
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaction of unsymmetrical α-bromo-1,3-diketones with N-substituted thioureas: regioselective access to 2-(N-arylamino)-5-acyl-4-methylthiazoles and/or rearranged 2-(N-acylimino)-3-N-aryl-4-methylthiazoles†
Ranjana Aggarwal *ab,
Shilpa Sharmaa,
Naman Jaina,
Dionisia Sanzc,
Rosa M. Claramuntc,
Patricia Delgadod and
M. Carmen Torralbae
*ab,
Shilpa Sharmaa,
Naman Jaina,
Dionisia Sanzc,
Rosa M. Claramuntc,
Patricia Delgadod and
M. Carmen Torralbae
aDepartment of Chemistry, Kurukshetra University, Kurukshetra, Haryana, India
bCSIR-National Institute of Science Communication and Policy Research, New Delhi, India. E-mail: ranjana67in@yahoo.com; ranjanaaggarwal67@gmail.com; Tel: +91 9896740740
cDepartamento de Química Orgánica y Bio-orgánica, Facultad de Ciencias, UNED, Avenida Esparta s/n, Las Rozas, E-28232, Madrid, Spain
dUnidad de Difracción de Rayos X – CAI de Técnicas Químicas, Facultad de Ciencias Químicas, UCM, E-28040 Madrid, Spain
eDepartamento de Química Inorgánica, Facultad de Ciencias Químicas, UCM, E-28040 Madrid, Spain
First published on 8th November 2024
Abstract
The present study reports some fascinating results of Hantzsch's [3 + 2] cyclic condensation of α-bromo-1,3-diketones, a tri-electrophilic synthon generated in situ by bromination of 1,3-diketones using the mild brominating reagent NBS with trinucleophilic N-substituted thioureas. Interestingly, out of a total of 20 combinations, 10 resulted in the exclusive formation of the desired 2-(N-arylamino)-5-acyl-4-methylthiazoles regioselectively, seven led to the formation of unexpected 2-(N-acylimino)-3-N-aryl-4-methylthiazoles through an interesting C–N acyl migration, and three furnished a mixture consisting of both products. The regioselectivity pattern of the two products may be attributed to a greater electrophilicity of the carbonyl carbon of the acetyl group than that of the acyl group towards both nitrogens of thiourea. The structures of the thiazole derivatives were unambiguously assigned using 1H-NMR, 13C-NMR, and rigorous heteronuclear 2D-NMR [(1H–13C) HMQC and (1H–13C) HMBC] spectroscopic techniques. The outcomes of the spectroscopic experiments were further concurred through X-ray crystallographic studies, and a plausible mechanism for acyl migration was proposed for the formation of the unexpected rearranged product.
Introduction
Among heterocycles, thiazoles are notably important due to their potential as bioactive compounds and versatile building blocks for natural products and pharmaceuticals. 2-Aminothiazoles and 2-iminothiazoles are important sub-units in a number of therapeutically active compounds with a wide range of pharmacological activities, such as antiviral,1 anticancer,2 antibacterial,3 antifungal,4 anticonvulsant,5 antimicrobial,6 anti-parkinsonian7 and anti-inflammatory activities,8 which can be illustrated well by the large number of drugs in the market containing this moiety. In this regard, the anticonvulsant riluzole, anti-parkinsonian talipexole, antischistosomal miridazole, anthelmintic tiabendazole, antibacterial sulfathiazole, antidepressant pramipexole, and anti-inflammatory fentiazac and fanetizole are well-cited examples.9 Moreover, the 2-aminothiazole ring has been applied in other fields, such as polymers,10 fungicides11 and antioxidants.12Pertaining to the immense therapeutic eminence, a number of synthetic attempts have been made by various research groups for the synthesis of 2-aminothiazole derivatives, including the cyclocondensation of amidines with phenacyl bromides13–15 (path I, Chart 1), N-substituted thiourea with β-ketoesters16–18 (path II, Chart 1) and propargyl bromides19 (path III, Chart 1), potassium thiocyanate with vinyl azides20 or oximes21 (path IV, Chart 1), thiourea with phenacyl bromides22,23 (path V, Chart 1) and aliphatic or aromatic amines with monothiodiketones24 (path VI, Chart 1). However, only a few reports have cited the use of NBS and N-substituted thiourea derivatives. Additionally, although occasionally refined, these methods suffer from demerits such as a longer reaction time, involvement of lachrymatory α-haloketone, low yields, and tedious workup methods.
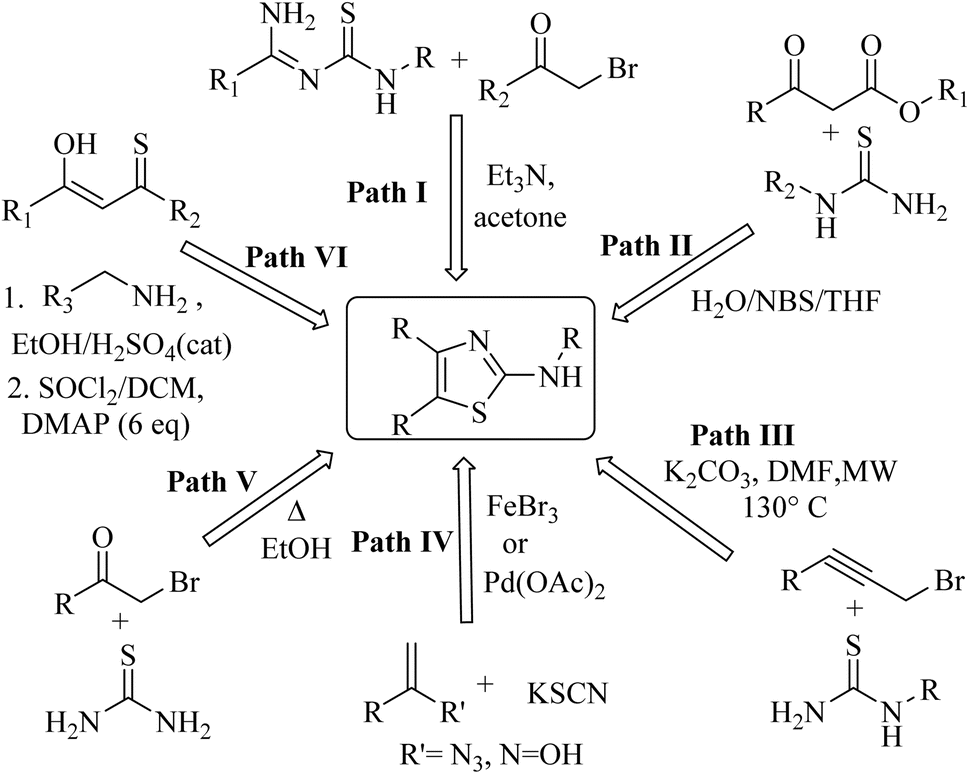 | ||
| Chart 1 Reported literature methods for 2-aminothiazole derivatives. | ||
Moreover, there is no existing literature report involving a regioselective study of the reaction between the unsymmetrical 1,3-diketones with thiourea derivatives.
However, a careful literature survey revealed that there are some limited methods to synthesis 2-iminothiazole derivatives, mainly dealing with the reaction of 2-chloro carbonyl compounds with aromatic amines and phenacyl bromides in two-step TEBA catalyzed aqueous medium (path I, Chart 2).25 Other synthetic methods involve the cyclocondensation of amines, and isocyanates with phenacyl bromides (path II, Chart 2),26,27 nitro epoxides (path III, Chart 2),28 and β-nitroacrylates (path IV, Chart 2).29 Additionally, the reaction of tetramethyl guanidine with isocyanate and 2-chloro-1,3-dicarbonyl compounds30 has been reported to yield a 2-iminothiazole derivative (path V, Chart 2). However, these methods suffer from drawbacks like a multi-step reaction sequence, long reaction time, use of catalysts, and low reaction yields. Also, synthons such as nitro epoxides, β-nitroacrylates and tetramethyl guanidine are not easily accessible, which makes the synthesis of 2-iminothiazole even more challenging.
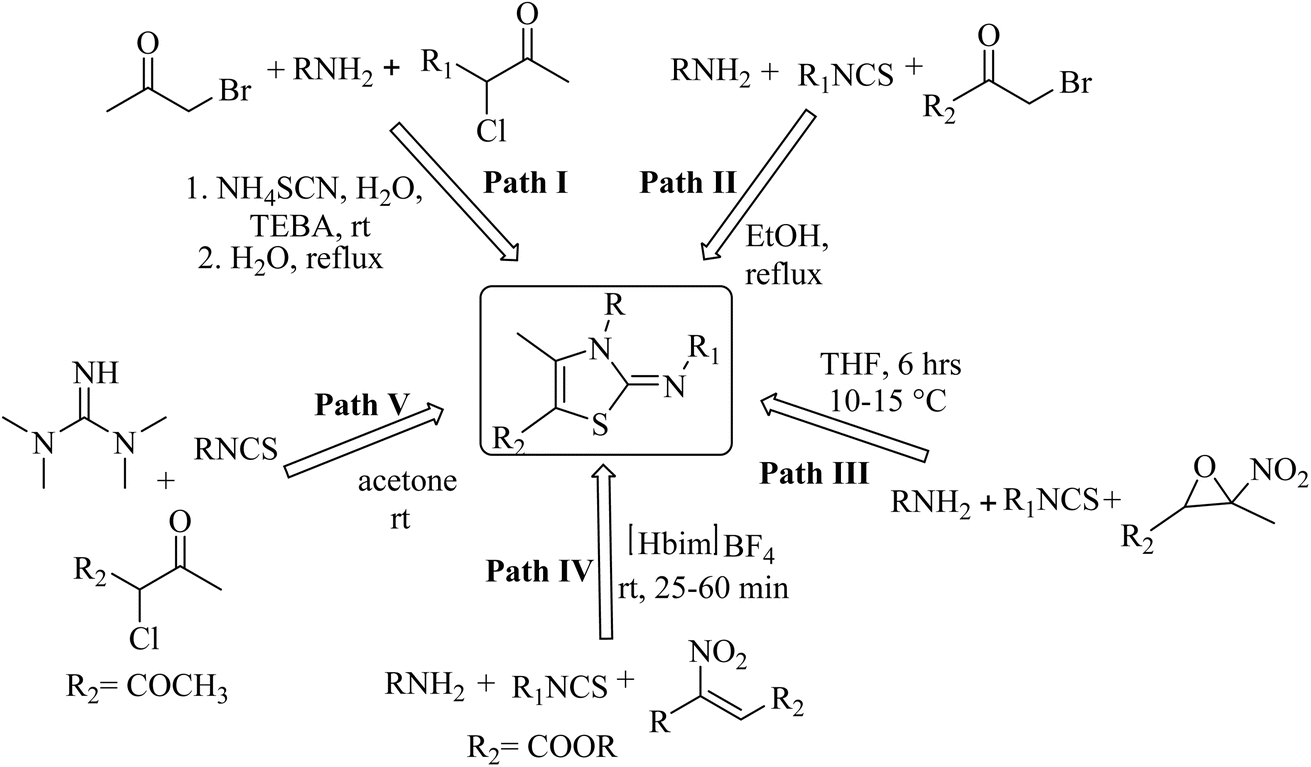 | ||
| Chart 2 Reported literature methods for 2-iminothiazole derivatives. | ||
Our research group has been actively engaged in heterocyclic synthesis using a regioselective reaction between α-bromo-1,3-diketones and various binucleophiles, such as 2-aminopyridine,31 tetrahydropyrimidine-2-thione,32 imidazole-2-thione,33 [1,2,4]triazole-3-thiol,34 4-amino-[1,2,4]triazole-3-thiol,35 thiazole-4-thiocarboxamide36 and thiosemicarbazide,37 employing various eco-friendly synthetic routes. Very recently, we have reported the solvent-free regioselective reaction of unsymmetrical diketones 1 with thioamide 3 derivatives, as the binucleophile, for thiazole synthesis,38 where only one regioisomer is obtained exclusively out of two possible regioisomers (Scheme 1).
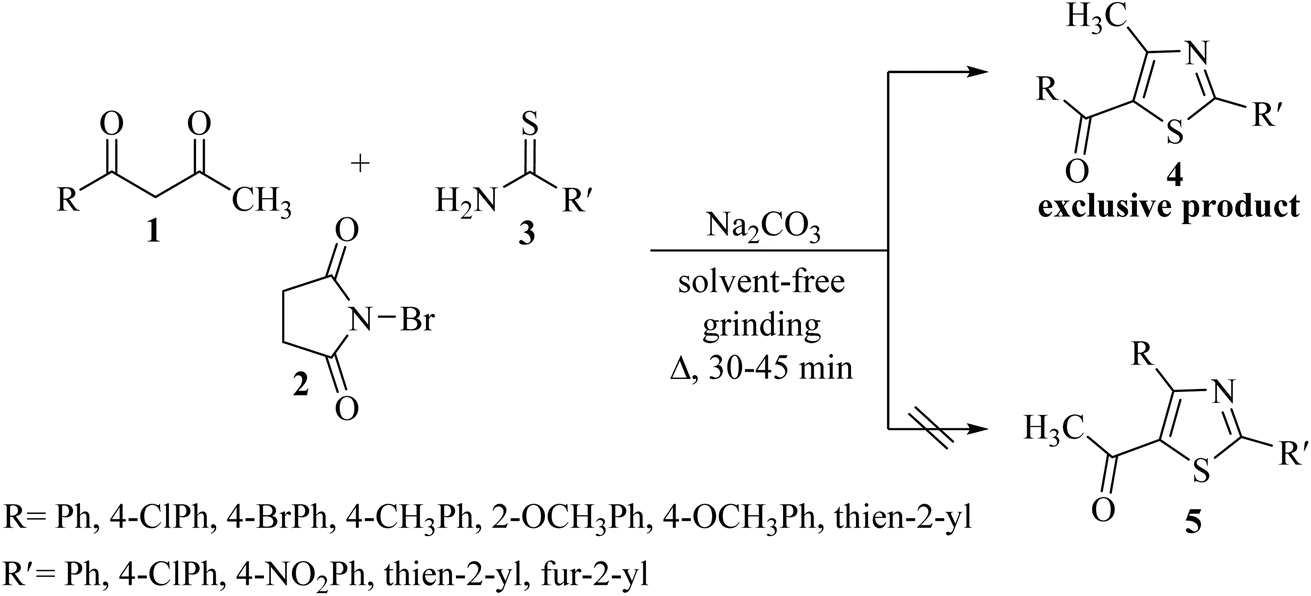 | ||
| Scheme 1 Previous work on the synthesis of thiazole derivatives from thioamides. | ||
This intrigued us to undertake the reaction of trinucleophilic N-substituted thiourea derivatives with trielectrophilic α-bromo-1,3-diketone, as regiochemical control of the reaction may generate further complexity in the product formation. α-Bromo-1,3-diketones 6 (generated in situ by bromination of 1,3-diketones 1 using the mild brominating reagent NBS 2) is a potential trielectrophile with sites A1, A2 and A3. In principle, its reaction with N-substituted thiourea derivatives 7 (a trinucleophile with reactive sites B1, B2 and B3) may result in the formation of four isomers: 2-(N-arylamino)-5-acyl-4-methylthiazoles 8, 2-(N-arylamino)-5-acetyl-4-arylthiazoles 9, 2-imino-3-(N-aryl)-5-acyl-4-methylthiazoles 10, and 2-imino-3-(N-aryl)-5-acetyl-4-arylthiazoles 11. This would fix the attack of more nucleophilic sulfur (B2) to displace bromine at A2 through nucleophilic substitution in the first step (Scheme 2).
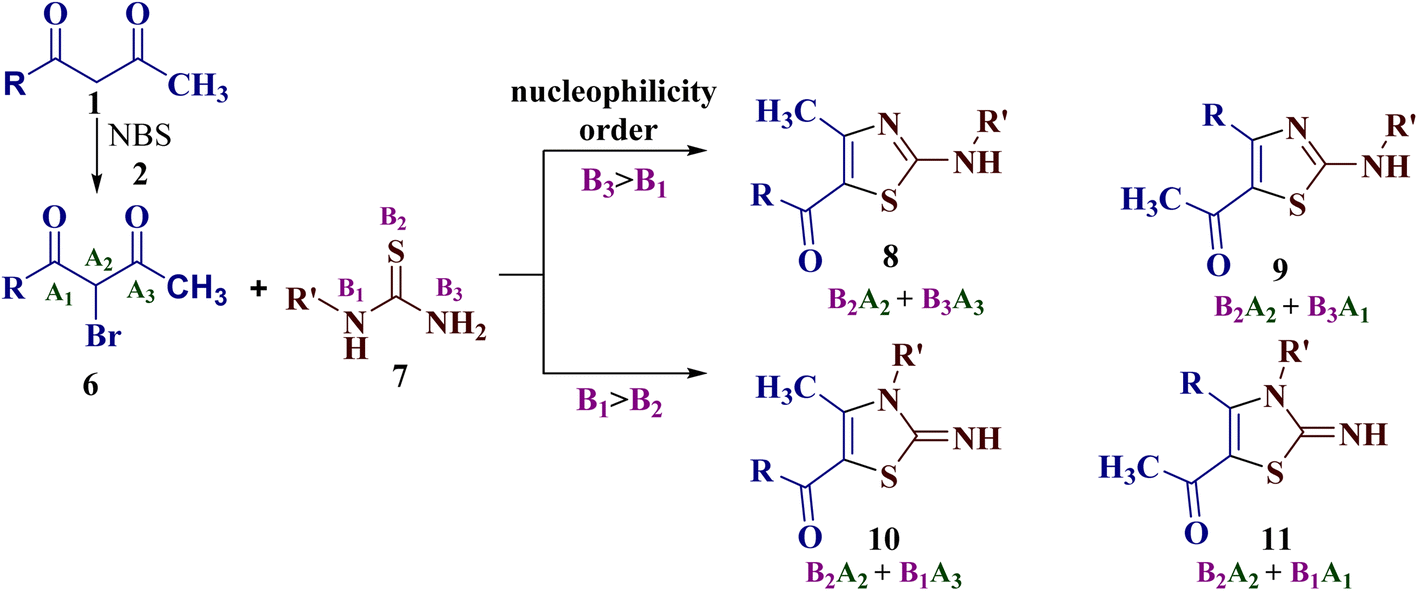 | ||
| Scheme 2 The four possible regioisomers 8, 9, 10 and 11 of the reaction of α-bromo-1,3-diketones 6 with N-substituted thiourea derivative 7. | ||
Despite the possibility of the insertion of an additional chemical handle, the reaction of α-bromo-1,3-diketones with a trinucleophile (such as substituted thioureas) remains unexplored. Keeping in mind our interest in the synthesis of 2-amino/imino thiazole derivatives,39–41 along with the quest to study the regioselectivity pattern in the reaction of 1,3-diketones with binucleophiles, we report here the synthesis of 2-(N-arylamino)-5-acyl-4-methylthiazole derivatives, along with the rearranged product 2-(N-acylimino)-3-N-aryl-4-methylthiazoles under a solvent-free synthetic approach using 1,3-diketones and N-substituted thiourea derivatives.
Results and discussion
For the reaction, unsymmetrical 1,3-diketones 1a–e were prepared by the sodium ethoxide-mediated Claisen condensation reaction of various acetophenones with ethyl acetate, according to the literature method.42 N-Bromosuccinimide 2 was chosen as a brominating agent due to its mild oxidizing ability, easy availability, low cost, and ease of application. N-Substituted thioureas 7 were prepared by HCl-catalyzed thiocyanation reaction of different anilines with ammonium thiocyanate in water at 70–80 °C.43In our earlier laboratory studies,44 the solvent-free organic synthesis has been a matter of practice45,46 to induce excellent regioselectivity, along with extraction of a considerable product yield (70–80%), ruling out of the possibility of solvolysis. Solid-phase synthesis has emerged as the preferred synthetic route in the last few years due to its associated merits, including a more efficient and regioselective product formation, the avoidance of separating lachrymatory α-halocarbonyl compounds, simple reaction conditions and reduced reaction times, environmentally benign approach, cost-effective method, and reduced or no use of organic solvents.
Exploiting the same synthetic strategy, the present reaction protocol was studied by implementing solid-phase synthesis. In this pretext, 1-phenylbutane-1,3-dione 1a (1.0 eq.) was pulverized with NBS 2 (1.0 eq.) in a dry mortar using a pestle to form α-bromo-1-phenylbutane-1,3-dione 6a, according to a literature procedure.47 The so-formed α-bromo-1-phenylbutane-1,3-dione 6a was treated with N-phenyl thiourea 7a (1.0 eq.). The resulting mixture was minced well, but the TLC did not confirm the completion of the reaction and the product could be recovered only in 50% yield.
As a mild base, sodium carbonate has been reported to be a good promoter in thiazole synthesis,38 and is helpful in accelerating the reaction by consuming hydrobromic acid produced within the reaction. Taking the lead, the present reaction protocol was improvised by the addition of an equivalent amount of sodium carbonate and smashing the reaction mixture well using a pestle and mortar at a temperature between 70 and 80 °C. A reaction mass developed into a sticky solid in about an hour, with TLC indicating the complete consumption of reactants 6a and 9a, along with the formation of a single product. The reaction content on dilution with water resulted in a fluffy solid formation at ambient temperature. The solid product was filtered, dried, and recrystallized using ethanol to give a thiazole derivative in 77.8% reaction yield. The so-obtained thiazole derivative was later characterized as 2-(N-phenylamino)-4-methyl-5-benzoylthiazole 8aa (Scheme 3).
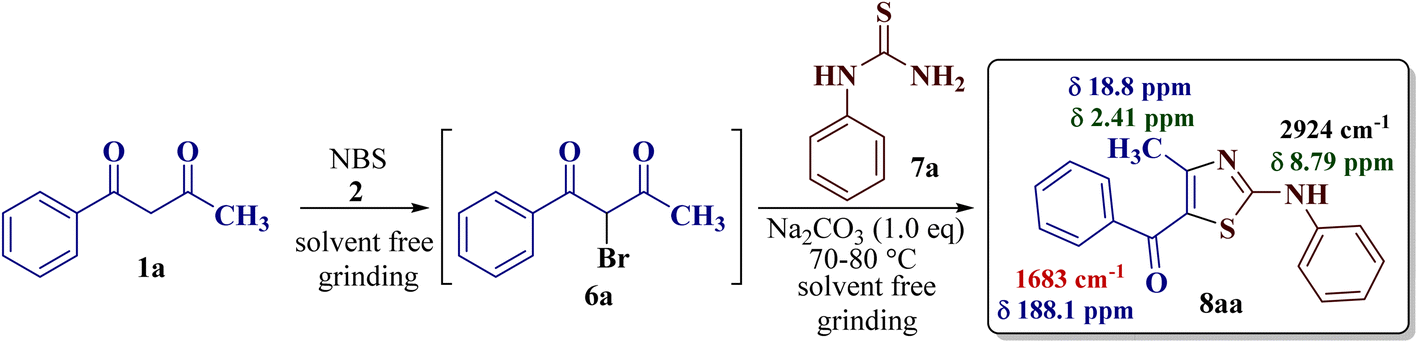 | ||
| Scheme 3 Reaction optimization between 1-phenylbutane-1,3-dione 1a with N-phenyl thiourea 7a to give 2-(N-phenylamino)-4-methyl-5-benzoylthiazole 8aa. | ||
The IR spectrum of compound 8aa revealed the presence of a strong absorption band at 1683 cm−1, which was attributed to a carbonyl group, and a sharp band corresponding to the N–H group was observed at 2924 cm−1. Also, the presence of a single peak at δ 2.41 ppm integrated to three protons of a methyl group and at δ 8.79 ppm integrated for the N–H proton, along with the multiplet of ten protons of two phenyl rings in region δ 7.16–7.75 ppm, in the 1H-NMR spectrum of compound 8aa indicated the complete exhaustion of the starting materials. The desired corresponding fourteen peaks in the 13C-NMR spectrum of compound 8aa demonstrated the successful construction of the thiazole ring. Further, the product formation of 8aa was established, comparing the melting point and other analytical data with the literature.19
To study the scope of the reaction, differently substituted 1,3-diketones 1a–e were employed to react with various N-substituted thiourea derivatives 7a–f containing electron-donating and electron-withdrawing groups under identical reaction conditions to obtain regioselective thiazoles (Scheme 4).
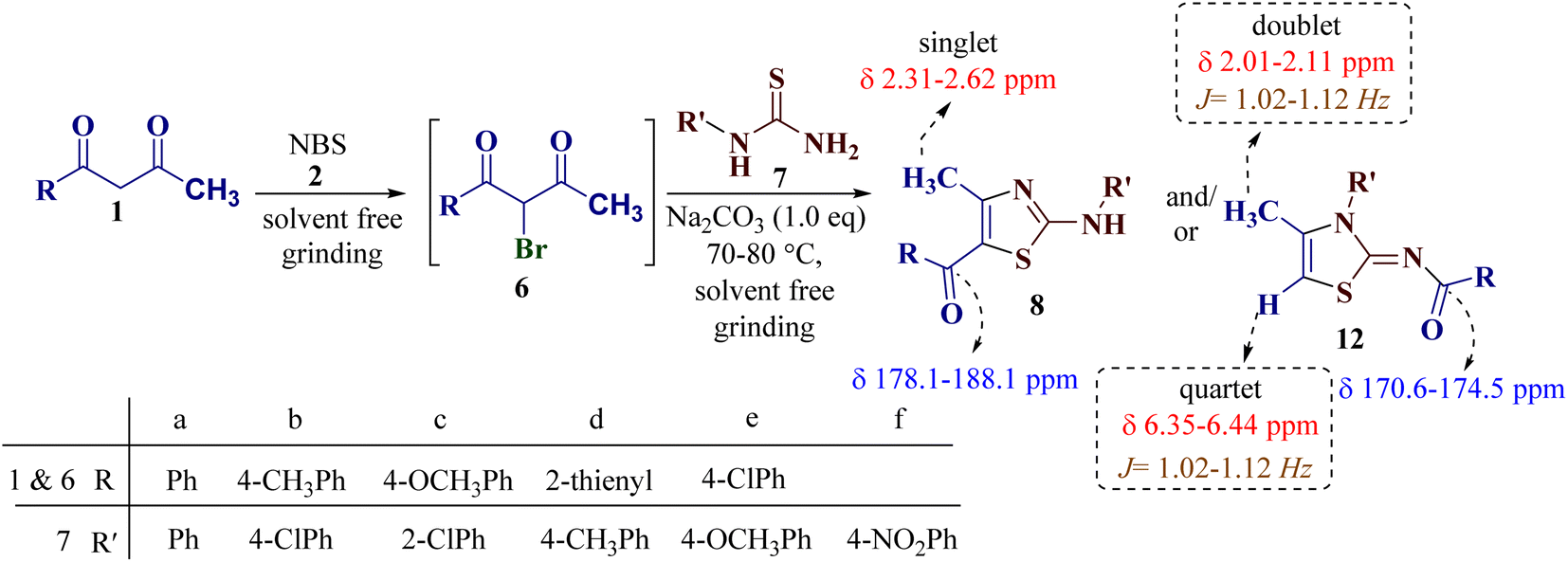 | ||
| Scheme 4 The general synthetic route to thiazole derivatives using 1,3-diketones 1a–e and N-substituted thiourea derivatives 7a–f. | ||
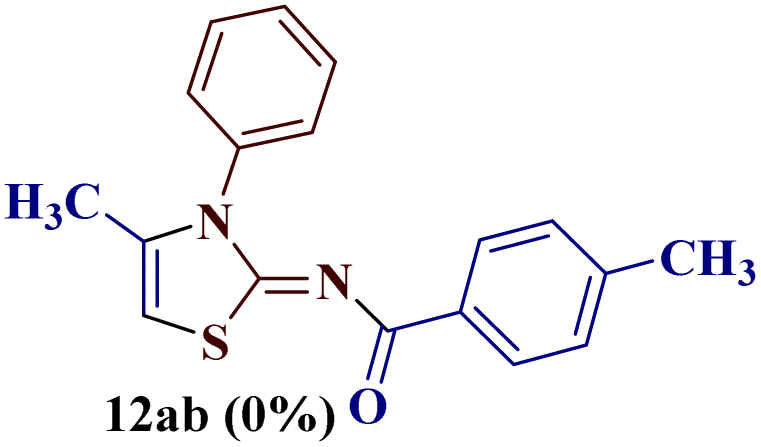
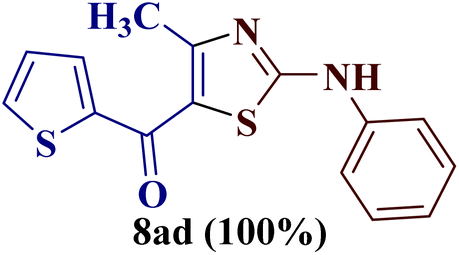
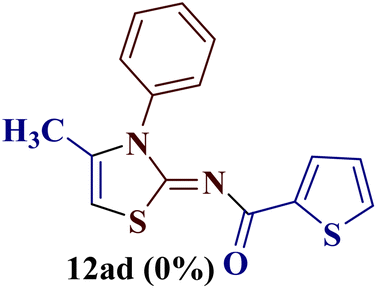
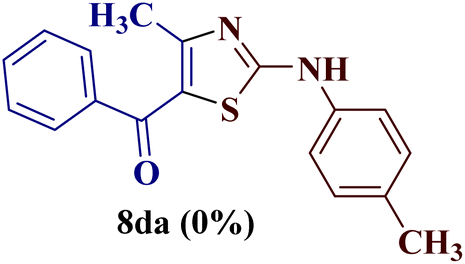
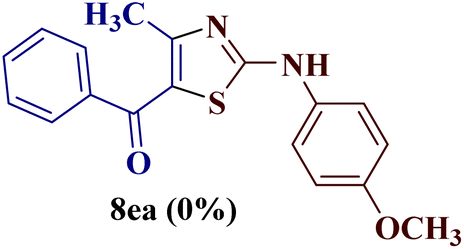
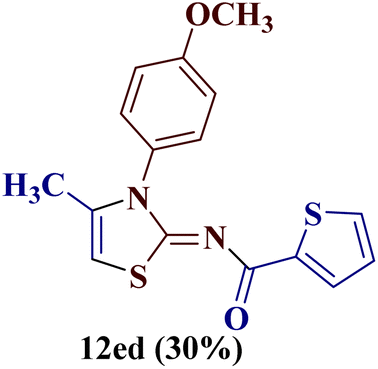
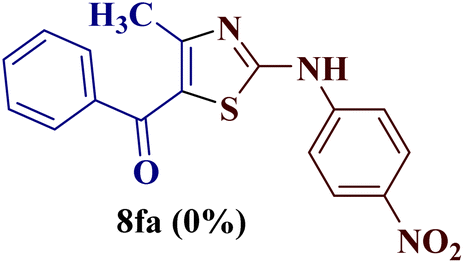
Encouraged by the success of the unsubstituted phenyl thiourea 7a with differently substituted 1,3-diketones 1a–d to afford 2-(N-arylamino)-5-acyl-4-methylthiazoles 8aa–ad, we envisaged exploring the reaction scope with other N-substituted thiourea 7b–f with 1,3-diketones 1a–e. It is interesting to note that the reaction outcomes were not uniform as per our expectations. Instead, various combinations resulted in the production of the anticipated product 2-(N-arylamino)-4-methyl-5-acyl thiazole 8, in some cases an unforeseen rearranged product 2-(N-acylimino)-4-methyl-3-(N-aryl)thiazole 12 and a combination of both in rest (Table 1).

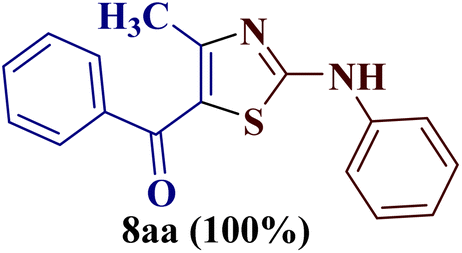
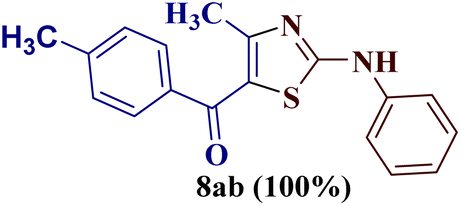
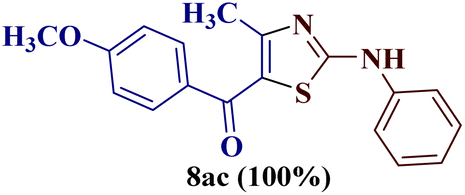
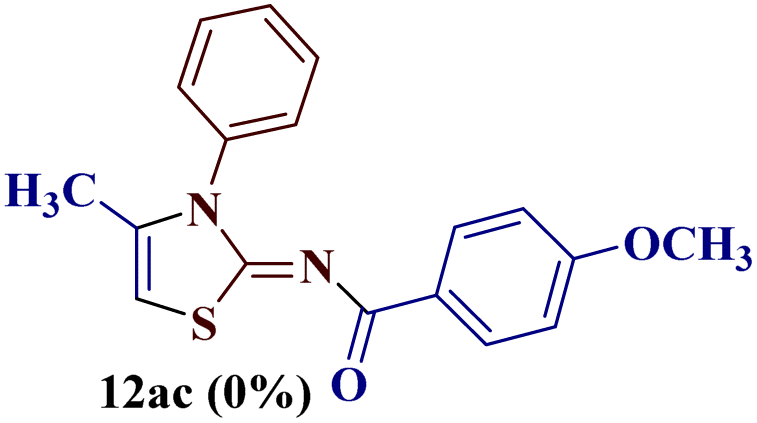
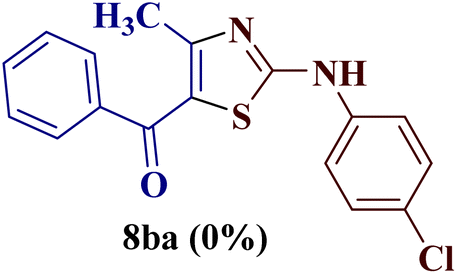

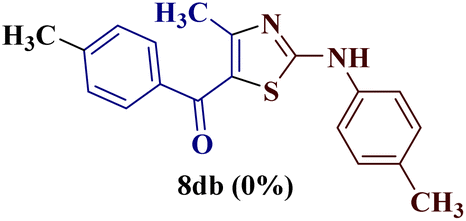
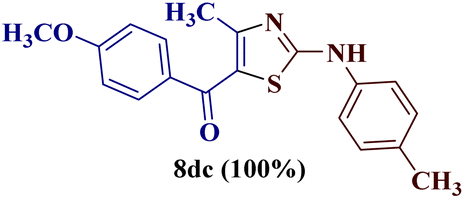

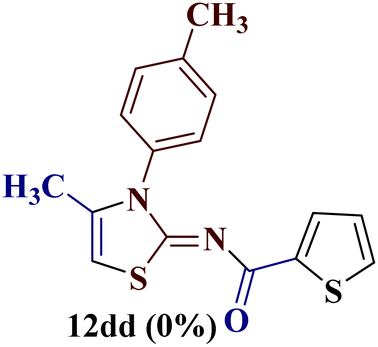
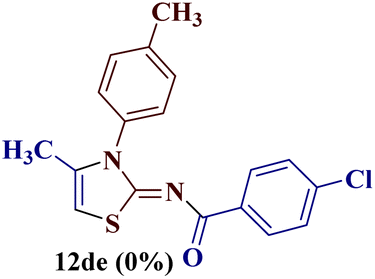
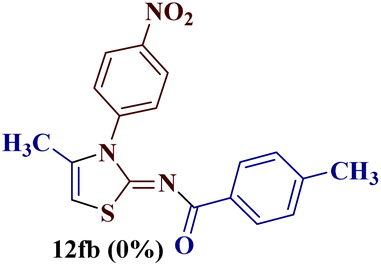
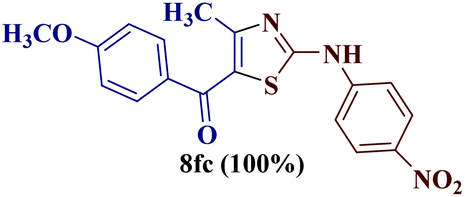
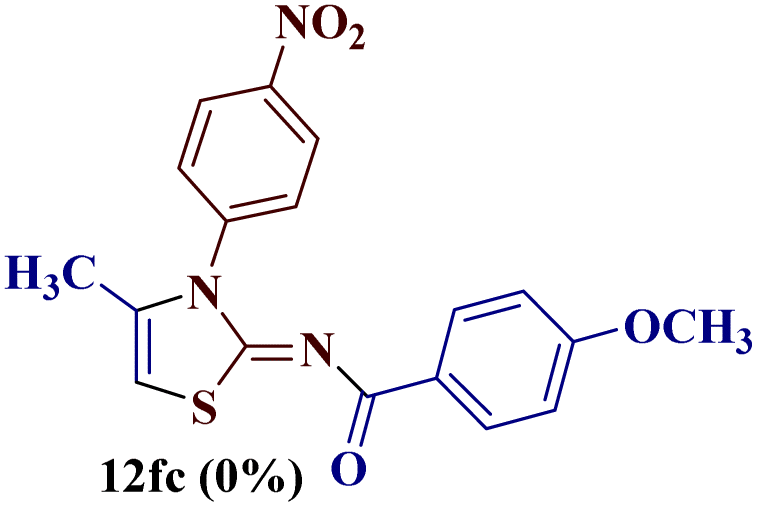

The condensation reaction of N-phenyl thiourea 7a with differently substituted 1,3-diketones 1a–d was attempted under identical conditions, which resulted in the formation of the corresponding 2-N-(4-methylphenyl)amino-5-acyl-4-methyl thiazoles 8aa–ad in quantitative yields (60–80%).
It is interesting to note that condensation of p-chlorophenyl thiourea 7b with 1-phenylbutane-1,3-dione 6a led to the formation of an unexpected product with interesting observations. The IR spectrum of the product did not display the significant peak for the NH stretch expected from the desired amino thiazole 8. Rather, it showed a stretching band at 1599 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N. The most striking feature in the 1H NMR spectra was a doublet of three proton intensity, which was attributed to the methyl group at δ 2.06 ppm with a coupling constant of J = 1.12 Hz, instead of a sharp peak of three proton intensity at the thiazole derivative with δ 2.67–2.41 ppm, as observed in isomer 8aa–ad. Also, a quartet of one proton intensity at δ 6.39 ppm was attributed to 5-H with a coupling constant value of J = 1.12 Hz, indicated the allylic cross-coupling of methyl protons with the 5-H in 1H-NMR spectrum. Similarly, the chemical shift of the methyl group shifted upfield to δ 15.1 ppm in the 13C-NMR spectrum, instead of δ 18.8–18.6 ppm in isomers 8aa–ad. Furthermore, HRMS studies supported thiazole ring formation by displaying the base peak at an m/z value of 329.8231.
N. The most striking feature in the 1H NMR spectra was a doublet of three proton intensity, which was attributed to the methyl group at δ 2.06 ppm with a coupling constant of J = 1.12 Hz, instead of a sharp peak of three proton intensity at the thiazole derivative with δ 2.67–2.41 ppm, as observed in isomer 8aa–ad. Also, a quartet of one proton intensity at δ 6.39 ppm was attributed to 5-H with a coupling constant value of J = 1.12 Hz, indicated the allylic cross-coupling of methyl protons with the 5-H in 1H-NMR spectrum. Similarly, the chemical shift of the methyl group shifted upfield to δ 15.1 ppm in the 13C-NMR spectrum, instead of δ 18.8–18.6 ppm in isomers 8aa–ad. Furthermore, HRMS studies supported thiazole ring formation by displaying the base peak at an m/z value of 329.8231.
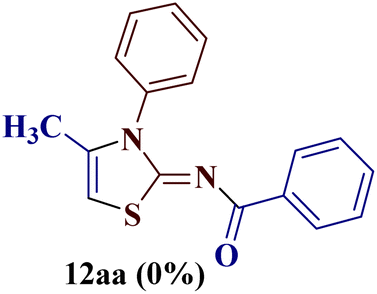
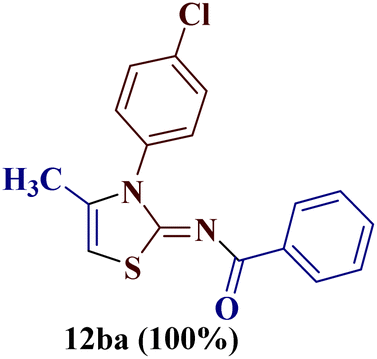
It has been proposed earlier that the reaction between the trielectrophilic α-bromo-1,3-diketone 8 and trinucleophilic N-substituted thiourea derivative 7 can lead to the formation of four closely resembling regioisomers (Scheme 1). However, the combined spectral data did not correspond to either of the proposed thiazole derivatives 8–11, as none of these justify the presence of the mutually correlated methyl group and vicinal 1-H proton in the product. Therefore, on the basis of the data, the compound was identified as 2-(N-benzoylimino)-3-N-(p-chlorophenyl)-4-methylthiazole 12ba, which is possible through an intramolecular rearrangement involving C–N acyl migration.
Formation of 2-(N-benzoylimino)-3-N-(p-chlorophenyl)-4-methylthiazole 12ba has also been reported in the literature through the aqueous mediated reaction of N-(4-chlorophenylcarbamothioyl)benzamide with 1-bromopropan-2-one (lit. mp 210 °C) by Wang et al.25 The melting point and the analytical data of the rearranged thiazole derivative were found to be in agreement with 2-(N-benzoylimino)-3-N-(p-chlorophenyl)-4-methylthiazole 12ba, thereby confirming the proposed structure.
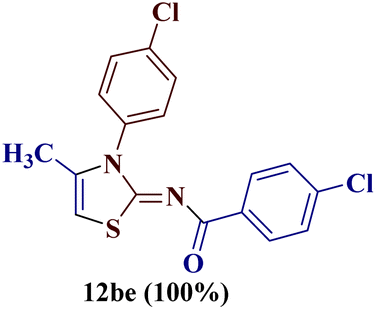
Similar results were obtained for the reaction of 7b with 1-(p-chlorophenyl)butane-1,3-dione 1e, where a quantitative amount of 2-(N-(p-chlorobenzoyl)imino)-3-N-(p-chlorophenyl)-4-methylthiazole 12be was observed.
However, the reaction of p-chlorophenyl thiourea 7b with 1-(p-methylphenyl)butane-1,3-dione 1b proceeded to give the expected 2-(N-(p-chlorophenylamino))-4-methyl-5-(p-methylbenzoyl)thiazole 8bb efficiently with a single peak for the three protons of the methyl group (4-CH3) at δ 2.39 ppm, along with eight aromatic protons of two p-substituted phenyl rings.
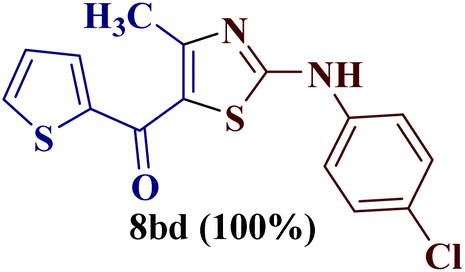
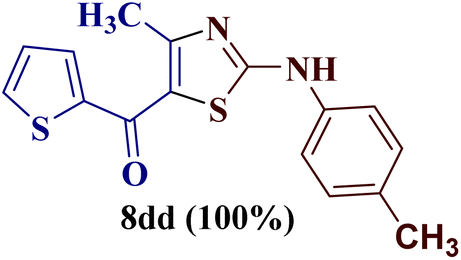
This type of observation was also consistent for 1-(thiophen-2-yl)butane-1,3-dione 1d to furnish 2-N-(p-chlorophenylamino)-4-methyl-5-(2-thienoyl)thiazole 8bd.
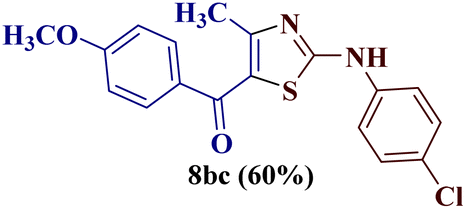
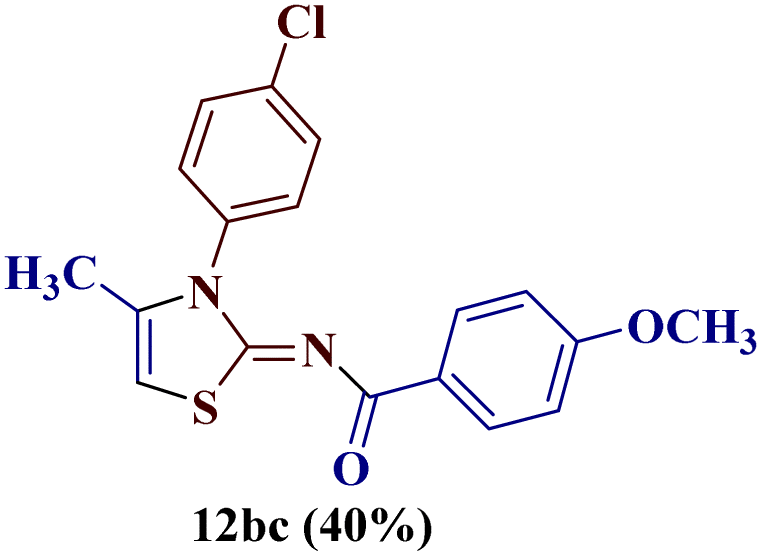
Reaction of p-chlorophenyl thiourea 7b with p-methoxyphenylbutane-1,3-dione 1c yielded a mixture of two products, as evident from TLC (Table 1). The two isomers 8bc and 12bc were distinguishable from each other due to the slightly different Rf value on TLC (EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pet ether, 1:4), and existed in a 60:40 ratio in a crude mixture, as indicated by the chemical shift values of the methyl group in 1H and 13C-NMR spectroscopy.
:pet ether, 1:4), and existed in a 60:40 ratio in a crude mixture, as indicated by the chemical shift values of the methyl group in 1H and 13C-NMR spectroscopy.
For a detailed structural investigation of the two isomers, the product mixture of the reaction between the N-(p-chlorophenyl)thiourea derivative 7b and 1-(4-methoxyphenyl)butane-1,3-dione 1c was separated through column chromatography using pet ether:ethyl acetate (4:1) as eluting medium. The two isomers 2-N-(p-chlorophenylamino)-4-methyl-5-(p-methoxybenzoyl)thiazole 8bc and 2-(N-(p-methoxybenzoyl)imino)-3-N-(p-chlorophenyl)-4-methylthiazole 12bc obtained after separation were individually analyzed through 1H and 13C-NMR spectroscopic techniques.
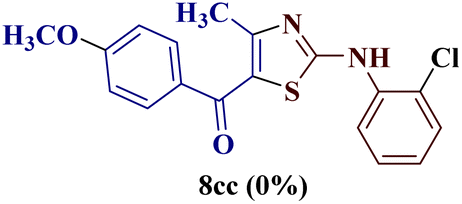
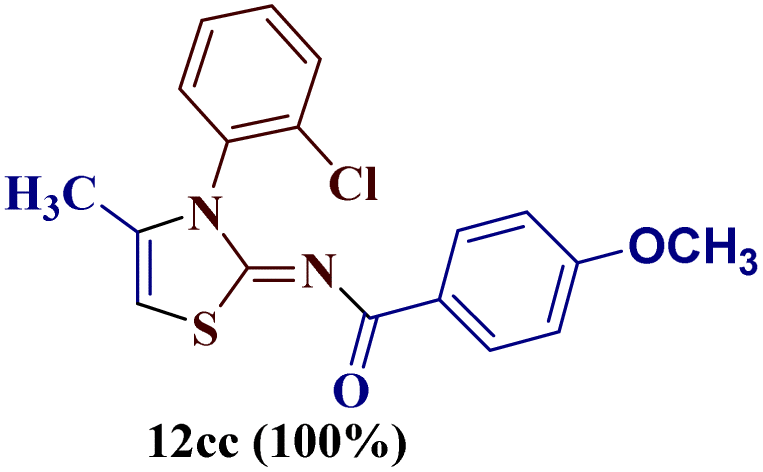
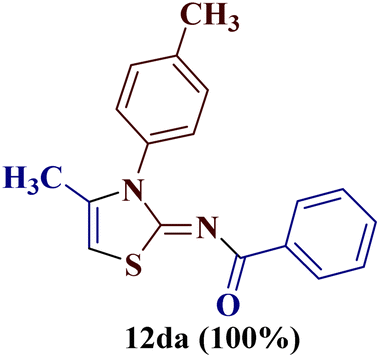
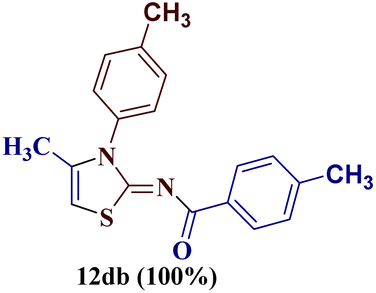
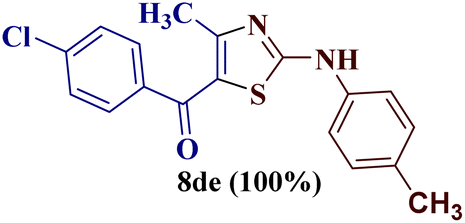
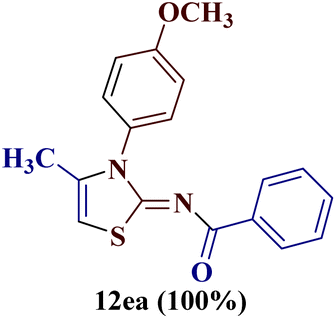
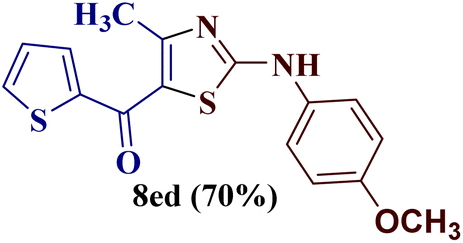
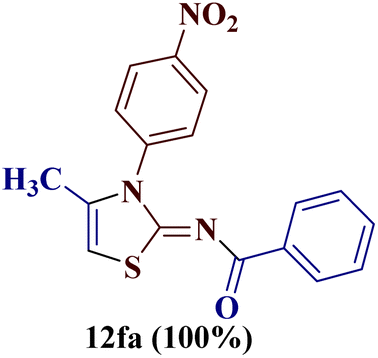
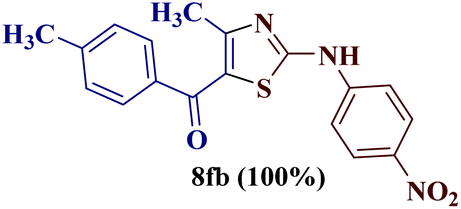
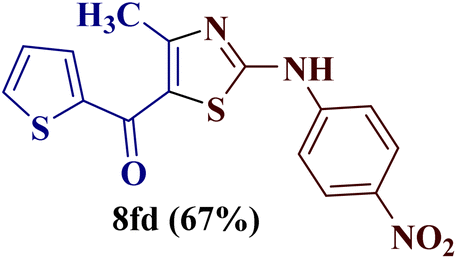
However, when the reaction was carried out between N-(o-chlorophenyl)thiourea derivative 7c and 1-(4-methoxyphenyl)butane-1,3-dione 1c, 2-imino thiazole derivative 12cc was obtained exclusively. The reaction of the N-(p-methylphenyl)thiourea derivative 7d with unsymmetrical 1,3-diketones 1a–e furnished 2-amino thiazoles in three combinations (8dc, 8dd and 8de), while 2-imino thiazoles were used in the remaining two combinations (12da and 12db). Furthermore, the reaction of N-(p-methylphenyl)thiourea derivative 7e with unsymmetrical 1,3-diketones 1a and 1d furnished 2-imino thiazole derivative exclusively in one case (12ea), while a mixture of 2-imino thiazole and 2-amino thiazole was used in the second case (8ed and 12ed). Finally, the reaction of N-(p-nitrophenyl)thiourea derivative 7f with unsymmetrical 1,3-diketones 1a–d lead to the formation of 2-amino thiazoles in two combinations (8fb and 8fc), 2-imino thiazole in one combinations (12fa) and a mixture of 2-imino thiazole and 2-amino thiazole in the remaining one case (8fd and 12fd).
Determination of regioisomeric structure by 2D-NMR studies
The chemical shift values obtained by the 1H and 13C spectral studies of the two isomers provided ample information about the structure. However, to gain better insight into the two structures 8bc and 12bc, two-dimensional NMR spectroscopic [(1H–13C) HMQC (1H–13C) HMBC and (1H–15N) HMBC] studies of the two isomers were conducted.The (1H–13C) HMBC of compound 2-(N-(p-chlorophenyl)amino)-4-methyl-5-(4-methoxybenzoyl)thiazole 8bc confirmed the direct correlation of the CO group at δ 186.9 ppm with the 2′′,6′′-H proton (δ 7.78–7.76 ppm) of the p-methoxy phenyl ring, indicating the presence of the (p-methoxy)benzoyl group. Also, the correlations between C-4 (δ 157.4 ppm) with C-5 (δ 120.6 ppm) and the methyl protons (δ 2.41 ppm) demonstrated the methyl group presence at the fourth position of the thiazole ring (Fig. 1).
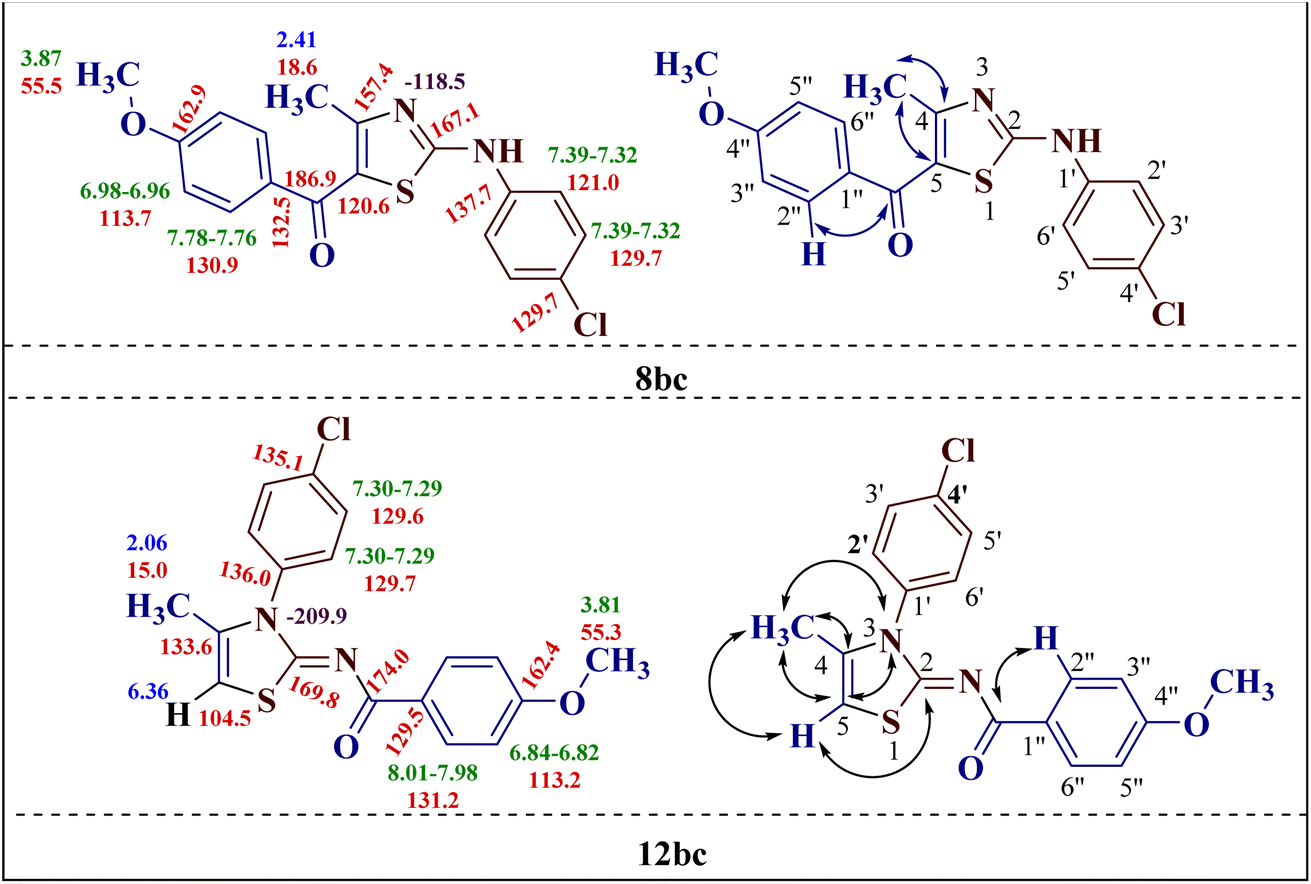 | ||
| Fig. 1 Important correlations illustrated by (1H–13C) HMQC and (1H–13C) HMBC experiments for compounds 8bc and 12bc, along with the 1H (in green), 4-CH3 and 5-H protons (in blue) and 13C (in red) chemical shifts. | ||
The (1H–13C) HMBC of compound 2-(N-(p-methoxybenzoyl)imino)-3-N-(p-chlorophenyl)-4-methylthiazole 12bc showed cross-coupling between the methyl protons at C-4 and vinylic hydrogen at C-5 with a coupling constant of 1.3 Hz, which splits their peaks to doublet and quartet, respectively. Additionally, the cross peak between the carbonyl carbon at 174.0 ppm with 2′′,6′′-H of the p-methoxy phenyl ring (δ 8.01–7.98 ppm) was observed, as in the other isomer 8bc. This observation confirms the formation of the unexpected 2-(N-(p-methoxybenzoyl)imino)-3-N-(p-chlorophenyl)-4-methylthiazole 12bc as a product. The formation of the rearranged product 12bc could be attributed to the acyl group cleavage and migration in the transition states of the reaction progression (Fig. 1).
Additionally, 2-(N-benzoylimino)-4-methyl-3-N-(p-methylphenyl)thiazole 12da and 2-(N-(p-methylphenyl)amino)-4-methyl-5-(p-methoxybenzoyl)thiazole 8dc were subjected to multinuclear NMR analysis. The combined correlation results, along with the complete assignments of the [(1H–13C) HMQC, (1H–13C) HMBC and (1H–15N) HMBC] experiments, are depicted in Fig. 2.
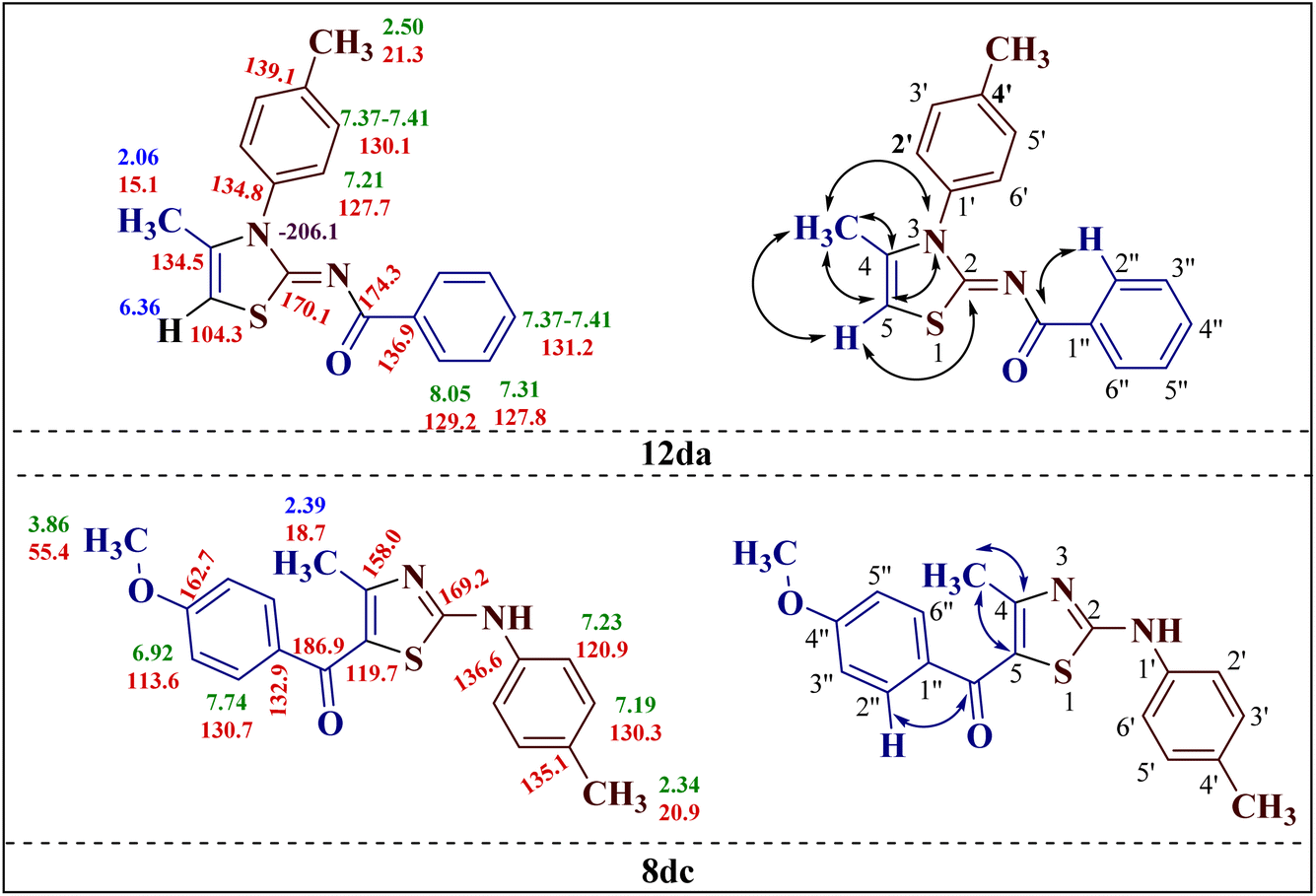 | ||
| Fig. 2 Important correlations illustrated by (1H–13C) HMQC and (1H–13C) HMBC experiments for compounds 12da and 8dc, along with the 1H (in green), 4-CH3 and 5-H protons (in blue) and 13C (in red) chemical shifts. | ||
Conformation of the regioisomeric structure by X-ray crystallographic studies
The structures 2-(N-arylamino)-4-methyl-5-acylthiazoles 8 and 2-(N-acylimino)-4-methyl-3-(N-aryl)thiazoles 12 were finally confirmed by X-ray crystallographic studies. For this, single crystals of compounds 8aa, 12da and 12bc were obtained using gradual dissipation of the solvent at ambient conditions.On examination, the single crystal structure of compound 8aa revealed that the compound crystallizes in the monoclinic C2/c space group (Fig. 3a) containing one molecule per asymmetric unit, which shows the presence of one ketonic group and one amine group. The ORTEP diagram for compound 8aa with 20% probability is shown in Fig. 3b.
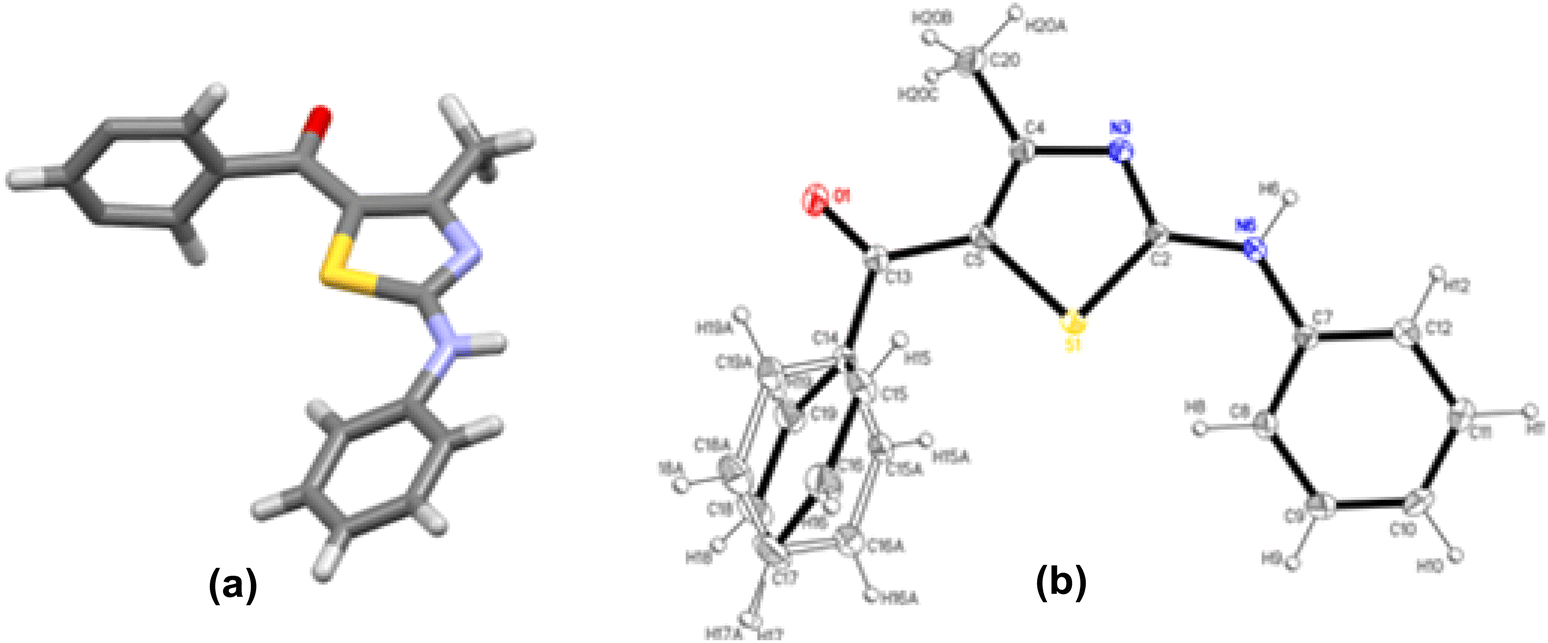 | ||
| Fig. 3 (a) C2/c space group for compound 8aa. (b) ORTEP plot with 20% probability for structure 8aa with all atoms labelled. | ||
The electronic delocalization on the main fragment is broken, as evidenced by the bond distances. Hence, the molecule is nonplanar with dihedral angles of 66.2(3)° and 28.9(3)° between the thiazole and both phenyl rings. The lower electronic delocalization induces differences in the thiazole bond distances, with the N3–C2 and C4–C5 bonds having an enhanced double bond character. The loss of electronic delocalization is also observed by the positional disorder found on the benzoyl ring, where the four carbon atoms are distributed over two positions with an occupancy of about 50%.
Further, the crystal packing of 8aa consists of zigzagged chains formed by intermolecular hydrogen bonds between the amine group (N6H6) of one molecule and the thiazole N3 atom of an adjacent molecule with a bond distance of 2.266(4) Å and an angle of 167.3(3)°, as shown in Fig. 4.
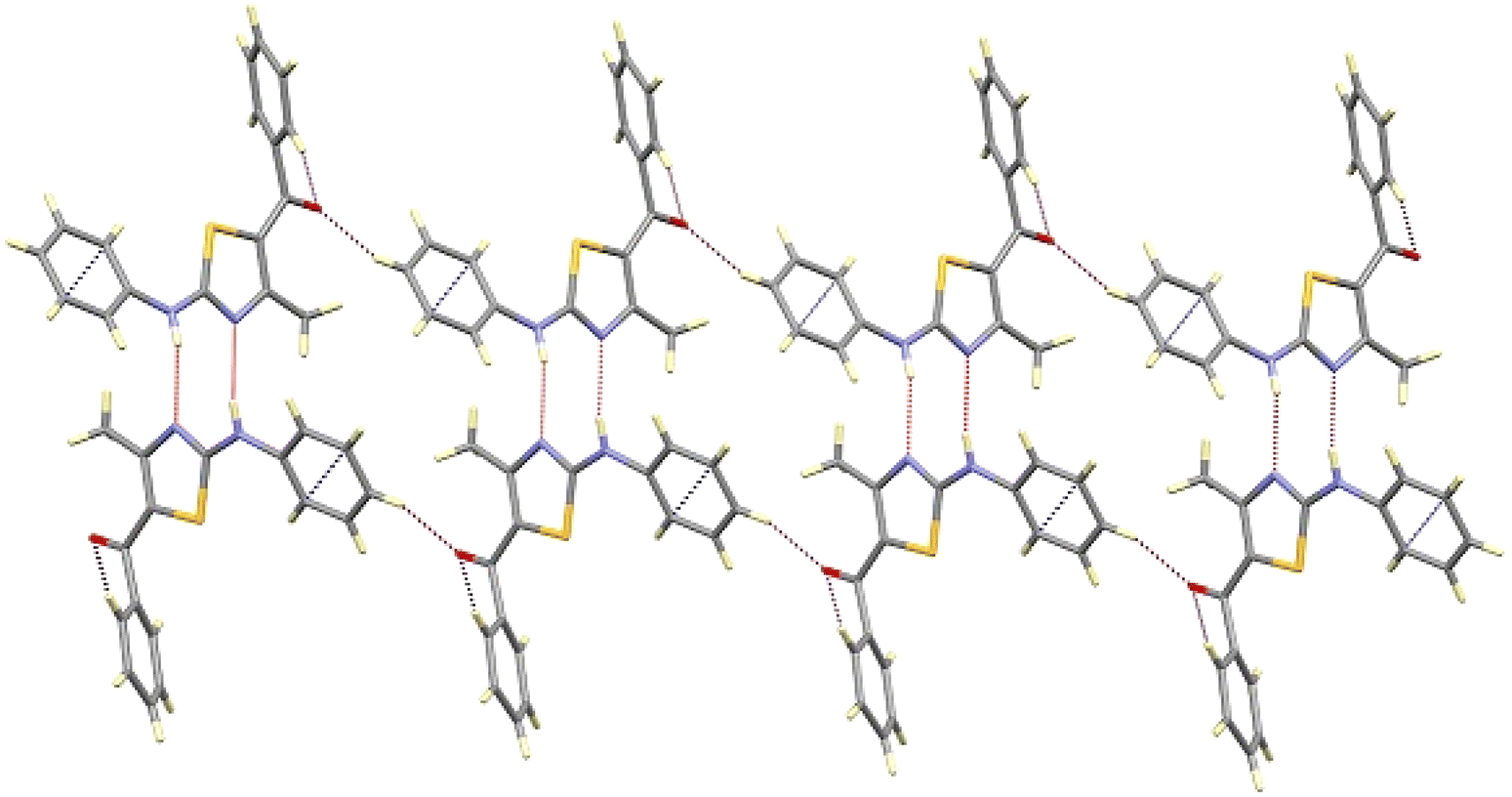 | ||
| Fig. 4 View of the chain of compound 8aa along the b-axis, showing the intermolecular hydrogen bonding. | ||
This interaction extends along the b-axis, and it is strengthened by a partial overlapping of the non-disordered phenyl rings of two neighbouring molecules (shortest C–C distance of about 3.3 Å). Those chains are interconnected through weak contacts between the ketonic groups and neighbouring aromatic hydrogen atoms, spreading out the interactions along the a-axis, as depicted in Fig. 5.
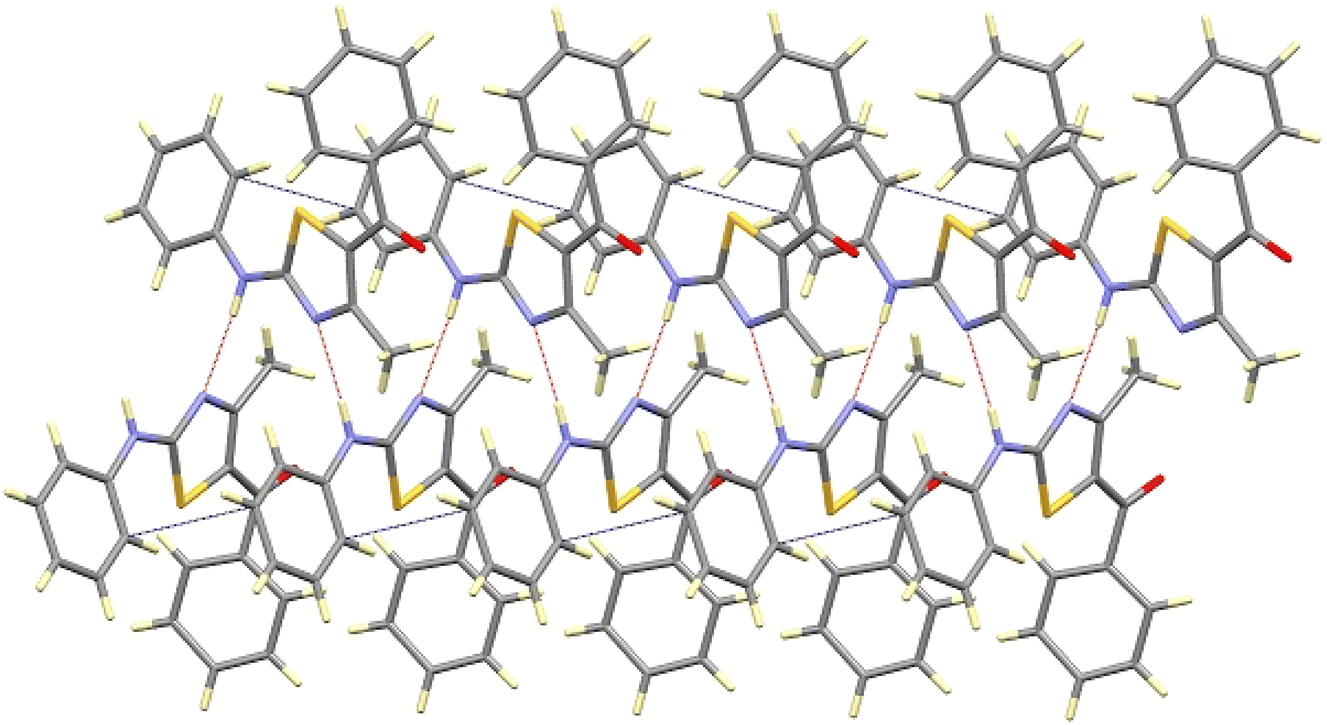 | ||
| Fig. 5 View of the interaction between the chains along the b-axis of compound 8aa. | ||
The X-ray studies of the compound 12da revealed that the compound crystallizes in orthorhombic Pnma space group (Fig. 6a) with the asymmetric unit formed by the C18H16N2OS moiety, being the rest of the molecule generated by symmetry. The ORTEP plot for molecule 12da is shown in Fig. 6b with 20% probability.
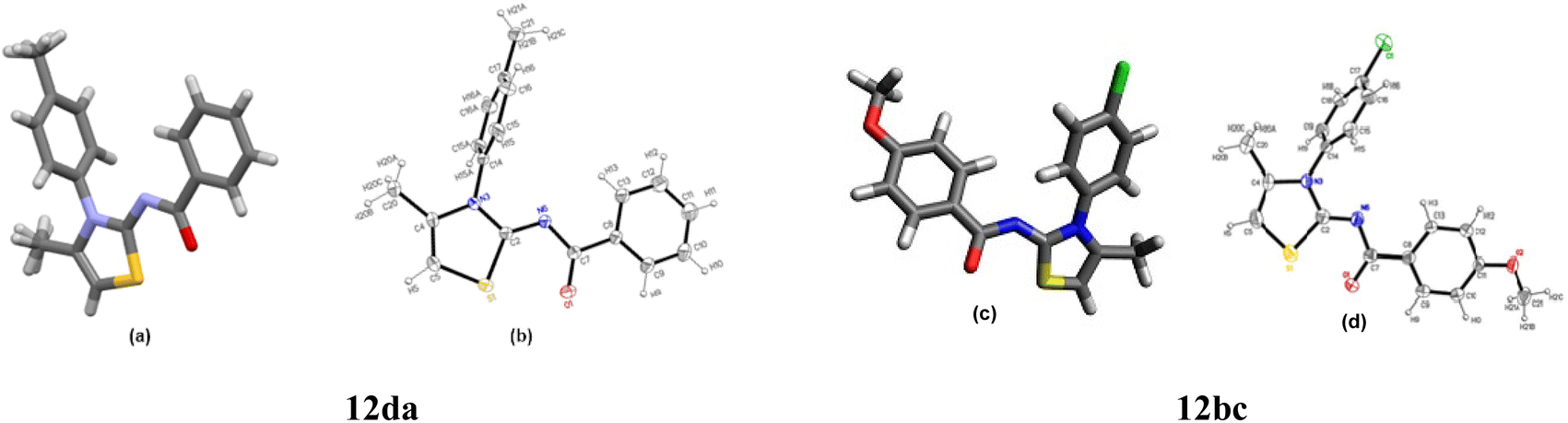 | ||
Fig. 6 (a) P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group for compound 12da. (b) ORTEP plot of compound 12da with 20% probability for the ellipsoids with their labelling scheme. (c) Pnma space group for compound 12bc. (d) ORTEP plot with 20% probability for structure 12bc with all atoms labelled. space group for compound 12da. (b) ORTEP plot of compound 12da with 20% probability for the ellipsoids with their labelling scheme. (c) Pnma space group for compound 12bc. (d) ORTEP plot with 20% probability for structure 12bc with all atoms labelled. | ||
The X-ray studies supported the P space group for compound 12bc with one molecule per asymmetric unit, and the ORTEP plot for compound 12bc with labelling scheme is shown in Fig. 6.
The compound 12da forms a planar fragment formed by the thiazole and phenylamide groups related to the electronic delocalization along them, as deduced by their bond distances and bond angles. The plane containing this fragment acts as a mirror plane that forces the p-methylphenyl ring, linked to N3, to be placed perpendicular to the rest of the molecule, as shown in Fig. 7. The molecular packing of compound 12da is quite similar to that described for 12bc, arranging the molecules in the bc plane too.
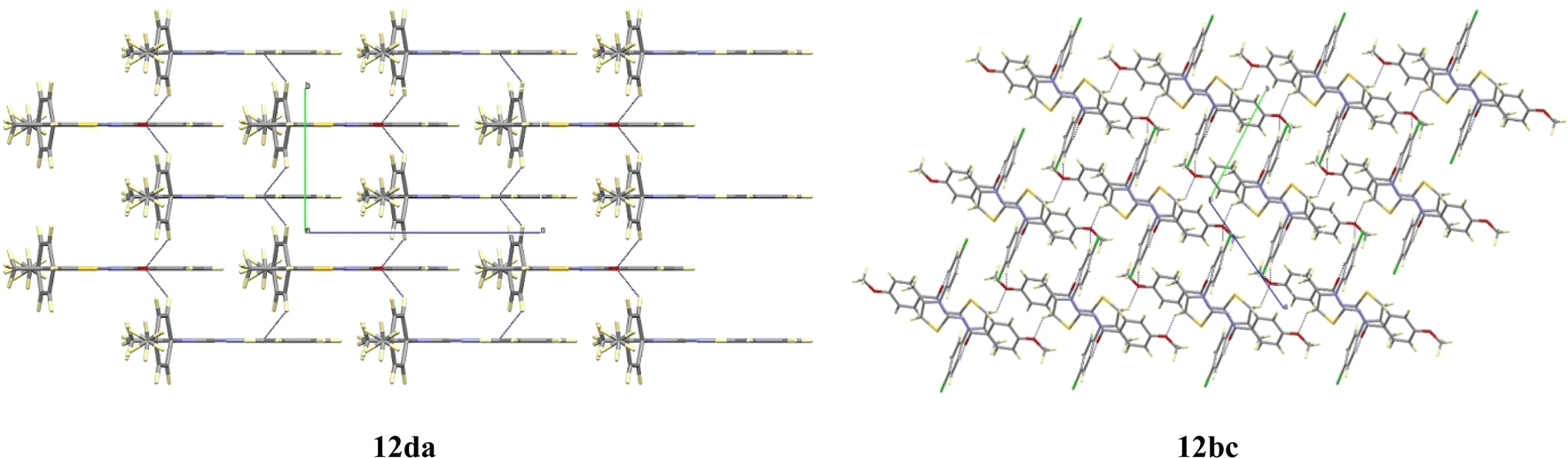 | ||
| Fig. 7 View of the crystal packing for compounds 12da and 12bc. | ||
Compound 12bc exhibits a lower planarity in the main fragment, as indicated by the dihedral angle of about 13° defined by the thiazole and the phenylamide moieties. This bending is attributed to the presence of the para-methoxy group. Moreover, the p-chlorophenyl substituent present on the N3 atom is found to be twisted 73.5(2)° relative to the thiazole plane. The molecular packing of compounds 12bc shows only weak contacts between the oxygen atoms and the aromatic rings that arrange the molecules in the bc plane, as shown in Fig. 7.
Although the molecular packing of compounds 12da and 12bc are similar, the two structures differ only in the weak contacts between the oxygen atoms and the aromatic rings that arrange the molecules in the bc plane.
Plausible mechanism
A possible reaction path for the reaction of trielectrophilic α-bromo-1,3-diketones 6 and trinucleophilic N-substituted thiourea derivatives 7 to furnish 2-(N-arylamino)-4-methyl-5-acylthiazoles 8 and 2-(N-acylimino)-3-N-aryl-4-methylthiazoles 12 is depicted in Scheme 5. Considering the bromine-bearing methylene carbon (electrophilic centre A2) of α-bromo-1,3-diketones 6 to be the most electrophilic and sulfur (nucleophilic centre B2) of the N-substituted thiourea derivatives 7 to be the most nucleophilic of all centres, the reaction is assumed to be triggered by the initial nucleophilic displacement of bromine from α-bromo-1,3-diketones 6 by the sulphur of thiourea derivatives 7, leading to the formation of the S-alkylated open chain intermediate 13. This alkylated open chain structure 13 having two different electrophilic carbonyl carbons from diketone (A1 and A3) and two nucleophilic nitrogens (B1 and B3) from the thiourea moiety may undergo different routes to ring closure, leading to the formation of 8 and 12.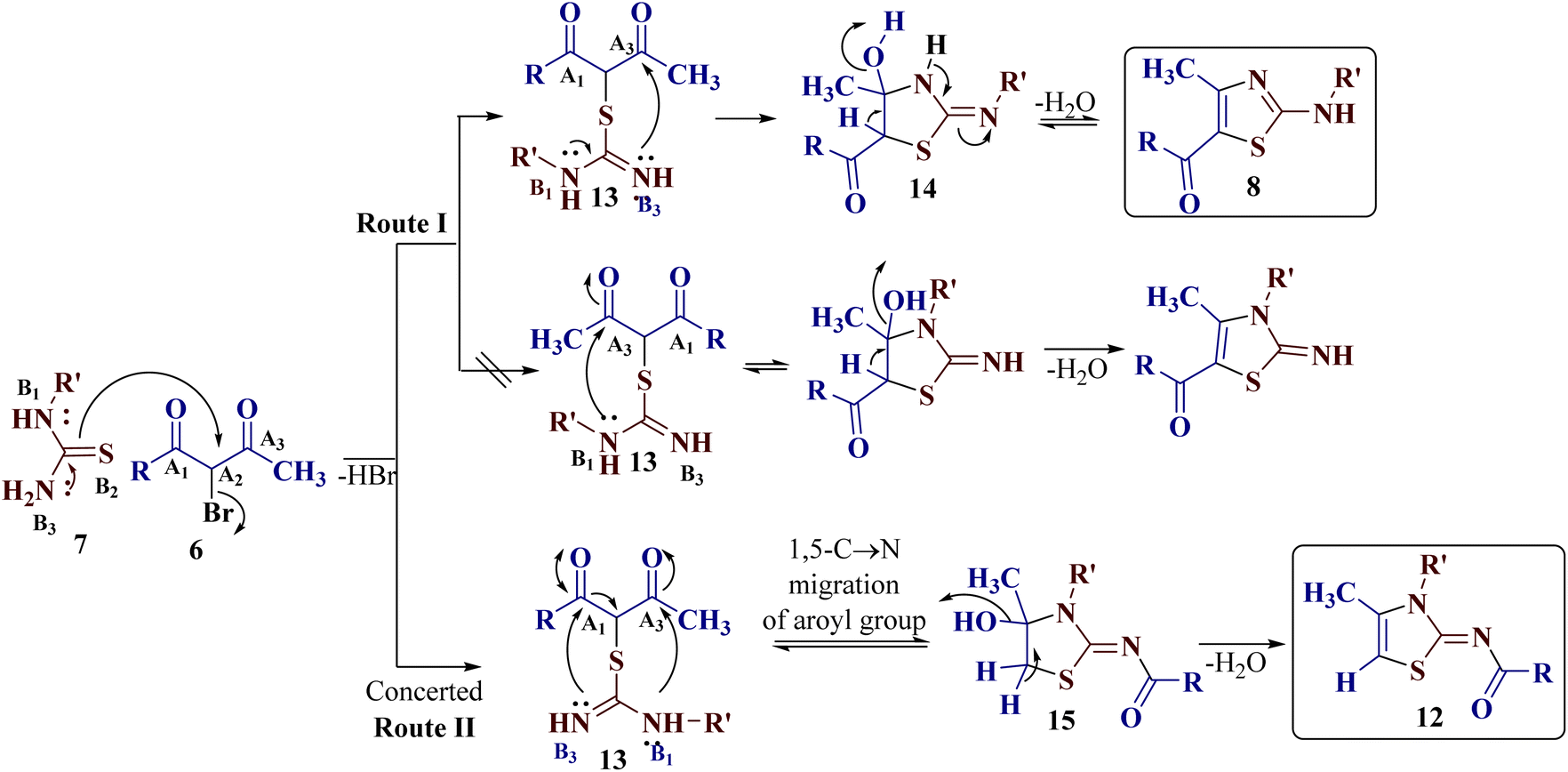 | ||
| Scheme 5 | ||
Synthesis of 2-(N-arylamino)-4-methyl-5-acylthiazoles 8 demands the nucleophilic attack of the unsubstituted imino group (nucleophilic centre B3) on the less sterically hindered acetyl group (electrophilic centre A3) to initially produce a cyclic intermediate 14, which undergoes dehydration to furnish 10 (route I, Scheme 5).
The formation of unexpected 2-(N-acylimino)-4-methyl-3-(N-aryl)thiazole derivatives 12 may be attributed through participation of a nucleophilic attack of a terminal or substituted amine nitrogen (nucleophilic centre B1) on the less substituted acetyl group (electrophilic centre A3), and the nucleophilic addition of an imine nitrogen (nucleophilic centre B3) to the aroyl group (electrophilic centre A1) in a concerted manner in the S-alkylated open chain structure 13. During the attack of the amino group (nucleophilic centre B3) on the acyl group (electrophilic centre A1), intramolecular C → N acyl migration takes place by C–C bond cleavage to furnish 2-imino-4-hydroxythiazole 15 as an intermediate, which on dehydration, affords 12 as the product (route II, Scheme 5).
It is proposed that competitive reaction routes are operative in cases where a mixture of both regioisomers has been achieved.
Although it is difficult to generalize the influence of a substituent on substituted thiourea on the reactivity pattern, and thus to control the regioselectivity in either direction of 8 or 12, attempts are underway to conclusively establish the reaction conditions (such as solvents, pH, energy source etc.) necessary to uniformly obtain 8 or 12.
Conclusion
In summary, the novel synthesis of 2-(N-arylamino)-5-acyl-4-methylthiazoles along with 2-(N-acylimino)-3-N-aryl-4-methylthiazoles has been achieved using a simple green protocol involving a sodium carbonate-mediated, one-pot, solvent-free synthetic approach. The present protocol offers a wide substrate scope, as differently substituted 2-aminothiazoles and 2-iminothiazoles were obtained in good to excellent yields with remarkable regioselectivity for the substrates substituted with electron-donating and -withdrawing groups. It was very interesting to observe that the reaction of N-substituted thioureas with differently substituted diketones did not follow any pattern to uniformly yield 2-aminothiazoles or 2-iminothiazoles, even upon repeating the reaction. Rather, different combinations abruptly led to the formation of the expected product 2-(N-arylamino)-4-methyl-5-acyl thiazole and/or an unexpected rearranged product 2-(N-acylimino)-4-methyl-3-(N-aryl)thiazole. The formation of the unexpected product has been supported by 1,5-C → N acyl group migration in the S-alkylated open chain transition state, as depicted in the proposed mechanism. The structures of the regioisomers have been established by intensive multinuclear NMR [(1H–13C) HMBC and (1H–13C) HMQC] spectroscopy and X-ray crystallographic studies.Experimental
General information
Commercial solvents and reagents were used as purchased. Melting points were determined in open capillaries on a digital melting point apparatus (MEPA), and were uncorrected. IR spectra were recorded on a Buck Scientific IR M-500 spectrophotometer using KBr pellets (νmax in cm−1). Analytical TLC was performed using Merck Kieselgel 60 F254 silica gel plates. Visualization was performed by UV light (254 nm). Elemental analysis was performed at the Sophisticated Analytical Instrumentation Facility, Panjab University, Chandigarh. All compounds gave C, H and N analysis results within ±0.5 of the theoretical values.NMR experiments
1H and 13C NMR spectra were recorded on a Bruker instrument at 400 and 100 MHz, respectively, using deuterochloroform and deuterodimethyl sulphoxide as solvents, and chemical shifts were recorded in ppm (δ) downfield from the internal standard tetramethylsilane (δ 0.00 ppm). The 2D correlation spectra, (1H–13C) gs-HMQC and (1H–13C) gs-HMBC were acquired on a Bruker DRX 400 (9.4 tesla, 400.13 MHz for 1H, 100.62 MHz for 13C, and 40.56 MHz for the 15N spectrometer with a 5-mm inverse-detection H–X probe equipped with a z-gradient coil, at 300 K), and processed using standard Bruker NMR software and in the non-phase-sensitive mode. Gradient selection was achieved through a 5% sine truncated shaped pulse gradient of 1 ms.Single crystal X-ray diffraction
X-ray data collection for compounds 8aa, 12bc and 12da were carried out at room temperature on a Bruker Smart CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) operating at 50 kV and 40 mA. Data were collected over a sphere of the reciprocal space by combination of five exposure sets. Each exposure was of 30 s covered 0.3 in ω. The cell parameters were determined, and refinement was performed by least-squares fit of all reflections. The 50 frames were recollected at the end of the data collections to monitor crystal decay, and no appreciable decay was observed.The structures were solved by direct methods and refined by full-matrix least-square procedures on F2 (SHELXL-97).1 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were included in their calculated positions and refined on the respective carbon atoms. Compound 8aa shows a positional disorder for the C atoms of the benzoyl ring over two positions. This disorder was modeled and refined with occupancies of about 50%. Mercury CSD 4.02 was used for molecular graphics.
CCDC 2312181–2312183 deposition numbers for compounds 8aa, 12bc and 12da contain the supplementary crystallographic data for this paper.
Synthesis
Unsymmetrical 1,3-diketones47 1a–e and N-substituted thioureas 7a–f were synthesized employing the literal cited approach, and NBS 2 was procured commercially.General method for the synthesis
1,3-Diketone 1a–e (1.0 mmol) was ground with N-bromo succinimide 2 (0.177 g, 1.0 mmol) with a dry pestle mortar until a thick paste was formed, and the reaction mass was subsequently combined with N-substituted thiourea derivative 7a–f (1.0 mmol) and sodium carbonate (0.108 g, 1.0 mmol). The reaction mixture was homogenized properly at room temperature for 5 min, transferred to a conical flask and heated to a temperature of 80 °C. The reaction progress was monitored with TLC. On completion of the reaction, water was added to the reaction mixture and filtered to get the coarse product, which was recrystallized from boiling ethanol.O); 1H NMR (400 MHz, CDCl3) δ: 8.79 (s, 1H, N–H), 7.75–7.16 (m, 10H, 2′,3′,4′,5′,6′,2′′,3′′,4′′,5′′,6′′-H), 2.41 (s, 3H, 4-CH3); 13C-NMR (100 MHz, CDCl3) δ: 188.1, 168.4, 158.9, 140.4, 138.9, 131.7, 129.8, 128.4, 128.2, 124.9, 120.4, 120, 18.8; HRMS (ESI): m/z calcd for C17H14N2OS: 294.0827; found: 295.0914 [M + 1]+; elemental analysis: calcd for C17H14N2OS: C, 69.36; H, 4.79; N, 9.52% found: C, 69.35; H, 4.75; N, 9.49%.O); 1H NMR (400 MHz, CDCl3) δ: 8.48 (s, 1H, N–H), 7.65–7.63 (d, 2H, J = 8.12 Hz, 2′′,6′′-H), 7.41–7.37 (t, 2H, J = 8.48 Hz, 2′,6′-H), 7.34–7.32 (d, 2H, J = 7.62 Hz, 3′′,5′′-H), 7.26–7.24 (d, 2H, J = 8.72 Hz, 3′,5′-H), 7.18–7.14 (t, 1H, J = 7.32 Hz, 4′-H), 2.41 (s, 6H, 4′′-CH3, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 187.9, 167.9, 158.5, 142.5, 139.1, 137.6, 129.7, 129.1, 128.5, 124.7, 120.4, 119.8, 21.6, 18.8; HRMS (ESI): m/z calcd for C18H16N2OS: 308.0893; found: 309.0952 [M + 1]+; elemental analysis: calcd for C18H16N2OS: C, 70.10; H, 5.23; N, 9.08% found: C, 70.05; H, 5.19; N, 9.05%.O); 1H NMR (400 MHz, CDCl3) δ: 8.91 (s, 1H, N–H), 7.76–7.74 (d, 2H, J = 9.32 Hz, 2′′,6′′-H), 7.41–7.33 (m, 4H, 2′,3′,5′,6′-H), 7.15–7.13 (t, 1H, J = 7.64 Hz, 4′-H), 6.94–6.92 (d, 2H, J = 9.24 Hz, 3′′,5′′-H), 3.86 (s, 3H, 4′′-OCH3), 2.41 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 186.9, 167.9, 162.7, 157.9, 139.1, 132.8, 130.8, 129.7, 124.7, 120.1, 119.9, 113.6, 55.5, 18.7; HRMS (ESI): calcd for C18H16N2O2S: 324.0932; found: 325.0812 [M + 1]+; elemental analysis: calcd for C18H16N2O2S: C, 66.65; H, 4.97; N, 8.64% found: C, 66.61; H, 4.93; N, 8.61%.O); 1H NMR (400 MHz, CDCl3) δ: 8.56 (s, 1H, NH), 7.78–7.77 (d, 1H, J = 3.6 Hz, 5′′-H), 7.64–7.62 (d, 1H, J = 4.8 Hz, 3′′-H), 7.44–7.40 (m, 2H, 2′,6′-H), 7.37–7.35 (d, 2H, J = 7.6 Hz, 3′,5′-H), 7.20–7.16 (t, 1H, J = 7.2 Hz, 4′-H), 7.14–7.11 (t, 1H, J = 4.8 Hz, 4′′-H), 2.67 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 178.1, 167.3, 159.9, 145.6, 138.9, 132.8, 131.8, 129.8, 127.7, 124.7, 119.6, 117.1, 18.6; HRMS (ESI): calcd for C15H12N2OS2: 300.0391; found: 301.0405 [M + 1]+; elemental analysis: calcd for C15H12N2OS2: C, 59.98; H, 4.03; N, 9.33% found: C, 59.93; H, 3.99; N, 9.00%.O); 1H NMR (400 MHz, CDCl3) δ: 7.76–7.75 (d, 1H, J = 3.32 Hz, 5′′-H), 7.65–7.63 (d, 1H, J = 4.56 Hz, 3′′-H), 7.38–7.32 (m, 4H, 2′,6′,3′,5′-H), 7.14–7.12 (t, 1H, J = 4.12 Hz, 2′′-H), 2.60 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 178.1, 167.4, 158.4, 145.2, 137.5, 133.5, 132.3, 130.4, 129.9, 128.0, 121.6, 117.1, 18.5; HRMS (ESI): calcd for C15H11ClN2OS2: 334.8360, found: 335.8432 [M + 1]+; elemental analysis: calcd for C15H11ClN2OS2: C, 53.81; H, 3.31; N, 8.37% found: C, 53.76; H, 3.26; N, 8.34%.O); 1H NMR (400 MHz, CDCl3) δ: 7.75–7.73 (d, 2H, J = 8.68 Hz, 2′′,6′′-H), 7.23–7.17 (m, 4H, 2′,6′,3′,5′-H), 6.93–6.91 (d, 2H, J = 8.68 Hz, 3′′,5′′-H), 3.86 (s, 3H, 4′′-OCH3), 2.40 (s, 3H, 4-CH3), 2.34 (s, 3H, 4′-CH3); 13C NMR (100 MHz, CDCl3) δ: 186.9, 169.2, 162.6, 158.1, 136.6, 135.1, 132.8, 130.8, 130.3, 120.8, 119.8, 113.6, 55.4, 20.9, 18.7; HRMS (ESI): calcd for C19H18N2O2S: 338.1089; found: 339.2143 [M + 1]+; elemental analysis: calcd for C19H18N2O2S: C, 67.43; H, 5.36; N, 8.28% found: C, 67.38; H, 5.32; N, 8.25%.O); 1H NMR (400 MHz, CDCl3) δ: 8.86 (s, 1H, N–H), 7.76–7.74 (d, 1H, J = 3.82 Hz, 5′′-H), 7.62–7.60 (d, 1H, J = 4.72 Hz, 3′′-H), 7.27–7.21 (m, 4H, 2′,6′,3′,5′-H), 7.12–7.09 (t, 1H, J = 4.38 Hz, 4′′-H), 2.59 (s, 3H, 4-CH3), 2.37 (s, 3H, 4′-CH3); 13C NMR (100 MHz, CDCl3) δ: 178.1, 169.4, 160.3, 145.8, 136.6, 135.5, 132.7, 131.7, 130.4, 127.7, 121.4, 116.4, 21.1, 18.9; HRMS (ESI): calcd for C16H14N2OS2: 314.0548, found: 315.1478 [M + 1]+; elemental analysis: calcd for C16H14N2OS2: C, 61.12; H, 4.49; N, 8.91% found: C, 60.92; H, 4.44; N, 8.77%.O); 1H NMR (400 MHz, DMSO, d6) δ: 10.46 (s, 1H, N–H), 7.68–7.66 (d, 2H, J = 7.6 Hz, 2′′,6′′-H), 7.47–7.41 (m, 4H, 2′,6′,3′′,5′′-H), 7.14–7.12 (d, 2H, J = 8.32 Hz, 3′,5′-H), 2.37 (s, 3H, 4′-CH3), 2.31 (s, 3H, 4-CH3); 13C NMR (100 MHz, DMSO, d6) δ: 185.6, 166.7, 159.3, 138.9, 137.3, 136.5, 132.1, 129.4, 129.3, 128.3, 118.8, 118.5, 20.4, 18.7; HRMS (ESI): calcd for C18H15ClN2OS: 342.8410, found: 343.8121 [M + 1]+, 345.9112 [M + 1 + 2]+ (3:1); elemental analysis: calcd for C18H15ClN2OS: C, 63.06; H, 4.41; N, 8.17% found: C, 63.01; H, 4.37; N, 5.14.O); 1H NMR (400 MHz, CDCl3) δ: 8.30–8.28 (d, 2H, J = 7.2 Hz, 3′,5′-H), 7.71–7.69 (d, 2H, J = 8.0 Hz, 2′′,6′′-H), 7.59–7.57 (d, 2H, J = 7.0 Hz, 2′,6′-H), 7.32–7.30 (d, 2H, J = 7.0 Hz, 3′′,5′′-H), 2.49 (s, 3H, 4′′-CH3), 2.47 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 187.9, 163.8, 158.0, 144.5, 143.2, 142.9, 136.9, 129.2, 128.7, 125.7, 117.1, 95.7, 21.7, 18.7; HRMS (ESI): calcd for C18H15N3O3S: 353.083, found: 354.0914 [M + 1]+; elemental analysis: calcd for C18H15N3O3S: C, 61.18; H, 4.28; N, 11.89% found: C, 61.13; H, 4.24; N, 11.86%.O); 1H NMR (400 MHz, DMSO, d6) δ: 10.39 (s, 1H, N–H), 7.75–7.73 (d, 2H, J = 7.88 Hz, 3′,5′-H), 7.60–7.58 (d, 2H, J = 6.94 Hz, 2′′,6′′-H), 7.27–7.25 (d, 2H, J = 6.90 Hz, 2′,6′-H), 6.97–6.95 (d, 2H, J = 7.86 Hz, 3′′,5′′-H), 3.89 (s, 3H, 4′′-OCH3), 2.41 (s, 3H, 4-CH3); 13C NMR (100 MHz, DMSO, d6) δ: 186.9, 167.1, 162.9, 157.4, 137.7, 132.5, 130.9, 129.7, 121.0, 120.6, 113.7, 55.5, 18.6; HRMS (ESI): calcd for C18H15N3O4S: 369.0783, found: 370.0865 [M + 1]+; elemental analysis: calcd for C18H15N3O4S: C, 58.53; H, 4.09; N, 11.38% found: C, 58.47; H, 4.05; N, 11.35%.O), 1599 (CN); 1H NMR (400 MHz, CDCl3) δ: 8.05–8.02 (d, 2H, J = 8.48 Hz, 2′′,6′′-H), 7.58–7.55 (d, 2H, J = 9.52 Hz, 3′′,5′′-H), 7.43–7.39 (m, 1H, 4′′-H), 7.35–7.26 (m, 4H, 2′,6′,3′,5′-H), 6.39 (q, 1H, J = 1.12 Hz, 5-H), 2.06 (d, 3H, J = 1.12 Hz, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 174.1, 170.6, 136.6, 135.8, 135.2, 133.8, 131.5, 129.8, 129.5, 129.2, 127.9, 104.8, 15.1; HRMS (ESI): calcd for C17H13ClN2OS: 328.8140, found: 329.8231 [M + 1]+, 331.9750 [M + 1 + 2]+ (3:1); elemental analysis: calcd for C17H13ClN2OS: C, 62.10; H, 3.99; N, 8.52% found: C, 61.9; H, 3.94; N, 8.52%.O), 1582 (CN); 1H NMR (400 MHz, CDCl3) δ: 7.97–7.95 (d, 2H, J = 6.72 Hz, 2′′,6′′-H), 7.59–7.57 (d, 2H, J = 6.6 Hz, 3′′,5′′-H), 7.31–7.27 (m, 4H, 2′,6′,3′,5′-H), 6.43 (q, 1H, J = 1.08 Hz, 5-H), 2.08 (d, 3H, J = 1.04 Hz, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 173.4, 170.2, 137.6, 135.7, 135.3, 134.0, 130.6, 129.8, 129.4, 128.2, 104.9, 15.0; HRMS (ESI): calcd for C17H12Cl2N2OS: 362.0067, found: 363.1058 [M + 1]+, 364.0078 [M + 1 + 2]+, 367.0098 [M + 1 + 4]+ (9:6:1); Elemental analysis: calcd for C17H12Cl2N2OS: C, 56.21; H, 3.33; N, 7.71% found: C, 56.01; H, 3.28; N, 7.58%.O); 1H NMR (500 MHz, CDCl3) δ: 7.96 (d, 2H, J = 11.0 Hz, 2′′,6′′-H), 7.65–7.63 (m, 1H, 3′-H), 7.54–7.47 (m, 2H, 4′,5′-H), 7.40–7.38 (m, 1H, 3′-H), 6.80 (d, 2H, J = 11 Hz, 3′′,5′′-H), 6.38 (q, 1H, J = 1.5 Hz, 5-H), 3.79 (s, 3H, 4′′-OCH3), 2.01 (d, 3H, J = 1.5 Hz, 4-CH3); 13C NMR (125 MHz, CDCl3) δ: 174.1, 169.4, 162.4, 135.3, 133.6, 132.9, 131.3, 130.9, 130.6, 130.3, 129.6, 128.0, 113.1, 104.2, 55.3, 14.4; elemental analysis: calcd for C18H15ClN2O2S: C, 60.25; H, 4.21; N, 7.81% found: C, 60.23; H, 4.18; N, 7.80%.O), 1597(CN); 1H NMR (400 MHz, CDCl3) δ: 8.06–8.04 (d, 2H, J = 7.62 Hz, 2′′,6′′-H), 7.41–7.37 (m, 3H, 3′,5′,4′′-H), 7.33–7.29 (m, 2H, 3′′,5′′-H), 7.22–7.21 (d, 2H, J = 6.46 Hz, 2′,6′-H), 6.38 (q, 1H, J = 1.2 Hz, 5-H), 2.50 (s, 3H, 4′-CH3), 2.06 (d, 3H, J = 1.2 Hz, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 174.4, 170.2, 139.2, 136.9, 134.8, 134.5, 131.3, 130.1, 129.3, 127.8, 127.7, 104.4, 21.4, 15.1; HRMS (ESI): calcd for C18H16N2OS: 309.0983, found: 310.0997 [M + 1]+; elemental analysis: calcd for C18H16N2OS: C, 70.10; H, 5.23; N, 9.08% found: C, 69.90; H, 5.17; N, 8.84%.O), 1589 (CN); 1H NMR (400 MHz, CDCl3) δ: 7.94–7.92 (d, 2H, J = 8.08 Hz, 2′′,6′′-H), 7.37–7.35 (d, 2H, J = 8.0 Hz, 3′′,5′′-H), 7.21–7.19 (d, 2H, J = 8.2 Hz, 3′,5′-H), 7.12–7.10 (d, 2H, J = 7.92 Hz, 2′,6′-H), 6.35 (q, 1H, J = 1.04 Hz, 5-H), 2.49 (s, 3H, 4′′-CH3), 2.33 (s, 3H, 4′-CH3), 2.05 (d, 3H, J = 0.96 Hz, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 173.9, 157.2, 141.6, 139.1, 134.4, 130.1, 129.3, 128.6, 127.8, 104.2, 21.6, 21.4, 15.1; HRMS (ESI): calcd for C19H18N2OS: 322.1140, found: 323.2149 [M + 1]+; elemental analysis: calcd for C19H18N2OS: C, 70.78; H, 5.63; N, 8.69% found: C, 70.6; H, 5.58; N, 8.55%.O), 1597 (CN); 1H NMR (400 MHz, CDCl3) δ: 8.07–8.04 (d, 2H, J = 7.44 Hz, 2′′,6′′-H), 7.39–7.37 (m, 1H, 4′′-H), 7.33–7.29 (m, 2H, 3′′,5′′-H), 7.25–7.24 (d, 2H, J = 5.76 Hz, 2′,6′-H), 7.09–7.06 (d, 2H, J = 6.76 Hz, 3′,5′-H), 6.37 (q, 1H, J = 1.08 Hz, 5-H), 3.92 (s, 3H, 4′-OCH3), 2.07 (d, 3H, J = 1.08 Hz, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 174.5, 170.4, 159.9, 136.9, 134.8, 131.4, 130.1, 129.3, 129.2, 127.9, 114.7, 104.3, 55.7, 15.2; HRMS (ESI): calcd for C18H16N2O2S: 324.0932, found: 325.0954 [M + 1]+; elemental analysis: calcd for C18H16N2O2S: C, 66.65; H, 4.97; N, 8.64% found: C, 66.59; H, 4.94; N, 8.59%.O), 1573 (CN); 1H NMR (400 MHz, CDCl3) δ: 8.49–8.46 (d, 2H, J = 7.26 Hz, 3′,5′-H), 8.01–7.99 (d, 2H, J = 7.8 Hz, 2′′,6′′-H), 7.61–7.58 (d, 2H, J = 8.15 Hz, 3′′,5′′-H), 7.44–7.41 (m, 1H, 4′′-H), 7.35–7.31 (m, 2H, 2′,6′-H), 6.44 (q, 1H, J = 1.08 Hz, 5-H), 2.09 (d, 3H, J = 0.84 Hz, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 174.5, 170.1, 147.9, 142.7, 136.3, 132.9, 131.8, 129.6, 129.2, 128.1, 124.8, 105.6, 15.1; HRMS (ESI): calcd for C17H13N3O3S: 339.0678, found: 340.0754 [M + 1]+; elemental analysis: calcd for C17H13N3O3S: C, 60.17; H, 3.86; N, 12.38% found: C, 60.12; H, 3.82; N, 12.35%.O); 1H NMR (400 MHz, CDCl3) δ: 7.78–7.76 (d, 2H, J = 6.82 Hz, 2′′,6′′-H), 7.39–7.32 (m, 4H, 2′,6′,3′,5′-H), 6.98–6.96 (d, 2H, J = 8.11 Hz, 3′′,5′′-H), 3.87 (s, 3H, 4′′-OCH3), 2.42 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 186.9, 167.1, 162.9, 157.4, 137.7, 132.5, 130.9, 129.7, 121.0, 120.6, 113.7, 55.5, 18.6; HRMS (ESI): calcd for C18H15ClN2O2S: 358.8400, found: 359.9975 [M + 1]+, 361.0876 [M + 1 + 2]+ (3:1); elemental analysis: calcd for C18H15ClN2O2S: C, 60.25; H, 4.21; N, 7.81% found: C, 60.05; H, 4.16; N, 7.67%.O), 1528 (CN); 1H NMR (400 MHz, CDCl3) δ: 8.01–7.98 (d, 2H, J = 8.88 Hz, 2′′,6′′-H), 7.57–7.56 (d, 2H, J = 7.62 Hz, 2′,6′-H), 7.30–7.28 (d, 2H, J = 7.6 Hz, 3′,5′-H), 6.84–6.82 (d, 2H, J = 8.92 Hz, 3′′,5′′-H), 6.36 (q, 1H, J = 1.3 Hz, 5-H), 3.81 (s, 3H, 4′′-OCH3), 2.06 (d, 3H, J = 1.3 Hz, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 174.0, 169.8, 162.4, 136.0, 135.1, 133.6, 131.2, 129.7, 129.6, 129.5, 113.2, 104.5, 55.3, 15.0; HRMS (ESI): calcd for C18H15ClN2O2S: 358.5769, found: 359.5876 [M + 1]+, 361.5768 [M + 1 + 2]+ (3:1); elemental analysis: calcd for C18H15ClN2O2S: C, 60.25; H, 4.21; N, 7.81% found: C, 60.05; H, 4.16; N, 7.67%.O); 1H NMR (400 MHz, CDCl3) δ: 7.73–7.72 (d, 1H, J = 3.76 Hz, 5′′-H), 7.61–7.59 (d, 1H, J = 4.94 Hz, 3′′-H), 7.32–7.30 (d, 2H, J = 6.72 Hz, 2′,6′-H), 7.10–7.07 (m, 1H, 4′′-H), 6.98–6.96 (d, 2H, J = 6.74 Hz, 3′,5′-H), 3.83 (s, 3H, 4′-OCH3), 2.57 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 178.1, 160.3, 157.8, 145.8, 132.8, 132.1, 131.7, 129.2, 127.8, 123.9, 115.1, 114.5, 55.6, 18.8; HRMS (ESI): calcd for C16H14N2O2S2: 330.0497, found: 331.2567 [M + 1]+; elemental analysis: calcd for C16H14N2O2S2: C, 58.16; H, 4.27; N, 8.48% found: C, 57.96; H, 4.22; N, 8.35%.O), 1585 (CN); 1H NMR (400 MHz, CDCl3) δ: 7.66–7.63 (m, 1H, 5′′-H), 7.41–7.39 (m, 1H, 3′′-H), 7.24–7.22 (m, 1H, 4′′-H), 7.08–7.05 (m, 2H, 3′,5′-H), 7.00–6.97 (m, 2H, 2′,6′-H), 6.35 (q, 1H, J = 1.16 Hz, 5-H), 3.83 (s, 3H, 4′-OCH3), 2.06 (d, 3H, J = 1.12 Hz, 4-CH3); HRMS (ESI): calcd for C16H14N2O2S2: 330.0497, found: 331.2564 [M + 1]+; elemental analysis: calcd for C16H14N2O2S2: C, 58.16; H, 4.27; N, 8.48% found: C, 57.96; H, 4.22; N, 8.35%.O); 1H NMR (400 MHz, CDCl3) δ: 8.32–8.30 (d, 2H, J = 7.6 Hz, 3′,5′-H), 7.84–7.83 (d, 1H, J = 3.8 Hz, 5′′-H), 7.72–7.71 (d, 1H, J = 5.0 Hz, 3′′-H), 7.63–7.61 (d, 2H, J = 7.2 Hz, 2′′,6′′-H), 7.21–7.18 (t, 1H, J = 4.6 Hz, 4′′-H), 2.69 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 186.3, 167.4, 158.4, 143.4, 132.7, 131.5, 126.9, 124.8, 116.2, 17.6; HRMS (ESI): calcd for C15H11N3O3S2: 345.0242, found: 346.1254 [M + 1]+; elemental analysis: calcd for C15H11N3O3S2: C, 52.16; H, 3.21; N, 12.17% found: C, 51.96; H, 3.16; N, 12.04%.O), 1590 (CN); 1H NMR (400 MHz, CDCl3) δ: 8.48–8.45 (d, 2H, J = 9.38 Hz, 3′,5′-H), 7.64–7.62 (d, 1H, J = 4.7 Hz, 5′′-H), 7.60–7.57 (d, 2H, J = 8.18 Hz, 2′,6′-H), 7.40–7.38 (d, 1H, J = 4.96 Hz, 3′′-H), 7.02–6.99 (m, 1H, 4′′-H), 6.44 (q, 1H, J = 1.28 Hz, 5-H), 2.11 (d, 3H, J = 1.02 Hz, 4-CH3); HRMS (ESI): calcd for C15H11N3O3S2: 345.0242, found: 346.1254 [M + 1]+; elemental analysis: calcd for C15H11N3O3S2: C, 52.16; H, 3.21; N, 12.17% found: C, 51.96; H, 3.16; N, 12.04%.Data availability
The analytical data (1H-NMR, 13C-NMR, HMQC and HMBC) of all compounds supporting the research have been included in the ESI. CCDC 2312181–2312183 deposition numbers for compounds 8aa, 12bc and 12da contain the supplementary crystallographic data for this paper. The X-ray crystallographic data CIF files in the pdf format have been uploaded separately.Conflicts of interest
The authors declare no competing interest.Acknowledgements
We are grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi, India for providing financial assistance through a Senior Research Fellowship (SRF) to Shilpa Sharma. We are also grateful to the Haryana State Council for Science and Technology (HSCST) for providing financial help to Project Fellow Naman Jain. We would like to thank the Spanish Ministerio de Economia, Industria y Competitividad (Ministerio de Economia, Industria y Competitividad, Gobierno de Espana) [CTQ2014-56833-R] for financial support to Dionisia Sanz and Rosa M. Claramunt.References
- K. M. Dawood, T. M. Eldebss, H. S. El-Zahabi and M. H. Yousef, Eur. J. Med. Chem., 2015, 102, 266–276 CrossRef PubMed.
- G. Turan-Zitouni, M. D. Altintop, A. Ozdemir, Z. A. Kaplancıkli, G. A. Çiftçi and H. E. Temel, Eur. J. Med. Chem., 2016, 107, 288–294 CrossRef CAS PubMed.
- J. R. Li, D. D. Li, R. R. Wang, J. Sun, J. J. Dong, Q. R. Du, F. Fang, W. M. Zhang and H. L. Zhu, Eur. J. Med. Chem., 2014, 75, 438–447 CrossRef CAS PubMed.
- C. B. Mishra, S. Kumari and M. Tiwari, Eur. J. Med. Chem., 2015, 92, 1–34 CrossRef CAS PubMed.
- H. A. Ghabbour, A. A. Kadi, K. E. H. El-Tahir, R. F. Angawi and H. El-Subbagh, Med. Chem. Res., 2015, 24, 3194–3211 CrossRef CAS.
- S. Bondock, T. Naser and Y. A. Ammar, Eur. J. Med. Chem., 2013, 62, 270–279 CrossRef CAS PubMed.
- S. Carradori, F. Ortuso, A. Petzer, D. Bagetta, C. De Monte, D. Secci, D. De Vita, P. Guglielmi, G. Zengin, A. Aktumsek, S. Alcaro and J. P. Petzer, Eur. J. Med. Chem., 2018, 143, 1543–1552 CrossRef CAS PubMed.
- S. Koppireddi, J. R. Komsani, S. Avula, S. Pombala, S. Vasamsetti, S. Kotamraju and R. Yadla, Eur. J. Med. Chem., 2013, 66, 305–313 CrossRef CAS PubMed.
- M. V. N. de Souza, J. Sulfur Chem., 2005, 26, 429–449 CrossRef CAS.
- L. Y. Wang, C. X. Zhang, Z. Q. Liu, D. Z. Liao, Z. H. Jiang and S. P. Yan, Inorg. Chem. Commun., 2003, 6, 1255–1258 CrossRef CAS.
- Z. Yan, A. Liu, M. Huang, M. Liu, H. Pei, L. Huang, H. Yi, W. Liu and A. Hu, Eur. J. Med. Chem., 2018, 149, 170–181 CrossRef CAS PubMed.
- I. Parasotas, K. Anusevicius, R. Vaickelioniene, I. Jonuskiene, M. Stasevych, V. Zvarych, O. Komarovska-Porokhnyavets, V. Novikov, S. Belyakov and V. Mickevicius, Arkivoc, 2018,(iii), 240–256 Search PubMed.
- M. Romero-Ortega, A. Aviles, R. Cruz, A. Fuentes, R. M. Gomez and A. Plata, J. Org. Chem., 2000, 65, 7244–7247 CrossRef CAS PubMed.
- P. Yogil, M. Ashid, N. Hussain, S. Khan and A. Joshi, Asian J. Chem., 2016, 28, 927–932 CrossRef.
- V. Facchinettia, M. M. Avellar, A. C. S. Nery, C. R. B. Gomes, T. R. A. Vasconcelos and M. V. N. de Souza, Synthesis, 2016, 48, 437–440 Search PubMed.
- M. Narender, M. S. Reddy, V. P. Kumar, B. Srinivas, R. Sridhar, Y. V. D. Nageswar and K. R. Rao, Synthesis, 2007, 22, 3469–3472 Search PubMed.
- G. Meng, M. Wang, A. Zheng, J. Dou and Z. Guo, Green Chem. Lett. Rev., 2014, 7, 46–49 CrossRef.
- C. L. Liu, Z. M. Lia and B. Zhonga, J. Fluorine Chem., 2004, 125, 1287–1290 CrossRef CAS.
- D. Castagnolo, M. Pagano, M. Bernardinia and M. Botta, Synlett, 2009, 13, 2093–2096 Search PubMed.
- B. Chen, S. Guo, X. Guo, G. Zhang and Y. Yun, Org. Lett., 2015, 17, 4698–4701 CrossRef CAS PubMed.
- X. Tang, J. Yang, Z. Zhu, M. Zheng, W. Wu and H. Jiang, J. Org. Chem., 2016, 81, 11461–11466 CrossRef CAS PubMed.
- F. Rahim, M. T. Javeda, H. Ullaha, A. Wadood, M. Taha, M. Ashraf, Q. ul-Ain, M. A. Khan, F. Khan, S. Mirza and K. M. Khan, Bioorg. Chem., 2015, 62, 106–116 CrossRef CAS PubMed.
- H. Bouherrou, A. Saidoun, A. Abderrahmani, L. Abdellaziz, Y. Rachedi, F. Dumas and A. Demenceau, Molecules, 2017, 22, 757–769 CrossRef PubMed.
- T. R. Swaroop, H. Ila and K. S. Rangappa, Tetrahedron Lett., 2013, 54, 5288–5292 CrossRef CAS.
- X. Wang, F. Wang, Z. Quan, Z. Zhan and M. G. Wang, Synth. Commun., 2006, 36, 2453–2460 CrossRef CAS.
- (a) R. Raja, D. Murugan, A. Sivasubramaniyan, J. George, P. Sakhtivel, P. Alagusundaram, M. R. Jayakumar and M. Saminathan, Synth. Commun., 2016, 46, 942–948 CrossRef CAS; (b) J. A. Shiran, A. Yahyazadeh, M. Mamaghani and R. Mehdi, J. Mol. Struct., 2013, 1039, 113–118 CrossRef.
- I. J. Barve, W. J. Chang, Y. T. Lin, T. U. Thikekar and C. M. Sun, ACS Comb. Sci., 2019, 21, 269–275 CrossRef CAS PubMed.
- E. Badali, A. Z. Halimehjani and A. Habibi, Sci. Rep., 2023, 13, 3079–3087 CrossRef CAS PubMed.
- G. S. Kumar, S. P. Ragini and H. Meshram, Tetrahedron Lett., 2003, 54, 5974–5978 CrossRef.
- I. Yavari, A. Malekafzali and S. Seyfi, J. Iran. Chem. Soc., 2014, 11, 285–288 CrossRef CAS.
- R. Aggarwal, G. Singh, D. Sanz, R. M. Claramunt, M. C. Torralba and M. R. Torres, Tetrahedron, 2016, 72, 3832–3838 CrossRef CAS.
- R. Aggarwal, N. Jain, S. Sharma, P. Kumar, G. P. Dubey, H. Chugh and R. Chandra, Sci. Rep., 2021, 11, 22135–22153 CrossRef CAS PubMed.
- R. Aggarwal, M. Hooda, P. Kumar, N. Jain, G. P. Dubey, H. Chugh and R. Chandra, J. Org. Chem., 2022, 87, 3952–3966 CrossRef CAS PubMed.
- R. Aggarwal, M. Hooda, P. Kumar and M. C. Torralba, Org. Biomol. Chem., 2022, 20, 584–595 RSC.
- R. Aggarwal, S. Sharma, M. Hooda, D. Sanz, R. M. Claramunt, B. Twamley and I. Rozas, Tetrahedron, 2019, 75, 130728 CrossRef CAS.
- R. Aggarwal, N. Jain, G. P. Dubey, S. Singh and R. Chandra, Biomacromolecules, 2023, 24, 4798–4818 CrossRef CAS PubMed.
- R. Aggarwal, M. Hooda, P. Kumar, S. Kumar, S. Singh and R. Chandra, Bioorg. Chem., 2023, 136, 106524 CrossRef CAS PubMed.
- R. Aggarwal, M. Hooda, N. Jain, D. Sanz, R. M. Claramunt, B. Twamley and I. Rozas, J. Sulfur Chem., 2022, 43, 12–21 CrossRef CAS.
- R. Aggarwal R, R. Kumar, D. Sanz and R. M. Claramunt, J. Heterocycl. Chem., 2014, 51, 598–603 CrossRef.
- N. Aggarwal, R. Kumar, P. Dureja and J. M. Khurana, Eur. J. Med. Chem., 2011, 46, 4089–4099 CrossRef CAS PubMed.
- G. Sumran and R. Aggarwal, J. Sulfur Chem., 2015, 36, 170–177 CrossRef CAS.
- B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, in Vogel's Textbook of Practical Organic Chemistry, Longman Scientific and Technical, England, 1991, pp. 634–635 Search PubMed.
- K. P. Bhusari, N. D. Amnerkar, P. B. Khedekar, M. K. Kale and R. Bhole, Asian J. Res. Chem., 2008, 1, 53–58 CAS.
- R. Aggarwal, S. Kumar, R. Sadana, A. Guzman and V. Kumar, Synth. Commun., 2021, 51, 3308–3324 CrossRef CAS.
- (a) R. Aggarwal, R. Kumar and V. Kumar, J. Sulfur Chem., 2007, 28, 617–623 CrossRef CAS; (b) R. Aggarwal, S. Kumar and S. P. Singh, J. Sulfur Chem., 2012, 33, 521–525 CrossRef CAS.
- S. Kumar, R. Aggarwal, V. Kumar, R. Sadana, B. Patel, P. Kaushik and D. Kaushik, Eur. J. Med. Chem., 2016, 123, 718–726 CrossRef CAS PubMed.
- I. Pravst, M. Zupan and S. Stavber, Green Chem., 2006, 8, 1001–1005 RSC.
- S. Murru, C. B. Singh, V. Kavala and B. K. Patel, Tetrahedron, 2008, 64, 1931–1942 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data (1H, 13C, HMBC, HMQC spectra and X-ray crystallographic studies) for the final compounds. CCDC 2312181–2312183. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05436a |
| This journal is © The Royal Society of Chemistry 2024 |