
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring dual cross-linked polymer-ionic liquid composites by blending co-crystallizable polymers for stretchable electronics†
Minjun Kim‡
 ,
Moonsung Park‡,
Hobin Seon,
Sohyun Choi,
Hee Joong Kim* and
Sangwon Kim*
,
Moonsung Park‡,
Hobin Seon,
Sohyun Choi,
Hee Joong Kim* and
Sangwon Kim*
Department of Polymer Science and Engineering, Program in Environmental and Polymer Engineering, Inha University, Incheon 22212, Republic of Korea. E-mail: heejoong@inha.ac.kr; sangwon_kim@inha.ac.kr
First published on 11th November 2024
Abstract
Facile adjustment of the behavior of dual cross-linked polymer-ionic liquid composites (PICs) for stretchable electronics was achieved via solution blending of two copolymers having the same monomer pairs. Two poly(docosyl acrylate-r-tert-butyl acrylate) (poly(A22-r-tBA)) copolymers with different molar ratios were synthesized and solution-cast with ionic liquids (ILs) to fabricate ternary PICs. The phase behavior and the thermal and structural properties of the composites were investigated by varying the mixing ratio, providing insights into the cross-linking mechanisms. The observed changes enabled systematic modulation of the stretchability, thermal stability, and self-healing capability of PICs, which are crucial attributes of wearable devices. Mechanically tough and conductive PICs were utilized in fabricating strain sensors capable of detecting human motion.
1. Introduction
Wearable electronics are poised to become mobile assistants, doctors, and constant companions for humans through the development of personalized electronic devices. For effective communication, these devices need to be conformally attached to human bodies. Considering the inherent difference in the mechanical properties between conventional, rigid electronics and soft, curvilinear human bodies, various methods have been devised to achieve stretchable and flexible electronic devices.1 Electronic conductivity is generally imparted by hybridizing the conductive components of various structures (e.g., Ag nanowires, carbon nanotubes, serpentine Au) with soft matrices or substrates. On the other hand, hydrogels and ion gels (or ionic liquid (IL) composites), which are network structures swollen with water and ILs, respectively, have been employed for soft electronics owing to their unique characteristics, encompassing intrinsic malleability and conformality to any given shape. ILs in the substrates are characterized by low vapor pressure and thermal/chemical stability. Furthermore, ILs impart ionic conductivity to the composites, making them applicable in various wearable applications such as sensors and light-emitting devices.2 Moreover, the mechanical and other functional properties of the composites can be readily tuned by adjusting the IL content and the type of network cross-linking.Various semicrystalline polymers have been employed as network scaffolds in the fabrication of polymer-IL composites (PICs). Examples include poly(vinylidene fluoride-co-hexafluoropropylene) (P(VDF-co-HFP)),3 polyamide,4 and polycaprolactone.5 The crystalline domains formed by the backbone chains constitute the physical cross-links, and the properties of the composite are determined by the crystalline morphology and the degree of crystallinity. Furthermore, interactions among side chains in comb-shaped polymers provide access to a wide spectrum of properties that significantly differ from those of linear polymers.6 The crystallization of long alkyl side chains and their applications have been extensively studied. Jordan et al. studied the dependence of side-chain crystallization on the content and chemical structure of randomly interspersed amorphous co-units in copolymers.7 A progressive decline in the crystallinity was observed as the number of incorporated amorphous units increased, which agreed with the trend observed in the copolymers with crystallizable main chains. However, the level of suppression was found to be much more gradual compared to main-chain crystallization, allowing the copolymers to retain their crystallinity over a wide range of compositions. The crystallization behavior of copolymers containing alkyl (meth)acrylates has been utilized in various applications. Wrigley and coworkers reported that crystallizable alkyl side chains in copolymers are capable of co-crystallizing with compounds having similar chemical structures, and this ability has enabled the common use of such copolymers as pour-point depressants in oils.8 Hattori et al. studied the microphase segregation in bulk random copolymers with octadecyl pendants and observed nanostructures with a domain spacing of 5–6 nm.9 Notably, the size and morphology were independent of the chain length. This differs from the phase behavior typically exhibited by linear block copolymers in bulk and thin-film states.10,11 Osada and Matsuda observed a shape memory effect in octadecyl acrylate-based hydrogels.12 The shape of a deformed gel was fixed through the reversible formation of crystalline aggregates, and the original shape of the gel was restored upon raising the temperature. The feasibility of introducing additional cross-links upon temperature variation has enabled the development of triple-shape materials with alkyl (meth)acrylates.13
Considering the wide spectrum of PIC applications, it would be desirable to devise general methods of tailoring the properties to individual applications, highlighting the need for facile adjustment of the material properties. Physical blending is a general method used to tune the properties of known polymers. This process poses significant commercial implications for the plastics industry.14 The widespread interest in physical blending is due in part to the fact that the process employs polymers prepared via established synthetic methods, while the new chemical structures obtained via a synthetic approach may as well be implemented to adjust the properties. Physical blending of existing materials is also anticipated to significantly reduce the time and effort required for developing PICs. It is important to note that there are only a limited number of previous studies on solution-blended PICs,15–17 and this scarcity is primarily due to the challenges in achieving homogeneous mixtures. Polymer blends exhibit the intrinsic tendency to undergo phase segregation due to their large molecular weights,18 leading to performance deterioration. Therefore, it is generally important to achieve homogeneity in blended PICs, while phase segregation can sometimes be beneficial for performance enhancement in PICs.19,20
A second network or cross-linking is often introduced to attain a double network or dual cross-linked network to overcome the limitations of a single network. One common issue with network structure is the trade-off relationship, where an enhancement in one property often entails the loss of another property. The introduction of the second network can generate synergistic effects or incorporate new functionalities that are not attainable with a single network. For example, dual cross-linking based on strong and weak hydrogen bonding caused the supramolecular network to exhibit exceptional mechanical properties, while the single networks based on each type of hydrogen bonding performed significantly worse.21 A significant enhancement in mechanical properties was attained via a double-network structure consisting of interpenetrated rigid and soft networks.22,23 Although the double-network structure is generally realized via two-step sequential polymerization, simple solution-blending may be used to incorporate multiple network precursors and cross-linking mechanisms. For example, poly(ethylene oxide) (PEO) homopolymers and triblock copolymers of PEO and poly(propylene oxide) (PPO) were solution-cast to fabricate self-standing PICs.15 The dual-dynamic network in PICs imparted mechanical toughness and enabled nearly complete self-healing within 2 min at room temperature. Similarly, the hydrogen bonding within the mixtures of poly(vinylpyrrolidone) (PVP) and semicrystalline poly(vinyl alcohol) (PVA) polymers contributed to the formation of self-healable PICs.16 Double-network PICs were obtained by blending methacrylate copolymers and semicrystalline P(VDF-co-HFP).17 The thermal reversibility associated with the Diels–Alder reaction and crystallization was utilized to achieve thermally induced self-healing at 100 °C. However, only limited studies on co-crystallizable materials in ion gels or composites have been reported. Furthermore, examples of polymer blends that comprise polymers with different contents of the same moieties (i.e., the same monomeric pair) are even more limited in PICs, despite their potential simplicity and efficiency compared to other blends.
In this study, mechanically tough and dual cross-linked PICs are prepared via solution blending of ILs and two poly(docosyl acrylate-r-tert-butyl acrylate) (poly(A22-r-tBA)) random copolymers that differ only in their compositions. While each polymeric precursor features a different cross-linking mechanism, the network in the blended system is characterized by enhanced crystallization due to potential intermolecular interactions. Previous studies suggest that the network structure and physicochemical properties of PICs derived from poly(A22-r-tBA) vary significantly with the polymeric composition.24,25 When the composition of A22 is 10 mol% or less, the reversible association of the A22 side chains forms cross-links in the composites, rendering it self-healable at room temperature. Meanwhile, network formation is mainly afforded by crystallization when the A22 composition is higher than 20 mol%, and the high crystallinity causes the specimens to fracture in a brittle manner during uniaxial tensile experiments. This difference suggests that the same docosyl side groups can contribute to two distinct cross-linking mechanisms. In this work, poly(A22-r-tBA) copolymers with different mechanical behavior (A22 composition = 8, 28 mol%) are solution-blended with ILs to fabricate ternary PICs. The blended system exhibits conditional miscibility. The phase and thermal behavior are investigated by adjusting the mixing ratios. Despite the marginal compositional window of the homogeneous states, the tunability of the thermal, structural, and mechanical properties of the resulting composites is demonstrated. We further explore the capability for self-healing and the application of PICs to strain sensors.
2. Experimental
2.1. Materials
Docosyl acrylate (A22, >95%, TCI chemicals) was dissolved in dichloromethane (>99.5%, Daejung Chemicals) and passed through an alumina column (neutral, Sigma-Aldrich) prior to use. tert-Butyl acrylate (tBA, >98%, Sigma-Aldrich) was passed through the same column prior to use. N,N′-Azobisisobutyronitrile (AIBN, 99%, Daejung Chemicals) was recrystallized using methanol and dried in a vacuum oven for 24 h. Toluene (HPLC grade, Daejung Chemicals), methanol (HPLC grade, Daejung Chemicals), chloroform (HPLC grade, Ducksan Chemicals), tetrahydrofuran (THF, HPLC grade, J. T. Baker®), 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([EMI][TFSI], >98%, Sigma-Aldrich), and methylene blue hydrate (>70%, TCI Chemicals) were used as received. Unless otherwise specified, other chemicals were purchased from general vendors and used as received.2.2. Polymer synthesis
Poly(docosyl acrylate-r-tert-butyl acrylate) (poly(A22-r-tBA)) random copolymers were synthesized via free radical polymerization. The monomers (A22 and tBA, total 200 mmol), AIBN (1.8 mmol), and toluene (70 mL) were added to a round-bottom flask with a magnetic stirring bar. The round-bottom flask was degassed via Ar bubbling, and the polymerization was carried out at 80 °C for 24 h. The resultant polymer solution was precipitated into methanol. The precipitated polymer was recovered and dried in a vacuum oven for 48 h. The resulting polymer was designated as TA-X, where X represents the feed ratio of A22 in mol%. In this study, TA-10 and TA-30 were used in fabricating PICs.2.3. Preparation of polymer-IL composite (PIC)
Desired amounts of TA-10 and TA-30 (6 g in total) were added to a round-bottom flask, followed by the addition of [EMI][TFSI] (14 g) and chloroform (42 g). The mixture was subsequently stirred at 50 °C for 3 h. The mixture was poured into a Petri dish (20 cm in diameter) and dried in a desiccator at room temperature. The resultant ternary composites are designated as PIC-#, where # represents the weight percentage (wt%) of TA-10 in the polymeric precursors. For example, PIC-80 represents the composite, in which the polymer components consist of 80 wt% TA-10 and 20 wt% TA-30.2.4. Characterization
The mole fraction of the synthesized polymers was analyzed by utilizing nuclear magnetic resonance (1H-NMR) (Avance 400 MHz, Bruker) using chloroform-d as a solvent. The molecular weight and dispersity (Đ) were obtained via size-exclusion chromatography (SEC) (Ultimate 3000, Thermo Scientific) with tetrahydrofuran (THF) as the mobile phase at a flow rate of 1 mL min−1. The results from the chromatography were analyzed based on polystyrene standards (SM-105, Shodex, 1.3–2600 kg mol−1). The crystalline structures of the composites were investigated using X-ray diffraction (XRD) (X'Pert powder diffractometer, PANalytical) over the range of 10°–80° with Cu Kα radiation (wavelength = 0.154 nm) at room temperature. To further gain insights into the structure of the specimens, small angle X-ray scattering (SAXS) (Rigaku SmartLab, λ = 0.154 nm) was performed at room temperature. Differential scanning calorimetry (DSC) (Jade, PerkinElmer) was conducted over the temperature range of −40 to 80 °C (±10 °C min−1) under nitrogen flow. Thermogravimetric analysis (TGA) (Q50, TA Instrument) was carried out under nitrogen flow at a heating rate of 10 °C min−1. The mechanical properties of the mixtures were analyzed at a strain rate of 5 mm min−1 using a universal testing machine (UTM) (AGS-X STD, Shimadzu). The mechanical toughness was calculated based on the area under the stress–strain curves. The temperature of the scratched specimens was maintained constant by using a heating stage (TMS 94, Linkam Scientific Instruments), and the self-healing behavior of the samples was monitored using an optical microscope (Optiphot-2 Pol, Nikon). The morphology of specimens was studied using scanning electron microscopy (SEM) (S-4300, Hitachi) equipped with energy dispersive X-ray spectroscopy (EDS). The optical transparency of the mixtures was evaluated by acquiring transmittance spectra over the wavelength range of 300–800 nm using an ultraviolet-visible spectrometer (Lambda 900, PerkinElmer). Electrical impedance spectroscopy (EIS) (SP-200, Bio-Logic) was utilized to measure the ionic conductivity of the composites (in the metal/mixture/metal structure) over the frequency range of 10 Hz to 3 MHz and to test the feasibility of sensor operation.3. Results and discussion
3.1. Polymer synthesis and preparation of polymer-ionic liquid composites
ILs exhibit a wide range of solvating characteristics.26 Poly(methyl)acrylate is a representative example that exhibits miscibility with [EMI][TFSI].27 The degree of phase separation of poly(meth)acrylates in ILs varies based on the length of the alkyl side chains, a behavior induced by numerous types of interactions. For example, the phase behavior of poly(n-butyl methacrylate) was rationalized based on the specific orientation of ILs around the side chains and the microphase segregation due to the alkyl chains.28 Poly(tert-butyl acrylate) homopolymers (PtBA, TA-0) in [EMI][TFSI] formed homogeneous solutions for all compositions at room temperature, and tBA was employed as the IL-philic component in the TA-X random copolymers. In contrast, the macrophase separation was observed in the mixtures of poly(docosyl acrylate) homopolymers (TA-100) and [EMI][TFSI], rendering A22 the IL-phobic unit.Two copolymers of poly(A22-r-tert-butyl acrylate) (TA-10, TA-30) with different compositions (A22 feed amounts = 10, 30 mol%) were synthesized via free radical polymerization (Table 1, Fig. S1 and S2†). Based on 1H-NMR analysis (Fig. S1†), the actual content of A22 in TA-10 and TA-30 was 9 and 28 mol%, respectively, indicating that the resultant copolymers possibly have a near-statistical sequence. The analyzed composition is consistent with the TGA data (Fig. S3†). Mixtures of TA-10, TA-30, [EMI][TFSI], and chloroform were solvent-cast and dried to form ternary PICs. The fabricated composites are denoted as PIC-#, where # represents the weight percentage (wt%) of TA-10 in the polymeric precursors (Table 2). The IL loading may affect the crystallinity and mechanical properties of the composites; however, the IL content in the specimens containing the two copolymers was similar (ca. 20 wt%) (Fig. S4† for TGA results). A minor decrease in IL loading was observed with increasing TA-30 content.
| FA22a | FA22b | Mnc (kg mol−1) | Đc | Tmd (°C) | χe (%) | |
|---|---|---|---|---|---|---|
| a Mole fraction of A22 (FA22) in copolymers, measured by 1H-NMR.b Determined by TGA.c Number average molecular weight (Mn) and dispersity (Đ) of copolymers, determined by SEC (THF).d Melting temperature (Tm).e Crystallinity (χ), measured by DSC. | ||||||
| TA-10 | 0.09 | 0.09 | 28.0 | 2.8 | — | 4.2 |
| TA-30 | 0.28 | 0.28 | 29.6 | 3.0 | 47.3 | 75.7 |
TA-10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TA-30a (wt%) :TA-30a (wt%) |
IL contentb (wt%) | Tmc (°C) | χc (%) | χd (%) | |
|---|---|---|---|---|---|
| a Weight percentage of copolymers used in fabricating PICs.b Ionic liquid (IL) content of PICs, calculated from TGA data.c Determined by DSC.d Theoretical weight-averaged values of χ for PICs, calculated based on χ of PIC-0 and PIC-100. | |||||
| PIC-0 | 0:100 |
17.5 ± 1.7 | 47.3 | 75.2 | — |
| PIC-80 | 80:20 |
20.2 ± 1.1 | 44.1 | 37.3 | 30.7 |
| PIC-90 | 90:10 |
21.3 ± 1.5 | 44.0 | 30.4 | 19.1 |
| PIC-95 | 95:5 |
23.2 ± 1.5 | 43.8 | 21.9 | 12.2 |
| PIC-100 | 100:0 |
33.0 ± 1.1 | — | 4.4 | — |
3.2. Miscibility of blended polymer-IL composites
The macrophase separation developed in solvent-cast PICs was studied using optical microscopy (Fig. S5†).29 While the crystallinity of TA-10 and TA-30 differed significantly (Table 1 and Fig. S6†), a critical limit above which the system remained homogeneous was identified. The films of PICs with high TA-10 loadings (≥80%) were smooth and homogeneous (Fig. 1a), and this homogeneity was retained even after thermal annealing at 65 °C for 24 h (Fig. S5†). Fig. 1b shows the quantitative transmission data for these clear films. The transmittance declined with increasing TA-30 content, possibly owing to an increase in the crystalline regions that scatter light. The effect of the copolymer composition on the crystallinity of PICs is discussed in the next section. The other composites (PIC-#, 10 ≤ # ≤ 70) exhibited separated regions with distinct optical clarities. 1H-NMR spectral analysis of the different regions confirmed that the polymeric component in each region corresponded to the precursors (either TA-10 or TA-30), indicating a nearly exclusive phase separation. | ||
| Fig. 1 (a) Photographs of polymer-IL composite (PIC) films (thickness = 0.3 mm). (b) Transmittance of PICs at a wavelength of 400 nm (black dot) and 600 nm (blue triangle). (c) Vial tilt test results after thermal annealing at 40 °C for 36 h. | ||
The polymer blends of TA-10 and TA-30 without ILs also showed similar and consistent phase behavior (Fig. S7†). The binary blends containing over 80% TA-10 had a uniform and clear appearance. A similar asymmetric dependence of the phase segregation on the composition has previously been reported for crystallizable mixtures of linear and branched poly(ethylene)s (LPE and BPE).30 In this prior study, macroscopic homogeneity was observed for high LPE contents, whereas phase separation was favored for blends with low LPE contents. The phase behavior of blends comprising chemically similar and crystallizable units may be complex, as crystallization develops during solidification.31 The phase separation and crystallization behavior of PE blends depend on the blend composition, the architecture of the precursors (e.g., degree of branching), and the processing conditions (e.g., isothermal crystallization temperature, rate of cooling). The subtlety associated with the phase separation is noted as it has been reported to occur over a wide range of length scales and arrangements. The phase separation may involve the formation of clusters on molecular scales,32 or each species in a mixture can undergo separate crystallization without complete macrophase segregation.31,33 The effect of incorporating crystallizable units into the side chain was studied with poly(n-alkyl acrylate) homopolymers and copolymers.34 This preceding study showed that phase segregation generally occurs when the components of the mixtures exhibit different crystallization behaviors (i.e., amorphous vs. semicrystalline). Moreover, homogeneous mixing in poly(n-alkyl acrylate) homopolymer blends occurs when the difference in the side chain length is less than 2–4 methylene units.
Given that wearable devices are physically attached to the human body, PIC needs to retain its mechanical integrity at normal human body temperature (≈37 °C). Based on this criterion, the ambient temperature selected for the tilt test was 40 °C (Fig. 1c and S8†). PIC-100 started to flow after 9 h at the elevated temperature but remained tilted at room temperature, consistent with the results of a previous study.25 In contrast, the blended specimens (PIC-80, PIC-90, and PIC-95) did not show any sign of flow even after 36 h at 40 °C. The enhancement in the thermal stability via the incorporation of TA-30 in PICs is attributable to the crystalline domains in TA-30 that maintain cross-links at the specified temperature. The result obtained with PIC-95 suggests that a TA-30 loading of only 5 wt% is sufficient to arrest the dynamics, allowing the specimen to retain its given shape.
3.3. Thermal and structural properties
The crystalline behavior of PICs was studied using DSC (Fig. 2a). Compared to the indistinct peak observed for PIC-100, a prominent melting peak was observed at 47.3 °C for PIC-0. The crystallinities (χ) of PIC-100 and PIC-0 are 4.4% and 75.2% (Table 2), respectively, similar to those of their respective polymer precursors (TA-10, TA-30) (Fig. S6†). The definition of crystallinity χ used in this study is based on the previous publication by Jordan and coworkers, and χ reflects the ratio of crystallized A22 units with respect to all A22 units.7 While the introduction of ILs is expected to increase crystallinity owing to improved polymer mobility, the previously reported effect of water on semicrystalline hydrogels suggests that ILs can also have the opposite effect and inhibit ordering.35 The characteristic features in the endotherm traces of PIC-100 and PIC-0 were retained to some degree in the blended specimens (PIC-80, PIC-90, and PIC-95), whereas the peak intensities and widths differed to various degrees. The peaks identified near 44 °C in the profiles of PIC-80, PIC-90, and PIC-95 appear to originate from PIC-0, given that the positions changed negligibly as the precursor ratio varied. The broadening of the melting peaks shown in Fig. 2a has been typically observed when the related crystalline units are diluted by amorphous units.7 This feature indicates a wider distribution of crystal sizes.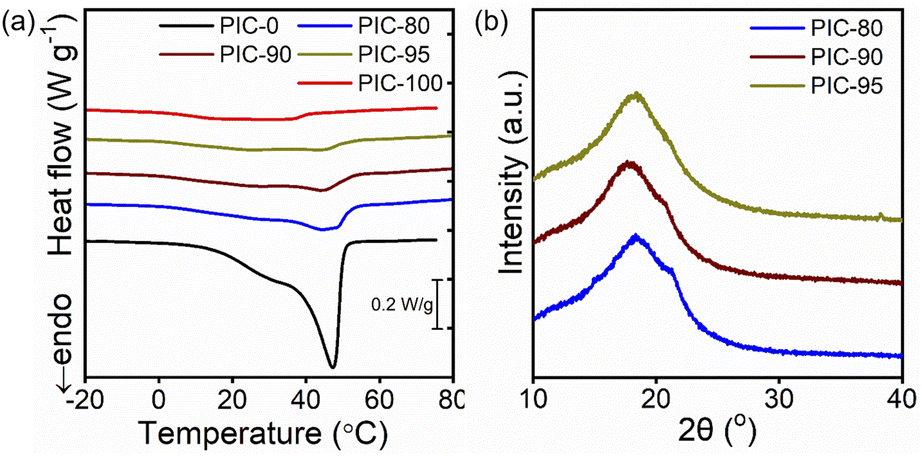 | ||
| Fig. 2 (a) DSC thermograms (second heating at 10 °C min−1) and (b) XRD profiles of PICs. | ||
Based on the DSC results, the crystallinity χ increased systematically in the composites with higher incorporation of highly crystalline TA-30 (Table 2). Notably, PIC-80, PIC-90, and PIC-95 exhibited χ values of 37.3%, 30.4%, and 21.9%, respectively, considering only 20, 10, and 5 wt% of TA-30 in the composites. As mentioned in the introduction section, the capability to maintain the periodic arrangement despite a large amount of the amorphous unit can be ascribed to the side-chain architecture. The theoretical crystallinities of the blended systems, which were calculated as the weight-averaged χ values of PIC-0 and PIC-100, are presented in Table 2 and are compared to the experimental values. Interestingly, the measured crystallinity values of blended PICs were higher than the theoretical values, suggestive of interactions between the two copolymers comprising the networks. Enhanced crystallinity has previously been observed for mixtures with side-chain architecture and was attributed to co-crystallization involving the alkyl side chains.8,36 For example, the hexagonal packing of n-octadecyl side chains in copolymers is enhanced by co-crystallization with small amounts of n-octadecanoic acid additives.36 Synergistic crystallization was also observed in polymers that undergo crystallization via the main chains; the DSC thermograms and scattering profiles obtained for the mixtures of PEs and poly(ethylene-co-vinyl acetate) copolymers deviate from the theoretical values, ascribable to co-crystallization.33 SEM analysis was conducted to investigate the interior morphology of the samples. The cross-sectional images of PICs in Fig. S9† corroborate the presence of crystalline domains dispersed within the amorphous regions. EDS analysis was conducted to identify the distributions of ILs. The EDS mapping images of C, F, and S presented in Fig. S10† indicate that the crystalline domains, consisting of A22 components, are depleted of ILS for all PIC specimens. On the other hand, ILs are homogeneously distributed in the amorphous regions.
XRD was used to investigate the atomic arrangements in the crystalline unit cells (Fig. 2b). All the specimens exhibited scattering at 2θ = 18°, attributable to amorphous halo.9,36 The XRD peak at 2θ = 22° has been commonly associated with hexagonal packing of the alkyl pendants in the side-chain architecture.6,35 The intensity of the peak at 2θ = 22° increased gradually with higher TA-30 loading, which is consistent with the DSC thermograms in Fig. 2a. The observation of the inflections in the spectra of all the samples suggests the formation of the same arrangements despite the blending of ILs and two copolymers. According to previous studies, additives in semicrystalline networks may have mixed impacts on the crystalline structure and layered packing.35,37 The SAXS results presented in Fig. S11† suggest the formation of lamellar domains driven by A22 side chains.
3.4. Mechanical properties, self-healing, and sensor operation
The mechanical properties of the PIC systems were studied through uniaxial tensile measurements. The stretchability of PICs changed systematically with variations in the precursor mixing ratio (Fig. 3a and Table S1†). PIC-0 fractured in a brittle manner (strain at break = 20%, Fig. S12†), whereas the strain at break for PIC-80, PIC-90, and PIC-95 was 1300, 2100, and 2400%, respectively. Although the stretchability of PIC-80, PIC-90, and PIC-95 was less than that of PIC-100, the mechanical toughness was mostly retained for the PIC systems (10.9–13.0 MJ m−3), comparable to that of PIC-100 (14.0 MJ m−3, Fig. S12†). The crystalline domains in PICs are anticipated to act as strong bonds while the nano-associations function as weak bonds; the combination of these cross-linking mechanisms may contribute to the high mechanical toughness observed in PICs.21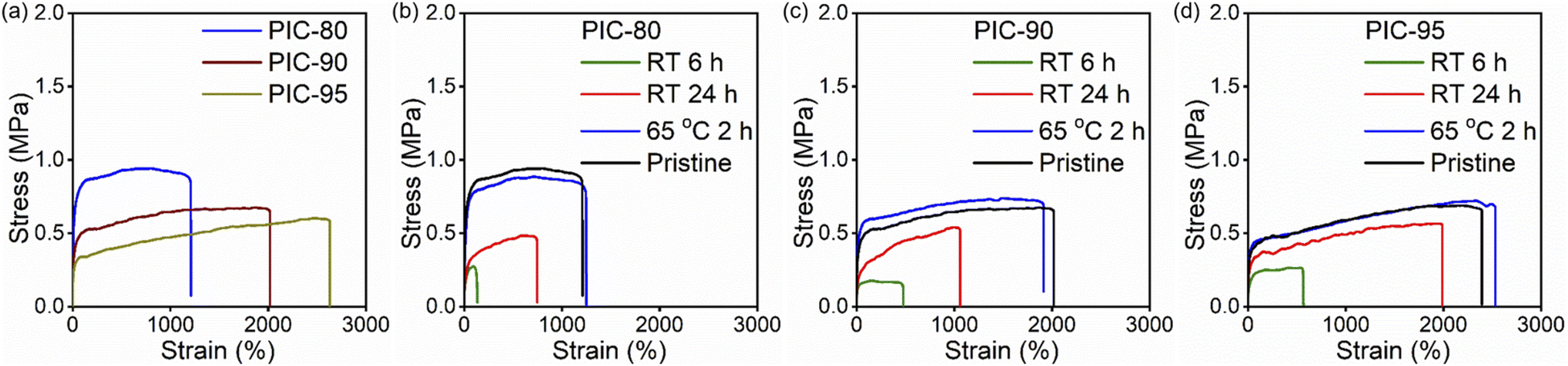 | ||
| Fig. 3 Stress–strain curves of (a) pristine and (b–d) self-healed PIC samples. Self-healing was conducted at room temperature for 6 h (green), 24 h (red), and at 65 °C for 2 h (blue). | ||
Self-healing refers to the restoration of the functional and mechanical properties of a material. Various methods of achieving self-healing have been intensively studied for prolonging the lifetime of products. Self-healing in macromolecules requires the polymeric diffusion across the damaged interface, allowing chain rearrangement and bond reformation. A previous study suggested that the width of the mixed region needs to reach approximately 100 nm to achieve a high degree of healing.38 Notably, Forrest and coworkers fabricated polystyrenes with a stepped-film geometry and found that the local dynamics near the damaged region differed from those in the bulk.39 They monitored the evolution of the surface topography and reported the formation of a thin layer at the top, exhibiting liquid-like behavior. The depressed glass transition temperature at the polymer–air interface enhances the segmental mobility and facilitates the healing process in the vicinity of the fractured regions.
The self-healing capability of PIC-80, PIC-90, and PIC-95 was evaluated by first cutting the specimens in half with a razor blade. The sliced specimens were placed in molds within a few minutes after cutting. The mechanical properties were evaluated via uniaxial extension as a function of the contact duration (Fig. 3b–d). The self-healing efficiency, η, is expressed as the ratio of the toughness of the self-healed samples to that of the pristine samples. Autonomous self-healing refers to the repair process without an external input, such as heat or light. PIC-80 and PIC-90, characterized by a crystallinity of χ > 30%, exhibited a low degree of autonomous repairability at room temperature (efficiency η < 40%, Fig. 3 and Table S1†). The sliced specimens could adhere to each other to some extent but were readily fractured at the damaged regions upon applying stress. On the other hand, the degree of self-healing for PIC-95 increased progressively over time, reaching η ≈ 75% after 24 h. Notably, the efficiency, η, for PICs did not improve significantly for contact durations longer than 24 h.
The autonomous self-healing ability of PICs at room temperature is attributed to the reversible association of A22 in TA-10, as suggested in our previous work.25 The associative nature of side chains in specific media has been studied by Terashima and coworkers.40 In their report, amphiphilic random copolymers comprising hydrophobic side chains associated to form micelles in water, which were utilized in achieving network structures in hydrogels. The self-healing capability stems from the chain-exchange property of the micelles, which enables the reversible formation of physical cross-links. In the PIC system, the mechanical slicing mainly involves the breaking of weak associates by A22, while the covalent bonds are preserved in general. The dynamics associated with A22 were evaluated based on the DSC analysis of TA-05 (Fig. S13†). The result suggests that the glass transition temperature associated with the A22 units is low, and the core dynamics required for A22 reaggregation are not expected to hinder the self-healing process. Upon contact, self-healing proceeds mostly via re-association of the fractured associates (Fig. S14†).
The complete reversibility of the physical cross-linking in PIC-80, PIC-90, and PIC-95 was examined by reprocessing the fractured specimens at elevated temperatures. It is not surprising to observe the limited role played by crystallization in the repair process when contact between the sliced specimens occurs below the melting temperature. This is evidenced by the decline in the self-healing capability of PICs with TA-30 incorporation, as described above. Notably, the addition of only 10 wt% TA-30 significantly compromised the self-healing capability at room temperature. Raising the temperature above the melting temperature (Tm ≈ 44 °C for PIC-80, PIC-90, and PIC-95) removed the restrictions on self-healing imposed by the crystalline domains. Reprocessed PICs were obtained by compression molding of the fractured specimens at 65 °C, and the recovery of the mechanical properties was evaluated by comparing the toughness of the reprocessed samples to that of the original samples. The similarity between the stress–strain curves of all the reprocessed and pristine samples (Fig. 3) suggests effective reformation of the cross-links due to the enhanced mobility of the polymeric segments (η ≈ 100%). Even PIC-80 and PIC-90, which show limited self-healing capabilities at room temperature, exhibited complete recovery of mechanical properties.
The self-healing capability of PICs was further examined by mending dyed specimens and subjecting the films to scratch tests. Dyed and undyed strips of PICs were put together with their respective counterparts for 24 h at room temperature (Fig. 4a–c and S15†). The PIC-95 specimen was able to withstand a weight of 50 g, whereas the healed PIC-80 sample with low η fractured readily when a weight was suspended from it. An optical microscope was used to monitor the evolution of scratches in the PIC films over time (Fig. 4d and e). The specimens stored at room temperature exhibited a limited degree of healing; the damaged region in PIC-80 remained nearly unchanged even after 24 h. In contrast, the gap caused by the scratch closed considerably in PIC-90 and PIC-95, although full recovery was not achieved within 24 h (Fig. 4d and S16†). A significant enhancement in the self-healing ability was observed at an elevated temperature (65 °C); for all specimens, the scratches nearly disappeared within 30 s (Fig. 4e and S17†). This behavior is consistent with the results of the uniaxial tensile measurement presented in Fig. 3.
 | ||
| Fig. 4 Dyed and non-dyed PIC-95 specimens (a) before and (b) after self-healing (RT, 24 h). (c) Self-healed PIC-95 withstood a weight of 50 g. (d and e) Optical microscope images of scratched PIC-80, PIC-90, and PIC-95 monitored after (d) 24 h at RT and (e) 30 s at 65 °C. The scale bars correspond to 100 μm. | ||
The incorporation of ILs confers ionic conductivity to PICs. To assess the electrochemical properties of PICs in this study, the impedance was measured as a function of the frequency via electrochemical impedance spectroscopy (EIS). Fig. 5 shows the Nyquist plots of PICs, indicating positive slopes (see Fig. S18† for Z′ and Phase Z vs. frequency). The high-frequency limits of the real part Z′ in the Nyquist plots were extrapolated, and the sample dimensions were used to calculate the ionic conductivities. PIC-80, PIC-90, and PIC-95 exhibited average ionic conductivities of 0.56, 0.40, and 0.35 mS cm−1, respectively.
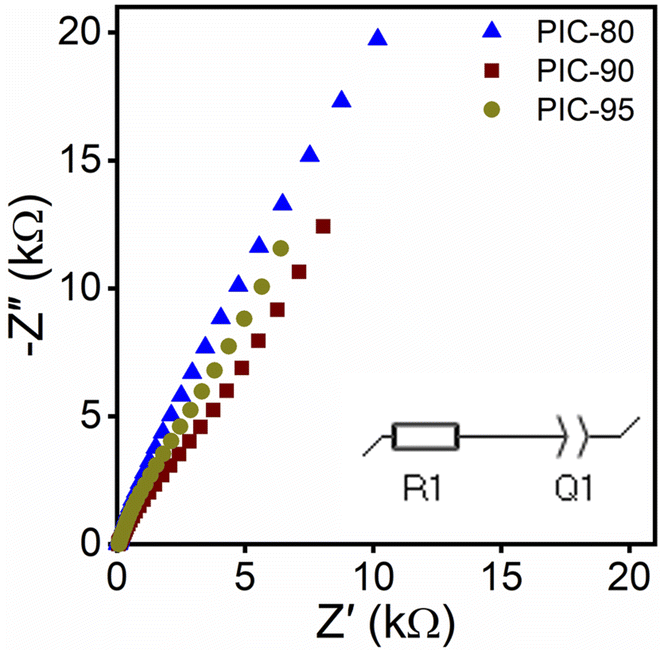 | ||
| Fig. 5 Nyquist plots of PICs. The inset shows the equivalent circuit model utilized in the extrapolation. R1 and Q1 correspond to the bulk resistance and constant phase element of the composite, respectively. | ||
Strain sensors convert mechanical deformation into electrical signals by monitoring the corresponding change in the resistance across the specimens. They are expected to play an integral role in health monitoring, human motion detection, and soft robotics. One of the well-known methods to fabricate stretchable strain sensors is to incorporate conductive components of various structures into soft polymer matrices.1 In addition to sensitivity and transparency, mechanical toughness is also considered an important aspect of strain sensors, subjected to the repetitive mechanical motions. Strain sensors also need to maintain the mechanical integrity and functionality over time. PICs have been also utilized in strain sensors as well as other stretchable electronics devices, such as transistors, energy storage devices, and actuators.2 To explore their potential application as strain sensors, a mechanical deformation representing human motion was applied to the fabricated composites. The corresponding changes in the resistance of the composites were monitored, and their relationships with the degree of mechanical deformation were investigated. Fig. 6a shows the PIC-95 strip attached to a finger, and Fig. 6b shows the relative resistance change (ΔR/R0) upon repetitive finger bending. R0 represents the resistance across the undeformed specimen. All the samples exhibited responses that closely reflected the motion of the finger. The resistance output was linearly related to the strain, which was applied at controlled levels using a motorized stage (Fig. 6c and S19†). The gauge factor (GF), derived from the slope of the ΔR/R0 vs. strain plot, provides the measure of the sensitivity of strain sensors. The GF values for PICs, estimated based on the linear dependence, were in the range of 2.66–3.06 (Table S2†). The comparison between this work and previous studies (Table S3 and Fig. S20†) demonstrates that PIC-95 exhibits a significantly higher toughness value compared to other strains sensors, while the gauge factors are comparable. The stability of the strain sensors was studied by observation of their behavior during aging. Compared to the as-fabricated specimens, after four weeks of storage at room temperature, no notable difference was observed in the properties of the sensors employing the PIC samples (Fig. 6d and S21†).
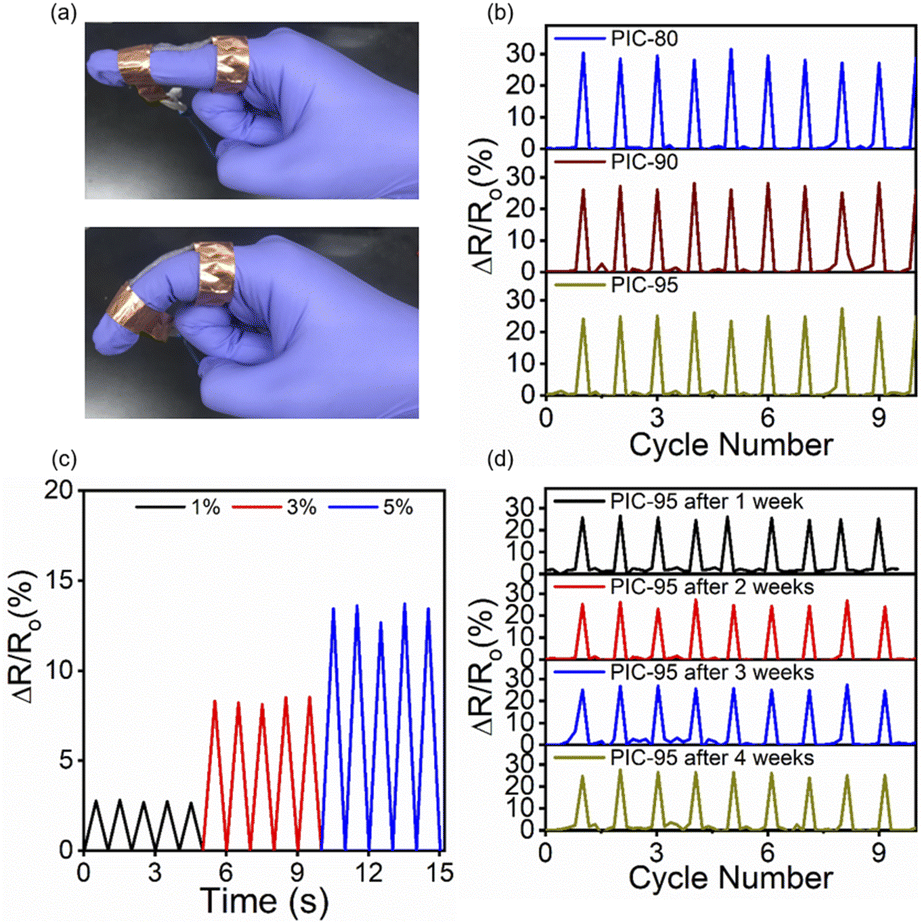 | ||
| Fig. 6 (a) Photographs of the PIC-95 strain sensor attached to a finger before and after bending. (b) Relative resistance changes for PICs during repeated bending and extension of the finger. (c) ΔR/R0 as a function of the imposed tensile strains for PIC-95. (d) Operation of the PIC-95 strain sensor over 4 weeks. | ||
4. Conclusion
Dual cross-linked PICs consisting of two poly(docosyl acrylate-r-tert-butyl acrylate) copolymers of different compositions (TA-10 and TA-30) and ILs were successfully fabricated. Most previous dual cross-linked PICs are based on different physical/chemical interactions, and their fabrication often requires a series of experimental steps. In contrast, this study utilized simple solution-blending of copolymers with the same monomer pairs. Notably, the side-chain architecture allowed the copolymers to adopt two different cross-linking mechanisms (crystallization and reversible association), leading to dual cross-linking in the ternary composites. Interestingly, the overall crystallinity of PICs was higher than the weighted averages of the precursor crystallinities, suggestive of additional interactions such as co-crystallization. The composites adopted homogeneous states over a range of mixing ratios (TA-10:TA-30 = 80:20, 90:10, 95:5), whereas the macrophase segregation was observed for the other mixtures. Despite the narrow compositional window for miscibility, the composites exhibited a wide spectrum of material performance. The enhancement in the thermal stability of PICs at 40 °C was achieved by incorporating small amounts of TA30. The stretchability increased significantly from 1300% to 2400% while retaining a mechanical toughness of ca. 11–13 MJ m−3. The autonomous self-healing efficiency at room temperature over 24 h ranged from 25% to 75%, whereas the mechanical properties were completely recovered after thermal annealing at 65 °C for 2 h. PICs show potential utility in stretchable electronics, particularly as strain sensors, owing to their high ionic conductivities (0.35–0.56 mS cm−1) and robust mechanical properties. The present findings demonstrate that the properties of composites can be precisely tuned via the simple methodology of blending co-crystallizable copolymers with ILs. These results highlight the versatility and effectiveness of this approach in developing advanced materials for various next-generation wearable electronic devices.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Inha University research grant. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (RS-2024-00431381).References
- J. Wang, M. F. Lin, S. Park and P. S. Lee, Mater. Today, 2018, 21, 508–526 CrossRef.
- H. Wang, Z. Wang, J. Yang, C. Xu, Q. Zhang and Z. Peng, Macromol. Rapid Commun., 2018, 39, 1800246 CrossRef PubMed.
- J. C. Jansen, K. Friess, G. Clarizia, J. Schauer and P. Izák, Macromolecules, 2010, 44, 39–45 CrossRef.
- Z. Liu, X. Wang, Z. Liu, S. Zhang, Z. Lv, Y. Cui, L. Du, K. Li, G. Zhang, M.-C. Lin and H. Du, ACS Appl. Mater. Interfaces, 2021, 13, 28164–28170 CrossRef CAS.
- T. T. Guan, X. H. Wang, Y. L. Zhu, L. Qian, Z. Y. Lu, Y. F. Men, J. Li, Y. T. Wang and J. Q. Sun, Macromolecules, 2022, 55, 5816–5825 CrossRef CAS.
- N. A. Platé and V. P. Shibaev, Comb-Shaped Polymers and Liquid Crystals, Plenum Press, New York and London, 1987 Search PubMed.
- E. F. Jordan Jr, B. Artymyshyn, A. Speca and A. Wrigley, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 3349–3365 CrossRef.
- E. F. Jordan Jr, S. Smith Jr, R. D. Zabarsky and A. Wrigley, J. Appl. Polym. Sci., 1978, 22, 1547–1567 CrossRef.
- G. Hattori, M. Takenaka, M. Sawamoto and T. Terashima, J. Am. Chem. Soc., 2018, 140, 8376–8379 CrossRef CAS.
- G. H. Fredrickson and E. Helfand, J. Chem. Phys., 1987, 87, 697–705 CrossRef CAS.
- S. Kim, P. F. Nealey and F. S. Bates, Nano Lett., 2014, 14, 148–152 CrossRef CAS.
- Y. Osada and A. Matsuda, Nature, 1995, 376, 219 CrossRef CAS PubMed.
- A. Argun, U. Gulyuz and O. Okay, Macromolecules, 2018, 51, 2437–2446 CrossRef CAS.
- D. R. Paul and C. B. Bucknall, Polymer Blends, Wiley-Interscience, 2000 Search PubMed.
- Y. Shi, Y. Wang, Y. Gu, L. Zheng, S. Ma and X. Xu, Chem. Eng. J., 2020, 392, 123645 CrossRef CAS.
- D. Weng, F. Xu, X. Li, S. Li, Y. Li and J. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 57477–57485 CrossRef CAS PubMed.
- Z. H. Tang, X. L. Lyu, A. Q. Xiao, Z. H. Shen and X. H. Fan, Chem. Mater., 2018, 30, 7752–7759 CrossRef CAS.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, CRC Press, 2nd edn, 2007 Search PubMed.
- M. Wang, P. Zhang, M. Shamsi, J. L. Thelen, W. Qian, V. K. Truong, J. Ma, J. Hu and M. D. Dickey, Nat. Mater., 2022, 21, 359–365 CrossRef CAS PubMed.
- R. Tamate, K. Hashimoto, T. Horii, M. Hirasawa, X. Li, M. Shibayama and M. Watanabe, Adv. Mater., 2018, 30, 1802792 CrossRef PubMed.
- J. Kang, D. Son, G. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- H. Y. Yoo, D. Son, H. Kim, K. G. Cho, M. Kim, K. H. Lee and S. Kim, Org. Electron., 2020, 84, 105788 CrossRef CAS.
- Y. Lee, M. Kim, H. Kim, K. H. Lee and S. Kim, ACS Appl. Polym. Mater., 2022, 4, 5821–5830 CrossRef.
- N. Winterton, J. Mater. Chem., 2006, 16, 4281–4293 RSC.
- M. A. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976–4983 CrossRef CAS PubMed.
- H. N. Lee and T. P. Lodge, J. Phys. Chem. B, 2011, 115, 1971–1977 CrossRef CAS PubMed.
- B. Crist and M. J. Hill, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 2329–2353 CrossRef CAS.
- M. J. Hill, P. J. Barham, A. Keller and C. C. A. Rosney, Polymer, 1991, 32, 1384–1393 CrossRef CAS.
- L. Mandelkern, R. Alamo, G. Wignall and F. Stehling, Trends Polym. Sci., 1996, 4, 377–383 CAS.
- J. Schelten, G. Wignall, D. Ballard and G. Longman, Polymer, 1977, 18, 1111–1120 CrossRef CAS.
- R. Alamo, R. Glaser and L. Mandelkern, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 2169–2195 CrossRef CAS.
- B. S. Kirkland, PhD thesis, University of Texas, 2007.
- A. Matsuda, J. P. Gong and Y. Osada, Polym. Gels Networks, 1998, 6, 307–317 CrossRef CAS.
- K. Inomata, Y. Sakamaki, T. Nose and S. Sasaki, Polym. J., 1996, 28, 986–991 CrossRef CAS.
- K. Inomata, Y. Sakamaki, T. Nose and S. Sasaki, Polym. J., 1996, 28, 992–999 CrossRef.
- L. N. Neumann, E. Oveisi, A. Petzold, R. W. Style, T. Thurn-Albrecht, C. Weder and S. Schrettl, Sci. Adv., 2021, 7, eabe4154 CrossRef CAS PubMed.
- Y. Chai, T. Salez, J. D. McGraw, M. Benzaquen, K. Dalnoki-Veress, E. Raphael and J. A. Forrest, Science, 2014, 343, 994–999 CrossRef CAS.
- S. Imai, M. Takenaka, M. Sawamoto and T. Terashima, J. Am. Chem. Soc., 2019, 141, 511–519 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05968a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |