
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombining de novo molecular design with semiempirical protein–ligand binding free energy calculation†
Michael Iff‡
a,
Kenneth Atz‡a,
Clemens Iserta,
Irene Pachon-Angona a,
Leandro Cotosa,
Mattis Hillekea,
Jan A. Hissa and
Gisbert Schneider*ab
a,
Leandro Cotosa,
Mattis Hillekea,
Jan A. Hissa and
Gisbert Schneider*ab
aETH Zurich, Department of Chemistry and Applied Biosciences, Vladimir-Prelog-Weg 4, 8093 Zurich, Switzerland. E-mail: gisbert@ethz.ch
bETH Zurich, Department of Biosystems Science and Engineering, Klingelbergstrasse 48, 4056 Basel, Switzerland
First published on 20th November 2024
Abstract
Semi-empirical quantum chemistry methods estimate the binding free energies of protein–ligand complexes. We present an integrated approach combining the GFN2-xTB method with de novo design for the generation and evaluation of potential inhibitors of acetylcholinesterase (AChE). We employed chemical language model-based molecule generation to explore the synthetically accessible chemical space around the natural product Huperzine A, a potent AChE inhibitor. Four distinct molecular libraries were created using structure- and ligand-based molecular de novo design with SMILES and SELFIES representations, respectively. These libraries were computationally evaluated for synthesizability, novelty, and predicted biological activity. The candidate molecules were subjected to molecular docking to identify hypothetical binding poses, which were further refined using Gibbs free energy calculations. The structurally novel top-ranked molecule was chemically synthesized and biologically tested, demonstrating moderate micromolar activity against AChE. Our findings highlight the potential and certain limitations of integrating deep learning-based molecular generation with semi-empirical quantum chemistry-based activity prediction for structure-based drug design.
Introduction
Computational de novo design encompasses the autonomous generation of new molecules with desired properties from scratch.1,2 Chemical language models (CLMs) are machine learning techniques designed to process and learn from molecular structures represented as sequences (e.g., simplified molecular input line entry system (SMILES)-strings3). CLMs have found numerous applications in the de novo design of novel bioactive molecules.4,5 Transfer learning, also known as fine-tuning, is one of the most prevalent applications of CLMs in the field of molecular design.6,7 Transfer learning in the context of CLMs can be conceptualized as a two-step process. In the first step, the CLM undergoes pre-training using a vast data set of bioactive molecules that is not specifically tailored for the task at hand. This initial phase focuses on developing a principal understanding of chemistry and acquiring knowledge about the characteristics of drug-like chemical space.7,8 In the second step, the pre-trained CLM is fine-tuned using a smaller data set comprising molecules that specifically represent the desired activity and property profile.9 This process refines the CLM's ability to generate molecules with the desired characteristics. Once trained, the CLM can generate virtual molecular libraries tailored to the specific task at hand.10,11Medicinal chemistry has historically relied on natural products with known bioactivities as starting points for molecular optimization.21 However, synthesizing structurally intricate (“complex”) natural products can pose formidable challenges, complicating structure–activity relationship studies.22 Computer-assisted de novo design of natural product mimetics has presented a promising approach to streamline synthesis efforts and to produce bioactive small molecules inspired by natural products.23 However, for CLM applications on complex template molecules (e.g., certain natural products) the structural features learned by the CLM during fine-tuning not only transfer knowledge that resembles the desired property profile but also the synthetic complexity. Adopting the synthetic complexity into newly generated molecules has limited applications of conventional CLMs to natural-product-inspired molecular design.24
A strong binding affinity of the investigated compound to its desired macromolecular target is a crucial requirement for hit and lead candidates in drug discovery.25 Computational binding affinity estimation supports the identification of useful molecular designs.26 However, its accuracy often depends on the target of interest and is influenced by a variety of factors, e.g., crystal water displacement, side chain and backbone flexibility on the protein, binding enthalpy, entropy, and conformational energies.27–29 Even when suitable structure-based binding affinity methods are identified, scoring a large library of drug-like molecules often results in a trade-off between prediction accuracy and computational cost.30,31 Prominent among the existing structure-based scoring models are free energy perturbation (FEP) approximations,32 geometric deep learning,33–37 semi-empirical quantum chemistry methods,38,39 machine-learned force fields,40 and purely statistics-driven models.41–43 While deep learning-based binding affinity prediction methods have shown promise in some computational drug design projects,26 challenges with generalization and their potential inability to capture the physical underpinnings of intermolecular interactions have attracted criticism,34,44–47 rendering physics-based approaches methods of choice for applications in drug discovery.48,49
In this study, we demonstrate the integration of a de novo design algorithm with a semi-empirical quantum chemistry (SEQM) method for the structural evaluation and selection of ligands from a virtual molecular library. We employed the recently developed DRAGONFLY (DRug-target interActome-based GeneratiON oF noveL biologicallY active molecules) algorithm15 to explore the natural-product inspired chemical space around Huperzine A (1),12,13 aiming to generate potential novel inhibitors of acetylcholinesterase (AChE) (Fig. 1). AChE is a clinically relevant drug target due to its ability to increase acetylcholine levels, which is beneficial in treating neurodegenerative diseases, where acetylcholine deficiency is a problem.50 The generated molecular library was then evaluated using GFN2-xTB, an SEQM method, to estimate the Gibbs free energy of protein–ligand interactions. Subsequently, the top-ranked compound (2) was synthesized and biologically evaluated (Fig. 1).
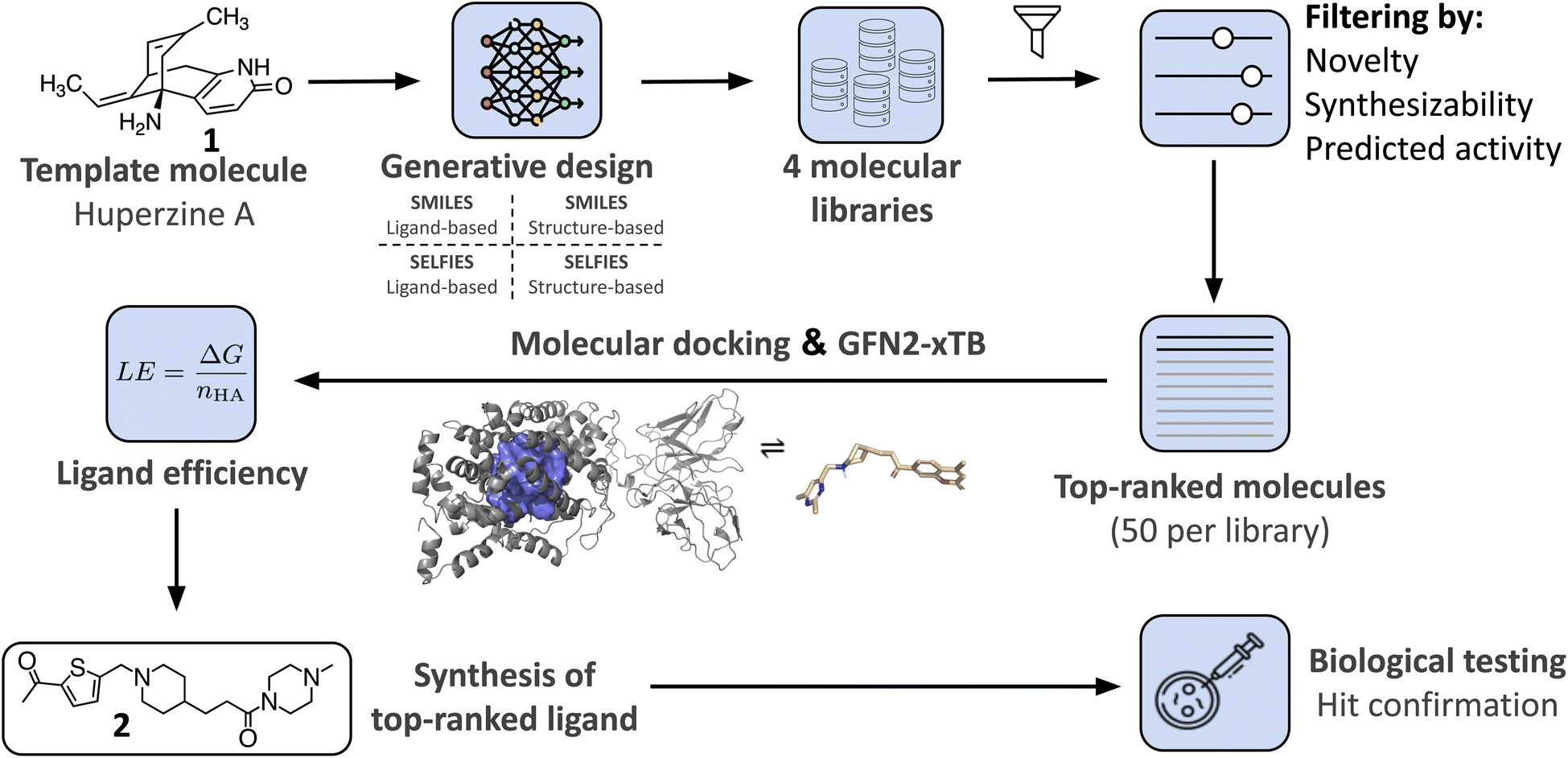 | ||
| Fig. 1 Overview of the research study. The natural product template Huperzine A12,13 and the structure of AChE14 are used as templates for the DRAGONFLY algorithm15,16 to design four natural product-inspired libraries. For the structure-based libraries, the co-crystal structure of Huperzine A bound to acetylcholinesterase (AChE) (PDB-ID:4EY5).17 Comparison of the four libraries to each other and ranking based on predicted retro-synthetic accessibility, novelty and predicted biological activity on AChE (QSAR score).15 Generation of ligand–protein binding poses of the top-200 molecules using MOE18 and GOLD.19 The highest scored binding pose that includes the key interactions is selected for subsequent Gibbs free energy calculations using GFN2-xTB.20 For each bound molecule, the ligand efficiency (i.e. Gibbs free energy divided by number of heavy atoms) is calculated, resulting in the amide 2 as the molecule with the highest score. Finally, design 2 was chemically synthesized and biologically tested. | ||
Methods
SQM GFN2-xTB
SEQM methods are quantum mechanical (QM) methods that use a simplified description of the electronic structure of a molecular system in order to reduce required computational effort compared to QM methods.51 SEQM methods have successfully been applied to infer protein–ligand binding energy estimations.38 They can be derived from the two most prominent QM approximations, namely Hartree-Fock (HF)52 and density functional theory (DFT),53 by applying systematic approximations which reduce computational effort by several orders of magnitude, albeit at the cost of accuracy.54 The foundational approximation is the use of a valence-only self-consistent field method, where only the valence electrons in a system follow a QM description and the inner shell electrons are approximated by a mean field.51 For this study, the GFN2-xTB method was chosen due to its proven accuracy in capturing essential electronic effects, such as polarization and charge transfer, while maintaining computational efficiency, which makes it suitable for large-scale protein-ligand binding studies.20The geometry, frequency, noncovalent, extended tight binding (GFN2-xTB) method was primarily designed for fast calculations of non-covalent interaction energies for macromolecular systems containing up to 1000 atoms, and has been used in numerous previous studies of drug-like molecules.20,55–57 GFN2-xTB follows a density functional tight binding (DFTB) theory, where the total energy is expanded by density fluctuations δp around a reference density p0 and δp is restricted to valence orbital space.20 It includes electrostatic interactions and exchange–correlation effects up to the second order and follows an element-specific parameter strategy (i.e., no parameters pertaining to pairs or combinations of elements are fitted). Like other GFN methods, it is designed with a focus on molecular properties that can be described at a low level of theory, namely geometries, vibrational frequencies, and non-covalent interactions. Chemical energies are not used in the training data and serve merely as cross-checks.58
The GFN-FF method was designed to provide a general force field which is fully automated for all atoms.59 In force field based methods, the electronic structure of a molecule is replaced by an interatomic interaction potential. As input, the GFN-FF method only requires the Cartesian coordinates and the elemental composition. The covalent-bonding information as well as atomic charges and bond orders are generated automatically.60
The Gibbs free energy of a protein-ligand complex can be calculated according to the scheme in Fig. 3. Thereby, the total binding free energy of a system is comprised of the energy of the system in vacuum (coined the molecular gas-phase energy E), the corresponding energy of the solvation of the system in a given solvent (Gsolv), and the thermostatic contribution to the free energy GTRVC. GTRVC takes translations, rotations, vibrations, and conformational degrees of freedom into consideration and can be approximated by the rigid rotor harmonic oscillator approximation at temperature 298.15 K (GRRHO).61 Alternative methods for approximating GTRVC are thermodynamic integration or free energy perturbation based on force-field molecular dynamics simulations.62 The association free energies of a protein-ligand system (ΔG) were obtained according to equation:
| ΔG = Gcomplex − Gprotein − Gligand | (1) |
De novo design
The recently published DRAGONFLY algorithm enables the creation of a virtual molecular library based on a single reference ligand or a single three-dimensional (3D) structure of a protein binding site.15 DRAGONFLY consists of a neural network architecture combining a graph transformer neural network (GTNN)63–66 and a CLM67 utilizing long-short-term memory (LSTM) cells. DRAGONFLY was applied to the Huperzine A template molecule in a ligand-based fashion, and to the crystal structure of AChE complexed with the template molecule Huperzine A (PDB-ID: 4EY5)68 in a structure-based fashion. For both templates, DRAGONFLY, trained on simplified molecular input line entry system (SMILES) strings3 and self-referencing embedded strings (SELFIES),69,70 was used to generate four molecular libraries (i.e., Ligand-SMILES, Ligand-SELFIES, Structure-SMILES, Structure-SELFIES), each containing 1.4 million strings.Scoring
These four molecular libraries were ranked according to three molecular properties: (i) quantitative structure–activity relationship (QSAR) score through ligand-based bioactivity prediction as described in ref. 15, (ii) novelty score based on structural and scaffold novelty,15 and (iii) retrosynthetic accessibility score (RAScore).71 The novelty score considers the Tanimoto similarity of the molecule to its closest neighbor in the ChEMBL database13 and the uniqueness of the molecular framework (“Murcko scaffold”),72 as well as the molecular graph (“skeleton scaffold”). Three different molecular descriptors were selected to calculate the QSAR score: (i) extended-connectivity fingerprints (ECFP),73 (ii) chemically advanced template search (CATS),74 and (iii) ultrafast shape recognition with pharmacophoric constraints (USRCAT).75 The arithmetic mean of the scores emerging from these three descriptors was then used as the QSAR score. Synthesizability was assessed using the retrosynthetic accessibility score (RAScore), a recently published metric that assesses the feasibility of synthesizing a given molecule yielding a numerical values between 0 and 1, where 1 means readily synthesizable and 0 unsynthesizable.71 All three ranking criteria were applied as described in ref. 15. Subsequently, molecules were excluded if they did not fulfill the following characteristics: molecular weight (MW) ≤ 500 g mol−1, RAScore ≥ 0.95, novelty score ≤ 0.65, and QSAR score ≥ 0.5 (i.e., predicted bioactivity lower than 1 μM). After applying these filters, a ranking was conducted using the QSAR score and novelty score in a weight ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The top 50 molecules were selected for each library and prepared for automated molecular docking.
:1. The top 50 molecules were selected for each library and prepared for automated molecular docking.
Molecular docking
Two AChE structures, PDB-ID: 1EVE76 and PDB-ID: 1ODC,77 were used for molecular docking. These two PDBs were selected because they contain synthetic drug-like small molecular ligands. The crystal structures were downloaded from the RCSB Protein Data Bank (https://www.rcsb.org/)78 and modified using Molecular Operating Environment (MOE) software (version 2022.02). Solvent molecules and small molecules not in the active site of the protein were removed using MOE software. Bound ligands were re-docked using Genetic Optimization for Ligand Docking (GOLD) software19 for ligand placement, and the force field-based scoring function GBVI/WSA dG was used for refinement of the protein pocket within 5 Å proximity to the placed ligand. The binding free energy was evaluated using the GoldScore79 method with efficiency at the default value, retaining 10 poses per ligand. The protein was ionized using the MOE function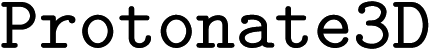 with default settings (T = 300 K, pH = 7.0, ionic strength I = 0.1 mol dm−3).
with default settings (T = 300 K, pH = 7.0, ionic strength I = 0.1 mol dm−3).
The top-50 ranked molecules from the data sets were divided into two groups based on their molecular weight. Molecules with ≤400 g mol−1 were docked into PDB-ID: 1EVE,76 and molecules with >400 g mol−1 were docked into PDB-ID: 1ODC.77 This division was necessary to maximize the likelihood of proper docking, as the crystal structure of 1EVE is optimized for smaller ligands similar in size to Huperzine A. Larger ligands often failed to dock appropriately into 1EVE due to steric clashes or inadequate fit within the binding pocket. Therefore, PDB-ID: 1ODC was selected for molecules exceeding 400 g mol−1 to ensure a better accommodation of larger structures and to improve docking accuracy across a wider molecular size range. RDKit (version 2022.09.01)81 was used to transform the SMILES-strings into a molecular format accessible for the automated docking. Thereby, a 3D conformation of the ligand was generated and hydrogen atoms were added. The molecule was then stored in SDF format. An identical procedure was performed to dock the molecules on the crystal structure of AChE complexed with template molecule Huperzine A (PDB-ID: 4EY5 (ref. 17)). The 200 binding poses were manually scanned for presence of key hydrogen bond interactions with crystal water in the binding site and orientation within the binding pocket. Key interactions for binding pose selection were hydrogen bond formation with the water molecule in the binding pocket by TYR121 and TYR334 for PDB-ID: 1EVE, and hydrogen bond formation with HIS 440 for PDB-ID: 1ODC. The binding pose with the highest calculated GoldScore that complies with the aforementioned criteria was selected. 3D representation of protein-ligand complexes were visualized using PyMOL software (Ver. 2.5.3).82
Free energy calculation
The GFN2-xTB method was used to calculate the total Gibbs free energy of the top-ranked docking pose. Solvation effects were included by the continuum model generalized Born (GB) with surface area (SA) contributions, with water as the implicit solvent. The Hessian was calculated on unoptimized structures obtained from the selected docking poses, and calculated charge information was extracted. The binding free energy relating to ligand–protein binding, ΔG, at 298.15 K in aqueous solution, was calculated as:| ΔGbind = Gcomplex − (Gligand + Gprotein) | (2) |
| G = E + Gsolv + GRRHO | (3) |
Furthermore, ligand efficiency (LE) was used to correct binding free energies for differences in molecule size between the two protein pockets (PDB-ID: 1ODC and PDB-ID: 1EVE). LE relates the Gibbs free energy of protein–ligand interaction to the number of heavy atoms (nHA) of the ligand as:
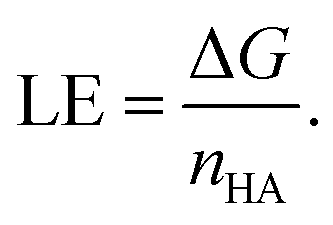 | (4) |
Chemical synthesis
The synthesis of 3-(1-((5-acetylthiophen-2-yl)methyl)piperidin-4-yl)-1-(4-methylpiperazin-1-yl)propan-1-one (2) began with esterification of commercially available piperidine-carboxylic acid 3 to afford the corresponding amine 4 in 43%. Concurrently, the aldehyde 5 was selectively reduced to a primary alcohol using sodium triacetoxyborohydride, obtaining the alcohol 6 in 87% yield. Intermediate 6 was then converted to an alkyl iodide by reaction with iodine, triphenylphosphine, and imidazole (49% yield). The careful evaporation in this step was critical due to the volatility of the halide 7. Compound 8 was formed through an N-alkylation between the piperidine-derivative 4 and the alkyl iodide 7, yielding 56%), which was later hydrolyzed in the presence of LiOH to obtain the corresponding carboxylic acid 9 in quantitative yield. Finally, an amide coupling between 9 and N-methylpiperazine afforded the desired compound 2 in 16% yield (Fig. 5; ESI S1†). Compound 2 was fully characterized by 1H NMR and electrospray (ESI) high-resolution mass spectrometry (HRMS) (ESI S1†).1H NMR (400 MHz, MeOD): δ 6.90 (t, J = 12.3 Hz, 1H), 6.24 (d, J = 3.8 Hz, 1H), 2.93 (s, 2H), 2.80–2.71 (m, 4H), 2.12 (d, J = 11.7 Hz, 2H), 1.72 (s, 3H), 1.67–1.55 (m, 6H), 1.51 (d, J = 8.5 Hz, 3H), 1.25 (t, J = 11.2 Hz, 2H), 0.92 (d, J = 9.2 Hz, 2H), 0.79–0.67 (m, 2H), 0.46 (dd, J = 16.6, 5.5 Hz, 3H).
13C NMR (101 MHz, MeOD): δ 193.08, 174.11, 152.59, 144.73, 134.77, 128.96, 58.23, 56.04, 55.54, 54.55, 46.36, 45.96, 42.31, 36.39, 32.98, 32.89, 31.34, 26.48.
HRMS (ESI): [M + H]+ m/z calcd for C20H32N3O2S: 378.2210, found: 378.2205.
Biological characterization
Biological activity of the synthesized compound 2 was determined by utilizing an enzymatic acetylcholinesterase (AChE) inhibition assay.84 In brief, enzymatic activity of recombinant human AChE was monitored by tracking the conversion of acetylthiocholine to 5-thio-2-nitro-benzoic acid spectrophotometrically. Compound 2 was tested at 1 μM, 10 μM, and 30 μM in triplicates. Obtained readings were normalized to a galanthamine control and final results were reported as % AChE inhibition. All tests were conducted by Eurofins Cerep (France) on a fee-for-service basis (Eurofins Cerep ref. 363). The inhibition assay was conducted in N = 3 repeats resulting in the following values: 1 μM: 97.7, 93.3, 93.3; mean = 94.8, 10 μM: 90.9, 89.7, 86.0; mean = 88.9 and 30 μM: 69.3, 68.5, 67.3; mean = 68.4.Results
DRAGONFLY15 was applied to the Huperzine A template molecule in a ligand-based fashion, and to the crystal structure of AChE (PDB-ID: 4EY5) in a structure-based fashion using models trained on SMILES-strings and SELFIES. The resulting four molecular libraries were analyzed by a variety of metrics relevant to drug discovery (Table 1). While the SELFIES molecular libraries resulted in a higher fraction of valid, unique, and novel molecules (80% vs. 61%), SMILES-string-based models achieved higher enrichment in retrosynthetic accessibility (74% vs. 65%) and predicted bioactivity (31% vs. 19%). Additionally, SELFIES-based models generated more molecules that fulfilled the defined novelty criteria (68% vs. 52%) as well as a higher fraction of novel atomic (93% vs. 90%) and skeleton scaffolds (98% vs. 97%). The observed differences mirrors earlier observations.15 The number of molecules that fulfilled all required criteria for further processing (i.e., retrosynthetic accessibility, predicted activity, and novelty) converged to a similar percentage, i.e., 4.4% for SMILES-strings and 4.0% for SELFIES. The novelty criterion was purposely set to a high cut-off, ensuring the novelty of the selected molecules. The remaining molecules that fulfilled all properties were ranked by novelty score (weighted 1×) and predicted bioactivity (weighted 4×), and the top 50 molecules of each library were selected for further processing.| SMILES-strings | SELFIES | |||
|---|---|---|---|---|
| Ligand | Structure | Ligand | Structure | |
| a Abbreviation: RA score: retrosynthetic accessibility score. Pred.: predicted. | ||||
| Valid, unique & novel/# | 847′546 | 782′901 | 1′125′792 | 1′079′221 |
| Valid, unique & novel/% | 60.5 | 55.6 | 80.4 | 77.1 |
| RA score ≥ 0.95/% | 72.3 | 74.1 | 62.0 | 64.6 |
| Novelty score ≥ 0.7/% | 49.4 | 51.7 | 66.8 | 67.5 |
| Pred. activity ≤ 1 μM | 30.5 | 28.8 | 19.0 | 19.1 |
| Pred. activity ≤ 1 μM, novelty ≥ 0.7 & RA score ≥ 0.95/% | 3.9 | 4.4 | 3.4 | 4.0 |
| Novel Murcko scaffolds/% | 90.3 | 87.8 | 93.0 | 89.9 |
| Novel skeleton scaffolds/% | 97.5 | 97.1 | 98.0 | 97.7 |
Furthermore, the overlap between the four libraries was analyzed at the level of whole molecules, as well as across different scaffold definitions, including skeleton scaffolds, Murcko scaffolds, and Bemis-Murcko scaffolds (Fig. 2). The generated chemical entities from the different runs showed a non-overlapping fraction ranging from a minimum of 96% (for Structure-SMILES) to a maximum of 98% (for Ligand-SELFIES). The smallest non-overlapping fraction for all scaffold types was observed for the Bemis-Murcko scaffolds, with a non-overlapping fraction of 64%.
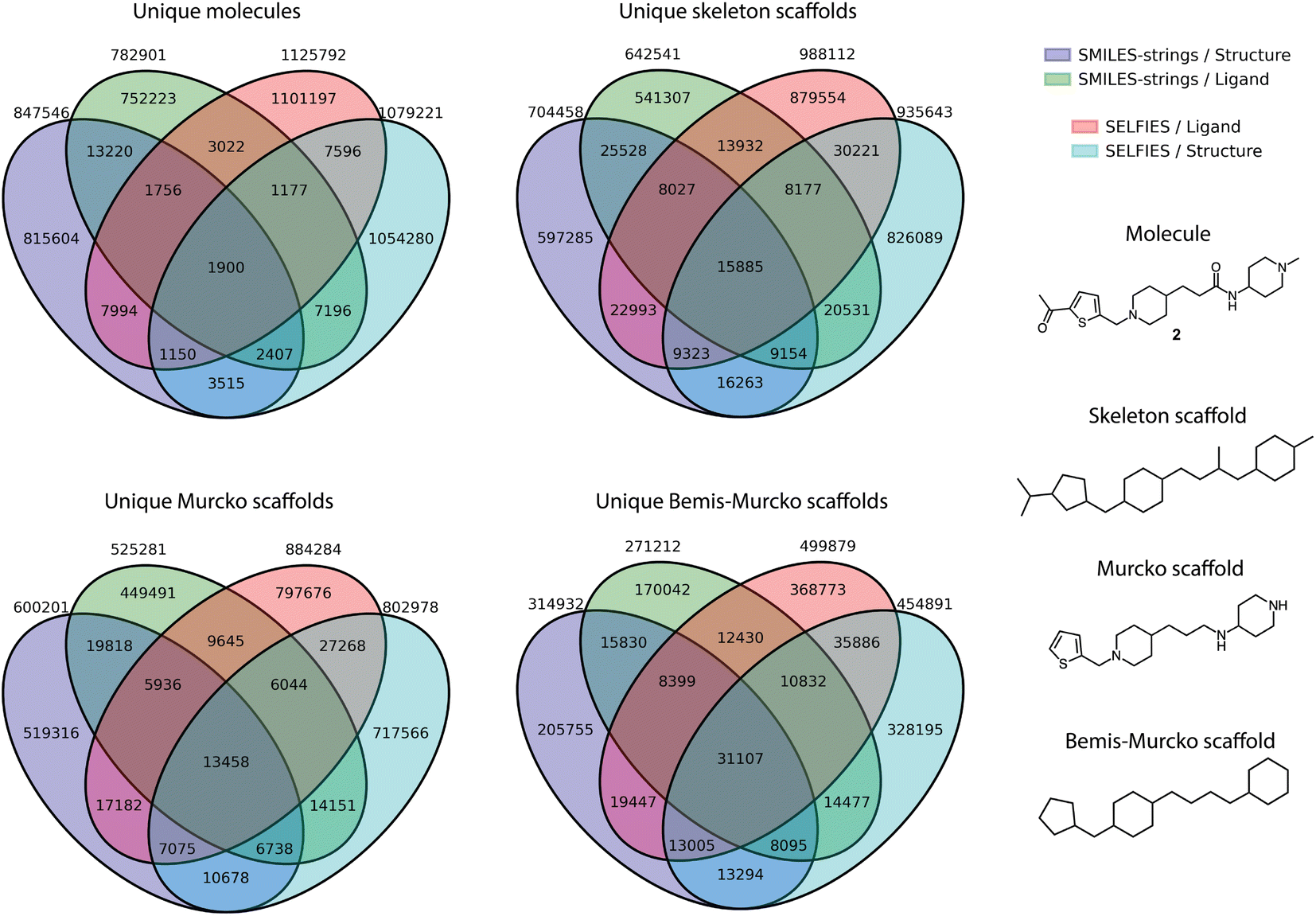 | ||
| Fig. 2 Comparison of four virtual compound libraries. Venn diagrams illustrate the overlap between the libraries (absolute numbers): Structure-SMILES (purple), Ligand-SMILES (green), Ligand-SELFIES (red), and Structure-SELFIES (blue). The top left plot shows whole molecules, the top right shows skeleton scaffolds, the bottom left represents Murcko scaffolds, and the bottom right shows Bemis–Murcko scaffolds. An example of the three scaffold abstractions for compound 2 is provided on the right. | ||
AChE-binding molecules are known to exhibit a positively charged protonated amine, i.e., often tertiary amines, enabling hydrogen bond interactions with two tyrosines (TYR), i.e., the so-called oxyanion hole, involving one water-mediated hydrogen bond to TYR124 and a direct hydrogen bond to TYR337.68,85 To allow for the formation of the water-mediated hydrogen bond, all water molecules except the one in the binding pocket near TYR124 were removed before molecular docking. Therefore, docking with GOLD using GBVI/WSA dG86 was applied to identify binding poses that exhibit these key hydrogen interactions. Automated protein–ligand docking with flexible side chains into the Huperzine A crystal structure (PDB-ID: 4EY5 (ref. 68)) yielded 52% (104 of 200) of top-ranking docked molecules exhibiting one key interaction with the water molecule in the oxyanion hole. Consequently, two different protein–ligand co-crystal structures were selected for automated docking, i.e., PDB-ID: 1EVE83 for molecules with ≤4 rings, and PDB-ID: 1ODC87 for molecules with >4 rings. Automated protein–ligand docking with flexible side chains using the two selected co-crystal structures yielded 100% of molecules exhibiting at least one key hydrogen interaction in the oxyanion hole. The mean GOLDScore of the selected top-200 ranked molecules was log(Ki) = −9.6 kcal mol−1 compared to the GOLDScore of Huperzine A docked to the AChE structure PDB-ID:4EY5, GOLD-Score = −8.6 kcal mol−1.
Using the top pose for each of the 200 molecules, binding free energies were calculated at two levels of theory, i.e., GFN-FF and GFN2-xTB, according to the schematic in Fig. 3. For both methods, water as an implicit solvent was used, and absolute point charges of proteins, ligands, and protein–ligand complexes were extracted and used as specified input. Subsequently, calculating Gibbs free energies (eqn (2)) for the three complexes enabled the computation of the respective ligand efficiency (ΔG/number of heavy atoms, see eqn (4)) for the 200 ligands, allowing for the evaluation of the predicted quality of the protein-ligand binding.88 To rank the top 200 molecules, the mean value between the ligand efficiency calculated through GFN2-xTB and the QSAR-based activity score was used, resulting in designed molecule 2 (Fig. 4). Molecule 2 (highlighted in orange) was selected since it showed the highest normalized QSAR score and showed competitive performance in both GFN–FF– and GF2-xTB-calculated ligand efficiencies. It yielded comparable calculated binding free energies to the Huperzine A template in both GFN-FF (−62.5 kcal mol−1 vs. −33.6 kcal mol−1) and GFN2-xTB (−19.2 kcal mol−1 vs. −11.1 kcal mol−1) calculations (Fig. 4).
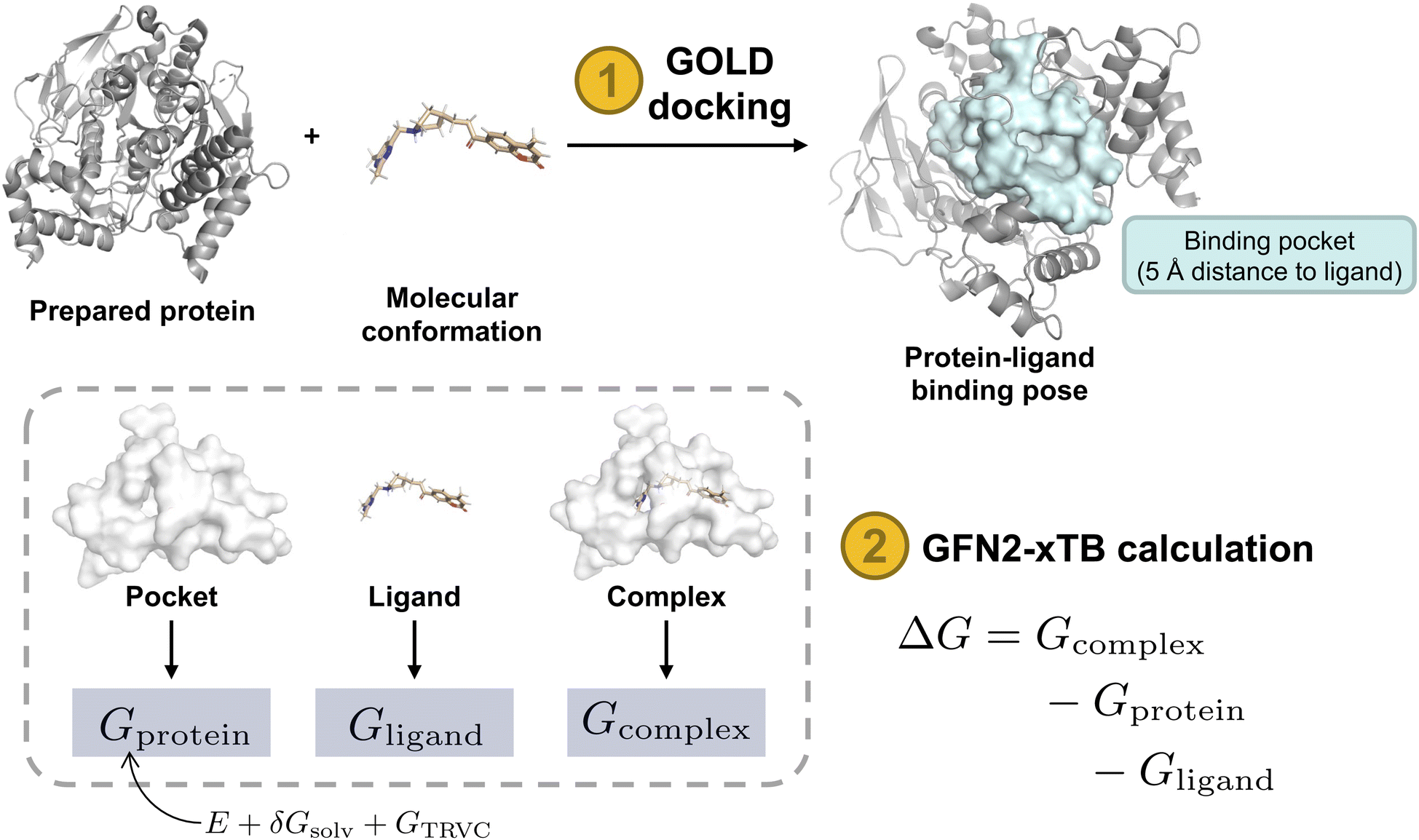 | ||
| Fig. 3 Schematic of Gibbs free energy calculation. Steps in orange and output in blue. Protein structure (PDB-ID: 1EVE). Protein surface shown in light blue and white indicates protein-ligand binding pocket in 5 Å distance to bound ligand with intact amino acid residues. Gx: total free energy of the respective system, ΔG GBSA: Gibbs free energy of protein–ligand interaction evaluated using GoldScore with pose optimization using GBVI/WSA dG, ΔG GFN2-xTB: Gibbs free energy of protein ligand interaction calculated from the total free energies of the three systems. E: total molecular gas-phase energy, Gsolv: solvation energy, GTRVC: thermostatic contribution to the free energy (approximated by the rigid rotor-harmonic oscillator, GRRHO).61,80 | ||
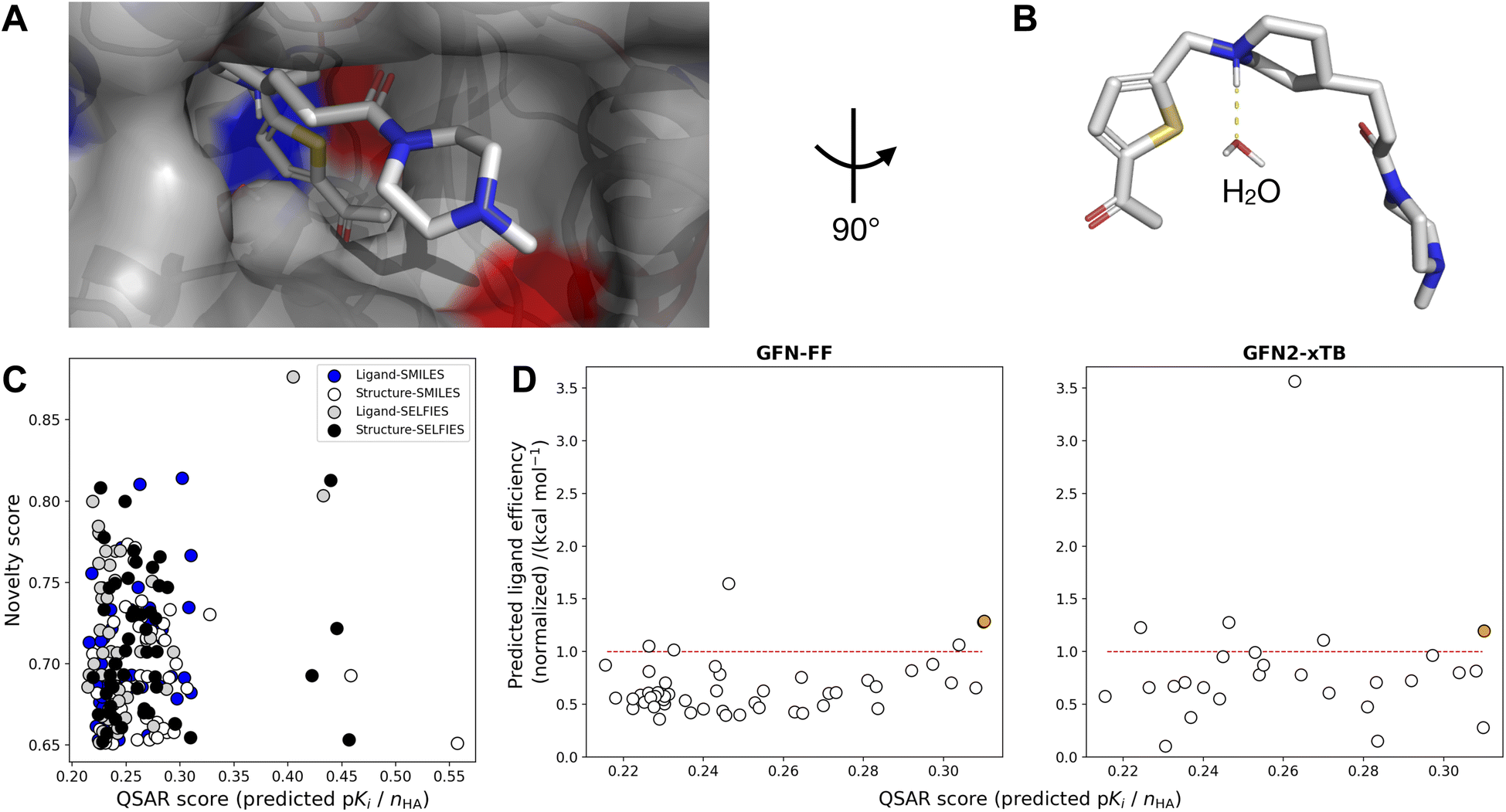 | ||
| Fig. 4 (A) Ligand binding pose of amide 2 bound to acetylcholinesterase (AChE) (PDB-ID: 1EVE),83 with nonpolar hydrogens omitted for clarity. Atom colors denote carbon (gray), nitrogen (blue), oxygen (red), and sulfur (yellow). The pocket surface is colored by positively (red) resp. negatively (blue) charged residues. (B) Close-up of water interaction without binding pocket. (C) Novelty score vs. QSAR score (normalized by number of heavy atoms) of the top-50 ranked compounds from four de novo generated libraries (Ligand-SMILES, Ligand-SELFIES, Structure-SMILES, Structure-SELFIES). (D) Predicted ligand efficiency (normalized wrt Huperzine A docked to PDB-ID: 4EY5) on two levels-of-theory (GFN-FF and GFN2-xTB) against ligand efficiency in terms of QSAR score (predicted pKi divided by number of heavy atoms), for Ligand-SMILES library. Points above the red line indicate better performance than Huperzine A. The point in orange indicates the compound (2) selected for synthesis. 1 (GFN-FF) resp. 23 (GFN2-xTB) outliers with negative normalized ligand efficiency values are not shown. | ||
The selected molecule (2) was successfully synthesized through a six-step convergent synthesis with the longest subsequent route of five steps and a total yield of 16% (Fig. 5) (ESI S1†). Subsequently, compound 2 was subjected to biological testing in an AChE inhibition assay, demonstrating low micromolar affinity. Specifically, compound 2 showed 31.6% (±0.8%) inhibition at 30 μM and 11% (±2%) inhibition at 10 μM (Table 2).
 | ||
| Fig. 5 Chemical Synthesis. Synthesis of computer-generated molecular design 2. The synthesis of compound 2 employed a convergent approach, starting from commercially available building blocks 3 and 4, and spanning a total of 6 steps. The longest sequential route involved 5 steps. The overall yield achieved for the synthesis of 2 was 16%. Conditions: (a) (I) AcCl, EtOH, 0 °C, 20 min, (II) H2SO4, EtOH, 80 °C, 5 min; (b) NaO(Ac)3BH, THF, 65 °C, 1 h; (c) I2, PPh3, imidazole, DCM, 25 °C, 16 h; (d) Cs2CO3, MeCN, 25 °C, 16 h; (e) LiOH, H2O/Dioxane, 25 °C, 16 h; (f) N-methylpiperazine, EDC HCl, HOBt, dry DFM, 25 °C, 18 h. | ||
| Compound (concentration) | AChE inhibition/% |
|---|---|
| Compound 2 (1 μM) | 5.2 (±0.8) |
| Compound 2 (10 μM) | 11.1 (±2.0) |
| Compound 2 (30 μM) | 31.6 (±0.8) |
| Huperzine A 1 (1 μM) | 98.1 (±0.7) |
Discussion
The integration of the DRAGONFLY algorithm with semi-empirical quantum chemistry methods resulted in the generation of a natural product-inspired molecular library. This study compared the utility of SMILES and SELFIES representations for molecular generation, highlighting that while SMILES-based models achieved higher enrichment in retrosynthetic accessibility and predicted bioactivity, SELFIES-based models excelled in generating structurally novel molecules. Ultimately, both representations proved valuable, with a convergence in the number of molecules meeting all required criteria for further processing. Furthermore, the overlap analysis of the four libraries, both at the whole molecule level and across different scaffold abstractions, concluded that the libraries are predominantly non-overlapping, underscoring the diversity of drug-like chemical space that can be addressed by molecular design methods.The SEQM method GFN2-xTB served as a scoring function in this study. By estimating binding free energies, the approach enabled the selection of potential AChE inhibitors from the virtual library. GFN2-xTB provided a balance between computational efficiency and estimated accuracy, making it suitable for the large-scale evaluation of protein–ligand interactions.
The top-ranked molecule identified through this integrated approach, i.e., compound (2), was successfully synthesized, confirming the DRAGONFLY de novo design method as useful for medicinal chemistry. Although the observed activity is modest, the novelty of the molecule underscores the potential of this combined de novo design and SEQM methodology for scaffold hopping. The new inhibitor may serve as a starting point for hit-to-lead optimization efforts.
In conclusion, the application of SEQM methods in deep-learning-based de novo molecular design presents an avenue for future hit identification in drug discovery, extending the toolkit available to medicinal chemists. The interactome-based machine learning model has shown its ability to generate novel synthesizable molecules that meet relevant criteria. However, it became apparent that the molecule scoring and selection approach employed here has limitations in its predictive power. The ligand docking and the binding free energy estimates of the inhibitory activity of the computer-generated molecules were overly optimistic. Given the proven scaffold-hopping potential of the molecule construction algorithm, future research will need to improve the molecule scoring and selection process. A potential extension of standard SEQM methods for this purpose might be to include correction terms for the affinity estimations, such as Δ-learning or heuristic approaches based on pharmacophore and shape similarity of the de novo designs to the template molecule.89–91
Data availability
A reference implementation of the Gibbs free energy calculation procedure based on GFN2-xTB and GFN-FF method20 is available at https://github.com/ETHmodlab/seqm_scoring (DOI: http://10.5281/zenodo.13959365, https://doi.org/10.5281/zenodo.13959364).Conflicts of interest
G. S. is a co-founder of inSili.com LLC, Zurich, and Xanadys LLC, Zurich, and a consultant to the pharmaceutical industry.Acknowledgements
This work was financially supported by the Swiss National Science Foundation (grant no. 205321_182176 and CRSII5_202245). C. I. acknowledges support from the Scholarship Fund of the Swiss Chemical Industry.Notes and references
- G. Schneider and U. Fechner, Nat. Rev. Drug Discovery, 2005, 4, 649–663 CrossRef CAS
.
- G. Schneider and D. E. Clark, Angew. Chem., Int. Ed., 2019, 58, 10792–10803 CrossRef CAS
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS
- W. Yuan, D. Jiang, D. K. Nambiar, L. P. Liew, M. P. Hay, J. Bloomstein, P. Lu, B. Turner, Q.-T. Le, R. Tibshirani, P. Khatri, M. G. Moloney and A. C. Koong, J. Chem. Inf. Model., 2017, 57, 875–882 CrossRef CAS PubMed
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed
- D. Merk, L. Friedrich, F. Grisoni and G. Schneider, Mol. Inf., 2018, 37, 1700153 CrossRef
- M. Moret, I. Pachon Angona, L. Cotos, S. Yan, K. Atz, C. Brunner, M. Baumgartner, F. Grisoni and G. Schneider, Nat. Commun., 2023, 14, 114 CrossRef CAS
- F. Grisoni, B. J. Huisman, A. L. Button, M. Moret, K. Atz, D. Merk and G. Schneider, Sci. Adv., 2021, 7, eabg3338 CrossRef CAS PubMed
- F. Grisoni and G. Schneider, J. Comput. Aided Mol. Des., 2019, 20, 35–42 Search PubMed
- M. Moret, L. Friedrich, F. Grisoni, D. Merk and G. Schneider, Nat. Mach. Intell., 2020, 2, 171–180 CrossRef
- M. A. Skinnider, R. G. Stacey, D. S. Wishart and L. J. Foster, Nat. Mach. Intell., 2021, 3, 759–770 CrossRef
- M. L. Raves, M. Harel, Y.-P. Pang, I. Silman, A. P. Kozikowski and J. L. Sussman, Nat. Struct. Mol. Biol., 1997, 4, 57–63 CrossRef CAS
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Félix, M. Magariños, J. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Marañón, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. Radoux, A. Segura-Cabrera, A. Hersey and A. Leach, Nucleic Acids Res., 2019, 47, D930–D940 CrossRef CAS PubMed
- H. Dvir, H. L. Jiang, D. M. Wong, M. Harel, M. Chetrit, X. C. He, G. Y. Jin, G. L. Yu, X. C. Tang, I. Silman, D. L. Bai and J. L. Sussman, Biochemistry, 2002, 41, 10810–10818 CrossRef CAS
- K. Atz, L. Cotos, C. Isert, M. Håkansson, D. Focht, M. Hilleke, D. F. Nippa, M. Iff, J. Ledergerber and C. C. Schiebroek, et al., Nat. Commun., 2024, 15, 3408 CrossRef CAS PubMed
- A. T. Müller, K. Atz, M. Reutlinger and N. Zorn, ICML’24 Workshop ML for Life and Material Science: from Theory to Industry Applications, 2024 Search PubMed
- J. Cheung, M. J. Rudolph, F. Burshteyn, M. S. Cassidy, E. N. Gary, J. Love, M. C. Franklin and J. J. Height, J. Med. Chem., 2012, 55, 10282–10286 CrossRef CAS PubMed
- Chemical Computing Group ULC, Molecular Operating Environment (MOE) 2019.01, Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite 910, Montreal, QC, Canada, H3A 2R7, 2021, 2019 Search PubMed
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed
- P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2017, 56, 7971–7974 CrossRef CAS
- D. Merk, F. Grisoni, L. Friedrich, E. Gelzinyte and G. Schneider, J. Med. Chem., 2018, 61, 5442–5447 CrossRef CAS PubMed
- D. Merk, F. Grisoni, L. Friedrich and G. Schneider, Commun. Chem., 2018, 1, 1–9 CrossRef
- T. Steinbrecher, Protein-Ligand Interactions, 2012, pp. 207–236 Search PubMed
- C. Isert, K. Atz and G. Schneider, Curr. Opin. Struct. Biol., 2023, 79, 102548 CrossRef CAS
- T. Steinbrecher, D. L. Mobley and D. A. Case, J. Chem. Phys., 2007, 127, 21 CrossRef PubMed
- L. Wang, Y. Wu, Y. Deng, B. Kim, L. Pierce, G. Krilov, D. Lupyan, S. Robinson, M. K. Dahlgren and J. Greenwood, et al., J. Am. Chem. Soc., 2015, 137, 2695–2703 CrossRef CAS PubMed
- T. B. Steinbrecher, M. Dahlgren, D. Cappel, T. Lin, L. Wang, G. Krilov, R. Abel, R. Friesner and W. Sherman, J. Chem. Inf. Model., 2015, 55, 2411–2420 CrossRef CAS PubMed
- C. E. Schindler, H. Baumann, A. Blum, D. Bose, H.-P. Buchstaller, L. Burgdorf, D. Cappel, E. Chekler, P. Czodrowski and D. Dorsch, et al., J. Chem. Inf. Model., 2020, 60, 5457–5474 CrossRef CAS
- T. Steinbrecher, C. Zhu, L. Wang, R. Abel, C. Negron, D. Pearlman, E. Feyfant, J. Duan and W. Sherman, J. Mol. Biol., 2017, 429, 948–963 CrossRef CAS PubMed
- G. A. Ross, C. Lu, G. Scarabelli, S. K. Albanese, E. Houang, R. Abel, E. D. Harder and L. Wang, Commun. Chem., 2023, 6, 222 CrossRef
- K. Atz, F. Grisoni and G. Schneider, Nat. Mach. Intell., 2021, 3, 1023–1032 CrossRef
- C. Isert, K. Atz, S. Riniker and G. Schneider, RSC Adv., 2024, 14, 4492–4502 RSC
- G. Corso, B. Jing, R. Barzilay, T. Jaakkola et al., International Conference on Learning Representations (ICLR 2023), 2023 Search PubMed
- C. Harris, K. Didi, A. R. Jamasb, C. K. Joshi, S. V. Mathis, P. Lio and T. Blundell, 2023, preprint, arXiv:2308.07413, DOI:10.48550/arXiv.2308.07413.
- M. Buttenschoen, G. M. Morris and C. M. Deane, 2023, preprint, arXiv:2308.05777, DOI:10.48550/arXiv.2308.05777.
- Y.-q. Chen, Y.-j. Sheng, Y.-q. Ma and H.-m. Ding, Phys. Chem. Chem. Phys., 2022, 24, 14339–14347 RSC
- A. Pecina, J. Fanfrlík, M. Lepšík and J. Řezáč, Nat. Commun., 2024, 15, 1127 CrossRef CAS PubMed
- O. T. Unke, M. Stöhr, S. Ganscha, T. Unterthiner, H. Maennel, S. Kashubin, D. Ahlin, M. Gastegger, L. M. Sandonas, A. Tkatchenko et al., arXiv, 2022, preprint, arXiv:2205.08306, DOI:10.48550/arXiv.2205.08306.
- A. Tosstorff, J. C. Cole, R. Taylor, S. F. Harris and B. Kuhn, J. Chem. Inf. Model., 2020, 60, 6595–6611 CrossRef CAS PubMed
- A. Tosstorff, J. C. Cole, R. Bartelt and B. Kuhn, ChemMedChem, 2021, 16, 3428–3438 CrossRef CAS PubMed
- A. Tosstorff, M. G. Rudolph, J. C. Cole, M. Reutlinger, C. Kramer, H. Schaffhauser, A. Nilly, A. Flohr and B. Kuhn, J. Comput. Aided Mol. Des., 2022, 36, 753–765 CrossRef CAS
- J. Sieg, F. Flachsenberg and M. Rarey, J. Chem. Inf. Model., 2019, 59, 947–961 CrossRef CAS
- L. Chen, A. Cruz, S. Ramsey, C. J. Dickson, J. S. Duca, V. Hornak, D. R. Koes and T. Kurtzman, PLoS One, 2019, 14, e0220113 CrossRef CAS PubMed
- J. Scantlebury, N. Brown, F. Von Delft and C. M. Deane, J. Chem. Inf. Model., 2020, 60, 3722–3730 CrossRef CAS PubMed
- M. Volkov, J.-A. Turk, N. Drizard, N. Martin, B. Hoffmann, Y. Gaston-Mathé and D. Rognan, J. Med. Chem., 2022, 65, 7946–7958 CrossRef CAS PubMed
- U. Ryde and P. Soderhjelm, Chem. Rev., 2016, 116, 5520–5566 CrossRef CAS PubMed
- M. Manathunga, A. W. Götz and K. M. Merz Jr, Curr. Opin. Struct. Biol., 2022, 75, 102417 CrossRef CAS PubMed
- U. Holzgrabe, P. Kapková, V. Alptüzün, J. Scheiber and E. Kugelmann, Expert Opin. Ther. Targets, 2007, 11, 161–179 CrossRef CAS PubMed
- A. S. Christensen, T. Kubař, Q. Cui and M. Elstner, Chem. Rev., 2016, 116, 5301–5337 CrossRef CAS PubMed
- V. Fock, Z. Phys., 1930, 61, 126–148 CrossRef
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef
- N. D. Yilmazer and M. Korth, Comput. Struct. Biotechnol. J., 2015, 13, 169–175 CrossRef CAS PubMed
- Y.-q. Chen, Y.-j. Sheng, Y.-q. Ma and H.-m. Ding, Phys. Chem. Chem. Phys., 2022, 24, 14339–14347 RSC
- C. Isert, K. Atz, J. Jiménez-Luna and G. Schneider, Sci. Data, 2022, 9, 273 CrossRef CAS
- S. Schmitz, J. Seibert, K. Ostermeir, A. Hansen, A. H. Göller and S. Grimme, J. Phys. Chem. B, 2020, 124, 3636–3646 CrossRef CAS PubMed
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2021, 11, e1493 CrossRef CAS
- J. D. Gale, L. M. LeBlanc, P. R. Spackman, A. Silvestri and P. Raiteri, J. Chem. Theory Comput., 2021, 17, 7827–7849 CrossRef CAS PubMed
- S. Spicher and S. Grimme, Angew. Chem., Int. Ed., 2020, 59, 15665–15673 CrossRef CAS PubMed
- S. Spicher and S. Grimme, J. Phys. Chem. Lett., 2020, 11, 6606–6611 CrossRef CAS PubMed
- D. L. Mobley and M. K. Gilson, Annu. Rev. Biophys., 2017, 46, 531–558 CrossRef CAS PubMed
- D. F. Nippa, K. Atz, R. Hohler, A. T. Müller, A. Marx, C. Bartelmus, G. Wuitschik, I. Marzuoli, V. Jost and J. Wolfard, et al., Nat. Chem., 2024, 16, 239–248 CrossRef CAS PubMed
- C. Isert, J. C. Kromann, N. Stiefl, G. Schneider and R. A. Lewis, ACS Omega, 2023, 8, 2046–2056 CrossRef CAS PubMed
- D. F. Nippa, K. Atz, A. T. Müller, J. Wolfard, C. Isert, M. Binder, O. Scheidegger, D. B. Konrad, U. Grether and R. E. Martin, et al., Commun. Chem., 2023, 6, 256 CrossRef CAS PubMed
- K. Atz, D. Nippa, A. Mueller, V. Jost, A. Anelli, M. Reutlinger, C. Kramer, R. E. Martin, U. Grether and G. Schneider, et al., RSC Med. Chem., 2024 Search PubMed
- F. Grisoni, M. Moret, R. Lingwood and G. Schneider, J. Chem. Inf. Model., 2020, 60, 1175–1183 CrossRef CAS PubMed
- J. Cheung, M. J. Rudolph, F. Burshteyn, M. S. Cassidy, E. N. Gary, J. Love, M. C. Franklin and J. J. Height, J. Med. Chem., 2012, 55, 10282–10286 CrossRef CAS PubMed
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Mach. Learn.Sci. Technol., 2020, 1, 045024 CrossRef
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle, K. M. Jablonka, R. F. Lameiro, D. Lemm, A. Lo, S. M. Moosavi, J. M. Nápoles-Duarte, A. Nigam, R. Pollice, K. Rajan, U. Schatzschneider, P. Schwaller, M. Skreta, B. Smit, F. Strieth-Kalthoff, C. Sun, G. Tom, G. F. von Rudorff, A. Wang, A. White, A. Young, R. Yu and A. Aspuru-Guzik, arXiv, 2022, preprint, arXiv:2204.00056, DOI:10.1016/j.patter.2022.100588.
- A. Thakkar, V. Chadimová, E. J. Bjerrum, O. Engkvist and J.-L. Reymond, Chem. Sci., 2021, 12, 3339–3349 RSC
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed
- M. Reutlinger, C. P. Koch, D. Reker, N. Todoroff, P. Schneider, T. Rodrigues and G. Schneider, Mol. Inf., 2013, 32, 133–138 CrossRef CAS PubMed
- A. M. Schreyer and T. Blundell, J. Cheminf., 2012, 4, 27 CAS
- G. Kryger, I. Silman and J. L. Sussman, Structure, 1999, 7, 297–307 CrossRef CAS PubMed
- E. H. Rydberg, B. Brumshtein, H. M. Greenblatt, D. M. Wong, D. Shaya, L. D. Williams, P. R. Carlier, Y.-P. Pang, I. Silman and J. L. Sussman, J. Med. Chem., 2006, 49, 5491–5500 CrossRef CAS PubMed
- S. K. Burley, H. M. Berman, G. J. Kleywegt, J. L. Markley, H. Nakamura and S. Velankar, Protein Crystallography: Methods and Protocols, 2017, 627–641 Search PubMed
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Proteins: Struct., Funct., Bioinf., 2003, 52, 609–623 CrossRef CAS PubMed
- S. Decherchi and A. Cavalli, Chem. Rev., 2020, 120, 12788–12833 CrossRef CAS PubMed
- G. Landrum, RDKit: Open-source cheminformatics, accessed September 2020, http://www.rdkit.org, 2022 Search PubMed.
- L. L. C. Schrödinger, The PyMOL Molecular Graphics System, Version 2.3.5, 2022, http://www.pyymol.org/pymol Search PubMed.
- G. Kryger, I. Silman and J. L. Sussman, Structure, 1999, 7, 297–307 CrossRef CAS PubMed
- G. L. Ellman, K. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed
- I. Silman and J. L. Sussman, Chem. Biol. Interact., 2008, 175, 3–10 CrossRef CAS PubMed
- C. R. Corbeil, C. I. Williams, P. Labute and J. Comp, Mol. Des., 2012, 26, 775–786 CAS
- E. H. Rydberg, B. Brumshtein, H. M. Greenblatt, D. M. Wong, D. Shaya, L. D. Williams, P. R. Carlier, Y.-P. Pang, I. Silman and J. L. Sussman, J. Med. Chem., 2006, 49, 5491–5500 CrossRef CAS PubMed
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discov., 2014, 13, 105–121 CrossRef CAS PubMed
- K. Atz, C. Isert, M. N. Böcker, J. Jiménez-Luna and G. Schneider, Phys. Chem. Chem. Phys., 2022, 24, 10775–10783 RSC
- L. Friedrich, R. Byrne, A. Treder, I. Singh, C. Bauer, T. Gudermann, M. Mederos y Schnitzler, U. Storch and G. Schneider, ChemMedChem, 2020, 15, 566–570 CrossRef CAS PubMed
- L. Friedrich, G. Cingolani, Y.-H. Ko, M. Iaselli, M. Miciaccia, M. G. Perrone, K. Neukirch, V. Bobinger, D. Merk, R. K. Hofstetter, O. Werz, A. Koeberle, A. Scilimati and G. Schneider, Adv. Sci., 2021, 8, 2100832 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05422a |
| ‡ These authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2024 |