15563 CrossRef CAS PubMed.
- R. Chand, A. Karmakar, S. Kundu and S. Neogi, Small, 2024, 2404085 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: optical images (Fig. S1), FTIR spectra (Fig. S2), PXRD patterns (Fig. S3), additional catalysis data (Fig. S4–S7) and structural parameters (Tables S1 and S2). CCDC 2367606–2367610. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05352d |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Jin-Zhong Gu
and
Jin-Zhong Gu