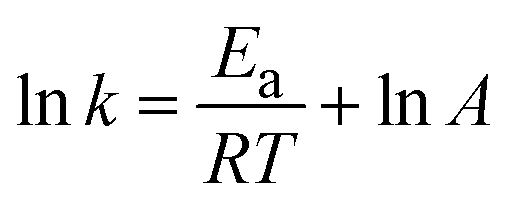DOI:
10.1039/D4RA05455E
(Paper)
RSC Adv., 2024,
14, 39511-39522
A facile approach for the synthesis of solketal, a fuel additive, from biowaste glycerol using transition metal-based solid acid catalysts†
Received
27th July 2024
, Accepted 7th October 2024
First published on 16th December 2024
Abstract
The rise in global biodiesel production leads to the surplus of glycerol as a byproduct, necessitating its industrial application in various chemicals to sustain the biodiesel industry economically. Solketal is one of the value-added chemicals in the biodiesel industry with numerous applications in the food, polymer, and chemical industries and as a fuel additive. Nowadays, researchers mainly focus on synthesizing solketal using biowaste glycerol obtained from the biodiesel industry due to its immense applications. In this work, the experimental data demonstrated that the incorporation of ZnAl2O4 plays a key role in establishing the surface sulfate groups and structural composites of the solid acid catalysts. Highly efficient heterogeneous acid SO42−/ZnAl2O4–ZrO2 and SO42−/ZnAl2O4–TiO2 catalysts were synthesized and applied for solketal synthesis. It was found that SZZ proved to be highly efficient and active towards 99.3% glycerol conversion with 98% solketal yield. In contrast, SZT showed 83% glycerol conversion with 81% solketal yield at optimized reaction conditions. SZZ showed excellent catalytic activity compared to SZT due to its better reusability, higher acidity, and good structural stability. Green metric parameters, like E-factor, PMI, and AE were studied, and it was found that SZZ is more efficient and the acetalization reaction of glycerol is environmentally friendly in nature.
1 Introduction
The continuous surge in population, urbanization, and industrialization over the past few decades has significantly hiked the energy demand by 2.3% annually. These rising demands lead to the rising consumption of fossil fuel reserves and cause environmental pollutions by increasing the emissions of greenhouse gases that pose a threat to the health of human beings and their surroundings.1 The research fraternity are trying to find environmentally benign and renewable alternative fuels to minimize reliance on fossil fuels. Biodiesel is a renewable alternative energy owing to its excellent fuel properties, including biodegradability, non-toxicity, and carbon neutrality, and is produced by a trans-esterification process.2,3 About 10% of crude glycerol byproduct is produced as waste in the biodiesel industry. The growth in the biodiesel industry is directly related to glycerol production as a waste that needs to be either removed or repurposed.4 The surplus crude glycerol raises concerns regarding waste management, leading to environmental challenges. The abundance of glycerol showed assorted applications in several fields, including food additives, pharmaceuticals, plastics, polymers, and cosmetics. However, the current utilization cannot swallow up the glut of glycerol.5
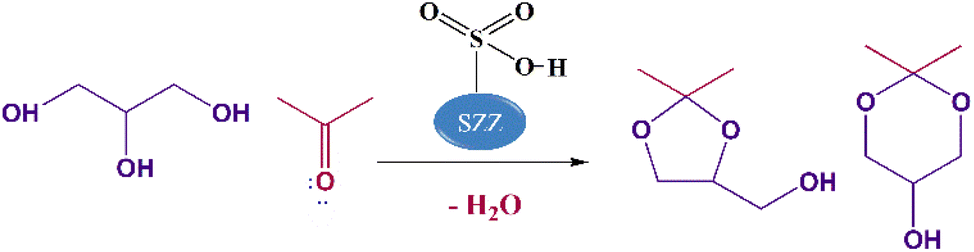
To ensure the profitable utilization of excess glycerol produced during the transesterification process and improve the sustainability of biodiesel production, an efficient catalytic process must be employed to valorize surplus glycerol. This involves the conversion of this crucial feedstock into various value-added compounds. These include hydrogenolysis for the production of propylene glycol,6 oxidation for the production of dihydroxyacetone,7 reforming for the production of syn gas,8 chlorination for the generation of epichlorohydrin,9 dehydration for the formation of acrolein,10 and transesterification for the formation of glycerol carbonate.11 Among these available methods, acetalization is the most promising method that involves the condensation of glycerol with acetone to produce five-membered cyclic solketal (2,2-dimethyl-1,3-dioxolan-4-yl methanol) and six-membered acetal (2,2-dimethyl-1,3-dioxan-4-yl methanol), as shown in Scheme 1. Solketal is considered a clean fuel additive capable of enhancing the transportation fuel's cold flow properties and oxidation stability and mitigating environment-polluting gases and particulate matter emissions.12 Moreover, solketal acts as an anti-knocking agent that helps increase the octane number of fuels by 2.5 points by minimizing the gum formation, when blended with regular gasoline. In addition, solketal finds application as a green solvent in several chemical syntheses, and serves as a plasticizer, surfactant, suspending agent in pharmaceutical industries, anti-freezing agent, and flavoring agent.13,14
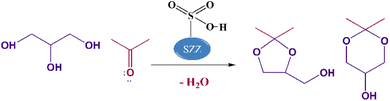 |
| | Scheme 1 Glycerol acetalization with acetone using the catalyst. | |
The acetalization of glycerol is an exothermic reaction facilitated by the acid catalyst. The yield of solketal is primarily influenced by the density and acid strength of the catalyst surface, which plays a crucial role in activating the carbonyl carbon of acetone.15 Solketal synthesis often employs both homogeneous and heterogeneous acid catalysts. Various homogeneous acid catalysts including H2SO4, p-toluene sulfonic acid, H3PO4, HCl, and SnCl2 have been employed by researchers.16–18 Homogeneous catalysts provide a remarkable advantage over heterogeneous catalysts by increasing the reaction efficiency and lowering the mass transfer resistance. Despite these benefits, some limitations are associated with catalyst recovery, waste generation during separation, and product purification from reaction effluent, cost, and corrosive nature.19 Shifting to a heterogeneous catalyst promises to enhance the long-term sustainability of the process. Researchers have focused on a heterogeneous catalyst for glycerol acetalization to address the drawback mentioned earlier. Heterogeneous acid catalysts proposed a broad spectrum, including acid clays,20 hetero-poly acids,21 ion exchange resins,22 sulfated metal–organic frameworks,23 mesoporous solid acid catalysts,24 mixed oxides,25 and beta zeolites.26 This catalyst shows remarkable catalytic activity in acidity, surface area, non-toxic behavior, facile separation from the reaction matrix, reduced natural corrosion, and less waste generation. All these characteristics collectively enhance the economic viability and attractiveness of the catalyst.
A SO42−/MxOy heterogeneous acid catalyst has been acknowledged as the most promising green catalyst, which showed better catalytic activity, good selectivity, and easy separation from the reaction matrix in a glycerol acetalization reaction.27,28 The conventional SO42−/MxOy solid acid catalyst primarily comprises an active component MxOy and promoter SO42−, emphasizing the synthesis of SO42−/ZrO2 and SO42−/TiO2 systems.29 SO42−/TiO2 initially shows good catalytic activity but faces some issues regarding rapid deactivation and a short lifespan. This is primarily attributed to the deactivation of the sulfur species and carbon deposition. Consequently, synthesizing acid catalysts for liquid reactions remains a challenge, requiring a focus not only on the high catalytic activity but also on the durability and stability of the catalyst.30 Similarly, SO42−/ZrO2 has drawn attention towards its catalytic activity due to its distinctive properties including robust acidic properties, good catalytic activity, and selectivity in solketal synthesis. However, this catalyst still has some shortcomings, such as lack of stability, poor reusability, and fast deactivation due to the leaching of surface sulfur or coke deposition.31 To resolve the above-mentioned drawbacks, we tried to study the synthesis and utilization of SO42−/ZrO2–MxOy and SO42−/TiO2–MxOy solid acid composites by the incorporation of SO42−/ZrO2 and SO42−/TiO2 with additional oxides of MxOy for the acetalization of glycerol. Hence, the utilization of ZnAl2O4 spinel-type oxide as MxOy in the synthesis of SO42−/ZrO2–MxOy and SO42−/TiO2–MxOy composites increases the catalytic activity, stability, and acidic strength of modified catalysts for solketal synthesis. Modified metal oxide has attracted significantly increased attention for catalyst synthesis over the past few decades. In this study, the effect of ZnAl2O4 spinel oxide on SO42−/ZrO2 and SO42−/TiO2 was studied, and the synthesized solid acid catalysts such as SO42−/ZnAl2O4–ZrO2, SO42−/ZnAl2O4–TiO2 were applied in the acetalization reaction of glycerol. The authors also conducted a comparative analysis of synthesized catalyst's activity. To our knowledge, this is the first study to apply a catalyst for solketal synthesis. No previous research has been performed for the comparative analysis of these modified catalysts. Here, a set of heterogeneous acid catalysts based on ZnAl2O4 and transition metal oxides (Zr and Ti) were synthesized by a wetness impregnation process used in solketal production due to its robust acidic strength, high catalytic activity, good selectivity towards the five-membered ring, and easy separation. Every reaction was meticulously observed using both these catalysts and the rate of glycerol conversion was thoroughly examined. A comprehensive analysis was also performed to analyze the relationship between the acidic properties, glycerol conversion rate, product yield percentage, and selectivity. Different characterization methods including FTIR spectroscopy, TGA, BET-surface area, XRD, and XPS analysis assessed the catalytic activity of the modified catalyst. Based on the environmental approach, the overall process for solketal synthesis is sustainable and green in nature.
1.1 Experimental procedure
1.2 Materials required
Glycerol was synthesized as a byproduct by a transesterification process during biodiesel production by our lab colleague and distilled before its application in the acetalization process. Acetone (99%, SRL), Zn (NO3)2·6H2O (extra pure AR grade, SRL), ZrOCl2·8H2O (98%, Merck), Al (NO3)3·9H2O (98%, Titan Biotech Ltd.), TiO2 (99%, Merck), H2SO4 (98%, Merck), etc., were obtained from the local vendors in Varanasi, India, and used without further purification.
1.3 Catalyst synthesis
The co-precipitation and wet impregnation techniques were used in the synthesis of solid acid heterogeneous catalysts, in particular SO42−/ZnAl2O4–ZrO2 and SO42−/ZnAl2O4–TiO2, named SZZ and SZT, respectively. At first, ZnAl2O4 was synthesized by a co-precipitation process using nitrate salt as a precursor. In this procedure, a required quantity of zinc nitrate and aluminum nitrate was taken and dissolved in deionized water separately (Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al = 1:2). At 30 °C, both the solutions were mixed and stirred for 3–4 h with the addition of ammonium hydroxide solution drop by drop to regulate the solution's pH within the range of 8–10. The resulting solution mixture was agitated additionally for 5–6 h to promote selective precipitation growth. Following filtration, the reaction mixture was rinsed with distilled water three to four times to remove any remaining impurities. The solid mixture was then placed in a hot air oven at 120 °C for 8 h and subsequently calcined at 700 °C for 5 h in an air muffle furnace to obtain the desired spinel. Then, the stipulated amount of ZnAl2O4 was immersed in deionized water and added to ZrO2. In order to augment the acidic strength of the synthesized catalyst, the mixture powder was immersed in 0.5 M H2SO4 for 5–6 h at 550 rpm by wetness impregnation. Afterward, the white powder underwent heat treatment in an oven overnight at 90 °C and was then calcined at 550 °C in an air muffle furnace for intervals of 3 h with a 10 °C min−1 heating rate to obtain SO42−/ZnAl2O4–ZrO2. Finally, the calcined catalyst was ground with mortar and pestle, stored in a desiccator to prevent moisture, and further applied for the acetalization reaction. Another acetalization catalyst, like SO42−/ZnAl2O4–TiO2, was also synthesized and tested similarly, as discussed above.
:Al = 1:2). At 30 °C, both the solutions were mixed and stirred for 3–4 h with the addition of ammonium hydroxide solution drop by drop to regulate the solution's pH within the range of 8–10. The resulting solution mixture was agitated additionally for 5–6 h to promote selective precipitation growth. Following filtration, the reaction mixture was rinsed with distilled water three to four times to remove any remaining impurities. The solid mixture was then placed in a hot air oven at 120 °C for 8 h and subsequently calcined at 700 °C for 5 h in an air muffle furnace to obtain the desired spinel. Then, the stipulated amount of ZnAl2O4 was immersed in deionized water and added to ZrO2. In order to augment the acidic strength of the synthesized catalyst, the mixture powder was immersed in 0.5 M H2SO4 for 5–6 h at 550 rpm by wetness impregnation. Afterward, the white powder underwent heat treatment in an oven overnight at 90 °C and was then calcined at 550 °C in an air muffle furnace for intervals of 3 h with a 10 °C min−1 heating rate to obtain SO42−/ZnAl2O4–ZrO2. Finally, the calcined catalyst was ground with mortar and pestle, stored in a desiccator to prevent moisture, and further applied for the acetalization reaction. Another acetalization catalyst, like SO42−/ZnAl2O4–TiO2, was also synthesized and tested similarly, as discussed above.
1.4 Catalyst characterization
Various characterization techniques have been performed to study the designed catalyst's physiochemical characteristics. The pre-calcined catalyst underwent thermal treatment ranging from 50 °C to 900 °C at a heating rate of 10 °C min−1 using a Thermogravimetric Analyzer TGA 4000 instrument in the presence of nitrogen gas with 20.0 ml min−1. The crystalline nature of the synthesized catalyst was examined using a Rigaku MiniFlex-300/600 Desktop X-ray Diffraction System. The diffractogram was scaled within a 2-theta range of 10°–80° with 0.02° step width at 5° min−1 scan rate. The obtained data matched with the JCPDS file number database to confirm the materials' phase. The BET surface area and BJH pore volume of the catalyst were determined by conducting N2 adsorption/desorption isotherm measurements using a BELLSORP MAX II surface area analyzer and BELCAT-II MicrotracBEL Corp. First, the catalyst underwent degassing using nitrogen gas for 18 h at 300 °C to remove any adsorbed impurities before the analysis. The stretching and bending vibrations of the catalyst sample surface were fetched using a Nicolet iS5 (Thermo Electron Scientific Instrument) mounted with a KBr crystal. The spectra were recorded within the wavenumber range of 4000–400 cm−1. The surface topology of the modified catalyst was executed using an EVO-SEM MA15/18, which was combined with an EDS (energy dispersive spectroscopy) unit using a 51N1000-EDS detector for elemental analysis by energy-dispersive X-ray spectroscopy (EDS) system. The manifestation of elements and their respective oxidation states was done by X-ray photoelectron spectrometer (XPS) analysis. The spectra were recorded using a K-α source of Thermo Fischer Scientific, with C 1s (i.e., 284.8 eV binding energy) as a reference peak for calibrating the other elements' binding energy of the catalyst. The surface acidity of the modified catalyst was analyzed by an N-butylamine titration method using neutral red as an indicator. First, 50 mg of catalyst was immersed in 20 ml of 0.05 M solution of N-butylamine in toluene and stirred at 30 °C for 24 h. Following this, the solid catalyst was separated from the reaction mixture via centrifugation, and the solution was titrated with trichloroacetic acid in a toluene solution by adding 2–3 drops of indicator.
1.5 Evaluation of the catalyst in solketal synthesis
The acetalization reaction between glycerol and acetone was executed in a 250 ml round-bottom flask associated with a reflux condenser and a mechanical stirrer (Tarson digital spinout). The reactor setup was immersed in a silica oil bath for continuous heating. Initially, 2.5 mmol and 25 mmol of glycerol and acetone were introduced along with a catalyst loading of 3 wt% (relative to glycerol) at the optimum reaction temperature of 70 °C for 120 min. Following the reaction completion, the resulting mixture was cooled and separated from the solid catalyst by centrifugation. The quantitative validation of the synthesized solketal was studied by gas chromatography-mass spectrometry (GCMS). The percentage conversion of glycerol, selectivity, and yield of solketal were determined by providing the following equation. Additionally, the verification of solketal synthesis was carried out by 1H and 13C NMR spectroscopy as well as FTIR spectroscopy.32,33| | |
% of glycerol conversion = (initial moles of glycerol − final moles of glycerol)/(initial moles of glycerol taken) × 100
| (1) |
| | |
% of selectivity = (no. of moles of solketal produced)/(total no. of moles of all products) × 100
| (2) |
| | |
% of solketal yield = (% of glycerol conversion × % of solketal selectivity)/100
| (3) |
| | |
Turnover frequency = (conversion of glycerol (%))/(catalyst wt% × molar mass of glycerol × reaction time)
| (4) |
The turnover frequencies for the SO42−/ZnAl2O4–ZrO2 and SO42−/ZnAl2O4–TiO2 catalysts were observed to be 0.19 mol g−1 h−1 and 0.17 mol g−1 h−1, respectively. The synthesized catalysts were applied for solketal synthesis, and conversion and selectivity were obtained, as shown in Table 1.
Table 1 Screening of catalysts for the synthesis of solketal with time
| Catalyst |
Conversion (%) |
Solketal selectivity (%) |
TOF (mol g−1 h−1) |
| SZZ |
99.3 |
99 |
0.19 |
| SZT |
83 |
96 |
0.17 |
2 Results and discussion
2.1 Characterizations of the synthesized catalyst
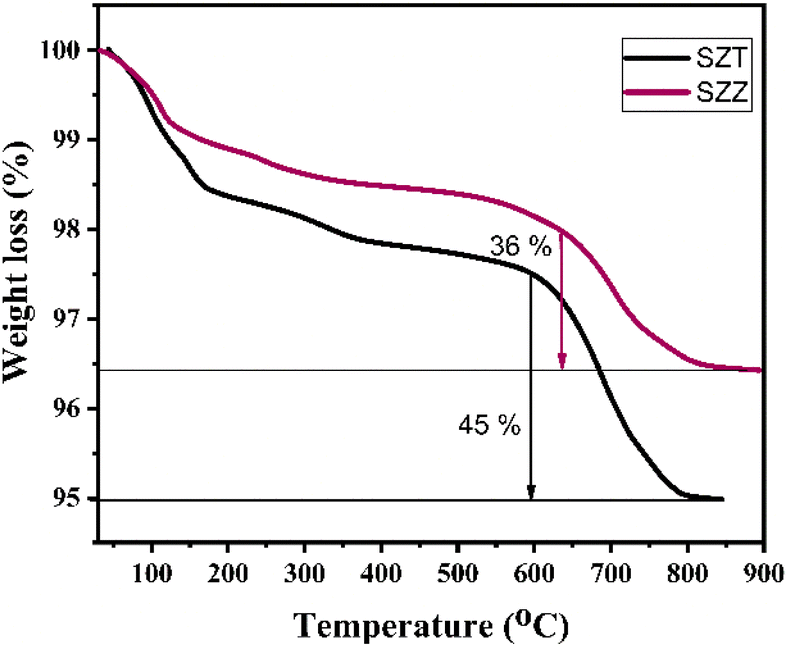
2.1.1 TGA studies. The temperature disintegration versus mass loss of the uncalcined catalysts SZZ and SZT was studied by the thermogravimetric analysis in the presence of a nitrogen atmosphere. In Fig. 1, three distinct phases of mass loss are evident as the temperature increases for the SZT catalyst. The initial phase of weight deduction, observed between 100 and 200 °C, may be due to the dehydration of the surface water molecule adsorbed by the surface of the catalyst. The 2nd phase was observed around 200–550 °C, possibly due to the evaporation of the crystalline water molecules or any organic impurities. The 3rd stage of weight reduction occurred within the temperature range of 500 °C to 850 °C due to the decomposition of the surface sulfate group. Therefore, the SZT catalyst showed 45% mass weight loss above 550 °C. Similarly, the TGA curve of the SZZ catalyst is shown here. In the SZZ case, the initial phase of weight loss for the catalyst, occurring below 200 °C, could be assigned to removing physically bound water molecules. The 2nd weight loss observed between 200 °C and 550 °C is attributed to the release of structural water molecules. The 3rd stage of mass reduction was ascertained at a higher temperature range between 500 and 900 °C; gradual decomposition of the sulfate group from the catalyst surface was observed.34,35 Based on the TGA graph, the weight loss of the sample was estimated to be 36%. The weight loss in the SZT catalyst above 550 °C was higher, which indicates that the catalyst is highly unstable above that temperature, whereas in the SZZ catalyst, the weight loss was estimated to be less (i.e., 36%) than that of the SZT catalyst. This happens because the SZZ catalyst surface was found to be more stable and decomposed to a less extent above that temperature.
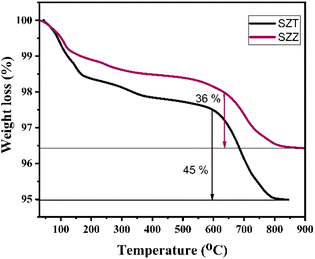 |
| | Fig. 1 TGA graph of the synthesized uncalcined SZZ and SZT catalysts. | |
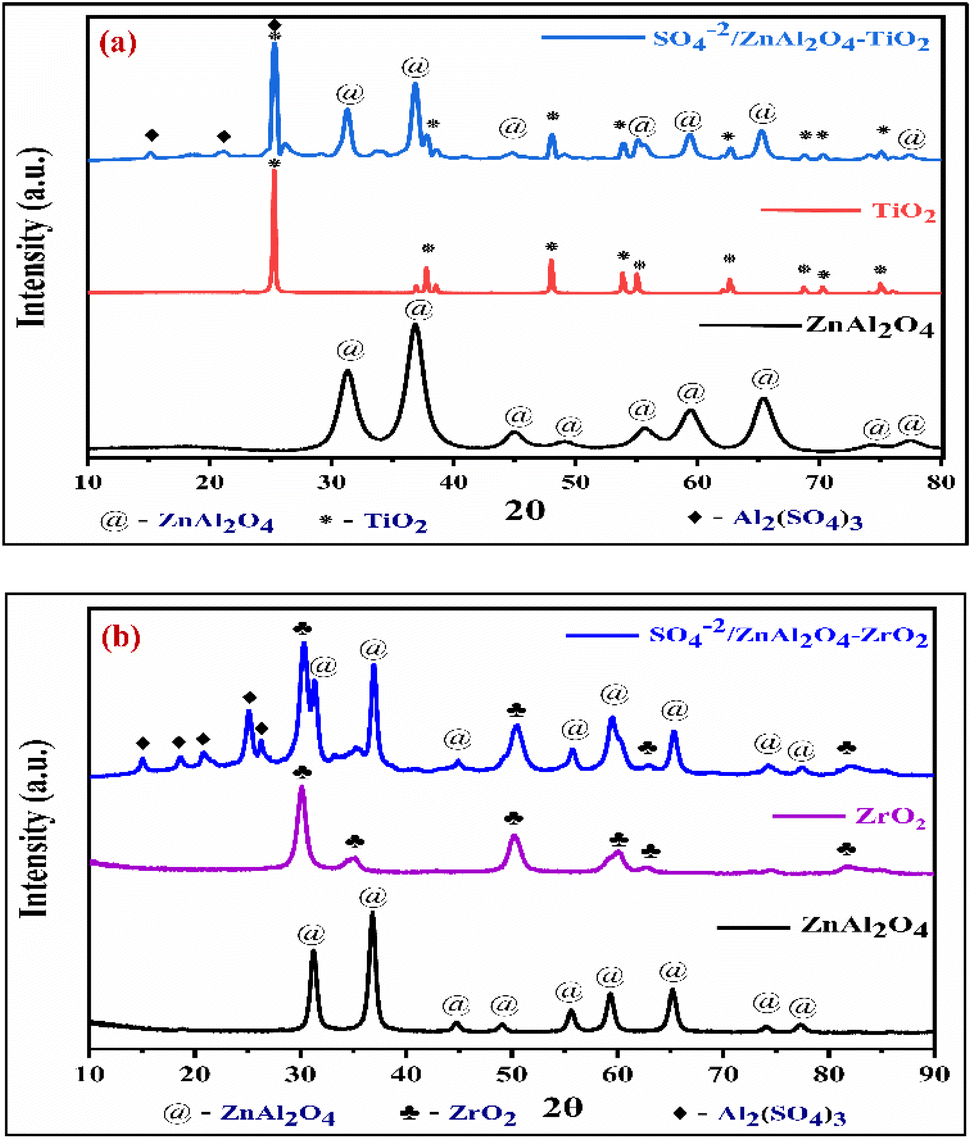
2.1.2 Powder X-ray diffraction studies. The XRD technique studied the purity of the phase, crystalline structure, and orientation of the designed catalyst. Fig. 2(a) shows the XRD graph of the SZT catalyst. The characteristic diffraction peaks appeared at 2θ values equal to 31.2°, 36.8°, 44.9°, 49.0°, 55.5°, 59.4°, 65.2°, 74.2°, and 77.3° corresponding to the miller indices values (220), (311), (400), (331), (422), (511), (440), (620), and (533) respectively, showing that the orientation planes of the cubic phase of ZnAl2O4 matched with the JCPDS card no. 05-0669. Other prominent peaks appear at 25.3°, 37.8°, 47.9°, 53.7°, 54.9°, 62.7°, 68.8°, 70.3°, and 75.1° with Miller indices (101), (004), (200), (105), (211), (204), (116), (220), and (215), respectively, all of which correspond to the anatase phase of TiO2 matched with the JCPDS no-65-5714. Due to sulphonation, some minor Al2(SO4)3 peaks are also observed at 2θ values of 15.1°, 21.1°, and 25.2°, matched with the JCPDS no-22-0021. This confirms the successful impregnation of the sulfate group on the SZT catalyst. Using Scherrer's equation, the crystalline size of the SZT catalyst was determined to be 23.03 nm:| | |
D = (0.9λ)/βcosθ
| (5) |
where D denotes the average crystallite size, β corresponds to the line broadening at FWHM, θ is the diffraction Bragg angle, and λ refers to the wavelength of the X-ray. Similarly, in Fig. 2(b), the XRD pattern of the SZZ catalyst is depicted. The predominant diffraction peaks of tetragonal ZrO2 were observed at 2θ of 30.1°, 35.2°, 50.3°, 60.2°, 62.7°, and 74.5° consisting of the hkl planes of (101), (110), (112), (211), (202), and (220) respectively matched with the JCPDS card no-88-1007. Some minor peaks of Al2(SO4)3 also appeared on the surface of the SZZ catalyst at 15.0°, 20.8°, and 25.1°, corresponding to the miller indices (012), (104), and (113) (JCPDS no-22-0021). The crystalline size of the SZZ catalyst was observed to be 46.29 nm using the above-mentioned Scherrer's equation.36 The diffraction peaks of SZZ catalysts are sharper and more intense than SZT catalysts, indicating that SZZ catalysts are more crystalline. The higher crystallinity might cause higher glycerol conversion to solketal.
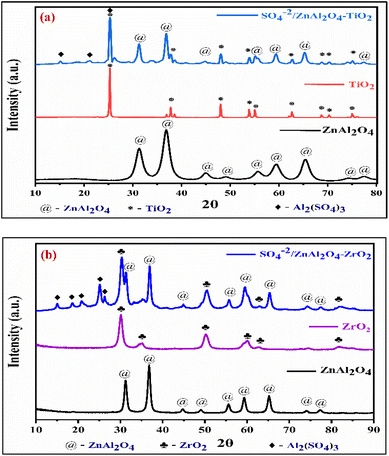 |
| | Fig. 2 XRD patterns of (a) SZT and (b) SZZ catalysts. | |
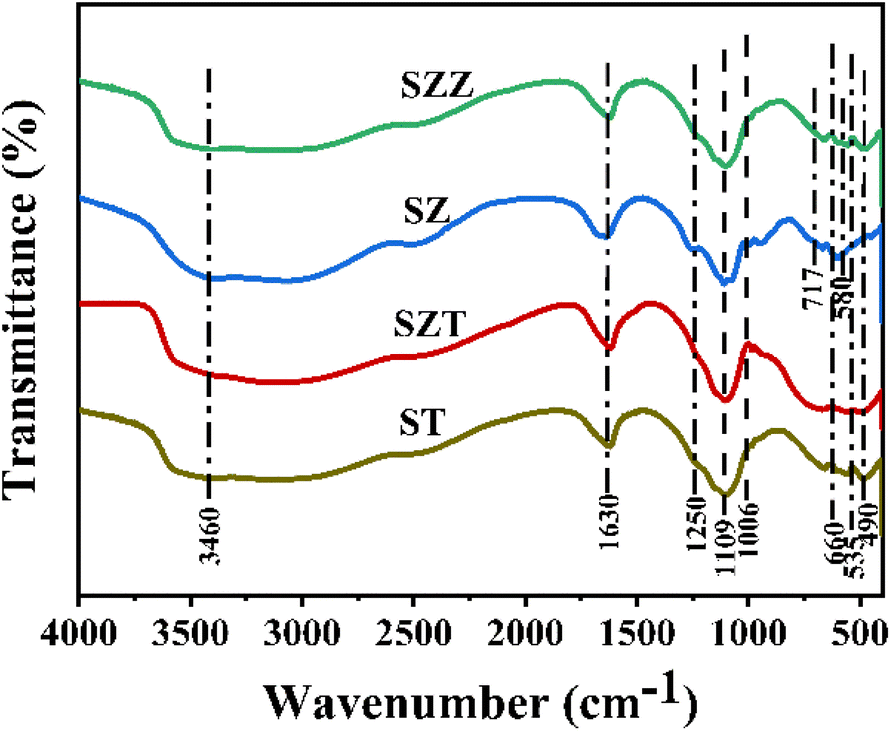
2.1.3 FTIR spectra studies. The existence of the functional group on the catalyst surface plays a crucial role in the glycerol conversion, which was studied by the FTIR analysis. Fig. 3 shows the FTIR spectra of the SZT and SZZ catalysts. The characteristic peaks observed between 500 cm−1 and 670 cm−1 indicate metal–oxygen and metal–oxygen–metal bonding. The low-frequency absorption band appeared at ∼490, 535, and 660 cm−1, corroborating the Zn–O–Al, Al–O, and Zn–O bonds in SZT and SZZ, respectively. The stretching vibration band of Zr–O was detected at 600 cm−1 and 725 cm−1 for SZ and SZZ catalysts.37 Similarly, the vibration mode of the Ti–O–Ti bond appeared in the range of 400–800 cm−1 in ST.38 The peaks originating at 1006 and 1102 cm−1 were depicted as the symmetric and asymmetric stretching of S–O bonds, respectively. Other peaks around 1250 cm−1 could be evident attributed to the stretching vibration of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, indicating the formation of a bidentate chelating structure between sulfate and metal ions. In addition, 1630 cm−1 and 3460 cm−1 peaks assigned the –OH bending and stretching vibrations of the adsorbed water molecule on the catalyst surface, respectively.39
O bonds, indicating the formation of a bidentate chelating structure between sulfate and metal ions. In addition, 1630 cm−1 and 3460 cm−1 peaks assigned the –OH bending and stretching vibrations of the adsorbed water molecule on the catalyst surface, respectively.39
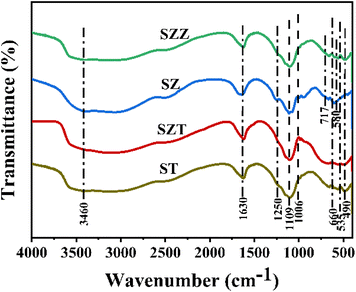 |
| | Fig. 3 FTIR spectra of ST, SZT, SZ, and SZZ catalysts. | |
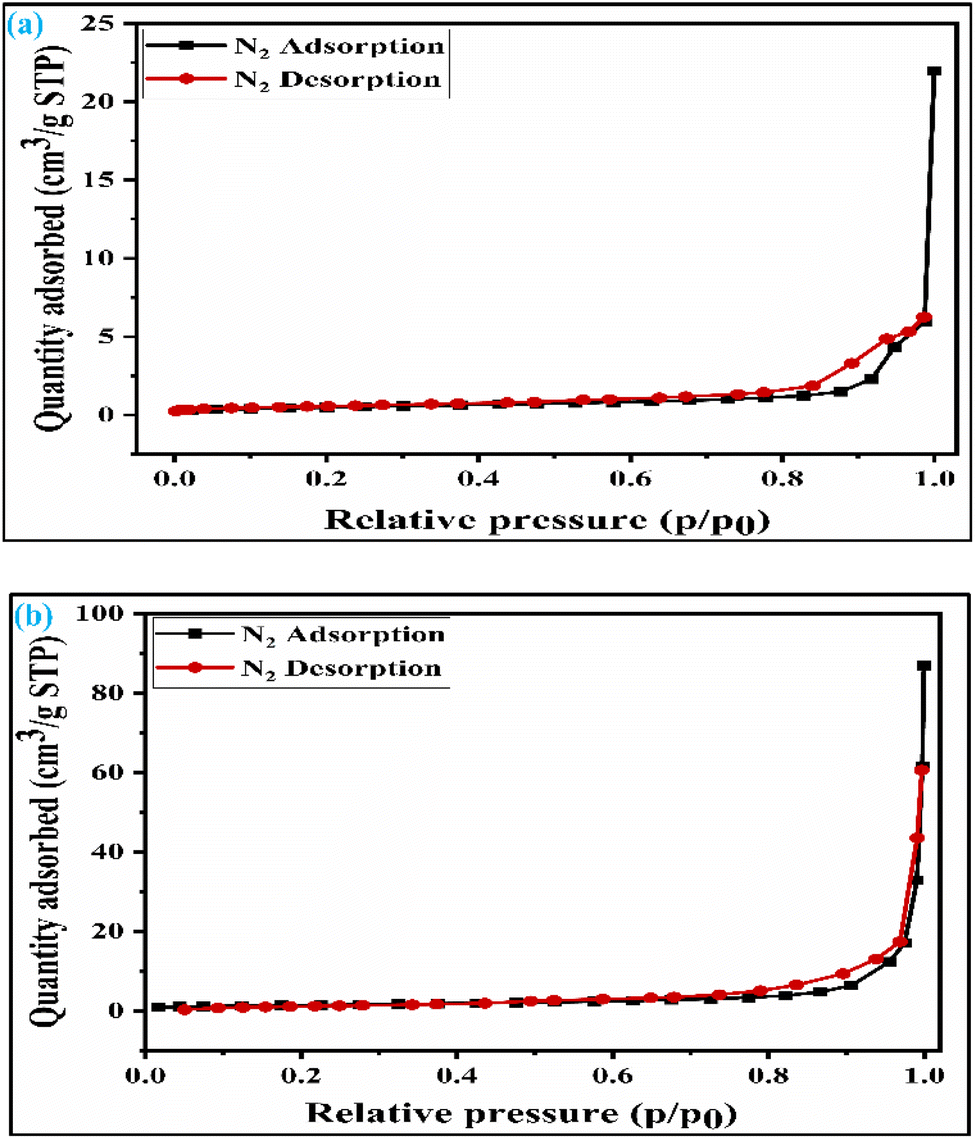
2.1.4 Studies of the surface area of the synthesized catalyst. The specific surface area of the synthesized catalyst plays a crucial role in the glycerol acetalization reaction. Acidity and surface area both affect the solketal synthesis. Nonetheless, acidity is a more prominent factor than surface area analysis. The surface area of both SZT and SZZ catalysts was tentatively quantified by BET nitrogen adsorption–desorption isotherm, and the obtained plot is illustrated in Fig. 4(a) and (b), respectively. Similarly, the pore diameter and pore volume were also elucidated by the BJH (Barrett–Joyner–Halenda) method. The surface area values, along with the pore diameter and pore volume of SZT and SZZ catalysts, are listed in Table 2. It was found that the calculated specific surface area and pore diameter of the SZT catalyst are 3.9 m2 g−1 and 31.8 nm, while those of the SZZ catalyst are 5.3 m2 g−1 and 39.4 nm, respectively. This indicates that SZZ has a higher surface area than that of SZT. Likewise, the pore diameter and pore size of SZZ were found to be larger than those of SZT. Both catalysts follow a type IV isotherm with an H1-type hysteresis loop. This signifies that the materials are mesoporous in nature (2–50 nm). The higher specific surface area of the SZZ catalyst facilitates the acetalization reaction of glycerol. It is recognized that the glycerol acetalization reaction with a catalyst is a surface adsorption phenomenon. As a result, the SZZ catalyst with a higher surface area boosted the glycerol conversion higher than the SZT catalyst.40
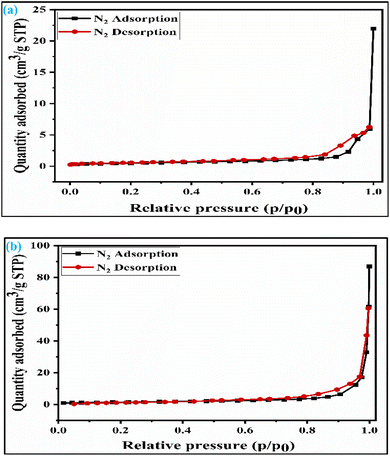 |
| | Fig. 4 BET isotherm of (a) SZT catalyst and (b) SZZ catalyst. | |
Table 2 Surface properties and acidic sites of the catalyst
| Catalyst |
Surface areaa (m2 g−1) |
Pore volumeb (cm3 g−1) |
Average pore diameterc (nm) |
Acidic sitesd (mmol g−1) |
| The specific surface area was determined using BET method. Pore volume was calculated by the amount of adsorbed nitrogen (p/p0 = 0.990). Average pore diameter was deliberated by using BJH method. Acidity was determined by N-butylamine titration. |
| SZZ |
5.3 |
0.05 |
39.4 |
2.87 |
| SZT |
3.9 |
0.01 |
31.8 |
2.45 |
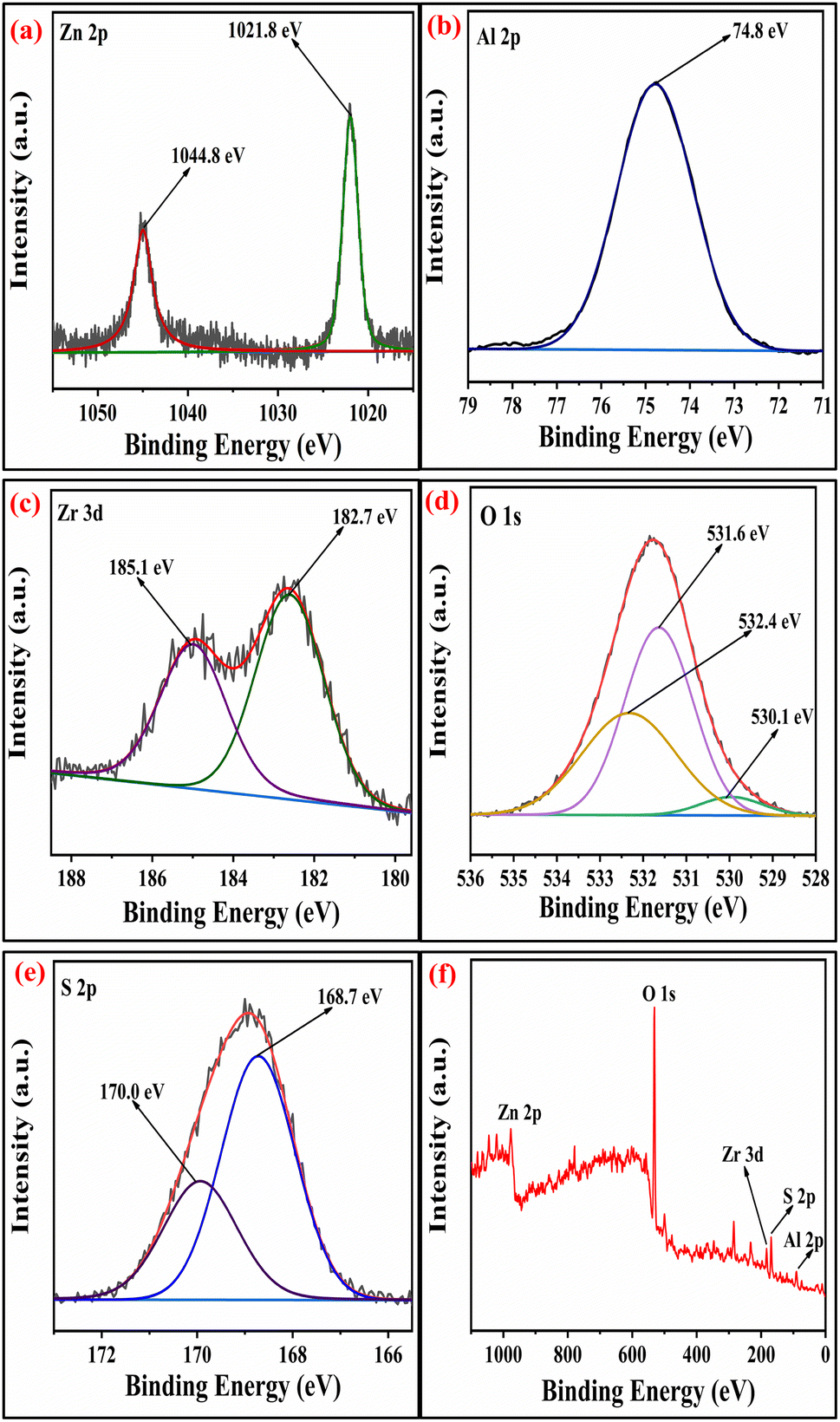
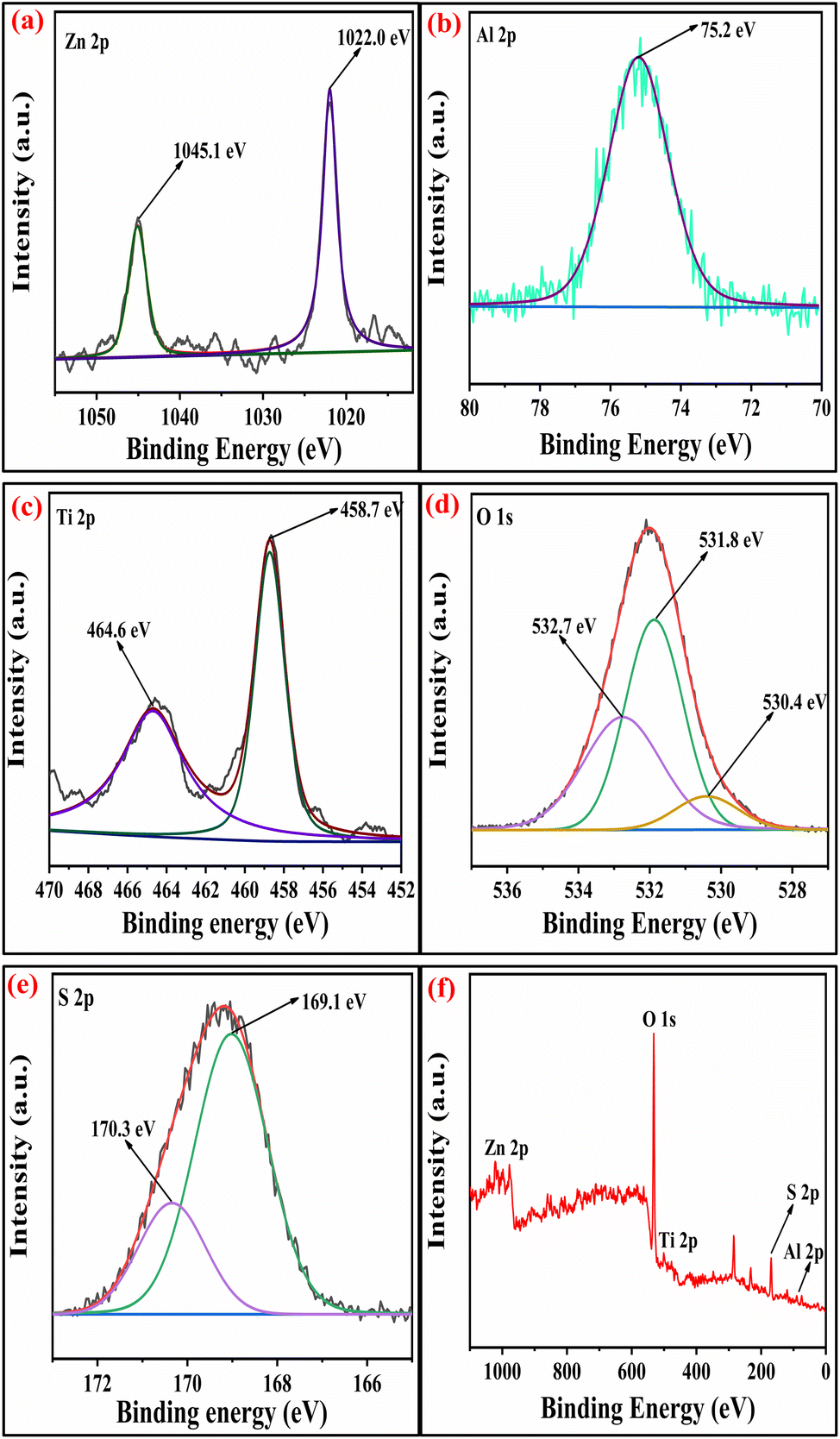
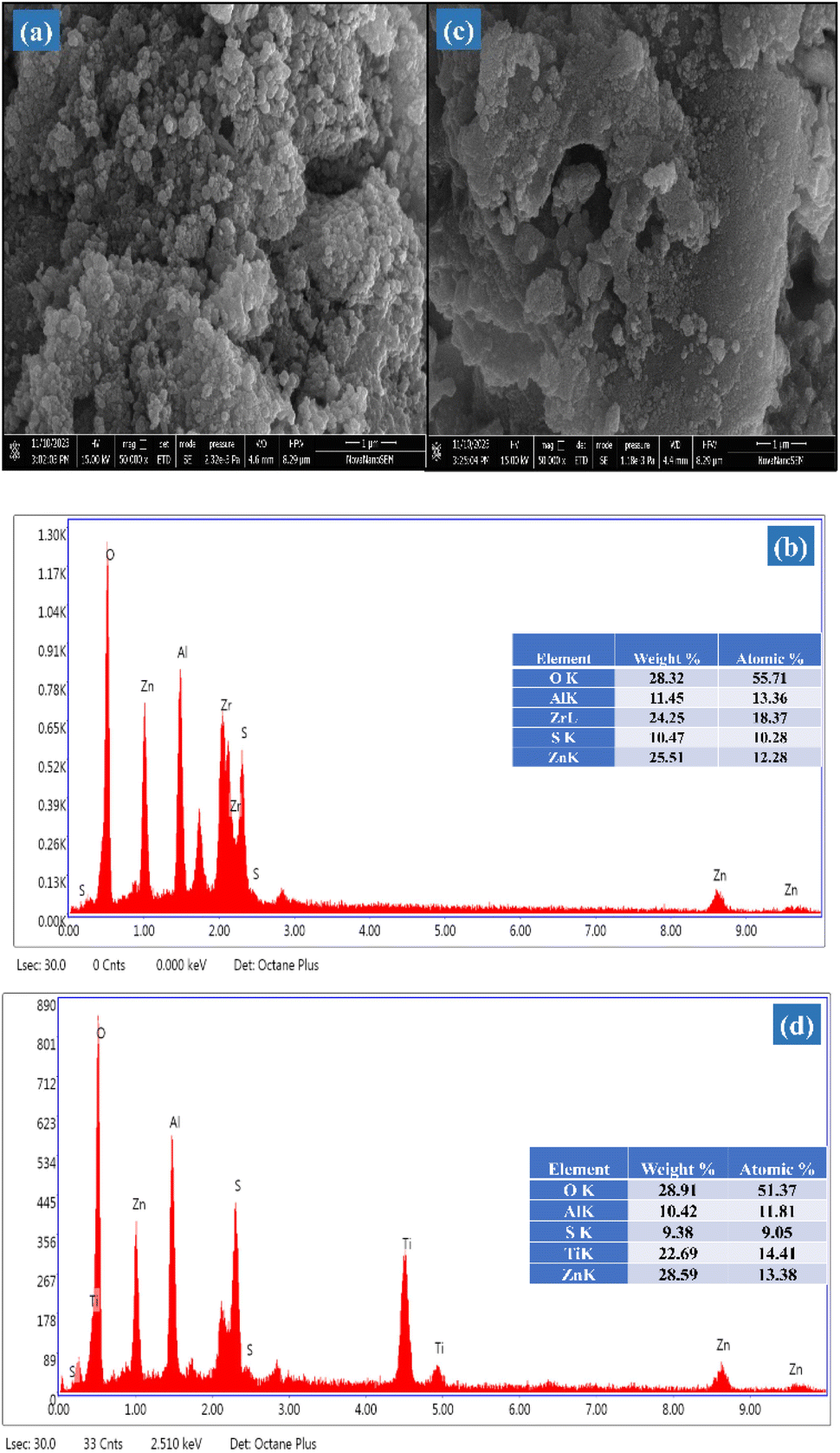
2.1.5 XPS studies. The surface analysis of the SZZ and SZT catalysts was performed by X-ray photoelectron spectroscopy (XPS). The XPS technique was employed to determine the material's oxidation state and chemical composition. Fig. 5 displays the XPS spectra of the SZZ catalyst that indicate the presence of Zn, Al, O, Zr, and S on the catalyst surface. The distinctive peaks that appeared at 183.5 eV and 185.9 eV corresponding to Zr 3d5/2 and Zr 3d3/2, respectively, corroborated that Zr is present in the (IV) oxidation state. The O 1s spectrum reveals three deconvoluted peaks corresponding to different binding energy states. The first peak appeared at 530.1 eV, which can be associated with lattice oxygen involved in metal–oxygen bonding, while the peak at 531.5 eV is assigned to the bridged oxygen of the –OH group. Moreover, the peaks observed at 532.5 eV correspond to the sulfate oxygen group. In the Zn 2p spectrum, the characteristic peak appeared at 1021.5 eV, which signifies Zn 2p3/2, while the peak at 1044.8 eV corresponds to Zn 2p1/2, which shows the existence of Zn in the +2-oxidation state, which was close to standard data. The peak appeared at 75.3 eV, which corresponds to the +3 oxidation state of Al. The S 2p spectra exhibit two distinct peaks, one appearing at 169.4 eV and the other at 170.2 eV. These peaks are attributed to the binding energies of S 2p3/2 and S 2p1/2, respectively, which were associated with the +6 oxidation state of the sulfur.41,42 The presence of Zn, Al, Ti, O, and S in the SZT catalyst is depicted in Fig. 6. The presence of aluminum in the +3 oxidation state is indicated by the peak observed at 75.2 eV in the spectra. The deconvoluted peak that appeared at a binding energy of 1045.8 eV resembles Zn 2p1/2, and 1022.7 eV corresponds to Zn 2p3/2, showing the +2-oxidation state of Zn. In the Ti spectrum, Ti 2p peaks are observed at binding energies of 459.1 eV and 464.5 eV associated with Ti 2p3/2 and Ti 2p1/2, respectively. These peaks exhibit a doublet splitting of 5.7 eV caused by the spin–orbit coupling of Ti 2p, which indicates the +4-oxidation state of titanium. In the O 1s spectrum, three characteristic peaks were observed: first at 530.4 eV corroborated to the lattice metal–oxygen bonding, second at 531.4 eV associated with the bridged oxygen of the hydroxyl group, and third at 532.3 eV linked to the sulfate oxygen group. In the S 2p spectra, the binding energies of S 2p3/2 and S 2p1/2 were obtained at 169.4 eV and 170.2 eV, assigned to the +6 oxidation state of the bidentate sulfur group.43 In general, the peak observed at lower binding energies is mentioned as the lattice oxygen [O lat], while the peak observed at higher binding energies corresponds to the adsorbed oxygen [O ads] on the catalyst surface. The comparison of the area ratios [O ads]/[O lat] allows for the evaluation of the relative quantities of two distinct oxygen species. A greater area ratio suggests a higher concentration of oxygen vacancies, whereas a smaller area ratio suggests fewer oxygen vacancies.44 In this context, the [O ads]/[O lat] area ratio for the SZZ (0.68) catalyst is higher than that of the SZT (0.65) catalyst. Consequently, SZZ exhibits a higher concentration of surface and unoccupied oxygen than those of SZT.
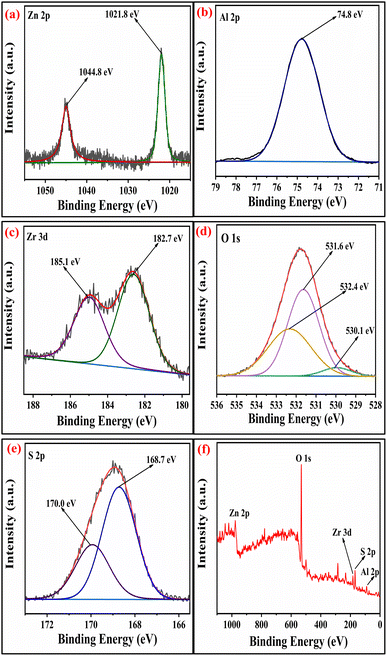 |
| | Fig. 5 XPS spectra of (a) Zn 2p, (b) Al 2p, (c) Zr 3d, (d) O 1s, (e) S 2p, and (f) survey peak of SZZ catalyst. | |
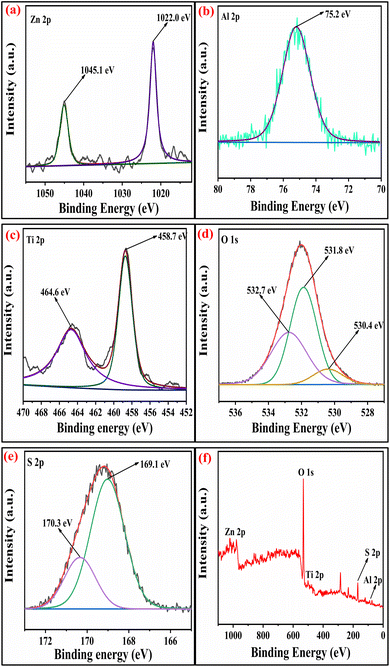 |
| | Fig. 6 XPS spectra of (a) Zn 2p, (b) Al 2p, (c) Ti 2p, (d) O 1s, (e) S 2p, and (f) survey peak of SZT catalyst. | |
2.1.6 Morphological study. The structural characteristics and surface topology of the SZZ and SZT catalysts were viewed using SEM (scanning electron microscope) techniques. Fig. 7(a) depicts the morphology of the SZZ catalyst, showing a homogeneous distribution of both grains in variable size. The bulk grain particles of ZrO2 are finely dispersed across the spherical surface of ZnAl2O4. This outcome clearly showed that both ZnAl2O4 and ZrO2 were successfully composited on the SO42−/ZnAl2O4–ZrO2. ZrO2 helps prevent the agglomeration of ZnAl2O4 particles and facilitates the dispersion on SZZ solid acid catalyst, improving the surface area that is highly suitable for acetalization reactions. The atomic ratio of the catalyst was cross-verified by EDX (energy-dispersive X-ray) spectroscopy. The EDX histogram of SZZ Fig. 7(b) reveals the existence of elements such as Zn, O, Zr, Al, and S on the catalyst surface, as indicated by their atomic and weight percentage.45,46 Furthermore, Fig. 7(c) depicts the surface morphology of SZT. It was found that the plate-like morphology of TiO2 was not homogenously mixed with spherical particles of ZnAl2O4, revealing that particles are not uniformly scattered on the SZT surface. As a result, the particle size of SZT is larger, which decreases the surface area. Consequently, SZT has lower catalytic activity than SZZ towards glycerol acetalization. The EDX histogram confirms the synthesis of SZT Fig. 7(d) on the basis of their atomic percentage present on designed catalyst surfaces such as Zn, Al, Ti, O, and S.47
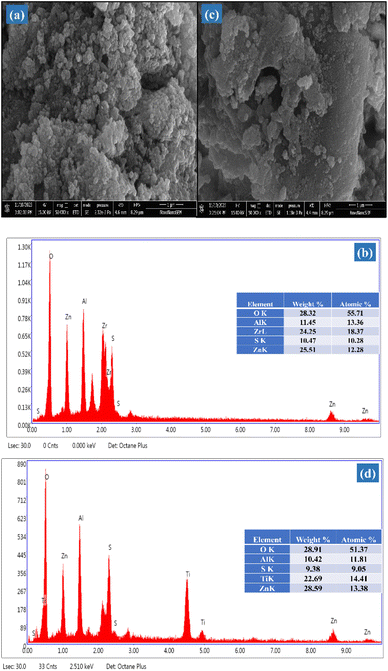 |
| | Fig. 7 SEM-EDX image of (a) & (b) SZZ and (c) & (d) SZT catalysts. | |
2.2 Acidity of catalysts
Synthesis catalyst's acidity and acidic strength are crucial in glycerol acetalization reactions. As a result, the catalyst's active site protonates the carbonyl carbon, increases the electrophilicity, and further proceeds the reaction. Here, we performed the N-butylamine titration process to estimate the overall acidity of the catalyst quantitatively, and the obtained results are illustrated in Table 2. The obtained result showed that SZZ had more acidic sites than SZT. Given that the acetalization reaction of glycerol with acetone relies on an acidic catalyst, the higher acidity of SZZ contributed to improved glycerol conversion.13
2.3 Influence of variable parameters on acetalization
The optimization process for glycerol conversion involved the various optimized parameters. Important variables including reaction temperature and time, glycerol and acetone molar ratios, and catalyst weight% were systematically optimized to achieve a better yield of solketal. To assess the individual impact of these factors on glycerol conversion, a series of acetalization reactions were performed by the one-variable-at-a-time (OVAT) method. The following section outlines the obtained results.
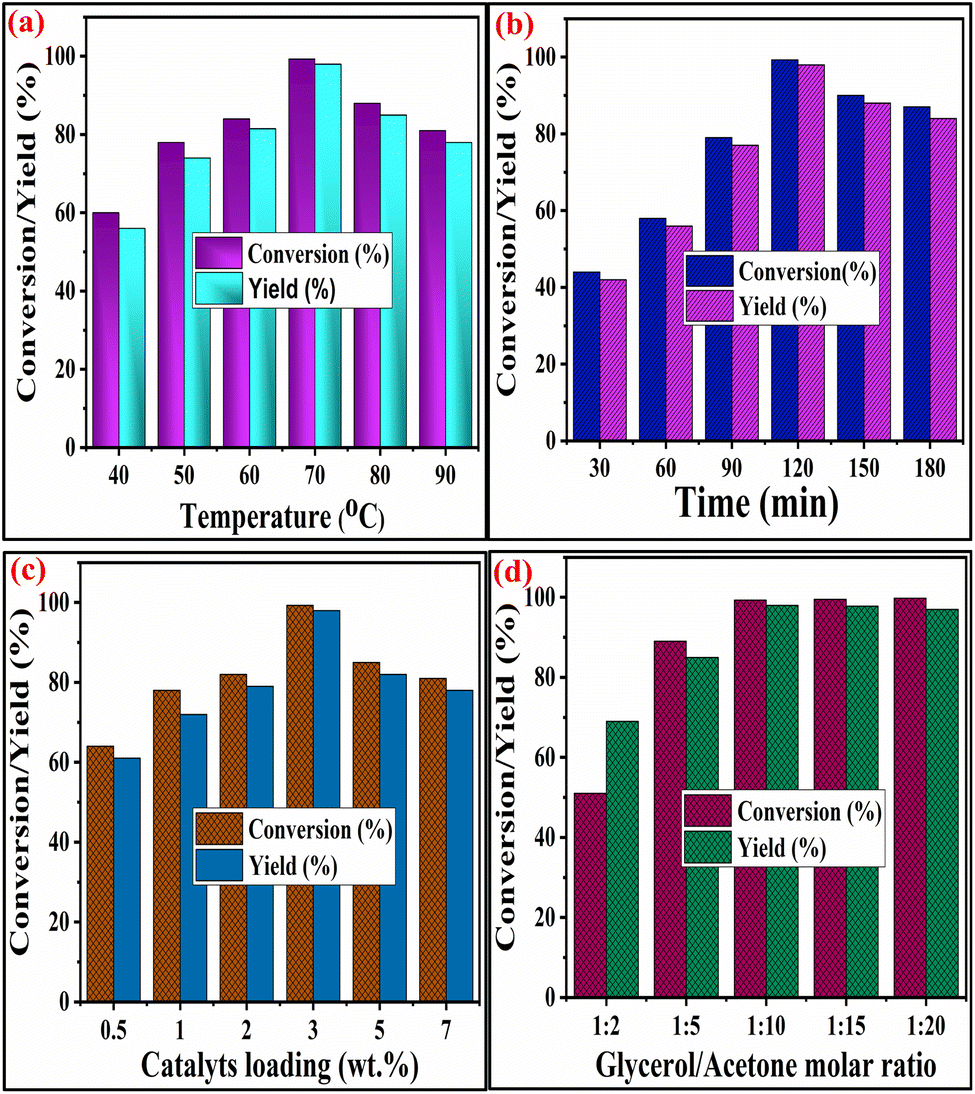
2.3.1 Optimization of temperature in solketal synthesis. The reaction temperature, one of the key factors influencing glycerol conversion, was also optimized by fluctuating the temperature from 40 °C to 80 °C. In contrast, all other parameters remained constant, as revealed in Fig. 8(a). The optimum temperature for the highest conversion of glycerol is 70 °C. This temperature provides a sufficient amount of kinetic energy to the reactants, which accelerates the glycerol acetalization reaction. It was found that both the glycerol conversion and percentage yield of solketal increased as the temperature increased from 40 °C to 70 °C. This temperature increase facilitates the collision between the reactant molecules, consequently promoting conversion. However, above 70 °C, the conversion and yield percentages began to decrease. This decline can be ascribed to the evaporation of the acetone molecule from the reaction mixture. Consequently, an incomplete acetalization reaction occurs due to the acetone's unavailability in the medium's liquid phase. It was noteworthy that acetalization is an exothermic reaction, with no need for further temperature for glycerol acetalization.48
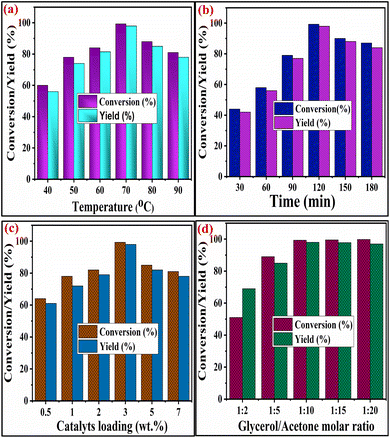 |
| | Fig. 8 Optimization of (a) reaction temperature (glycerol-to-acetone molar ratio = 1/10, catalyst wt% = 3, time = 120 min), (b) reaction time (glycerol-to-acetone molar ratio = 1/10, catalyst wt% = 3, reaction temperature = 70 °C), (c) catalyst weight percentage (glycerol-to-acetone molar ratio = 1/10, time = 120 min., reaction temperature = 70 °C) and (d) glycerol-to-acetone molar ratio (reaction temperature = 70 °C, catalyst wt% = 3, time = 120 min.). | |
2.3.2 Influence of reaction time. The conversion of glycerol and solketal yield was also affected by the duration of the reaction. The optimization of the reaction time for the conversion of glycerol to solketal is illustrated in Fig. 8(b). It was found that as the reaction time increased from 30 min to 120 min, the percentage conversion of glycerol increased from 44% to 99.3%. This rise in conversion can be ascribed to the increased number of reacting species (glycerol and acetone), which aid in forming new bonds following the cleavage of existing ones. However, beyond 120 minutes spent on reaction time, the conversion of glycerol decreased. This reduction in glycerol conversion may be attributed to the hydrolysis of the synthesized product, facilitated by water forming during the acetalization process.49
2.3.3 Influence of catalyst weight percentage. The acetalization reaction for solketal synthesis is highly influenced by the catalyst loading percentage of SZZ, and the obtained glycerol conversion and solketal yield are depicted in Fig. 8(c). In the absence of a catalyst, no glycerol conversion was detected. However, with the increase in the catalyst loading percentage (based on glycerol weight) from 0.5 to 3.0 wt%, there was a gradual increase in the conversion of glycerol from 61% to 99.3%, respectively. This increase can be accredited to the increased availability of the active sites on the catalyst's surface, thereby facilitating the interaction and collision between the acetone and glycerol molecules. Additionally, with an extension in the loading of catalyst from 5 wt%, both glycerol conversion and solketal yield began to decrease, potentially due to the hydrolysis of the synthesized solketal.50
2.3.4 Influence of the ratio of acetone to glycerol reactants. The acetalization reaction of acetone with glycerol is reversible. A higher reactant concentration is necessary to drive the equilibrium towards the product side. In this process, when the glycerol-to-acetone molar ratio was set at 1/2, the conversion of glycerol was notably very low (i.e., 51%). However, a further increase in the reactant molar ratio from 1/4 to 1/10 showed a remarkable improvement in conversion and was found to be 99.3% in Fig. 8(d). With the further increase in reactant molar ratio (above 1/10), a marginal variation was observed in glycerol conversion. This result could be attributed to glycerol in acetone's increased accessibility that helps shift the equilibrium toward the product formation side. These outcomes suggest that minimal changes were found in glycerol conversion with the augmentation of the reactant molar ratio (i.e., acetone and glycerol).51
2.5 Reaction kinetics
To study the kinetics of the glycerol acetalization reaction using SZZ heterogeneous catalysts at different reaction temperatures from 50 °C to 70 °C for 90 to 120 min reaction time, 1/10 glycerol-to-acetone molar ratio, and 3 wt% catalyst loading. In the absence of a catalyst, no significant rate constant was observed. The glycerol acetalization reaction follows the pseudo-first-order kinetics. As we take acetone in excess amount (1:10), it would shift the reaction equilibrium towards the product formation side. Due to its abundance, the concentration of acetone remains unchanged during the reaction, and the rate of acetalization reaction is only influenced by the concentration of glycerol. Therefore, the reaction rate can be written as follows.| |
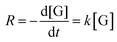 | (6) |
where 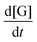 is the change in glycerol concentration per time t (min), [G] is the concentration of glycerol, and k is the rate constant. The rate constant for the first-order reaction was calculated by using glycerol conversion at different time intervals in eqn (7):X shows the conversion of glycerol at time t.
is the change in glycerol concentration per time t (min), [G] is the concentration of glycerol, and k is the rate constant. The rate constant for the first-order reaction was calculated by using glycerol conversion at different time intervals in eqn (7):X shows the conversion of glycerol at time t.
The reaction's activation energy was calculated using the Arrhenius equation at different temperatures (50–70 °C).
| |
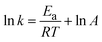 | (8) |
where
A is the pre-exponential factor,
R is the gas constant (8.314 × 10
−3 kJ K
−1 mol
−1), and
T is the reaction temperature.
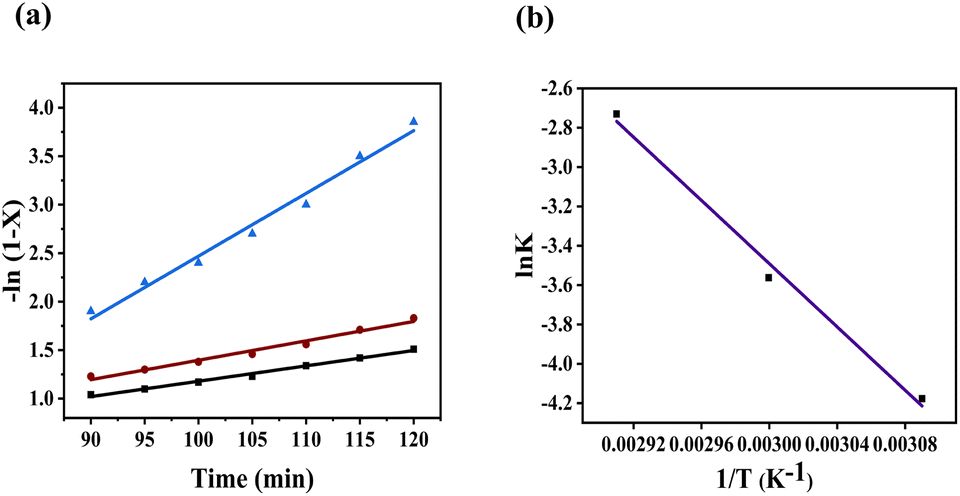
Fig. 9(a) shows the plot of −ln(1 −
X)
vs. reaction time (
t) at three different temperatures. This plot shows good agreement with the linear aggression model that followed the pseudo-first-order kinetics. The reaction's activation energy was calculated using the Arrhenius equation by plotting between ln
K vs. 1/
T using
eqn (8), as shown in
Fig. 9(b). The obtained activation energy of this catalyst was observed to be 66.8 kJ mol
−1.
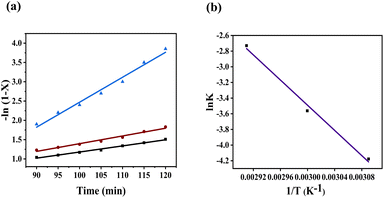 |
| | Fig. 9 (a) Kinetic plot −ln(1 − X) vs. time (min) and (b) Arrhenius plot lnK vs. 1/T. | |
2.6 Plausible reaction mechanism
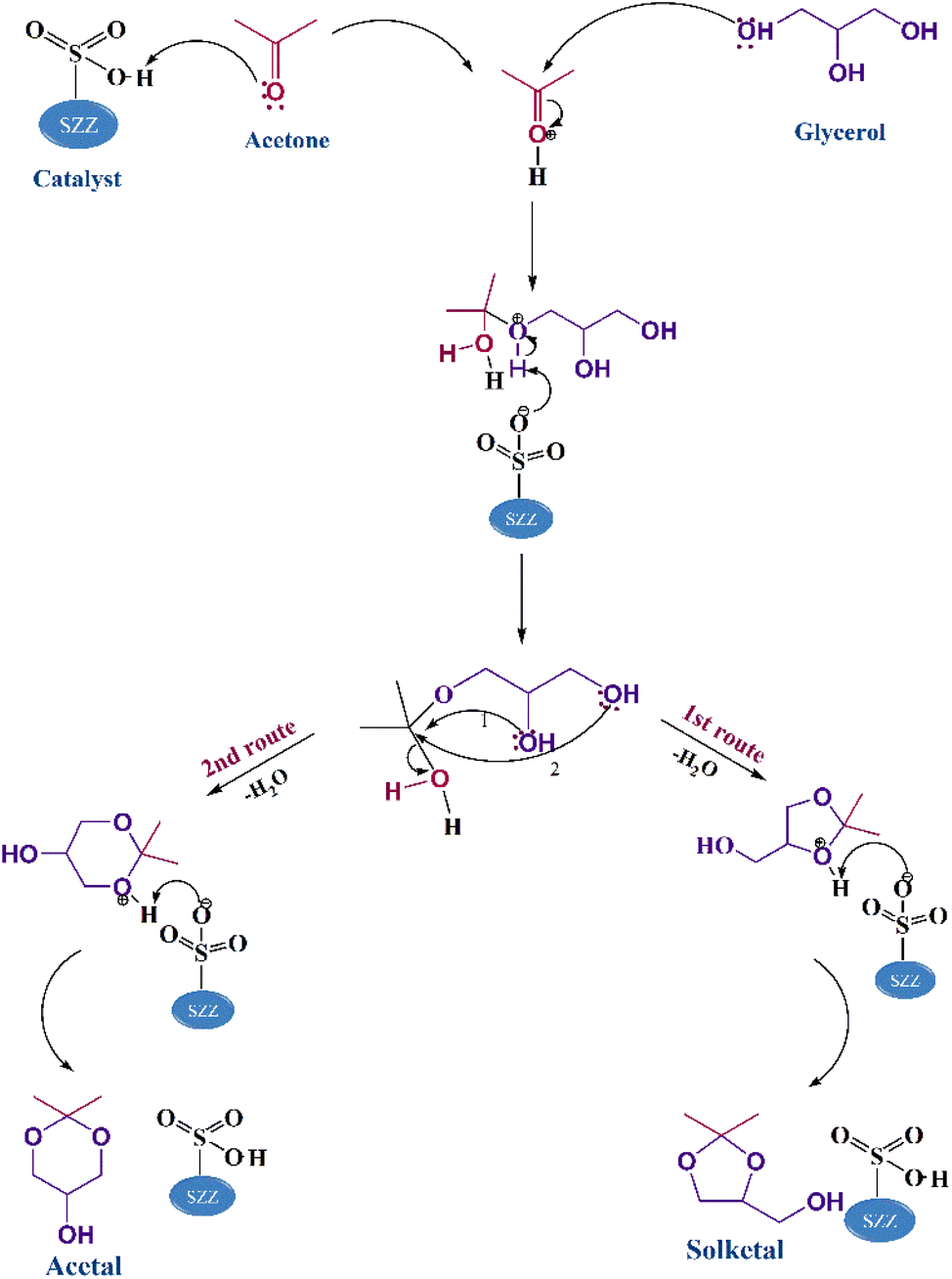
The mechanist pathway of acetalization reaction using SO42−/ZnAl2O4–ZrO2 acid catalyst is designed by drawing upon the previously documented literature. The Langmuir–Hinshelwood (L–H) model was selected as the most effective in describing the mechanism of this reaction, which goes through several phases illustrated in Scheme 2. This reaction mechanism follows the molecular diffusion technique. At first, reactant molecules (glycerol and acetone) are diffused on the catalyst surface, where they are adsorbed into the pores. In the first phase, acetone's carbonyl carbon undergoes protonation by the SZZ catalyst's acidic sites, enhancing the electrophilicity of carbonyl carbon. Subsequently, the second phase involves glycerol's hydroxyl group, which acts as a nucleophile and initiates an attack on the most electrophilic carbon center of the carbonyl group. The third phase is attended by deprotonation, leading to a highly unstable hemiacetal intermediate. This intermediate undergoes cyclization by eliminating water molecules and engaging in an SN2 nucleophilic substitution via two pathways. A five-membered solketal is created if the intermediate is attacked by the glycerol's secondary –OH group. Conversely, cyclization occurs by forming six-membered acetals by attacking the primary –OH group. Notably, kinetic parameters favor the five-membered cyclic solketal over the six-membered acetal, making the solketal a major product in this reaction pathway.52
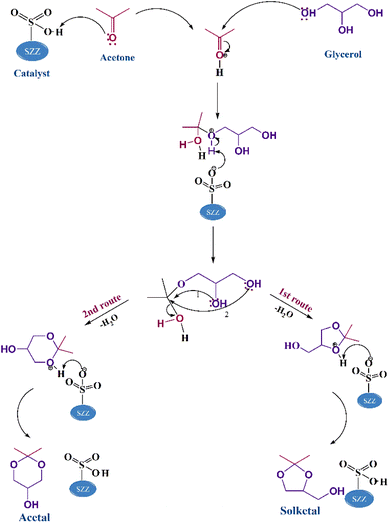 |
| | Scheme 2 Mechanistic demonstration of the acetalization of glycerol using the SZZ catalyst. | |
2.7 Characterization of the derived product
2.7.1 GCMS studies. Gas chromatograph-mass spectroscopy is a robust analytical method employed to identify the composition of products within an analytical mixture. The qualitative and quantitative investigation of the synthesis product was performed using the GC-MS studies, as illustrated in Fig. S2.† The retention peak confirmed the production of five-membered solketal, which was found at 3.98 min while others demonstrated the formation of six-membered acetal at 4.31 min. A very small amount of unreacted glycerol was found at a peak at 4.78 min.
2.7.1 FTIR spectroscopy analysis. Solketal synthesis was also verified by FTIR spectroscopy. The FTIR spectra of both glycerol and synthesized solketal are shown in Fig. S4.† Fig. S4(a)† shows the FTIR spectrum of glycerol. The hydroxyl group's O–H stretching is allocated to a broad peak at 3408 cm−1, while the glycerol absorption peak, which falls between 2885 and 2942 cm−1, signifies the C–H stretching. The peak assigned to the C–O–H bending vibration appeared at 1419 cm−1. The peaks with low intensity at 1111 cm−1 were assigned to the stretching vibration of C–O in 1° alcohol, whereas the peak at 913 cm−1 was ascribed to the bending vibration of the O–H group within the glycerol moiety.The FTIR spectrum of the synthesized solketal is depicted in Fig. S4(b).† A noticeable peak was observed at ∼3427 cm−1, assigned to the hydroxyl bonds (–OH) of solketal molecules. The intensity of this band is comparatively lower than that of glycerol, indicating that glycerol is converted into solketal. Within the range of wave numbers between 2881 and 2987 cm−1, regarded as stretching of the C–H methyl bonds, another peak was observed at 1376 cm−1, which signifies the “umbrella” trend associated with the ketone's methyl group. The band at 1212 cm−1 represents the dioxolane –C–O bond. The asymmetric bands found at 1155 and 1116 cm−1 are accredited to the –C–O–C–O–C– bonds, while the peaks at 906 and 788 cm−1 are allocated to the symmetric vibration of the same bond within a solketal ring. Additionally, the band at 1042 cm−1 is attributed to the –C–C–OH bond in the 4th position of the alcoholic group.53 Several characteristic bands, such as 1045, 1156, 1216, and 1380 cm−1, were not found in glycerol FTIR spectra, which indicates the synthesis of solketal from glycerol.
2.7.1 Study of the green parameters of the catalyst. As per the postulates of green chemistry, minimizing waste generation in chemical reactions signifies a more environmentally friendly process. As a result, a sustainable process should generate less amount of waste. Therefore, investigating waste generation during the catalytic cycle was important to ascertain the process's overall environmental impact and eco-friendliness. The degree of ‘greenness’ of the acetalization reaction catalyzed using the SZZ catalyst was assessed by considering various key green parameters. These parameters, environmental factor, process mass index, and atom economy were calculated using the provided equations.| | |
E-factor = mass of the produced waste (g)/mass of the desired product (g)
| (9) |
| | |
Process mass index = total mass used in acetalization process (g)/mass of product (g)
| (10) |
| | |
Atom economy (AE) = mass of the isolated product/total mass of all the substance produced
| (11) |
The E-studies serve as an indicator of the environmental compatibility of the solketal synthesis. A lower E-value reflects the lower waste production during the process. Similarly, PMI is defined as the ratio of the total mass of the reactant employed in the acetalization process to the mass of the resulting isolated product. Atom economy, however, defines the mass of the isolated product relative to the total mass of all the products produced in the synthesis process. The only water as a byproduct generated is benign and serves as a solvent in numerous chemical processes. Similarly, excess acetone was recovered using a rotatory evaporator and further used in the acetalization process. Notably, the reusability of the catalyst and solvent is not considered in the green metrics calculation. E-factor, PMI, and atom economy were calculated to be 0.81, 1.82 g−1, and 100%, respectively, demonstrating minimum waste generation during the glycerol acetalization process.54
2.7.1 Reusability studies of the catalyst. Catalyst reusability plays a significant role in the acetalization reaction of solketal synthesis. The activity and reusability of the catalyst were studied by performing the batch reaction, and it was found that the catalyst can be used for up to the 5th catalytic cycle. Throughout the repeated cycle, both the conversion percentage and solketal yield remain unaffected, as shown in Fig. S5.† However, beyond the 5th catalytic cycle, a sudden decline in glycerol conversion and solketal yield was observed. The observed catalytic activity loss could be due to some mass loss of sulfur content or pore blockage and surface poisoning by glycerol and acetone during the recycling process. To ascertain the cause of catalyst deactivation, a leaching test was conducted using the hot filtration technique. After a 90 minute reaction period, the catalyst was segregated from the reaction mixture by filtration under hot conditions, during which the glycerol conversion was found to be >90%. Subsequently, the reaction was prolonged without the catalyst for an additional 180 min and showed a slight increase in glycerol conversion (Fig. S6†). This examination demonstrated that the filtrate did not contain a significant amount of soluble active catalyst. Consequently, it demonstrated the catalyst's heterogeneous nature. The XRD and FTIR study of the recycled catalyst is presented in Fig. S7 and S8.† Similarly, the SEM image of the 5th recycled catalyst is displayed in Fig. S9.† The similarity in the plots and image of the reused catalyst with the fresh catalyst validates its catalytic stability.
2.8 Comparative analysis of the synthesized catalyst with other catalysts
The heterogeneous acid catalysts for glycerol acetalization under various reaction conditions that have been reported by several studies are tabulated in Table 3. Catalysts such as P-25, Amberlyst-15,55 and SO42−/CeO2–ZrO2 (ref. 56) showed appreciable glycerol conversion, but the time duration for solketal synthesis is very high. In contrast, the SZZ catalyst showed good catalytic activity and gave 99.3% glycerol conversion at 120 min. Similarly, Fe (NO3)3·9H2O showed a good conversion under moderate reaction conditions. Still, it required a higher amount of acetone (i.e., 1:20 M of glycerol and acetone) and is reusable only up to the 3rd reaction cycle.57 In contrast, the SZZ catalyst utilizes a lower glycerol-to-acetone molar ratio (1:10) and shows excellent catalytic stability till the 5th reaction cycle. Likewise, MoM3/SiO2,58 and montmorillonite59 catalyst showed very poor catalytic activity and used solvents such as toluene and CH3CN and gave low conversion of glycerol. Here, the synthesized catalyst exhibited notable catalytic activity in the solketal synthesis under solvent-free conditions. In this reaction, the synthesized catalyst is facile and more efficient for the acetalization process. A comparative analysis indicates that the SZZ catalyst exhibits outstanding catalytic activity throughout the reaction when compared to the reported catalysts. The catalyst's thermal stability and reusability established it as an effective heterogeneous acid catalyst for its broad potential applications.
Table 3 Comparative study of the catalytic activity of the synthesized SZZ catalyst with the reported catalysts employed in solketal synthesis
| Catalyst |
Catalyst loading |
Reaction time (min) |
Reaction temperature (°C) |
Gly:Ace molar ratio |
Solvent |
Glycerol conversion (%) |
Endurance capacity |
Ref. |
| 20% MoO3–ZrO2 |
0.03 (gm) |
10 |
50 |
1:8 |
Free |
89 |
5th reaction cycle |
2 |
| MOF-808-SO4 (DF) defective/sulfonated |
0.01 (gm) |
30 |
78 |
1:7 |
Free |
78 |
4th reaction cycle |
4 |
| GS-SO3H |
5 (wt%) |
240 |
RT |
1:4 |
Free |
91 |
5th reaction cycle |
60 |
| SO42−/SnO2 |
5 (wt%) |
240 |
RT |
1:1 |
Free |
95 |
5th reaction cycle |
61 |
| Zr–S |
0.3 (wt%) |
60 |
40 |
1:6 |
Free |
80 |
4th reaction cycle |
62 |
| SO42−/CeO2–ZrO2 |
9 (wt%) |
480 |
100 |
1:3 (benzaldehyde) |
Toluene |
91.8 |
Not reported |
56 |
| Acidic carbon |
3 (wt%) |
60 |
100 |
1:2 |
Free |
82 |
5th reaction cycle |
63 |
| Fe(NO3)3·9H2O |
0.3 (mol%) |
60 |
25 |
1:20 |
Free |
90 |
3rd reaction cycle |
57 |
| Zr–S-400 |
0.6 (wt%) |
60 |
40 |
1:6 |
Free |
80 |
4th reaction cycle |
62 |
| PSF/K–SiO2 |
5 (wt%) |
90 |
25 |
1:10 |
Free |
86.3 |
5th reaction cycle |
64 |
| MoM3/SiO2 |
10 (wt%) |
480 |
100 |
1:1(benzaldehyde) |
Toluene |
72 |
Not reported |
58 |
| Montmorillonite |
0.7 (wt%) |
30 |
50 |
1:4.1 |
Free |
54 |
3rd reaction cycle |
59 |
| Montmorillonite |
0.7 (wt%) |
30 |
50 |
1:4.1 |
CH3CN |
94 |
3rd reaction cycle |
59 |
| DT-851 sulfonic acid resin |
5 (wt%) |
120 |
58 |
1:20 |
Free |
95 |
10th reaction cycle |
65 |
| P-25 |
0.8 (wt%) |
1440 |
30 |
1:13 |
Free |
87 |
Not reported |
55 |
| Amberlyst-15 |
0.8 (wt%) |
1440 |
30 |
1:13 |
Free |
64 |
Not reported |
55 |
| SZZ (present work) |
3.0 (wt%) |
120 |
70 |
1:10 |
Free |
99.3 |
5th reaction cycle |
3 Conclusions
This study provided a more environment-friendly route to produce solketal in laboratories using the byproduct glycerol obtained from biodiesel plants via a transesterification process. We have successfully synthesized heterogeneous acid SZT and SZZ catalysts and examined their catalytic performance in glycerol acetalization reactions. A comparative study between SZT and SZZ catalysts was performed for solketal synthesis. Several characterization processes such as SEM-EDX, XRD, FTIR spectroscopy, TGA, BET-surface area, XPS, and the N-butylamine titration method revealed sulfur impregnation on the catalyst's surface. All characterizations consistently demonstrated that the SZZ catalyst exhibits a higher catalytic activity than that of the SZT catalyst. The catalyst's heightened activity relies on its elevated acidity, which is a pivotal factor in gaining a substantial percentage of solketal. The higher acidity of the SZZ catalyst was very helpful for the acetalization reaction of glycerol to solketal. The solketal produced was subjected to quantitative and qualitative analyses through techniques including 1H and 13C NMR spectroscopy, FTIR spectroscopy, and gas chromatography. The acidity of the synthesized catalyst and the conditions of the reaction played a significant role in influencing both the conversion of glycerol and the solketal yield. The higher TOF of the SZZ catalyst makes it more desirable for catalytic activities than the SZT catalyst. To obtain an excellent solketal yield, the reaction conditions were optimized. Greenness parameters such as E-factor, PMI, and AE were employed to show that the synthesis route is environmentally benign and eco-friendly in nature. In summary, the SZZ catalyst proves to be a cost-effective and highly active catalyst, and its reusability is demonstrated up to the 5th catalytic cycle. The repeated utilization of the SZZ catalyst in solketal synthesis implies the economic feasibility of the process on an industrial scale, and the cost-effectiveness indicates the sustainability of the solketal market.
Data availability
The additional data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are acknowledged to Indian Institute of Technology (BHU), Varanasi India, for providing Central Instruments Facilities. The authors also show their sense of gratitude to the Ministry of Human Resource Development (MHRD), Government of India for providing financial assistance.
References
- T. Roy, S. Sahani and Y. Chandra Sharma, J. Clean. Prod., 2020, 247, 119166 CrossRef.
- H. Huang, J. Mu, M. Liang, R. Qi, M. Wu, L. Xu, H. Xu, J. Zhao, J. Zhou and Z. Miao, Mol. Catal., 2024, 552, 113682 CrossRef.
- A. Vivian, L. Soumoy, L. Fusaro, S. Fiorilli, D. P. Debecker and C. Aprile, Green Chem., 2021, 23, 354–366 RSC.
- B. Dashtipour, S. Dehghanpour and M. Sharbatdaran, Polyhedron, 2024, 247, 116733 CrossRef.
- X. Li, Y. Jiang, R. Zhou and Z. Hou, Appl. Clay Sci., 2019, 174, 120–126 CrossRef.
- S. Liu, Z. Yu, C. Lu, Y. Wang, F. Sun, Z. Sun, Y. Liu, C. Shi and A. Wang, Fuel, 2023, 334, 126763 CrossRef.
- L. Shen, X. Chen, X. Chen, Y. Qin, S. Shao, G. Zhu and H. Yin, J. Chem. Technol. Biotechnol., 2023, 98, 179–187 CrossRef.
- M. N. Nor Shafiqah, H. Mohamed, J. Panpranot and S. Zainal Abidin, Chem. Eng. Sci., 2024, 285, 119567 CrossRef.
- Ł. Madej and A. A. Kiss, J. Chem. Technol. Biotechnol., 2023, 98, 2110–2121 CrossRef.
- A. Sandid, J. Esteban, C. D’Agostino and V. Spallina, J. Clean. Prod., 2023, 418, 138127 CrossRef.
- G. Pradhan and Y. C. Sharma, J. Clean. Prod., 2021, 315, 127860 CrossRef.
- S. B. Putla, P. Subha, B. Swapna, N. Singh and P. Sudarsanam, Catal. Commun., 2024, 186, 106827 CrossRef.
- K. Saikia, K. Rajkumari, N. S. Moyon, S. Basumatary, G. Halder, U. Rashid and S. L. Rokhum, Fuel Process. Technol., 2022, 238, 107482 CrossRef.
- P. Sudarsanam, B. Mallesham, A. N. Prasad, P. S. Reddy and B. M. Reddy, Fuel Process. Technol., 2013, 106, 539–545 CrossRef.
- G. Vicente, J. A. Melero, G. Morales, M. Paniagua and E. Martín, Green Chem., 2010, 12, 899–907 RSC.
- S. Zaher, L. Christ, M. Abd El Rahim, A. Kanj and I. Karamé, Mol. Catal., 2017, 438, 204–213 CrossRef.
- M. J. da Silva, F. de Ávila Rodrigues and A. A. Júlio, Chem. Eng. J., 2017, 307, 828–835 CrossRef.
- S. R. Churipard, P. Manjunathan, P. Chandra, G. V. Shanbhag, R. Ravishankar, P. V. C. Rao, G. Sri Ganesh, A. B. Halgeri and S. P. Maradur, New J. Chem., 2017, 41, 5745–5751 RSC.
- V. Rossa, Y. D. S. P. Pessanha, G. C. Díaz, L. D. T. Câmara, S. B. C. Pergher and D. A. G. Aranda, Ind. Eng. Chem. Res., 2017, 56, 479–488 CrossRef.
- I. Zahid, M. Ayoub, B. Bin Abdullah, M. H. Nazir, M. A. Kaimkhani and F. Sher, Sustainability, 2021, 13, 1–17 Search PubMed.
- P. Ferreira, I. M. Fonseca, A. M. Ramos, J. Vital and J. E. Castanheiro, Appl. Catal., B, 2010, 98, 94–99 CrossRef.
- J. Esteban, M. Ladero and F. García-Ochoa, Chem. Eng. J., 2015, 269, 194–202 CrossRef.
- Y. Jiang, R. Zhou, B. Ye and Z. Hou, J. Ind. Eng. Chem., 2022, 110, 357–366 CrossRef.
- I. B. Laskar, K. Rajkumari, R. Gupta and L. Rokhum, Energy Fuels, 2018, 32, 12567–12576 CrossRef.
- S. B. Umbarkar, T. V. Kotbagi, A. V. Biradar, R. Pasricha, J. Chanale, M. K. Dongare, A. S. Mamede, C. Lancelot and E. Payen, J. Mol. Catal. Chem., 2009, 310, 150–158 CrossRef.
- H. Hussein, C. Aprile and M. Devillers, Appl. Catal., A, 2023, 667, 119444 CrossRef.
- P. S. Reddy, P. Sudarsanam, G. Raju and B. M. Reddy, J. Ind. Eng. Chem., 2012, 18, 648–654 CrossRef.
- C. Metcalfe and O. Kesler, Fuel Cells, 2020, 20, 730–740 CrossRef.
- Y. Li, X. D. Zhang, L. Sun, J. Zhang and H. P. Xu, Appl. Energy, 2010, 87, 156–159 CrossRef.
- W. Shi, Catal. Lett., 2013, 143, 732–738 CrossRef.
- A. Kumar, Priyanka, J. Mangalam, V. Yadav and T. Goswami, React. Kinet. Mech. Catal., 2022, 135, 1929–1944 CrossRef.
- K. Saikia, K. Rajkumari, N. S. Moyon, S. Basumatary, G. Halder, U. Rashid and S. L. Rokhum, Fuel Process. Technol., 2022, 238, 107482 CrossRef.
- H. Hussein, C. Aprile and M. Devillers, Appl. Catal., A, 2023, 667, 119444 CrossRef.
- W. Hua, Y. Xia, Y. Yue and Z. Gao, J. Catal., 2000, 196, 104–114 CrossRef.
- B. M. Reddy, P. M. Sreekanth, P. Lakshmanan and A. Khan, J. Mol. Catal. Chem., 2006, 244, 1–7 CrossRef.
- I. Dosuna-Rodríguez, C. Adriany and E. M. Gaigneaux, Catal. Today, 2011, 167, 56–63 CrossRef.
- K. Saravanan, B. Tyagi, R. S. Shukla and H. C. Bajaj, Appl. Catal., B, 2015, 172–173, 108–115 CrossRef.
- K. Wijaya, A. R. Putri, S. Sudiono, S. Mulijani, A. Patah, A. C. Wibowo and W. D. Saputri, Catalysts, 2021, 12, 1492 CrossRef.
- J. Wang, A. Wang, X. Tian, H. Wang, M. Xu and L. Yang, Appl. Clay Sci., 2017, 135, 596–602 CrossRef.
- G. Pradhan, S. Jaiswal and Y. C. Sharma, Mol. Catal., 2022, 526, 112332 CrossRef.
- E. Huízar-Padilla, H. Guillén-Bonilla, A. Guillén-Bonilla, V. M. Rodríguez-Betancourtt, A. Sánchez-Martínez, J. T. Guillen-Bonilla, L. Gildo-Ortiz and J. Reyes-Gómez, Sensors, 2021, 21, 2362 CrossRef.
- J. Wang, A. Wang, Y. Liao, L. Shi and L. Yang, React. Kinet. Mech. Catal., 2023, 136, 2123–2145 CrossRef.
- P. Yan, H. Wang, Y. Liao and C. Wang, Fuel, 2023, 342, 127685 CrossRef.
- J. Hu, W. Zhao, R. Hu, G. Chang, C. Li and L. Wang, Mater. Res. Bull., 2014, 57, 268–273 CrossRef.
- A. Wang, J. Wang, C. Lu, M. Xu, J. Lv and X. Wu, Fuel, 2018, 234, 430–440 CrossRef.
- A. Kumar, Priyanka, J. Mangalam, V. Yadav and T. Goswami, React. Kinet. Mech. Catal., 2022, 135, 1929–1944 CrossRef.
- A. Q. Wang, J. X. Wang, H. Wang, Y. N. Huang, M. L. Xu and X. L. Wu, RSC Adv., 2017, 7, 14224–14232 RSC.
- X. Li, L. Zheng and Z. Hou, Fuel, 2018, 233, 565–571 CrossRef.
- I. B. Laskar, K. Rajkumari, R. Gupta and L. Rokhum, Energy Fuels, 2018, 32, 12567–12576 CrossRef.
- J. A. Vannucci, N. N. Nichio and F. Pompeo, Catal. Today, 2021, 372, 238–245 CrossRef.
- B. Saini, A. P. Tathod, S. K. Saxena, S. Arumugam and N. Viswanadham, ACS Sustain. Chem. Eng., 2022, 10, 1172–1181 CrossRef.
- A. Ghosh, A. Singha, A. Auroux, A. Das, D. Sen and B. Chowdhury, Catal. Sci. Technol., 2020, 10, 4827–4844 RSC.
- I. Zahid, M. Ayoub, B. B. Abdullah and M. Hamza Nazir, Glycerol Derivatives as Fuel Additive: Synthesis of Solketal From Glycerol and Acetone With Various Acid Clay Catalysts, 2020 Search PubMed.
- T. Roy, A. K. Ágarwal and Y. C. Sharma, Waste Manage., 2021, 135, 243–255 CrossRef.
- J. Hidalgo-Carrillo, R. C. Estévez-Toledano, F. J. López-Tenllado, F. M. Bautista, F. J. Urbano and A. Marinas, J. Taiwan Inst. Chem. Eng., 2021, 125, 297–303 CrossRef.
- R. M. Kulkarni and N. Arvind, Heliyon, 2021, 1, 7 Search PubMed.
- M. J. da Silva, A. A. Rodrigues and P. F. Pinheiro, Fuel, 2020, 276, 118164 CrossRef.
- S. B. Umbarkar, T. V. Kotbagi, A. V. Biradar, R. Pasricha, J. Chanale, M. K. Dongare, A. S. Mamede, C. Lancelot and E. Payen, J. Mol. Catal. Chem., 2009, 310, 150–158 CrossRef.
- M. N. Timofeeva, V. N. Panchenko, V. V. Krupskaya, A. Gil and M. A. Vicente, Catal. Commun., 2017, 90, 65–69 CrossRef.
- A. Ghosh, A. Singha, A. Auroux, A. Das, D. Sen and B. Chowdhury, Catal. Sci. Technol., 2020, 10, 4827–4844 RSC.
- B. Mallesham, P. Sudarsanam and B. M. Reddy, Catal. Sci. Technol., 2014, 4, 803–813 RSC.
- J. A. Vannucci, N. N. Nichio and F. Pompeo, Catal. Today, 2021, 372, 238–245 CrossRef CAS.
- M. Gonçalves, R. Rodrigues, T. S. Galhardo and W. A. Carvalho, Fuel, 2016, 181, 46–54 CrossRef.
- R. Zhou, Y. Jiang, H. Zhao, B. Ye, L. Wang and Z. Hou, Fuel, 2021, 291, 120207 CrossRef.
- J. Yang, N. Li, W. J. Ma, J. H. Zhou and H. Z. Sun, Adv. Mater. Res., 2014, 830, 176–179 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
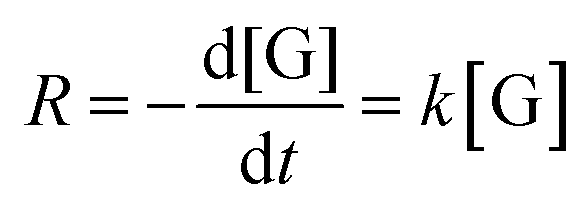
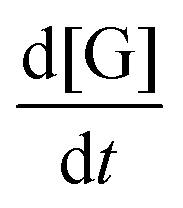 is the change in glycerol concentration per time t (min), [G] is the concentration of glycerol, and k is the rate constant. The rate constant for the first-order reaction was calculated by using glycerol conversion at different time intervals in eqn (7):
is the change in glycerol concentration per time t (min), [G] is the concentration of glycerol, and k is the rate constant. The rate constant for the first-order reaction was calculated by using glycerol conversion at different time intervals in eqn (7):