
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation and development of novel synthetic approaches for synthesis of euxanthone and derived dyes†
M. Mustafa Cetin *
*
Faculty of Engineering and Natural Sciences, Kadir Has University, Cibali, Istanbul, 34083, Turkiye. E-mail: mustafamcetin@yahoo.com
First published on 7th November 2024
Abstract
The historical dye Indian yellow, derived from euxanthic acid formed from 1,7-dihydroxyxanthone (euxanthone) and methyl (tri-O-acetyl-α-D-glucopyranosyl bromide) uronate, has significantly influenced the art world due to its vibrant color and unique production process. Studying Indian yellow is important for its historical relevance and impact on various art forms, as well as the challenges in its synthetic production. Herein, this work investigates the synthesis of the two main components, a novel method for obtaining euxanthone, and attempts to produce euxanthic acid and Indian yellow. All key intermediates and desired compounds have successfully been synthesized with good to high isolated yields, and characterized using different analytical and spectroscopic techniques. A proposed mechanism for euxanthone synthesis via 2,6,2′,5′-tetramethoxybenzophenone formation is also offered. During this process, 2,7-dihydroxyxanthone has also been synthesized, revealing an equilibration reaction that produced three isomeric tetramethoxybenzophenones, confirmed by both GC/MS and NMR. Following the synthesis of euxanthone and clarification of the equilibration, the production of Indian yellow via euxanthic acid formation has further been explored.
Introduction
Natural products and organic dyes play significant roles in cultural heritage and human health, from historical paints to medicinal applications for various diseases.1–14 They have been used to relieve ailments, produce medicines from plant parts,4,15–26 dye wool, and color historical paints for restoration.1–15,17–19,27–37 Naturally occurring xanthones (Fig. 1) are notable organic dyes utilized in both pharmaceuticals and art.1–38 Xanthones, or 9H-xanthen-9-one (Fig. 1A), possess a dibenzo-γ-pyrone structure and are not found in their unsubstituted form in nature. They are typically colored due to hydroxyl groups and are structurally similar to γ-pyrone derivatives, such as flavonoids and chromones (Fig. 1A–D).39–41 While unsubstituted xanthone is absent in nature, various oxygenated derivatives have been isolated from plant and animal sources, including euxanthone and mangostin from higher plants and lichexanthone from fungi.39,40 Natural xanthone derivatives have been classified by Denisova-Dyatlova et al.40 and studied for their chemotaxonomy and biogenesis pathways.42 In recent years, interest in natural xanthones has surged due to their promising pharmacological properties, including antidepressant, antimicrobial, antiviral, antifungal, antibacterial, antioxidant, antimalarial, anti-inflammatory, antithrombotic, and anticancer effects, largely attributed to their hydroxyl group positioning.39,40 Thus, the study of both natural and synthetic xanthones suggests their potential as medicinal compounds.40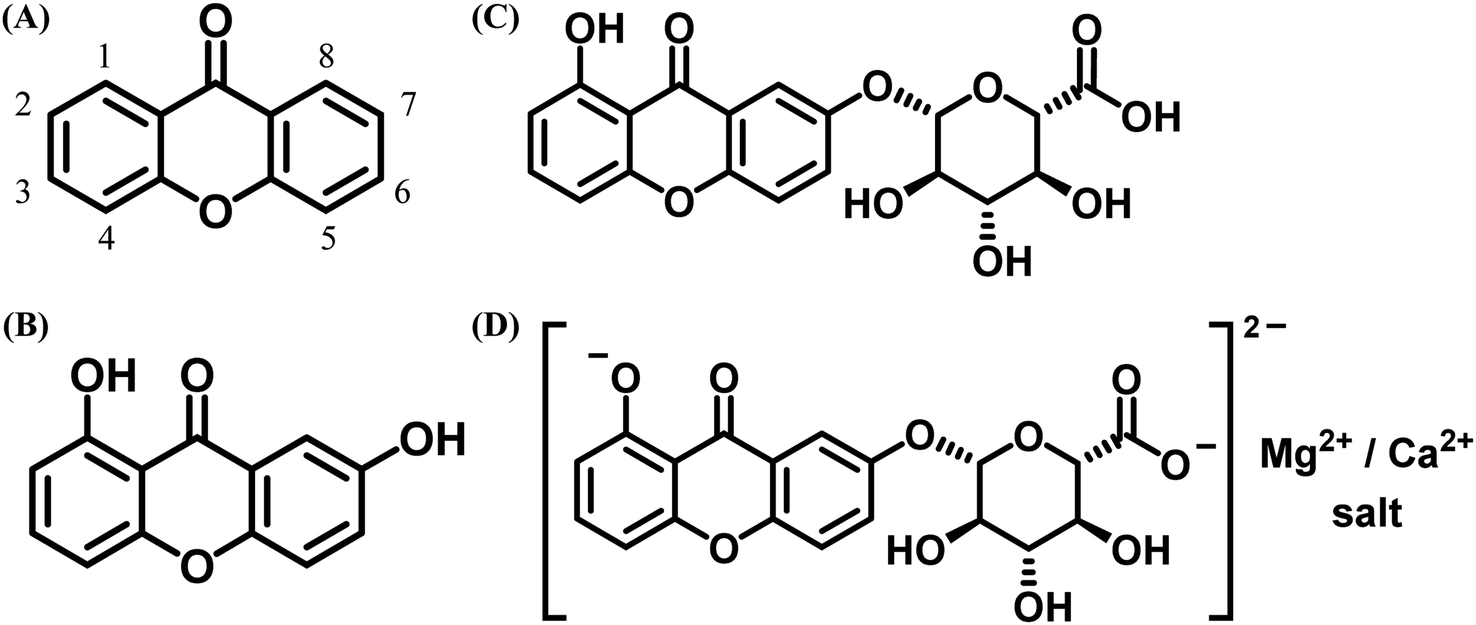 | ||
| Fig. 1 Chemical structures: (A) dibenzo-γ-pyrone (xanthone) with numbering; (B) 1,7-dihydroxyxanthone (euxanthone), pale yellow needles from the heartwood of platonia insignis; (C) euxanthic acid, pale yellow needles with a β-type glycoside linkage to euxanthone,39,43 and (D) Indian yellow, an O-glycoside of euxanthone and glucuronic acid, which increases water solubility, making it widely used as a yellow pigment in oil and watercolor painting on walls, doors, and frameworks.39,44,45 | ||
Indian yellow (Fig. 1D) and euxanthone (Fig. 1B), both part of the xanthone family, have intrigued artists, historians, and scientists for centuries, offering potential applications in pharmaceuticals, dyes, and materials science.38,39,44,46–51 Indian yellow is noted for its intense orange-yellow color and its historical significance in art and medicine.38,39 euxanthone and its derivatives are used as colorants in textiles and in fluorescence applications, including microscopy and organic photovoltaics (OPVs), due to their properties as organic semiconductors. They can also function as photocatalysts for various photochemical reactions, making further research into their potential essential. However, producing Indian yellow from euxanthone and glucuronic acid presents challenges, including ethical concerns with traditional methods,1,52–54 limited raw material availability, labor-intensive processes, color and quality variability due to factors like animal diet and production methods,55,56 chemical instability, and a lack of modern production techniques. These issues pose difficulties for the preservation of artworks using Indian yellow, requiring careful handling to prevent degradation.57 Addressing these challenges requires balancing historical preservation with the development of sustainable, ethical production methods. Continued research and innovation are crucial for ensuring the availability and longevity of Indian yellow pigment through euxanthone and glucuronic acid.
Given the existing literature on xanthones,1–57 particularly Indian yellow from euxanthone and glucuronic acid, synthesizing these natural compounds has become a crucial research focus. Various synthetic techniques for euxanthone and its derivatives have been documented.58,59 Synthetic protocols can be categorized into six types: Michael–Kostanecki (e.g., 1,3-dihydroxyxanthone),19 Ullmann (e.g., euxanthone),60 Robinson–Nishikawa (e.g., 1,3-dihydroxyxanthone),61–64 Asahina–Tanase (e.g., 3-methoxyxanthone),39 Tanase (e.g., 3,8-dihydroxy-1-methoxyxanthone),39 and Friedel–Crafts (e.g., gentisin).64 Common analytical methods for separation and purification include solvent extraction (e.g., Soxhlet), chromatography (e.g., paper, thin-layer, and column), and recrystallization.48,65 Key techniques for structural determination of xanthones are Fourier Transform Infrared (FTIR),65–68 UV/Vis absorption,69 Nuclear Magnetic Resonance (NMR),70 photo-induced luminescence (PL) imaging,38,47,50,65,70–83 X-ray crystallography (XRD), mass spectrometry (MS), and high-performance liquid chromatography (HPLC).30,84
Taking into account the synthetic protocols, analytical and structural determination methods as well as separation and purification information provided by the abovementioned literature, this study aims to synthesize euxanthone and explore methods to produce it and euxanthic acid in sufficient quantities for Indian yellow synthesis, suitable for art conservation studies. The synthesis, characterization, and purification of euxanthone, along with the attempted synthesis of Indian yellow, have been reported.
Results and discussion
The primary goal of this work is to investigate existing synthetic protocols for xanthones to obtain euxanthone (1,7-dihydroxyxanthone), develop a novel synthetic approach, and attempt to synthesize its derived dye, Indian yellow, particularly for its significance in medicinal chemistry and art studies. Various methods are documented for synthesizing naturally occurring xanthones, including Michael–Kostanecki,19 Ullmann,60 Robinson–Nishikawa,61–64 Asahina–Tanase,39 Tanase,39 and Friedel–Crafts.64 This study focuses on producing euxanthone and euxanthic acid in sufficient quantities for Indian yellow synthesis. Initially, the focus was on synthesizing euxanthone and then methyl(tri-O-acetyl-α-D-glucopyranosyl bromide) uronate, both essential for producing Indian yellow. The preparation of intermediates and desired compounds has been reported (Scheme 1), along with challenges encountered during synthesis and explanations for some anomalous findings.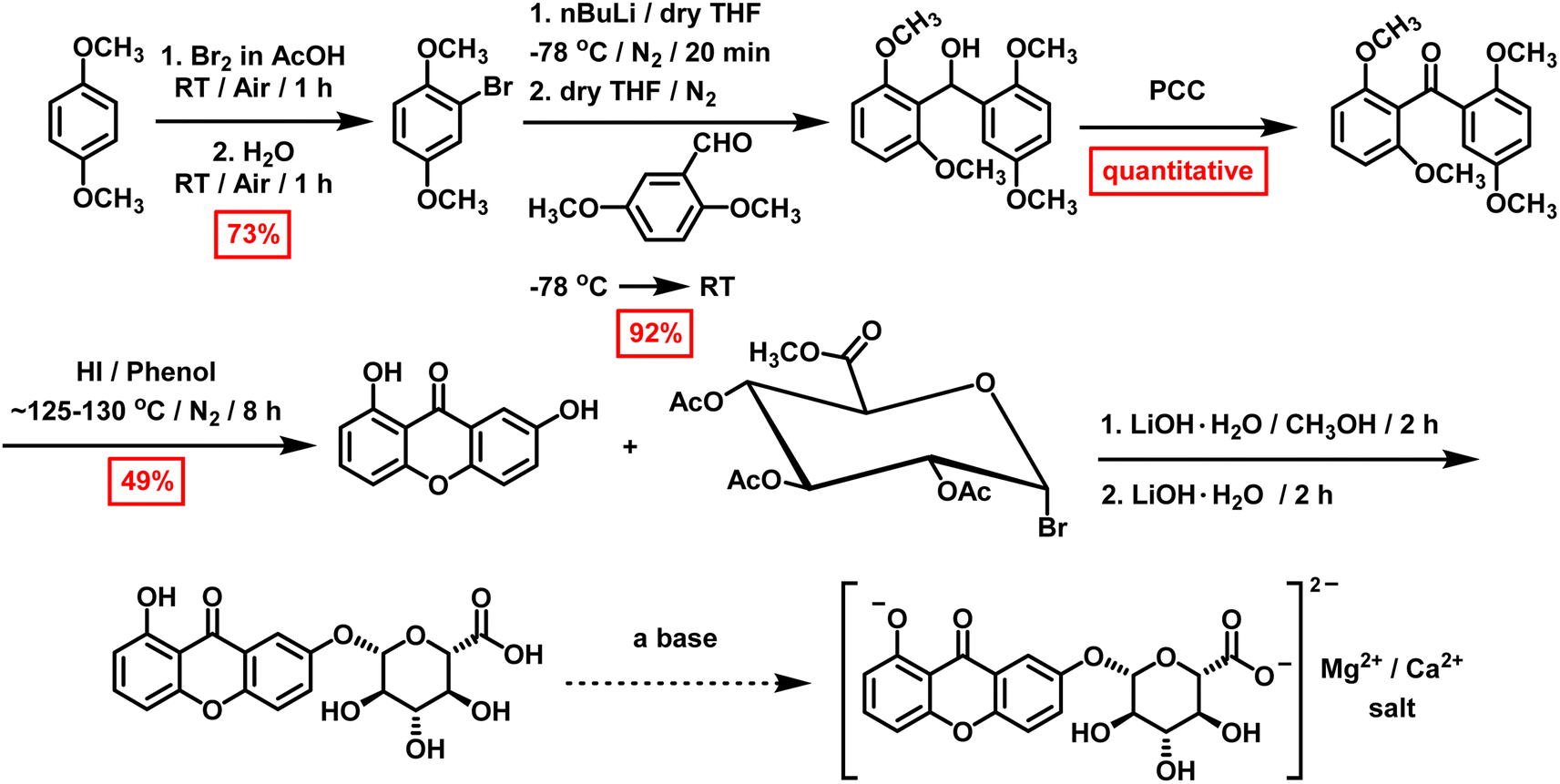 | ||
| Scheme 1 The new synthetic protocol for the synthesis of euxanthone and Indian yellow dye. | ||
As noted in several literature protocols where the synthesis of euxanthone was claimed,19,39,60–64,85 the synthesis of euxanthone begins with 2,6-dihydroxybenzoic acid methyl ester (Scheme S1†). This ester precursor was obtained with a 76% isolated yield by reacting 2,6-dihydroxybenzoic acid with methanol in the presence of concentrated sulfuric acid. This compound was then used in various existing protocols19,39,60–64,85 to synthesize euxanthone. In the first attempted protocol (Scheme S2†),85 the methyl ester and hydroquinone were refluxed; however, only 1,3-dimethoxybenzene, m-methoxyphenol, and CO2 were identified as decomposition products in the 1H-NMR and GC/MS results. Following this, the protocol was modified, heating the methyl ester and hydroquinone mixture to 225 °C (Scheme S3†). Although euxanthone was obtained in trace amounts (0.45%) after chromatographic purification, this protocol could not be reproduced despite multiple revisions. A similar approach was attempted using a microwave reactor (Scheme S4†) for 30 minutes (300 W, 250 °C, 293 psi, high stirring), but the desired euxanthone was not obtained, even after altering the conditions (Table S1†).
After the unsuccessful outcomes and decomposition of the methyl ester in the first protocol, another method was adopted18 using 2-hydroxy-5-methoxybenzoic acid, resorcinol, and freshly fused zinc chloride (Scheme S5†). Euxanthone was expected to crystallize as yellow needles after purification; however, while the GC/MS results indicated the correct molecular weight, the 1H-NMR spectrum (Fig. S8 and S9†) revealed signals for 2,7-dihydroxyxanthone, likely due to an equilibration reaction of the benzophenone intermediate (Schemes S14 and S15†). Thus, euxanthone was not obtained via this protocol either. Substituting zinc chloride with polyphosphoric acid (PPA) or phosphorus oxychloride (POCl3) was also explored. In the case of POCl3, the GC/MS showed two peaks at 12.06 and 12.42 min with an m/z value of 302; however, euxanthone was not detected, and a new set of products was observed instead (vide infra).
Based on the previous synthetic protocols, it was concluded that these methods were not viable for producing euxanthone in sufficient quantities. Subsequently, other protocols3,86 were employed (Schemes S6 and S7†). The first step involved synthesizing 2-hydroxy-6-methoxy-2′,5′-dimethoxybenzophenone and 2,6-dimethoxy-2′-hydroxy-5′-methoxybenzophenone by reacting 2,6-dimethoxybenzoic acid with oxalyl chloride in anhydrous benzene at room temperature (Scheme S7†). However, the resulting yellow oil was primarily ethyl 2,6-dimethoxybenzoate, not the expected benzophenone derivatives. Repeating the reaction yielded similar results, suggesting that the ether solvent and AlCl3 might form a Lewis acid–base adduct or that AlCl3 decomposed 2,6-dimethoxybenzoyl chloride, or both. Additionally, temperature and solvent effects likely contributed to the formation of unexpected products, as indicated by GC/MS and 1H-NMR analyses, which showed rings and ethyl groups instead of methoxy and hydroxy groups. To address these issues, ether was replaced with dichloromethane (CH2Cl2), and the reaction was performed at approximately 10 °C with AlCl3 added in small portions (Scheme S8†). Following proper workup and purification, colorless crystals of 2,6,2′,5′-tetramethoxybenzophenone—a key intermediate for euxanthone synthesis—were obtained (Fig. S13, S14 and S16†) with a 73% isolated yield.
Alternatively (Scheme S9†),87–90 a solution of 1,4-dimethoxybenzene in glacial acetic acid (AcOH) was treated dropwise with a bromine solution in AcOH at room temperature, yielding 2-bromo-1,4-dimethoxybenzene (73%). Next, 2.5 M n-butyl lithium in hexane was added to this intermediate in dry THF at −78 °C. After stirring for 20 minutes, 2,6-dimethoxybenzaldehyde was introduced, and the mixture was allowed to warm to room temperature. Following proper workup and purification, 2,6,2′,5′-tetramethoxybenzophenone was obtained as a waxy solid (92%) after oxidizing the reaction mixture with pyridinium chlorochromate (PCC) in CH2Cl2, improving the isolated yield.
This key intermediate was then used to produce euxanthone (Scheme S10†) via a revised protocol,3 involving the dissolution of 2,6,2′,5′-tetramethoxybenzophenone in phenol and the addition of hydroiodic acid, followed by heating for 8 hours. After the required coupling and removal of methoxy groups, the desired product crystallized as yellow needles with an isolated yield of 49%, confirmed by instrumental spectra (Fig. S1–S4 and S15†). The new protocol was repeated, first increasing the yield by 10% with the same conditions, then by another 10% after lowering the temperature to 125–130 °C, leading to high purity. A reaction mechanism for euxanthone formation was proposed (Scheme 2), illustrating a Friedel–Crafts acylation (an electrophilic aromatic substitution). The process involves replacing an aryl hydrogen atom with an acyl moiety from the acyl chloride, resulting in an alkyl aryl ketone. The acyl chloride first reacts with a Lewis acid catalyst (AlCl3) to form an acylium intermediate, which then releases a chloride ion to produce the acylium ion. This ion is electrophilic and undergoes nucleophilic attack by π-system of the aromatic ring. Deprotonation at the carbon bearing the acyl group restores aromaticity. In the next step, ring closure occurs in the presence of strong acid (HI), followed by demethylation with HI, yielding the desired euxanthone product.
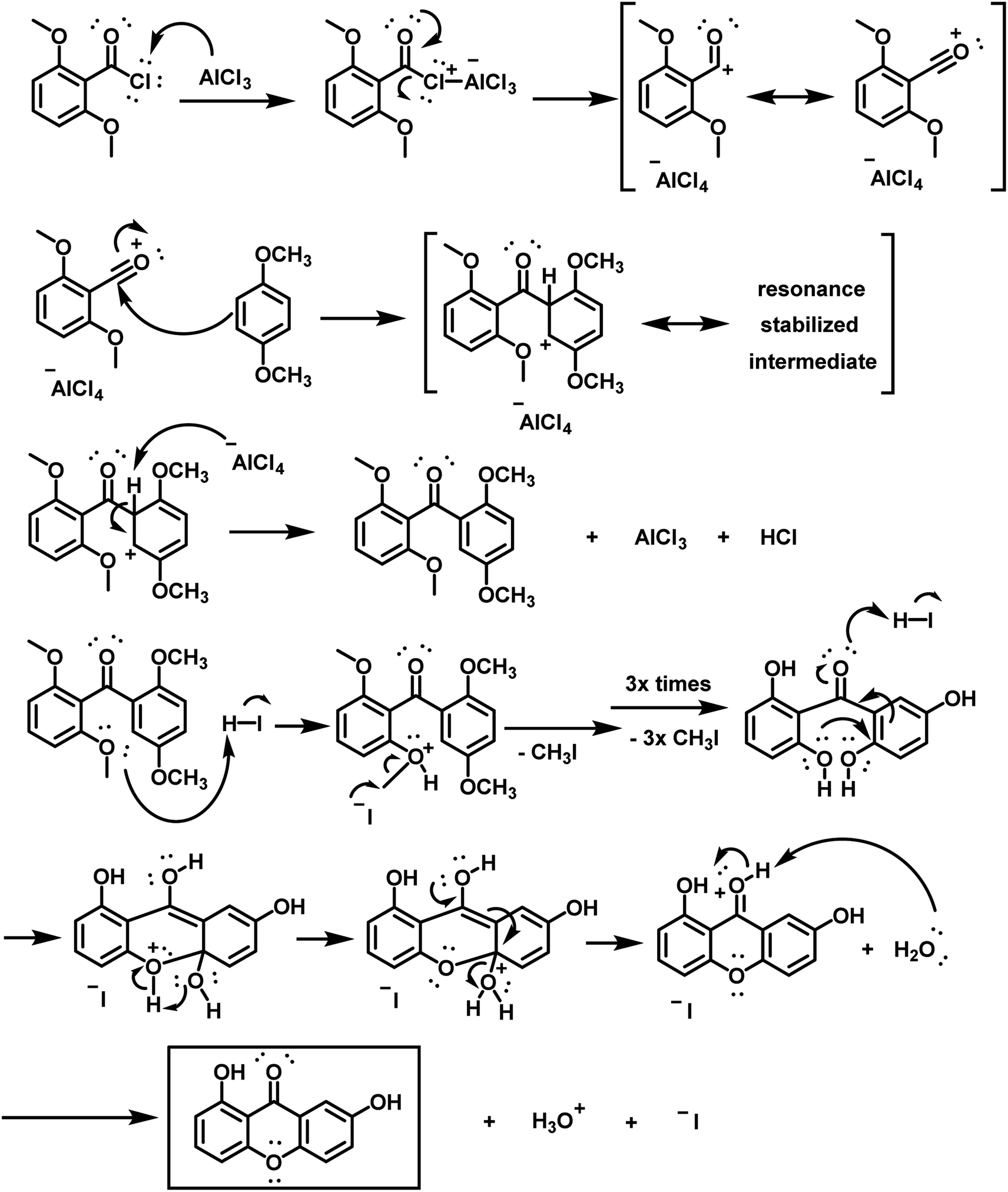 | ||
| Scheme 2 A proposed mechanism for the synthesis of the desired compound euxanthone. | ||
A high-yielding synthetic protocol for euxanthone has been developed, but new findings reveal an equilibration reaction involving benzophenone intermediates. During attempts to synthesize euxanthone, multiple m/z = 302 peaks were observed in the GC/MS spectra (Fig. S17–S19†) and NMR results (Fig. S10–S12†), indicating isomeric products. Notably, the derived compound 2,7-dihydroxyxanthone emerged unexpectedly, suggesting an equilibration reaction (Schemes S14 and S15†) not previously discussed in literature. To investigate this, the synthesis of 2,6,2′,5′-tetramethoxybenzophenone was repeated using 2,6-methoxybenzoic acid, 1,4-dimethoxybenzene, and polyphosphoric acid (PPA). Thin-Layer Chromatography (TLC) revealed three structural isomers (Scheme 3), confirmed by GC/MS and NMR: 2,5,2′,5′-tetramethoxybenzophenone (isomer 1) leading to 2,7-dihydroxyxanthone, 2,4,2′,5′-tetramethoxybenzophenone (isomer 2) leading to 2,6-dihydroxyxanthone, and 2,4,2′,4′-tetramethoxybenzophenone (isomer 3) leading to 3,6-dihydroxyxanthone. GC/MS distinguished these isomers by retention times of 14.12, 15.70, and 16.52 min for isomers 1, 2, and 3, respectively (Fig. S17–S19†), while the key intermediate had a retention time of 13.60 min. The 1H NMR spectra of the isomers were distinct (Fig. S10–S12†), particularly isomer 2, which showed (Fig. S11†) characteristic singlets for methoxy groups and a signal at 6.40 ppm for the 3-position proton. Isomers 1 and 3 were similar, differing only in the singlet for isomer 3 at 6.40 ppm, indicating protons at the 3- and 3′-positions on the phenyl rings (Fig. S12†).
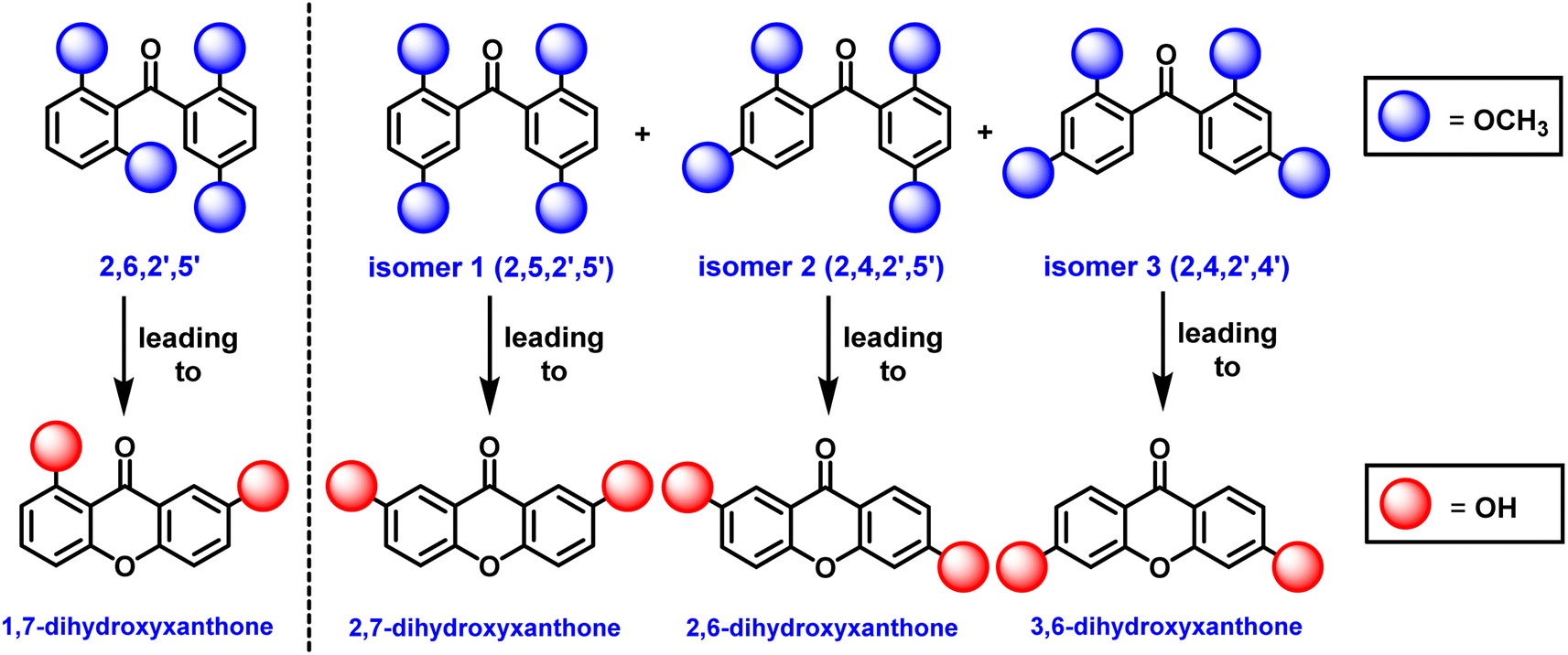 | ||
| Scheme 3 A new discovery of an equilibrium reaction resulting with three isomers (1, 2, and 3) of 2,6,2′,5′-tetramethoxybenzophenone leading to the euxanthone (left of dashed-line). | ||
To determine if the key intermediate 2,6,2′,5′-tetramethoxybenzophenone was involved in the equilibration, a reaction was also conducted using this compound with PPA. Following similar steps as before, three fractions were collected via TLC and analyzed by GC/MS and NMR. The analyses confirmed the same structural isomers, with retention times of 14.07, 15.31, and 16.07 min, all having an m/z value of 302.
These results indicate that the Friedel–Crafts reaction is reversible, and the desired benzophenone, 2,6,2′,5′-tetramethoxybenzophenone, is not favored at equilibrium. Its synthesis is only feasible at lower temperatures, while other isomers are produced at room temperature or higher under acidic conditions. Initial attempts to synthesize euxanthone using high temperatures and acidic conditions resulted in low yields or undesirable products. This was confirmed when 2,6,2′,5′-tetramethoxybenzophenone was obtained (Fig. S13 and S14†) at low temperatures without other isomers. Additionally, these equilibration results raise concerns about earlier protocols that claimed to synthesize euxanthone without adequate product characterization.
Once euxanthone was obtained and the equilibration reaction elucidated, the synthesis of the derived dye Indian yellow via euxanthic acid formation and deprotonation was investigated. The first step involved preparing methyl tetra-O-acetyl-β-D-glucopyranuronate (Scheme S11†) by reacting glucuronolactone with acetic anhydride in the presence of methanol, sodium hydroxide, and pyridine, following an existing protocol.91 After careful crystallization from 2-propanol, the desired β-anomer was obtained in high yield (91%), confirmed by 1H-NMR (Fig. S5†) and GC/MS, which matched with the literature.91 After protecting the hydroxyl groups, the anomeric acetyl group (β) was converted to a bromo group (α)92 to obtain methyl (tri-O-acetyl-α-D-glucopyranosyl bromide) uronate (Scheme S12†). This was achieved using a solution of HBr (27%) in acetic acid, successfully synthesizing the product, which was structurally confirmed by 1H-NMR (Fig. S6†) and GC/MS. Although this intermediate decomposed after two days at room temperature, repeating the synthesis with commercial HBr (33%) yielded a high isolated yield (92%). Finally, Indian yellow synthesis was attempted using an adopted protocol91 with methyl (tri-O-acetyl-α-D-glucopyranosyl bromide) uronate and euxanthone in the presence of LiOH·H2O in methanol. However, TLC analysis showed only starting materials, and GC/MS and 1H-NMR confirmed the absence of the desired Indian yellow dye despite several modifications to the approach.
Conclusions
This work primarily focused on investigating synthetic protocols for 1,7-dihydroxyxanthone (euxanthone) and developing a novel approach to produce it, along with its derived dye, Indian yellow. Based on existing literature, key components and desired compounds were synthesized, purified, and characterized. Despite some unsuccessful attempts, the key precursor 2,6,2′,5′-tetramethoxybenzophenone was obtained with a 92% yield. Using this intermediate, euxanthone was synthesized as yellow needles with a 49% yield, confirmed by 1D/2D NMR and GC/MS. The new and optimized protocol improved yield by 20–25% with high purity. A mechanistic proposal involving Friedel–Crafts acylation was presented. During the euxanthone synthesis attempts, 2,7-dihydroxyxanthone was coincidentally synthesized, leading to the discovery of an equilibration reaction producing three isomers with m/z = 302, identified as 2,5,2′,5′-, 2,4,2′,5′-, and 2,4,2′,4′-tetramethoxybenzophenones. All isomers were confirmed by GC/MS and NMR. Initial high-temperature syntheses resulted in low yields and undesirable products due to isomerization. Analysis indicated that the Friedel–Crafts reaction is reversible and that 2,6,2′,5′-tetramethoxybenzophenone forms only at lower temperatures, contrary to previous protocols. This equilibration detail was not addressed in earlier studies, which lacked thorough product characterization. Following the successful euxanthone synthesis and equilibration clarification, attempts to synthesize Indian yellow from euxanthic acid using euxanthone and methyl (tri-O-acetyl-α-D-glucopyranosyl bromide) uronate resulted only in the recovery of starting materials, despite various modifications. Further exploration of alternative methods to produce euxanthic acid and Indian yellow in sufficient quantities for art conservation studies is ongoing in our laboratories.Experimental section
Experimental: general methods
Synthetic procedures and structural determination data
The detailed synthetic procedures and structural characterization data for the intermediates and desired compounds are presented in the ESI.† Some important details and structural determination data for the intermediates and desired product(s) are presented below.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
This manuscript and related documents regarding writing, reviewing, editing, data analysis, and other details have been prepared by M. Mustafa Cetin (MMC).Conflicts of interest
There are no conflicts to declare. The author also declares no competing financial interest.Acknowledgements
The author thanks to Prof M. Scott Goodman for his endless support in the discussion of the obtained results, sage advice, insightful criticism and discussion, and patient encouragement during this work. The author also thanks to the personnel in the Department of Chemistry at the State University of New York (SUNY) College at Buffalo and Office of Undergraduate Research for their assistance in the collection of the data and use of facilities. Financial support from the Kadir Has University (MMC_BAF and MMC_SEED) and the SUNY Research Foundation is gratefully acknowledged. This research was also supported by SUNY College at Buffalo and Kadir Has University computer laboratories and facilities. Special thanks go to the Fulbright–International Institute of Education for providing me this opportunity and supporting my education and career. Finally, the author greatly thanks to his wife Mine and sons Metehan Arda and Mert Kaan for willingly giving him time to complete this work.Notes and references
- J. Bailkin, J. Mater. Cult., 2005, 10, 197 CrossRef.
- C.-N. Lin, H.-K. Hsieh, S.-J. Liou, H.-H. Ko, H.-C. Lin, M.-I. Chung, F.-N. Ko, H.-W. Liu and C.-M. Teng, J. Pharm. Pharmacol., 1996, 48, 887 CrossRef CAS PubMed.
- C.-N. Lin, M.-I. Chung, S.-J. Liou, T.-H. Lee and J.-P. Wang, J. Pharm. Pharmacol., 1996, 48, 532 CrossRef CAS PubMed.
- K. Likhitwitayawuid, W. Chanmahasathien, N. Ruangrungsi and J. Krungkrai, Planta Med., 1998, 64, 281 CrossRef CAS PubMed.
- S. F. Kouam, D. B. Yapna, K. Krohn, B. T. Ngadjui, J. Ngoupayo, M. I. Choudhary and B. Schulz, J. Nat. Prod., 2007, 70, 600 CrossRef CAS PubMed.
- S. Ngouela, B. L. Ndjakou, D. N. Tchamo, F. Zelefack, E. Tsamo and J. D. Connolly, Nat. Prod. Res., 2005, 19, 23 CrossRef CAS PubMed; Z. Y. Xiao, W. K. P. Shiu, Y. H. Zeng, Q. Mu and S. Gibbons, Pharm. Biol., 2008, 46, 250 CrossRef.
- Z. Y. Xiao, W. K. P. Shiu, Y. H. Zeng, Q. Mu and S. Gibbons, Pharm. Biol., 2008, 46, 250 CrossRef CAS.
- Y.-H. Duan, Y. Dai, G.-H. Wang, X. Zhang, H.-F. Chen, J.-B. Chen, X.-S. Yao and X.-K. Zhang, J. Nat. Prod., 2010, 73, 1283 CrossRef CAS PubMed.
- M. Naidu, C.-Y. K. Kuan, W.-L. Lo, M. Raza, A. Tolkovsky, N.-K. Mak, R. N.-S. Wong and R. Keynes, Neurosci., 2007, 148, 915 CrossRef CAS PubMed.
- S. F. Kouam, S. N. Khan, K. Krohn, B. T. Ngadjui, D. G. W. F. Kapche, D. B. Yapna, S. Zareem, A. M. Y. Moustafa and M. I. Choudhary, J. Nat. Prod., 2006, 69, 229 CrossRef CAS PubMed.
- M. Gallorini, S. Carradori, D. I. S. P. Resende, L. Saso, A. Ricci, A. Palmeira, A. Cataldi, M. Pinto and E. Sousa, Int. J. Mol. Sci., 2022, 23, 13319 CrossRef CAS PubMed.
- C. Tantapakul, W. Maneerat, T. Sripisut, T. Ritthiwigrom, R. J. Andersen, P. Cheng, S. Cheenpracha, A. Raksat and S. Laphookhieo, J. Agric. Food Chem., 2016, 64, 8755 CrossRef CAS PubMed.
- N. Tocci, G. Simonetti, F. Diodata D'Auria, S. Panella, A. T. Palamara, A. Valletta and G. Pasqua, Appl. Microbiol. Biotechnol., 2011, 91, 977 CrossRef CAS PubMed.
- Y. Fromentin, N. Gaboriaud-Kolar, B. N. Lenta, J. D. Wansi, D. Buisson, E. Mouray, P. Grellier, P. M. Loiseau, M.-C. Lallemand and S. Michel, Eur. J. Med. Chem., 2013, 65, 284 CrossRef CAS PubMed.
- R. A. Finnegan and J. K. Patel, J. Chem. Soc., Perkin Trans. 1, 1972, 1896 RSC.
- A. Aubreville, Flore Forestière Soudano-Guinéenne, A.O.F.-Cameroun-A.E.F. Société D’éditions Géographiques, Maritimes et Colonials, Paris, 1950, pp. 1597–1601 Search PubMed.
- H. D. Locksley and 1. G. Murray, J. Chem. Soc. C, 1969, 1567 RSC.
- H. D. Locksley, I. Moore and F. Scheinmann, J. Chem. Soc. C, 1966, 430 RSC.
- H. D. Locksley, 1. Moore and F. Scheinmann, J. Chem. Soc. C, 1966, 2186 RSC.
- H. D. Locksley, I. Moore and F. J. Scheinmann, J. Chem. Soc. C, 1966, 2265 RSC.
- H. D. Locksley, I. Moore and F. Scheinmann, Tetrahedron, 1967, 23, 2229 CrossRef CAS.
- H. D. Locksley and I. G. Murray, J. Chem. Soc. C, 1971, 1332 RSC.
- K. R. Gustafson, J. W. Blunt, M. H. G. Munro, R. W. Fuller, T. C. McKee, J. H. Cardellina, J. B. McMahon, G. M. Cragg and M. R. Boyd, Tetrahedron, 1992, 48, 10093 CrossRef CAS.
- R. W. Fuller, J. W. Blunt, J. L. Boswell, J. H. Cardellina and M. R. Boyd, J. Nat. Prod., 1999, 62, 130 CrossRef CAS PubMed.
- L. A. Mitcher, Isolation, Separation and Purification of Antibiotics, ed. G. Weinstein and G. Wagman, Elsevier Science Publishers, Amsterdam, 1977, p. 463 Search PubMed.
- V. Peres, T. J. Nagem and F. F. de Olivera, Phytochem., 2000, 55, 683 CrossRef CAS PubMed.
- P. W. Westerman, S. P. Gunasekera, M. Uvais, S. Sultanbawa and R. Kazlauskas, Org. Magn. Reson., 1977, 9, 631 CrossRef CAS.
- L. Kato, C. M. A. de Oliveira, I. Vencato and C. Lauriucci, J. Chem. Crystallogr., 2005, 35, 23 CrossRef CAS.
- N. Khamto, K. Utama, S. Tateing, P. Sangthong, P. Rithchumpon, N. Cheechana, A. Saiai, N. Semakul, W. Punyodom and P. Meepowpan, J. Chem. Inf. Model., 2023, 63, 2104 CrossRef CAS PubMed.
- O. Otłowska, M. Ślebioda, M. Wachowiakc and M. Śliwka-Kaszyńska, RSC Adv., 2015, 5, 48786 RSC.
- C. M. de Fonjaudran, A. Acocella, G. Accorsi, D. Tamburini, G. Verri, A. Rava, S. Whittaker, F. Zerbetto and D. Saunders, Dyes Pigm., 2017, 144, 234 CrossRef.
- M.-C. Song, F. Nigussie, T.-S. Jeong, C.-Y. Lee, F. Regassa, T. Markos and N.-I. Baek, J. Nat. Prod., 2006, 69, 853 CrossRef CAS PubMed.
- E. R. Sukandar, S. Kaennakam, K. Rassamee, T. Ersam, P. Siripong and S. Tip-pyang, J. Nat. Prod., 2019, 82, 1312 CrossRef CAS PubMed.
- S. Fuse, K. Matsumura, K. Johmoto, H. Uekusa, H. Tanaka, T. Hirose, T. Sunazuka, S. Ōmura and T. Takahashi, Chem.–Eur. J., 2016, 22, 18450 CrossRef CAS PubMed.
- J. Shen and J.-S. Yang, Chem. Pharm. Bull., 2006, 54, 126 CrossRef CAS PubMed.
- C.-N. Lin, S.-J. Liou, T.-H. Lee, Y.-C. Chuang and S.-J. Won, J. Pharm. Pharmacol., 1996, 48, 539 CrossRef CAS PubMed.
- B. K. Sharma, A. M. Shaikh and R. M. Kamble, J. Chem. Sci., 2015, 127, 2063 CrossRef CAS.
- N. S. Baer, A. Joel, R. L. Feller and N. Indictor, in Artists' Pigments: a Handbook of Their History and Characteristics, ed. R. L. Feller, National Gallery of Art, Washington, 1986, pp. 17–36 Search PubMed.
- J. C. Roberts, Chem. Rev., 1961, 61, 591 CrossRef CAS.
- O. A. Denisova-Dyatlova and V. I. Glyzin, Russ. Chem. Rev., 1982, 51, 1007 CrossRef.
- M. E. Perel'son, Yu. N. Sheinker and L. A. Savina, The Spectra and Structure of Coumarins, Chromones, and Xanthones, Meditsina, Moscow, 1975 Search PubMed.
- I. Carpenter, H. D. Locksley and F. Scheinmann, Phytochemistry, 1969, 8, 2013 CrossRef CAS.
- C. Neuberg and W. Z. Neimann, Biol. Chem., 1905, 44, 114 CrossRef CAS.
- A. Cosentino, T. McArdle, U. Andrejczuk, D. Savino, J. Lipscher, F. Maulana, M. Douma and S. E. Smith, Pigments through the Ages: Indian Yellow, https://www.webexhibits.org/pigments/indiv/history/indianyellow.html, accessed Sep 5, 2024 Search PubMed.
- A. Pollard, Thorpe's Dictionary of Applied Chemistry, ed. M. A. Whiteley, Longmans, Green and Company, London, 4th edn, 1946, vol. VII, p. 502 Search PubMed.
- D. J. Ross, New Zealand J. Sic. Technol. B, 1950, 32(3), 39 (Chem. Abstracts, 1952, 46, 4539) Search PubMed.
- S. R. Dalal, S. Seathne and R. C. Shah, J. Indian Chem. Soc., 1953, 30, 457 (Chem. Abstracts, 1955, 49, 1656) CAS.
- W. J. McMaster, A. I. Scott and S. Trippett, J. Chem. Soc., 1960, 4628 RSC.
- F. E. King, T. J. King and L. C. Manning, J. Chem. Soc., 1957, 563 RSC.
- J. H. Townsend, Stud. Conserv., 1993, 38, 231 CAS.
- M. C. Beach, Bundi Fort: a Rajput World, The Marg Foundation, Mumbai, India, 2016 Search PubMed.
- J. Stenhouse, London Edinburgh Philos. Mag. J. Sci., 1844, 25, 32 Search PubMed.
- V. Finlay, Colour: Travels through the Paintbox, London: Sceptre, London, 2002 Search PubMed.
- J. F. L. Merimee, The Art of Painting in Oil and in Fresco Being a History of the Various Processes and Materials Employed, from its Discovery, by Hubert and John Van Eyck, to the Present Time, London: Whittaker & Co; London, 1839 Search PubMed.
- N. E. Artz and E. M. Osman, Biochemistry of Glucuronic Acid, Academic Press Inc., New York, 1950 Search PubMed.
- R. Nietzki, A. Collin and W. Richardson, Chemistry of the Organic Dyestuffs, London: Gurney & Jackson, London, 1892 Search PubMed.
- S. Saxena and A. S. M. Raja, Natural Dyes: Sources, Chemistry, Application and Sustainability Issues, in, Roadmap to Sustainable Textiles and Clothing. Textile Science and Clothing Technology, ed. S. Muthu, Springer, Singapore, 2014 Search PubMed.
- P. K. Grover, G. D. Shah and R. C. Shah, Chem. Ind., 1955, 62 CAS.
- P. K. Grover, G. D. Shah and R. C. Shah, J. Chem. Soc., 1955, 3982 RSC.
- F. Ullmann and L. Panchaud, Adv. Cycloaddit., 1906, 350, 108 Search PubMed.
- H. Nishikawa and R. J. Robinson, J. Chem. Soc., Trans., 1922, 121, 839 RSC.
- K. Hoesch, Ber., 1915, 48, 1122 CrossRef CAS.
- J. Houben, ibid., 1926, 59, 2878 Search PubMed.
- C. V. Rao and T. R. Seshadri, Proc. Indian. Acad. Sci. A, 1953, 37, 710 (Chem. Abstracts, 1954, 48, 10017) CrossRef.
- A. J. Birch, J. Baldas, J. R. Hlubucek, T. J. Simpson and P. W. Westerman, J. Chem. Soc., Perkin Trans. 1, 1976, 898 RSC; K. Janssens, G. Van der Snickt, F. Vanmeert, S. Legrand, G. Nuyts, M. Alfeld, L. Monico, W. Anaf, W. De Nolf, M. Vermeulen, J. Verbeeck and K. De Wael, Non-Invasive and Non-Destructive Examination of Artistic Pigments, Paints, and Paintings by Means of X-Ray Methods, in Analytical Chemistry for Cultural Heritage. Topics in Current Chemistry Collections, ed. Mazzeo, R., Springer, Cham, 2017 Search PubMed.
- J. L. Mass and A. N. Shugar, Handheld XRF for Art and Archaeology, Leuven University Press, Leuven, Belgium, 2012 Search PubMed.
- M. Manfredi, E. Barberis, A. Rava, E. Robotti, F. Gosetti and E. Marengo, Anal. Methods, 2015, 7, 2313 RSC.
- B. Brunetti, C. Miliani, F. Rosi, B. Doherty, L. Monico, A. Romani and A. Sgamellotti, Top. Curr. Chem., 2016, 374, 1 CrossRef CAS PubMed.
- S. Whittaker, Photo-induced fluorescence imaging to characterise and map organic colorants, MA thesis, Courtauld Institute of Art (Conservation of Wall Painting Department), 2013.
- Y. Hatsuda and S. Kuyama, S. J. Agr. Chem. Soc. Japan, 1954, 28, 989 (Chem. Abstracts, 1956, 50) CAS.
- E. Isacco and J. Darrah, Artibus Asiae, 1993, 53, 470 CrossRef.
- B. W. Singer, R. Smith and D. J. Gardiner, Non-destructive analysis of pigments in Indian miniature paintings including the novel use of a diamond-ATR-FTIR technique. in 8th International Conference on “Non-destructive Investigations and Microanalysis for the Diagnostics and Conservation of the Cultural and Environmental Heritage, ed. Parisi C., Buzzanca G. and Paradisi A., Brescia: Associazione Italiana Prove non Distruttive Monitoraggio Diagnostica, Lecce, Italy, 2005 Search PubMed.
- G. Verri, Anal. Bioanal. Chem., 2009, 394, 1011 CrossRef CAS PubMed.
- G. Accorsi, G. Verri, A. Acocella, F. Zerbetto, G. Lerario, G. Gigli, D. Saunders and R. Billinge, Chem. Commun., 2014, 50, 15297 RSC.
- A. Daveri, M. Vagnini, F. Nucera, M. Azzarelli, A. Romani and C. Clementi, Microchem. J., 2016, 125, 130 CrossRef CAS.
- S. Bellei, A. Nevin, A. Cesaratto, V. Capogrosso, H. Vezin, C. Tokarski, G. Valentini and D. Comelli, Anal. Chem., 2015, 87, 6049 CrossRef CAS PubMed.
- C. Clementi, F. Rosi, A. Romani, R. Vivani, B. G. Brunetti and C. Miliani, Appl. Spectrosc., 2012, 66, 1233 CrossRef CAS PubMed.
- D. Comelli, A. Nevin, A. Brambilla, I. Osticioli, G. Valentini, L. Toniolo, M. Fratelli and R. Cubeddu, Appl. Phys. A, 2012, 106, 25 CrossRef CAS.
- D. Comelli, V. Capogrosso, C. Orsenigo and A. Nevin, Heritage Sci., 2016, 4, 21 CrossRef.
- G. Accorsi, G. Verri, M. Bolognesi, N. Armaroli, C. Clementi, C. Milianid and A. Romanic, Chem. Commun., 2009, 3392 RSC.
- M. Thoury, J. K. Delaney, E. R. de la Rie, M. Palmer, K. Morales and J. Krueger, Appl. Spectrosc., 2011, 65, 939 CrossRef CAS PubMed.
- A. Cesaratto, C. D'Andrea, A. Nevin, G. Valentini, F. Tassone, R. Alberti, T. Frizzid and D. Comellia, Anal. Methods, 2014, 6, 130 RSC.
- F. Rosi, C. Grazia, F. Gabrieli, A. Romani, M. Paolantoni, R. Vivani, B. G. Brunetti, P. Colomban and C. Miliani, Microchem. J., 2016, 124, 856 CrossRef CAS.
- D. L. A. de Faria, H. G. M. Edwards, V. Careaga, N. Walt and M. S. Maier, Forensic Sci. Int., 2017, 271, 1 CrossRef CAS PubMed.
- G. N. Patel and K. N. Trivedi, Synth. Commun., 1989, 19, 1641 CrossRef CAS.
- A. J. Quillinan and F. Scheinmann, J. Chem. Soc., Perkin Trans. 1, 1973, 1239 Search PubMed.
- G. Guillaumet, M. Hretani and G. Coudert, Tetrahedron Lett., 1988, 29, 475 CrossRef CAS.
- B. M. Trost and M. G. Saulnier, Tetrahedron Lett., 1985, 26, 123 CrossRef CAS.
- E. Gonzales-Cantalapiedra, M. Ruiz, B. Gómez-Lor, B. Alonso, D. García-Cuadrado, D. J. Cárdenas and A. M. Echavarren, Eur. J. Org. Chem., 2005, 2005, 4127 CrossRef.
- M. B. Andrus, E. J. Hicken and J. C. Stephens, Org. Lett., 2004, 6, 2289 CrossRef CAS PubMed.
- G. N. Bollenback, W. L. Long, D. G. Benjamin and J. A. Lindquist, J. Am. Chem. Soc., 1955, 77, 3310 CrossRef CAS.
- C.-H. Liu, H. Liu, X.-Y. Han, B. Wu, B.-H. Zhong and Z.-H. Gong, Synth. Commun., 2005, 35, 701 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06475e |
| This journal is © The Royal Society of Chemistry 2024 |