
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability and magnetic properties of transition metal (V, Cr, Mn, and Fe) doped cobalt oxide clusters: a density functional theory investigation†
Nguyen Thi Mai *a,
Tran Dang Thanh*a,
Do Hung Manha,
Nguyen Thi Ngoc Anha,
Ngo Thi Lanab,
Phung Thi Thuc and
Nguyen Thanh Tunga
*a,
Tran Dang Thanh*a,
Do Hung Manha,
Nguyen Thi Ngoc Anha,
Ngo Thi Lanab,
Phung Thi Thuc and
Nguyen Thanh Tunga
aInstitute of Materials Science, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Vietnam. E-mail: maint@ims.vast.ac.vn; thanhxraylab@yahoo.com
bInstitute of Science and Technology, TNU – University of Sciences, Thai Nguyen, 250000, Vietnam
cUniversity of Science and Technology of Hanoi, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Vietnam
First published on 11th November 2024
Abstract
Con−1TMOn−2+ (n = 6–8), (TM = V, Cr, Mn, and Fe) clusters are investigated using density functional theory calculations. The transition metal atoms preferentially replace one Co atom at sites where the number of metal–oxygen bonds is maximized, forming more stable structures. The evaporation of a Co atom is the most fragile dissociation channel for both pure and doped species. Bare cobalt oxide clusters exhibit parallel spin ordering, whereas both parallel and antiparallel spin ordering are observed in the doped species. Notably, a ferromagnetic-to-ferrimagnetic transition occurs in the V-doped clusters, while the ferromagnetic behavior is enhanced in the Fe-doped species.
1 Introduction
Cobalt oxides are notable species known for their intriguing bulk properties, which make them highly relevant for applications in electronics, magnetism, and catalysis.1–3 Selected cobalt oxides can stabilize the magnetic moments of embedded ferromagnetic nanoparticles, surpassing the superparamagnetic limit observed in isolated particles.4 Electrodes composed of both pure and doped cobalt oxides demonstrate exceptional capacity and cyclability, even at high charging rates.5,6 Furthermore, cobalt oxides exhibit unique catalytic behaviors, particularly in water-splitting reactions.7Recent years have witnessed significant interest in both experimental and theoretical investigations into the structure, stability, magnetism, and bonding of cobalt oxide clusters. These tiny pieces of matter, though governed by the same fundamental principles as bulk materials, exhibit properties that nonmonotonically vary with their size and composition.8–16 For instance, studies on electronic and geometric structures of cobalt oxide clusters have revealed spontaneous ferromagnetic properties at various sizes, which are uncommon in larger dimensions.17,18 The ferromagnetic variation with the size was verified in the neutral clusters of Co6On/Co7On (n = 3–6), increasing from 16 or 15 μB (n = 4, 3) to 18 or 17 μB (n = 5, 4) and then decreasing to 16 or 15 μB at n = 6, 5.19 Specifically, the total magnetic moments in Co3On+ (n = 1–3) clusters go from 6 to 12 μB as the number of unpaired electrons in Co increases,11 a trend that is also observed with a rising number of Co atoms.18 Nevertheless, Aguilera-del-Toro et al., reveals that the magnetism of cation clusters is greater than that of corresponding neutral ones in almost cases.18 Except Co6O8+ cluster showing the antiferromagnetism, the ferromagnetic behavior is observed in all ConOm+ cations clusters (n = 3–6 and m = 3–8) through the use of combined infrared vibrational spectroscopy and density functional theory (DFT) calculations.20 Another aspect, while bulk cobalt oxide crystals possess a cubic rock-salt structure, a combined ion mobility mass spectrometry and computational study of (CoO)n (n = 2–7) suggests a geometrical transition from monocyclic ring shapes to compact cube/tower structures at Co6O6+.21
Not only size but their chemical and physical properties can also be strongly influenced by the number of constituent elements, doped elements, and their positions within the cluster. In this regard, cobalt oxide clusters, ConOm+ (m = 1–4 and n = 2, 3), and their Cr-doped counterparts produced by laser vaporization sources have been photofragmented and examined for their thermodynamic stability.22,23 Among studied clusters, the incorporation of Cr dopants significantly impacts their geometric structures, as observed in CoCrO3+, Co2CrO2+, Co2CrO4+, and Co3CrO4+. This is supported by dissociation behavior that aligns well between experimental data and density-functional-theory (DFT) calculations. Furthermore, the presence of Cr dopant can either enhance or suppress the total magnetic moment in CoxCryOm+ (x + y = 2, 3 and 1 < m < 4) clusters, influenced by chemical bonds between Cr/Co and O.11 That work demonstrated that the number of metal–oxygen bonds significantly governed the local spin magnetic moments, a higher oxygen concentration resulting in a lower localized magnetic moments on the Cr atom. Notably, the total magnetic moments are notably influenced by their geometric structure. Ferromagnetism enhancement in cobalt oxide Co3O4 was reported when replacing Co2+ ions by Mn2+ ions,24 with the concentration of Mn2+ ions playing a key role in the increase of saturation magnetization, coercivity, and remanent magnetization. On the other hand, Ignatiev and coworkers demonstrated the ferromagnetic ordering independence on transition metal impurities (Fe, Mn, and Ni) in cobalt oxide crystals.25
It could be conjectured that interesting properties may be revealed in larger cluster. Meanwhile, the clusters of ConOn−2+ with n = 6–8 contain a wealth of structural and bonding information. There are still a lot of unknowns regarding how a 3d transition metal atom interacts with Co and O atoms in this cluster. The energy required for common dissociation channels has not yet been examined to determine the kinetic stability of clusters. The presence of unpaired 3d electrons on transition metal atoms, especially the geometry, composition, number, and positions of bonds among atoms, is the primary factor driving the spin moment variation in the cluster. Moreover, understanding the size- and constituent-dependent magnetism of doped cobalt oxide clusters is crucial, as it may guide future Stern–Gerlach experiments aimed at tailoring the functionality of potential cluster-based multiferroics. Therefore, further efforts to achieve a comprehensive understanding of the geometrical and electronic structures as well as the magnetic properties of doped cobalt oxide clusters, are essential.
Inspired by the potential for future photodissociation and magnetic deflection experiments, this work systematically explores the impact of transition metal (TM = V, Cr, Mn, and Fe) on manipulating the geometric structure and magnetic properties of larger oxygen-deficient cobalt oxide clusters ConOn−2+ (n = 6–8) using DFT calculations. The doped geometric structures at the lowest energy state and their stability were determined by employing binding energy and dissociation energy. The DFT results indicate that a ferromagnetic-to-ferrimagnetic transition can be observed upon doping and that magnetic enhancement or reduction is strongly influenced by the geometric structure and the specific TM element. These theoretical findings provide valuable guidance for future experimental investigations.
2 Method
The DFT calculations were applied to investigate the stable geometric structures of Con−1TMOn−2+ (TM = V, Cr, Mn, and Fe; n = 6–8) clusters using Gaussian09 and GaussView packages.26,27 The DFT approach was selected due to its shown accuracy in previous studies involving cobalt oxide clusters.18,20,22,23,28,29 The hybrid functional B3LYP, in combination with the 6-31+G(d) basis set, was chosen for all calculations in this work based on previous investigations.22 Furthermore, by evaluating the binding energy using various combinations of functions and basis sets, we further validate the correctness and dependability of our calculations. The results were compared with available experimental results and are presented in Table S1.† Notably, the calculated binding energy of the TM-O dimer with B3LYP/6-31+G(d) was in best agreement with the experimental values. A self-consistent approach was employed with the convergence criteria of 2 × 10−5 hartree for energy and 5.0 × 10−3 Å for displacement.Initially, a large number of feasible input structures for Con−1TMOn−2+ were manually constructed by replacing a Co atom with a transition metal atom at all possible positions within the ConTMOn−2+ cluster using GaussView package. The vibrational frequencies and spin multiplicities were also considered. Subsequently, electronic configuration analyses were performed using the natural bonding orbital (NBO) and spin-density distribution. The magnetic moments, including total (TMMs) and local values (LMMs), were defined as the difference between spin-up and spin-down electrons occupying the molecular/atomic orbitals of the cluster/atom. It is important to note that each stoichiometry has several distinct geometries and spin configurations that meet the convergence criteria. In the following sections, only the lowest-energy isomers are discussed.
3 Results and discussions
3.1 Optimized geometric structures and spin configurations
The optimized geometric structures of ConOn−2+ and Con−1TMOn−2+ (n = 6–8 and TM = V, Cr, Mn, and Fe) clusters, and corresponding spin multiplicities are shown in Fig. 1 and ESI.†30 The abbreviation notation of n−1.TM.n−2 defines the cluster structure, where n the number of Co atom, respectively, while the superscript denotes the spin multiplicity.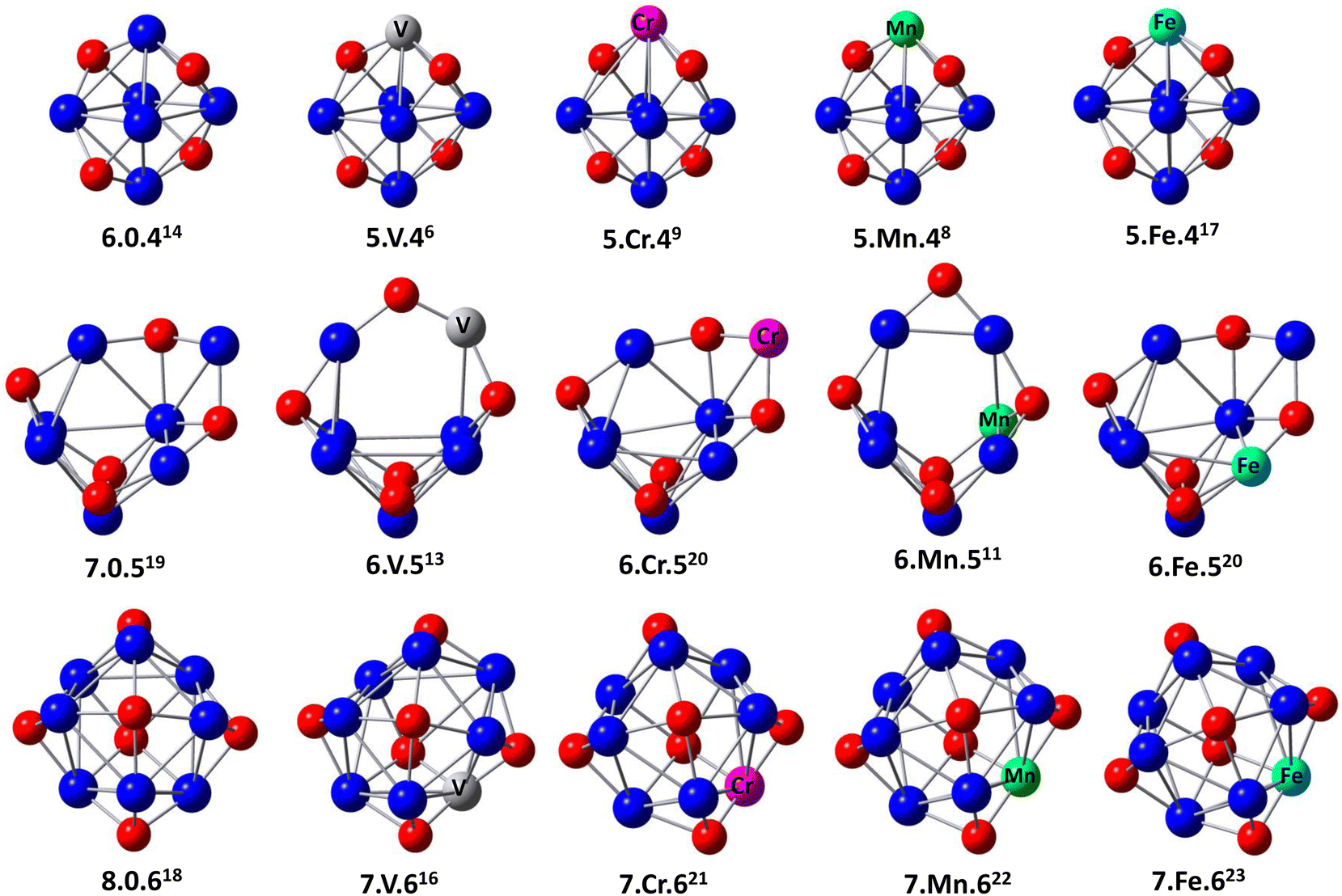 | ||
| Fig. 1 The optimized structures and spin multiplicities (superscript) of the lowest-energy isomers ConOn−2+ and Con−1TMOn−2+ (n = 6–8 and TM = V, Cr, Mn, and Fe) clusters. The structures are labeled as n.TM.n−2+, where n is defined as the number of Co atom, respectively. TM is labeled for transition metal atoms. The red, blue, dark gray, pink, green, and cyan spheres represent O, Co, V, Cr, Mn, and Fe atoms. | ||
Co6O4+ clusters have an octahedron-like structure, containing oxygen atoms occupying hollow sites, where the faces capped with an O atom are adjacent to those without an O atom. The Co7O5+ cluster features capped octahedral Co7 clusters, with each oxygen atom binding to three adjacent Co atoms to form triangular faces. A complex, tower-like structure is observed in the Co8O6+ cluster, which is composed of two connected pyramids, with one O atom at the top and four Co atoms in each pyramid vertex. These findings are in perfect agreement with the previous calculations reported by Aguilera-del-Toro and Mai.18,22
The process of substituting a transition metal atom with a cobalt one in ConOn−2+ clusters generates a large number of different structures and spin configurations. Most of the stable isomers Con−1TMOn−2+ negligibly change compared to those of ConOn−2+ clusters. This picture confirms that the initial frameworks of Co6O4+, Co7O5+, and Co8O6+ clusters are stable and highly symmetrical in structure. The oxygen atoms prefer to bind atomically to metal atoms in both pure and doped species until the number of oxygen–metal bonds is maximized to construct the sturdiest structures.23 Nevertheless, only the lowest relative energy isomers of Con−1TMOn−2+ clusters were discussed. In the ground state, V, Cr, Mn, and Fe are positioned at the top of the octahedron-like structure. The computed spin configurations for Co5VO4+, Co5CrO4+, Co5MnO4+, and Co5FeO4+ are sextets, nonet, octet, and 17-et, respectively.
In comparison with the Co7O5+ cluster, V and Mn doping disturbs the frame shape, causing an O atom to bind with two metals instead of three. Nevertheless, the Co6CrO5+ and Co6FeO5+ clusters retain the structure of their pure counterparts. The 6.V.5+ and 6.Mn.5+ clusters have lower spin configurations of 13-et and 11-et, respectively, while the 6.Cr.5+ and 6.Fe.5+ clusters exhibit higher spin configurations of 20-et.
For larger clusters, the ground-state structure of Co8O6+ indicates an 18-et spin state. The dopant atoms preferentially locate at the bridge of Co8O6+ and enhance the spin state to 21-et for 7.Cr.6+, 22-et for 7.Mn.6+, and 23-et for 7.Fe.6+, except for the 16-et spin state observed in the 7.V.6+ cluster.
It is noteworthy that oxygen prefers to occupy the O-TM-O sites in Con−1TMOn−2+. This preference can be attributed to the significantly larger binding energies of 3d transition metal–oxygen bonds. Specifically, the binding energies for metal–oxygen bonds are 6.50 ± 0.19 eV for V–O, 4.05 ± 0.18 eV for Cr–O, 4.18 ± 0.43 eV for Mn–O, 4.38 ± 0.21 eV for Fe–O, and 3.94 ± 0.16 eV for Co–O,31,32 whereas those for metal–metal ones are 1.43 eV for Co–Co,33 1.53 eV for Co–V,34 0.82 eV for Co–Cr,35 0.96 eV for Co–Mn.36
In addition to the results obtained using B3LYP/6-31+G(d), similar behaviors are also observed with other functionals/basis sets, as shown in the ESI.†30 These theoretical findings align closely with experimental results, reinforcing the observation that higher binding energies of the TM-O dimers cause oxygen atoms to preferentially form bonds with TM atoms over Co ones.
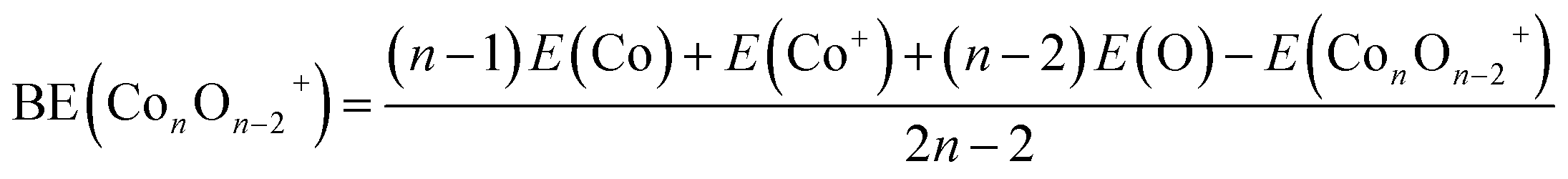 | (1) |
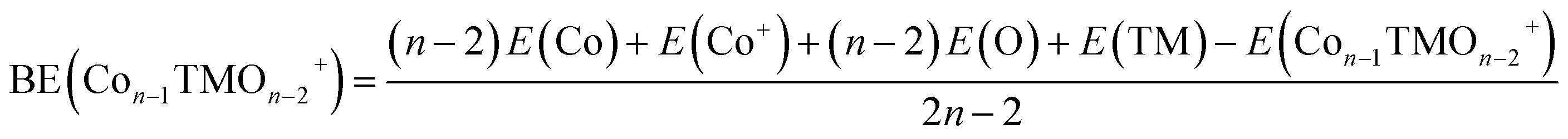 | (2) |
3.2 Cluster stability
The average binding energy per atom (BE) and dissociation energy (DE) are valuable parameters for determining cluster intrinsic stability.18,22,28,29,37–39 The BE in a cluster is considered as the total binding energy of the cluster divided by the number of atoms in the cluster. The corresponding BE values of ConOn−2+ and Con−1TMOn−2+ are defined and calculated by eqn (1) and (2), respectively. A higher BE generally indicates a more stable cluster, as it implies that each atom is more strongly bound to the cluster. Conversely, a lower BE suggests weaker interatomic bonds and, consequently, lower cluster stability. By comparing the BEs of different clusters, one can infer the relative stability and the effects of different compositions or sizes on the overall stability of the clusters. In this regard, doping significantly enhances the stability of cobalt oxide clusters. The stability enhancement is strongest for V-doped species and decreases gradually with the following order Con−1VOn−2+ > Con−1FeOn−2+ > Con−1CrOn+ − 2 > Con−1MnOn−2+, as shown in Table 1 and Fig. 2(a). This concept is now expanded to include V, Mn, and Fe-doped systems. Meanwhile, the relationship between BE and cluster size is also influenced by the presence of a dopant. For pure cobalt oxide species, the change of BE with size does not follow a specific trend, where Co7O5+ with a BE of 3.40 eV is the most stable one. Excepting for Con−1MnOn−2+, the doped clusters exhibit a monotonically increasing tendency in the BE with increasing cluster size. Since the geometric structures of pure clusters remain largely unchanged after doping, the most plausible explanation for this observation lies in the difference in the binding energy between TM-O and Co–O dimers: 6.50 ± 0.19 eV for V–O, 4.05 ± 0.18 eV for Cr–O, 4.38 ± 0.21 eV for Fe–O, 4.18 ± 0.43 eV for Mn–O, and 3.94 ± 0.16 for Co–O.31,32 A similar argument has been made for Cr-doped cobalt oxide clusters, successfully interpreting the increased binding energy in Cr-doped cobalt oxide clusters compared with pure oxide counterpart.11,23| Clusters | BE | DE(TM) | DE(Co) | DE(O) | DE(CoO) |
|---|---|---|---|---|---|
| Co6O4+ | 3.22 | — | 3.77 | 5.51 | 4.37 |
| Co5VO4+ | 3.52 | 6.76 | 3.02 | 8.03 | 5.85 |
| Co5CrO4+ | 3.39 | 5.41 | 3.47 | 8.08 | 6.44 |
| Co5MnO4+ | 3.35 | 5.05 | 3.05 | 6.35 | 5.94 |
| Co5FeO4+ | 3.44 | 5.92 | 3.95 | 7.24 | 6.13 |
| Co7O5+ | 3.40 | — | 4.20 | 6.10 | 5.37 |
| Co6VO5+ | 3.61 | 6.79 | 3.21 | 6.98 | 4.59 |
| Co6CrO5+ | 3.46 | 4.97 | 3.58 | 6.19 | 4.59 |
| Co6MnO5+ | 3.41 | 4.53 | 3.12 | 5.59 | 3.96 |
| Co6FeO5+ | 3.48 | 5.24 | 3.70 | 6.39 | 3.88 |
| Co8O6+ | 3.36 | — | 2.30 | 5.51 | 4.37 |
| Co7VO6+ | 3.65 | 6.45 | 2.99 | 6.39 | 4.33 |
| Co7CrO6+ | 3.50 | 4.33 | 2.93 | 6.95 | 4.03 |
| Co7MnO6+ | 3.41 | 3.03 | 2.13 | 5.19 | 3.25 |
| Co7FeO6+ | 3.52 | 4.57 | 2.97 | 6.86 | 4.00 |
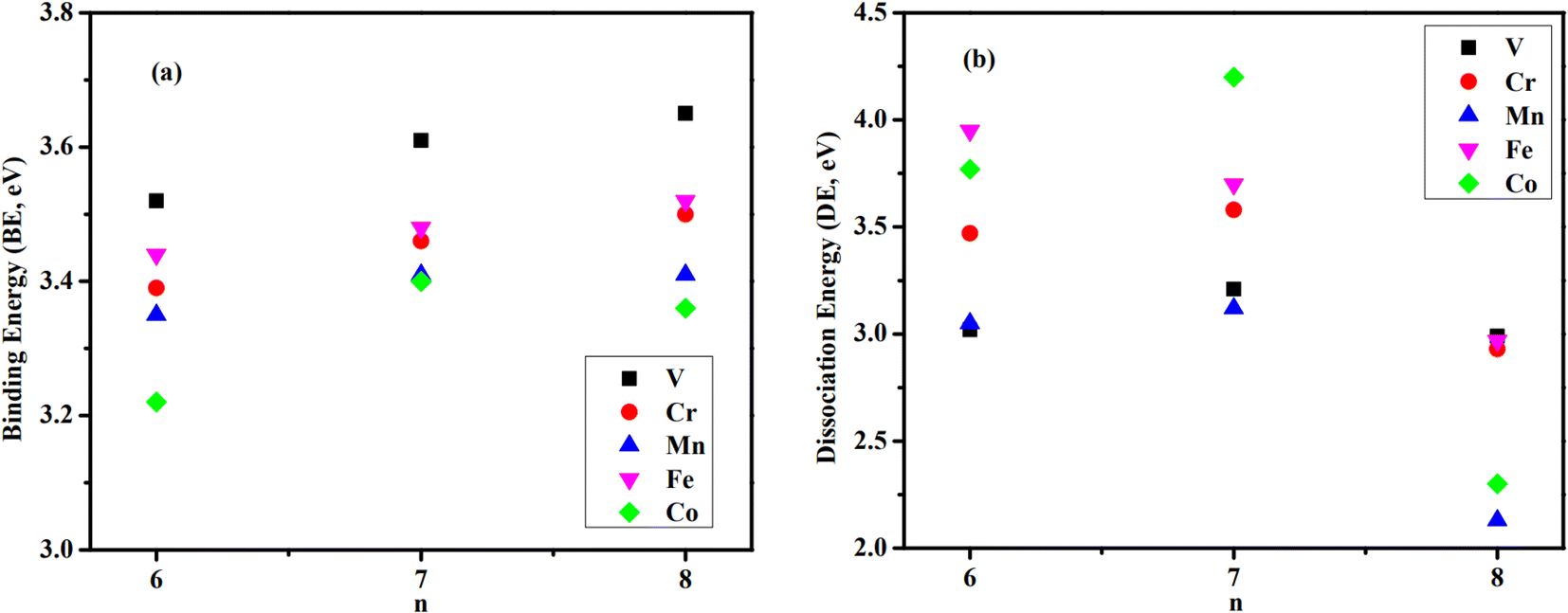 | ||
| Fig. 2 Binding energies (BE, in eV) (a) and lowest dissociation energies (DE, in eV) (b) of Con−1TMOn−2+ clusters correspond to the decay of a Co atom (n = 6–8, TM = V, Cr, Mn, Fe, and Co). | ||
While the BE measures how each atom on average bonds to the cluster, DE indicates the cluster's stability by quantifying the energy required to break it into smaller fragments or individual atoms.40–42 Under certain circumstances, clusters absorb energy and are promoted to an excited (electronic and/or vibrational) state. Instantly, the excited cluster goes through various transition states and finally enters the exit channel before dissociating into smaller fragments. The minimum energy required to trigger dissociation is referred to DE and in calculations is defined as the difference in energy between the parent cluster and the sum of the energies of its fragments. The DE associated with the most fragile dissociation channel represents the strength of the weakest bonds within the cluster and is a crucial parameter for assessing intrinsic stability. A higher DE for the most facile dissociation channel indicates the cluster is less likely to fragment under the same external stimuli, exhibiting greater stability. If there are no barriers in the dissociation reaction, the evaporative rate constant strongly depends on DE. Consequently, the channel with the lower DE is likely to correspond to the preferred evaporation pathway. By comparing the dissociation energies of different clusters, one can infer their relative stability and resilience under various conditions, i.e., collision-induced and photo-induced dissociation.43,44
Fundamentally, a parent cluster can dissociate into daughter clusters via possible decay channels. The daughter associated with the channel that requires lower DE and/or appears more frequently as one of the fragments is often identified with enhanced stability. In most cases, dissociation involving dimer or trimer fragments rarely took place.20,21 Additionally, since the ionization energy of larger species is lower than that of smaller ones, the charge often resides on the remaining clusters rather than on the fragment during the dissociation process.22,23 With this picture in mind, we calculated DEs of bare and doped cobalt oxide clusters in the four most likely dissociation channels, including the loss of a Co atom, a TM atom, an O atom, and CoO dimer, as per the following equations:
| Con−1TMOn−2+ → Con−2TMOn−2++ Co | (3) |
| Con−1TMOn−2+ → Con−1On−2+ + TM | (4) |
| Con−1TMOn−2+ → Con−1On−3+ + O | (5) |
| Con−1TMOn−2+ → Con−2On−3+ + CoO | (6) |
The calculated results are listed in Table 1 and Fig. 2(b). The energies are computed based on the parent and daughter clusters' lowest energy structures and spin states at 0 K. Potential reverse barriers along the dissociation pathway have not been taken into account. For both pure and doped species, the loss of a Co atom is identified as the most fragile dissociation channel, while the evaporation of an oxygen atom is the least likely. At first glance, this result appears reasonable, considering that the Co–O bond is the weakest. The evaporation of larger fragments, such as the CoO dimer, is also unlikely to occur, as discussed above. In particular, the preferred decay channel of Co6O4+, Co7O5+, and Co8O6+ is via the decay of a Co atom, with corresponding DEs of 3.77, 4.20, and 2.30 eV, respectively. Nevertheless, when a TM atom is introduced, although the most fragile channel remains unchanged, the relative stability of clusters is altered.
For Co5TMO4+ clusters, it requires only 3.02, 3.05, and 3.47 eV to release a Co atom from Co5VO4+, Co5MnO4+, and Co5CrO4+, respectively, making these species remarkably less stable than their pure counterpart. The only exception is Co5FeO4+, which is relatively more stable than the pure one, requiring at least 3.95 eV to decay into Co4FeO4+ and Co. While the Co dissociation channel occurs more easily for V, Cr, and Mn-doped species, their CoO/O dissociation channels require greater dissociation energy compared with Co6O4+ and even higher than the energy required to dissociate the TM atom. This finding suggests the likely formation and relative stable of Co4TMO4+ rather than that of Co4TMO3+, Co5TMO3+, or Co5O4+.
On the other hand, introducing the dopant remarkably destabilizes the Co7O5+ cluster. The required DE to evaporate a Co atom decreases from 4.20 eV to 3.70, 3.58, 3.21, and 3.12 eV when substituting one Co atom with Fe, Cr, V, and Mn atoms, respectively. The loss of CoO dimer is the second most preferred channel after the evaporation of a Co atom. Interestingly, the DE for the channel involving the loss of CoO from Co6FeO5+ is only 3.88 eV, which is significantly lower than other n = 7 species and comparable to that of the most fragile channel (evaporating a Co atom at 3.70 eV). This implies that the resulting daughters, Co5FeO5+ and Co5FeO4+, are relatively more stable than Co6FeO4+ and Co6O4+, consistent with above-calculated DEs for Co5FeO4+ and Co6O4+. The evaporation of a TM atom requires more energy than a Co atom or a CoO dimer, but it is more likely to occur than the evaporation of an O atom. The destabilization of Co7O5+ due to doping could be attributed to the structural distortion in the n = 7 series. Doping with V or Mn alters the structure of Co7O5+ such that an outer O atom binds with two metal atoms instead of three. This structural change likely accounts for the considerably lower DEs of Co6VO5+ and Co6MnO5+ compared to those of other n = 7 species. In contrast, Co6CrO5+ and Co6FeO5+ retain the structure of their pure counterpart, resulting in higher DEs than those of V and Mn-doped clusters.
The loss of Co remains the most fragile dissociation channel for n = 8 species. Doping with a TM atom enhances the stability of Co8O6+ except for Co7MnO6+. In particular, the corresponding DEs for Co8O6+, Co7VO6+, Co7CrO6+, Co7MnO6+, and Co7FeO6+ are 2.30, 2.99, 2.93, 2.13, and 2.97 eV, respectively. With 2.99 eV required to dissociate Co7VO6+ is identified as the most stable one while Co7MnO6+ is the most fragile species since it needs only 2.13 eV to decay into Co6MnO6+ and a Co atom. The formation of Co7O6+, Co6MnO5+, and Co7MnO5+ from the dissociation of Co7MnO6+ is relatively favored with only 3.03, 3.25, and 5.19 eV, respectively. In the case of the two dissociation channels which exhibit nearly equal dissociation energies, the thermodynamic entropy for the dissociation reaction of the Con−1TMOn−2+ clusters is calculated to determine the preferred channel, as listed in Table S3.† The fact that the increase in entropy gradient through the dissociation channels leading to the evaporation of an O atom and CoO molecule is greater than through the dissociation channel leading to the evaporation of Co and TM ones. The dissociation involving a Co atom becomes the most thermodynamically favorable pathway. This picture is completely consistent with the dissociation energy calculations mentioned previously.
3.3 Magnetic properties
The 3d transition metal atoms (V, Cr, Mn, and Fe) are spontaneous magnetic elements owing to their unpaired 3d electrons. Experiments on Co, and Fe clusters with fewer than around 1000 atoms demonstrate that the ferromagnetic state is preferred.45–48 The total magnetic moments of transition metal clusters composed of at least two transition metal elements tend to be stronger due to the favored parallel magnetic coupling with larger per-atom spin polarization.18 The local magnetic moment on each atom of the ground-state ConOn−2+ and Con−1TMOn−2+ clusters is calculated and shown in Fig. 3 and the ESI.†30 Previous DFT calculations have shown that the majority of Co oxide clusters exhibit the ferromagnetic state.22,49 This finding has been recently confirmed by Stern–Gerlach experiments,19 confirming the role of oxidation on the magnetic response of the oxide clusters. Our calculation results support the ferromagnetic of ConOn−2+ as shown in Fig. 3. As the number of Co atoms in the cluster increases, its magnetism does not comply with a monotonously increasing trend but fluctuates regardless of the size of the cluster. This is in line with the study of De Knijf et al.19 The reduction in spin multiplicity from 19-et to 18-et corresponding to from Co7O5+ to Co8O6+ was similarly observed in the study by Aguilera-del-Toro et al.,18 this phenomenon may partially stem from increasing the paired electrons due to direct electron exchange between 2p-O and 3d-Co orbitals. The ferromagnetic nature of the ConOn−2+ clusters still remain in the presence of TM atoms, nevertheless, the enhancement and decrease strongly depend on the spin contribution of doping atoms. Fig. 3 that there is a reduction in the spin moment of V-doped clusters (n = 6, 7, and 8). This change is explained by the decrease in the localized electron on the V atom during oxidation as well as forming the Co–O–V bond. Consequently, the 3d34s2 valence electron shell of the V atom is partially filled while the unpaired electrons on the Co change almost slightly due to the low oxygen concentration in the clusters. Taking a glance that the V atom decreases the total spin moment of ConOn−2+ clusters due to the antiparallel orientation of local spins in the V atom compared to other metal atoms, resulting in a ferrimagnetic state. Conversely, the presence of the Fe atom enhances the magnetism because the number of unpaired electrons on the Fe atom (3d64s2) is larger than that on the Co atom (3d74s2). As a result, replacing a Co atom by a Fe atom tends to increase the magnetic moment. The size-dependent improved magnetism is also observed in Con−1CrOn−2+ and Con−1MnOn−2+ clusters, typically, with n = 6, the magnetism significantly goes down, n = 8 the magnetism goes up, and n = 7 the decrease or increase is manipulated by the Cr and Mn atoms. Our study have demonstrated that the magnetism in cobalt oxide strongly depend on the number of metal–oxygen bonds and geometrical shape. In addition, it is significantly governed by the size, composition, and oxygen concentration, including the oxygen-deficient, oxygen-balanced, and oxygen-rich states. This work is a crucial premise for predicting the magnetic properties of cobalt oxide clusters before synthesizing and conducting practical applications.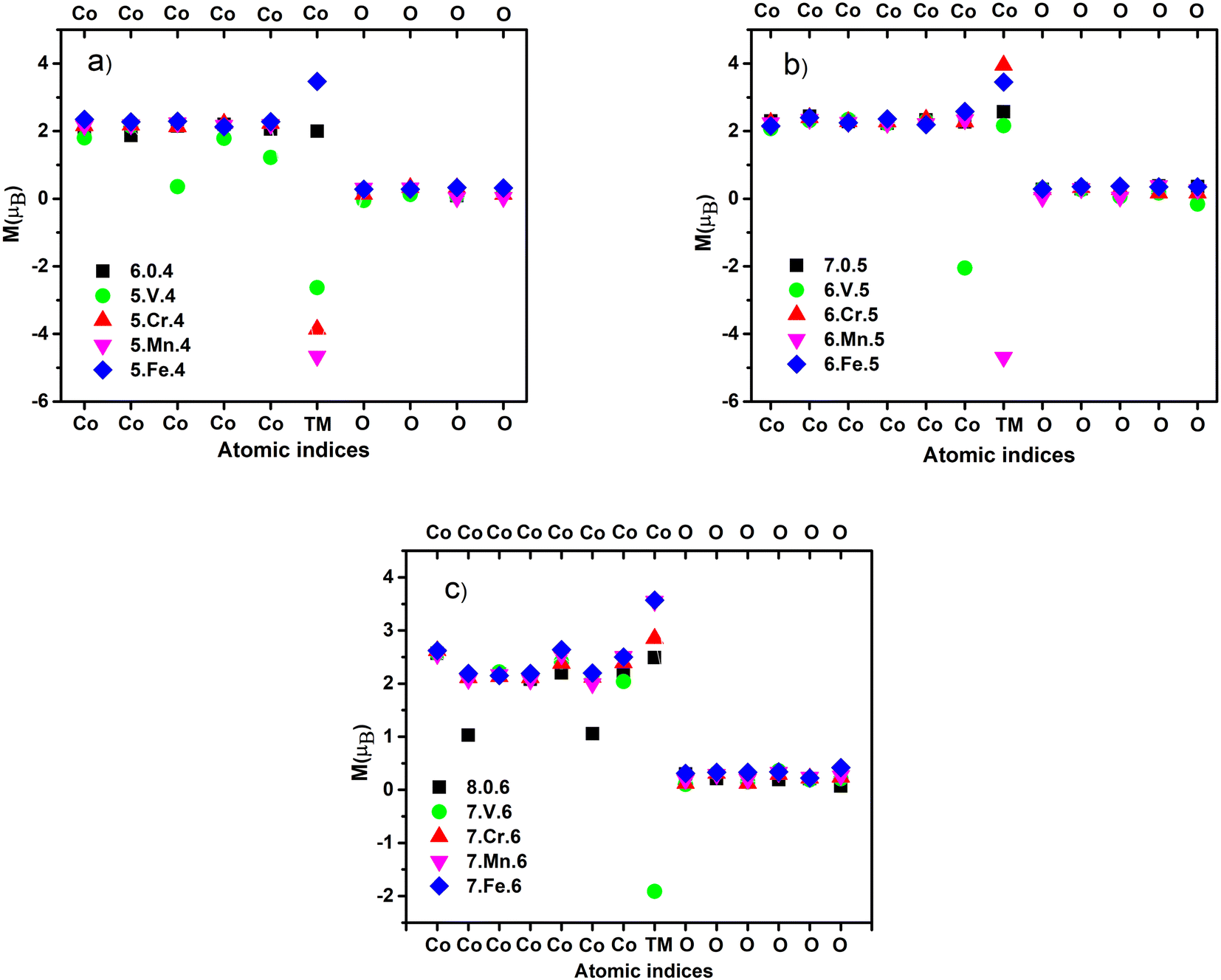 | ||
| Fig. 3 Calculated local magnetic moment [M (μB)] of the ground-state Co5TMO4+ and Co6O4+ (a); Co6TMO5+ and Co7O5+ (b); Co7TMO6+ and Co8O6+ (c) (TM = V, Cr, Mn and Fe) clusters. | ||
We calculated the local magnetic moment [M (μB)] and spin distribution on each atom for the ground-state clusters. One sees that the magnetic magnitude of the atoms in the un-doped clusters all has positive values, however negative values are observed in the doped clusters, giving rise to the change in magnetic behavior of the doped clusters. Generally, the calculated results point out that the magnetic properties are driven by the bond formation between atoms, and geometrical and electronic structures of clusters, being consistent with several previous articles.10–12,18 Pham et al.,11 reported that the total magnetic moments of (x + y = 2, 3 and 1 ≤ m ≤ 4) clusters were manipulated by the number of metal–oxygen bonds and the symmetry in structure. Additionally, Torres et al.50 suggested that the antiparallel spin ordering may emerge as the metal–oxygen bonds are enforced and concurrently the metal–metal bonds are weakened.29 In this context, our calculations reveal that the binding energy of V–O (3.14 eV) is two times larger than that of Co–O (1.59 eV), but the V–Co bond (0.77 eV) is smaller than the Co–Co bond (0.87 eV). The opposite propensity of local spin moment in V atom was found for three types of clusters studied. Albeit, analogous argument is in line with Cr- and Mn-doped Co5TMO4+ clusters, the ferromagnetic or antiferromagnetic spin alignment in the Cr and Mn atoms compared to others upon increasing the number of Co and O atoms reflects the complexity in the spin moment evolution. This can be highly related to the unpaired 3d electrons of TM, which do not participate in the formation of bonds, and electronic states contributed from oxygen and metal atoms in cluster.11 For instance, the ground state spin of Mn atom in Co6MnO5+ cluster maintains an opposite direction of the other atoms like as that in Co5MnO4+ cluster, while the spin is triggered to align in the same direction as the atoms in cluster Co7MnO6+.In the Co5TMO4+ (TM = Co, V, Cr, Mn, and Fe) clusters, the magnetic magnitude in the Co6O4+ cluster is mainly contributed by Co atoms with the magnitude of around 2 μB shown in Fig. 3(a). Redistribution of the magnetic moment occurs in the Co5VO4+, whereas the magnitude significantly decreases in two Co atoms. For the Co5CrO4+, Co5MnO4+, and Co5FeO4+, the local magnetic contribution of Co and O atoms is in analogy to those in the pure cluster. Since the Cr, Mn and Fe have the number of unpaired 3d electrons more than that in the Co, these doped atoms offer a higher magnetic magnitude than the Co atom. Specifically, the magnitude of Cr and Mn atoms is above twofold the that of Co atom. Nevertheless, excepting Fe, the Cr, Mn and V indicate negative values, confirming that these three atoms have an antiparallel magnetic moment orientation. It can be hence concluded that the doping of V, Cr and Mn causes the detriment about the magnetization. A similar propensity is unraveled in the Co6TMO5+ clusters. Excluding the Mn atom, which maintains a negative value, the three atoms V, Cr and Fe receive a positive value in magnetism. In contrast to the Co5VO4+ and Co7VO6+ clusters, the M (μB) of V atom has a value greater than 0 in the Co6VO5+ clusters. Herein, the compensation rule is observed in this cluster in which the M (μB) of a Co and V atoms gets opposite equal values, leading to mutual cancellation. Therefore, the presence of Fe and Cr atoms gives rise to magnetic enhancement because the magnetic moments of Fe and Cr are greater than that of Co. The behavior of V in the Co7VO6+ occurs analogically to that in the Co5VO4+ and, no negative value is found in other atoms in the Co7TMO6+ clusters. Notwithstanding, it can be seen that there are two Co atoms with magnitudes less than half of the remaining Co atoms in the Co8O6+, therefore its magnetization is lower than that of the Co7O5+.
To provide insight into the magnetism behavior of Con−1TMOn−2+ clusters, the local spin distribution was computed. Fig. 4 is a result describing the local spin density distribution on each atom of Con−1TMOn−2+ clusters (TM = V, Cr, Mn, and Fe), which was plotted at a density of 0.04 using the spin distribution analysis. The presence of metal–oxygen or metal–metal bonds can lead to the existence of localized spin moments in parallel or antiparallel ordering in some certain conditions of cobalt oxide clusters. In our work, parallel spin sates are assigned to yellow electron clouds (α spin) in Fig. 4, whereas antiparallel spin sates are labeled to blue ones (β spin). Obviously, all doped atoms have a certain spin contribution to the atomic cluster. Whereas, the local spin density is principally distributed on the transition metal atoms of Co, Cr, Mn and Fe. The spin density on the doped atoms is larger than the Co atoms, as a result, they significantly tune the magnetic properties of the cluster. This is in excellent consistency with the above calculation results. In clusters of Con−1VOn−2+, Co5Cr/MnO4+, Co6MnO5+, the V, Cr and Mn significantly contribute β spins which have an antiparallel orientation with the other atoms in clusters, giving rise to decreased total magnetic moment. Therefore, the transition from the ferromagnetic to ferrimagnetic states is observed in these clusters. Conversely, the parallel magnetic couplings are found in the other clusters because the Cr, Mn and Fe atoms offer α spins.
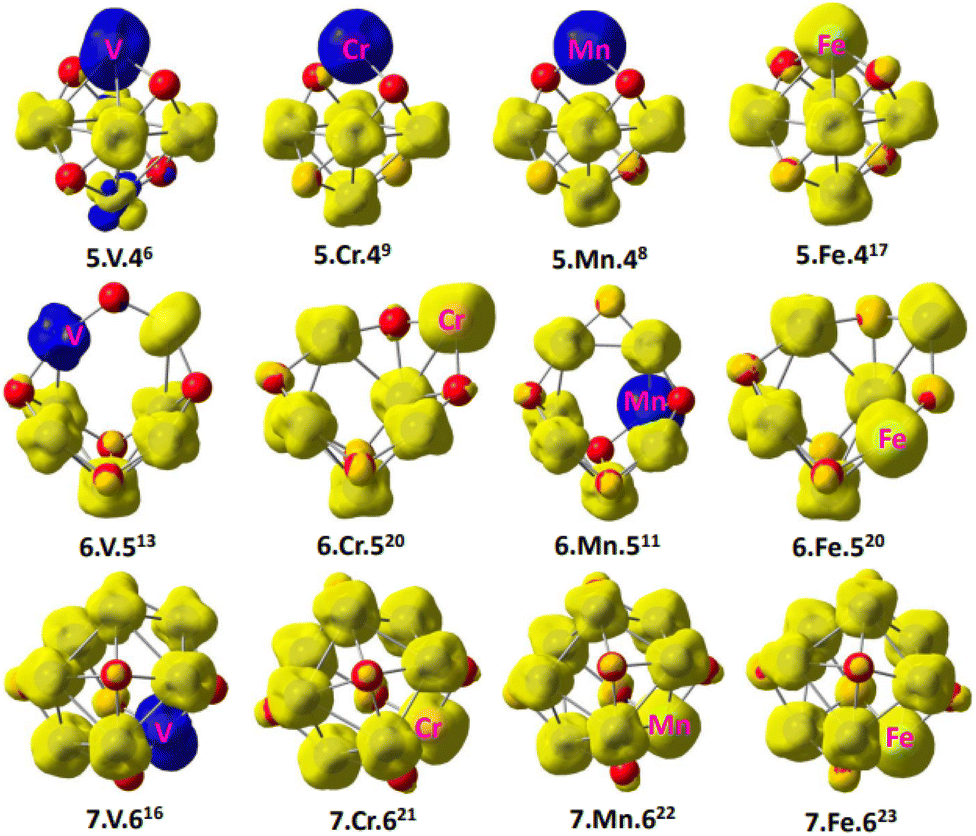 | ||
| Fig. 4 The local spin distribution of Con−1TMOn−2+ (n = 6–8 and M = V, Cr, Mn, and Fe), the yellow color stands for α spin while the blue one represents β spin. | ||
4 Conclusion
The hybrid functional B3LYP in combination with 6-31+G(d) basis set was applied to investigate the geometric structure and magnetic properties of Con−1TMOn−2+ (n = 6–8) (TM = V, Cr, Mn, and Fe) clusters. The presence of the TM atoms enhances the stability of atomic clusters, whereas the TM atom preferentially replace Co atom at the sites that the number of metal–oxygen bonds is maximal and negligibly change the initial framework. In addition, the calculated results indicate that the magnetic properties are tunable under the impact of around electronic environment, geometric structure and TM atom. The V-doped clusters undergo a the ferromagnetic–ferrimagnetic state transition. The ferromagnetic state is enhanced in Fe-doped clusters. These findings have brought out an attractive research orientation in catalytic application using magnetic cobalt oxide-based materials.Author contributions
Nguyen Thi Mai: conceptualization, data curation, formal analysis, investigation, validation, writing-original draft, review and editing. Tran Dang Thanh: conceptualization, funding acquisition, data curation, validation, writing-original draft, review and editing. Do Hung Manh: data curation, formal analysis, investigation, validation, review and editing. Nguyen Thi Ngoc Anh: data curation, formal analysis, investigation, validation, review and editing. Ngo Thi Lan: data curation, formal analysis, investigation, validation, writing-original draft, review and editing. Phung Thi Thu: data curation, formal analysis, investigation, validation, writing-original draft, review and editing. Nguyen Thanh Tung: conceptualization, data curation, formal analysis, investigation, validation, writing-original draft, review and editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the financial support of the Vietnam Academy of Science and Technology under the grant number: NCXS01.04/22-24.Notes and references
- C. N. Rao and B. Raveau, Transition Metal Oxides, Wiley, New York, 1998 Search PubMed.
- P. A. Cox, Transition Metal Oxides, Clarendon, Oxford, 1992 Search PubMed.
- S. Gupta, R. Fernandes, R. Patel, M. Spreitzer and N. Patel, A review of cobalt-based catalysts for sustainable energy and environmental applications, Appl. Catal., A, 2023, 661, 119254 CrossRef CAS.
- V. Skumryev, S. Stoyanov, Y. Zhang, G. Hadjipanayls, D. Givord and J. Nogues, Beating the superparamagnetic limit with exchange bias, Nature, 2003, 423, 850–853 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nano-sized transition-metal oxides as negative-electrode materials for lithium-ion batteries, Nature, 2000, 407, 496–499 CrossRef CAS.
- Q. Liu, et al., Approaching the capacity limit of lithium cobalt oxide in lithium ion batteries via lanthanum and aluminium doping, Nat. Energy, 2018, 3, 936 CrossRef CAS.
- A. Alivisatos, Semiconductor clusters, nanocrystals, and quantum dots, Science, 1996, 271, 933–937 CrossRef CAS.
- Y. Zhang, W. Li, J. Wang, J. Jin, Y. Zhang, J. Cheng and Q. Zhang, Advancement in utilization of magnetic catalysts for production of sustainable biofuels, Front. Chem., 2023, 10, 1106426 CrossRef PubMed.
- M. Zhang, H. M. Guo, J. Lv, J. F. Jia and H. S. Wu, The 3d transition-metals doping tunes the electronic and magnetic properties of 2D monolayer InP3, J. Magn. Magn. Mater., 2021, 533, 168026 CrossRef CAS.
- N. T. Lan, N. T. Mai, N. V. Dang and N. T. Tung, DFT investigation of pyramidal Au9M2+ and Au19M (M = Sc–Ni): similarities and differences of structural evolution, electronic and magnetic properties, Commun. Phys., 2023, 33, 63–72 CrossRef.
- H. T. Pham, N. T. Cuong, N. M. Tam, V. D. Lam and N. T. Tung, Structure, magnetism, and dissociation energy of small bimetallic cobalt-chromium oxide cluster cations: A density-functional-theory study, Chem. Phys. Lett., 2016, 643, 77–83 CrossRef CAS.
- N. T. Lan, N. T. Mai, N. T. Cuong, P. T. H. Van, D. D. La, N. M. Tam, S. T. Ngo and N. T. Tung, Density Functional Study of Size-Dependent Hydrogen Adsorption on AgnCr (n = 1–12) Clusters, ACS Omega, 2022, 7(42), 37379–37387 CrossRef CAS.
- N. T. Mai, N. T. Lan, N. T. Cuong, N. M. Tam, S. T. Ngo, T. T. Phung, N. V. Dang and N. T. Tung, Systematic Investigation of the Structure, Stability, and Spin Magnetic Moment of CrMn Clusters (M = Cu, Ag, Au, and n = 2–20) by DFT Calculations, ACS Omega, 2021, 6, 20341 CrossRef CAS.
- N. T. Lan, N. T. Mai, D. D. La, N. M. Tam, S. T. Ngo, N. T. Cuong, N. V. Dang, T. T. Phung and N. T. Tung, DFT investigation of Au9M nanoclusters (M = Sc–Ni): The magnetic superatomic behavior of Au9Cr, Chem. Phys. Lett., 2022, 793, 139451 CrossRef CAS.
- S. Lee, A. Halder, G. A. Ferguson, S. Seifert, R. E. Winans, D. Teschner, R. Schlogl, V. Papaefthimiou, J. Greeley, L. A. Curtiss and S. Vajda, Subnanometer cobalt oxide clusters as selective low temperature oxidative dehydrogenation catalysts, Nat. Commun., 2019, 10, 954 CrossRef PubMed.
- S. K. Sharma, H. T. Ahangari, B. Johannessen, V. B. Golovko and A. T. Marshall, Au cluster-derived electrocatalysts for CO2 reduction, Electrocatalysis, 2023, 14, 611–623 CrossRef CAS.
- A. W. Peters, K. Otake, A. E. Platero-Prats, Z. Li, M. R. DeStefano, K. W. Chapman, O. K. Farha and J. T. Hupp, Site-directed synthesis of cobalt oxide clusters in a metal–organic framework, ACS Appl. Mater. Interfaces, 2018, 10, 15073 CrossRef CAS.
- R. H. Aguilera-del-Toro, F. Aguilera-Granja, A. Vega and L. C. Balbás, Structure, fragmentation patterns, and magnetic properties of small cobalt oxide clusters, Phys. Chem. Chem. Phys., 2014, 16, 21732–21741 RSC.
- K. De Knijf, J. van der Tol, P. Ferrari, S. Scholiers, G. L. Hou, P. Lievens and E. Janssens, Influence of oxidation on the magnetism of small Co oxide clusters probed by Stern–Gerlach Deflection, Phys. Chem. Chem. Phys., 2023, 25, 171 RSC.
- C. N. van Dijk, D. R. Roy, A. Fielicke, T. Rasing, A. C. Reber, S. N. Khana and A. Kirilyuk, Structure investigation of CoxOy+ (x = 3-6, y = 3-8) clusters by IR vibrational spectroscopy and DFT calculations, Eur. Phys. J. D, 2014, 68, 357 CrossRef.
- K. Ota, K. Koyasu, K. Ohshimo and F. Misaizu, Structures of cobalt oxide cluster cations studied by ion mobility mass spectrometry, Chem. Phys. Lett., 2013, 588, 63–67 CrossRef CAS.
- N. T. Mai, S. T. Ngo, P. Lievens, E. Janssens and N. T. Tung, Photofragmentation patterns of cobalt oxide cations ConOm+(n = 5-9, m = 4-13): From oxygen-deficient to oxygen-rich species, J. Phys. Chem. A, 2020, 124, 7333–7339 CrossRef CAS PubMed.
- N. T. Tung, N. M. Tam, M. T. Nguyen, P. Lievens and E. Janssens, Influence of Cr doping on the stability and structure of small cobalt oxide clusters, J. Chem. Phys., 2014, 141, 044311 CrossRef.
- A. Nirmalesh Naveen, Subramanian Selladurai, Tailoring structural, optical and magnetic properties of spinel type cobalt oxide (Co3O4) by manganese doping, Phys. B, 2015, 457, 251–262 CrossRef.
- P. A. Ignatiev, N. N. Negulyaev, D. I. Bazhanov and V. S. Stepanyuk, Doping of cobalt oxide with transition metal impurities: Ab initio study, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 235123 CrossRef.
- Gaussian, https://gaussian.com/glossary/g09/ Search PubMed.
- P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev., 1964, 136(3B), B864 CrossRef.
- N. F. Shen, Y. B. Wang, S. Chen and J. L. Wang, Structural and magnetic properties of bimetallic Con−1 Cr clusters with density functional theory, Front. Phys. China, 2009, 4, 408–414 CrossRef.
- N. T. Tung, E. Janssens and P. Lievens, Dopant dependent stability of Co n TM+(TM= Ti, V, Cr, and Mn) clusters, Appl. Phys. B: Lasers Opt., 2014, 114, 497–502 CrossRef CAS.
- ESI†.
- Comprehensive Handbook of Chemical Bond Energies, ed. Y. R. Luo, CRC Press, BocaRaton, FL, 2007 Search PubMed.
- Handbook of Chemistry and Physics, Internet Version, ed. D. R. Line, CRC Press, Boca Raton, FL, 2005 Search PubMed.
- J. Lv, F.-Q. Zhang, J.-F. Jia, X.-H. Xu and H.-S. Wu, J. Mol. Struct.: THEOCHEM, 2010, 955, 14–22 CrossRef CAS.
- S. Datta, M. Kabir, T. Saha-Dasgupta and A. Mookerjee, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 085418 CrossRef.
- N. Shen, Y. Wang, S. Chen and J. Wang, Front. Phys. China, 2009, 4, 408 CrossRef.
- G. L. Gutsev, M. D. Mochena, P. Jena, C. W. Bauschlicher and H. Partridge III, J. Chem. Phys., 2004, 121, 6785 CrossRef CAS PubMed.
- A. Kant and B. Strauss, Dissociation energies of diatomic molecules of the transition elements. II. Titanium, chromium, manganese, and cobalt, J. Chem. Phys., 1964, 41(12), 3806–3808 CrossRef CAS.
- S. Datta, M. Kabir, T. Saha-Dasgupta and A. Mookerjee, Structure, reactivity, and electronic properties of V-doped Co clusters, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80(8), 085418 CrossRef.
- N. T. Tung, E. Janssens, S. Bhattacharyya and P. Lievens, Photofragmentation of mass-selected vanadium doped cobalt cluster cations, Eur. Phys. J. D, 2013, 67, 1–6 CrossRef.
- S. Molek, T. D. Jaeger and M. A. Duncan, Photodissociation of vanadium, niobium, and tantalum oxide cluster cations, J. Chem. Phys., 2005, 123, 144313 CrossRef PubMed.
- S. Molek, Z. D. Reed, A. M. Ricks and M. A. Duncan, Photodissociation of chromium oxide cluster cations, J. Phys. Chem. A, 2007, 111, 8080 CrossRef.
- S. Molek, C. Anfuso-Cleary and M. A. Duncan, Photodissociation of iron oxide cluster cations, J. Phys. Chem. A, 2008, 112, 9238 CrossRef.
- J. Dibble, S. T. Akin, S. Ard, C. P. Fowler and M. A. Duncan, Photodissociation of Cobalt and Nickel Oxide Cluster Cations, J. Phys. Chem. A, 2012, 116, 5398 CrossRef.
- B. Freas, B. I. Dunlap, B. A. Waite and J. E. Campana, The role of cluster ion structure in reactivity and collision-induced dissociation: Application to cobalt/oxygen cluster ions in the gas phase, J. Chem. Phys., 1987, 86, 1276 CrossRef.
- D. M. Cox, D. J. Trevor, R. L. Whetten, E. A. Rohlfing and A. Kaldor, Magnetic behavior of free-iron and iron oxide clusters, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 7290 CrossRef CAS.
- K. Kimura, Magnetic properties of iron: from clusters to bulk, Phys. Lett. A, 1991, 158, 85–88 CrossRef CAS.
- J. P. Bucher, D. C. Douglass and L. A. Bloomfield, Magnetic properties of free cobalt clusters, Phys. Rev. Lett., 1991, 66, 3052 CrossRef CAS.
- A. Heimermann and C. van Wullen, Magnetic moments of small cobalt clusters revisited: The contribution of 3d and 4s electrons, Int. J. Mass Spectrom., 2019, 438, 135 CrossRef CAS.
- M. N. Daud, Structural, Electronic and Magnetic Properties of Stoichiometric Cobalt Oxide Clusters (CoO)nq (n = 3–10; q = 0; +1): A Modified Basin-Hopping Monte Carlo Algorithm with SpinPolarized DFT, J. Theor. Comput. Chem., 2019, 1, 1950003 CrossRef.
- M. B. Torres, A. Aguado, F. A. Granja, A. Vega and L. C. Ballás, Structural, Vibrational, and Magnetic Properties of FeCoOn0/+(n = 1–6) Bimetallic Oxide Clusters, J. Phys. Chem. C, 2015, 119, 11200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05482b |
| This journal is © The Royal Society of Chemistry 2024 |