
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDrug release system based on a composite polycaprolactone nanofiber membrane with dual functionality of shape memory effect and antibacterial ability†
Le Thi Lea,
Hue Thi Nguyena,
Ha Thi Thu Buia,
Huy Quang Tran ab and
Thuy Thi Thu Nguyen*a
ab and
Thuy Thi Thu Nguyen*a
aPhenikaa University Nano Institute (PHENA), Phenikaa University, Hanoi 12116, Vietnam. E-mail: thuy.nguyenthithu@phenikaa-uni.edu.vn; Tel: +84924926886
bFaculty of Biomedical Sciences, Phenikaa University, Hanoi 12116, Vietnam
First published on 27th August 2024
Abstract
In this study, a multifunctional composite membrane based on polycaprolactone nanofibers having controlled drug release, shape memory effect, and antibacterial ability was successfully prepared by the electrospinning technique. The addition of graphene oxide (GO), zinc oxide nanoparticles (ZnO NPs), polyethylene glycol (PEG), and berberine (BBR) strongly affected the morphology, crystalline degree, melting temperature, and shape memory performance of the composite membrane, thanks to the physical crosslinking network formed by the hydrogen bonding or van der Waals interactions between the components. As a result, the recovery ratio of the composite membrane reached a higher value (76.3% ± 0.7%) than that of the PCL fiber membrane (22.8% ± 0.7%). The additional components significantly improved the wettability of the composite membrane, leading to a high amount of BBR released (42.7 wt%) during 40 hours, as well as effective antibacterial ability. Besides, the BBR release can be feasibly controlled by modulating the deformation ratio of the composite membrane, whereby the higher deformation ratio resulted in a higher BBR release. Therefore, it is suggested that the prepared composite nanofiber membrane is a potential smart material used in biomedical applications, such as wound dressing and drug release systems.
1 Introduction
Shape memory polymers (SMPs) are classified as emerging stimuli-responsive material, capable of switching between temporary and permanent shapes upon the application of external stimuli, such as heat, light, pH, electricity, magnetic field, or moisture. The permanent shape is determined by a chemical or physical network based on intermolecular interactions, crystalline phases, or molecular crosslinking. Meanwhile, the temporary shape is fixed by the weak interactions between molecular chains or chain segments of the reversible phase (switching segments), which are crystalline, liquid crystalline and amorphous phases.1 Thermally induced SMPs are commonly studied, whose shape memory mechanism involves heating above their transition temperature, including glass and melting temperatures for thermoset-SMPs and thermoplastic-SMPs, respectively.2 The thermoset-SMPs are chemically cross-linked polymers, and their shape memory behavior occurs at glass temperature (Tg). At or above Tg, the mobility of the polymeric chains increases, turning materials from a rigid state into a rubbery state. Therefore, they can be deformed to a temporary shape corresponding to a change in the molecular conformation and entropy by the application of an external stress.3 After that, the materials are cooled down below their Tg to fix the temporary shape and subsequently reheated above the Tg to induce polymeric chain mobility as well as to release the stored entropic energy, which is the driving force of the initial shape recovery. In the case of thermoplastic-SMPs, the actuation of shape memory phenomena can be performed at a transition temperature of either Tg or Tm, depending on their chemical nature and structure, processing, and programming parameters.4 In comparison with thermoset-SMPs, thermoplastic-SMPs have the advantages of appropriate adjustment of transition temperature (near the human-body temperature range) and of being more stretchable, degradable, and easily processed into various structures, such as films, foams, and micro/nanofibers.Recently, SMPs with fibrous structures produced by electrospinning technique have been increasingly investigated because of their interesting characteristics extending their potential applications in biomedical fields. Along with high-surface-area per volume unit, high porosity, nanoarchitecture favor for cell attachment and proliferation, the electrospun nanofibers show enhanced shape memory behaviors compared to other structures. Zhang et al.5 reported that the microfiber shape memory polyurethane (SMPU) film had a higher shape recovery rate and ratio because the high specific surface area of microfibers resulted in quick heat transfer, subsequently promoting the shape recovery. By reduction of the fiber diameter of the electrospun polyether urethane mat from the micro- to the nano-size, the molecular orientation in the fibers changed, leading to the improvement of the shape memory performance, however, decrease in shape-fixity.6 Some advanced shape memory composites in fibrous or bulk forms are being fabricated by incorporating various nanomaterials, such as graphene,7,8 graphene oxide,9,10 ZnO,11–14 MgO,15 clay,16 and multi-walled carbon nanotubes.17 Addition of GO at a content of 4 wt% in SMPU nanofibrous mat notably improved the mechanical strength, surface wettability, thermal stability, and shape memory effect because hydrophilic GO sheet played a role as crosslinking points that prevented the stretch and contraction of SPMU chains and reached the final structure more quickly.9 Meanwhile, the SMPU/nano ZnO composite had good memory properties due to the formation of hydrogen bonds between –OH groups of nano ZnO and –NHCOO– groups of polyurethane, promoting the self-assembly and inducing the micro-phase separation.11
Owing to the above-mentioned characteristics, the nanofibers-based shape memory scaffolds have great potential for use in biomedical applications. Due to the shape memory property, deformed nanofiber SMP scaffolds can recover their original shape using internal/external stimulation for repairing bone defects with minimally invasive surgical implantation. It is reported that18,19 the shape memory capability of nanofiber SMP scaffolds electrospun from a blend of chitosan, gelatin, and shape memory polyurethane can benefit the wound healing process through pre-programming shape recovery that assists in closing the wound.20 In addition, the shape memory effect can be combined with drug delivery to perform multifunction, including the release of therapeutic drugs over an extended period, providing nanoarchitecture for cell adhesion and proliferation, and wound healing. Bil et al.21 fabricated two types of electrospun shape memory nanofibers (PU-PLLA and PU-PLLA/PEG) incorporated with rapamycin for the design of implantable devices for self-fitting and drug delivery for cardiovascular system regeneration. Both electrospun scaffolds had a transition temperature that was close to body temperature, Rf and Rr values above 90%, and controlled delivery of rapamycin over 45 days with effective therapeutic dosage. A sandwich structure membrane composed of electrospun shape memory polyurethane nanofibers in the top and bottom layers and berberine (BBR) incorporated nanofibers in the middle layer was developed as a drug delivery system.22 80 wt% of BBR was released from the sandwich structure membrane in 144 h. Interestingly, the release of BBR can be accelerated by stretching and fixing the membrane in different deformation ratios.
Polycaprolactone is a biodegradable polymer with excellent mechanical properties and biocompatibility, endowing its widespread use in biomedical applications. However, the PCL process poor shape memory effect and a high transition temperature of above 60 °C. The aim of this study was to improve the shape memory capability of electrospun PCL nanofiber scaffold at lower temperatures using inorganic and organic additive components, including GO, ZnO nanoparticles, polyethylene glycol, and BBR. The effect of these components on the shape memory performance of the PCL nanofiber scaffold was explained based on the mechanism of molecular interactions and crystalline structure. In addition, the release of BBR as a model drug from these composite nanofiber scaffolds was investigated in relation to their deformation shape. The antibacterial activity of the scaffolds was examined to evaluate their potential capacity in extensive biomedical applications, such as wound dressing and drug delivery.
2 Materials and method
2.1 Materials
BBR chloride (pharmaceutical primary standard), polycaprolactone (Mw = 48.000 g mol−1), and polyethylene glycol (Mw = 4.000 g mol−1) were supplied from Sigma Aldrich. N,N-Dimethylformamide (DMF purity ≥99.5%) and chloroform (purity ≥99.5%) were purchased from Samchun Chemicals, Korea. Titan Biotech (India) provided nutrient agar. Bi-distilled water through a Milli-Q® system was used. All the chemicals were used without further purification.Two bacterial strains, including Escherichia coli and Methicillin-resistant Staphylococcus aureus (MRSA) represented Gram-negative and Gram-positive bacterium, respectively, were provided by the Department of Bacteriology at the National Institute of Hygiene and Epidemiology Vietnam.
2.2 Preparation of electrospun composite membranes
ZnO nanoparticles (ZnO NPs) and graphene oxide sheets (GO) were prepared according to the procedures reported in our previous studies.23,24The electrospun PCL membrane containing polyethylene glycol (PEG), GO, ZnO NPs, and BBR was fabricated using the electrospinning method. Firstly, 0.012 g of ZnO NPs were dispersed in 7.6 g of chloroform by ultrasonic for 15 minutes. Then, 1.9 g PCL and 0.1 g PEG corresponding to a 95/5 weight ratio of PCL/PEG were added into the above ZnO NP suspension. The mixture was stirred for 2 hours at room temperature using a magnetic stirrer. Besides, 0.02 g of GO was dispersed in 0.4 g of N,N-dimethylformamide (DMF) using an ultrasonic bath for 1 hour to ensure its uniform dispersion. Subsequently, the GO suspension and 0.02 g of berberine (BBR) were mixed with the above PCL/PEG/ZnO NPs mixture to obtain a 20 wt% PCL/PEG solution containing 0.6 wt% ZnO, 1 wt% GO, and 1 wt% BBR compared to the PCL/PEG amount. Finally, the mixture was stirred using magnetic stirring at 35 °C for at least 2 hours until a homogeneous PCL/PEG/ZnO NPs/GO/BBR (PCL/PZG/BBR) mixture was obtained.
To prepare the PCL/PZG/BBR nanofibrous membrane, the above PCL/PZG/BBR mixture was subsequently subjected to processing using an electrospinning system, which involved the utilization of a 5 mL syringe connected to a steel needle with an inner diameter of 24 gausses. The solution was administered at a flow rate of 1.0 mL h−1. A high-voltage power supply was used to establish a voltage of 9 kV while maintaining a working distance of 20 cm between the needle's tip and a roller collector. The electrospinning process was conducted for 6 hours to obtain the PCL/PEG/ZnO/GO membrane under the ambient condition with relative humidity <60% and a temperature of 27 °C ± 3 °C.
Electrospun PCL fiber membranes containing one or more components of GO, ZnO NPs, PEG, and BBR were fabricated using a similar procedure.
2.3 Characteristics of electrospun composite membranes
The scanning electron microscopy (SEM) images were taken using a JSM-6510LV instrument to observe the morphology of the prepared composite membranes. The diameter distribution of nanofibers was conducted by measuring the diameter of approximately 50 randomly selected nanofibers from SEM images using the ImageJ software.The melting properties of the composite membranes were examined by differential scanning calorimetry (DSC 204F1 Phoenix, Netzsch). The tests were performed by heating and cooling the sample several times at a rate of 10 °C per minute, over a temperature range of 25 °C to 80 °C. DSC data extracted from the second cycle were reported. The degree of crystallinity was calculated using eqn (1):
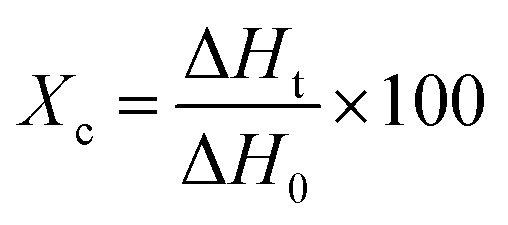 | (1) |
The chemical characteristics of the composite membranes were analyzed using an Infrared Spectrometer (Nexus 670 from Nicolet) with wavenumbers ranging from 500 to 4000 cm−1 and a Raman spectrometer (MacroRAM, Horiba) in the wavelength range of 500–3000 cm−1.
2.4 Shape memory performance of the composite membranes
Each composite membrane was cut into uniform rectangle shapes with original dimensions of 5 mm width, 10 mm length, and 0.1 mm thickness. Thermal-induced shape memory performance of these samples was evaluated in hot circulating air in an oven by following steps: (a) the sample with the origin length L0 (mm) was heated at 55 °C for 10 minutes in an oven, (b) the heated sample was rapidly stretched to obtain a deformation shape with a new length L1 (mm), (c) the deformed sample was fixed by cooling it at 5 °C in a fridge for 5 minutes to obtain temporary shape with the length of L2 (mm), and (d) the sample was recovered by reheating at 55 °C for 60 minutes in the oven and the final length was recorded as L3 (mm).The fixity ratio (Rf) and recovery ratio (Rr) were calculated according to eqn (2) and (3), respectively:
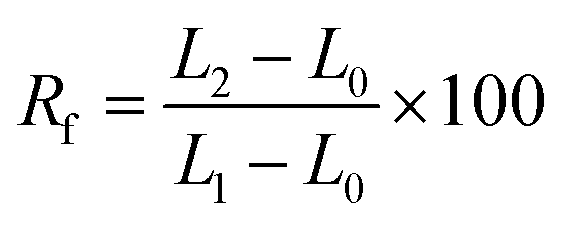 | (2) |
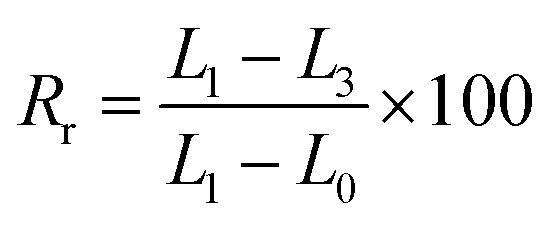 | (3) |
The shape memory test of each sample was repeated at least three times and the average data of the fixity and recovery ratios are reported.
The shape memory capacity of samples deformed at different ratios of elongation (0%, 20%, and 50%) was also investigated. The deformed samples were marked according to their elongation ratios as PCL/PZG/BBR0, PCL/PZG/BBR20, and PCL/PZG/BBR50. The length of deformed, temporary, and recovered samples was measured using a digital camera of the fluorescence stereomicroscope (M165 FC, Leica).
2.5 BBR release behavior of the composite membranes
In order to determine the quantity of BBR released from the composite membranes, a calibration curve of UV-vis absorbance versus BBR concentration was built as follows: BBR solutions with serial concentrations from 1 to 50 μg mL−1 were prepared by diluting the BBR stock solution (0.2 mg mL−1) in phosphate-buffered saline (PBS, pH = 7.4). Then, the absorbance of these BBR solutions was measured using a UV-vis spectrophotometer (6850 UV/vis, Jenway) at a maximum wavelength of 421 nm for building the BBR calibration curve.The composite membranes were cut into rectangular pieces containing the same amount of BBR in each sample. The cut samples were separately placed into 10 mL bottles containing 3 mL PBS. Then, these bottles were placed in a shaker PTR-35 vertical multifunction under room temperature with constant agitation at 30 rpm. At each time interval, the UV-vis absorbance of the solution in each bottle was measured at the wavelength of 421 nm and the quantity of BBR released from composite membranes was calculated based on the calibration curve. The percentage of BBR released at each time interval was determined using eqn (4) as follows:
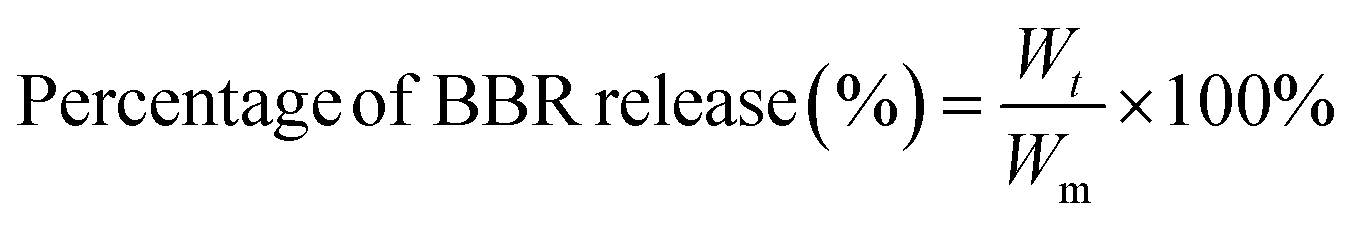 | (4) |
The BBR release behavior of the composite membranes deformed at different ratios of elongation (0%, 20%, and 50%) was also evaluated by the same procedure. All the experiments were repeated three times.
2.6 Assessment of antibacterial activity of the composite membranes
The composite membranes stretched at various ratios were tested for antibacterial activity against two representative pathogens causing infections, namely Methicillin-resistant Staphylococcus aureus (MRSA) and Escherichia coli O157:H7 (E. coli O157:H7), by the agar diffusion method. A bacterial suspension with a concentration of 107 CFU mL−1 was spread evenly on the surface of nutrient agar contained in a sterile plastic Petri dish. The composite membranes with different deformation ratios were gently placed on the agar surface. Subsequently, the Petri dishes were covered and incubated at 37 °C for 24 h. The dimensions of the inhibitory zones encircling the membranes were identified using a digital camera of the fluorescence stereomicroscope (M165 FC, Leica) and the average results are reported.Electrospun PCL membranes containing one or more components of BBR, GO, and ZnO NPs were fabricated with a similar procedure.
3 Discussion
3.1 Morphology of the composite membranes
The notable difference in surface morphology of the electrospun PCL, PCL/GO, PCL/ZnO/GO (marked as PCL/ZG), PCL/PEG/ZnO/GO (marked as PCL/PZG), PCL/BBR, and PCL/PZG/BBR membranes was distinguished in SEM images shown in Fig. 1. The PCL fibers (Fig. 1a) appeared with not uniform shape, having an average width size of 2190 nm. There was adhesion between the fibers, creating an interconnected network. In addition, branched fibers with a significantly smaller size were observed, probably due to the split of the main jet during the electrospinning process. It is reported that several crucial parameters decide the morphology of electrospun nanofibers, which are polymer concentration, solvent type, applied voltage, and distance from the needle to the collector.25 Yarin et al.26 studied the electrospinning process of PCL in acetone at a voltage of 4–10 kV and distance between the needle-and-collector in the range of 15–70 mm and observed branching jets emanating from the primary jet. This phenomenon was attributed to the undulating surface of the cylindrical jet becoming unstable, forming the branches from the primary jet under an electrical field. Similarly, the electrospinning parameters applied in this study (a solvent mixture of chloroform and DMF with a ratio of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, a PCL concentration of 20 wt%, a voltage of 9 kV, a needle-and-collector distance of 20 cm, and a feed rate of 1.0 mL h−1) might cause the secondary jets that induce the formation of branched fibers. Using the same solvent mixture of chloroform and DMF with different ratios, Zargarian et al.27 received wide distributions of fiber diameter when electrospinning the PCL solution at a concentration of 12 wt%, a voltage of 17 kV, a needle-and-collector distance of 13 cm, and a flow rate of 1.0 mL h−1.
:1, a PCL concentration of 20 wt%, a voltage of 9 kV, a needle-and-collector distance of 20 cm, and a feed rate of 1.0 mL h−1) might cause the secondary jets that induce the formation of branched fibers. Using the same solvent mixture of chloroform and DMF with different ratios, Zargarian et al.27 received wide distributions of fiber diameter when electrospinning the PCL solution at a concentration of 12 wt%, a voltage of 17 kV, a needle-and-collector distance of 13 cm, and a flow rate of 1.0 mL h−1.
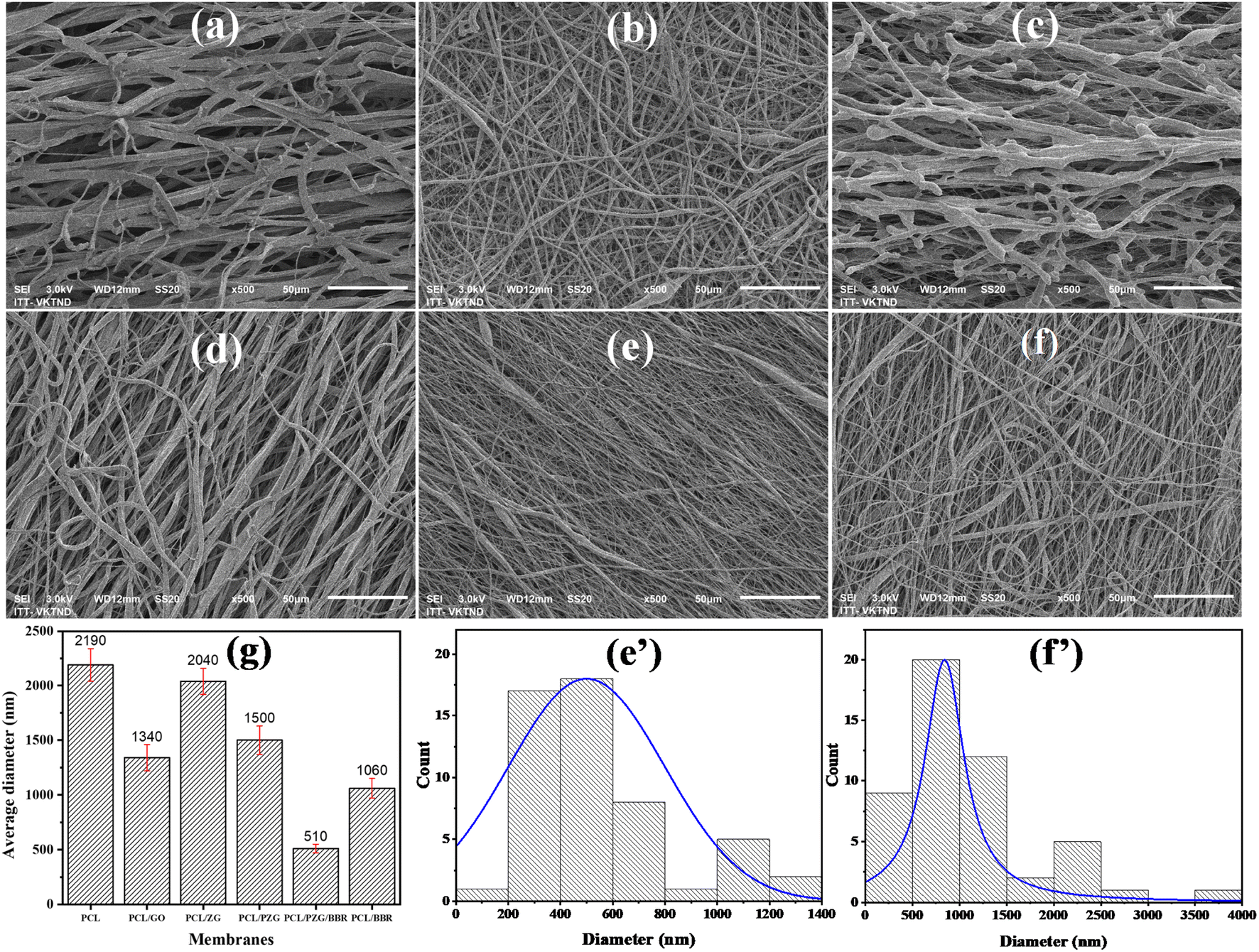 | ||
| Fig. 1 SEM images of electrospun (a) PCL, (b) PCL/GO, (c) PCL/ZG, (d) PCL/PZG, (e) PCL/PGZ/BBR, and (f) PCL/BBR fibers, average diameter of the fibers (g), and diameter distributions of electrospun (e′) PCL/PGZ/BBR and (f′) PCL/BBR fibers. | ||
In the case of the electrospun PCL/GO membrane, a broad distribution of fiber diameter was obtained in the presence of very fine branched fibers at a size of several hundred nanometers, resulting in an average diameter of 1340 nm. It is found that the reduction of solution viscosity and the increase in the conductivity caused a decrease in the diameter of PCL fibers with GO addition. Besides, the variation in surface tension of the PCL/GO solution was attributed to the diameter variation of the electrospun PCL/GO fibers.28 There was no aggregation of GO sheets on the surface of the PCL/GO membrane, indicating good compatibility of GO in the PCL solution. However, the addition of ZnO NPs significantly changed the morphology of PCL/GO fibers, forming discontinuous fibers sticking together (Fig. 1c). It is possible that the physical interaction between ester groups of PCL and hydroxyl groups of ZnO NPs hindered the entanglement between polymer chains,29 leading to a reduction in the ability to produce continuous fibers. Interestingly, more uniform, continuous, and smaller size fibers were obtained when adding PEG into the PCL/GO/ZnO NPs solution. Similar to PCL, PEG could form hydrogen bonding with ZnO NPs, where the proton acceptor oxygen of PEG bonded with the proton donor hydroxyl of ZnO NPs. Thus, PEG chains could reduce the PCL–ZnO NPs interaction and induce the dispersion of the inorganic particles and the organic phase,30 forming a better fiber morphology.
The SEM images and fiber diameter distribution of PCL/PZG/BBR (Fig. 1e and e′) and PCL/BBR (Fig. 1f and f′) reveal that the incorporation of BBR led to the formulation of thinner nanofibers with an average diameter of 1054 ± 97 nm and 500 ± 42 nm, respectively. Berberine chloride is a quaternary ammonium salt, which may increase the solution conductivity and thus increase the jet stretching under the electrical field, leading to a decrease in the fiber diameter of the composite membranes.31 The Influence of the fiber diameter on the shape memory performance was investigated by Sauter et al.32 It was reported that the reduction of the fiber diameter from the micro to the submicron, and finally to the nanoscale increased molecular orientation along the direction of the fiber due to radial geometrical confinement. A high molecular orientation in nanoscale fibers led to an increase in stress generation that facilitated a high shape recovery behavior (Rr increased). Meanwhile, the shape-fixity ratio (Rf) was reduced when the fiber diameter decreased to the nanoscale because higher stress generation induced higher retracting forces. Similarly, drug release behavior was also strongly affected by the fiber diameter. Chen et al.33 prepared 5-fluorouracil-loaded polylactide (PLA) with fiber diameter in the range of 350 nm and 1020 nm to discover the influence of fiber diameter on drug delivery. The results showed that the drug was released faster from the fine fibers than the large fibers, attributed to the fact that the fine fibers had a higher specific surface area and the drug diffused more easily from inside the fiber, which was beneficial for drug release. A similar conclusion has been drawn in the case of ampicillin trihydrate-loaded PLA.34 However, some studies reported a reverse phenomenon, for example, larger doxorubicin (DOX)-loaded PLA nanofibers released DOX faster than larger ones;35 burst release percentage for donepezil and curcumin was increased by the increase of fiber diameter.36 Therefore, the drug release behavior is complex and depends not only on fiber morphology but also other factors, such as the interactions between the drug, solvent and polymer matrix, the degree of swelling and degradation rate of the polymer matrix. In our study, the release rate of BBR from PCL/PZG/BBR nanofibers was faster than that of PLA/BBR nanofibers, attributing to the above-mentioned factors, in addition, the hydrophilicity degree of PCL/PZG/BBR nanofibers might be higher because of PEG composition (ESI data 1†).
3.2 Chemical characteristics of the composite membranes
The interaction between functional groups of components in the composite membranes was studied from their IR spectra as shown in Fig. 2. The characteristic peaks of PCL membrane appeared at 1176 cm−1 (–CH2– deformation), 1240 cm−1 (–C–O–C– stretching), 1720 cm−1 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), and 2865 and 2944 cm−1 (–CH2– stretching).37 Interestingly, when adding GO and ZnO NPs components in the PCL membrane, the vibration of the –CH2– deformation and –CO stretching bands slightly shifted to 1165 cm−1 and 1722 cm−1, respectively, accompanied by the significant decrease in the intensity of typical peaks of PCL (Fig. 2a1 and a2). This change might be attributed to the interaction through the hydrogen bonding between the oxygen-containing functional groups of PCL with hydroxyl groups of GO sheets and ZnO NPs.27,38 Meanwhile, the IR spectrum of PCL/PZG showed an increase in the intensity of –CH2– and –C–O–C– bands compared to the bands in PCL/ZG due to the supplementation of the PEG component. However, there was an absence of the absorption band of –OH stretching vibration which is specific to the PEG molecule and an increase in the intensity of the –CO stretching band in the IR spectrum of PCL/PZG, indicating that PEG molecules preferred to interact with GO and ZnO NPs components,39,40 reducing the interaction between –CO groups of PCL with others. It was reported in our previous study that the IR spectrum of pure BBR revealed the existence of –OH (3414 cm−1), –O–CH3 (2844 cm−1), CN+ (1633 cm−1), and –CC– bonds (1567 and 1506 cm−1).41 These characteristic bands of BBR either coincided with those of PCL or disappeared in the IR spectra of PCL/BBR and PCL/PZG/BBR membranes. The BBR component also reduced the intensity of typical vibration bands of both PCL and PCL/PZG membranes (Fig. 2b1 and b2). The change in the intensity of IR absorption bands may be attributed to changes in the polymer chain configuration as well as phase transitions.42 The bands at 1720 cm−1 and 1176 cm−1 assigned to the stretching of –CO and deformation of –CH2– groups, respectively, contained in the crystalline regions of PCL. It is reported that the decrease in the intensity of these bands indicated a decrease in the crystalline phase.42–44 A similar result occurred when BBR was added to the PCL matrix.
O stretching), and 2865 and 2944 cm−1 (–CH2– stretching).37 Interestingly, when adding GO and ZnO NPs components in the PCL membrane, the vibration of the –CH2– deformation and –CO stretching bands slightly shifted to 1165 cm−1 and 1722 cm−1, respectively, accompanied by the significant decrease in the intensity of typical peaks of PCL (Fig. 2a1 and a2). This change might be attributed to the interaction through the hydrogen bonding between the oxygen-containing functional groups of PCL with hydroxyl groups of GO sheets and ZnO NPs.27,38 Meanwhile, the IR spectrum of PCL/PZG showed an increase in the intensity of –CH2– and –C–O–C– bands compared to the bands in PCL/ZG due to the supplementation of the PEG component. However, there was an absence of the absorption band of –OH stretching vibration which is specific to the PEG molecule and an increase in the intensity of the –CO stretching band in the IR spectrum of PCL/PZG, indicating that PEG molecules preferred to interact with GO and ZnO NPs components,39,40 reducing the interaction between –CO groups of PCL with others. It was reported in our previous study that the IR spectrum of pure BBR revealed the existence of –OH (3414 cm−1), –O–CH3 (2844 cm−1), CN+ (1633 cm−1), and –CC– bonds (1567 and 1506 cm−1).41 These characteristic bands of BBR either coincided with those of PCL or disappeared in the IR spectra of PCL/BBR and PCL/PZG/BBR membranes. The BBR component also reduced the intensity of typical vibration bands of both PCL and PCL/PZG membranes (Fig. 2b1 and b2). The change in the intensity of IR absorption bands may be attributed to changes in the polymer chain configuration as well as phase transitions.42 The bands at 1720 cm−1 and 1176 cm−1 assigned to the stretching of –CO and deformation of –CH2– groups, respectively, contained in the crystalline regions of PCL. It is reported that the decrease in the intensity of these bands indicated a decrease in the crystalline phase.42–44 A similar result occurred when BBR was added to the PCL matrix.
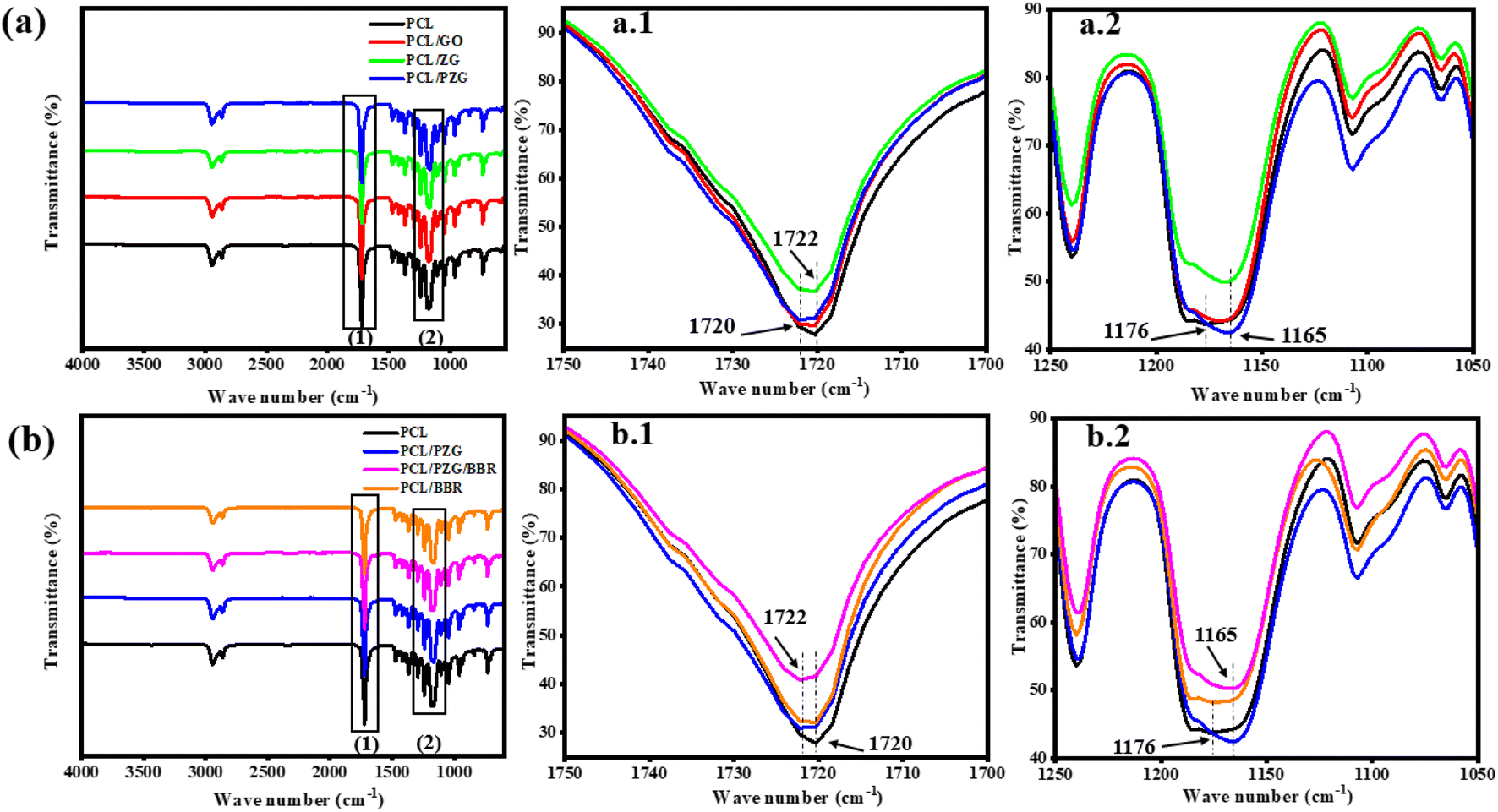 | ||
| Fig. 2 FTIR spectra of PCL fiber membrane and the composite fiber membranes in the wavenumber ranges from 500 to 4000 (a and b) 1700 to 1750 cm−1 (a.1 and b.1), and 1050 to 1250 cm−1 (a.2 and b.2). | ||
The effect of each component on the crystalline phase of the composite membranes was analyzed using Raman spectroscopy as shown from the spectra in Fig. 3. The assignment of vibration bands in Raman spectra of PCL and PCL/GO membranes is listed in Table 1. It can be observed that the characteristic bands referred to the crystalline fraction of PCL significantly enhanced in number and intensity, while the vibration band characterized by the amorphous phase disappeared when GO was added to the PCL membrane. These results are consistent with other studies reporting that GO acted as a nucleation agent increasing the crystallization of PCL.45 However, compared with the PCL/GO membrane, all the vibration bands of the PCL/ZG membrane were reduced in their intensity, indicating a decrease in the crystalline degree of the PCL matrix. Augustine et al. explained that the formation of the complex between PCL and ZnO NPs restricted the mobility of PCL chains, and thus inhibited the crystallization of PCL in the composite.38 In the case of the PCL/PZG membrane, the typical vibration bands of PEG and PCL molecules overlapped. As mentioned above, PEG chains could reduce the PCL–ZnO NPs interaction, resulting in not only better morphology but also a higher crystalline degree of the PCL/PZG fibers compared to that of PCL/ZG fibers. Meanwhile, the addition of BBR into PCL and PCL/PZG membranes slightly reduced the intensity of all typical bands, indicating a decrease in the crystalline phase.
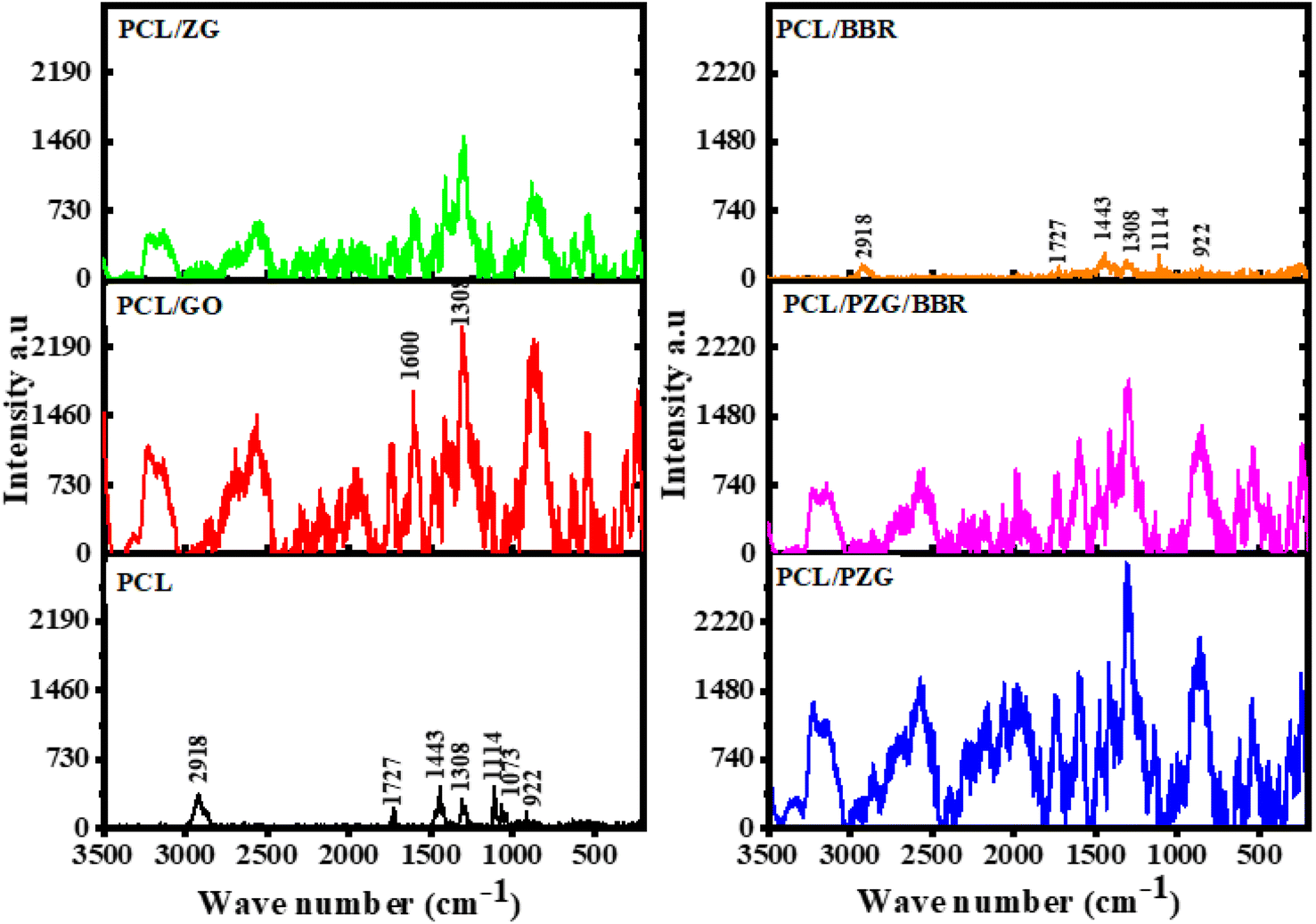 | ||
| Fig. 3 Raman spectra of the PCL fiber membrane and the composite fiber membranes. | ||
| Raman bands of PCL (cm−1) | Raman bands of PCL/GO (cm−1) | Assignment |
|---|---|---|
| 922 | 875 (broad band) | ν(C–COO) crystalline |
| 1073 | — | ν(COC) amorphous |
| 1114 | 1140 | ν(COC) crystalline |
| 1308 | 1308 | ω(CH) crystalline and amorphous |
| D band of GO | ||
| 1421 and 1443 | 1416 and 1483 | δ(CH2) crystalline |
| — | 1606 | G band of GO |
| 1727 | 1732 and 1745 | ν(CO) crystalline |
| 2918 | 2569, 3136, and 3227 | ν(CH) crystalline |
3.3 Shape memory property
The thermally activated shape memory behavior of electrospun PCL composite membranes could be performed at their melting temperature, at which switching segments have sufficient mobility to recover their original shape from a deformed shape. Therefore, DSC analysis was used to determine the transition temperature of the composite membranes for shape memory performance (Fig. 4 and Table 2). The DSC thermogram of the electrospun PCL membrane showed a transition peak, which indicated the melting temperature of PCL fibers at 65.4 °C. The incorporation of GO, ZnO NPs, and PEG components slightly decreased the melting peak of the PCL/PZG membrane to 64.1 °C and broadened its melting temperature in the range of 39 to 74 °C. Moreover, calculating from the heat of fusion of the membranes, the degree of crystallinity of the PCL/PZG membrane was higher than that of the PCL membrane (Table 2). This is attributed to GO acting as a nucleation agent.45 Additionally, it is reported that PEG and PCL could mutually promote the formation of thicker and more perfect crystals in the blend.46 Interestingly, two overlapped melting peaks were observed in the DSC thermogram of PCL/BBR and PCL/PZG/BBR membranes (Fig. 4c and d) accompanied by the reduction in the crystalline degree, which was also identified by IR and Raman analyses. A possible explanation is that BBR might interfere with the crystalline packing of the PCL segments, causing a micro-phase separation and a larger range of melting temperatures. Therefore, in this study, the recovery temperature for the shape memory test of the composite membranes was chosen to be 55 °C lower than the melting peak of the PCL membrane.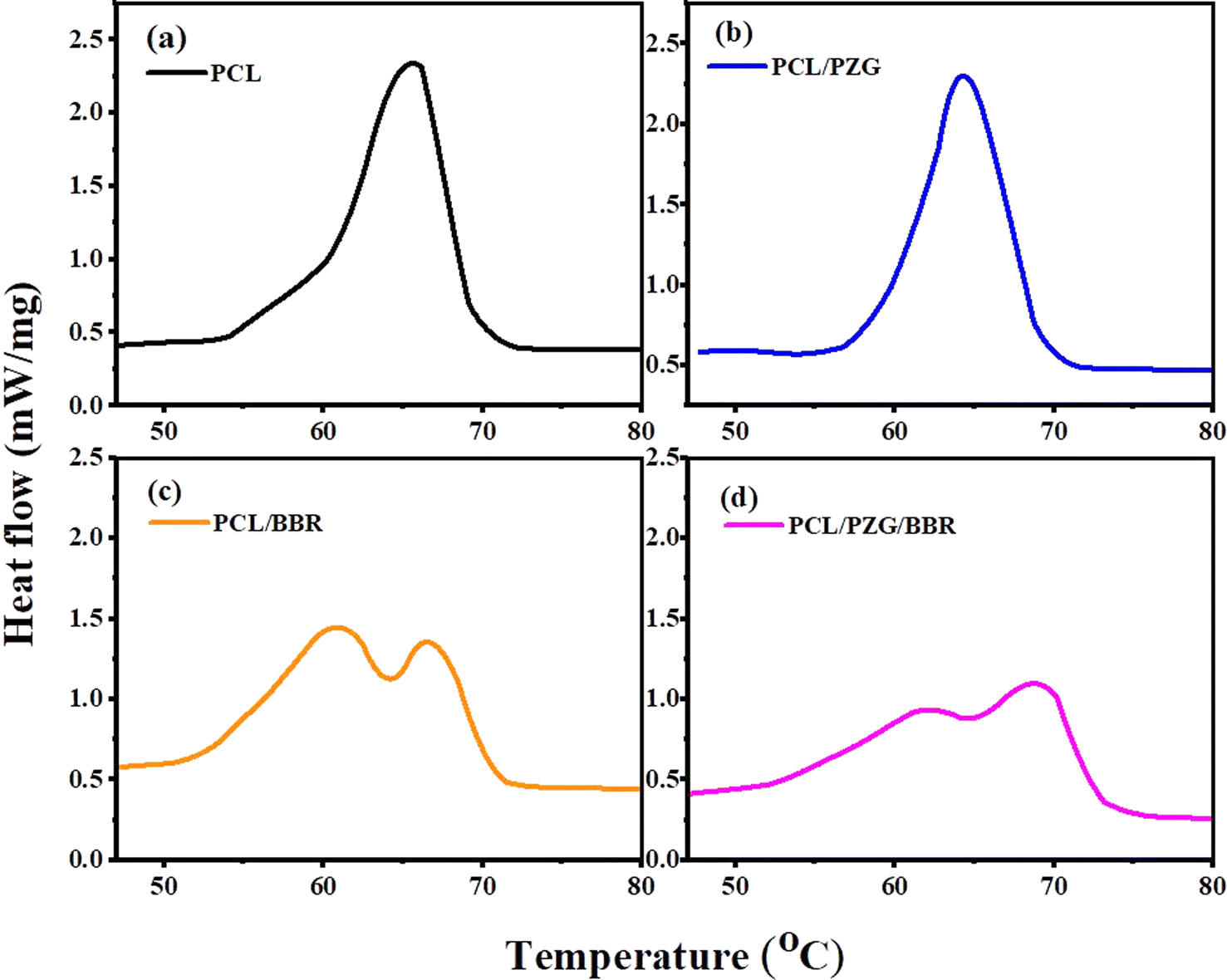 | ||
| Fig. 4 DSC curves of (a) PCL, (b) PCL/PZG, (c) PCL/BBR, and (d) PCL/PZG/BBR membranes. | ||
| Samples | Melting temperature peak (°C) | Melting temperature range (°C) | Heat of fusion ΔHt (J g−1) | Crystalline degree Xc (%) |
|---|---|---|---|---|
| PCL | 65.4 | 53 ÷ 73 | 78.31 | 57.5 |
| PCL/PZG | 64.1 | 39 ÷ 74 | 91.94 | 67.6 |
| PCL/BBR | 61.1 and 66.5 | 40 ÷ 72 | 71.14 | 52.3 |
| PCL/PZG/BBR | 62.5 and 69.0 | 39 ÷ 76 | 65.66 | 48.2 |
PCL-based SMPs have been widely studied because of the flexible adjustment of their phase transition temperature and shape memory behavior for biomedical applications. These materials were prepared by blending PCL with different polymers or covalently crosslinking PCL molecules to improve their shape memory characteristics.47,48 In this study, the shape memory behavior of electrospun PCL membrane incorporated with GO sheets, ZnO NPs, PEG, and BBR was examined (Fig. 5, Table 3, and ESI data 2†). The PCL membrane showed a high Rf (95.3% ± 2.6%) but a low Rr (22.8% ± 0.7%). When the deformed shape of the PCL membrane was fixed at 5 °C, the switching segments (amorphous phase) stopped moving and a stable new shape was maintained. Because the fixed sample was reheated at a temperature of 55 °C, lower than the melting temperature range of the PCL membrane, the switching segments did not have enough energy to elastically return to their original shapes, resulting in a low recovery ratio. The addition of GO, ZnO NPs, or BBR into the PCL membrane exhibited a positive impact on the shape memory characteristics of the composite membrane with an increase in both Rf and Rr values. The Rf value of PCL nanofiber membrane cumulatively increased from 22.8 ± 0.7 to 40.8 ± 0.8, 51.6 ± 3.2, 58.6 ± 3.7, and 76.3 ± 0.7 after the addition of GO, ZnO, PEG, and BBR components, respectively. These components contain polar chemical groups forming interaction points with PCL molecular chains, acting as additional physically cross-linked sites. This physical crosslinking reduced molecular chain slippage during deformation, keeping a high fixity ratio. Besides, the physical crosslinking strengthened hard domains (crystalline phase), causing more energy to be stored in the temporary shape, which improved the recovery performance.12,49,50 The presence of PEG in the PCL/PZG membrane led to an increase in Rr value but a reduction in Rf value. This can be explained by the plasticizing effect of PEG that improved the chain flexibility, leading to an enhancement in the recovery ability of the deformed sample.28 Besides, the addition of PEG induced the formation of the crystalline phase of the blend,29 which is responsible for energy storage and release during the thermal-activated shape memory program. The higher crystallinity was likely to form a harder network to produce a greater recovery energy in the case of the PCL/PZG membrane.30 Various studies indicated that the shape memory performance of polymers gradually augmented as the crystallinity increased51,52 because the crystal regions acted as network nodes that are responsible for the memorization of the original permanent shape of SPMs. The higher crystallinity resulted in more network nodes that hindered the chain mobility, leading to more entropic energy being stored in the SPM. When the deformed state of the SPM was reheated, that stored energy was released to regenerate the chain mobility, which could be the main driving force to regain the original shape. Therefore, higher crystallinity could improve shape memory performance. Additionally, the crosslinking density caused by the chemical or physical bonds also generates the network nodes that determine the shape memory performance. According to Fulati et al.,53 the shape recovery performance of PCL-based SMPs was more affected by the crosslinking density than the degree of crystallinity. In our study, the significant increase in the recovery ratio of PCL/PZG fiber membrane might be attributed to the increase of both crystallinity and physical crosslinking. With the addition of the BBR component, the recovery ratio of PCL/BBR and PCL/PZG/BBR notably enhanced although the degree of crystallinity reduced, indicating the dominant effect of the crosslinking density. As the impact of all components, the PCL/PZG/BBR membrane showed high values of Rf (94.27% ± 2.8%) and Rr (76.3% ± 0.7%), indicating a significant improvement in shape memory performance compared with the PCL membrane. Razqui et al.54 reported that the physical form of materials, such as film, foam, or micro/nanofibers, remarkably affected its shape memory behavior. Thanks to the porous morphology, the electrospun nanofiber membrane could be more adaptable to the deformed shape, resulting in a high fixity ratio. On the other hand, the higher orientation of the polymer chains during the electrospinning process produced better crystal morphology, attributed to a better recovery ratio.
 | ||
| Fig. 5 Photographs showing the shape memory behavior of the PCL fiber membrane and the composite membranes. (A) The original shape, (B) the fixed shape, (C) the recovered shape. | ||
| Membranes | Fixity ratio (%) | Recovery ratio (%) | Membranes | Fixity ratio (%) | Recovery ratio (%) |
|---|---|---|---|---|---|
| PCL | 95.3 ± 2.6 | 22.8 ± 0.7 | PCL/ZG | 98.1 ± 0.4 | 51.6 ± 3.2 |
| PCL/BBR | 96.2 ± 1.5 | 40.5 ± 4.75 | PCL/PZG | 95.2 ± 1.5 | 58.6 ± 3.7 |
| PCL/GO | 98.0 ± 0.6 | 40.8 ± 0.8 | PCL/PZG/BBR | 94.3 ± 2.8 | 76.3 ± 0.7 |
3.4 Drug release profiles
The release behavior of BBR was investigated relating to the composition and deformation ratio of the composite membranes (Fig. 6). The PCL/BBR membrane showed an extremely low release rate of BBR for the first 8 hours, releasing 5.5 wt% of the total BBR incorporated in the membrane. This result can be accounted for by the hydrophobic property of the PCL/BBR membrane surface, forming a barrier layer for water permeation and BBR diffusion in the initial state of release. After that, the membrane was wetted and BBR was gradually released, achieving a release percentage of 28.6 wt% at an equilibrium state after 40 hours. The addition of GO sheets, ZnO NPs, and PEG components significantly improved the wettability of the composite membrane, leading to a burst release of BBR in the first 2 hours and a higher amount of BBR released (42.7 wt%) during 40 hours.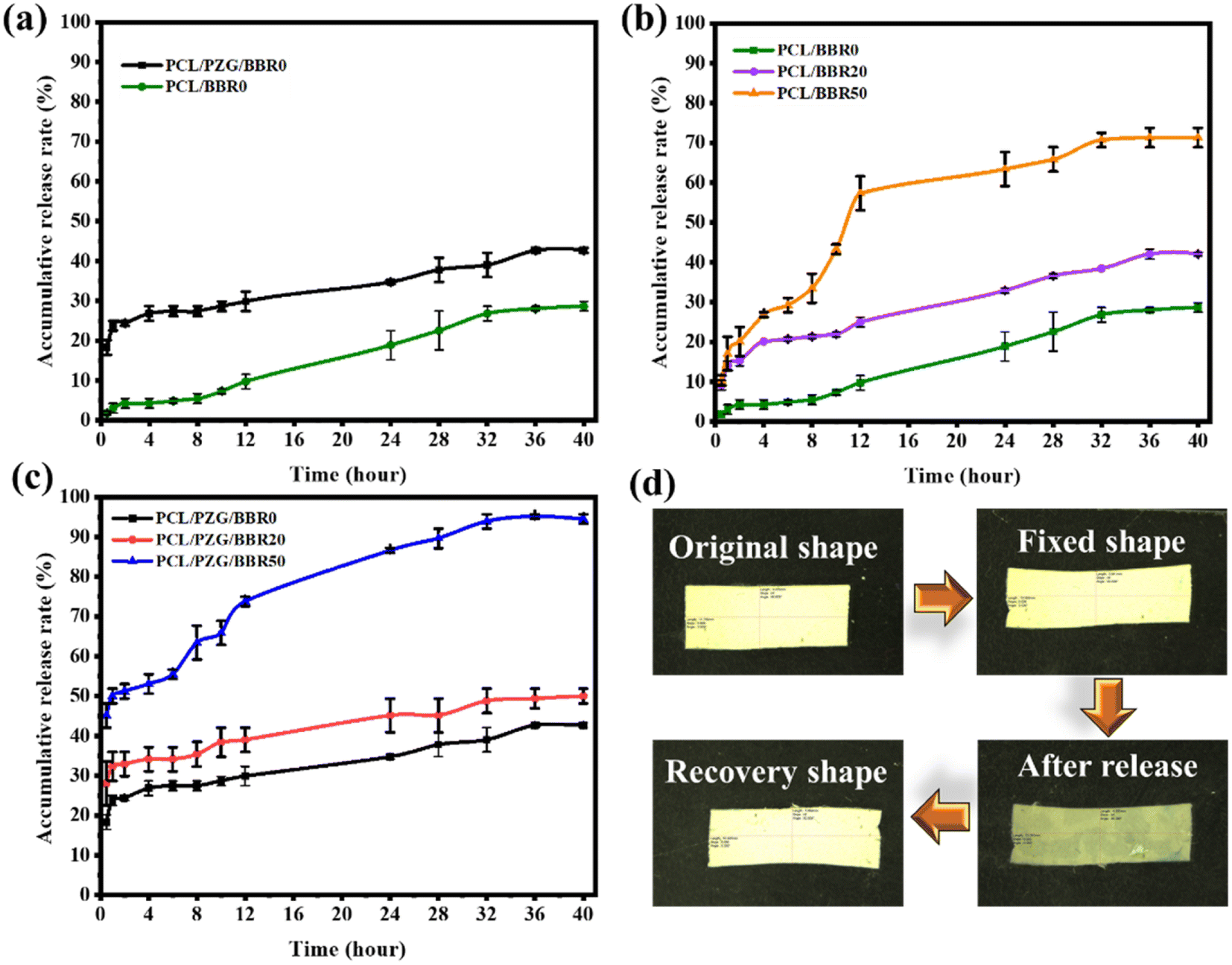 | ||
| Fig. 6 Drug release curves of (a) PCL/BBR and PCL/PZG/BBR membranes, (b) PCL/BBR at different deformation ratios, (c) PCL/PZG/BBR at different deformation ratios, and (d) photography pictures of PCL/PZG/BBR20 during release and shape memory tests. | ||
Fig. 6b and c show the BBR release behavior of the PCL/BBR and PCL/PZG/BBR membranes relating to the stretching deformation ratios of 0%, 20%, and 50%. It can be observed that the higher deformation ratio caused the larger release of BBR from membranes. Briefly, the percentage of BBR release from PCL/BBR50 and PCL/PZG/BBR50 was 2.5 and 2.3 folds that of BBR release from PCL/BBR0 and PCL/PZG/BBR0 at the equilibrium state, respectively. As expected, the stretching deformation of the composite membrane during the shape memory program can speed up the release of the drug. It can be explained that the polymer chains and BBR molecules were reoriented in the stretching direction, accompanied by a decrease in the fiber diameter. Hence, the diffusion pathway of BBR was shortened, inducing the release of BBR.22 Consequently, the drug release can be feasibly controlled by modulating the deformation ratio. The PCL/PZG/BBR membrane with a high deformation ratio would be effective in treatments requiring a high concentration of BBR over a long period. This assertion was examined by the antibacterial activity of the composite membranes at different deformation ratios.
3.5 Antibacterial activity of the composite membranes
The antibacterial activity of the composite membranes at deformation ratios of 0%, 20%, and 50% against MRSA and E. coli O157:H7 was evaluated based on the size of the inhibitory zone as shown in Fig. 7 and Table 4. It can be observed that the PCL/PZG/BBR membrane exhibited a larger size of inhibitory zones compared with the PCL/BBR membrane at the same deformation ratio. This is attributed to the addition of GO and ZnO NP components, which themselves possess antimicrobial action.55,56 Although GO and ZnO nanoparticles were reported to be effective antibacterial agents, their antibacterial activity depended on the polymer matrix containing the nanoparticles. Electrospun zein/GO nanofibers containing 1.0 wt% of GO displayed strong bactericidal activity against E. coli and S. aureus with the mean diameter of inhibition zones of 17.51 ± 0.76 and 19.18 ± 0.88 mm, respectively. This may be attributed to the fact that zein nanofibers possess a favorable surface for cell adherence.57 In the case of electrospun PLA/poly(butylene carbonate)/GO nanofibers containing 0.06 wt% of GO, the zone of inhibitions against E. coli and S. aureus were 1.208 ± 0.352 and 1.364 ± 0.327 mm, respectively.58 | ||
| Fig. 7 Photographs of antibacterial test of PCL/BBR and PCL/PZG/BBR membranes against (a) E. coli O157:H7 and (b) MRSA. | ||
| Bacterial pathogen | Size of the inhibitory zones (mm) | |||||
|---|---|---|---|---|---|---|
| PCL/BBR | PCL/PZG/BBR | |||||
| 0% | 20% | 50% | 0% | 20% | 50% | |
| E. coli O157:H7 | 0.091 ± 0.024 | 0.107 ± 0.033 | 0.131 ± 0.015 | 0.118 ± 0.033 | 0.133 ± 0.012 | 0.156 ± 0.004 |
| MRSA | 0.16 ± 0.02 | 0.176 ± 0.022 | 0.182 ± 0.020 | 0.168 ± 0.014 | 0.184 ± 0.009 | 0.201 ± 0.016 |
While the antibacterial ratios of polyacrylonitrile/GO 1.0 wt% nanofiber membranes against E. coli and S. aureus were 85.9% and 95.9%, respectively.59 These studies indicate that GO had higher antibacterial performance against Gram-positive bacteria than Gram-negative bacteria due to the difference in the structure of microorganism membranes. Meanwhile, ZnO nanoparticles incorporated in electrospun PCL/chitosan/curcumin nanofibers showed different effects on the antibacterial activity against E. coli and S. aureus, that is the increase in the antibacterial effectiveness against E. coli while a decrease in the antibacterial performance against S. aureus.60 In our study, GO and ZnO helped to improve the antibacterial activity of the PCL/BBR nanofiber membrane. The electrospun PCL/PZG/BBR showed a higher antibacterial activity against MRSA than E. coli O157:H7.
Additionally, the PCL/PZG/BBR membrane released a significantly higher BBR concentration than the PCL/BBR membrane during 24 hours, therefore it provided a better antibacterial ability. According to the BBR release profiles, the higher deformation ratio of the composite membranes caused the higher BBR release, as a result, the larger size of the inhibitory zone was obtained. At the deformation ratio of 50%, 63.4% and 86.6 wt% of total BBR incorporated in PCL/BBR50 and PCL/PZG/BBR50 membranes were released for 24 hours, achieving the inhibitory zone of 0.131 mm and 0.156 mm against E. coli O157:H7 and 0.182 and 0.201 against MRSA, respectively. This result is consistent with other studies reporting that BBR had higher antibacterial activity against Gram-positive bacteria (MRSA) than against Gram-negative bacteria (E. coli).61 Xia et al.62 reported that BBR significantly damaged the cell wall and membrane of MRSA, causing bacterial death.
4 Conclusions
In conclusion, an improvement in the shape memory performance of the electrospun PCL membrane was achieved by the incorporation of GO sheets, ZnO NPs, PEG, and BBR components. It was examined that the additional components formed the physical crosslinking network between PCL molecules through hydrogen bonding or van der Waals interactions that tuned the morphology, crystalline degree, and melting temperature of the composite nanofiber scaffold, thereby enhancing its shape memory performance. The composite PCL/PZG/BBR membrane had smaller nanofibers with an average diameter of 500 ± 42 nm, broader melting temperature in the range of 39–76 °C, high shape memory performance with fixity ratio of 94.27% ± 2.8% at 5 °C and recovery ratio of 76.3% ± 0.7% at 55 °C. Because the composite PCL/PZG/BBR membrane showed more wettability, it released a higher amount of BBR (42.7 wt%) during 40 hours, providing better antibacterial effectiveness compared to the PCL/BBR membrane. Furthermore, the release of BBR could be programmed by modulating the deformation ratio, and higher BBR release was achieved by a higher deformation ratio. Thanks to its multifunction, including shape memory effect, controlled drug release, and antibacterial ability, the composite PCL/PZG/BBR membrane is a prospective smart material in biomedical applications such as wound dressing and drug release systems.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.† The raw data files are available from the corresponding author upon reasonable request.Author contributions
Thuy Thi Thu Nguyen designed this research and drafted the manuscript. Le Thi Le and Hue Thi Nguyen carried out experiments. Le Thi Le and Ha Thi Thu Bui performed the data analysis and designed the figures. Huy Quang Tran revised the manuscript. The authors read and approved the final manuscript.Conflicts of interest
The authors have no competing interests or personal relationships that could have appeared to influence the work reported in this paper.References
- M. Behl and A. Lendlein, Mater. Today, 2007, 10, 20–28 CrossRef.
- P. Mora, H. Schäfer, C. Jubsilp, S. Rimdusit and K. Koschek, Chem.–Asian J., 2019, 14, 4129–4139 CrossRef PubMed.
- S. Pisani, I. Genta, T. Modena, R. Dorati, M. Benazzo and B. Conti, Int. J. Mol. Sci., 2022, 23, 1290 CrossRef PubMed.
- M. Imran Khan, M. M. Zagho and R. A. Shakoor, in Smart Polymer Nanocomposites, ed. D. Ponnamma, K. K. Sadasivuni, J. J. Cabibihan and M. A. A. Al-Maadeed, Springer Cham, vol. 1, 2017, pp. 281–301 Search PubMed.
- J. N. Zhang, Y. M. Ma, J. J. Zhang, D. Xu, Q. L. Yang, J. G. Guan, X. Y. Cao and L. Jiang, Mater. Lett., 2011, 65, 3639–3642 CrossRef.
- T. Sauter, K. Kratz, M. Heuchel and A. Lendlein, Mater. Des., 2021, 202, 109546 CrossRef CAS.
- H. J. Yoo, S. S. Mahapatra and J. W. Cho, J. Phys. Chem. C, 2014, 118, 10408–10415 CrossRef CAS.
- A. Kausar, E-Polym., 2022, 22, 165–181 CrossRef CAS.
- L. Tan, L. Gan, J. Hu, Y. Zhu and J. Han, Composites, Part A, 2015, 76, 115–123 CrossRef CAS.
- X. J. Zhang, Q. S. Yang and J. S. Leng, J. Intell. Mater. Syst. Struct., 2020, 31, 2152–2164 CrossRef.
- Y. Wang, P. Zhang, Y. Zhao, R. Dai, M. Huang, W. Liu, H. Liu, S. He and C. Zhu, Polym. Compos., 2020, 41, 2094–2107 CrossRef.
- M. Abbasi-Shirsavar, M. Baghani, M. Taghavimehr, M. Golzar, M. Nikzad, M. Ansari and D. George, J. Intell. Mater. Syst. Struct., 2018, 30, 116–126 CrossRef.
- V. K. Kotharangannagari and K. Krishnan, Mater. Des., 2016, 109, 590–595 CrossRef.
- H. Koerner, J. Kelley, J. George, L. Drummy, P. Mirau, N. S. Bell, J. W. P. Hsu and R. A. Vaia, Macromolecules, 2009, 42, 8933–8942 CrossRef CAS.
- A. Leonés, L. Peponi, S. Fiori and M. Lieblich, Polymers, 2022, 14, 2657 CrossRef.
- X. Feng, G. Zhang, S. Zhuo, H. Jiang, J. Shi, F. Li and H. Li, Compos. Sci. Technol., 2016, 129, 53–60 CrossRef CAS.
- F. Zhang, Z. Zhang, Y. Liu, H. Lu and J. Leng, Smart Mater. Struct., 2013, 22, 085020 CrossRef CAS.
- N. Nahavandizadeh and M. Rezaei, Polym.-Plast. Technol. Mater., 2020, 59, 1562–1573 CAS.
- S. Saghati, R. Rahbarghazi, S. Fathi Karkan, S. Nazifkerdar, A. B. Khoshfetrat and H. Tayefi Nasrabadi, J. Res. Clin. Med., 2022, 10, 30 CrossRef.
- L. Tan, J. Hu, H. Huang, J. Han and H. Hu, Int. J. Biol. Macromol., 2015, 79, 469–476 CrossRef.
- M. Bil, E. Kijeńska-Gawrońska, E. Głodkowska-Mrówka, A. Manda-Handzlik and P. Mrówka, Mater. Sci. Eng., C, 2020, 110, 110675 CrossRef PubMed.
- X. Yin, P. Tan, H. Luo, J. Lan, Y. Shi, Y. Zhang, H. Fan and L. Tan, Mater. Sci. Eng., C, 2020, 109, 110541 CrossRef PubMed.
- H. T. Nguyen, T. N. Pham, L. T. Le, T. K. Nguyen, A. T. Le, T. Q. Huy and T. T. T. Nguyen, RSC Adv., 2023, 13, 6027–6037 RSC.
- T. T. T. Nguyen, H. T. Nguyen, H. T. Trinh, T. T. T. Bui, A. T. Le and T. Q. Huy, Macromol. Res., 2022, 30, 124–135 CrossRef CAS.
- R. Abdulhussain, A. Adebisi, B. R. Conway and K. Asare-Addo, J. Drug Delivery Sci. Technol., 2023, 90, 105156 CrossRef CAS.
- A. L. Yarin, W. Kataphinan and D. H. Reneker, J. Appl. Phys., 2005, 98, 064501 CrossRef.
- S. S. Zargarian and V. Haddadi-Asl, Iran. Polym. J., 2010, 19, 457–468 Search PubMed.
- S. Ramazani and M. Karimi, Polym. Compos., 2016, 37, 131–140 CrossRef.
- E. S. Permyakova, A. M. Manakhov, P. V. Kiryukhantsev-Korneev, D. V. Leybo, A. S. Konopatsky, Y. A. Makarets, S. Y. Filippovich, S. G. Ignatov and D. V. Shtansky, Polymers, 2022, 14, 5364 CrossRef PubMed.
- N. Sened, L. Djahnit, K. El-Miloudi and M. A. Lopez-Manchado, Polym. Sci., Ser. A, 2021, 63, 855–864 CrossRef CAS.
- L. Ma, Y. Yu, H. Liu, W. Sun, Z. Lin, C. Liu and L. Miao, Sci. Rep., 2021, 11, 1027 CrossRef CAS.
- T. Sauter, K. Kratz, M. Heuchel and A. Lendlein, Mater. Des., 2021, 202, 109546 CrossRef CAS.
- A. C. Chen, X. B. Huang, X. M. Cai, J. Lu, J. Yuan and J. Shen, Fibers Polym., 2012, 13, 1120–1125 CrossRef.
- T. E. Boncu and N. Ozdemir, Beilstein J. Nanotechnol., 2022, 13, 245–254 CrossRef CAS PubMed.
- A. Doustgani, J. Ind. Text., 2017, 47, 71–88 CrossRef CAS.
- S. Aydin, I. Kabaoglu, E. Guler, F. Topal, A. N. Hazar-Yavuz, C. Ekentok, E. Tatar, F. Gurbuz, O. Gunduz and M. E. Cam, Macromol. Mater. Eng., 2022, 307, 2100855 CrossRef.
- P. Ranjbarvan, M. Soleimani, A. Samadi Kuchaksaraei, J. Ai, R. Faridi Majidi and J. Verdi, Microsc. Res. Tech., 2017, 80, 495–503 CrossRef PubMed.
- R. Augustine, H. N. Malik, D. K. Singhal, A. Mukherjee, D. Malakar, N. Kalarikkal and S. Thomas, J. Polym. Res., 2014, 21, 347 CrossRef.
- C. Li, M. Xiang and L. Ye, RSC Adv., 2016, 6, 72193 RSC.
- A. Badry, A. A. E. H. Ibrahim, M. I. Said, A. A. E. Nasr, M. A. Mohamed, A. K. Hassan and M. M. Safwat, BMC Vet. Res., 2023, 19, 1 CrossRef PubMed.
- P. R. Vuddanda, V. M. Rajamanickam, M. Yaspal and S. Singh, BioMed Res. Int., 2014, 2014, 951942 CrossRef PubMed.
- K. Philipson, J. N. Hay and M. J. Jenkins, Thermochim. Acta, 2014, 595, 74–82 CrossRef.
- C. A. Kelly, K. L. Harrison, G. A. Leeke and M. J. Jenkins, Polym. J., 2013, 45, 188–192 CrossRef.
- B. Zhu, J. Li, Y. He, N. Yoshie and Y. Inoue, Macromol. Biosci., 2003, 3, 684–693 CrossRef.
- A. Kołodziej, E. Długoń, M. Świętek, M. Ziąbka, E. Dawiec, M. Gubernat, M. Michalec and A. Wesełucha-Birczyńska, J. Compos. Sci., 2021, 5, 20 CrossRef.
- J. Dong, T. Yuan, X. T. Yu, L. Y. Jia and Z. Mei, High Perform. Polym., 2012, 24, 441–446 CrossRef CAS.
- M. E. Pekdemir, E. Öner, M. Kök and I. N. Qader, Iran. Polym. J., 2021, 30, 633–641 CrossRef CAS.
- S. Pandini, T. Riccò, A. Borboni, I. Bodini, D. Vetturi, D. Cambiaghi, M. Toselli, K. Paderni, M. Messori, F. Pilati, F. Chiellini and C. Bartoli, J. Mater. Eng. Perform., 2014, 23, 2545–2552 CrossRef CAS.
- S. Rana, J. W. Cho and L. P. Tan, RSC Adv., 2013, 3, 13796–13803 RSC.
- X. Qi, X. Yao, S. Deng, T. Zhou and Q. Fu, J. Mater. Chem. A, 2014, 2, 2240–2249 RSC.
- R. B. Cunha, F. R. Pe, P. Agrawal, G. F. Brito and T. J. A. Melo, Smart Mater. Struct., 2023, 32, 085016 CrossRef.
- D. Nie, X. Yin, Z. Cai and J. Wang, Polymers, 2022, 14, 1569 CrossRef CAS.
- A. Fulati, K. Uto and M. Ebara, Polymers, 2022, 14, 4740 CrossRef CAS.
- I. Razquin, A. Iregui, M. Cobos, J. Latasa, A. Eceiza, K. González, L. Martin, A. J. Müller, A. González and L. Irusta, Polymer, 2023, 282, 126160 CrossRef CAS.
- H. Mohammed, A. Kumar, E. Bekyarova, Y. Al-Hadeethi, X. Zhang, M. Chen, M. S. Ansari, A. Cochis and L. Rimondini, Front. Bioeng. Biotechnol., 2020, 8, 465 CrossRef.
- S. V. Gudkov, D. E. Burmistrov, D. A. Serov, M. B. Rebezov, A. A. Semenova and A. B. Lisitsyn, Front. Phys., 2021, 9, 641481 CrossRef.
- H. Asadi, A. Ghaee, J. Nourmohammadi and A. Mashak, Int. J. Polym. Mater., 2020, 69, 173–185 CrossRef CAS.
- X. Gu, Y. Li, R. Cao, S. Liu, C. Fu, S. Feng, C. Yang, W. Cheng and Y. Wang, AIP Adv., 2019, 9, 065315 CrossRef.
- J. H. Li, H. Zhang, W. Zhang and W. Liu, J. Biomater. Sci., Polym. Ed., 2019, 30, 1620–1635 CrossRef CAS.
- P. Mosallanezhad, H. Nazockdast, Z. Ahmadi and A. Rostami, Front. Bioeng. Biotechnol., 2022, 10, 1027351 CrossRef PubMed.
- M. Čerňáková and D. Košťálová, Folia Microbiol., 2002, 47, 375–378 CrossRef PubMed.
- S. Xia, L. Ma, G. Wang, J. Yang, M. Zhang, X. Wang, J. Su and M. Xie, Infect. Drug Resist., 2022, 15, 1933–1944 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05618c |
| This journal is © The Royal Society of Chemistry 2024 |