DOI:
10.1039/D4RA05697C
(Review Article)
RSC Adv., 2024,
14, 33864-33905
Recent progress in therapeutic applications of fluorinated five-membered heterocycles and their benzo-fused systems
Received
6th August 2024
, Accepted 18th October 2024
First published on 25th October 2024
Abstract
Heterocyclic derivatives grafted with fluorine atom(s) have attracted the attention of scientists due to the unique physicochemical properties of the C–F bond. The inclusion of fluorine atom(s) into organic compounds often increases their lipophilicity and metabolic stability, enhancing their bioavailability and affinity for target proteins. Therefore, it is not surprising to find that more than 20% of the medications on the market contain fluorine, and nearly 300 fluorine-containing drugs have been officially approved for use as medicines. In this review article, we are interested in classifying and describing the reports comprising varied therapeutic activities of the directly fluorinated five-membered heterocycles and their fused systems during the last two decades. These therapeutic activities included antiviral, anti-inflammatory, enzymatic inhibitory, antimalarial, anticoagulant, antipsychotic, antioxidant, antiprotozoal, histamine-H3 receptor, serotonin receptor, chemokine receptor, prostaglandin-D2 receptor, and PBR inhibition activities. In many cases, the activities of fluorinated azoles were almost equal to or exceeded the potency of reference drugs.
 Ashraf A. Abbas | Professor Ashraf A. Abbas was born in Egypt in September 1968 and he is presently Professor of Organic Chemistry, Department of Chemistry, Faculty of Science, Cairo University, Giza, Egypt since 2009. He graduated from Cairo University in 1990. He received his M.Sc. and PhD degrees in 1994 and 1997, respectively, from Cairo University. He spent six months in the Fakultat fur Chemie, universitat of Konstanz (Germany) on a DAAD fellowship (1996) to finish his PhD thesis and one year at Tokyo Institute of technology (TIT, Japan) as UNESCO fellow (postdoctoral fellowship) from Oct. 2001 to Sept. 2002. He received several research prizes in chemistry; (1) he was awarded the prize of Prof. Dr Mohamed Abdel Salam in chemistry (2001) for young scientists provided by Academy of Scientific Research and Technology (Egypt). (2) He was awarded the Cairo university encouragement prize in Chemistry in 2004. (3) He was awarded the Third Word Academy of Science (TWAS) prize in chemistry for young scientists in 2004 provided by ICTP-Strada, Triesta-Italy, (4) he was awarded the State Award in Chemistry, Egypt, 2005. He has published many papers in the field of synthesis and applications of macrocycles and bis-heterocycle chemistry. |
 Thoraya A. Farghaly | Thoraya Abd Elreheem Farghaly was born in Cairo, Egypt in 1974. She received her BSc (1996); M.Sc. (2002) and PhD (2005) degrees from University of Cairo. She is Professor of organic chemistry in the Chemistry Department, Faculty of Science, University of Cairo. She works at Umm Al-Qura university from 2014 till now. She joined the scientific school of Prof. A. S. Shawali in 1997 and she has published more than 220 papers in the area of the chemistry of hydrazonoyl halides, heterocyclic chemistry and bioactive heterocyclic compounds. |
 Kamal M. Dawood | Kamal M. Dawood graduated from Cairo University, Egypt in 1987, and received his PhD in 1995 from Cairo University. In 1997 he was awarded the UNESCO Fellowship at TIT for one year and in 1999 he was awarded the JSPS Fellowship for two years and in both fellowships he worked with Professor T. Fuchigami at the Tokyo Institute of Technology (TIT) in the field of “Anodic Selective Fluorination of Heterocycles'. Further, he was awarded the Alexander von Humboldt (AvH) Fellowship at Hanover University in 2004–2005 with Prof. A. Kirschning (in the area of polymer-supported palladium-catalyzed cross-coupling reactions) and as AvH three short visits in 2007, 2008 and 2012 with Prof. P. Metz at TU-Dresden (in the field of Metathesis Reactions in Domino Processes). Since May 2007 – to date, he has been appointed as a full Professor of Organic Chemistry, Faculty of Science, Cairo University. He worked as Professor of Organic Chemistry at Chemistry Department, Kuwait University from Sept. 2013 till Aug. 2017. He received many National Awards: Cairo University Award in Chemistry (2002), the State Award in Chemistry (2007), Cairo University Award for Academic Excellence (2012) and Cairo University Merit Award (2017). He has published more than 160 scientific papers, reviews, and book chapters in distinguished international journals. There are about 3900 citations of his work (Scopus h-index 33). |
1. Introduction
Approximately 85% of bioactive chemicals are expected to consist of heterocyclic moieties, therefore researchers have been motivated to include heterocyclic structures in synthetic medications.1 Conversely, adding fluorine atom(s) to medications throughout the latter part of the 20th century offered an additional essential method for designing pharmaceuticals.2–4 Fluorinated drugs have experienced significant growth since the introduction of the first fluorocorticosteroid and fludrocortisone in 1954.5 Currently, 20% of the drugs on the market are fluorinated, and approximately 30% of fluorinated drugs are considered blockbuster pharmaceuticals.6 Almost 300 pharmaceuticals containing fluorine have been officially approved for use as medications.7 The incorporation of the small size highly electronegative fluorine atom(s) into organic molecules was attributed to the unique physicochemical characteristics of the C–F bond,8 comprising the chemical and physical properties of the entire molecules such as polarity, high bond strength, and little steric hindrance, acidity, basicity, solubility, and hydrogen bonding interactions.9–13 In addition, the inclusion of fluorine atom(s) in organic compounds often increases their lipophilicity and metabolic stability, enhancing their bioavailability and binding affinity with target proteins.14–17
It was reported that fluorine-substituted thrombin inhibitors have C–F…C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O interactions greatly affected the protein–ligand interactions and significantly enhanced the binding affinities. Fluorinated thrombin inhibitors showed binding affinity five times stronger than the nonfluorinated analogues. X-ray structure analysis showed that the F atom is in remarkably close contact with the H–Cα–CO moiety of Asn98 of thrombin.18–20 In addition, the close amide-NH⋯F interaction between fluorine and amide residues in proteins was very predominant and established by X-ray crystallography.20–22
O interactions greatly affected the protein–ligand interactions and significantly enhanced the binding affinities. Fluorinated thrombin inhibitors showed binding affinity five times stronger than the nonfluorinated analogues. X-ray structure analysis showed that the F atom is in remarkably close contact with the H–Cα–CO moiety of Asn98 of thrombin.18–20 In addition, the close amide-NH⋯F interaction between fluorine and amide residues in proteins was very predominant and established by X-ray crystallography.20–22
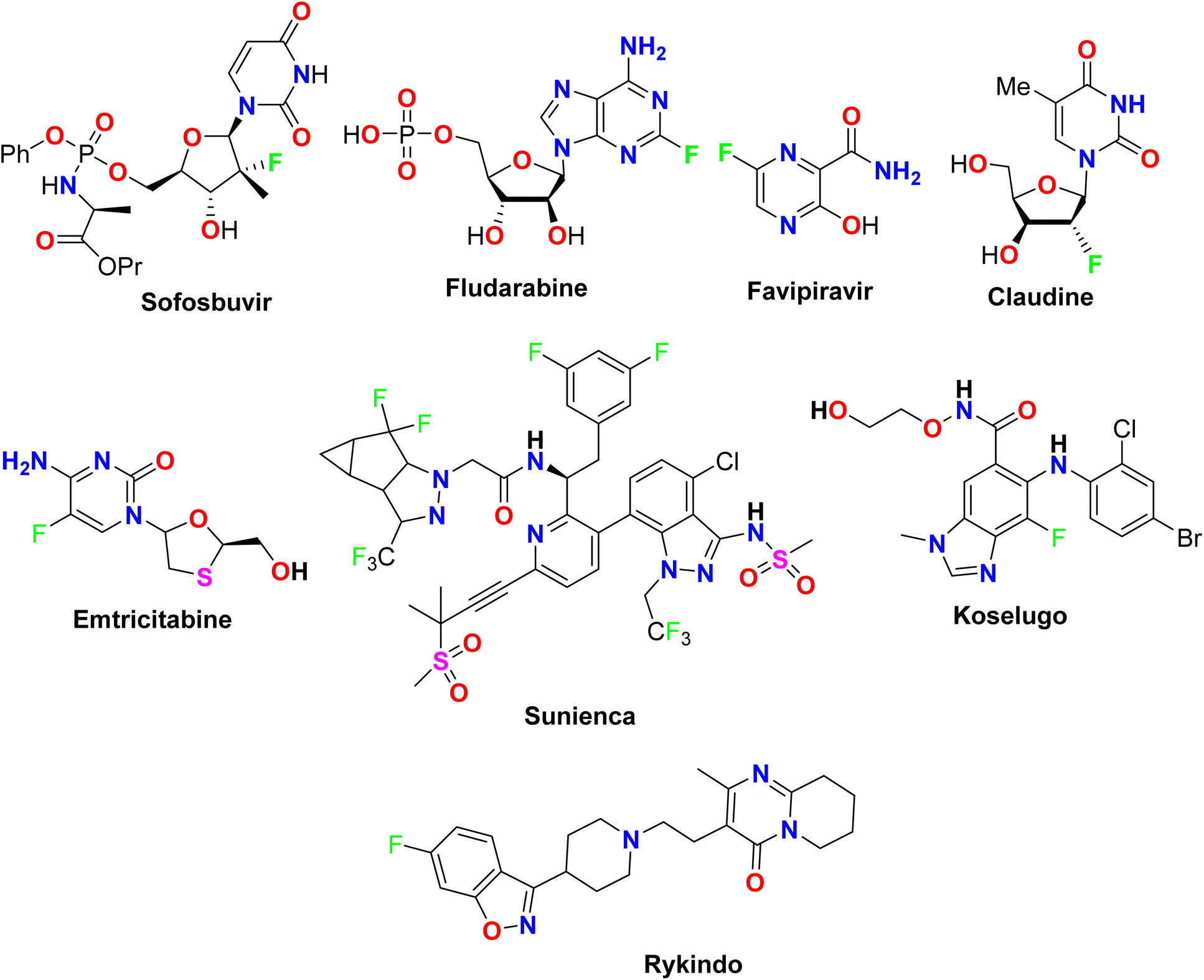
The presence of fluorine atom(s) in heterocyclic scaffolds made them more biologically potent compounds where several commercial drugs approved by the FDA were found to possess directly fluorinated heterocyclic ingredients as described in Fig. 1. For example, Favipiravir (or Avigan) was approved in 2014 as an antiviral drug used to treat influenza in Japan including A(H1N1), A(H5N1), and the recently emerged A(H7N9) avian virus.23 In addition, some fluorinated nucleosides were also approved as antiviral drugs, such as Sofosbuvir (Sovaldi) for the treatment of hepatitis C, and Claudine for the treatment of hepatitis B virus.24 Furthermore, Fludarabine was approved as an immunosuppressant drug.25 Some fluorine-containing heterocycles were also approved as anti-HIV drugs, such as Emtricitabine which was approved by FDA in 2003 as a NRTI (Nucleoside reverse transcriptase inhibitor),26,27 and Lenacapavir (Sunlenca, GS-6207), was approved in 2022, as potent HIV CA (human immunodeficiency virus capsid protein) inhibitor with picomolar-range potency.28 Koselugo (Selumetinib): for treatment of neurofibromatosis type 1, a genetic disorder that causes tumors to grow on nerves.29 Rykindo (Risperidone) was approved by the FDA in 2023 as an antipsychotic drug for the treatment of schizophrenia.30
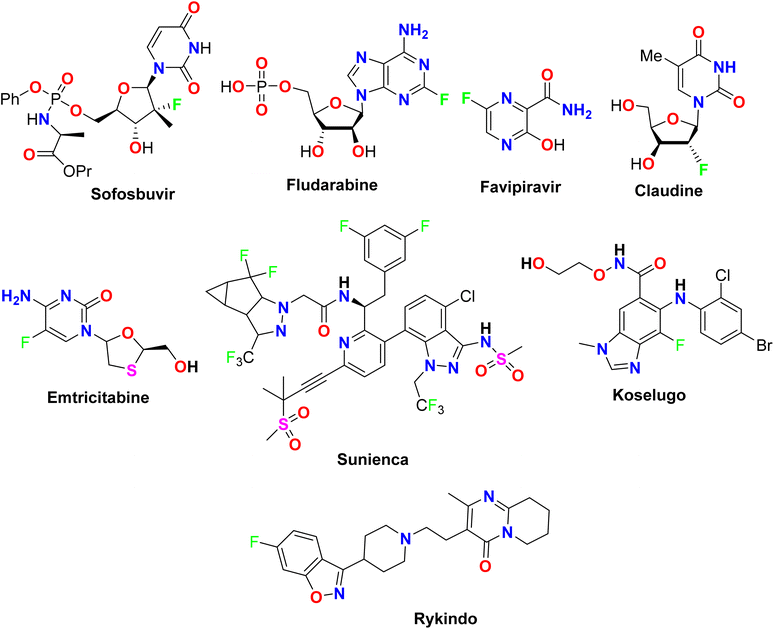 |
| | Fig. 1 The structure of some FDA-approved fluorinated medications. | |
Furthermore, the therapeutic potency of some non-fluorinated heterocycles of potent therapeutic effects was compared with their fluorinated analogues, to confirm the marked influence of replacing a hydrogen atom with a fluorine atom (Fig. 2).31–33 For example, the anti-viral drugs Emtricitabine (II) and its 5-fluoro analogue of Lamivudine (for treatment of HIV) (I) are examples of this effect, where II is a more potent HIV-1 inhibitor four-to ten-fold times than I.31,32 The 4-fluorinated indole IV is about 50-fold HIV-1 inhibitor than the non-fluorinated indole III.33,34 Similarly, the γ-secretase VI had extraordinary potency compared with its analogue V.35
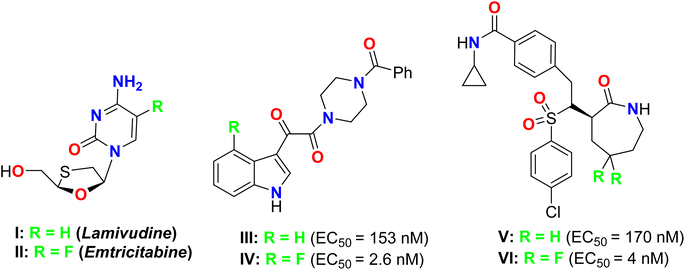 |
| | Fig. 2 Some medicinally active fluorinated heterocycles and their non-fluorinated analogues. | |
Thus, medicinal chemists and drug developers are highly fascinated with fluorine-based small molecules in drug discovery and are focusing their efforts on overcoming the challenges associated with the insertion of fluorine atom(s) into small organic molecules. Advancements in technology have made it easier to incorporate fluorine into new small molecules. Fluorine atom(s) have been inserted into heterocyclic rings via either chemical or electrochemical synthetic routes. Chemical fluorination of heterocyclic systems was reported employing either nucleophilic or electrophilic fluorinating agents such as F2, SF4, XeF2, Et2NSF3 (DAST), or N-fluoropyridinium triflates.36–38 On the other hand, the electrochemical fluorination protocol involved the use of the ionic liquids; alkyl ammonium fluoride salts (Et3N·nHF, or Et4NF·nHF (n = 3, 4, 5)) as stable fluoride ion sources.39–44 The late-stage fluorination of heterocycles was reported as a straightforward promising tool involving metal-catalysed procedures to enhance the C–F bond formation of small molecules with potential industrial applications.45
The aforementioned interesting historical background compiled with our ongoing insightful evaluations of bioactive heterocycles,46–51 inspired us to focus our attention on the recent reports comprising varied therapeutic activities of the directly fluorinated five-membered heterocycles and their fused systems during the last two decades from 2004 till the end of 2024. The heterocycles incorporating only fluoroaryl or fluoroalkyl groups are not considered in this review article. The current review article is expected to serve as an interesting pool for researchers interested in medicinal chemistry and pharmaceutics for the possibility of synthesis of drug-like small fluorinated molecules.
2. Antiviral activity of fluorinated heterocycles
2.1. Antiviral activity fluorinated furans
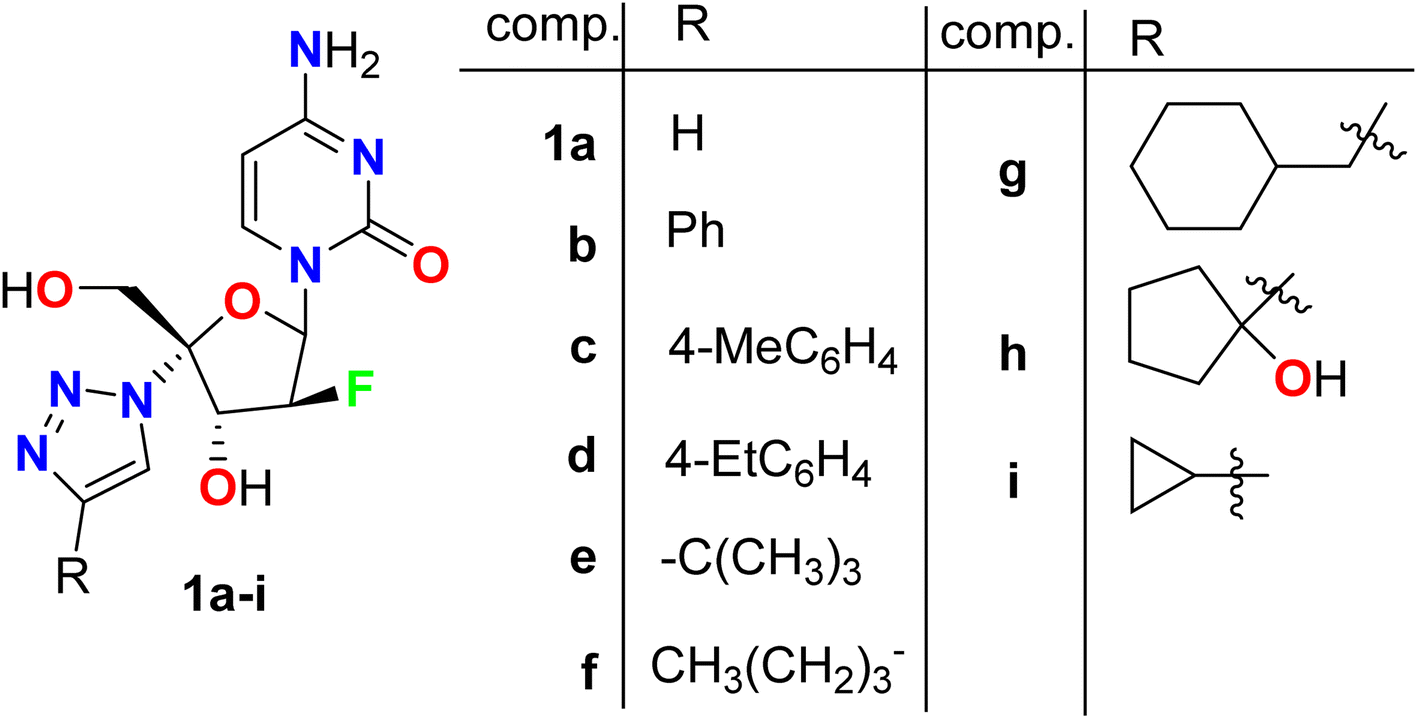
A series of triazole-based fluoro-arabinofuranoside molecular hybrids 1a–i were synthesized screened for their anti-HIV-1 activity (Fig. 3). The inhibitory activity (EC50) was determined by an anti-HIV (wild-type) replication assay. Most of these fluorinated derivatives demonstrated potent anti-HIV-1 activity. Interestingly, compounds 1a, 1b and 1e showed high potent antiviral activity with EC50 values of 0.09 μM, 0.083 μM, and 0.08 μM, respectively, with almost equipotent or better activity than Zidovudine reference anti-HIV drug (EC50 = 0.084 μM). SAR study disclosed that the unsubstituted triazole or the presence of phenyl group at the triazole-C-4 position maintained the potent antiviral activity (compounds 1a and 1b), however presence of electron donating substituents at the phenyl ring (1c and 1d) led to a dramatic decrease of anti-HIV potency. Among the other derivatives that R was alkyl group, the presence of the bulky t-butyl group (compound 1e) showed the highest antiviral activity, while alkyl substituents resulted in reduced antiviral activity. In addition, the anti-HBV activity of compounds 1b, 1c, and 1i was also investigated against the production of HBsAg (HB surface antigen) and HBeAg (HB e antigen) by ELISA assay, and the EC50 was determined. Compound 1c showed high activity against HBsAg production with EC50 = 0.01 μM but compound 1b possessed the most potent activity in inhibition of HBeAg production with EC50 = 0.25 μM. Therefore, these fluorinated hybrids were considered to have great potential as new anti-HIV drugs.52
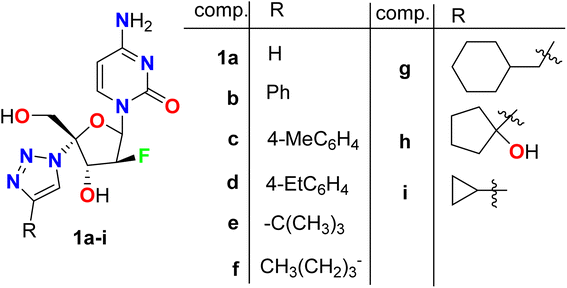 |
| | Fig. 3 Structure of triazole-based fluoro-arabinofuranoside derivative 1a–i. | |
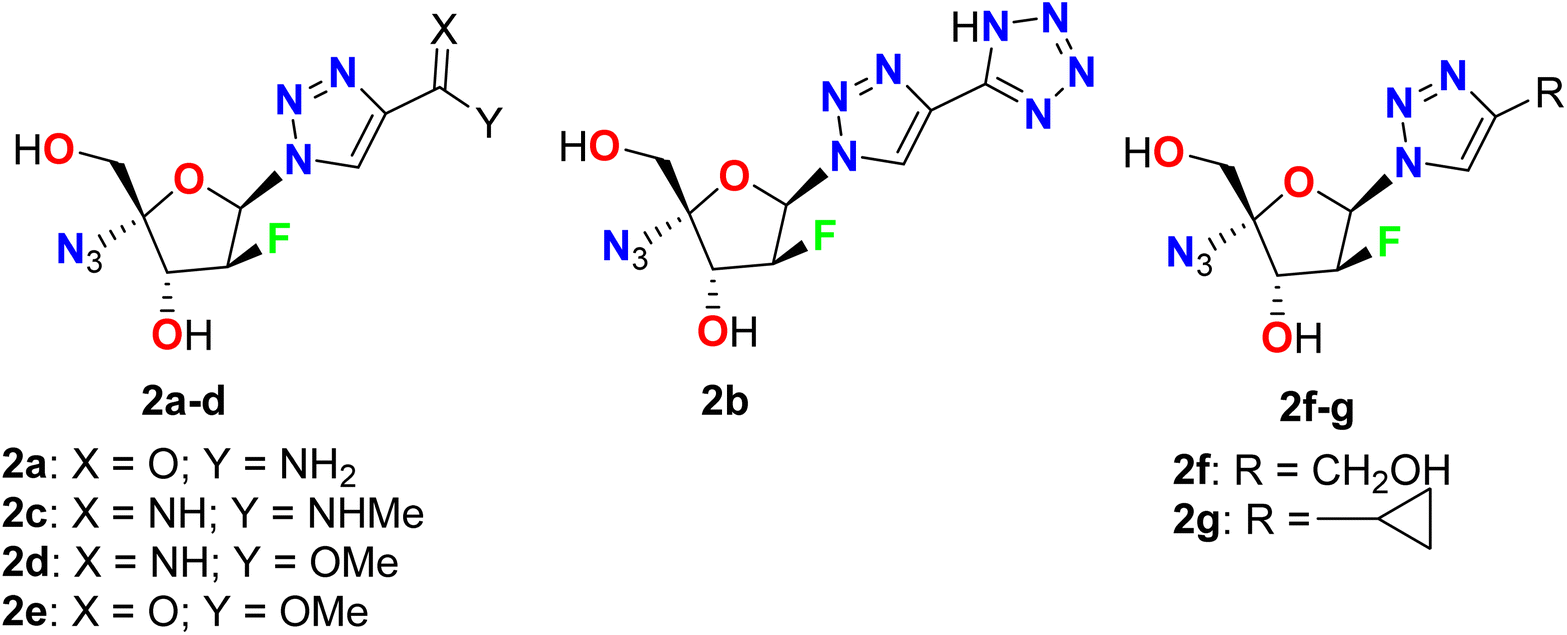
Liu et al. described the synthesis and in vitro anti-HBV activity of a series of 1,2,3-triazole-based fluoro-arabinofuranoside derivatives 2a–g in the cellular model (Fig. 4). All the tested compounds showed inhibitory activities comparable to the positive control, lamivudine at 20 μM. The most promising anti-HBV activity and low cytotoxicity in the cell model were reported for the amide-substituted candidate 2a, where this compound retained significant activity against lamivudine-resistant HBV mutants with 45.3% and 21.9% inhibition of HBsAg and HBeAg, compared with Lamivudine (3TC-treated) that showed 47.7% and 22.1% inhibitions, respectively, at 20 μM concentration, on 9 days. On the other hand, both the liver and serum DHBV DNA levels (53.3% and 67.4%, respectively) were decreased markedly upon treatment with 2a, in duck HBV (DHBV)-infected duck models. SAR study revealed that the high activity of 2a has relied on the presence of the amide group at the triazole ring that can interact with the viral DNA polymerase through hydrogen bonding between the amide group and dGMP, as well as the π–π stacking interactions between the 1,2,3-triazole planar heterocyclic and the adjacent DNA base. Besides, the azido group participated in a hydrophobic interaction with the side chains of Val84, Phe88, Leu180, and Met204. These developed studies provided strong support for the application of compound 2a as a potential alternative therapy for the treatment of HBV infection.53
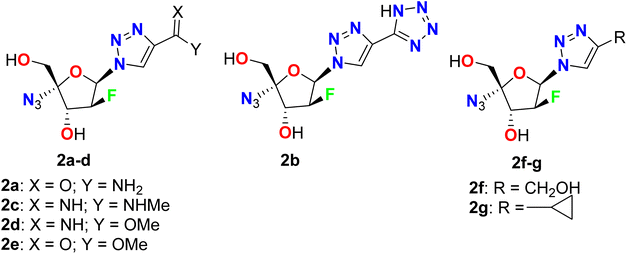 |
| | Fig. 4 Structure of 1,2,3-triazole-based fluoro-arabinofuranoside derivatives 2a–g. | |
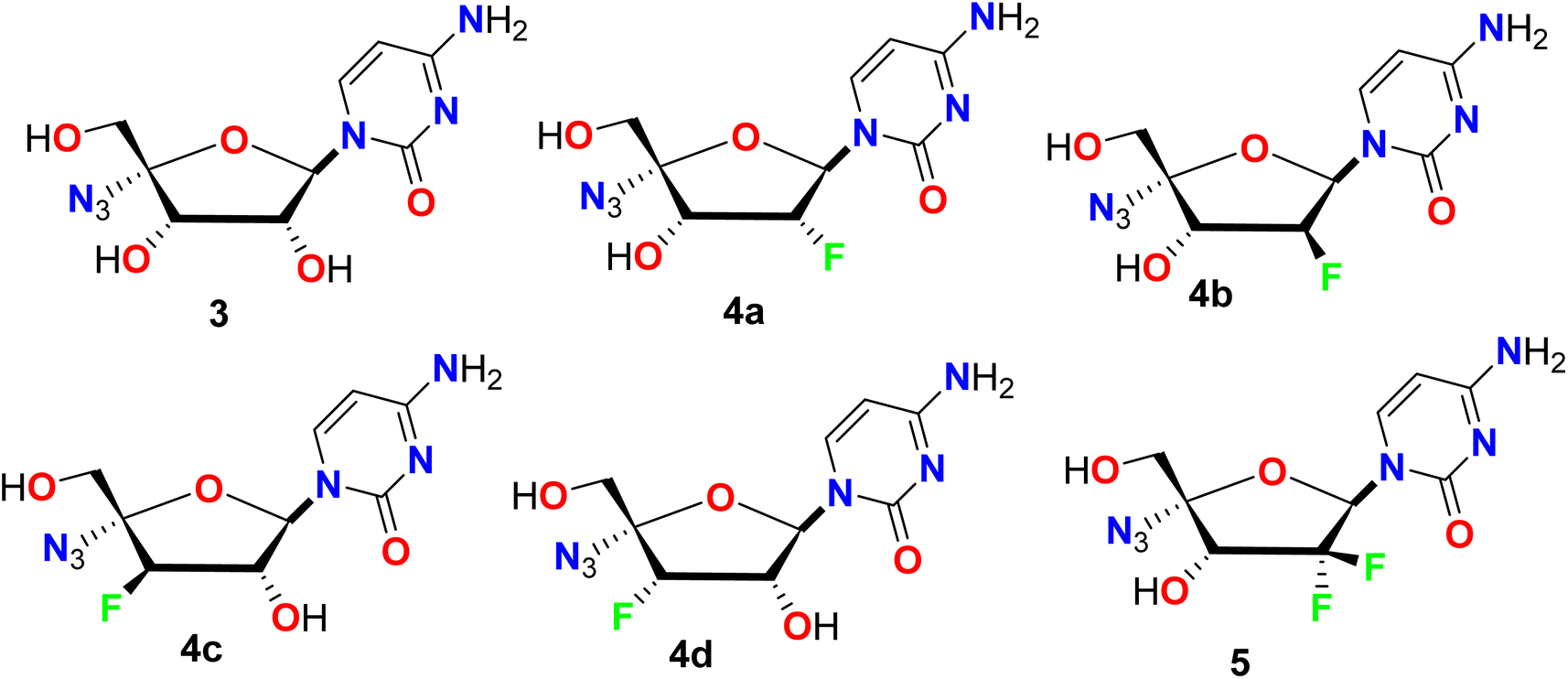
Smith et al. described a synthetic route to some fluorinated furano-nucleoside hybrids 3–5 and screened their potency as inhibitors of the RNA polymerase encoded by hepatitis C virus (HCV) in the subgenomic replicon assay system using the 2209-23 cell line (Fig. 5). Among the synthesized compounds in this series, the derivatives 4b and 5 proved their highest antiviral potency in the HCV replicon system with EC50 values of 24 nM and 66 nM, respectively. The derivative 4b exhibited more than a 50-fold enhancement in the antiviral potency when compared to the parent non-fluorinated nucleoside analogue 3.54
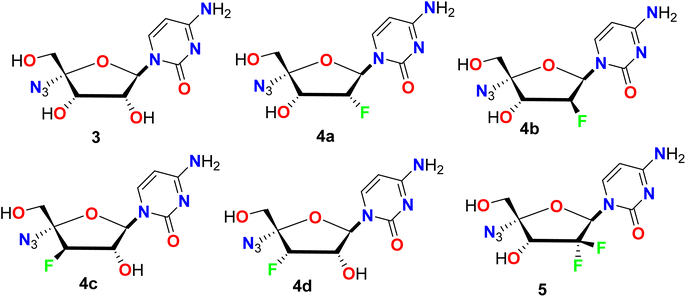 |
| | Fig. 5 Structure of fluorinated furano-nucleoside hybrids 3–5. | |
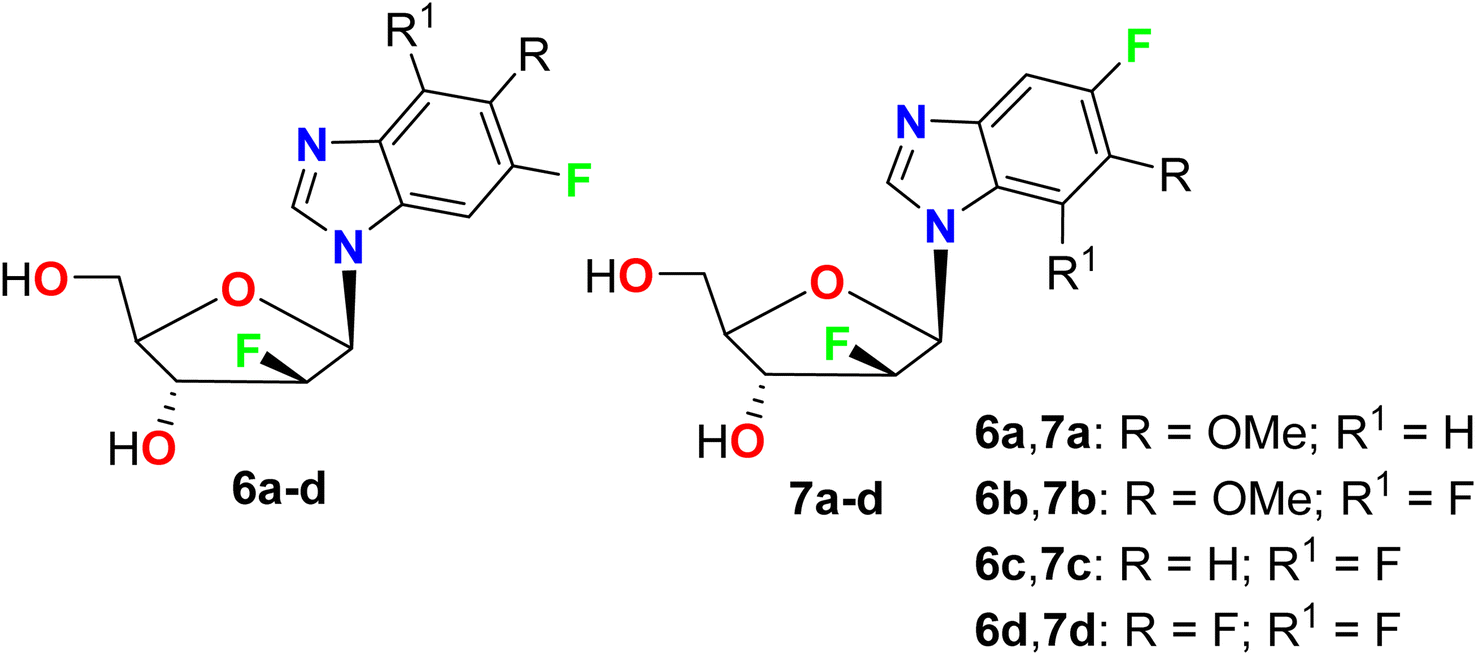
Synthesis of the regioisomeric N1- and N3-nucleoside fluorinated furanosyl nucleosides 6a–d and 7a–d was reported by Kharitonova et al. and their inhibitory activity against the herpes simplex virus type 1 (HSV-1) in vitro was performed by the method of cytopathic effect (CPE) inhibition assay (Fig. 6). The cytotoxicity was also studied using the Vero-E6 (African green monkey kidney) cells at the maximal concentration of 1000 μg mL−1 for 72 hours. Compounds 6–7 demonstrated inhibition of the development of virus-induced CPE in a wide concentration range 6–8 times lower than CD50, where compounds mixtures 6a and 6b had CD50 values of 245 and 487.5 μg mL−1, respectively.55
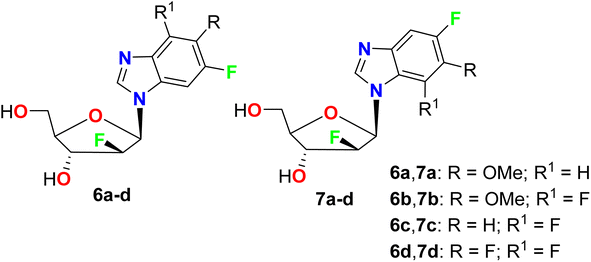 |
| | Fig. 6 Structure of fluorinated furanosyl nucleosides 6a–d and 7a–d. | |
2.2. Antiviral activity fluorinated pyrroles
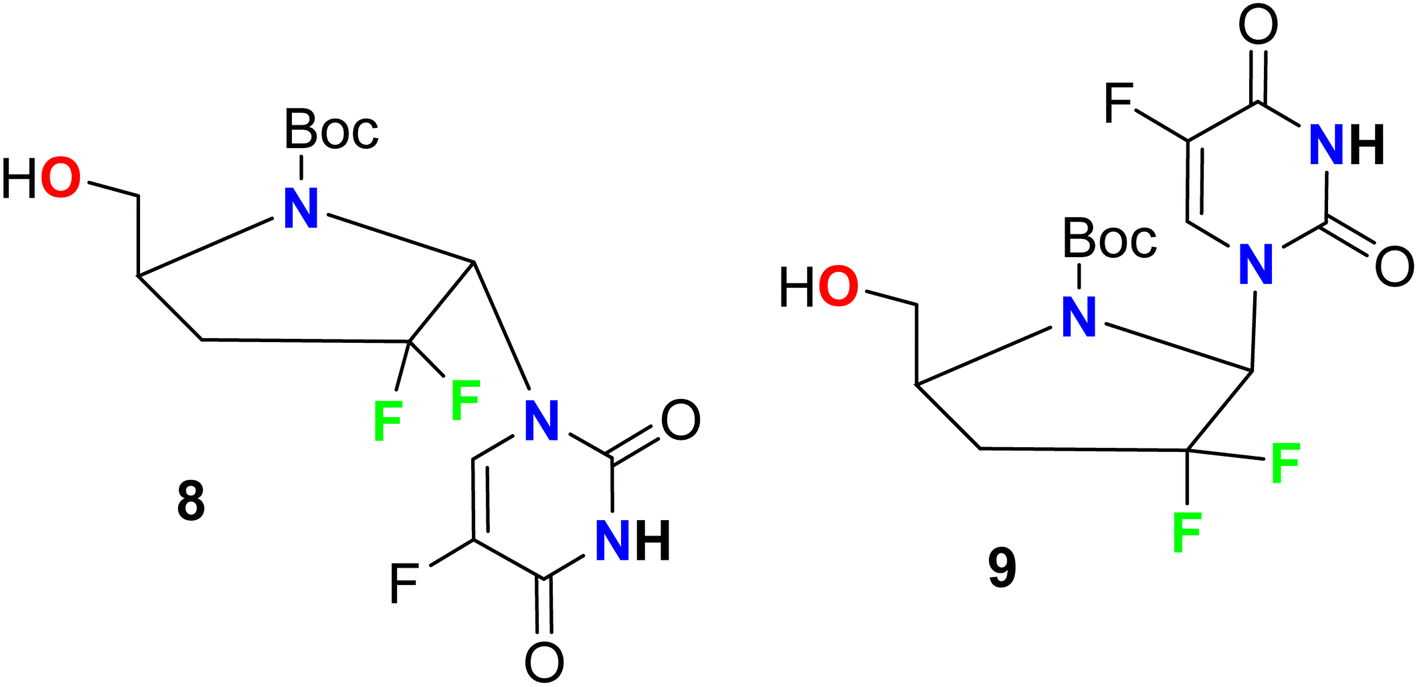
Ferrero et al. reported the synthesis of two fluorinated pyrrole-based hybrids 8 and 9 and their anti-HIV-1 activity was investigated (Fig. 7). The bioassay was carried out using AZT (zidovudine) as a reference standard with human peripheral blood mononuclear cell (PBM) protocol. Most of the tested compounds showed moderate activities against HIV-1LAI compared with AZT and the fluorinated derivatives 8 and 9 showed the best activity in the series with EC50 values 36.9 μM and 44.5 μM, respectively, compared with AZT (EC50 = 0.0017 μM).56
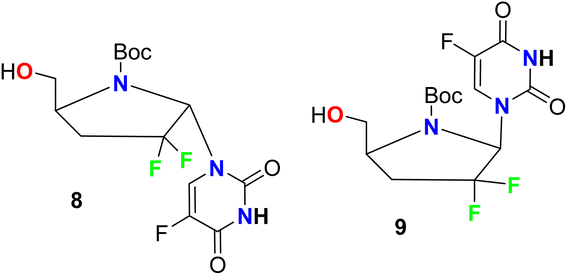 |
| | Fig. 7 Structure of fluorinated pyrrole-based hybrids 8 and 9. | |
2.3. Antiviral activity fluorinated isoxazoles
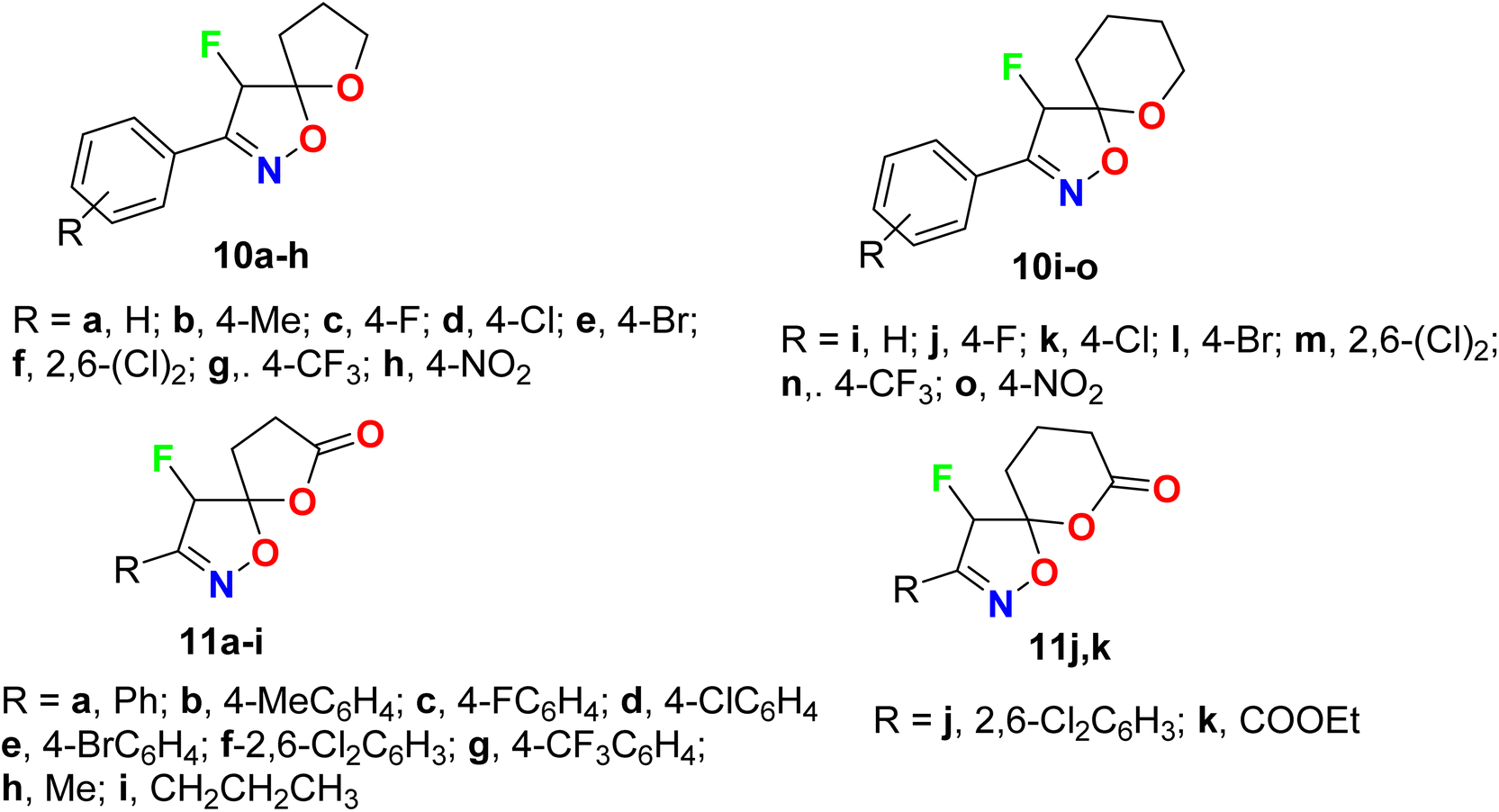
A wide range of the fluorinated spiro-isoxazoline derivatives 10a–o and 11a–k were synthesized and evaluated for their in vitro antiviral activity against human cytomegalovirus (HCMV) (Fig. 8). The viral inhibitory bioassay was carried out, in the presence of GFP (green fluorescent protein, using quantitative fluorescence microscopy. Four derivatives 10d, 10i, 10n, and 10o showed significant anti-HCMV properties with IC50 values of 9.47 μM, 11.2 mM, 10.47 μM, and 2.54 mM, respectively. Thus, compounds 10d and 10n presented significant activity against HCMV with IC50 9.47 μM and 10.47 μM, compared with ganciclovir (reference drug for treatment of HCMV) with IC50 = 4.96 μM. The cytotoxicity of the most active compounds 10d and 10n on HFF (human foreskin fibroblasts) cells, at the double value of the IC50 concentrations, was measured, where the tested compounds exhibited almost 100% cell viability without any significant cytotoxicity.57
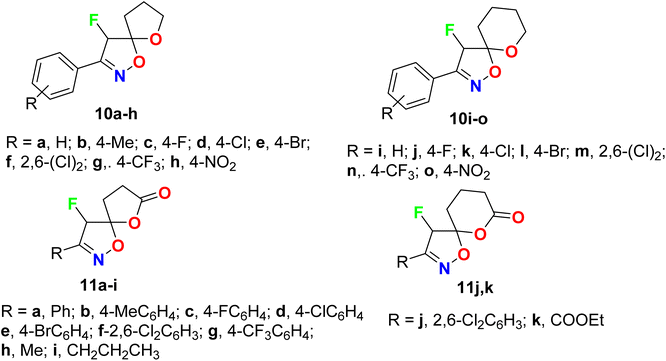 |
| | Fig. 8 Structure of fluorinated spiro-isoxazoline derivatives 10a–o and 11a–k. | |
2.4. Antiviral activity of fluorinated pyrazoles
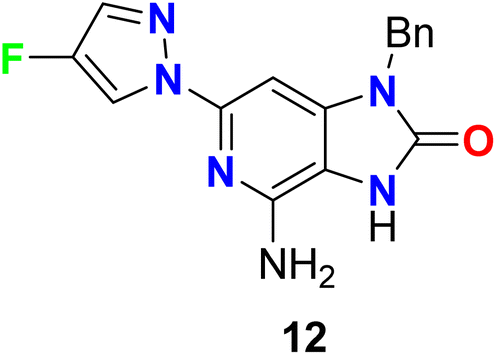
The 4-fluoropyrazole hybrid 12 was reported as Toll-Like Receptor 7 (TLR-7) modulator for treatment of viral infections (Fig. 9). By PBL/HCV replicon bioassay, compound 12 had selectivity modulated the TLR7 receptor activity over other known Toll-like Receptors with EC50 0.47 μM.58
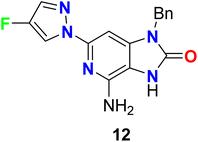 |
| | Fig. 9 Structure of 4-fluoropyrazole hybrid 12. | |
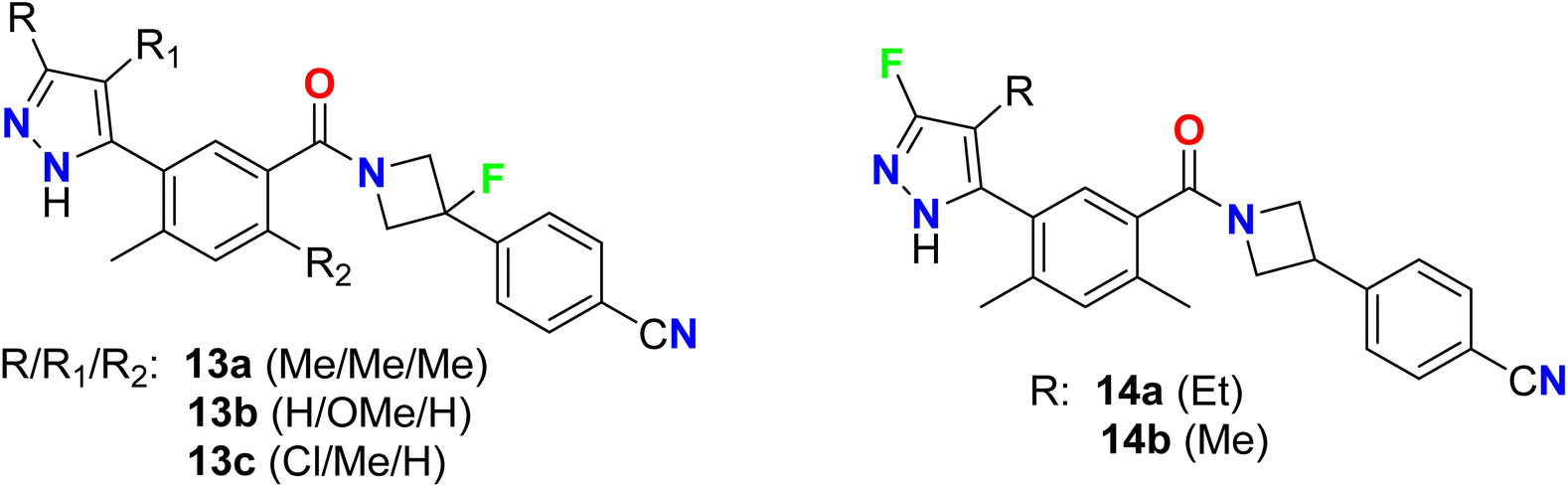
Some fluorinated azetidine-based pyrazole molecular hybrids 13a–c, and 14a–b were designed by Oslob et al. and were evaluated for their antiviral activities (Fig. 10). The antiviral activity was assessed using the HCV lb replicon system at ten three-fold dilutions. The fluorinated azetidine derivatives 13a–c displayed good inhibitory activity against the HCV genotype-lb (HCV-GT-lb) with EC50 values of 0.45, 0.74, and 0.23 μM, respectively. However, the fluorinated pyrazole analogue 14a showed promising inhibitory action with EC50 0.083 μM, 5-fold to 9-fold activity better than the fluorinated azetidine analogues.59
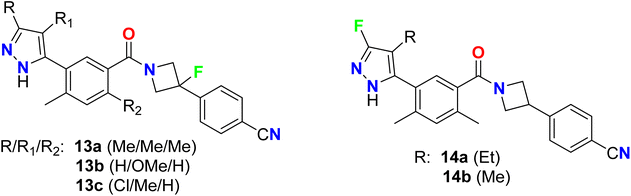 |
| | Fig. 10 Structure of 4-fluorinated azetidine-based pyrazole molecular hybrids 13 and 14. | |
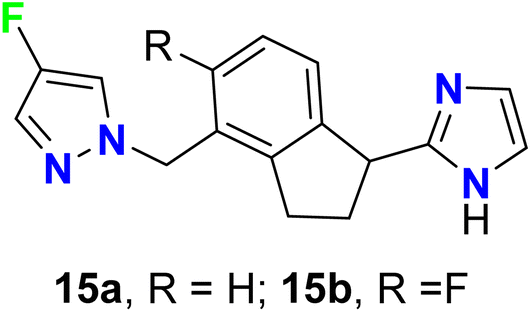
Roberts et al. described two imidazole-based 4-fluoropyrazole derivatives 15a–b as potent, partial agonists of the α1A adrenergic receptor (Fig. 11). Compounds 15a–b showed good selectivity over the α1B, α1D, and α2 sub-types. Both compounds 15a–b proved to be selective for α1A receptor over the other α sub-types, where they had EC50 values 9 nM and 17 nM, and their intrinsic efficacies (α1A Emax) were 83% and 60%, respectively. Furthermore, compound 15b had the best pharmacological properties with a binding activity α1A Ki = 5 nM.60
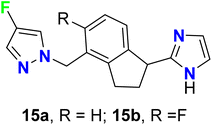 |
| | Fig. 11 Structure of imidazole-based 4-fluoropyrazole derivatives 15a–b. | |
2.5. Antiviral activity of fluorinated indazoles
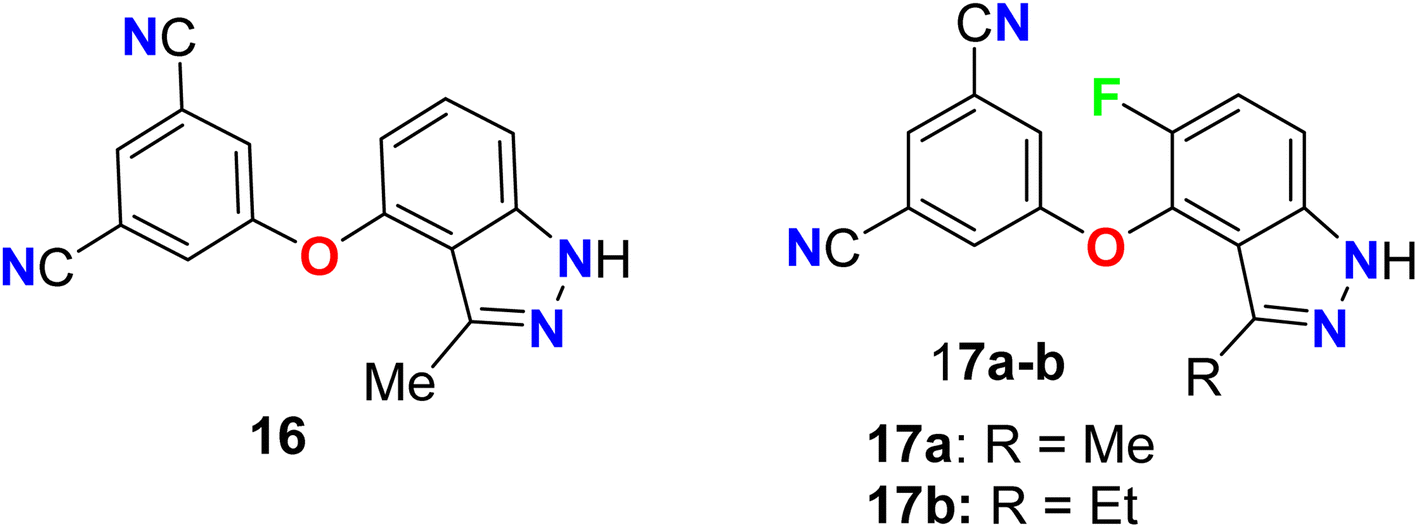
Two 5-fluoroindazole derivatives 17a–b (Fig. 12) were synthesized and examined for their anti-HIV activity. The lack of resilience to mutations in the reverse transcriptase (RT) enzyme, for the treatment of HIV, was a main obstacle related to NNRTIs (non-nucleoside reverse transcriptase inhibitors). The two derivatives were assigned as NNRT inhibitors and showed excellent metabolic stability and mutant resilience compared with the known inhibitors efavirenz and capravirine. Both compounds showed the presence of fluorine atom in compound 17a–b greatly improved their potency, against the wild-type reverse transcriptase enzyme, nearly 7-fold and 13-fold (IC values of 50 nM and 25 nM) better than the non-fluorinated derivative 16 (IC50 = 332 nM), respectively. Thus, SAR proved the importance of the presence of fluorine atom at position-5 as well the presence of ethyl group at position-3 instead of methyl group. Compounds 17a and 17b demonstrated also promising potency against the clinically relevant K103N and Y181C RT mutations, particularly compound 17b had the best potency against Y181C with an IC50 value of 32 nM much better than both efavirenz and capravirine, reference drugs of HIV, with IC50 values 40 nM and 61 nM, respectively.61
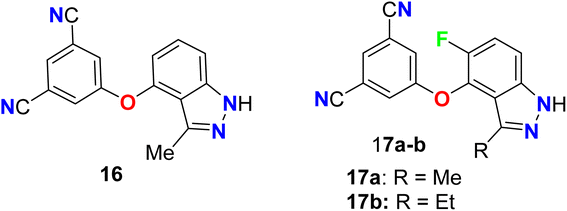 |
| | Fig. 12 Structure of 5-fluoroindazole derivatives 17a–b. | |
2.6. Antiviral activity of fluorinated isoindolines
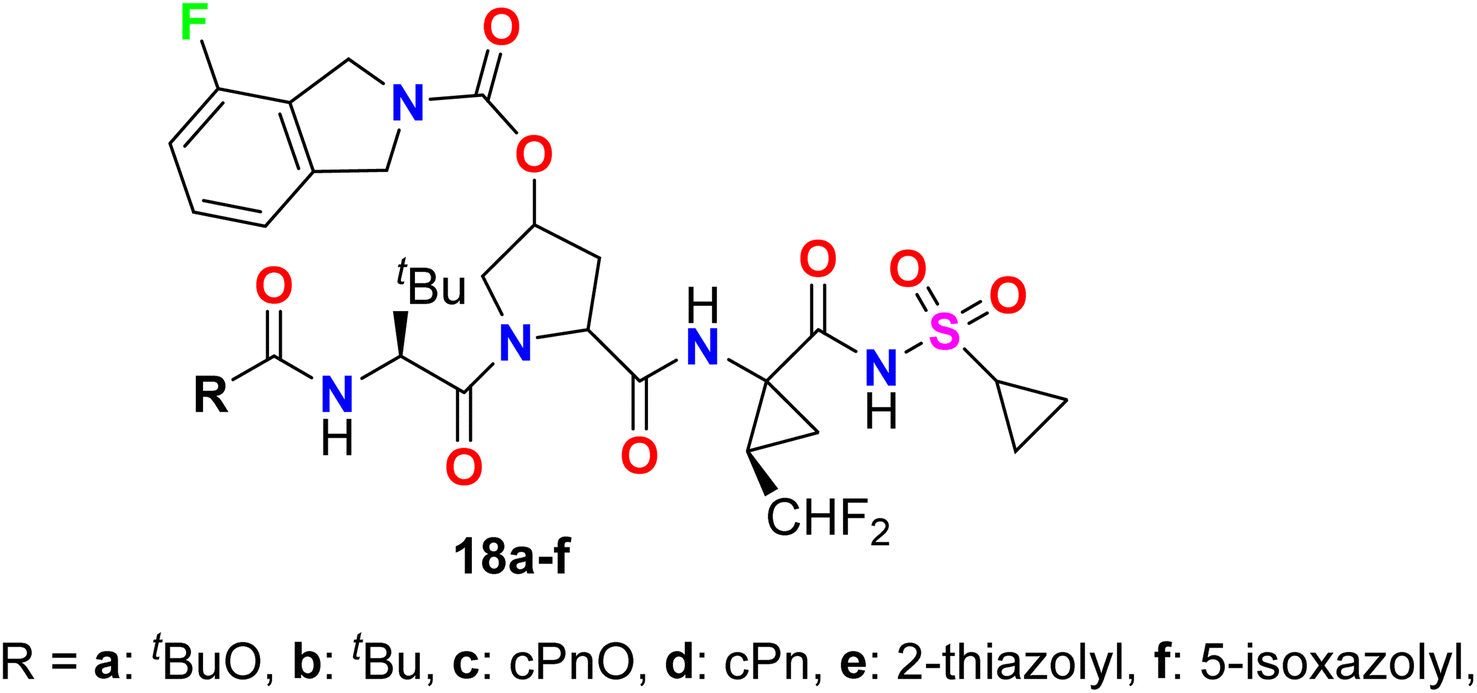
The 4-fluoroisoindoline derivatives 18a–f were invented and synthesized by Gai et al. and studied their inhibitory activity against hepatitis C virus (HCV) NS3-NS4A protease (Fig. 13). The activity of reported hybrids as inhibitors of HCV replication (cell-based assay) in replicon-containing Huh-7 cell lines was studied. Most of the invented compounds showed promising activities in both enzyme inhibition and cell-based replicon assays for HCV. The antiviral potency of the invented compounds on HCV replicon RNA levels in Huh-7 cells was calculated by comparing the ratio of HCV/GAPDH in the cells exposed to the compound versus cells exposed to the DMSO vehicle (negative control).62
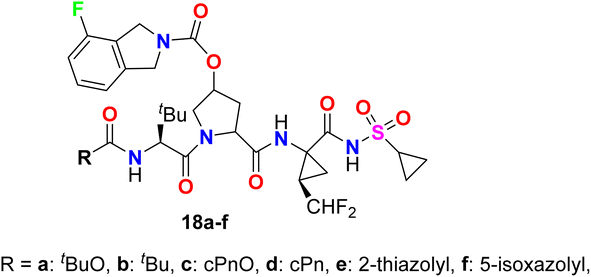 |
| | Fig. 13 Structure of 4-fluoroisoindoline derivatives 18a–f. | |
2.7. Antiviral activity of fluorinated indoles
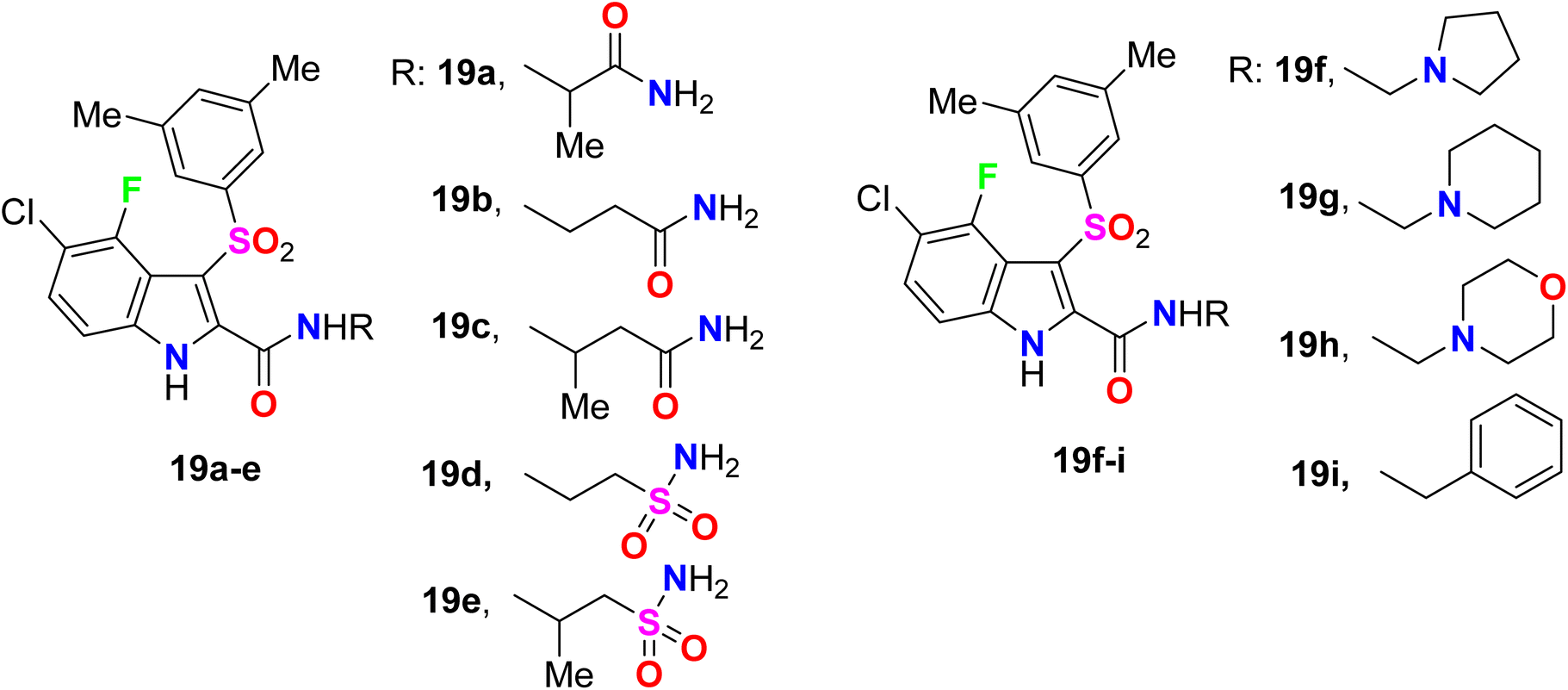
Piscitelli et al. reported the antiviral activity of some fluorinated indole-carboxamide derivatives 19a–i against the HIV-1 WT in human T-lymphocyte (CEM) (Fig. 14). All the examined compounds demonstrated high potent inhibition of the HIV-1 replication in human T-lymphocyte (CEM) cells at low concentrations and were weakly cytostatic. The antiviral activity of the fluorinated derivatives 19a–e was highly potent with EC50 values ranging between 2.0–4.6 nM against the HIV-1 WT. The other fluoro derivatives 19f–i showed also good antiviral activity with EC50 values ranged between 2.5–5.8 nM against the HIV-1 WT when compared to efavirenz standard (ED50 = 1.5 nM).63,64
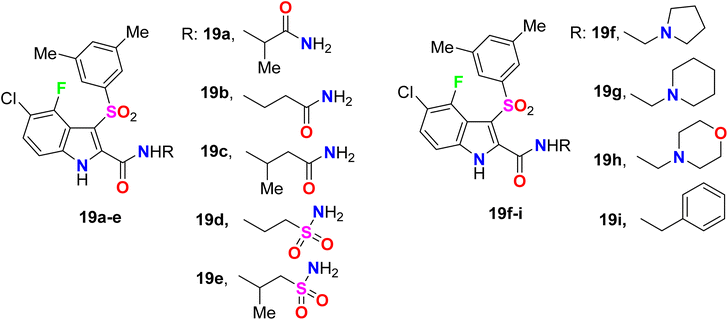 |
| | Fig. 14 Structure of fluorinated indole-carboxamide derivatives 19a–i. | |
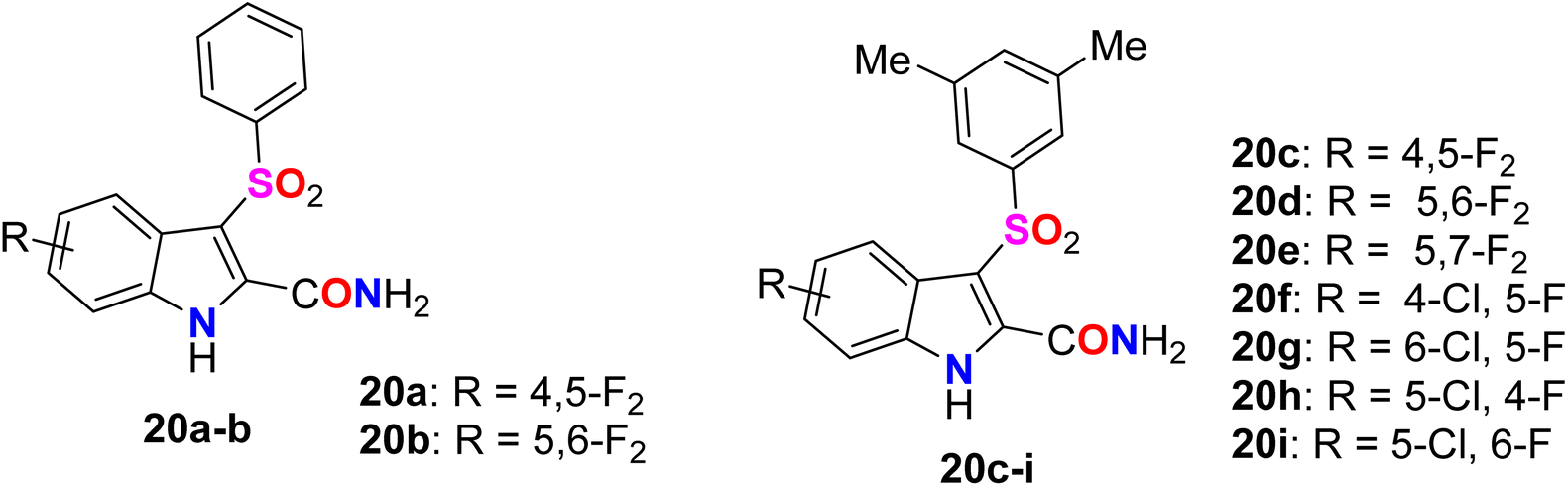
A series of benzenesulfonyl fluorinated-indolecarboxamide derivatives 20a–i were synthesized and their inhibitory activity against wild-type HIV-1 non-nucleoside reverse transcriptase (NNRT) was evaluated in MT-4 and C8166 cells using MTT assay (Fig. 15). Compounds 20a–i were found to possess potent activity against HIV-1 WT without any cytotoxicity up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM. The 4-fluoroindole derivative 20h was the most potent in MT-4 and C8166 cells with ED50 values of 0.5 nM and 0.8 nM, respectively. Compound 20i proved to have the most potent antiviral activity against the HIV-1 WT, Y181C, and K103N-Y181C resistant strains in MT-4 Cells, with ED50 values of 0.5 nM, 4 nM, and 300 nM, compared with efavirenz reference with ED50 values 3 nM, 10 nM, and 200 nM, respectively. Therefore, compound 20i was reported as a promising candidate for further development of NNRT inhibitors.65
000 nM. The 4-fluoroindole derivative 20h was the most potent in MT-4 and C8166 cells with ED50 values of 0.5 nM and 0.8 nM, respectively. Compound 20i proved to have the most potent antiviral activity against the HIV-1 WT, Y181C, and K103N-Y181C resistant strains in MT-4 Cells, with ED50 values of 0.5 nM, 4 nM, and 300 nM, compared with efavirenz reference with ED50 values 3 nM, 10 nM, and 200 nM, respectively. Therefore, compound 20i was reported as a promising candidate for further development of NNRT inhibitors.65
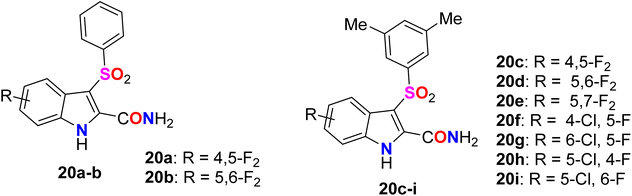 |
| | Fig. 15 Structure of benzenesulfonyl fluorinated-indolecarboxamide derivatives 20a–i. | |
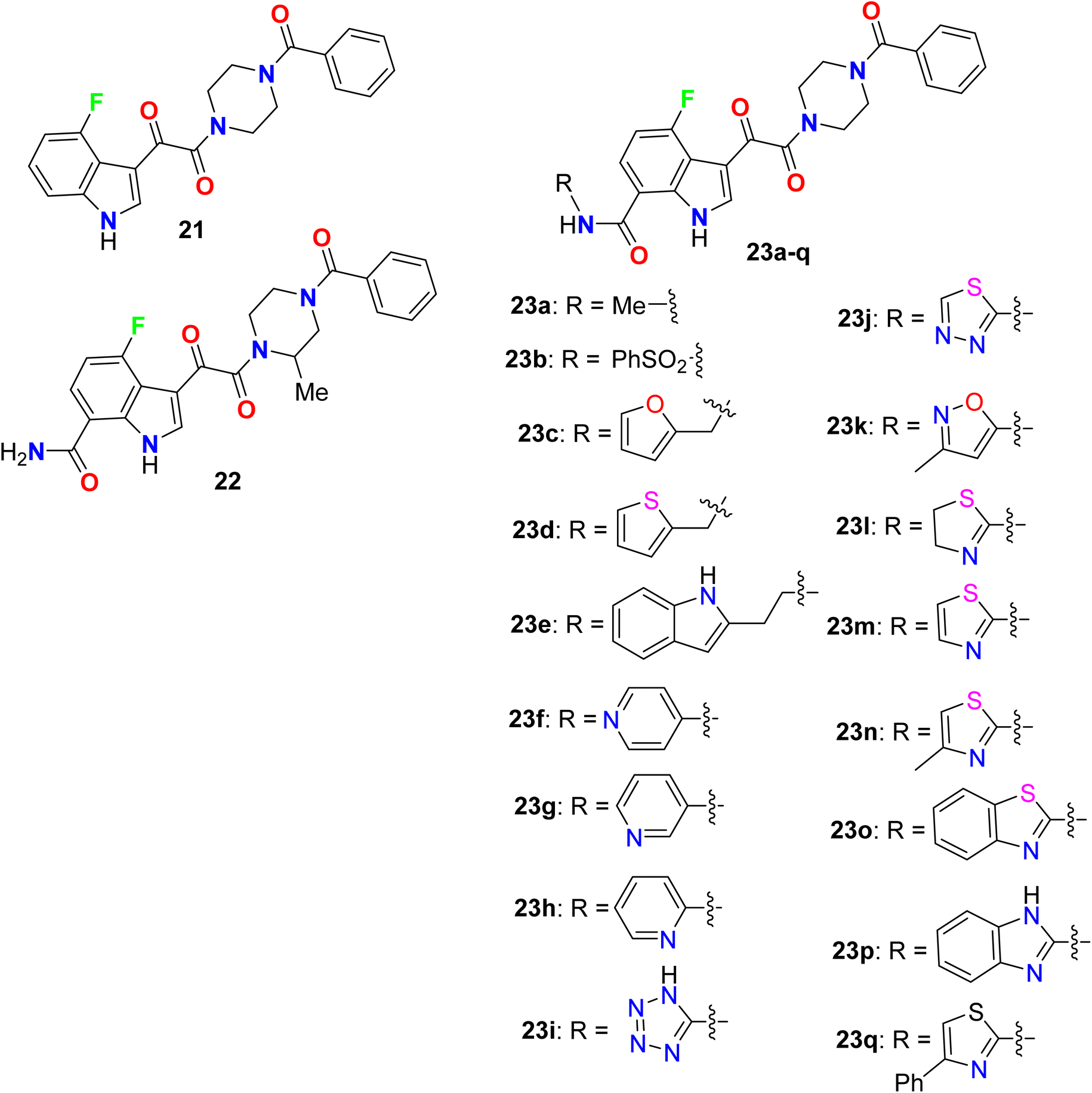
Yeung et al. described the synthesis of a series of 7-substituted carboxamides-4-fluoro indole and screened out their inhibition of HIV-1 activity (Fig. 16). All compounds provided higher inhibition potency than the lead compound 21 in vitro for human liver microsomal (HLM) stability in the primary cell-based assay with EC50 in nanomolar scale, and the fluorinated derivatives 23a (of alkylamide series) and 23b (of sulfonamide series), presented promising inhibitions with EC50 values of 0.29 nM and 0.52 nM. Among the fluorinated indoles having a primary amide group at C-7, compound 22 showed the highest inhibition activity with EC50 = 0.14 nM. The fluorinated indole series having a heteroaryl-carboxamide group at C-7, particularly compounds 23l–n and 23p demonstrated extraordinary antiviral activity in picomolar scale with EC50 values of 0.02 nM, 0.057 nM, 0.0058 nM and 0.04 nM, respectively. The most active derivatives among each series were further investigated in vivo for oral exposure in rats and in vitro for Caco-2 permeability and human liver microsomal (HLM) stability. Phenylsulfonamide 23b and tetrazolylamide 23i provided greater HLM stability but showed poor permeability and poor oral exposure in rats. The thiazol-2-acrylamide derivative 23m exhibited high permeability but was unstable in HLM, compound 23a, however, displayed good metabolic stability and permeability with high oral exposure in rats. The SAR studies revealed that the presence of heteroatom(s) in an arylmethyl group away from the C-7 carboxamide nitrogen led to enhanced potency (e.g. compounds 23c–e). In addition, the presence of hetaryl carboxamides such as pyridyl, tetrazolyl, thiadiazolyl, isoxazolyl, thiazolyl, benzothiazolyl, and benzimidazolyl (23h–j and 23k–q), resulted in subnanomolar to picomolar potency of antiviral activity. The antiviral potency of heterocycles having N-atom closer to the amide nitrogen was more potent than those with far N-atom from the amide group (for example 2-pyridyl (23h) > 3- and 4-pyridyl (23f, 23g)), and those having the thiazol-2-yl group 23m, 23n and exhibited half-maximal inhibition at picomolar concentrations.66,67
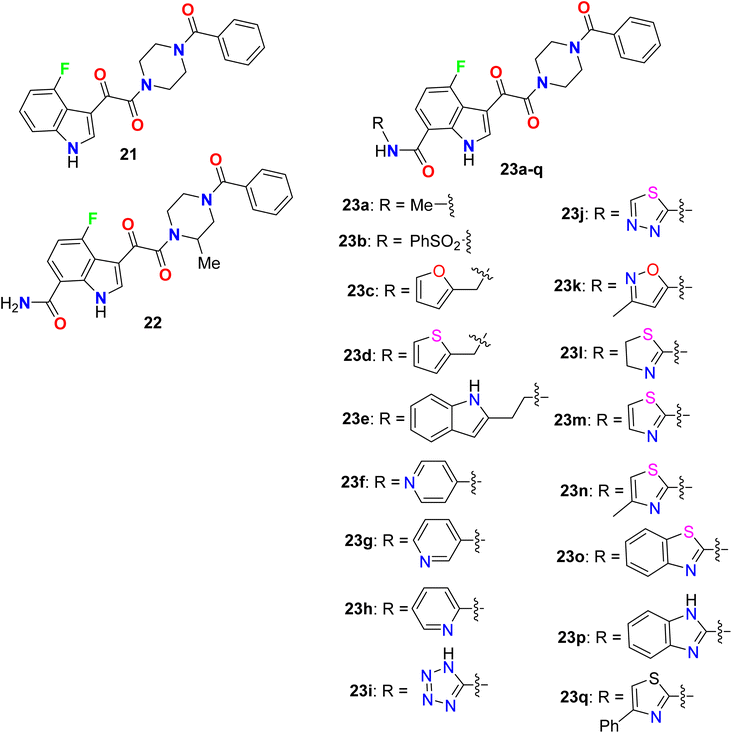 |
| | Fig. 16 Structure of 7-substituted carboxamides-4-fluoro indole derivatives 21–23. | |
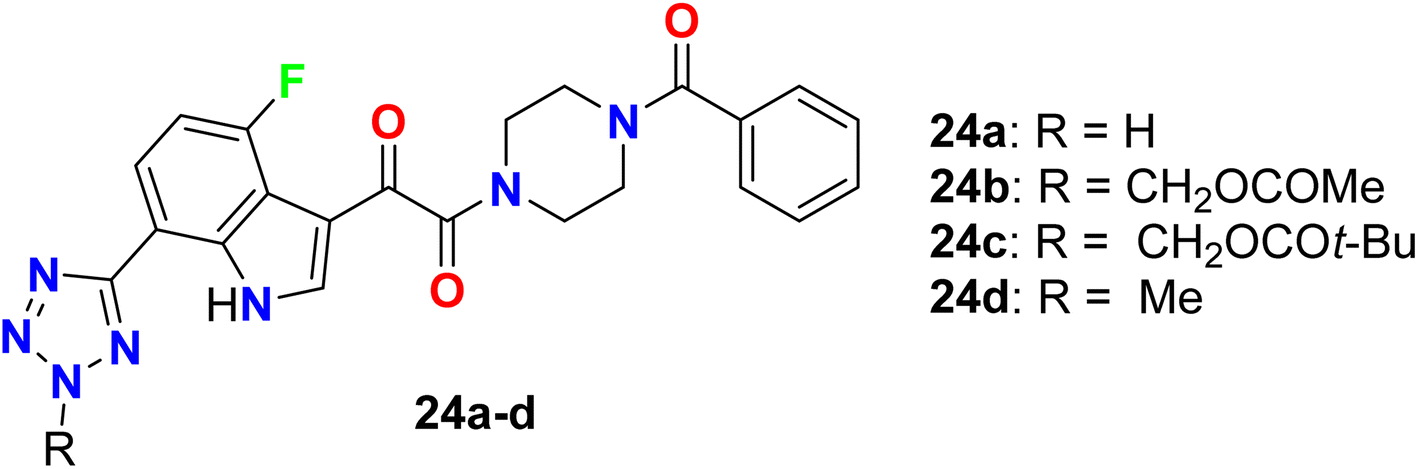
The antiviral activity of the tetrazole-based 4-fluoroindole hybrids 24a–d as inhibitors of HIV-1 attachment was reported (Fig. 17). The antiviral activity of the synthesized compounds was conducted in the single-cycle infectivity assay against HIV-JRFL pseudotyped virus. The reported compounds 24a–d showed potent inhibition with EC50 values ranging between 20–190 nM, and compound 24d had the highest potency with EC50 20 nM. Compounds 24b–d were examined also in vivo as potential oral prodrugs in rats at 30 and 120 min post-dosing, where compound 24b was found to have a great enhancement of the plasma concentration, but compound 24c proved to be ineffective in improving the plasma concentration. Interestingly, oral dosing of compound 24d provided an extraordinary increase in the plasma concentration in rats. Thus, these examples had the potentials to act as a prodrug for HIV-1 inhibition.68
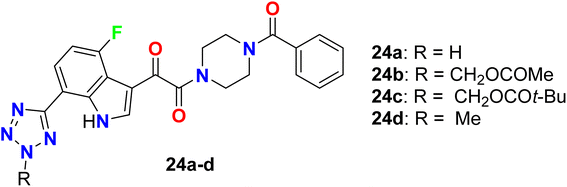 |
| | Fig. 17 Structure of tetrazole-based 4-fluoroindole hybrids 24a–d. | |
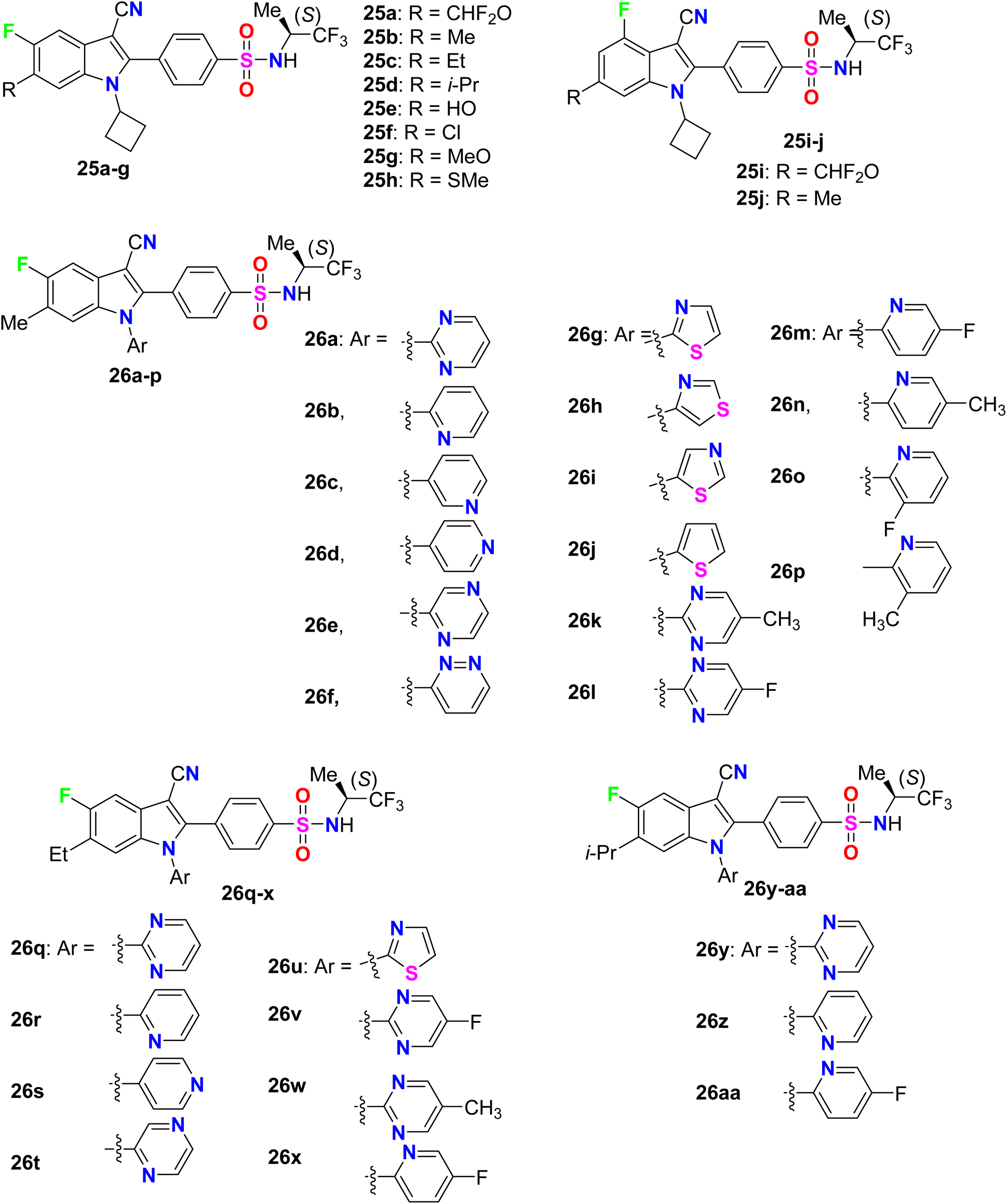
A series of N-cyclobutyl 4-fluoro- and 5-fluoroindole-3-carbonitrile derivatives 25a–j were designed, synthesized, and evaluated for their inhibition of HCV replicon activity (Fig. 18). All the 4-fluro- and 5-fluoroindole-3-carbonitrile series 25a–j showed high potency (EC50 = 4–460 nM), particularly compound 25c provided the highest activity (EC50 = 4 nM). SAR revealed that the 5-fluoroindoles had better activity when compared with their 4-fluoroindole analogues. For example, compound 25b (EC50 = 7 nM) had 22-fold better activity than its isomer 25j (EC50 = 153 nM), and compound 25a (EC50 = 7 nM) had 2.6-fold better activity than its isomer 25i (EC50 = 18 nM). Also, the more lipophilic groups greatly enhanced the inhibitory potency, for example; compound 25b having a 6-Me group (EC50 = 7 nM) had 58.6-fold higher activity than 25f having a 6-OH group (EC50 = 410 nM). Furthermore, another series of N-(heteroaryl) 5-fluoroindole-3-carbonitrile derivatives 26a-aa were evaluated for their inhibition of HCV replicon activity targeting NS4B and to evaluate their drug metabolism and pharmacokinetics (DMPK) properties. Most of the fluorinated derivatives 26a-aa exhibited high potency with EC50 = 2–32 nM. Interestingly, compound 26q demonstrated excellent potency in the cell-based HCV 1b replicon with EC50 = 2 nM, with more than 5000-fold selectivity concerning cellular GAPDH. Compound 26q proved to have acceptable pharmacokinetic properties with oral bioavailability values of 78%, 62%, and 18% in dogs, rats, and monkeys, respectively. Compound 26q had also favorable tissue distribution properties with a liver-to-plasma exposure ratio of 25 in rats.69
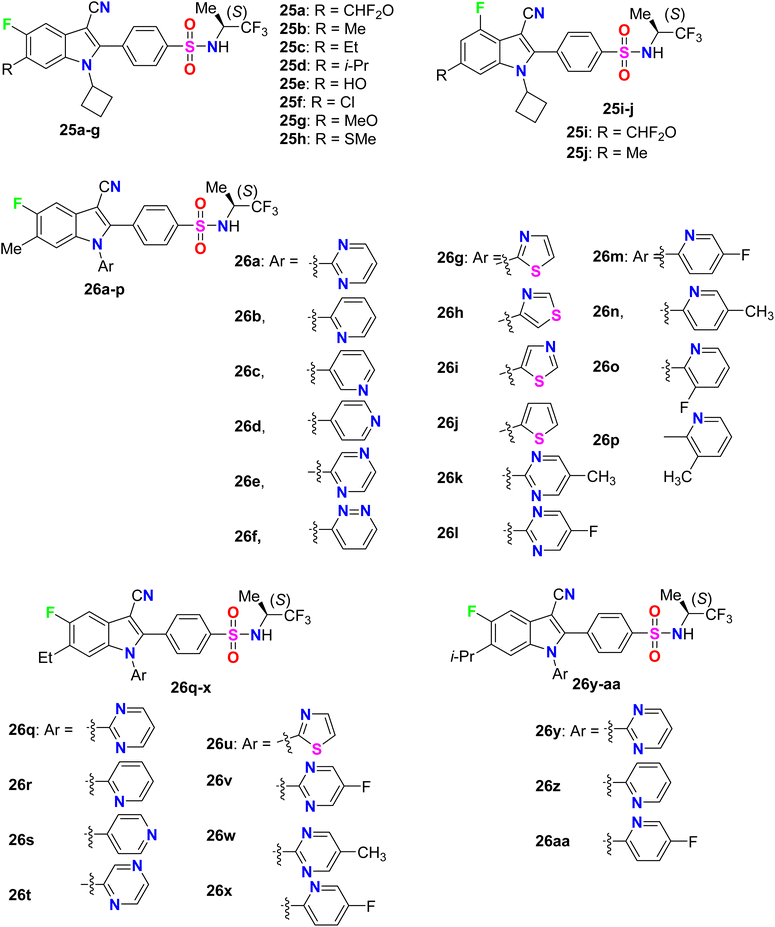 |
| | Fig. 18 Structure of 4-fluoro- and 5-fluoroindole-3-carbonitrile derivatives 25–26 derivatives. | |
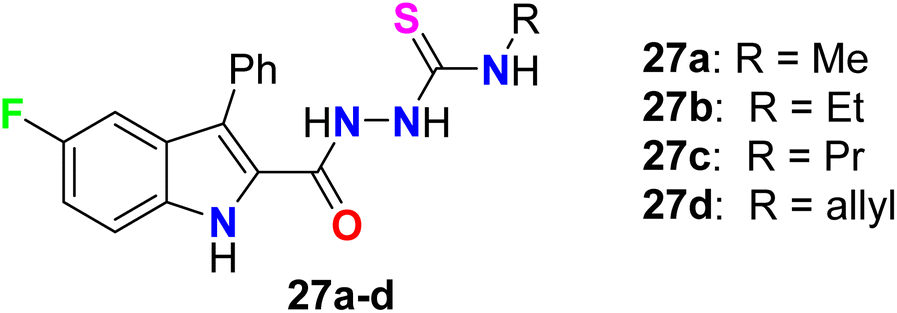
It was reported by Cihan-Üstündağ et al. that the synthesized 5-fluoroindole-thiosemicarbazide derivatives 27a–d provided significant antiviral activities against CVB4 (Coxsackie B4) various (Fig. 19). All the derivatives displayed interesting inhibition of CVB4 virus in Hela and Vero cell lines with EC50 values ranging between 0.4–2.1 μg mL−1. The thiosemicarbazide derivative 27b had the most potent activity with EC50 equal to 0.87 and 0.4 μg mL−1 in Hela and Vero cell lines, respectively. Furthermore, compounds 27b–d could inhibit replication of two other RNA viruses but with higher values of EC50 in comparison with the CVB4 virus, particularly Sindbis virus (EC50 2.3–4 μg mL−1, in Vero cells) and respiratory syncytial virus (EC50 3.2–6.5 μg mL−1, in Hela cells). Thus, compound 27b was considered as a promising scaffold for development of antiviral drugs.70
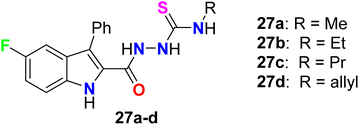 |
| | Fig. 19 Structure of 5-fluoroindole-thiosemicarbazide derivatives 27a–d. | |
2.8. Antiviral activity fluorinated benzimidazoles
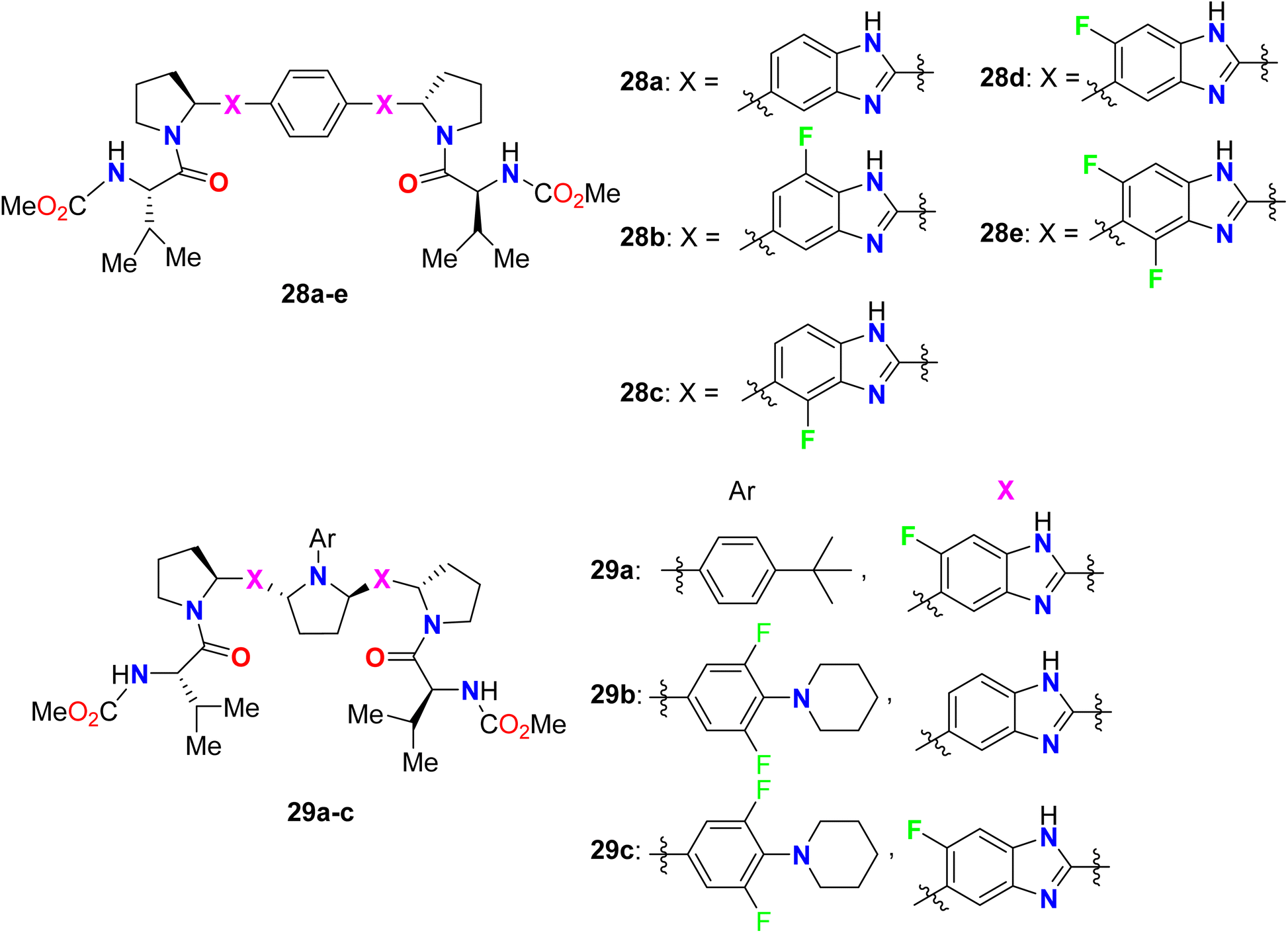
The bis-(fluorobenzimidazole) derivatives 28b–e (Fig. 20) were assigned as potent, broad-genotype in vitro inhibitory activity HCV genotypes 1–6 replicons. The fluorinated benzimidazoles 28b–e provided highly potent activity against most HCV genotypes (1a, 1b, 2b, 4a) with EC50 values ranging between 0.008–0.57 nM, much better than the non-fluorinated benzimidazole 28a. In particular, compound 28d was the most potent one against all HCV genotypes. The other series of bis-(fluorobenzimidazole) derivatives 29a–c showed also excellent inhibitory results against all HCV replicon subtypes, especially compound 22 had fascinating activity in the picomolar scale against all wild-type replicons (1a, 1b, 2a, 2b, 3a, 4a and 6a) with EC50 values ranged between 0.007–0.015 nM. The inhibition potency of the fluorobenzimidazoles 29a–c was also high against the genotype 1a NS5A variants M28T, Q30R, Y93C, and Y93H, particularly compound 29a demonstrated the best activity EC50 values 0.004, 0.005, 0.005, and 0.059 nM, respectively.71
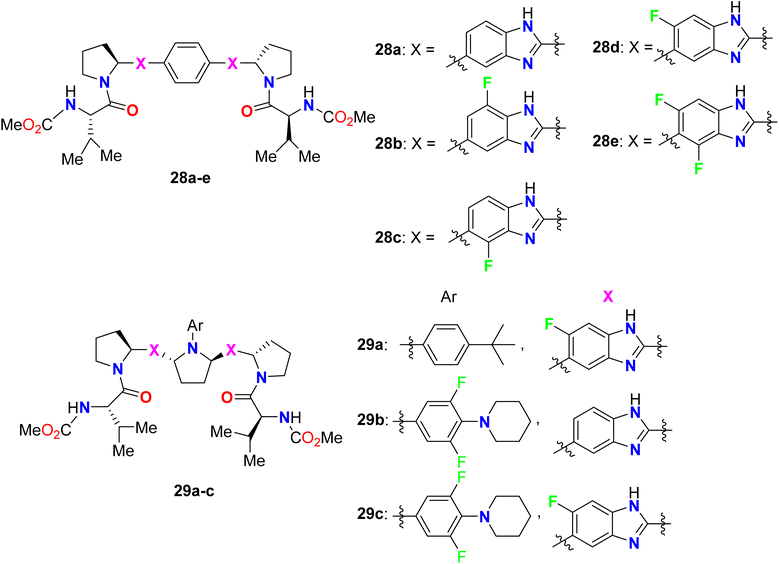 |
| | Fig. 20 Structure of bis-(fluorobenzimidazole) derivatives 28–29. | |
3. Anti-inflammatory activity of fluorinated heterocycles
3.1. Anti-inflammatory activity of fluorinated pyrazoles
Dressen et al. described the synthesis of the 4-fluorpyrazole molecular hybrid 30 and evaluated its potency as a human bradykinin (B1 and B2) receptor antagonist for the treatment of pain and inflammation. The bioassay was carried out using a fluorescent imaging plate reader (FLIPR) utilizing IL-1â stimulated IMR-90 human lung fibroblast cells. Compound 30 showed promising activity with an IC50 value of 23 nM against B1 with high selectivity over B2 (Fig. 21).72
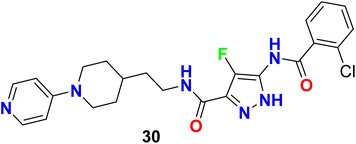 |
| | Fig. 21 Structure of 4-fluorpyrazole derivative 30. | |
Two series of fluorinated-pyrazole heterocyclic hybrids 31 and 32 were invented, patented, and screened as human bradykinin (BK B2) receptor antagonists (Fig. 22). Most of the reported derivatives were found to possess a wide range of properties such as high selectivity, low toxicity, low drug–drug interaction, good metabolic stability, good bioavailability, good stability in microsomal degradation assay as well as good solubility. Most of the assigned compounds demonstrated IC50 values of ≤50 nM.73,74
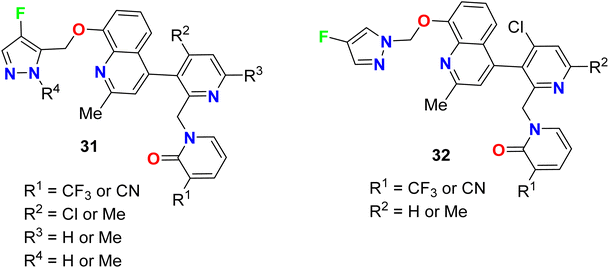 |
| | Fig. 22 Structure of fluorinated-pyrazole heterocyclic derivatives 31 and 32. | |
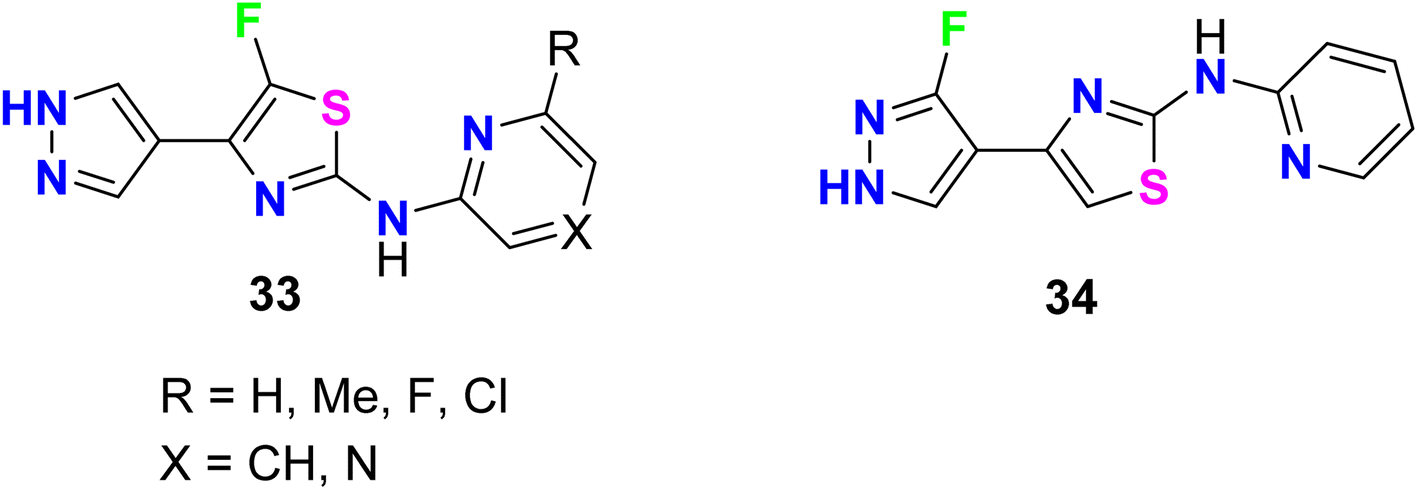
A series of 5-fluorothiazole 33 and 3-fluorpyrazole 34 heterocycles were invented, patented, and screened out as modulators of mGluR4 (metabotropic glutamate receptors-subtype 4) for the treatment of central nervous system disorders (Fig. 23). The activity of the invented compounds was tested on recombinant human mGluR4a receptors by detecting variations in intracellular Ca2+ concentration using Fluorometric Imaging Plate Reader (FLIPR). The assigned compounds showed positive allosteric modulator effect at mGluRA via enhancing the activity of the receptor with EC50 values were less than 100 nM.75
 |
| | Fig. 23 Structure of 5-fluorothiazole 33 and 3-fluorpyrazole 34 heterocycles. | |
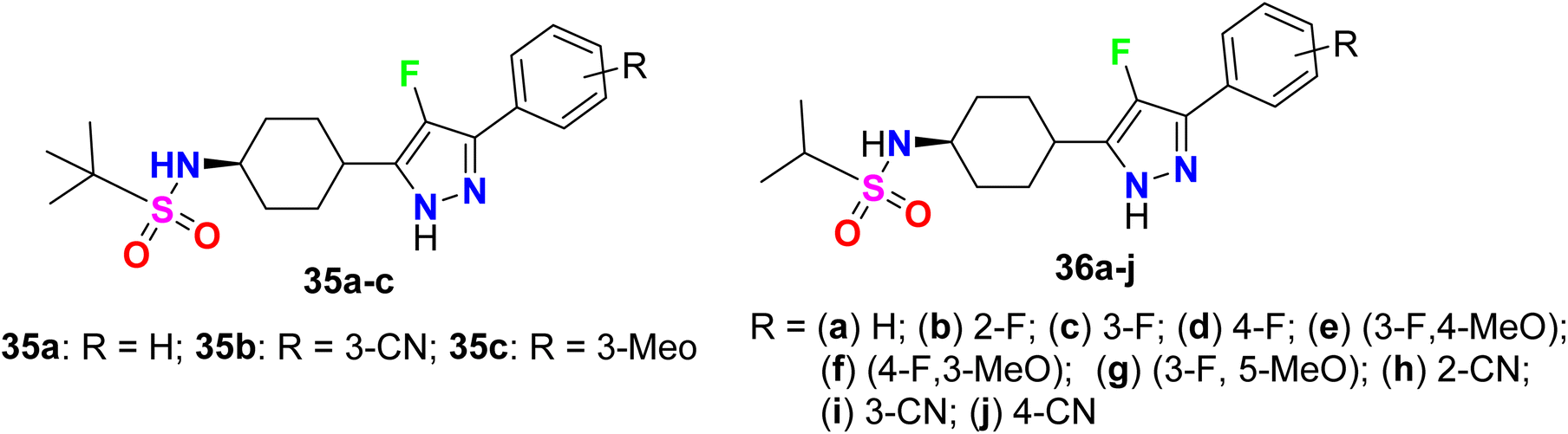
A series of 4-fluoropyrazole scaffolds 35a–c and 36a–j were patented by Sakagami and his co-workers as NPYY5 receptor antagonists (Fig. 24). All the invented compounds exhibited NPYY5 receptor antagonistic activity to be useful in the medication of obesity, depression, and sexual disorders. The reported compounds had little inhibition on drug-metabolizing enzymes, had good metabolic stability and water solubility as well as low toxicity, and were sufficiently safe for use in medication. The invented fluorinated pyrazoles showed a good binding affinity for the mouse NPYY5 receptor with IC50 values varying between 0.22 nM to 2.2 nM.76
 |
| | Fig. 24 Structure of 4-fluoropyrazole scaffolds 35a–c and 36a–j. | |
The guanidine-based 4-fluoropyrazole derivatives 37a, 37b were reported to have good inhibitory activity of F1F0-ATPase synthase enzyme for treatment of the inflammatory disease (Fig. 25). The bioactivity experiment measured the ability of compounds 37a, 37b to inhibit the ATP synthesis, as well as the cytotoxicity in Ramos cells. The biological activity results disclosed that both compounds 37a, 37b inhibited F1F0-ATPase activity in synthesizing ATP with IC50 values <10 μM. In addition, cytotoxicity in Ramos cells was also measured and both compounds presented EC50 < 10 μM.77
 |
| | Fig. 25 Structure of 4-fluoropyrazole derivatives 37a, 37b. | |
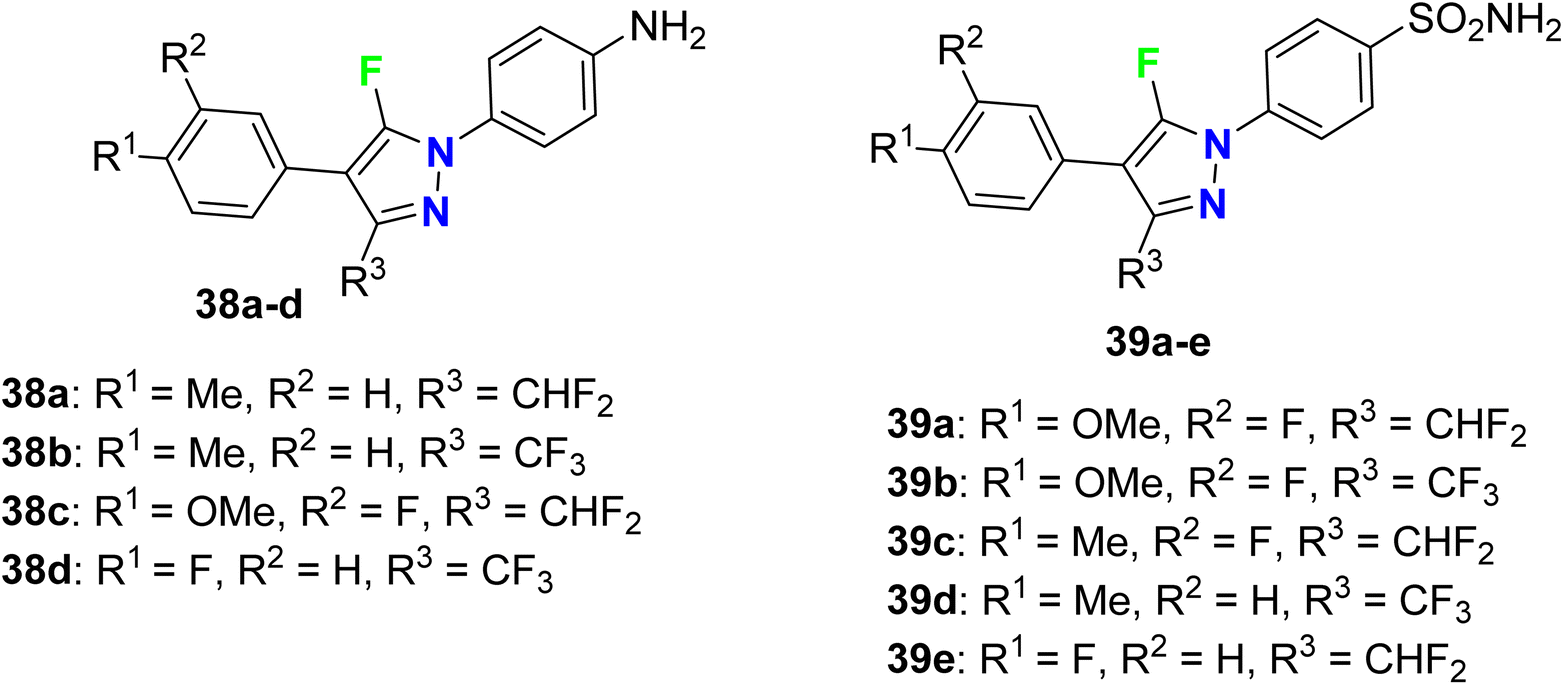
Synthesis and anti-inflammatory activity of a series of 5-fluoropyrazole molecular hybrids 38a–d and 39a–e (Fig. 26) were invented and patented by Mingchun et al. The bioassay study employed RAW 264.7 cells that were cultured in a high-sugar DMEM complete medium containing 10% FBS (fetal bovine serum). The COX-2 protein expression level was up-regulated, confirming the success of the cell inflammation (P < 0.001). After mixing celecoxib, as positive medicine, with the synthesized compounds in equal concentration, the COX-2 protein expression was significantly decreased compared with the neat celecoxib. Compounds 38a–d and 39a–e showed more remarkable COX-2 protein inhibitory activity (Δ P < 0.05) than the positive drug celecoxib. Thus, the patented fluoropyrazoles 38a–d and 39a–e showed anti-inflammatory activity through a potent inhibition effect on the key protein COX2 in a rheumatoid arthritis model and had good potential for further drug development.78
 |
| | Fig. 26 Structure of 5-fluoropyrazole derivatives 38a–d and 39a–e. | |
3.2. Anti-inflammatory activity of fluorinated indazoles
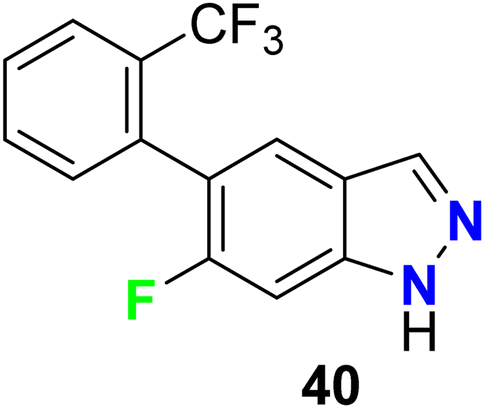
The 6-fluoroindazole scaffold 40 was developed as a selective antagonist of the TRPA1 (transient receptor potential A1) cation channel (Fig. 27). The in vitro study using an antagonist mode FLIPR calcium cation imaging assay in 1536-well format, revealed that compound 40 possessed a potent and selective antagonist of hTRPA1 with IC50 0.043 μM and 98% inhibition. Thus, compound 40 had moderate oral bioavailability in rodents and exhibited in vivo anti-inflammatory activity.79
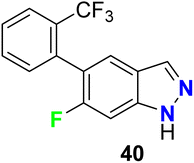 |
| | Fig. 27 Structure of 6-fluoroindazole scaffold 40. | |
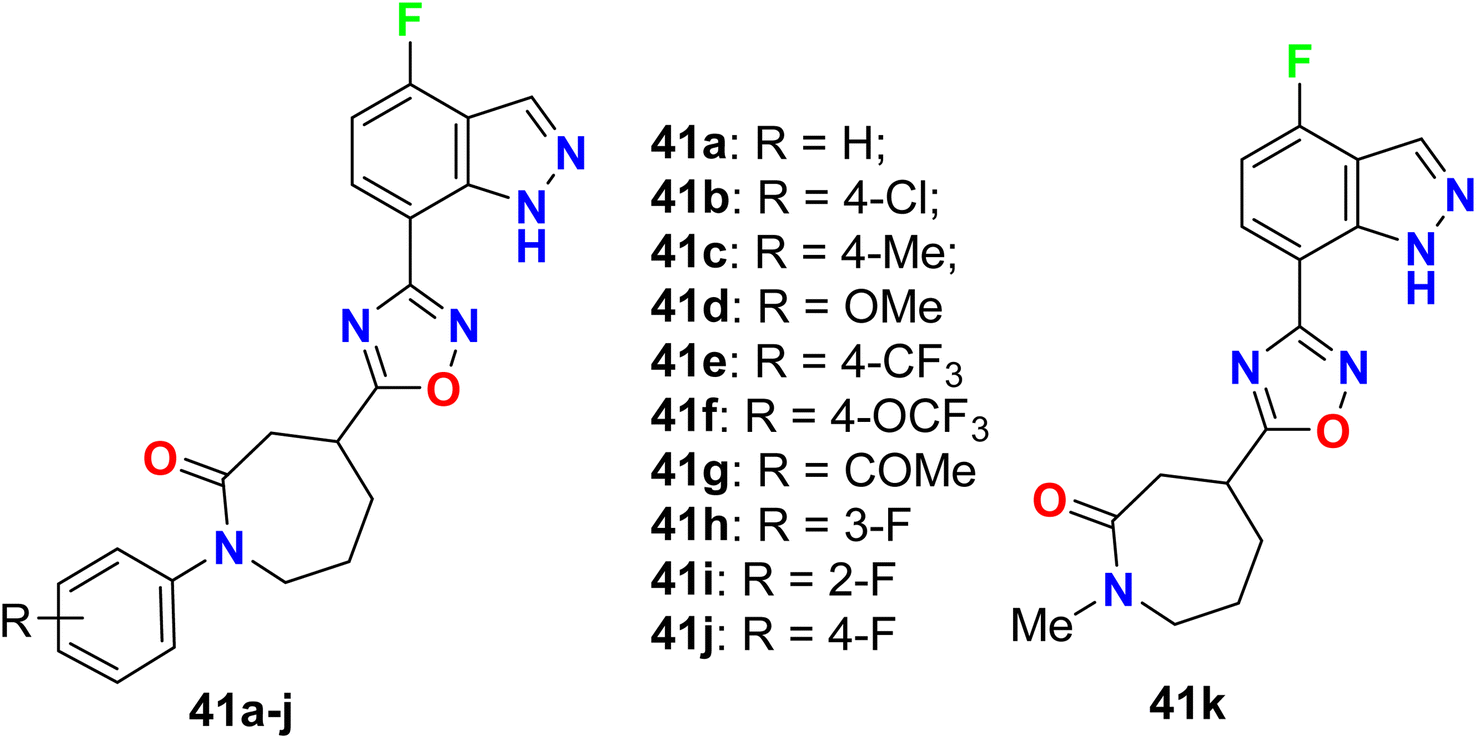
The 4-fluoroindazole derivatives 41a–k were reported as selective cannabinoid receptor (CB2) agonists for inflammation treatment without psychiatric side effects (Fig. 28). All compounds showed good to high potency as selective CB2 agonists. SAR investigations confirmed compound 41j as the most potent one showing high selectivity for CB2 versus CB1 (CB2: EC50 = 21.0 nM, Emax = 87%, compared with CB1 EC50 > 30 μM, and CB1/CB2 ratio >1428) with promising in vivo pharmacokinetic (PK) properties. In addition, compound 41j displayed significant efficacy in the analgesic model of rodent inflammatory pain. Therefore, compound 41j could be useful as a lead structure for treating inflammatory pain after further studies.80
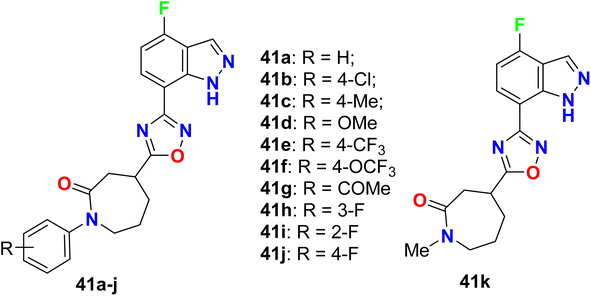 |
| | Fig. 28 Structure of 4-fluoroindazole derivatives 41a–k. | |
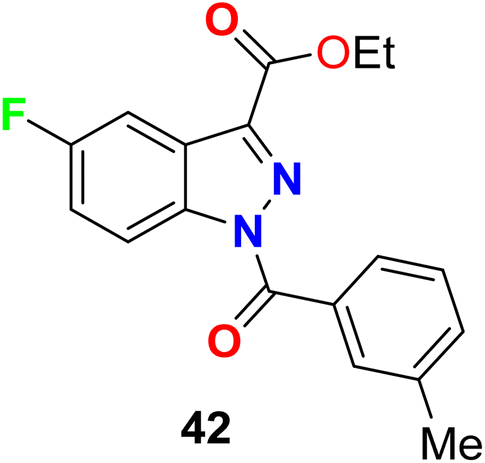
The 5-fluoroindazole derivative 42 (Fig. 29) was synthesized and evaluated for its inhibitory activity against human neutrophil elastase (HNE), for treatment of pulmonary diseases. Compound 42 showed good inhibitory potency, good stability, and selectivity for HNE over other serine proteases, with an IC50 value of 0.1 μM.81
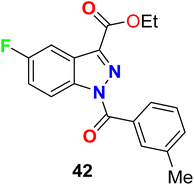 |
| | Fig. 29 Structure of 5-fluoroindazole derivative 42. | |
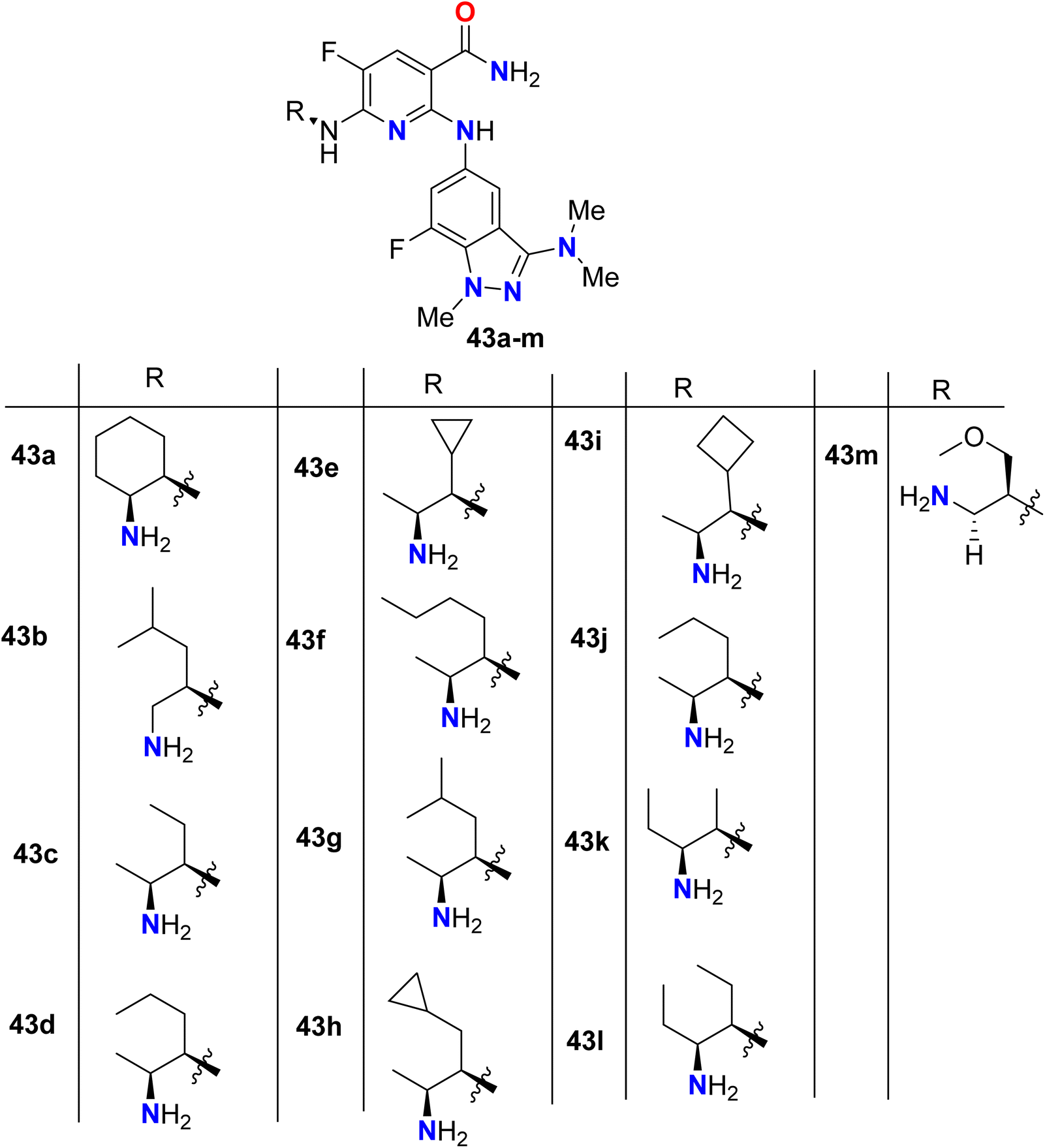
Some 7-fluoroindazole derivatives 43a–m were patented and their biological activity as inhibitors of human spleen tyrosine kinase (Syk) were screened, for possible treatment of inflammatory disorders (Fig. 30). All the tested compounds showed excellent human Syk kinase inhibitory potency with IC50 ranging between 10 nM and 50 nM. Evaluating the inhibitory activity of TNFα production was also reported, and most of the patented compounds 43a–g, 43k, 43m provided IC50 values about 65 nM.82
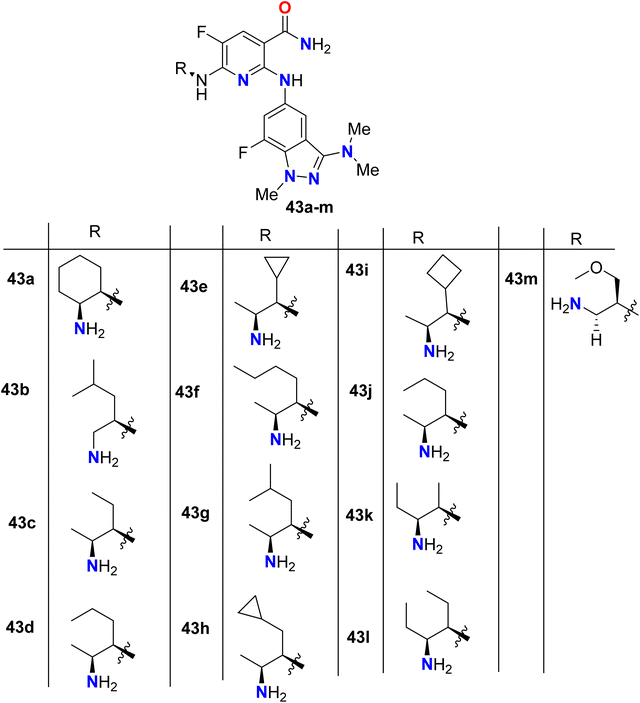 |
| | Fig. 30 Structure of 7-fluoroindazole derivatives 43a–m. | |
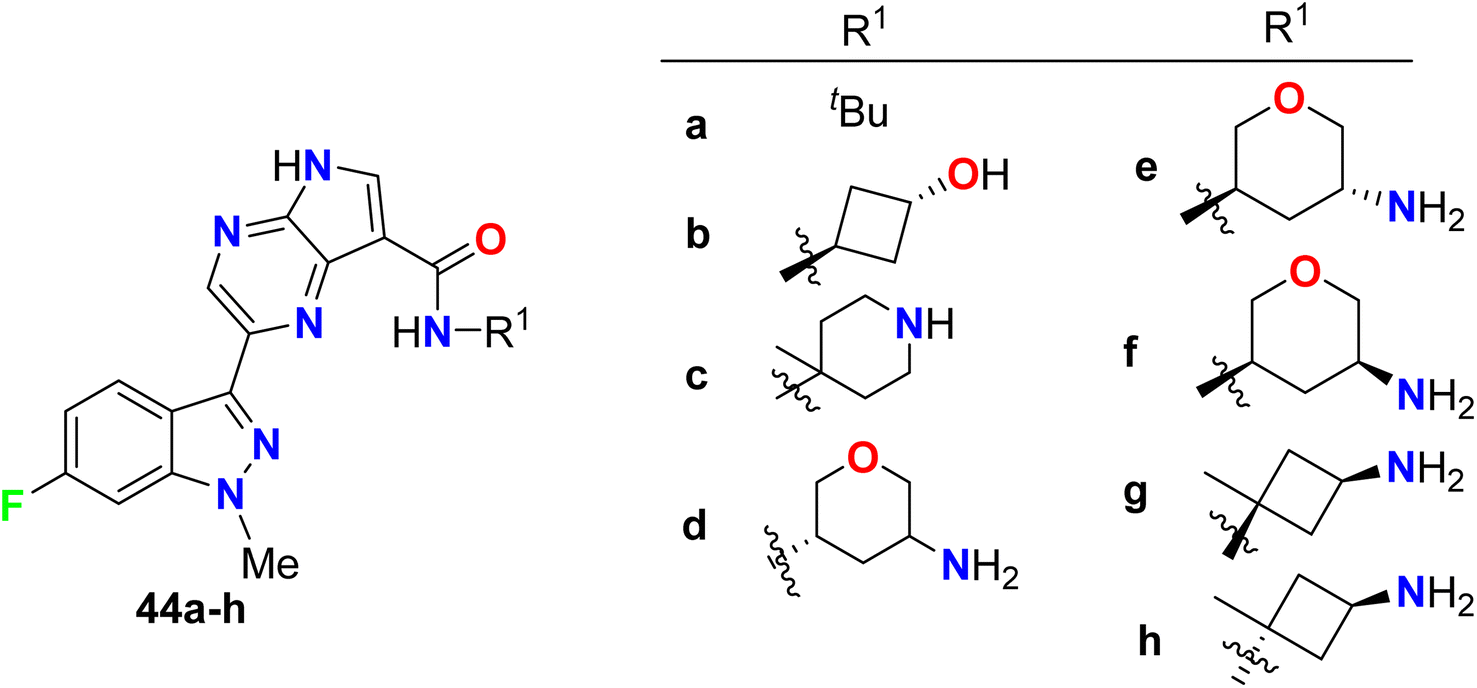
Synthesis of the 6-fluoroindazole molecular hybrids 44a–h was described by Padilla et al. and screened their inhibitory activity of Syk enzyme in addition to the selectivity of Syk/JAK and human whole blood (HWB) assay was studied (Fig. 31). The synthesized compounds were found to be potent and selective Syk inhibitors with IC50 values ranging between 4 nM to 64 nM, where structure 44g was the most potent one with IC50 = 4 nM, with high selectivity for Syk kinase (3/386) over the JAK (Janus kinase) family. The fluorinated compounds 44a–h showed also very good potency in Ramos B cell and HWB potency assays with IC50 values 0.151–3.70 μM and IC50 values 0.376–1.43 μM respectively.83
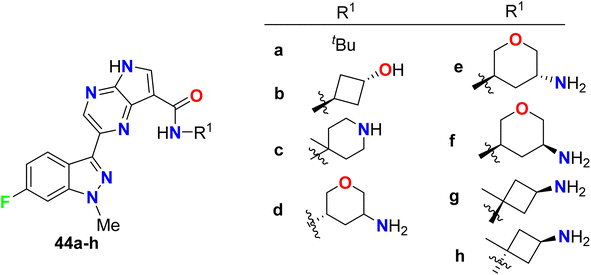 |
| | Fig. 31 Structure of 6-fluoroindazole molecular hybrids 44a–h. | |
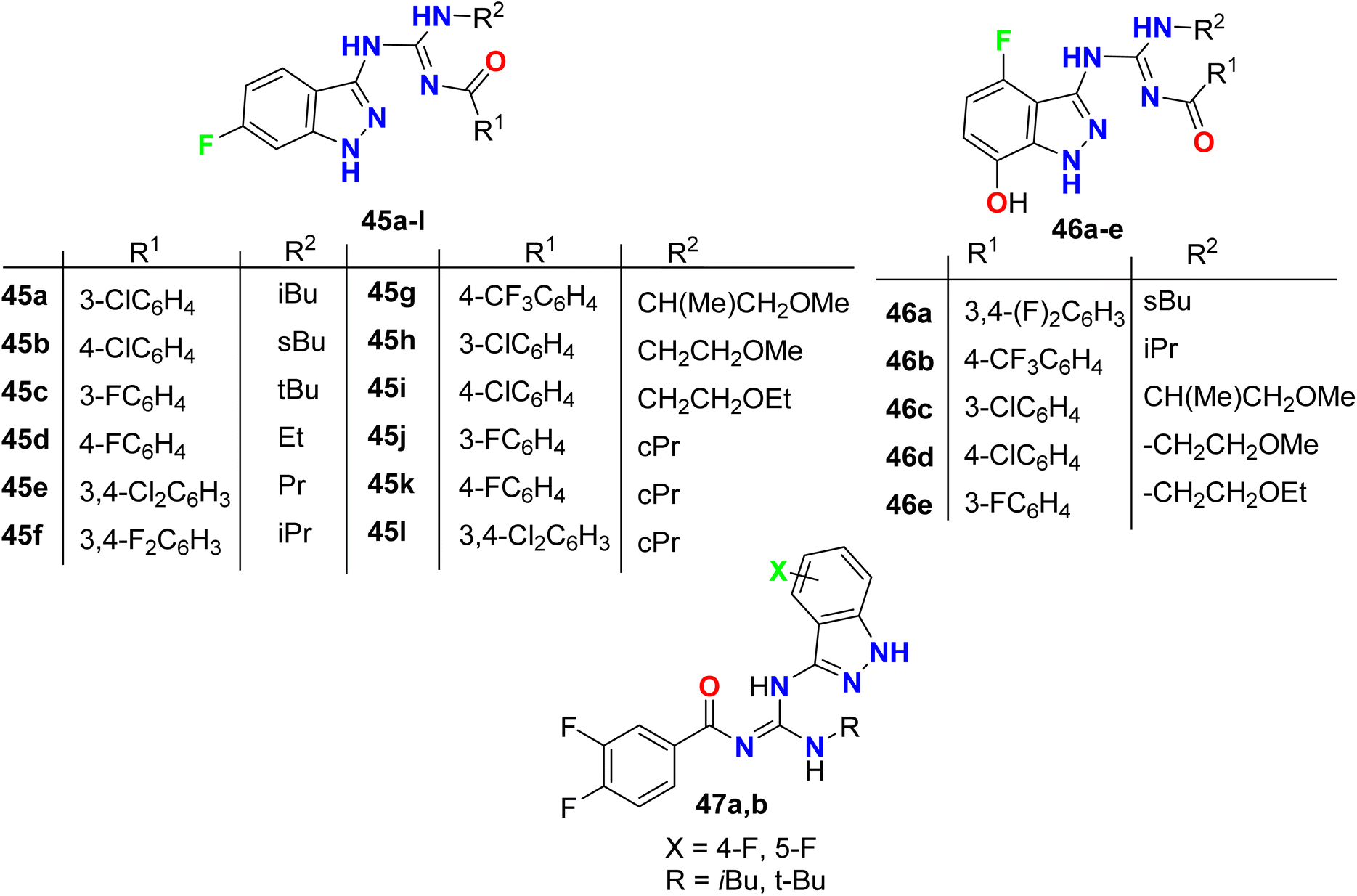
Hurd et al. described the synthetic routes to the monofluorinated 3-guanidyl-indazole structures 45–47 as shown in Fig. 32. All the reported compounds were tested for their activity against FiF0-ATPase by measuring their capability to inhibit ATP synthesis. In addition, the cytotoxicities of the indazole derivatives in Ramos cells (B lymphocyte cell line) were also assessed. The bioactivity results showed that most of the constructed fluorinated indazole scaffolds demonstrated potent inhibition of F1F0-ATPase activity with IC50 values <5 μM as well as cytotoxicity in Ramos cells with EC50 <5 μM.84
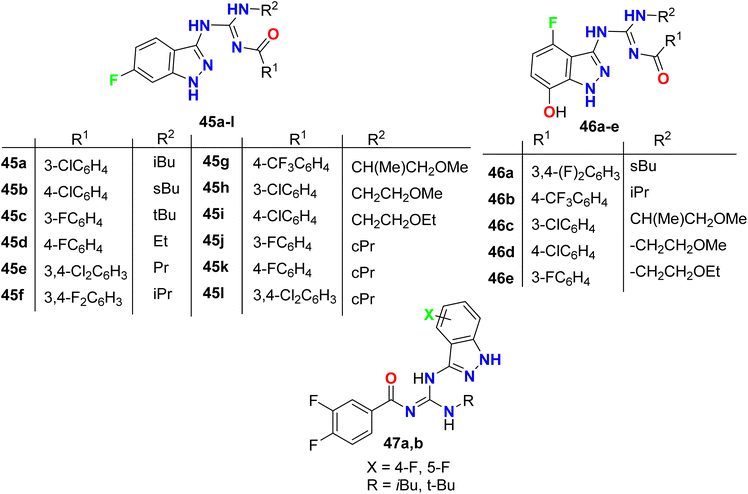 |
| | Fig. 32 Structure of monofluorinated 3-guanidyl-indazole structures 45–47. | |
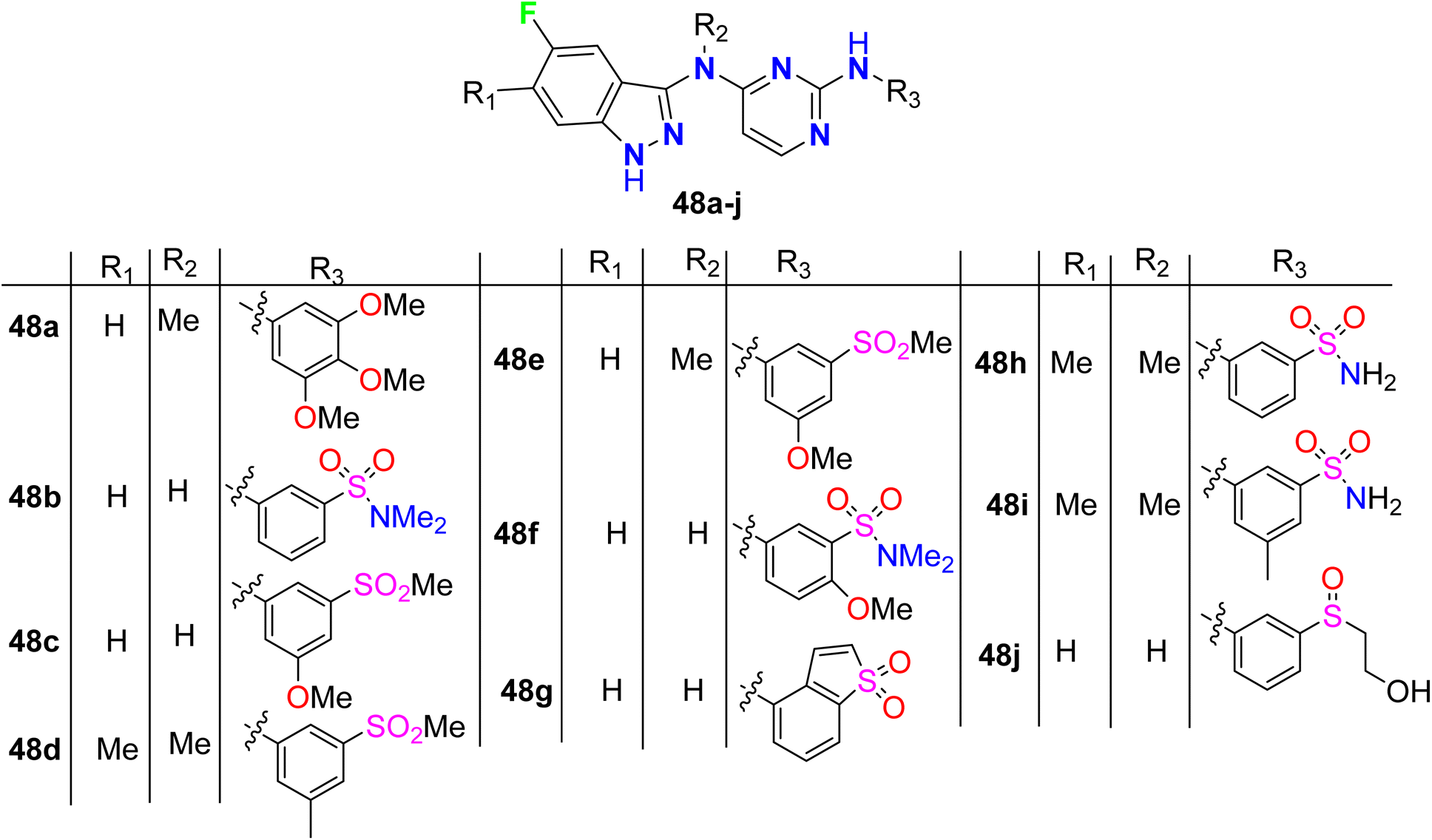
The 5-fluroindazole derivatives 48a–j were invented and characterized as inhibitors of RIP2 kinase (receptor-interacting protein 2) that were useful for the treatment of inflammatory diseases (Fig. 33). Most of the designed compounds showed promising inhibitory action against RIP2 kinase with pIC50 values < 8. 5-Fluoroindazole 48a (R1 = H, R2 = Me, R3 = 3,4,5-triMeOC6H2) interestingly displayed the best inhibition of the RIP2 kinase with pIC50 value of 6.0.85
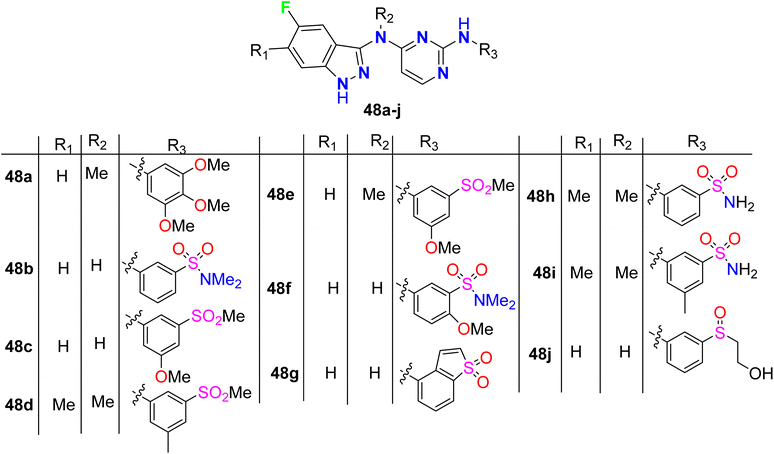 |
| | Fig. 33 Structure of 5-fluroindazole derivatives 48a–j. | |
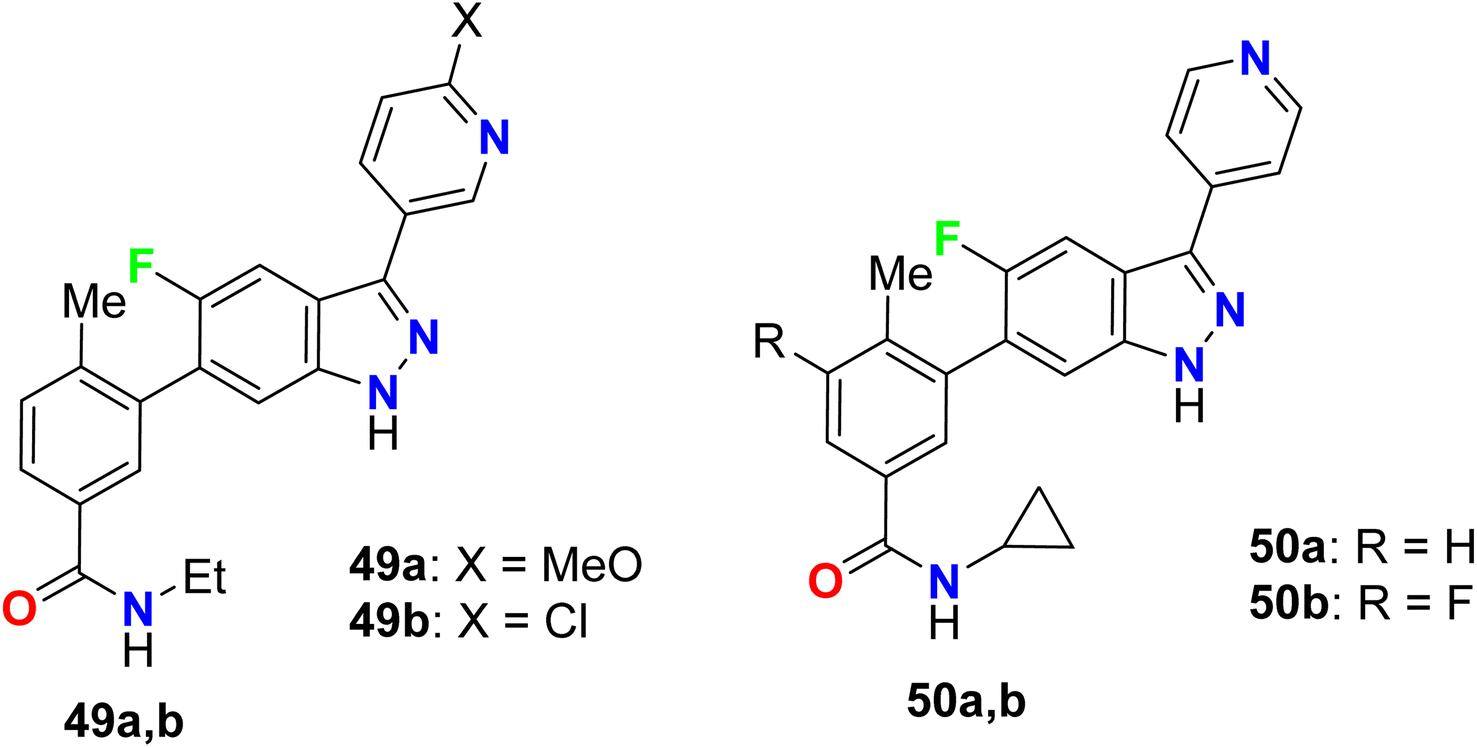
The 5-indazole derivatives 49a, 49b, and 50a, 50b were patented as inhibitors of the p38 kinase and were useful for the treatment of inflammatory diseases (Fig. 34). The inhibitory activity of the invented fluorinated compounds against p38 kinase was determined by in vitro fluorescence anisotropy kinase binding assay 1 and all compounds had IC50 values of <10 μM.86
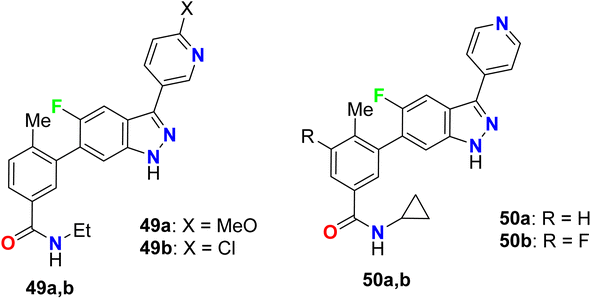 |
| | Fig. 34 Structure of 5-indazole derivatives 49 and 50. | |
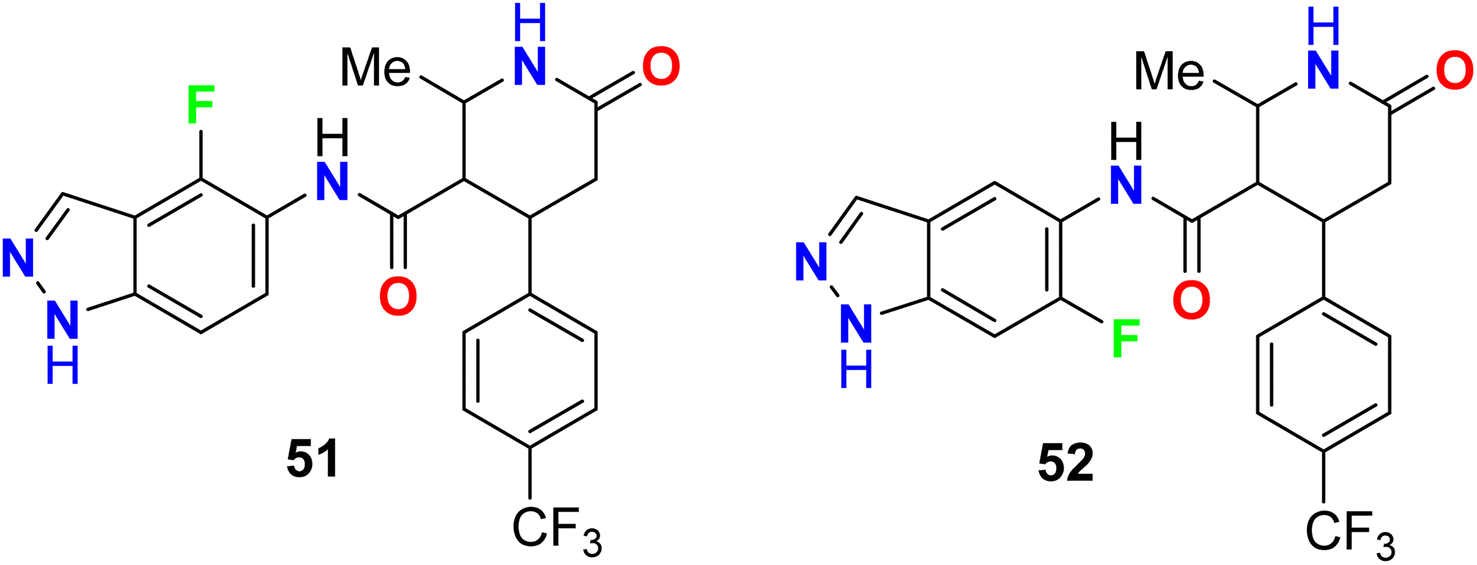
Two examples of the fluorinated indazoles 51 and 52 were synthesized and evaluated as inhibitors of Rho kinase (ROCK1) activity (Fig. 35). The in vitro bioassay results showed that the presence of fluorine at C4 (compound 51) displayed low potency with IC50 of 2500 nM. However, the presence of fluorine at C6 (compound 52) significantly enhanced the ROCK1 inhibitory potency with an IC50 value of 14 nM as well as a dramatic increase of oral bioavailability (61%) was reported for 6-fluoroindazole 52. The experiments showed also good in vivo results, where compound 52 dramatically reduced mean arterial pressure in spontaneously hypertensive rats after oral administration.87,88
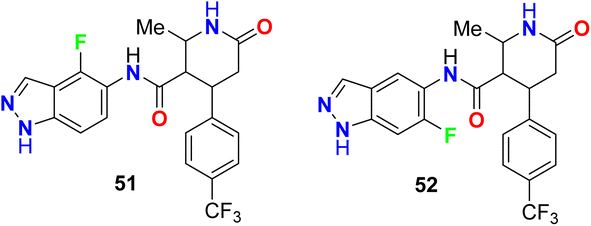 |
| | Fig. 35 Structure of fluorinated indazoles 51 and 52. | |
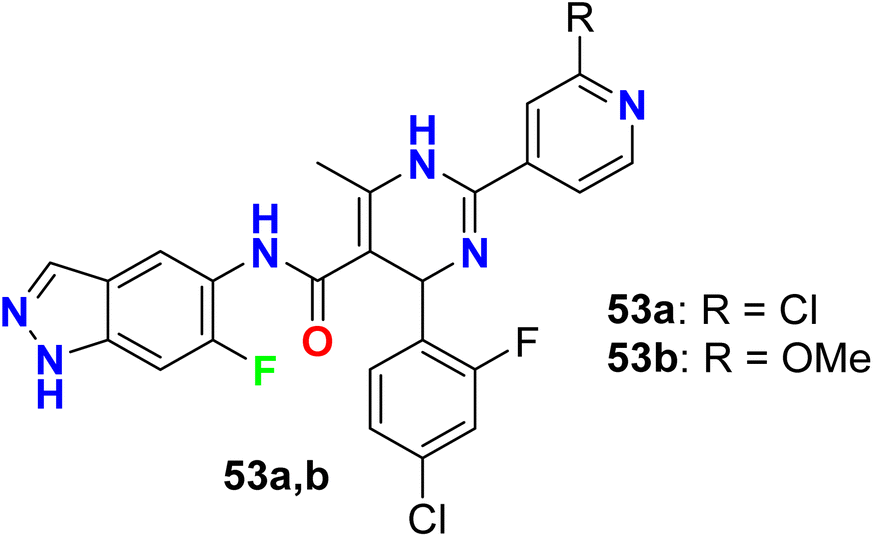
Two further examples of 6-fluoroindazoles 53a, 53b were found to have promising inhibitory activity of ROCK1 with IC50 values of 7 and 6 nM, respectively (Fig. 36). These compounds were also examined for P450 properties and rat PK (pharmacokinetics) studies. Both compounds 53a and 53b showed good oral bioavailability 49% and 53%, respectively, and both compounds showed improved P450 profile (2.1–5.3 μM at all isozymes tested; CYP2C9, CYP2D6, and CYP3A4). Compound 53a was tested for in vivo efficacy studies in a spontaneously hypertensive rat (SHR) model of hypertension, where at 30 mg kg−1 (po) compound 53a induced a 25 mmHg (t = 3 h) drop in arterial blood pressure. Thus Compound 53a demonstrated a good potency in vivo experiments.89
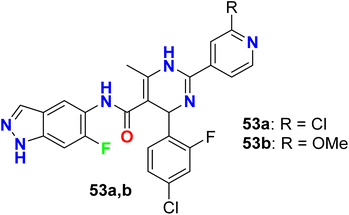 |
| | Fig. 36 Structure of 6-fluoroindazoles 53a, 53b. | |
Some fluorinated indazole derivatives 54 (Fig. 37) were patented as estrogen receptors (ER) modulators to be useful for the treatment for treating diseases that are dependent upon estrogen receptors. The bioassay results of all the invented derivatives for ER-α in cell western assay (SPl) showed inhibitory activity with IC50 values < 100 nM.90
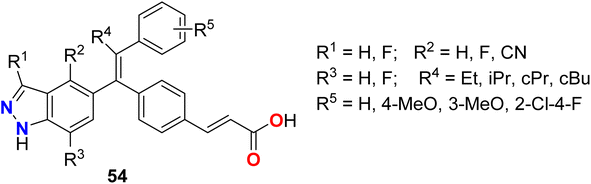 |
| | Fig. 37 Structure of fluorinated indazole derivatives 54. | |
6-Fluoroindazole scaffolds 55 (Fig. 38) were invented and evaluated for their positive allosteric modulators (PAM) activity using human α7nAChR stable expressing cells. The invented compounds exhibited promising α7 nAChR PAM activity at 10 μM with α7PAM% varied between 189–4639, particularly compound 55 (R1 = H, R2 = cHex, X = CH) had the highest PAM activity active with 4639%. These compounds might be used as therapeutic agents to cure diseases involving the cholinergic properties of CNS.91
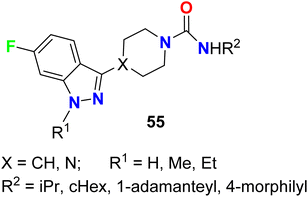 |
| | Fig. 38 Structure of 6-fluoroindazole scaffolds 55. | |
3.3. Anti-inflammatory activity of fluorinated benzimidazoles
Calderini et al. invented some fluorinated pyrazole-based heterocycles 56–57 and evaluated their biological activities as inhibitors of PDK1 (pyruvate dehydrogenase kinase 1) for treating inflammatory diseases (Fig. 39). Thus, the fluoropyrazole derivatives 56 and fluorobenzimidazole derivatives 57 were tested for inhibition of PDK1 using a flash-plate system with 384 wells/micro-titration assay. Interestingly all the fluorinated derivatives exhibited high potency with IC50 values varying between 1 nM to 0.1 μM.92
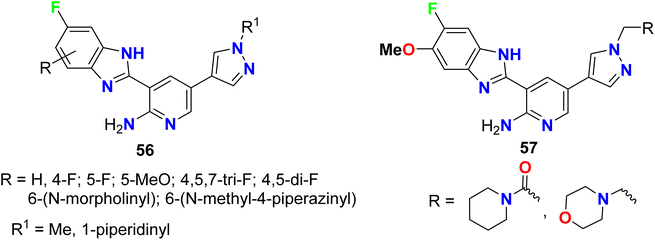 |
| | Fig. 39 Structure of pyrazole-based heterocycles 56–57. | |
3.4. Anti-inflammatory activity of fluorinated benzothiazoles
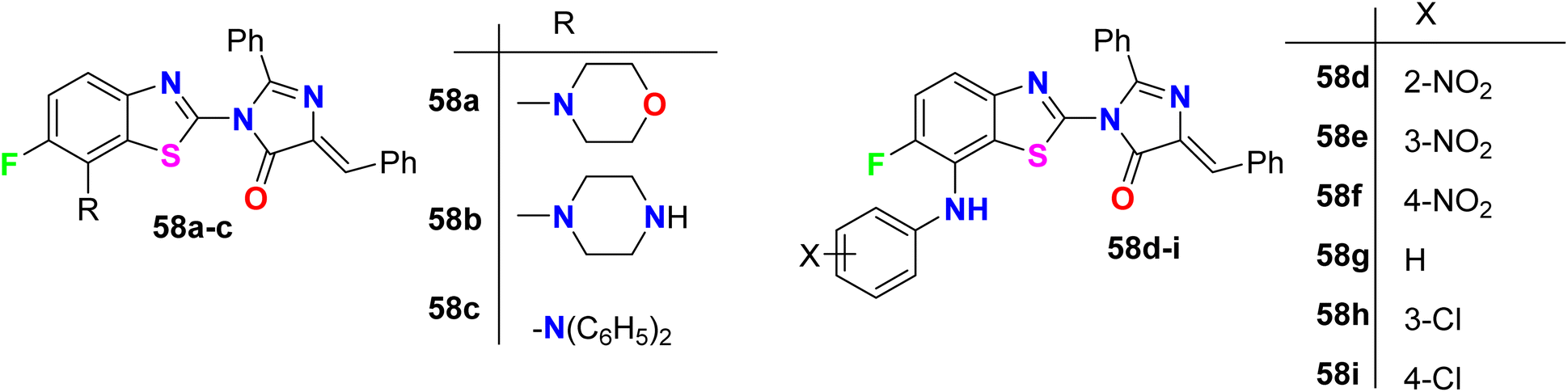
The 6-fluorobenzothiazole derivatives 58a–i (Fig. 40) were assembled by Sathe et al. and then screened for their in vitro anti-inflammatory activity using the technique of inhibition of albumin denaturation. The tested compounds showed in moderate to high range of activity from 20.40–79.93% of inhibition compared with the anti-inflammatory drug ibuprofen (93.87%). Compound 58h, specifically, had the best inflammation inhibition with 79.93%.93
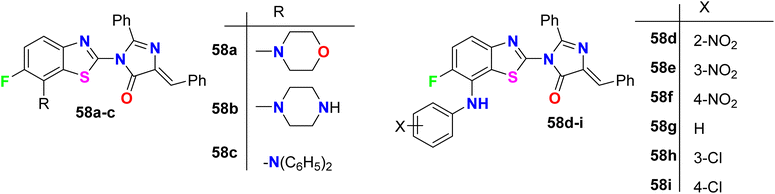 |
| | Fig. 40 Structure of 6-fluorobenzothiazole derivatives 58a–i. | |
3.5. Anti-inflammatory activity of fluorinated indoles
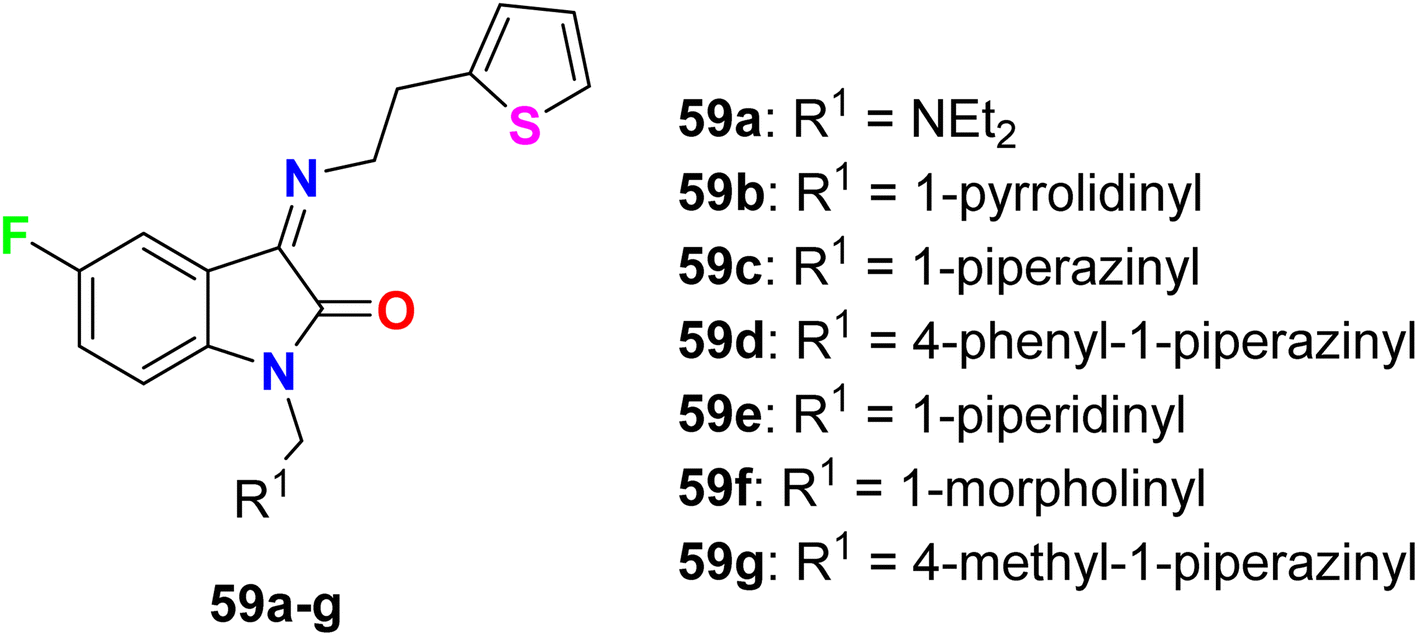
Varpe et al. reported the synthesis of 5-fluoroisatin derivatives 59a–g and evaluated their anti-inflammatory activity (Fig. 41). The anti-inflammatory activity was examined in vitro by measuring the inhibition % of denaturation of Bovine Serum Albumin (BSA). All compounds showed good inhibition %, especially the piperazine-based 5-fluoroisatin 59d at 100 μg mL−1. Compound 59d showed the highest anti-inflammatory activity with 80.08% inhibition compared with the standard drug diclofenac sodium with 89.38% inhibition, and compounds 59c and 59g showed 72.56 and 62.38% inhibition. Docking analysis disclosed that the compounds exhibited good interactions with the COX-2 enzyme binding site.94
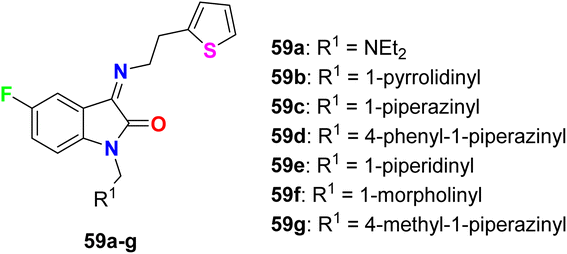 |
| | Fig. 41 Structure of 5-fluoroisatin derivatives 59a–g. | |
3.6. Anti-inflammatory activity of fluorinated benzofurans
The anti-inflammatory effects of the fluorinated benzofuran derivatives 60–63 in macrophages and in the air pouch model of inflammation, were investigated (Fig. 42). Most compounds suppressed the lipopolysaccharide-stimulated inflammation by inhibiting the expression of cyclooxygenase-2 and nitric oxide synthase-2 and decreased the secretion of the tested inflammatory mediators. Their IC50 values ranged between 1.2–9.04 μM for interleukin-6; between 1.5–19.3 μM for Chemokine (C–C) ligand 2; between 2.4–5.2 μM for nitric oxide; and between 1.1–20.5 μM for prostaglandin E2. Three fluorinated benzofuran compounds significantly inhibited cyclooxygenase activity. Most of these compounds showed anti-inflammatory effects in the zymosan-induced air pouch model.95
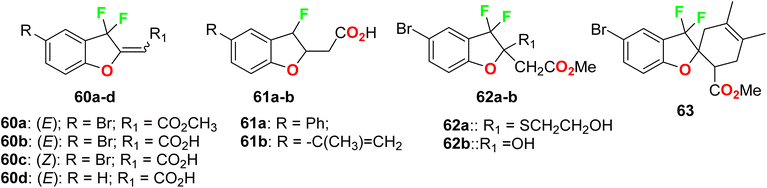 |
| | Fig. 42 Structure of fluorinated benzofuran derivatives 60–63. | |
4. Enzymatic inhibitory activity of fluorinated heterocycles
4.1. Enzymatic inhibitory activity of fluorinated thiophenes
Fuchigami designed and synthesized the monofluorinated 3-thiolanones (cis/trans isomers) 64a, 64b and evaluated their in vitro human type II phospholipase A2 (hPLA2) inhibitory activity for possible treatment of inflammatory diseases (Fig. 43). Interestingly, the cis/trans mixture of 64a, 64b showed substantial inhibitory action against PLA2 with IC50 = 0.2 μM much better than the manoalide reference drug with IC50 = 0.34 μM. The pure cis isomer of 64b (IC50 = 0.21 μM) was found to present higher activity than its trans isomer 64a (IC50 = 1.27 μM).96
 |
| | Fig. 43 Structure of monofluorinated 3-thiolanones (cis/trans isomers) 64a, 64b. | |
4.2. Enzymatic inhibitory activity of fluorinated pyrroles
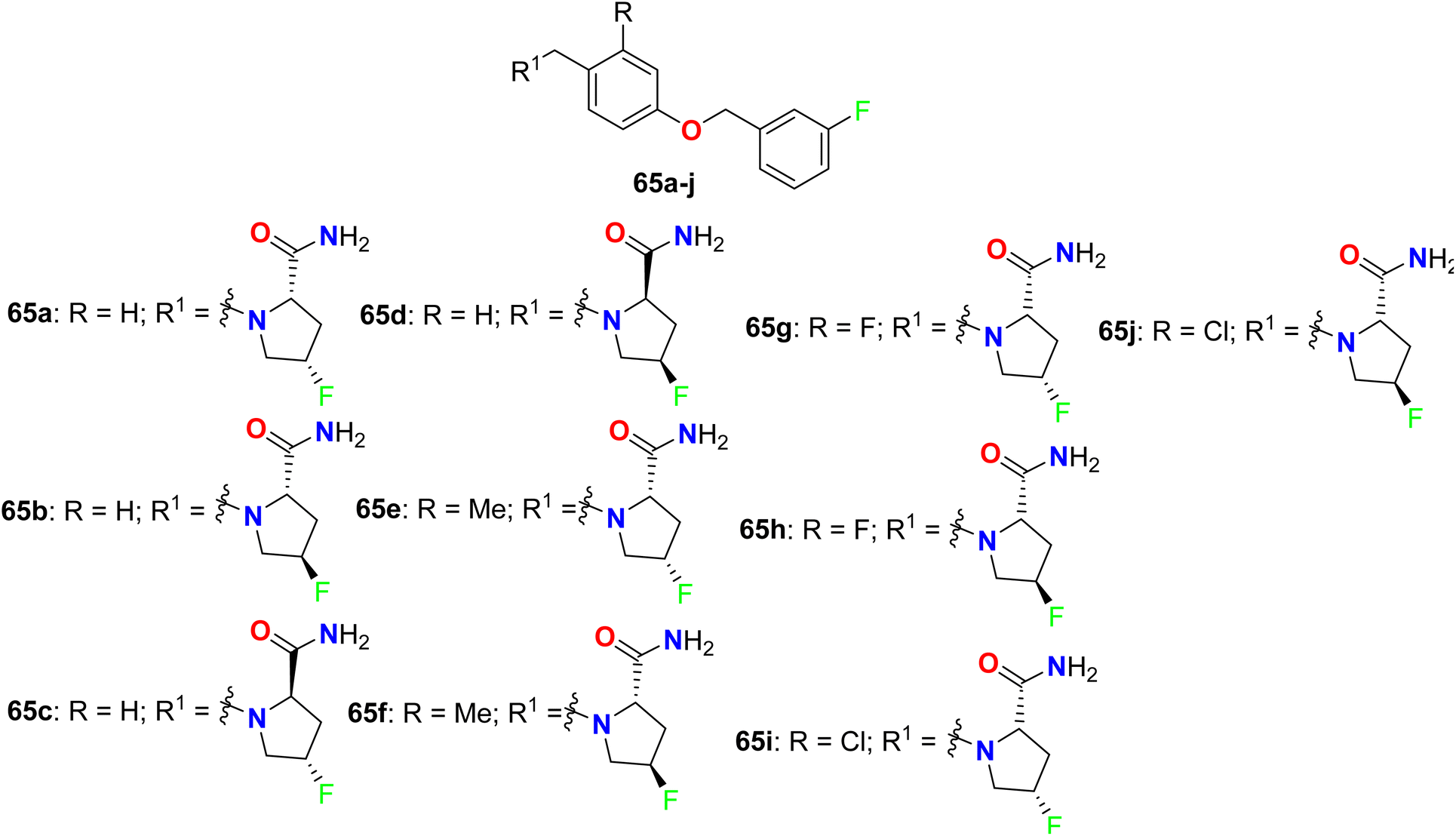
Some chiral fluorinated pyrrolidine hybrids 65a–j (Fig. 44) were synthesized and in vitro evaluated as MAO-B/MAO-A (monoamine oxidase B/A) inhibitors that were useful for Parkinson's disease (PD) in clinics. The biological experiments disclosed that compound 65a was the most potent, selective MAO-B inhibitor with 10-fold activity more than that of the safinamide drug, where compound 65a had IC50 = 0.019 μM, with selectivity index (SI) = 2440 of MAO-A/MAO-B, compared with safinamide (IC50 = 0.163 μM, with SI = 172 for MAO-A/MAO-B). SAR study revealed also that 4S chiral F-substituent on the pyrrolidine ring were more potent MAO-B inhibitors than those with 4R ones, but replacing 2S-carboxamide with 2R-carboxamide on the pyrrolidine ring led to a lowering of MAO-B inhibitory activity. Molecular docking results confirmed that the enhanced hydrophobic interaction of 65a increased the activity against MAO-B. Thus, 65a was a promising drug candidate for the treatment of PD.97
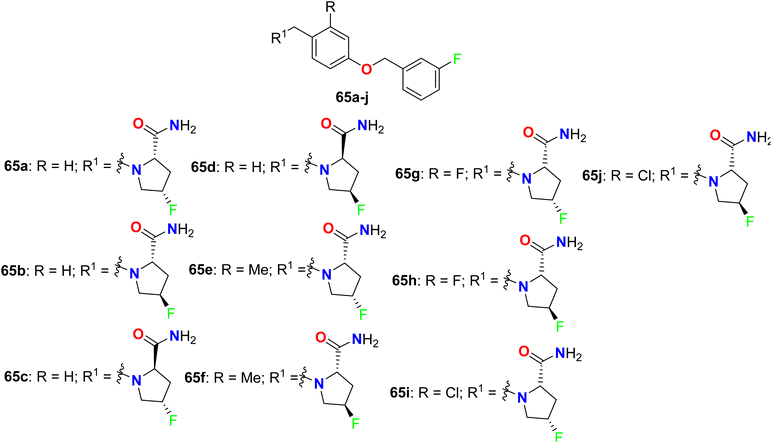 |
| | Fig. 44 Structure of chiral fluorinated pyrrolidine hybrids 65a–j. | |
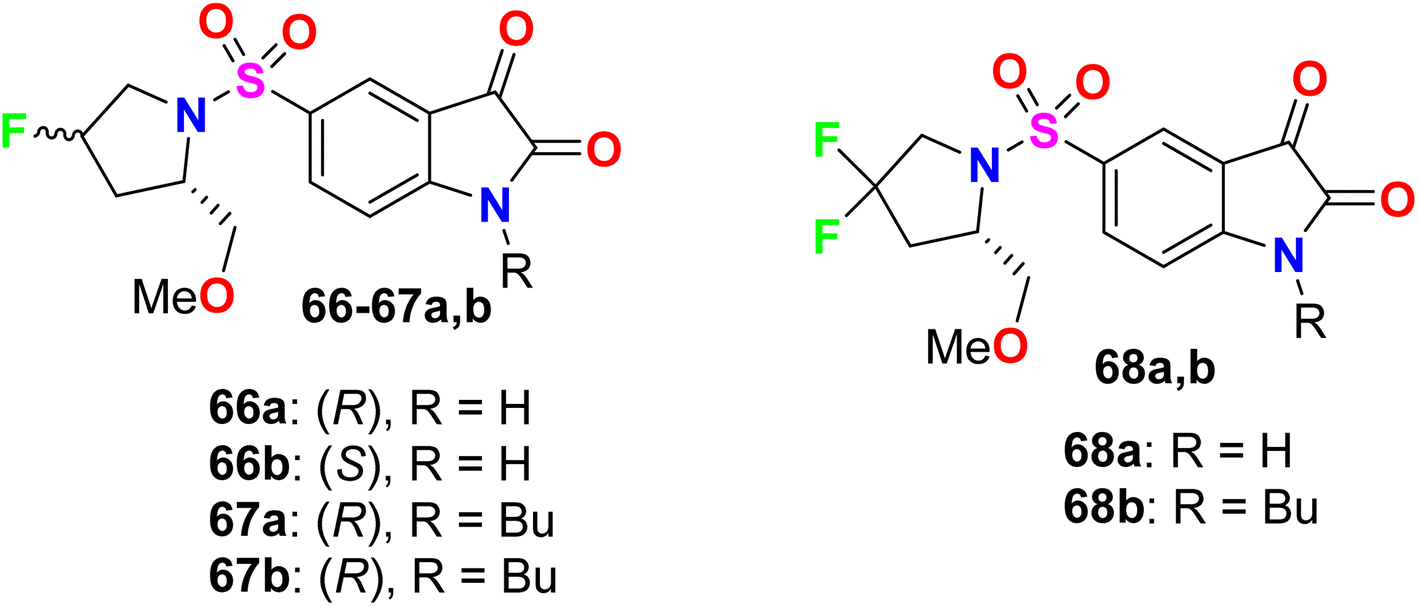
Some fluorinated pyrrolidine-based isatin compounds 66a, 66b, 67a, 67b, and 68a, 68b were reported by Limpachayaporn et al. and their in vitro inhibitory actions against caspases-3 and -7 were recorded (Fig. 45). The bioassay study showed that all compounds disclosed high inhibitory activity with IC50 values ranging between 0.362–2.57 μM (for caspase-3) and 0.178–14.9 μM (for caspase-7). The 4,4-difluorinated compound 68b presented the best enzyme inhibition results with IC50 values of 0.362 μM and 0.178 μM for caspases-3 and -7, respectively.98
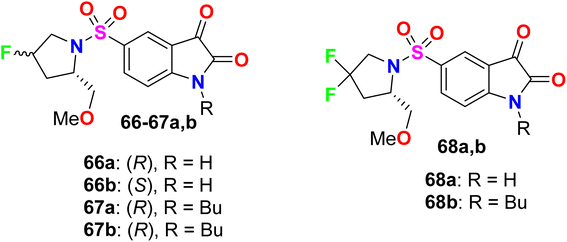 |
| | Fig. 45 Structure of fluorinated pyrrolidine-based isatin derivatives 66–68. | |
Wang et al. reported a series of directly fluorinated pyrroloindole structures 69a–z (Fig. 46) as potent acetylcholinesterase (AChE) inhibitors for possible treatment of Alzheimer's disease (AD). The bioassay experiments disclosed that most of the fluorinated heterocycles had potent AChE inhibitory activity, particularly compound 69a (R = H, R1 = Me, X = NCO2Me) displayed promising inhibitory activity against AChE with IC50 value 16.0 μM. The presence of bulky groups (such as Boc or Ts) of N-nucleophiles was essential for interaction with AChE. Therefore, compound 69a was a potential candidate for the treatment of AD.99
 |
| | Fig. 46 Structure of fluorinated pyrroloindole derivatives 69a–z. | |
4.3. Enzymatic inhibitory activity of fluorinated indoles
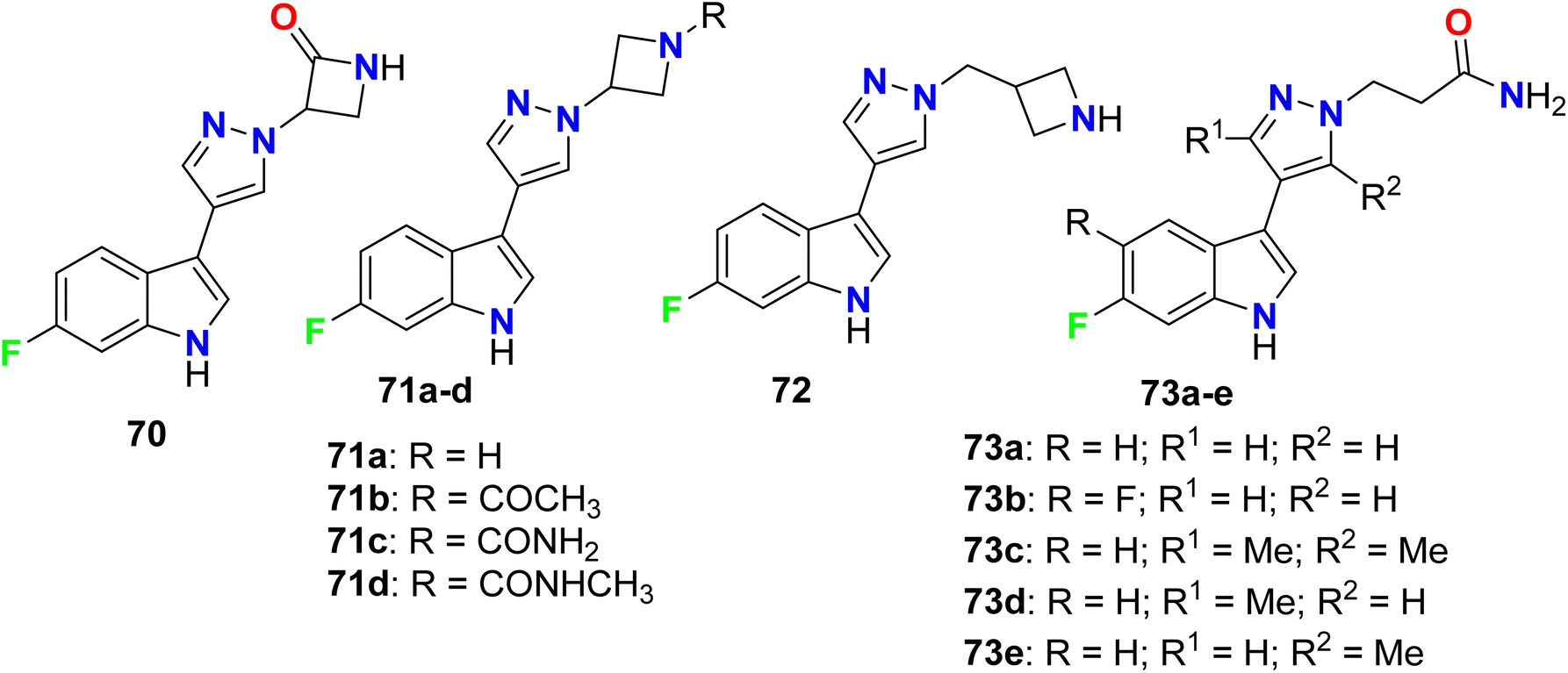
Crosignani et al. described a series of 6-fluoroindole derivatives 70–73 and were screened as tryptophan 2,3-dioxygenase enzymatic (TDO2) inhibitors to be useful in the treatment of cancers (Fig. 47). Most of the invented compounds greatly inhibited the enzymatic activity of human TDO2 with IC50 values ranging between 1 μM and 10 μM, and some compounds such as 71a, 72, and 73a had significant inhibitory activity with IC50 < 1 μM.100
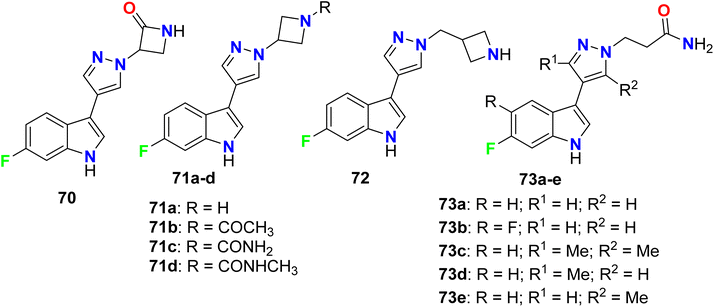 |
| | Fig. 47 Structure of 6-fluoroindole derivatives 70–73. | |
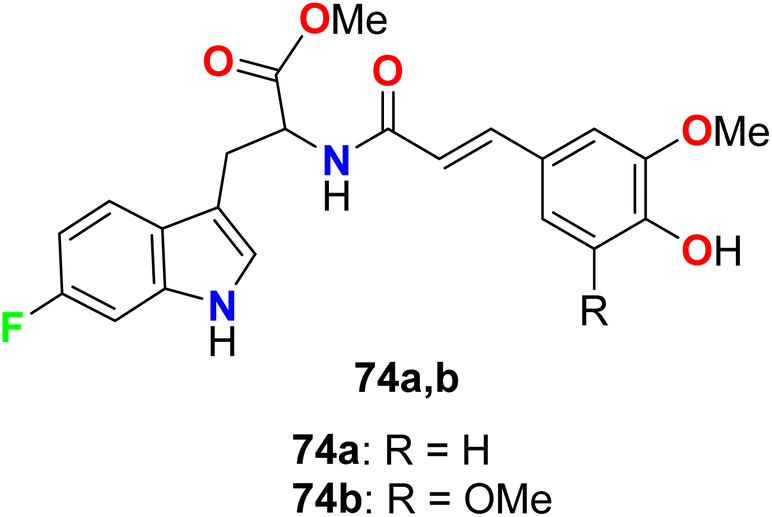
The 6-fluorotryptophan derivatives 74a, 74b (Fig. 48) were synthesized and screened for their in vitro tyrosinase inhibitory activity. The in vitro inhibitory activity assay was evaluated on mushroom tyrosinase using L-tyrosine as a substrate. The bioassay results disclosed that compounds 74a, 74b were highly effective as tyrosinase inhibitors much better than the hydroquinone reference standard.101
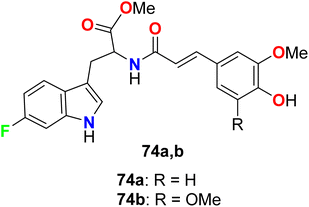 |
| | Fig. 48 Structure of 6-fluorotryptophan derivatives 74a, 74b. | |
Three fluorinated indole hybrids 75a–c (Fig. 49) were synthesized and screened as S1P (sphingosine-1-phosphate) receptor agonists by an in vivo peripheral lymphocyte reduction assay. The reported derivatives 75a–c proved to be promising selective S1P1 agonists, where compounds 75a and 75b both showed pEC50 value > 11 (for S1P1) compared with pEC50 < 5 (for S1P3). Compound 75a showed similar activity as the fingolimod reference standard. On the other hand, 6-fluoroindole molecule 75c provided less S1P1 agonist potency when compared with 4- and 5-fluoroindole derivatives 75a and 75b.102
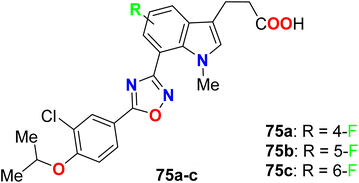 |
| | Fig. 49 Structure of fluorinated indole derivatives 75a–c. | |
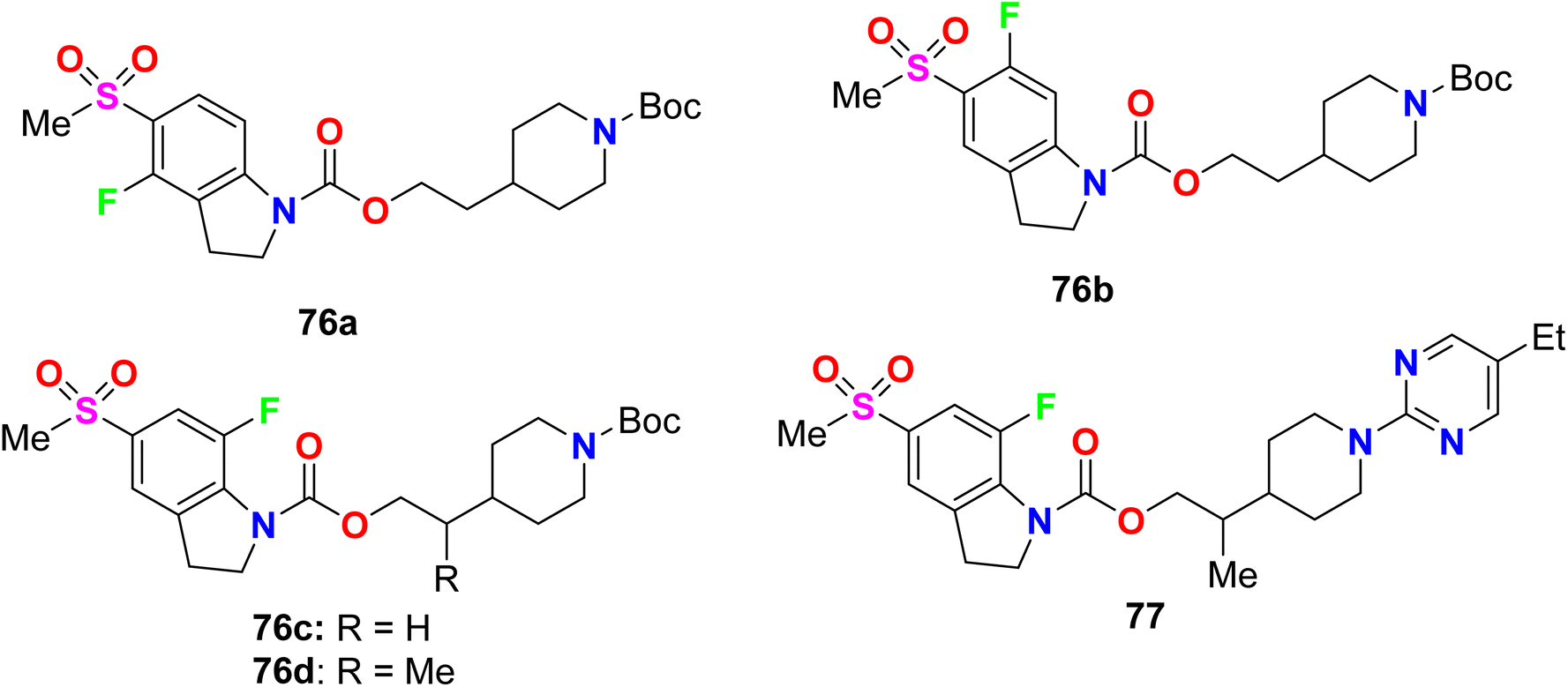
Some fluorinated indole hybrids 76a–d, and 77 were synthesized and evaluated for their potency as GPR119 agonist activity. GPR119 (G-protein-coupled receptor) is a target for anti-diabetic agents (Fig. 50). The bioassay study was carried out utilizing a cAMP reporter assay in CHO (Chinese hamster ovary) cells stably expressing human GPR119. All compounds displayed a nanoscale activity and the most active compounds were 76d and 77 with EC50 values of 6.8 and 3.9 nM, respectively. The marked increase in agonist potency of compound 77 might be due to the participation of carbonyl function in the carbamate spacer in interactions with GPR119 as a hydrogen bond acceptor.103
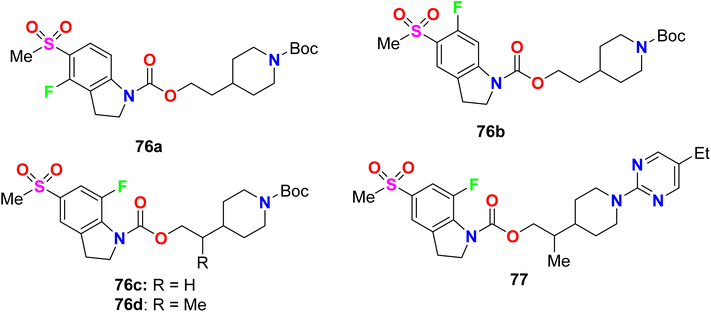 |
| | Fig. 50 Structure of fluorinated indole derivatives 76–77. | |
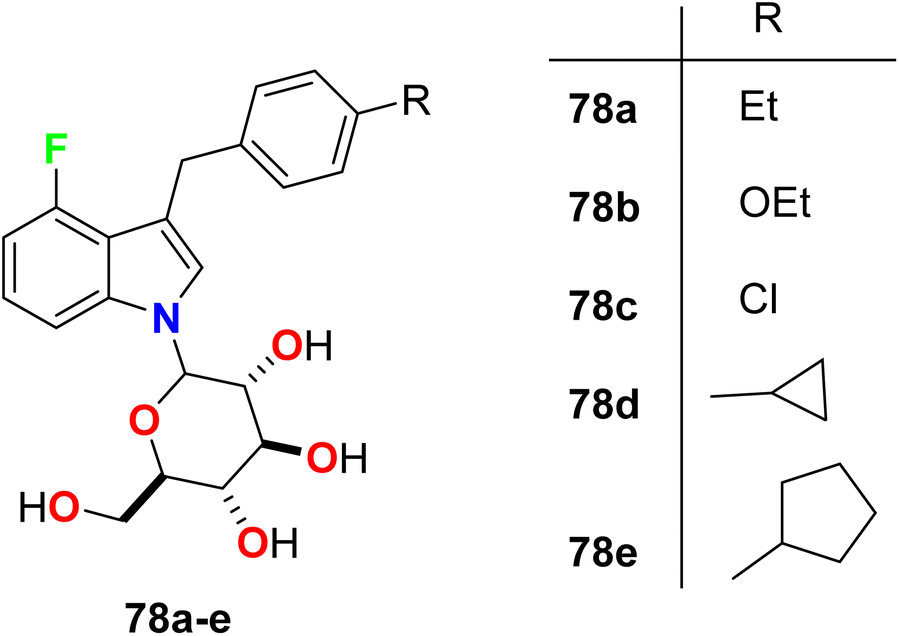
Nomura et al. constructed some fluorinated-indole-N-glucosides 78a–e (Fig. 51) and evaluated their inhibition effects on hSGLT activity (human sodium-glucose co-transporter) as anti-hyperglycemic agents. The selectivity of the highly active transporters of hSGLT1 or hSGLT2 was also determined. The synthesized 4-fluoroindole hybrids 78a–e were found to have substantial potency and selective hSGLT2 inhibitor with IC50 values ranged between 1.4 to 24 nM with high anti-hyperglycemic activity in high-fat diet-fed KK mice. Compound 78d was the most potent and most selective inhibitor of hSGLT2 both in vitro and in vivo with IC50 = 1.4 nM, and could be considered as lead compound for further developments.104
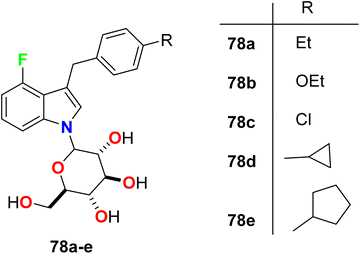 |
| | Fig. 51 Structure of fluorinated-indole-N-glucosides 78a–e. | |
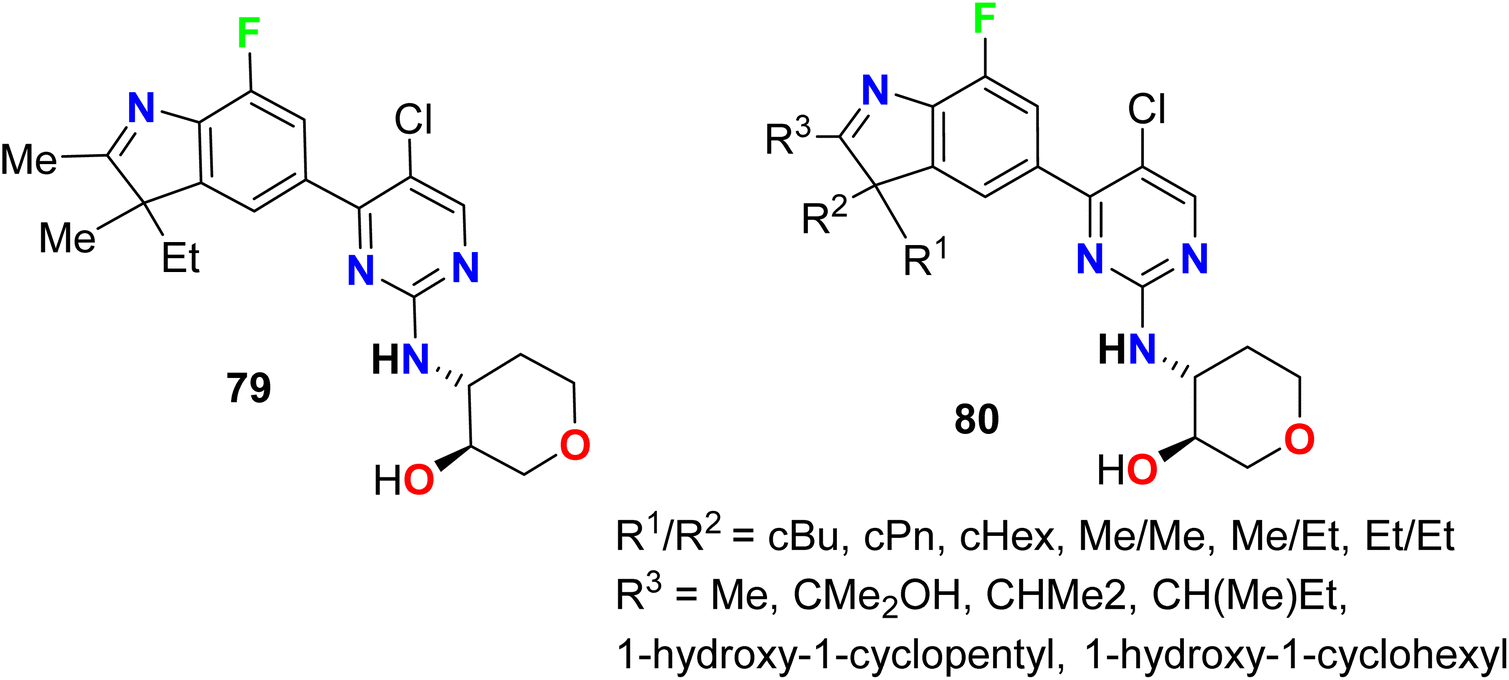
A series of patented fluorinated indoline hybrids 79–80 were synthesized and evaluated as CDK4/CDK6 (cyclin-dependent kinases 4 and 6) inhibitors (Fig. 52). The inhibitory effect of the invented compounds 79–80 on CDK4/CDK6 enzymes was measured using the EnVision multi-mode detection platform to detect the fluorescence values at 665 nm and 620 nm excited at 337 nm in HTRF mode. The IC50 values of the tested compounds on the inhibition of CDK4 enzyme activity was found to range between 0.9 nM–9.6 nM, however, the IC50 values for inhibition of CDK6 enzyme activity ranged between 7.3 nM and 1038 nM. Particularly, compound 79 showed the best inhibition against both CDK4 and CDK6 enzymes with IC50 values of 0.9 nM and 7.3 nM, respectively.105
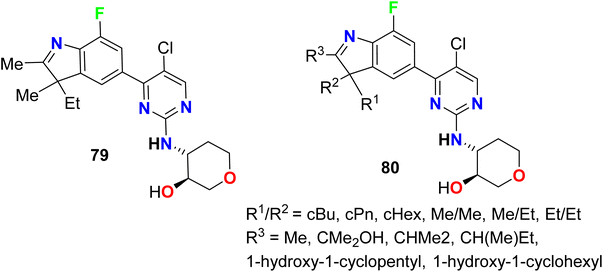 |
| | Fig. 52 Structure of fluorinated indoline hybrids 79–80. | |
4.4. Enzymatic inhibitory activity of fluorinated benzofurans
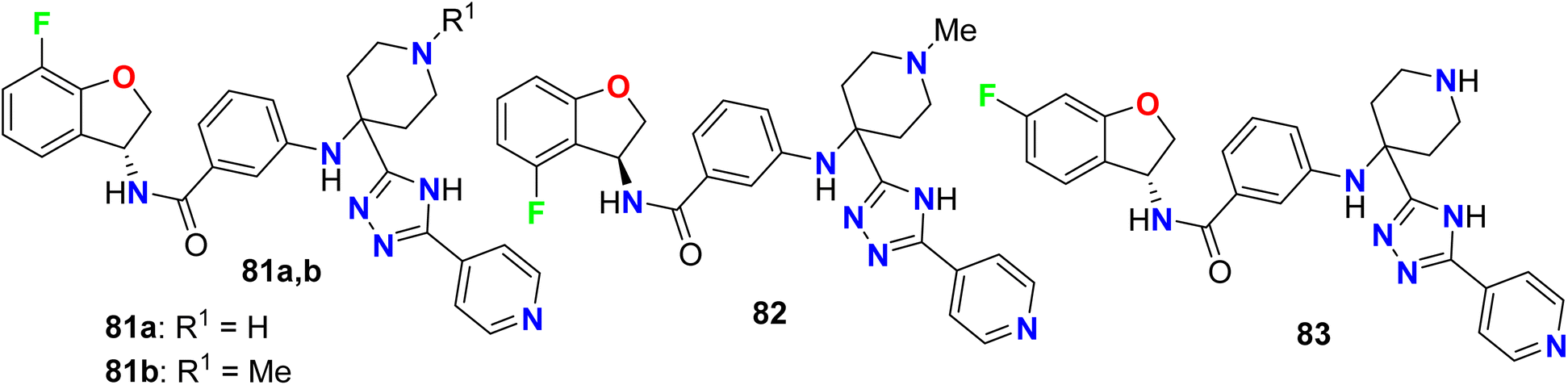
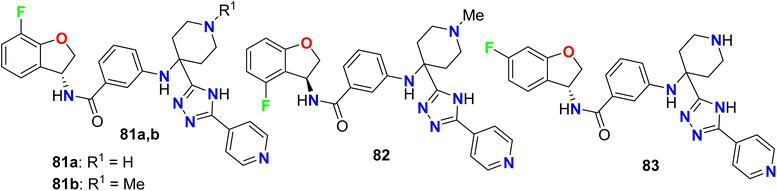
A series of fluorinated dihydrobenzofuran scaffolds 81–83 were patented and evaluated for their enzymatic inhibitory activity against G protein-coupled receptor kinase (GRK) (Fig. 53). The in vitro and in vivo inhibition of GRK2 and/or GRK3 activity was examined to reduce the tumor growth. Most of the compounds were selective inhibitors for GRK2, and showed excellent GRK2 enzymatic inhibition potency with IC50 values in nanomolar scale varied between 0.53 nM to 9.17 nM and the most active compound was 83 (IC50 = 0.53 nM).106
 |
| | Fig. 53 Structure of fluorinated dihydrobenzofuran scaffolds 81–83. | |
4.5. Enzymatic inhibitory activity of fluorinated pyrazoles
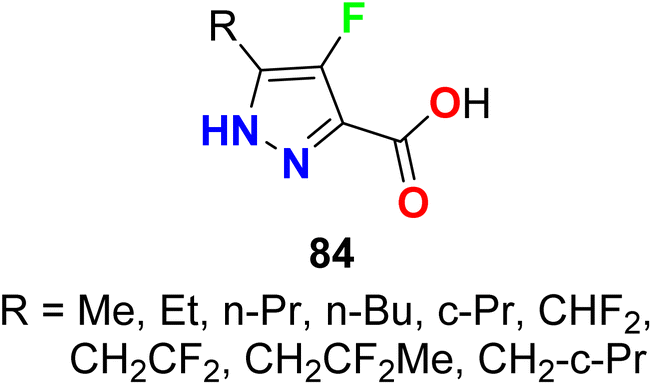
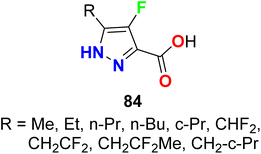
The fluorinated pyrazol-3-caarboxylic acid derivatives 84 were reported as antilipolytic agents and against the human receptor RUP25 for the treatment of dyslipidemia (Fig. 54). The in vitro biological activity was evaluated using the cAMP Whole Cell method and most of the compounds showed good activity against hRUP25, particularly compound 84 (R = Bu) had an IC50 value of 0.9 μM.107
 |
| | Fig. 54 Structure of fluorinated pyrazol-3-caarboxylic acid derivatives 84. | |
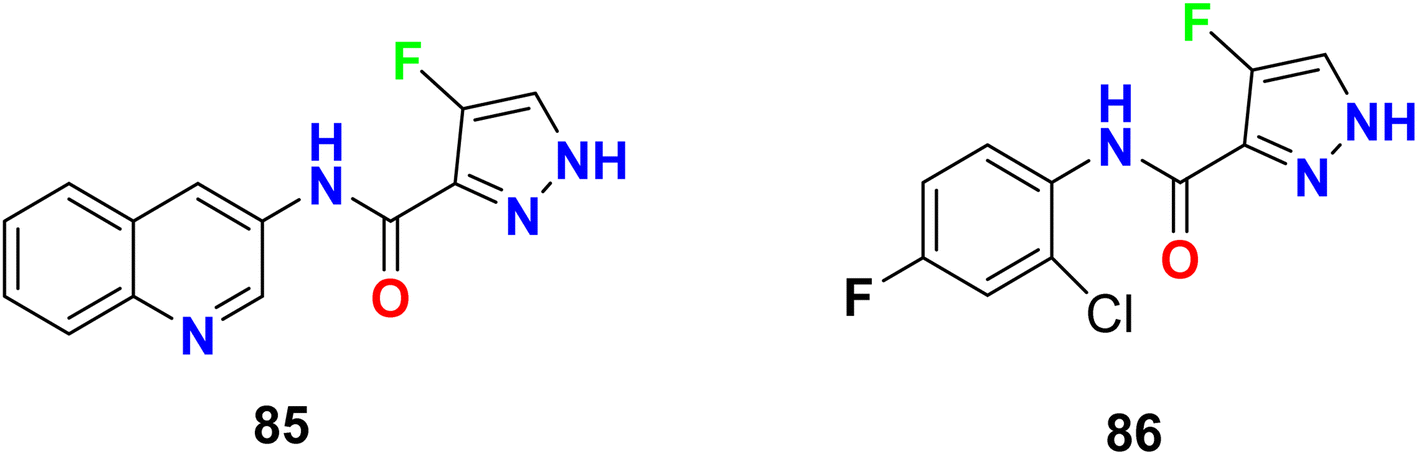
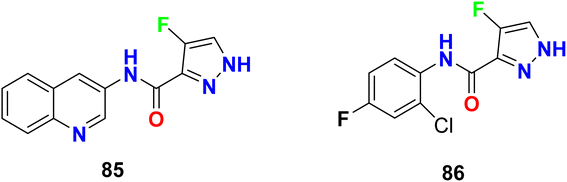
In addition, Pelcman et al. patented the fluoropyrazole-carboxamides 85–86 (Fig. 55) as inhibitors of 15-lipoxygenase activity, that catalyzes arachidonic acid oxygenation at position 15, that were useful in the treatment of inflammatory diseases. The tested compounds exhibited inhibition of 15-lipoxygenase activity with an IC50 value of 10 μM.108,109
 |
| | Fig. 55 Structure of fluoropyrazole-carboxamides derivatives 85–86. | |
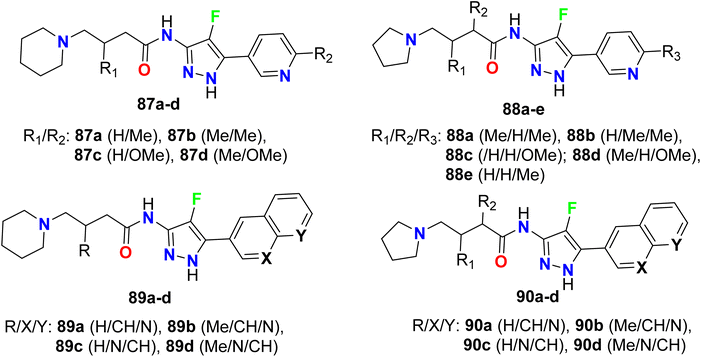
The fluoropyrazole-carboxamides 87a–d–90a–d (Fig. 56) were reported to display a high inhibitory activity of a7 nicotinic acetylcholine receptor (a7nAChR), ligand-dependent ionic channel in muscle fiber membrane. The bioassay was conducted using a FLIPR (Fluorescent Imaging Plate Reader) screening protocol employing the stable recombinant GH4C1 cell line expressing a7nAChR. Most of the fluoropyrazoles exhibited high inhibitory potency with EC50 values ranging between 10 nM and 10 μM.110
 |
| | Fig. 56 Structure of fluoropyrazole-carboxamides derivatives 87–90. | |
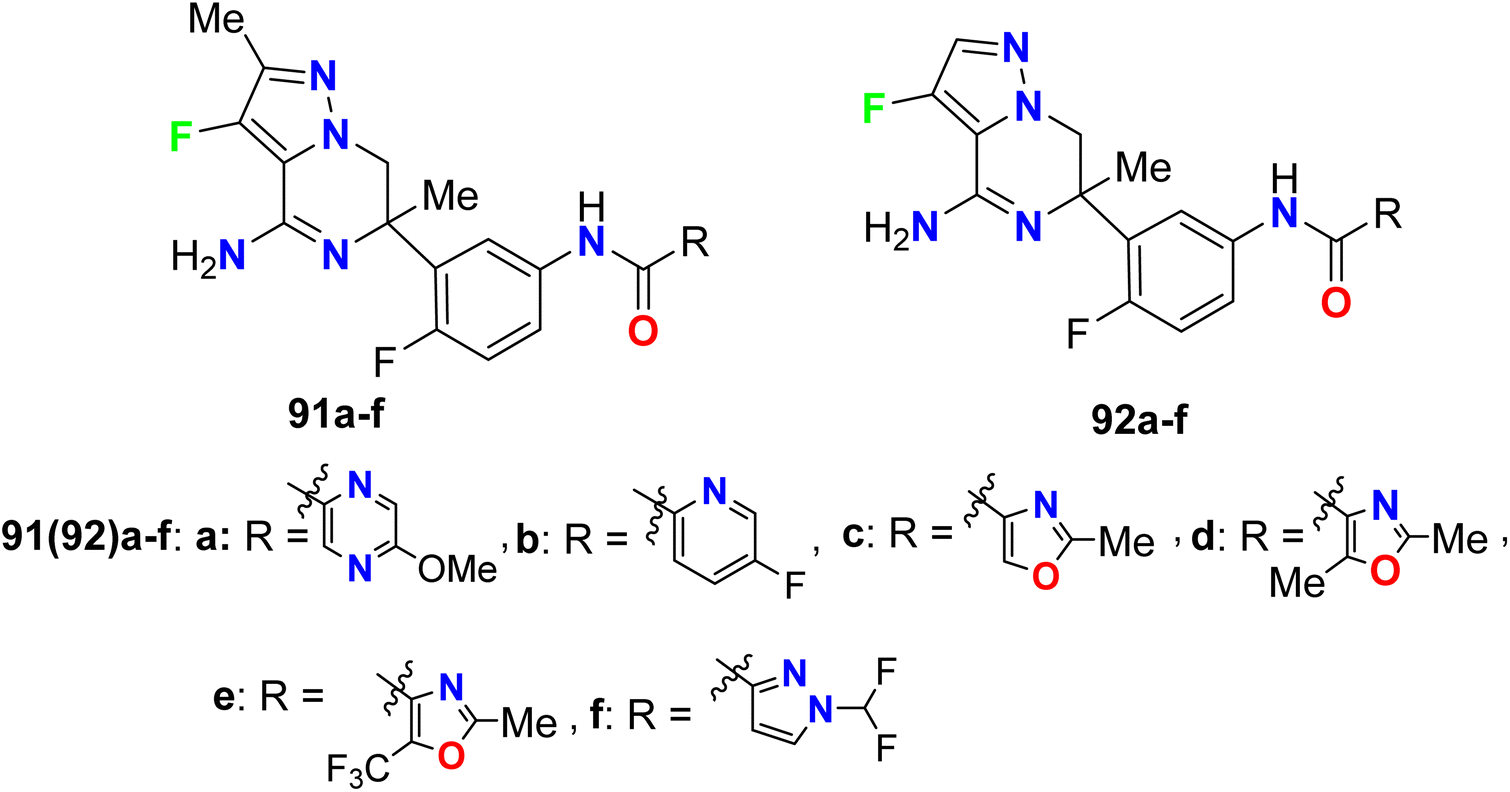
Trabanco-Suarez et al. patented the synthesis of many fluorinated pyrazole-based heterocyclic hybrids 91a–f and 92a–f and examined their activities as inhibitors of BACE1 (β-site APP-cleaving enzyme 1), for the treatment of Alzheimer's Disease (AD) (Fig. 57). The biological results were based on the FRET (Fluorescence Resonance Energy Transfer) assay, where the candidate for this assay was an APP (amyloid precursor protein). The examined compounds showed inhibition of FRET with pIC50 values 6.90–7.23.111
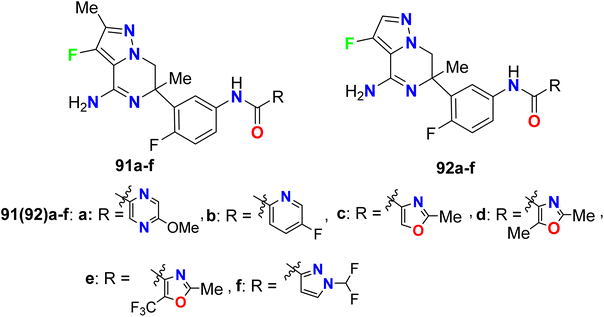 |
| | Fig. 57 Structure of fluorinated pyrazole-based heterocyclic hybrids 91–92. | |
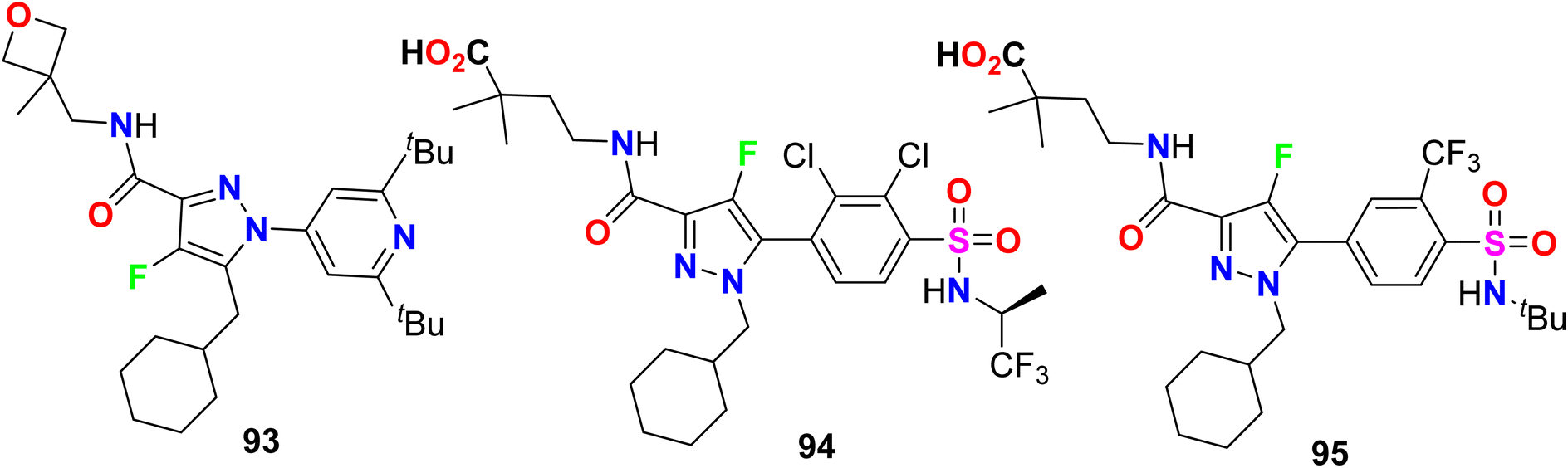
Gege et al. invented and patented some fluorinated pyrazole molecular hybrids 93–95 (Fig. 58) as modulators of the activity of human RAR-related orphan receptors gamma (RORγ) that are expressed in immune cells (Th17 cells) and useful in the regulation of circadian rhythms, for the treatment of the RORγ mediated chronic inflammatory. Applying the Gal4 reporter gene assay, the invented compounds displayed promising inhibitory action against RORγ with IC50 values < 10 nM. The pIC50-values of the tested compounds were 6.2–8.3 for fluorescence resonance energy transfer (FRET), firefly (FF) and renilla normalized (REN-norm).112
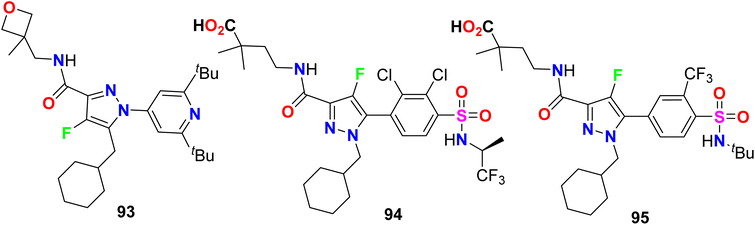 |
| | Fig. 58 Structure of fluorinated pyrazole derivatives 93–95. | |
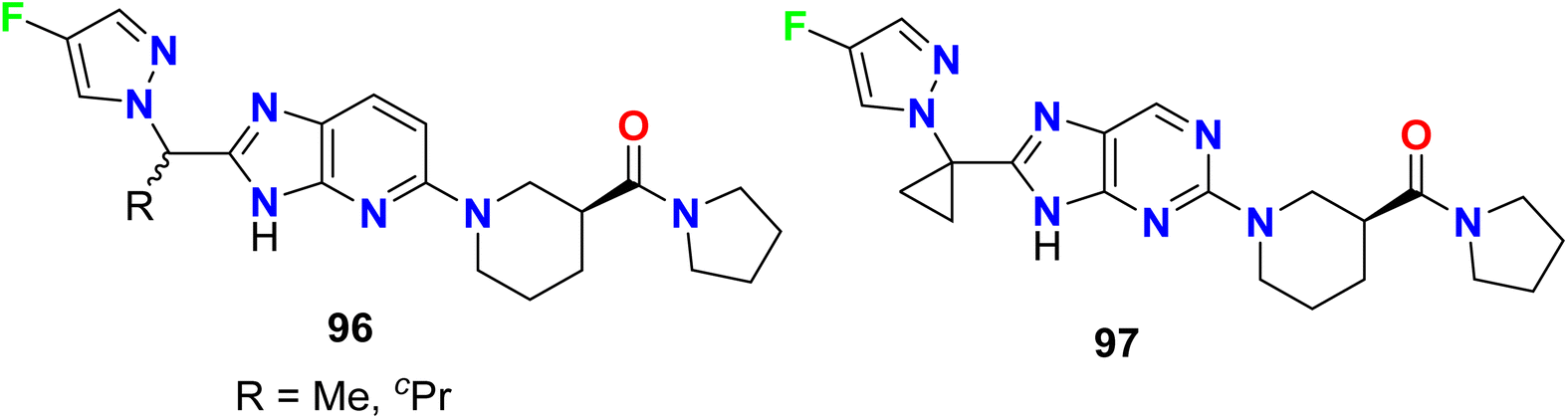
The fluorinated pyrazole-based heterocyclic hybrids 96–97 were patented by Ahn et al. as good inhibitors of the activity of the diacylglycerol-acyltransferase-2 (DGAT2). The in vitro inhibition activity against DGAT2 was recorded in a nanomolar scale with IC50 values ranging within 90–128 nM (Fig. 59).113
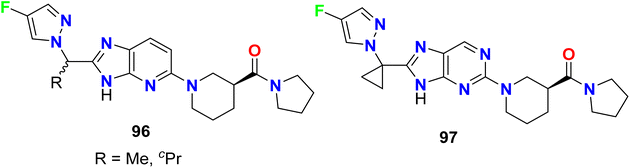 |
| | Fig. 59 Structure of fluorinated pyrazole derivatives 96–97. | |
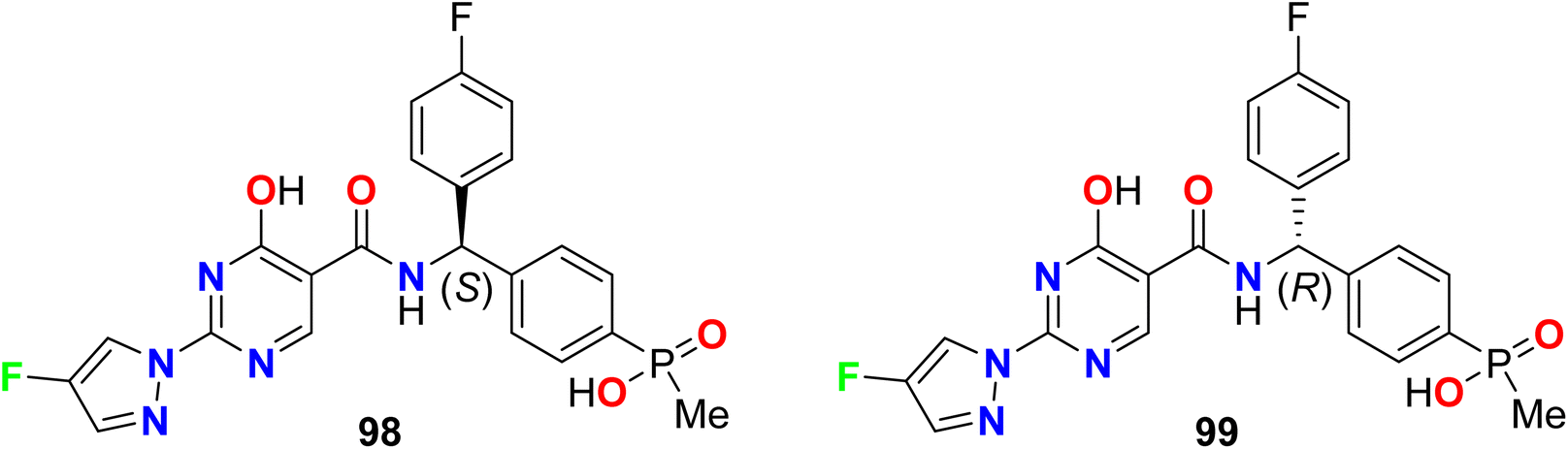
Zhou et al. reported two fluorinated pyrazole-based molecular hybrids 98 and 99 to be useful as inhibitors of hypoxia induced factor (HIF) prolyl hydroxylase (PHD2) for treatment of anemia. The reported invented compounds 98 and 99 displayed significant inhibition activities against HIF PHD2 with IC50 values of 0.5 nM and 20 nM, respectively (Fig. 60).114,115
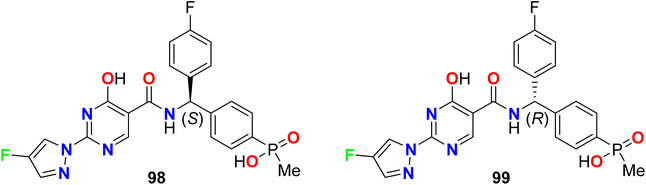 |
| | Fig. 60 Structure of fluorinated pyrazole derivatives 98–99. | |
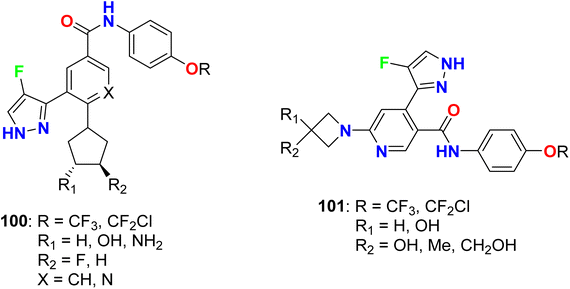
Dodd et al. patented several fluoropyrazole hybrids 100 and 101 (Fig. 61) and evaluated their inhibition of the tyrosine kinase enzymatic activity of ABL1 (Abelson protein). The experimental bioassay measured the ABL kinase activity in a radiometric filter binding (Radio) and microfluidic mobility shift (Caliper) assays. The invented hybrids revealed high potency in the nanomolar scale with IC50 values ranging between 1 to 7 nM (Radio-ABL1) and 0.2–1.2 nM for (Caliper ABL1) inhibition, respectively.116,117
 |
| | Fig. 61 Structure of fluoropyrazole derivatives 100–101. | |
4.6. Enzymatic inhibitory activity of fluorinated indazoles
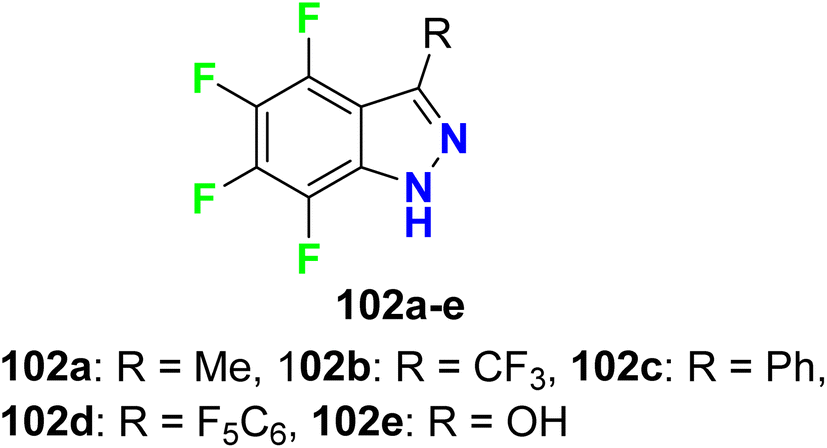
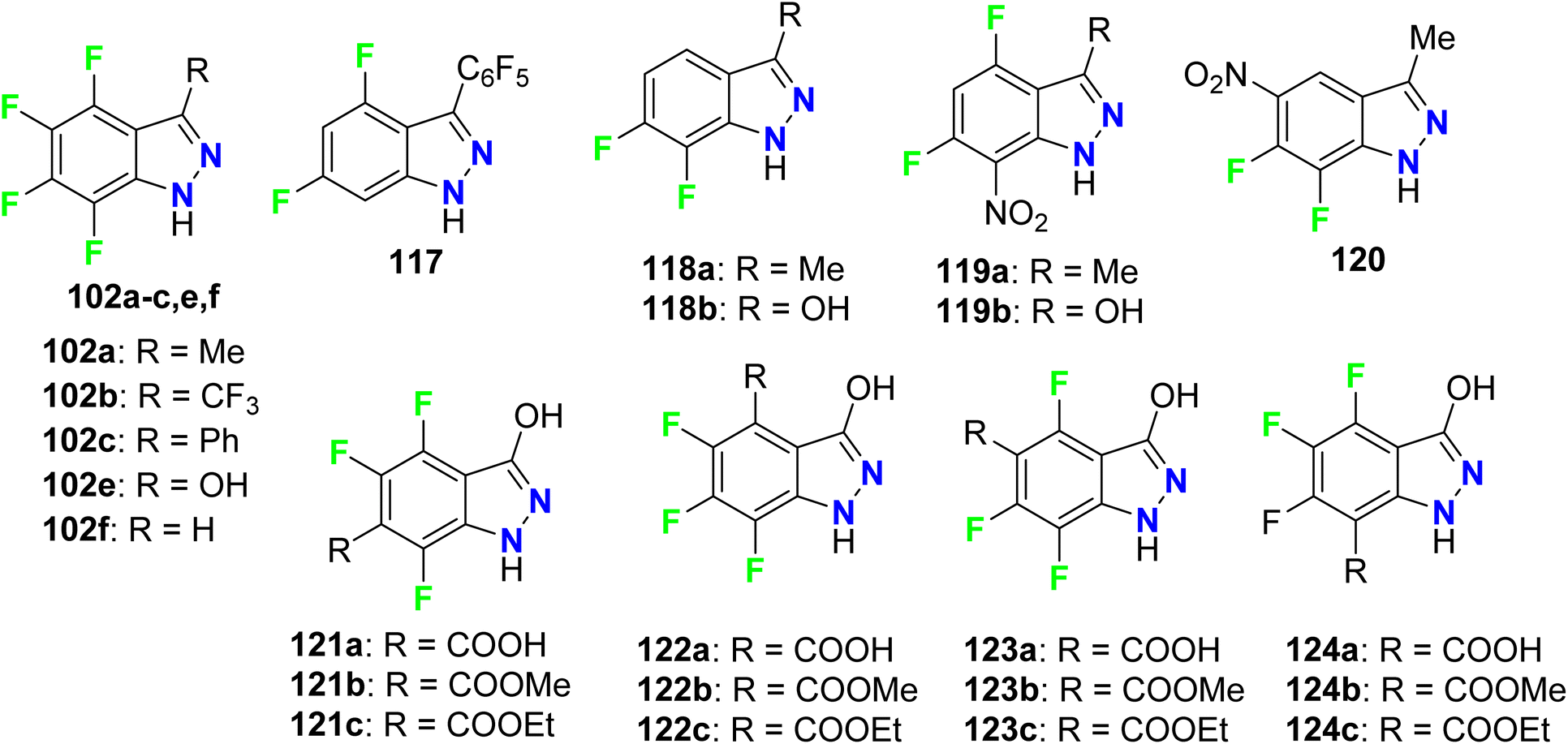
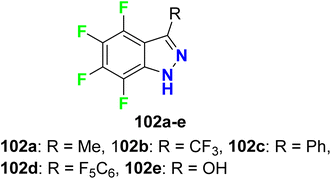
Claramunt et al. designed several fluorinated indazoles 102 and examined their activity as selective inhibitors of two nitric oxide synthase enzymes; inducible nitric oxide synthase (iNOS) and neuronal nitric oxide synthase (nNOS), that were useful for the regulation of blood pressure, neurotransmission, and the immune response. SAR study confirmed that R-substituents and the number and location of fluorine atoms greatly influenced the NOS inhibition process. The increasing number of fluorine atoms increased the inhibitory potency and nNOS selectivity, where fluorine atoms might form hydrogen bonds with the active center. The tetrafluoro-indazole derivative 102a (R = Me) displayed the best inhibitory activity among the tested compounds, where it inhibited iNOS by 63% and nNOS by 83%. Surprisingly, compound 102d (R = perfluorophenyl) showed the highest selective inhibition of nNOS activity by 80% but did not affect iNOS activity. The other derivatives exhibited partial selectivity for inhibition of iNOS and nNOS. Selective inhibition of nNOS was reported to be of great therapeutic importance for treating diabetes, neurodegenerative disorders, sepsis, and arthritis (Fig. 62).118
 |
| | Fig. 62 Structure of f fluorinated indazoles derivatives 102. | |
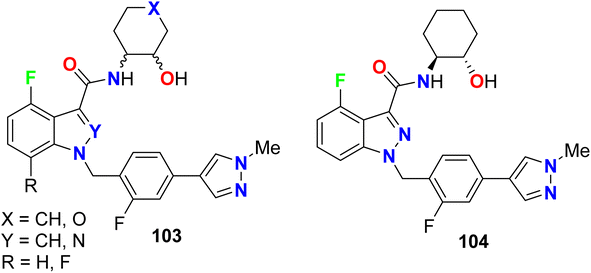
Ballard and his research group designed a series of fluorinated indazole derivatives 103–104 (Fig. 63) that were assigned as M1-PAM (muscarinic receptor positive allosteric modulators) for the treatment or prophylaxis of Alzheimer's disease. The assay disclosed that those derivatives had potent modulator activity at the acetylcholine muscarinic receptor expressed in CHO (Chinese hamster ovary) cells by measuring the intracellular calcium with a Fluorometric Imaging Plate Reader System (FLIPR). The EC50 (concentration effective in producing 50% of the maximal response) of hM1 of the fluorinated pyrazoles ranged between 0.025–0.266 μM, particularly compound 104 was the most efficient with EC50 = 0.025 μM. Thus, these compounds were assigned as possible neurogenic agents.119
 |
| | Fig. 63 Structure of f fluorinated indazoles derivatives 103–104. | |
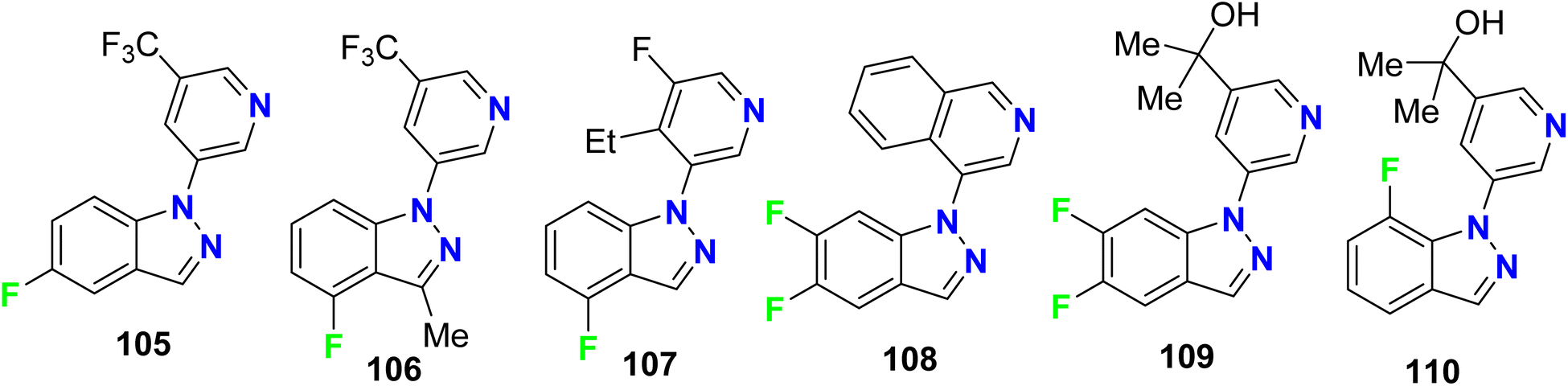
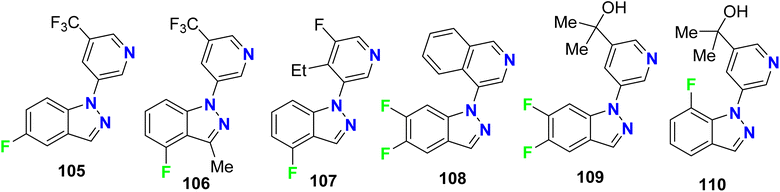
Hoyt et al. designed several fluorinated indazole hybrids 105–110 (Fig. 64) and reported them as selective inhibitors of aldosterone synthase enzyme (CYP 11B2) with little effect on steroid-11-β-hydroxylase (CYP1 1B1). The study employed V79-human cell assay and the studied compounds demonstrated potent inhibition activity of CYP 11B2 with IC50 values ranging between 2 nM and 250 nM, some compounds reached 60-fold selectivity for inhibition of CYP 11B2 better than CYP 11B1. For example, compounds 105, 106, 108–110 demonstrated inhibitions of CYP 11B2 with IC50 values of 83, 17, 1.9, 23, and 1.2 nM compared with 1911, 275, 107, 305 and 36.4 nM for of CYP 11B1 with selectivities of 23-, 16-, 56-, 13-, and 30-folds for CYP 11B2CYP 11B1.120
 |
| | Fig. 64 Structure of fluorinated indazoles derivatives 105–110. | |
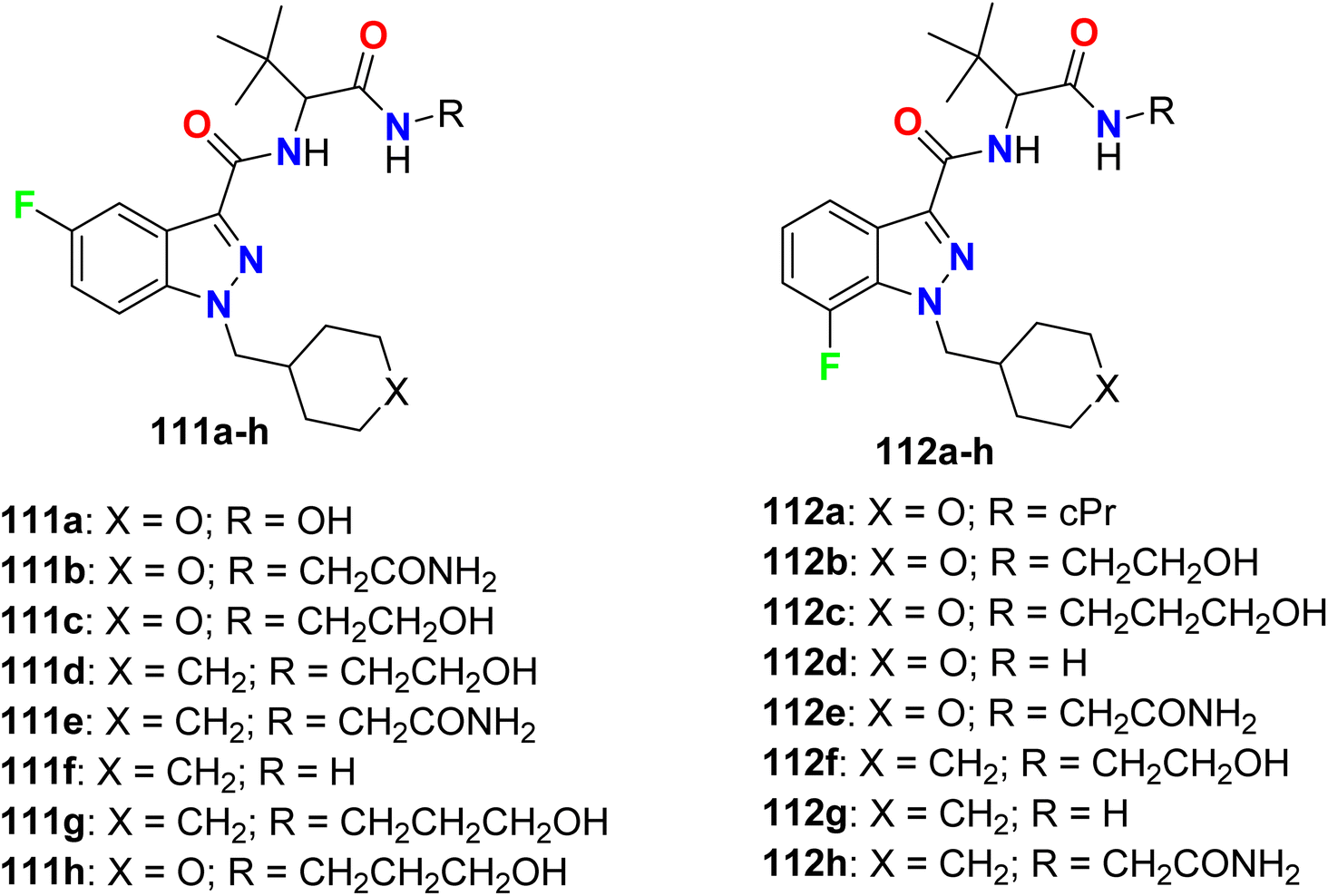
Some 5-fluoro- and 7-fluoroindazole derivatives 111a–h and 112a–h (Fig. 65) were patented as potent inhibitors of human cannabinoid receptor-1 (hCB1) receptors and their binding affinity Ki varied from 0.08–51 nM. Ki value (inhibitor constant) measure the potency of an inhibitor and it is the concentration necessary to produce half maximum inhibition. It was reported that compounds having N-cyclohexylmethylene moiety in general were more potent than those having N-pyranylmethylene moiety. In addition, the 7-fluoroindazole derivatives had better binding affinity than those of 5-fluoro isomers. The best binding affinity was recorded for compound 112f (Ki 0.087 nM).121
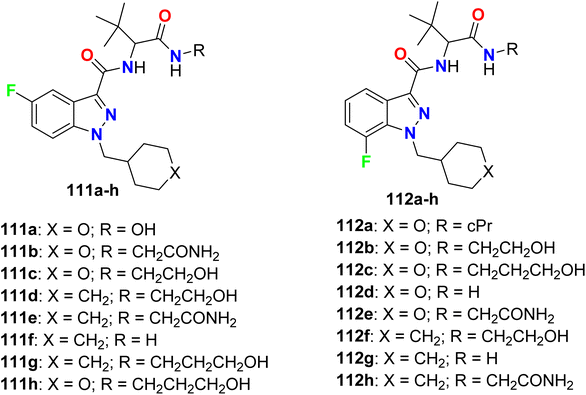 |
| | Fig. 65 Structure of 5-fluoro- and 7-fluoroindazole derivatives 111 and 112. | |
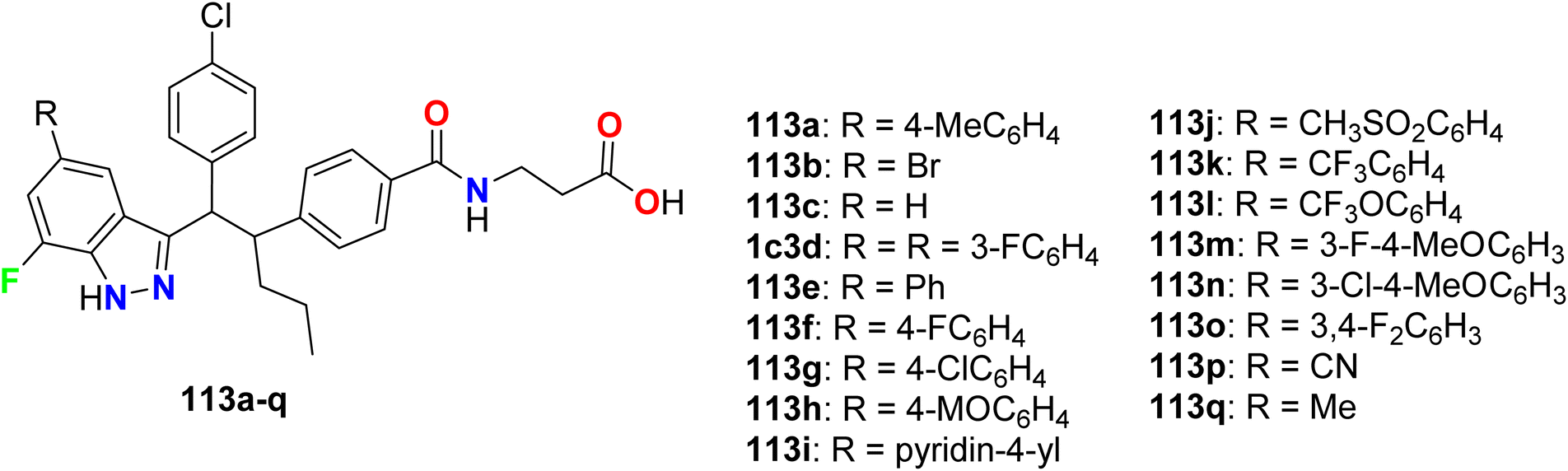
Some 7-fluoroindazole derivatives 113a–q (Fig. 66) were found to possess glucagon receptor antagonist activity to be useful for the treatment of type-2 diabetes mellitus via in vitro bioassay study. The results revealed that most of the invented fluorinated compounds had good to excellent inhibitory activity with IC50 values ranging between 1 nM and 500 nM. The most active compounds were 113a, 113d, and 113q with IC50 = 0.8–2.6 nM and these fluorinated derivatives were useful as glucagon antagonists.122
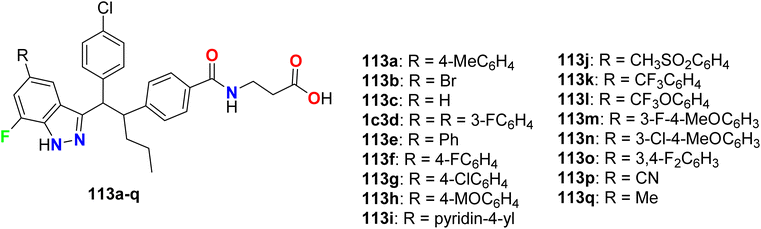 |
| | Fig. 66 Structure of 7-fluoroindazole derivatives 113. | |
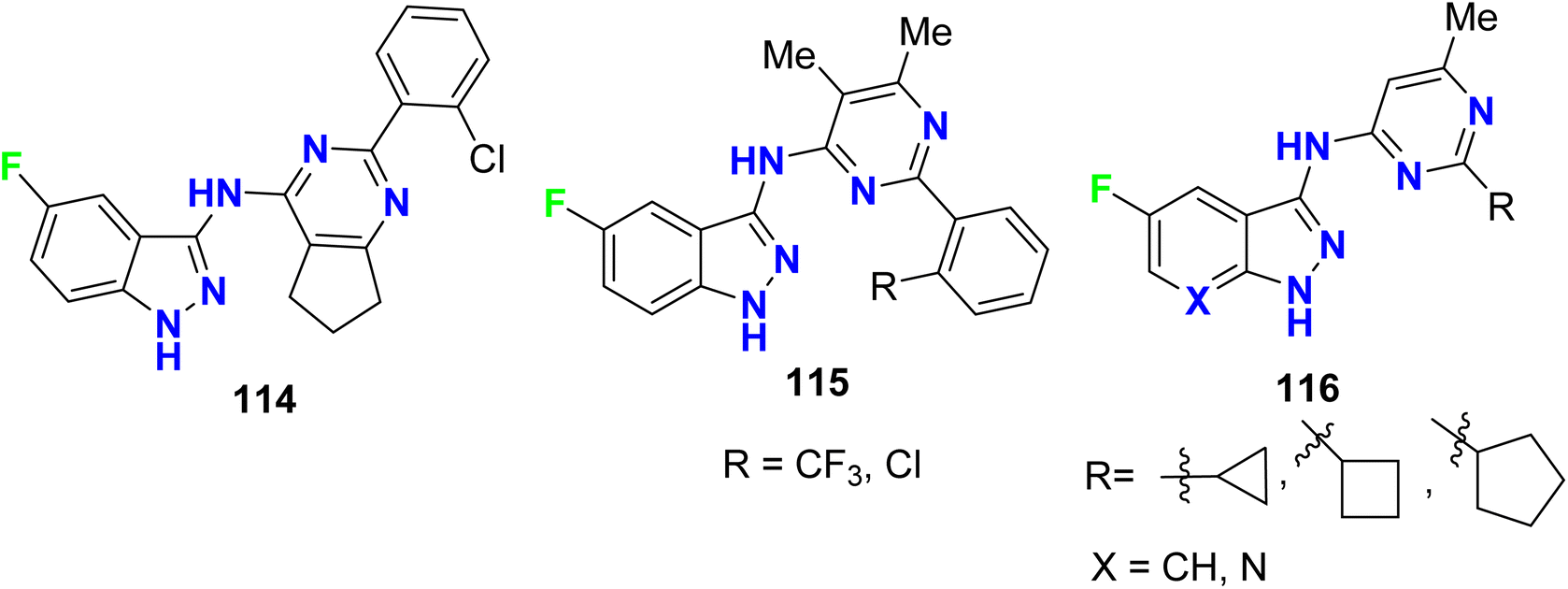
The fluoroindazole compounds 114–116 (Fig. 67) were designed and evaluated as inhibitors of glycogen synthase kinase-3 (GSK-3) using a standard coupled enzyme system. The designed compounds were screened for their ability to inhibit the phosphorylation of tyrosine (TYR) residues through the use of western blotting of Jurkat cells dosed with the reported compounds. The phosphorylation of the specific TYR residues tested were GSK3α (TYR 279) and GSK3β (TYR 216). The synthesized compounds exhibited moderate to high inhibitory activities of both GSK3α and GSK3β.123
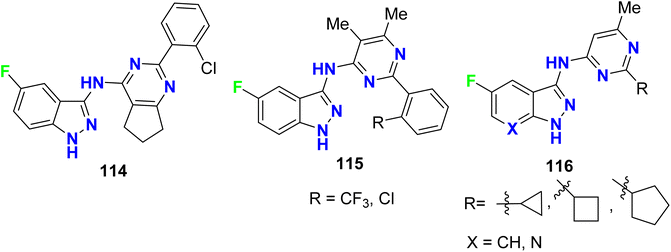 |
| | Fig. 67 Structure of fluoroindazole derivatives 114–116. | |
Claramunt et al. also reported the inhibitory action of the fluorinated indazoles 102, 117–127 against both iNOS and nNOS. The bioassay results declared that the most potent iNOS inhibitors were arranged in descending order as follows: 102f (98.7%) > 120 (95.6%) > 119a (92%) > 119b (91%)> 118b (89.1%) > 102a (83%), however, the most potent inhibitors of nNOS were 119a (90.0%) and 120 (85.4%). Most of the evaluated indazoles had better selective iNOS inhibitors than nNOS, for example, at a concentration of 1 mM, compounds, 117 (10-fold), 119b (6-fold), and 102f (2-fold) showed the highest iNOS selective inhibitors. The indazoles having a CO2H or CO2R groups, 122a–c, 123a–c and 124a–c, were moderate iNOS inhibitors and very weak nNOS ones. Among all fluorinated indazole derivatives compound 102f, had an outstanding iNOS inhibitory action, and was a promising candidate for further drug development (Fig. 68).124
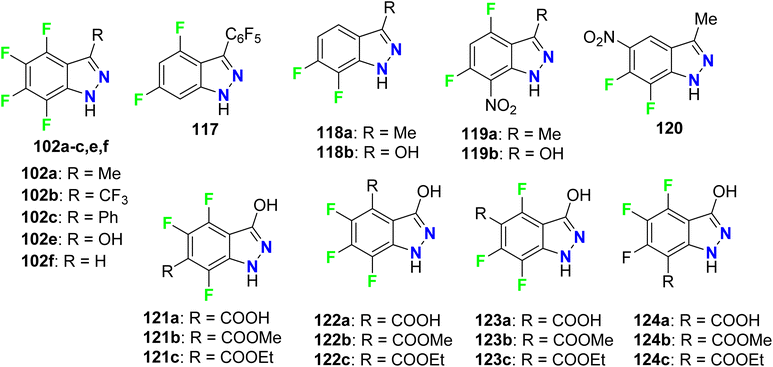 |
| | Fig. 68 Structure of fluorinated indazoles derivatives 102, 117–124. | |
4.7. Enzymatic inhibitory activity of fluorinated benzothiazoles
Diabetes mellitus type II is a metabolic disease characterized by insulin resistance and high blood glucose levels. The polyol pathway was significantly activated under hyperglycemic conditions, resulting in a series of stress conditions and, eventually, cellular damage. The first enzyme of the polyol pathway implicated in the onset of long-term diabetic complications was aldose reductase.
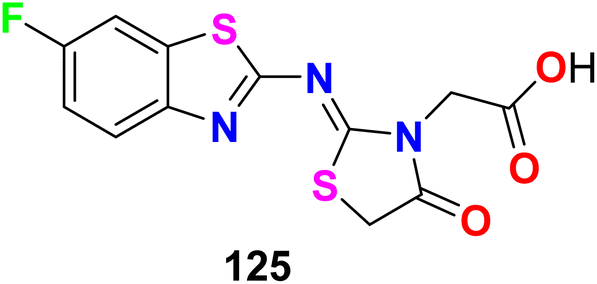
The fluorinated benzothiazole hybrid 125 was synthesized and evaluated for its inhibitory activity against aldose reductase (ALR2/ALR1) and human protein tyrosine phosphatase-1B (PTP1B) enzymes (Fig. 69). The fluorinated benzothiazole hybrid 125 exhibited aldose reductase inhibitory activity with IC50 values of 46 nM (for ALR2) and 6021 nM (for ALR1), compared with Epalrestat reference (IC50 = 227 nM) using rat lens ALR2. Interestingly, compound 125 displayed a potent selective inhibition of ALR2 with optimal selectivity index 131. Moreover, compound 125 was found to have potent protein tyrosine phosphatase 1B (PTP1B) inhibitory activity with IC50 = 58.79 μM. Thus, compound 125 was a dual ALR2/PTP1B inhibitor and had promising capability for treating diabetes mellitus. Docking study proved that compound 125 adopts two hydrogen bond (HB) interactions with OH– and SH– groups of Tyr48 and Cys298, in addition to a salt bridge interaction between the carboxylate moiety and the positively charged nicotinamide ring of NADP+.125
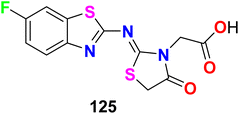 |
| | Fig. 69 Structure of fluorinated benzothiazole derivatives 125. | |
5. Miscellaneous bioactivity of fluorinated heterocycles
5.1. Antimalarial activity of fluorinated pyrazoles
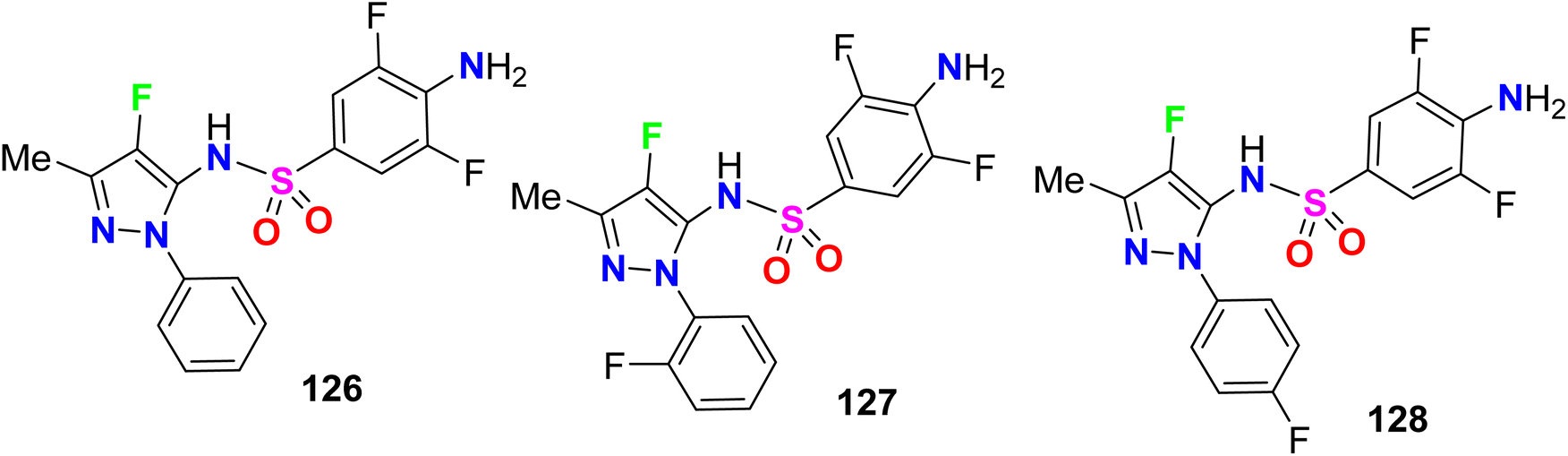
Some fluorinated pyrazole sulfonamides were synthesized 126–128 (Fig. 70) to determine their potential ligands with selective affinities for human and plasmodium dihydrofolate reductase (DHFR). With AutoDock Vina and the Molecular Operating Environment (MOE), binding affinities and interactions of the fluorinated sulfonamides were benchmarked with antimalarial drugs. The binding affinity scores from both programs identified three sulfonamides with strong interactions that were comparable to or greater than the current antimalarial drugs. Compound 128 displayed strong hydrogen bonding interactions with quadruple mutated P. falciparum DHFR active site residues and an estimated binding energy of −7.6 kcal mol−1. The presented fluorinated heterocyclic sulfonamides were promising pharmacophores and competitors to current folate pathway inhibitors. The molecular docking simulations supported the fluorinated sulfonamide ligands as promising molecules that could be used to target critical enzymes like DHFR in the Plasmodium folate pathway. The estimated binding affinity of fluorinated sulfonamides docked with human and P. falciparum DHFR isoforms were comparable to the benchmark antimalarial drugs like artesunate and stronger than sulfadoxine and pyrimethamine. The fluorinated pyrazolesulfonamide derivatives 126–128 matched the binding affinity of artesunate to within 0.25 kcal mol−1. The fluorinated sulfonamide framework for drug design had the potential to mitigate the impacts caused by DHFR mutations. The fluorinated sulfonamides modeled also had good drug-like characteristics based on the Lipinski rule of 5, and thence became important pharmacophores for the development of potent antimalarial drugs.126
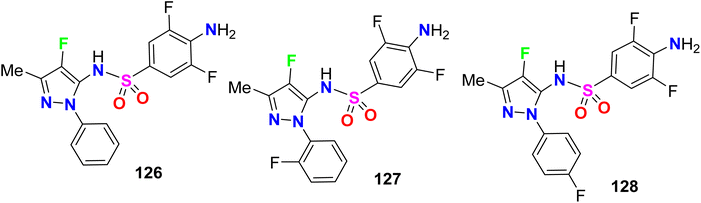 |
| | Fig. 70 Structure of fluorinated pyrazole sulfonamides derivatives 126–128. | |
5.2. Anticoagulant activity of fluorinated pyrazoles
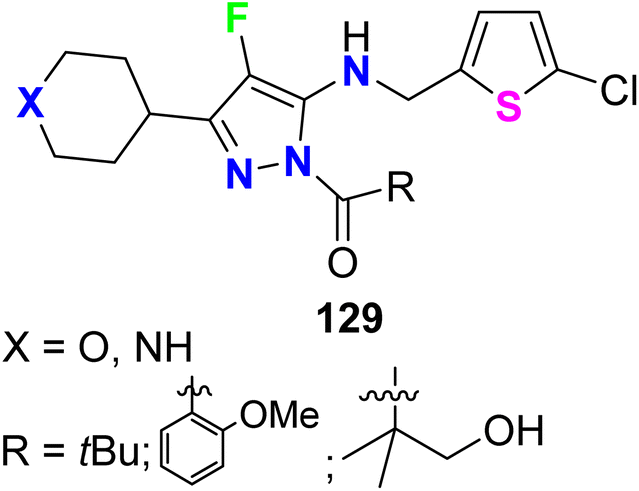
Recently, a series of N-acylpyrazole derivatives 129 (Fig. 71) was prepared with fluorine substitution at position-4 of the pyrazole ring and investigated their inhibition against thrombin in vitro to be useful as effective anticoagulants. It was reported that the efficacy in the thrombin generation assay (TGA) increases with increasing the potency in the thrombin enzyme assay, however, the fluorinated pyrazoles 129 consistently imparted decreased stability against all plasma species studied. All the fluorinated pyrazole derivatives 129 showed promising thrombin inhibition with IC50 values ranging from 0.004 μM to 0.06 μM with ETP (TGA) varied between 2.3 μM and 46 μM. Thus, the 4-fluoropyrazoles 129 were recommended as ideally safe and effective anticoagulants.127
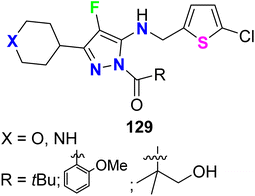 |
| | Fig. 71 Structure of N-acylpyrazole derivatives 129. | |
5.3. Antipsychotic activity of fluorinated indoles
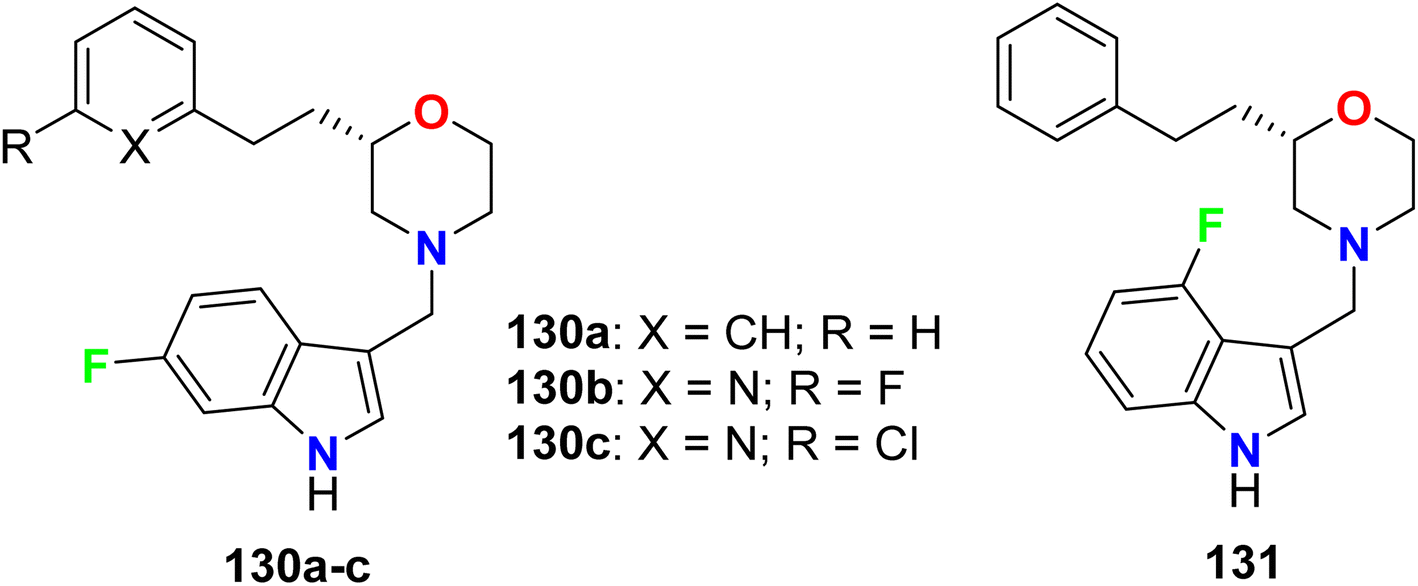
Four fluoroindole derivatives 130a–c and 131 (Fig. 72) were synthesized and investigated as potent and selective dopamine receptor 4 (D4R) antagonists for the treatment of the diseases of central nervous system. All compounds displayed high activities in the nanomolar scale, and the 6-fluoroindole derivatives 130a and 130c were the most active against D4 with Ki values of 5.2 nM and 3.3 nM, also had IC50 values of 18.9 and 11.9 nM, respectively. On the other hand, the 6-fluoroindole derivatives 130a were also active and selective against the dopamine receptors D2L and D2S with 78% and 76% inhibitions, respectively.128
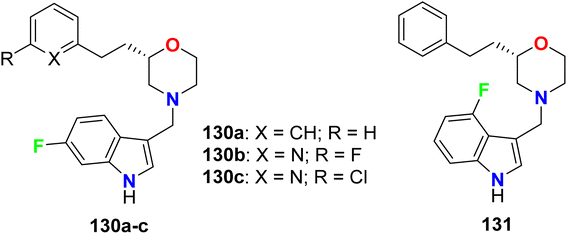 |
| | Fig. 72 Structure of fluoroindole derivatives 130a–c and 131. | |
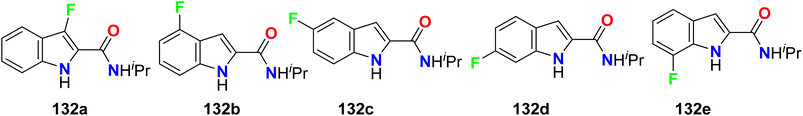
Five fluorinated indol-2-carboxamide derivatives 132a–e (Fig. 73) were assigned as NAM (negative allosteric modulators) of D2R (dopamine D2 receptor). Specifically, the 5-fluoroindole derivative 132c (KB = 91 μM) exhibited reduced affinity. In comparison, the 4-fluoro analogue 132b (KB = 18 μM, α = 0.07) and 7-fluoro analogue 132e (KB = 18 μM, α = 0.11) (where KB is the equilibrium dissociation constant of the allosteric ligand and α is the binding cooperativity parameter between the orthosteric and allosteric ligand), both displayed enhanced binding affinity and displayed high negative cooperativity with the dopamine D2R.129
 |
| | Fig. 73 Structure of fluorinated indol-2-carboxamide derivatives 132a–e. | |
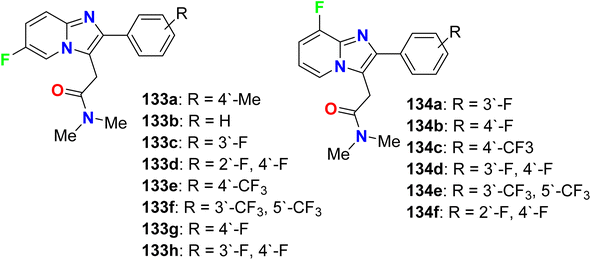
Two series of (6-fluoro- and 8-fluoro-) imidazo[1,2-a]pyridine derivatives 133-h and 134a–f (Fig. 74) were reported as a gamma-aminobutyric acid antagonist (GABA-A) receptors, to be useful as potential antipsychotic agents. These fluorinated derivatives showed high affinity and positive allosteric modulator properties at the GABA-A receptor and lack of hepatotoxicity when compared with zolpidem as a reference drug. Using rat brain tissue, the fluorinated compounds (133a–h and 134a–f) were tested for affinity for the benzodiazepine site of the GABA-A receptor by measuring the competitive displacement of [3H]-flunitrazepam. SAR study revealed that 6-fluorinated derivatives were much more effective than their 8-fluorinated analogues. It was also found that compound 133a had the highest affinity with (Ki = 25 nM), with 1.8-fold higher affinity compared with the reference drug zolpidem (Ki = 44 nM). Other 6-fluorinated derivatives 133g (Ki = 55.6 nM), 133h (Ki = 57.5 nM), and 133e (Ki = 68.0 nM) showed acceptable affinities. A notable decrease in the affinity was observed after the introduction of another fluorine atom at ortho-position (133d), or its replacement into meta-position (133c). The presence of two sterically trifluoromethyl groups (133f) caused a complete loss of activity. In addition, compound 133g demonstrated good antipsychotic-like activity in rats with EC50 = 408 nM compared with zolpidem drug (EC50 = 240 nM).130
 |
| | Fig. 74 Structure of 6-fluoro- and 8-fluoro-imidazo[1,2-a]pyridine derivatives 133 and 134. | |
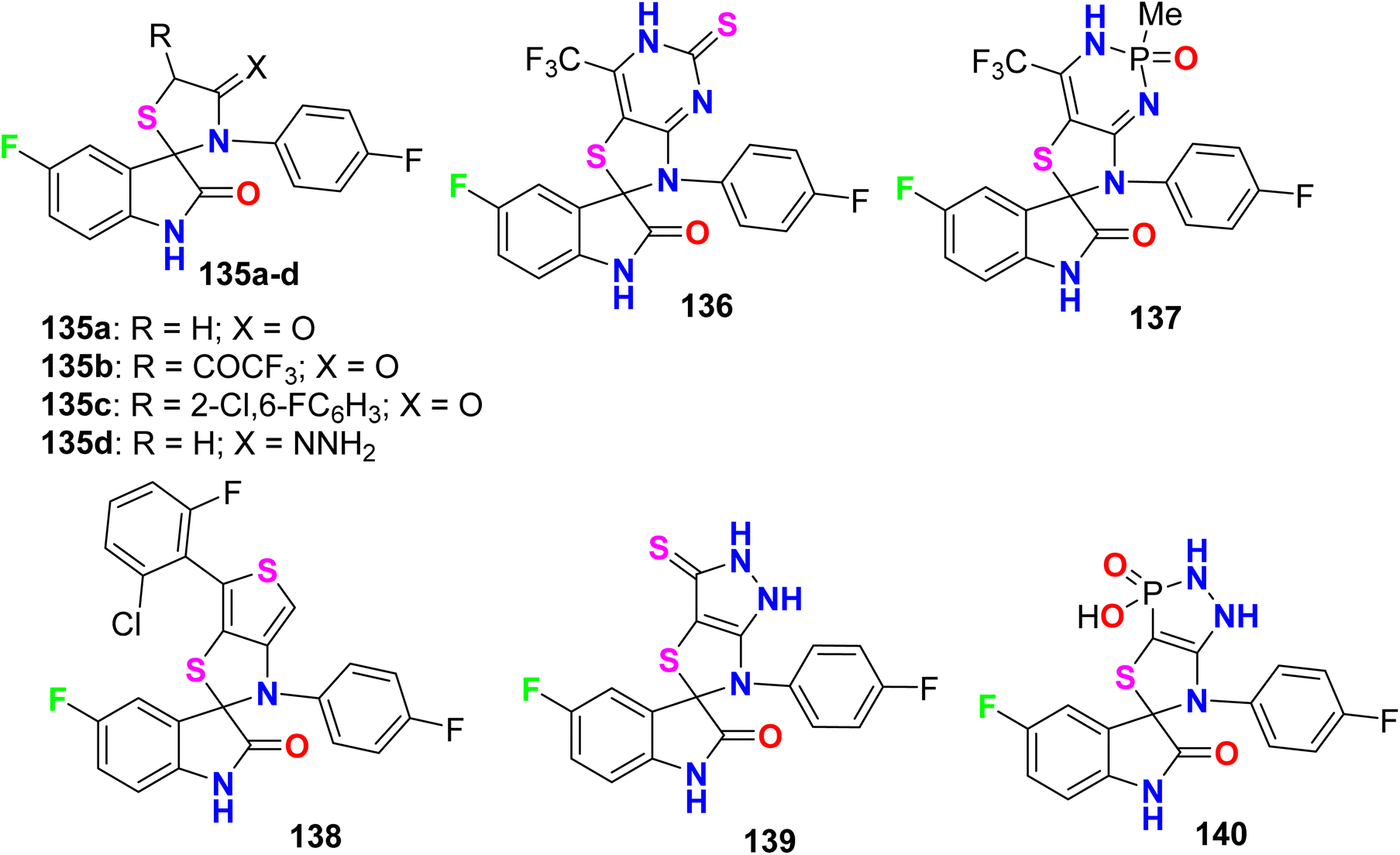
5.4. Antioxidant activity of fluorinated indolinones
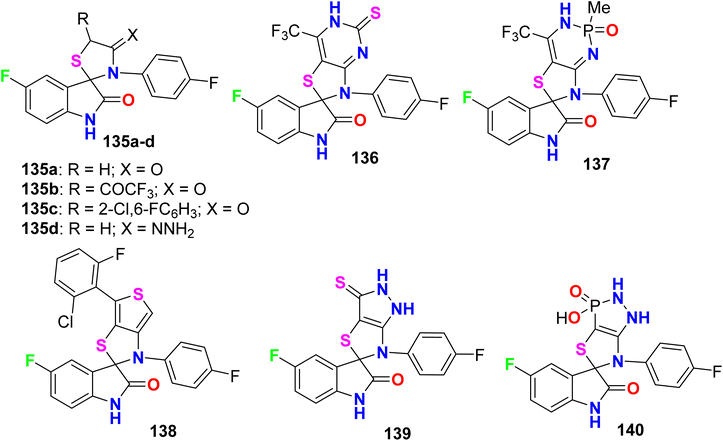
Some fluorinated spiro[istin-thiazolidine] derivatives 135–140 (Fig. 75) were synthesized and evaluated for their antioxidant activity by the 1,1-diphenyl-2-picrylhydrazyl (DPPH) protocol. The results of scavenging the stable DPPH radical were calculated at 150, 300, and 450 μM concentrations, where the fluorinated compounds scavenged 49–78% of the DPPH radicals. Compounds 136 and 137 proved to have potent antioxidant activities of 77.45% and 77.78% compared with ascorbic acid (55.2%), at 450 μM concentration, respectively.131
 |
| | Fig. 75 Structure of fluorinated spiro[istin-thiazolidine] derivatives 135–140. | |
5.5. Antiprotozoal activity of fluorinated benzothiazoles
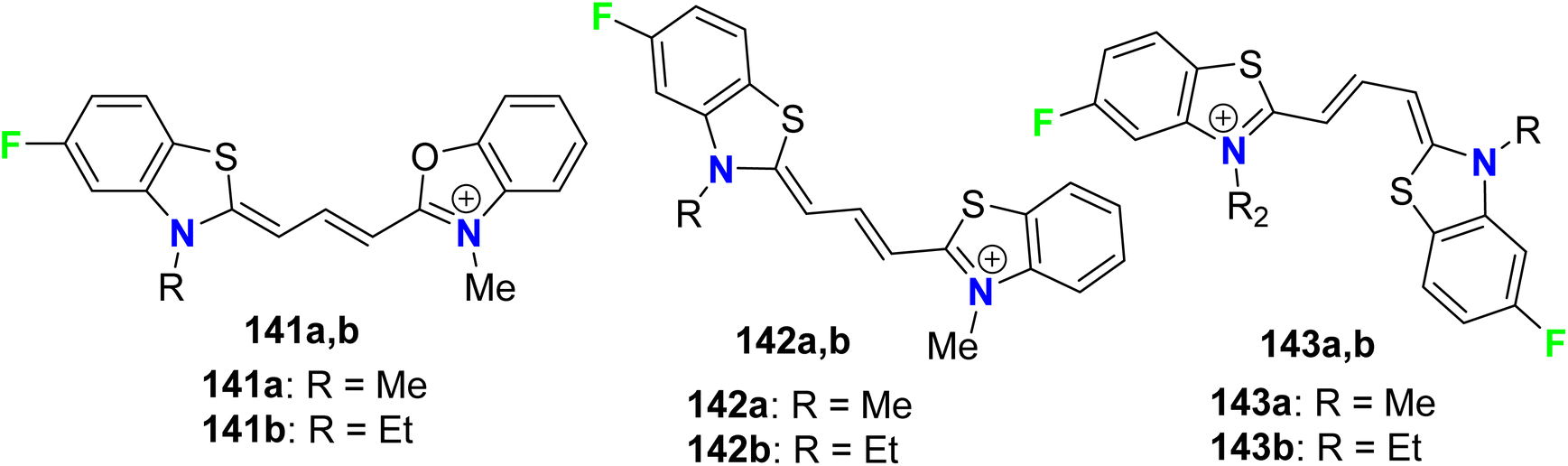
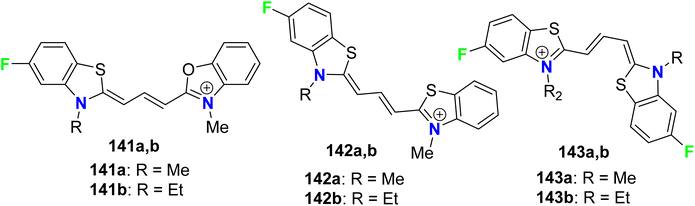
Some fluorinated cyanine dyes having benzoxazolyl or benzothiazolyl moieties 141a, 141b–143a, 143b were synthesized and screened for their in vitro antiprotozoal activities (Fig. 76). The in vitro bioassay was conducted using P. falciparum (K1), Trypanosoma cruzi, T. brucei rhodesiense, and L. donovani. The antimalarial activities of the fluorinated cyanines incorporating benzoxazolyl and benzothiazolyl groups 141a, 141b, and 142a, 142b against P. falciparum (K1) demonstrated high antimalarial activity with IC50 values ranged between 2–7 nM with selectivity index (SI) varied between 32–586, where both compounds 143a, 143b both had the most antiprotozoal activity with IC50 = 2 nM.132
 |
| | Fig. 76 Structure of fluorinated cyanine dyes benzoxazolyl or benzothiazolyl 141a, 141b–143a, 143b. | |
5.6. Histamine-H3 receptor activity of fluorinated benzimidazoles
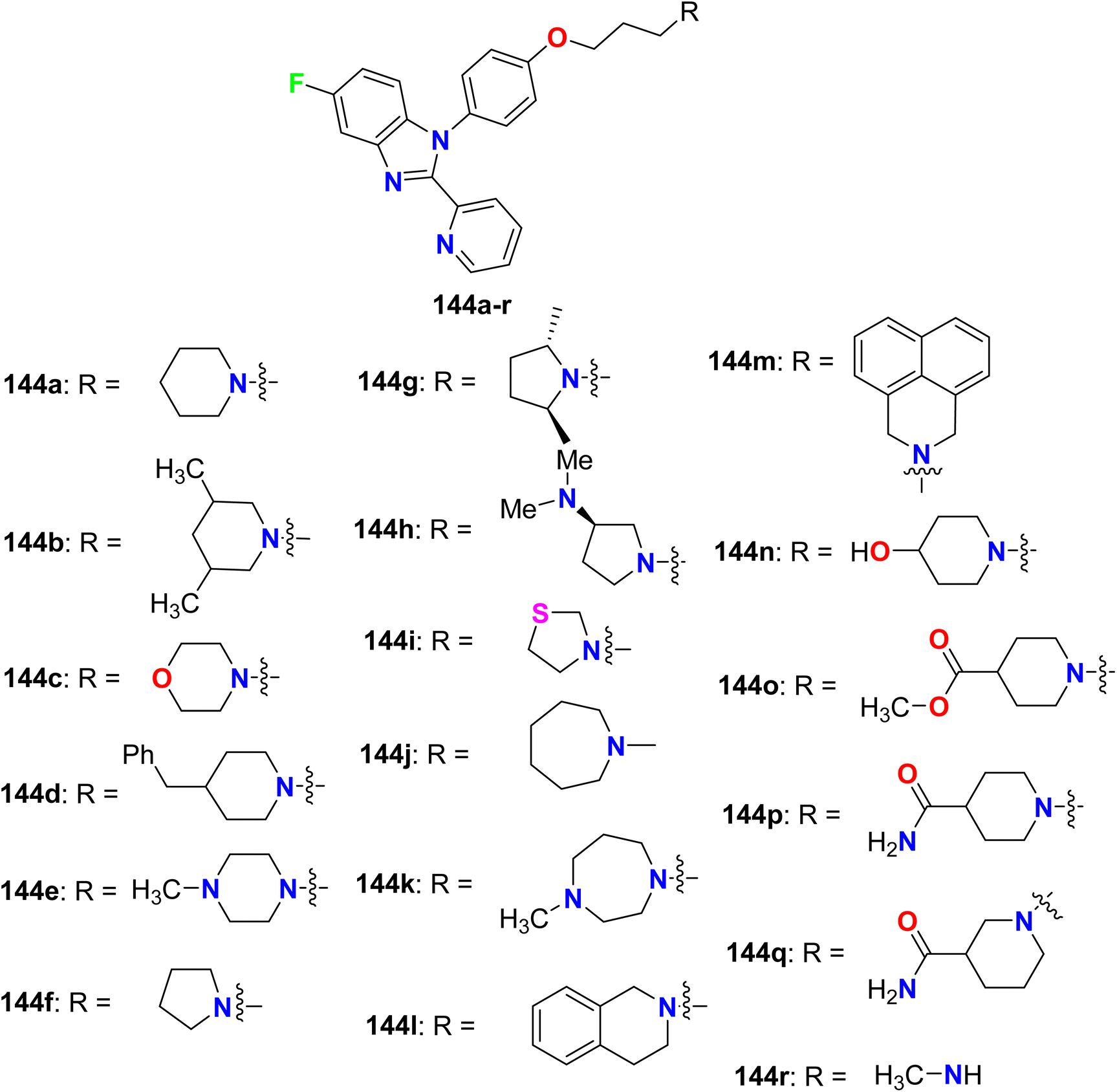
1,2-Disubstituted-5-fluorobenzimidazole derivatives 144a–r (Fig. 77) were reported by Aslanian et al. and were evaluated for histamine H3 receptor antagonists. The results showed that most of the derivatives had good to excellent H3 binding affinity, particularly compounds 144a (having piperidinyl group) and 144f (having pyrrolidinyl group) were identified as the most potent H3 antagonists with Ki value of 1.2 nM for both. Due to the excellent activity of compound 144a at the human H3 receptor, it was further profiled and tested against the mouse H3 receptor with Ki of 1.8 nM. In addition, the in vitro human cAMP assay with an AlphaScreen cAMP assay kit, and showed functional activity with 0.1 nM. Furthermore, compound 144a presented a reasonable oral pharmacokinetic profile in a high throughput rat pharmacokinetic assay.133
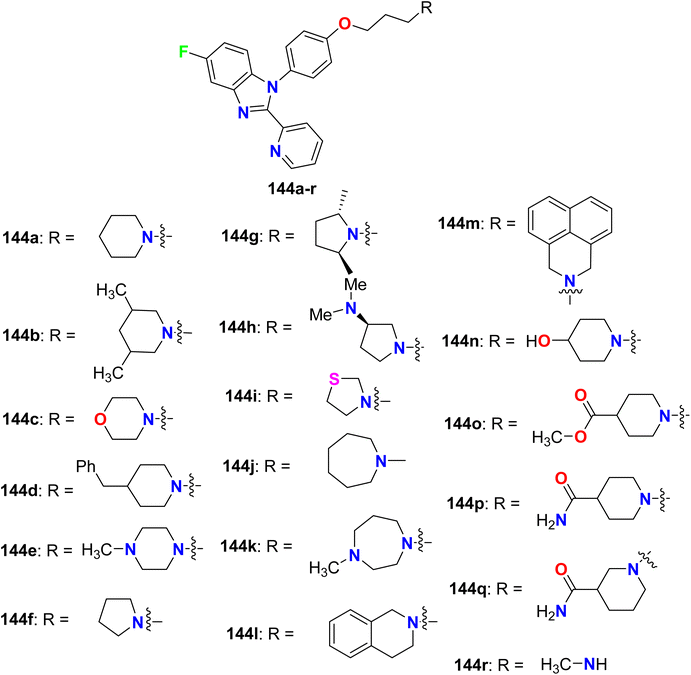 |
| | Fig. 77 Structure of 1,2-disubstituted-5-fluorobenzimidazole derivatives 144a–r. | |
5.7. Serotonin receptor affinity of fluorinated indoles and indazoles
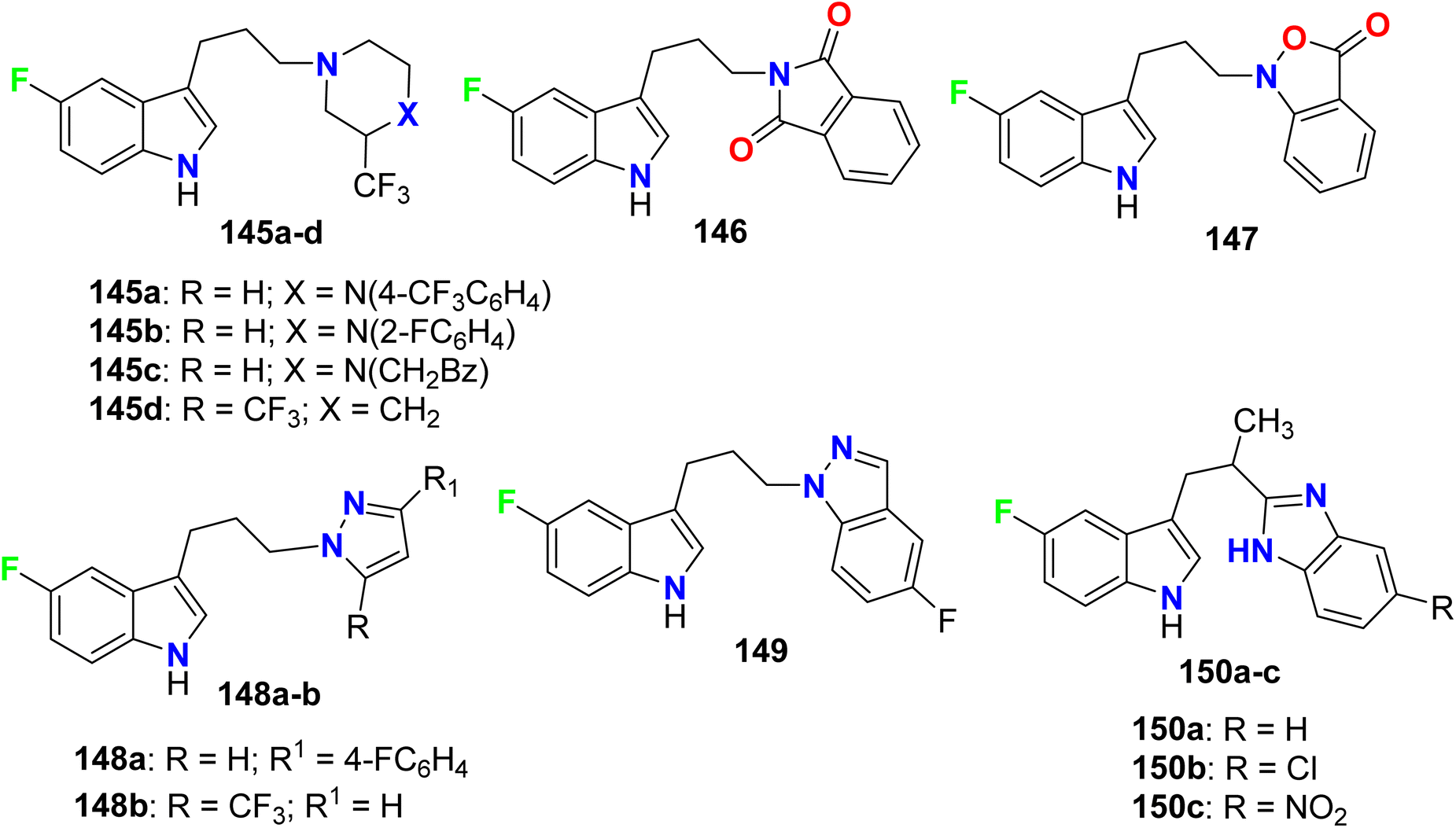
A series of 5-fluoroindole derivatives 145–150 (Fig. 78). were synthesized by Ojeda-Gómez et al. and investigated their in vitro affinities for the serotonin transporter (SERT). Among the synthesized compounds, the derivatives 145a–c presented good binding affinities (Ki = 33.0, 48.0, and 17 nM, respectively). The rest compounds showed moderate or no binding affinities.134
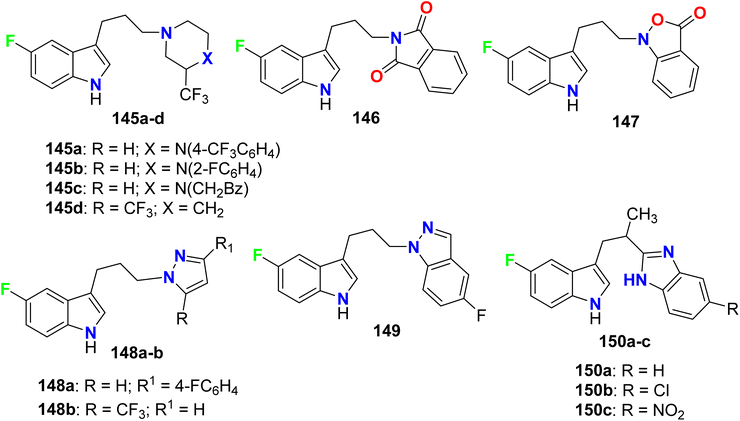 |
| | Fig. 78 Structure of 5-fluoroindole derivatives 145–150. | |
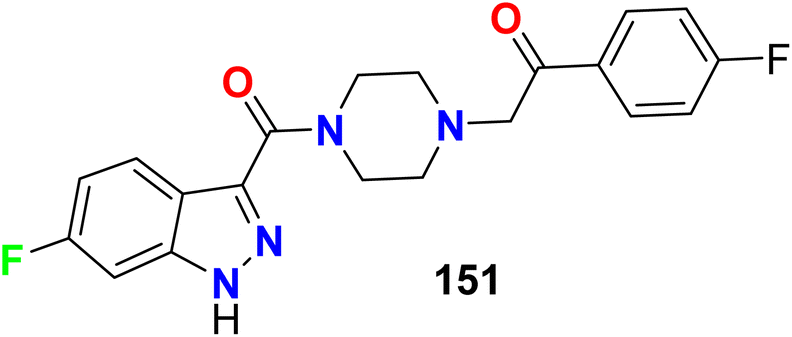
The 5-HT receptors were divided into seven main groups (5-HT1–7). The 6-fluoroindazole derivative 151 was reported as a modulator of the human serotonin (5-HT2A) receptor, radio-ligand binding assay of compound 151 for human 5-HT2A serotonin receptor was conducted using the 5-HT2 agonist [125I]DOI (2,5-dimethoxy-4-iodo-phenylisopropylamine) as radio-ligand. Compound 151 showed DOI binding affinity with an IC50 value of 2.3 nM (Fig. 79).135
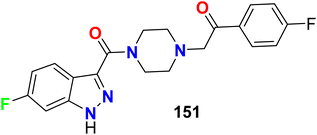 |
| | Fig. 79 Structure of 6-fluoroindazole derivative 151. | |
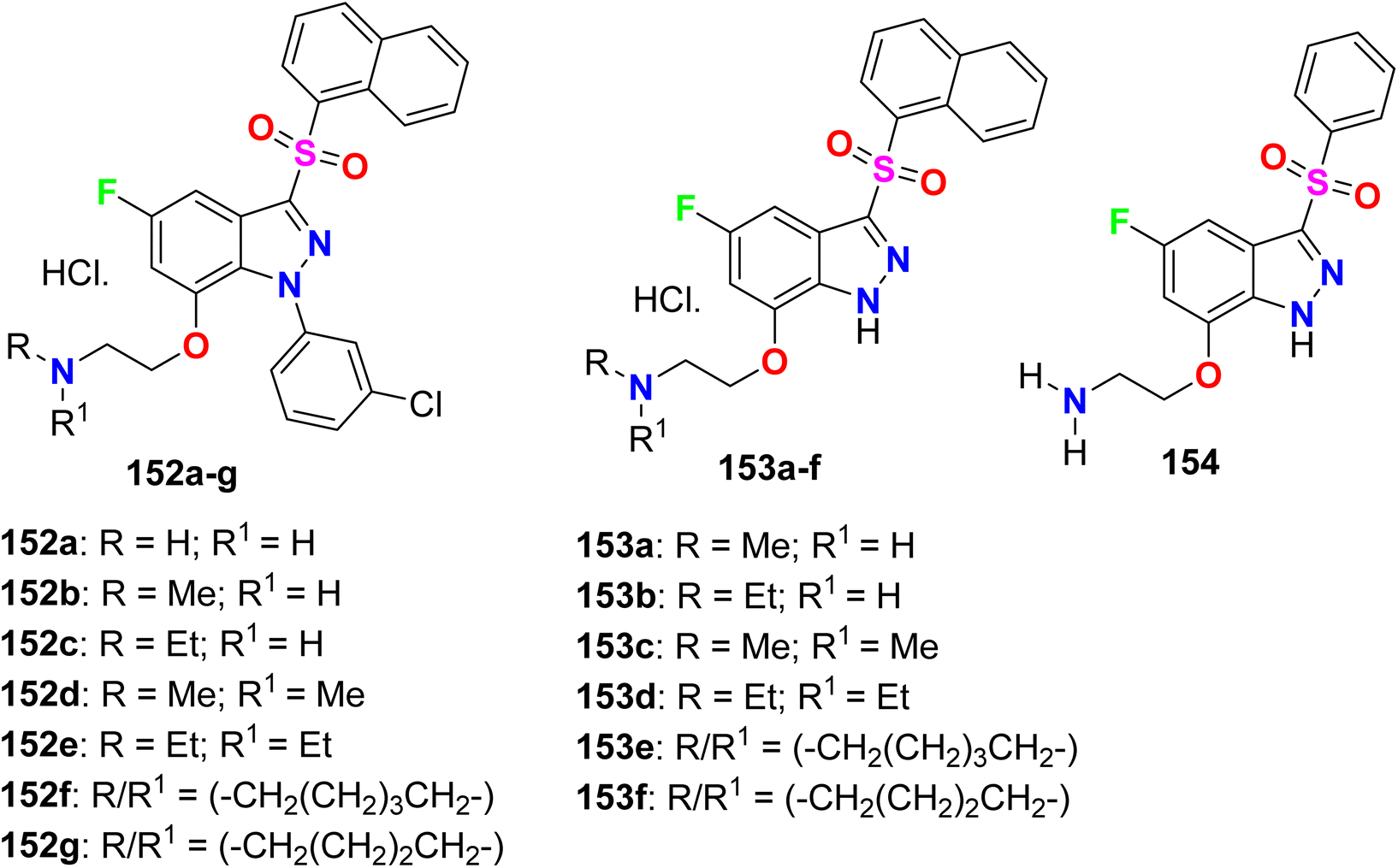
A series of fluorinated indazole derivatives 152–154 (Fig. 80) having naphthylsulfonyl group were designed and their binding affinity for 5-HT6 (5-hydroxytryptamine-6) receptor, for the treatment of central nervous system disorders, was patented. Compounds 152–154 demonstrated significant 5-HT6 affinity and selectivity. Interestingly, all compounds showed promising binding affinity (Ki) in a nanomolar scale ranging between 0.01 nM ∼45 nM, particularly compounds 153a, 153b, 153e, and 153f presented the highest binding affinity for 5-HT6 receptor with Ki values of 0.01–10 nM.136
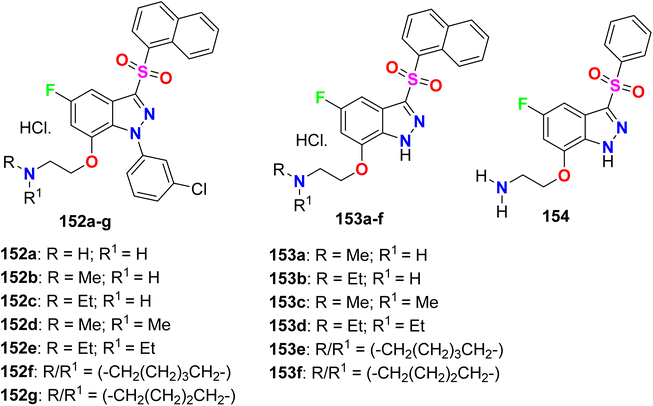 |
| | Fig. 80 Structure of fluorinated indazole derivatives 152–154. | |
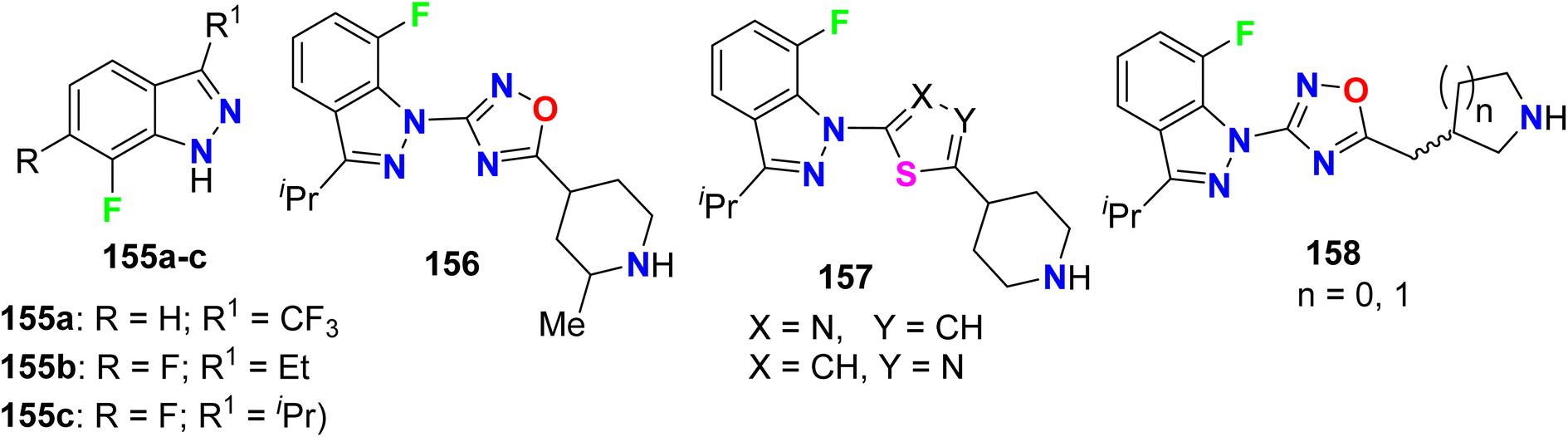
Several derivatives of fluorinated indazole 155–158 were invented and their agonist activity for the 5-HT4 (5-hydroxytryptamine-4) receptor was screened out (Fig. 81). All compounds 155–158 possessed excellent 5-HT4 receptor agonist activity in the nanomolar scale with IC50 values <40 nM and most of the studied compounds had EC50 ranging between 3.7 nM and 53.0 nM. In addition, the intrinsic activities (IA) of the tested fluorinated indazoles varied between 26–125%, and the best result was for compound 155a (IA = 125%).137
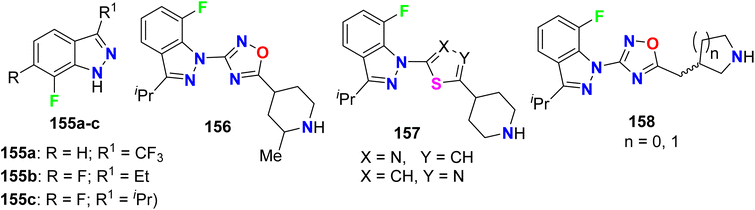 |
| | Fig. 81 Structure of fluorinated indazole derivatives 155–158. | |
5.8. Chemokine receptor activity of fluorinated pyrazoles and indazoles
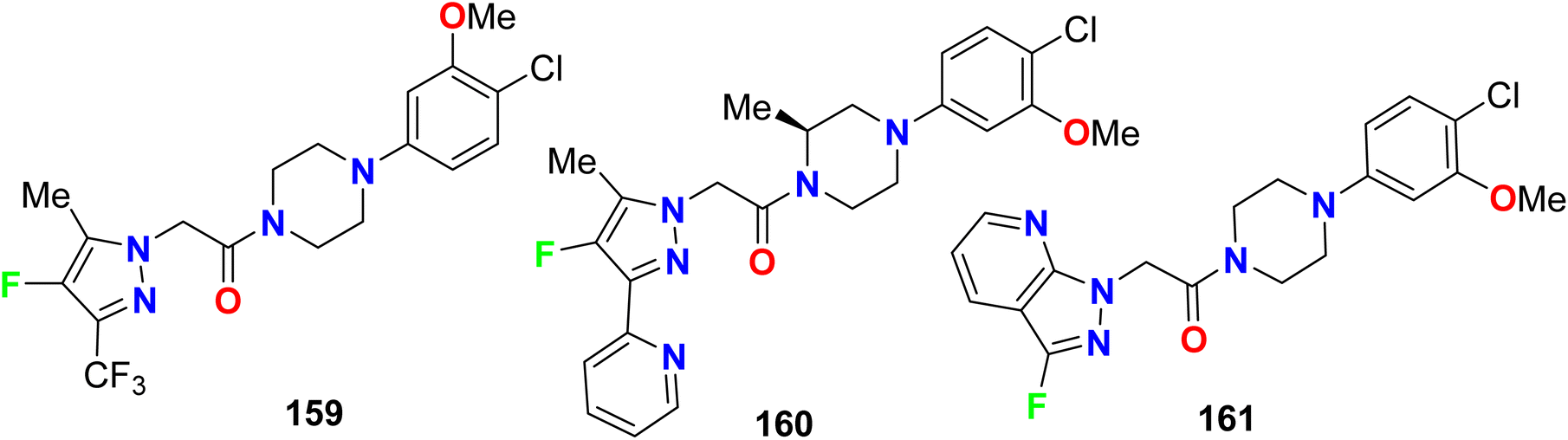
The piperazine-based 4-fluoropyrazole molecular hybrids 159–161 (Fig. 82), were synthesized and patented as antagonists of C–C chemokine receptor-1 (CCR1) and had in vivo anti-inflammatory activity. The chemotaxis assay showed good CCR1 antagonistic activity of both compounds 159 and 160 with IC50 < 0.5 μM but compound 161 had an IC50 value of 1.0 μM.138–141
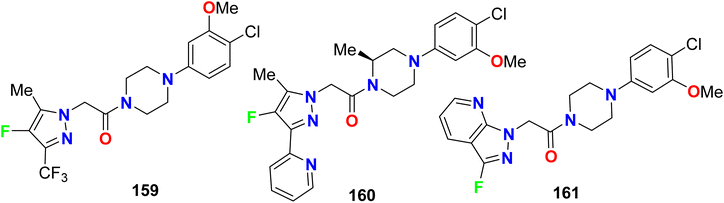 |
| | Fig. 82 Structure of piperazine-based 4-fluoropyrazole derivatives 159–161. | |
The 6-fluorininated indazole scaffolds 162–163 (Fig. 83) were synthesized and evaluated as allosteric human CCR4 antagonists. The [35S]GTPγS (i.e. guanosine 5′-O-[gamma-thio]triphosphate) binding assay, that measures the level of G-protein activation following agonist occupation of GPCR (G-protein-coupled receptor), was studied in vitro. Compounds 162 and 163 demonstrated human CCR4 antagonistic activity in the range: 6.0 ≤ GTPγS pIC50 < 8.1.142,143
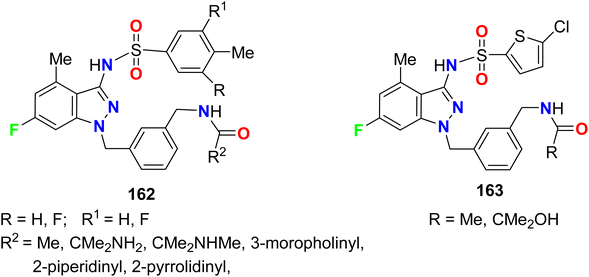 |
| | Fig. 83 Structure of piperazine-based 4-fluoropyrazole derivatives 159–161. | |
5.9. Prostaglandin-D2 receptor activity of fluorinated indazoles
The 6-fluoro-1-pyrrolinyl-indazole derivatives 164–166 (Fig. 84) were described and examined as antagonists of the Prostaglandin D2 (PGD2) receptor. The in vitro binding assay of human CRTH2 (chemoattractant receptor-homologous molecule expressed on T-helper type-2 cells) was measured, where activation of CRTh2 receptors occurs via prostaglandin D2 (PGD2). The results showed that most of the tested compounds displayed the high inhibitory activity of CRTH2 with Ki values <100 nM, especially compound 166 presented a Ki value of 2.8 nM for the CRTH2 receptor.144,145
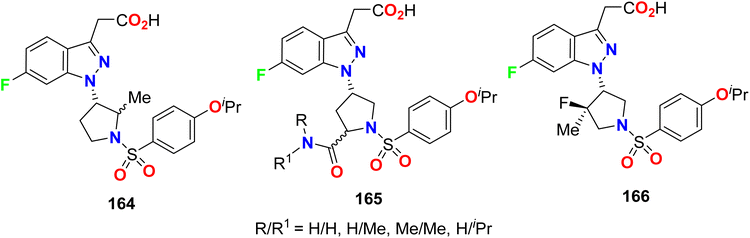 |
| | Fig. 84 Structure of 6-fluoro-1-pyrrolinyl-indazole derivatives 164–166. | |
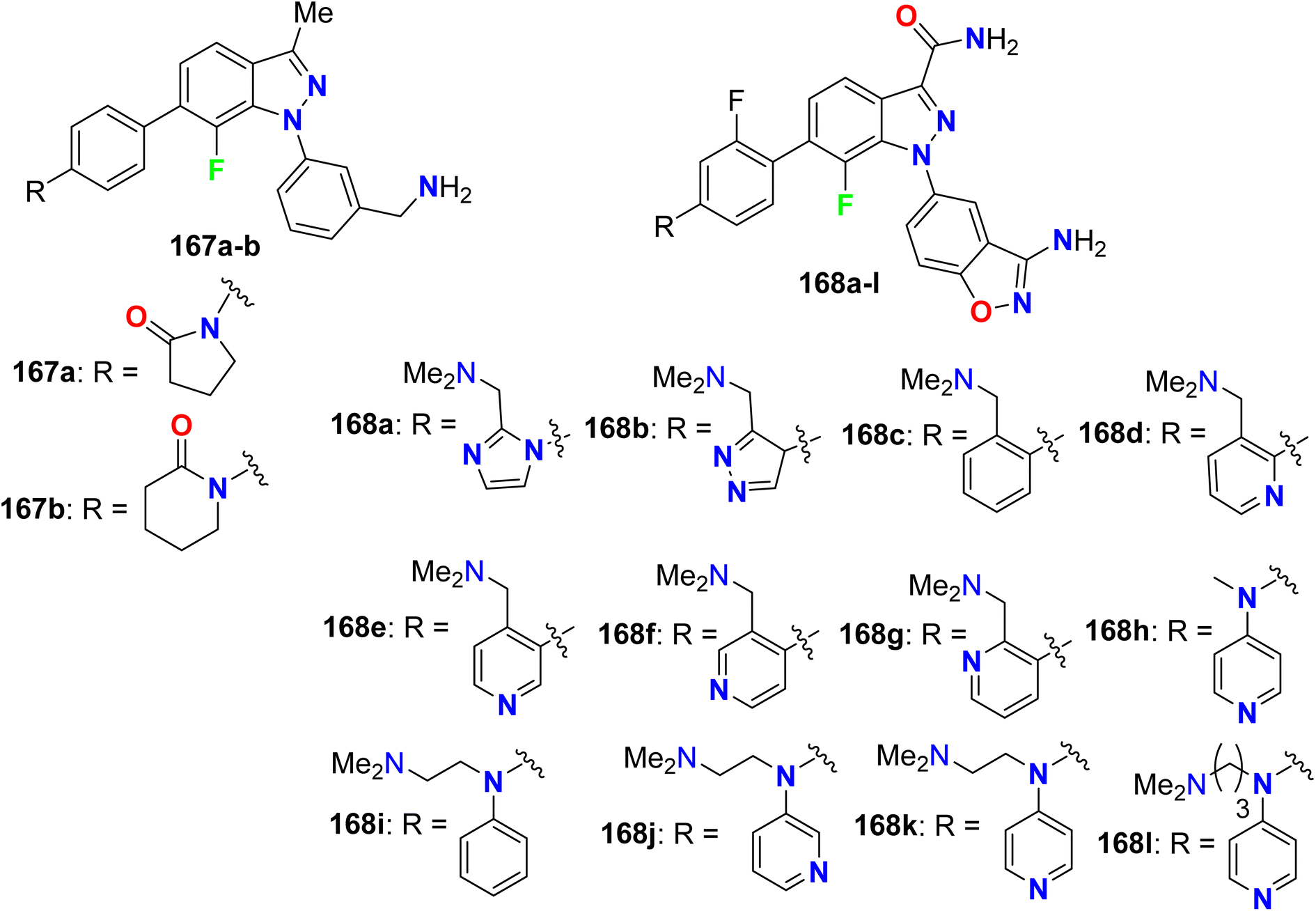
5.10. Xa-factor inhibitory activity of fluorinated indazoles
A series of 7-fluoroindazole derivatives 167a–b and 168a–l were described as potent and selective inhibitors of factor Xa, that were useful as anticoagulant agents (Fig. 85). The 7-fluoroindazoles 167a–b were highly active against factor Xa, where their potencies (fXa Ki) were 223 and 124 nM compared with their non-fluorinated analogues Ki > 14400 and 6850 nM, respectively, and their selectivities of versus trypsin were about 4-fold and 10-fold, respectively. In addition, most of the 7-fluoroindazole series 168a–l disclosed significant factor Xa potency (Ki) ranged between 1.4–15 nM, particularly compound 168b was the most potent where the presence of 1H-pyrazole-moiety caused a great enhancement of the factor Xa Ki to be 1.4 nM. The SAR study revealed that the factor Xa inhibitory potencies of 7-fluoroindazole derivatives 167a and 167bc were about 60-fold greater (ΔΔG ≈ 2.4 kcal mol−1) compared with their corresponding non-fluorinated indazoles. The structure of a factor Xa cocrystal having 7-fluoroindazoles proved the hydrogen bonding between the 7-fluoro atom and the N–H of Gly216 in the peptide backbone.146
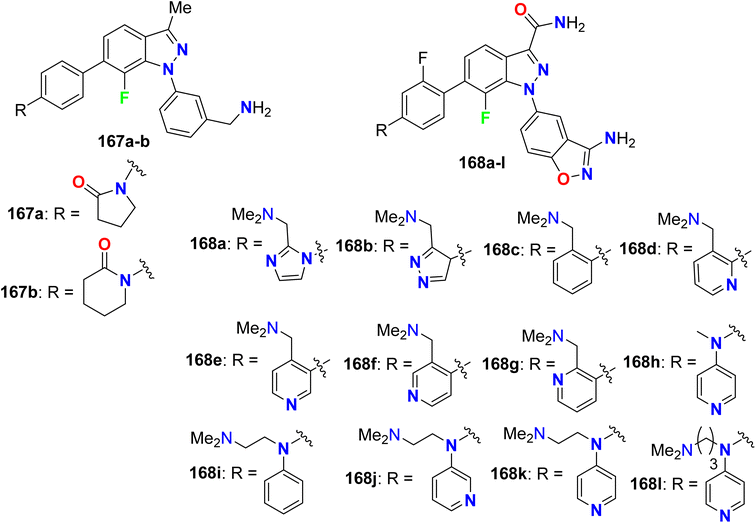 |
| | Fig. 85 Structure of 7-fluoroindazole derivatives 167a–b and 168a–l. | |
5.11. PBR inhibition activity of fluorinated imidazo[1,2-a]pyridine
Synthesis of some fluorinated imidazo[1,2-a]pyridine heterocycles 169–170 (Fig. 86) were patented and evaluated for their binding affinity towards PBR (peripheral benzodiazepine receptor), where PBR is a mitochondrial protein participated in the cholesterol transport regulation from the outer to the inner mitochondrial membrane. The in vitro binding studies disclosed that compounds 169a, 169b, and 170 had good inhibition potencies of for the PBR binding site with IC50 values of 38 nM, 32 nm, and 800 nM, respectively.147
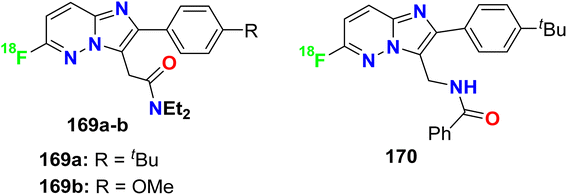 |
| | Fig. 86 Structure of fluorinated imidazo[1,2-a]pyridine heterocycles 169–170. | |
6. Conclusions
In this review article, we collected all reports covering the biological activities of directly ring-fluorinated five-membered heterocycles and their benzo-fused systems. This review covered antiviral, anti-inflammatory, enzymatic inhibitory, antimalarial, anticoagulant, antipsychotic, antioxidant, antiprotozoal, histamine-H3 receptor, serotonin receptor, chemokine receptor, prostaglandin-D2 receptor and PBR inhibition activities of the fluorinated heterocycles. Most of the reported fluorinated derivatives demonstrated potent antiviral activity such as anti-HIV-1, anti-HBV, hepatitis C virus (HCV), and inhibition of CVB4 virus with IC50 values better than the reference drugs. On the other side, the fluorinated azoles showed good anti-inflammatory inhibitory potency with good stability and selectivity for HNE over other serine proteases, with IC50 values of 0.1 μM. In addition, some fluorinated pyrazole-based heterocycles exhibited high inhibitory potency with IC50 values varied between 1 nM to 0.1 μM against PDK1 (pyruvate dehydrogenase kinase (1) for treating inflammatory diseases. In the case of enzymatic inhibitory activity, the tested derivatives of fluorinated azoles showed potent activity with IC50 that exceeded that of the reference drug. It is important to refer to some derivatives of fluorinated pyrazoles that were recommended as ideally safe and effective anticoagulants due to their promising thrombin inhibition with IC50 values ranging from 0.004 μM to 0.06 μM with ETP (TGA) varied between 2.3 μM and 46 μM. As a result, it is not surprising that fluorine is found in more than 20% of all medications on the market. The current review article is expected to be a valuable resource for researchers interested in medicinal chemistry and pharmaceutics looking into the possibility of synthesizing drug-like small fluorine molecules.
Data availability
This is a review article and there is no available data.
Conflicts of interest
The authors declare no conflicts of interest.
References
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- E. Henary, S. Casa, T. L. Dost, J. C. Sloop and M. Henary, Pharm., 2024, 17, 281 CAS.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- C. Zhang, ACS Omega, 2022, 7, 18206–18212 CrossRef CAS PubMed.
- J. Fried and E. F. Sabo, J. Am. Chem. Soc., 1954, 76, 1455–1456 CrossRef CAS.
- D. O'Hagan and J. Fluor, Chem, 2010, 131, 1071–1081 Search PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- T. T. Simur, T. Ye, Y. J. Yu, F. L. Zhang and Y. F. Wang, Chin. Chem. Lett., 2022, 33, 1193–1198 CrossRef CAS.
- Fluorine in Heterocyclic Chemistry, ed. V. Nenajdenko, Springer, 2014, vol. 1, 2 Search PubMed.
- Fluorinated Heterocyclic Compounds: Synthesis, Chemistry and Applications, ed. V. A. Petrov, Wiley, New York, 2009 Search PubMed.
- Fluorinated Heterocycles, ed. A. Gakh and K. L. Kirk, ACS, Washington, 2009 Search PubMed.
- V. P. Reddy, Organofluorine compounds in biology and medicine, Chapter 9, Organofluorine Compounds as Anticancer Agents, Elsevier, 2015, pp. 265–300 Search PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- H. J. Bohm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Muller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- X. G. Hu and L. Hunter, Beilstein J. Org. Chem., 2013, 9, 2696–2708 CrossRef PubMed.
- N. Sheikhi, M. Bahraminejad, M. Saeedi and S. S. Mirfazli, Eur. J. Med. Chem., 2023, 260, 115758 CrossRef CAS PubMed.
- W. K. Hagman, J. Med. Chem., 2008, 51, 4359–4369 CrossRef PubMed.
- J. A. Olsen, D. W. Banner, P. Seiler, B. Wagner, T. Tschopp, U. Obst-Sander, M. Kansy, K. Muller and F. Diederich, ChemBioChem, 2004, 5, 666–675 CrossRef CAS PubMed.
- J. A. Olsen, D. W. Banner, P. Seiler, U. O. Sander, A. D'Arcy, M. Stihle, K. Muller and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 2507–2511 CrossRef CAS PubMed.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- P. Zhou, J. Zou, F. Tian and Z. Shang, J. Chem. Inf. Model., 2009, 49, 2344–2355 CrossRef CAS PubMed.
- Y. X. Du and X. P. Chen, Clin. Pharmacol. Ther., 2020, 108, 242–247 CrossRef CAS PubMed.
- B. K. Kim, J. Oh, S. Y. Kwon, W. Hyeok and H. Lee, J. Hepatol., 2009, 51, 829–834 CrossRef PubMed.
- P. Fernandez-Calotti, R. Gamberale, M. Costas, J. S. Avalos, J. Geffner and M. Giordano, Int. Immunopharmacol., 2006, 6, 715–723 CrossRef CAS PubMed.
- Q. Meng, N. Liu, B. Huang, P. Zhan and X. Liu, Expert Opin. Ther. Pat., 2015, 25, 1477–1486 CrossRef CAS PubMed.
- D. C. Liotta and G. R. Painter, Acc. Chem. Res., 2016, 49, 2091–2098 CrossRef CAS PubMed.
- D. Ding, S. Xu, X. Zhang, X. Jiang, S. Cocklin, A. Dick, P. Zhan and X. Liu, Expert Opin. Drug Discovery, 2023, 18, 5–12 CrossRef CAS PubMed.
- World Health Organization, "International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 62", WHO Drug Information, 2009, 23, 3, p. 261 Search PubMed.
- R. J. Hamilton, Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition, Jones & Bartlett Learning, 2015, pp. 434–435 Search PubMed.
- L. H. Wang, J. Begley, R. L. St. Claire III, J. Harris, C. Wakeford and F. S. Rousseau, AIDS Res. Hum. Retroviruses, 2004, 20, 1173–1182 CrossRef CAS PubMed.
- J. Y. Feng, J. Shi, R. F. Schinazi and K. S. Anderson, FASEB J., 1999, 13, 1511–1517 CrossRef CAS PubMed.
- N. A. Meanwell, O. B. Wallace, H. Fang, H. Wang, M. Deshpande, T. Wang, Z. Yin, Z. Zhang, B. C. Pearce, J. James, K. S. Yeung, Z. Qiu, J. J. K. Wright, Z. Yang, L. Zadjura, D. L. Tweedie, S. Yeola, F. Zhao, S. Ranadive, B. A. Robinson, Y. F. Gong, H. G. Wang, W. S. Blair, P. Y. Shi and R. J. Colonno, Bioorg. Med. Chem. Lett., 2009, 19, 1977–1981 CrossRef CAS PubMed.
- J. P. Begue and D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, Wiley, Hoboken. 2008 Search PubMed.
- E. A. Kitas, G. Galley, R. Jakob-Roetne, A. Flohr, W. Wostl, H. Mauser, A. M. Alker, C. Czech, L. Ozmen, P. David-Pierson, D. Reinhardt and H. Jacobsen, Bioorg. Med. Chem. Lett., 2008, 18, 304–308 CrossRef CAS PubMed.
- P. L. Kalar, S. Agrawal, S. Kushwaha, S. Gayen and K. Das, Curr. Org. Chem., 2023, 27, 190–205 CrossRef CAS.
- R. Chatterjee, R. Dandela and J. Fluor, Chem, 2023, 268, 110133 CAS.
- R. Szpera, D. F. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew Chem. Int. Ed. Engl., 2019, 58, 14824–14848 CrossRef CAS PubMed.
- M. D. Markus, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- K. M. Dawood, Tetrahedron, 2004, 60, 1435–1451 CrossRef CAS.
- K. M. Dawood and T. Fuchigami, J. Org. Chem., 2004, 69, 5302–5306 CrossRef CAS PubMed.
- K. M. Dawood and T. Fuchigami, J. Org. Chem., 2001, 66, 7691–7695 CrossRef CAS PubMed.
- K. M. Dawood, H. Ishii and T. Fuchigami, J. Org. Chem., 2001, 66, 7030–7034 CrossRef CAS PubMed.
- T. Fuchigami, S. Higashiya, Y. Hou and K. M. Dawood, Rev. Heteroatom Chem., 1999, 19, 67–78 Search PubMed.
- M. G. Campbell and T. Ritter, Org. Process Res. Dev., 2014, 18, 474–480 CrossRef CAS PubMed.
- A. A. Abbas, T. A. Farghaly and K. M. Dawood, RSC Adv., 2024, 14, 19752–19779 RSC.
- A. A. Abbas and K. M. Dawood, Expert Opin. Drug Discovery, 2022, 17, 1357–1376 CrossRef CAS PubMed.
- K. M. Dawood, T. A. Farghaly and M. A. Raslan, Curr. Org. Chem., 2020, 24, 1943–1975 CrossRef CAS.
- K. M. Dawood and A. A. Abbas, Expert Opin. Ther. Pat., 2020, 30, 695–714 CrossRef CAS PubMed.
- H. Behbehani, K. M. Dawood and T. A. Farghaly, Expert Opin. Ther. Pat., 2018, 28, 5–29 CrossRef CAS PubMed.
- D. M. Mohamed, N. A. Kheder, M. Sharaky, M. S. Nafie, K. M. Dawood and A. A. Abbas, RSC Adv., 2024, 14, 24992–25006 RSC.
- J. Wu, W. Yu, L. Fu, W. He, Y. Wang, B. Chai, C. Song and J. Chang, Eur. J. Med. Chem., 2013, 63, 739–745 CrossRef CAS PubMed.
- Y. Liu, Y. Peng, J. Lu, J. Wang, H. Ma, C. Song, B. Liu, Y. Qiao, W. Yu, J. Wu and J. Chang, Eur. J. Med. Chem., 2018, 143, 137–149 CrossRef CAS PubMed.
- D. B. Smith, G. Kalayanov, C. Sund, A. Winqvist, T. Maltseva, V. J. P. Leveque, S. Rajyaguru, S. L. Pogam, I. Najera, K. Benkestock, X. X. Zhou, A. C. Kaiser, H. Maag, N. Cammack, J. A. Martin, S. Swallow, N. G. Johansson, K. Klumpp and M. Smith, J. Med. Chem., 2009, 52, 2971–2978 CrossRef CAS PubMed.
- M. I. Kharitonova, K. V. Antonov, I. V. Fateev, M. Y. Berzina, A. L. Kaushin, A. S. Paramonov, S. K. Kotovskaya, V. L. Andronova, I. D. Konstantinova, G. A. Galegov, V. N. Charushin and A. I. Miroshnikov, Synthesis, 2017, 49, 1043–1052 CrossRef CAS.
- S. Martínez-Montero, S. Fernández, Y. S. Sanghvi, E. A. Theodorakis, M. A. Detorio, T. R. Mcbrayer, T. Whitaker, R. F. Schinazi, V. Gotor and M. Ferrero, Bioorg. Med. Chem., 2012, 20, 6885–6893 CrossRef PubMed.
- P. Das, S. Boone, D. Mitra, L. Turner, R. Tandon, D. Raucher and A. T. Hamme, RSC Adv., 2020, 10, 30223–30237 RSC.
- P. Jones, D. C. Pryde and T. D. Tran, WO Pat., PCT WO 2007093901, 2007 Search PubMed.
- J. D. Oslob, R. S. McDowell, R. Johnson, H. Yang, M. Evanchik, C. A. Zaharia, H. Cai and L. W. Hu, WO Pat., PCT WO 2014008197, 2014 Search PubMed.
- L. R. Roberts, J. Bryans, K. Conlon, G. McMurray, A. Stobie and G. A. Whitlock, Bioorg. Med. Chem. Lett., 2008, 18, 6437–6440 CrossRef CAS PubMed.
- L. H. Jones, G. Allan, O. Barba, C. Burt, R. Corbau, T. Dupont, T. Knöchel, S. Irving, D. S. Middleton, C. E. Mowbray, M. Perros, H. Ringrose, N. A. Swain, R. Webster, M. Westby and C. Phillips, J. Med. Chem., 2009, 52, 1219–1223 CrossRef CAS PubMed.
- Y. Gai, Y. S. Or, Z. Wang, WO Pat., PCT WO 2009076173A2, 2009 Search PubMed.
- F. Piscitelli, A. Coluccia, A. Brancale, G. La Regina, A. Sansone, C. Giordano, J. Balzarini, G. Maga, S. Zanoli, A. Samuele, R. Cirilli, F. L. Torre, A. Lavecchia, E. Novellino and R. Silvestri, J. Med. Chem., 2009, 52, 1922–1934 CrossRef CAS PubMed.
- G. La Regina, A. Coluccia, A. Brancale, F. Piscitelli, V. Gatti, G. Maga, A. Samuele, C. Pannecouque, D. Schols, J. Balzarini, E. Novellino and E. Silvestri, J. Med. Chem., 2011, 54, 1587–1598 CrossRef CAS PubMed.
- G. La Regina, A. Coluccia, F. Piscitelli, A. Bergamini, A. Sinistro, A. Cavazza, G. Maga, A. Samuele, S. Zanoli, E. Novellino and M. Artico, J. Med. Chem., 2007, 50, 5034–5038 CrossRef CAS PubMed.
- K. S. Yeung, Z. Qiu, Q. Xue, H. Fang, Z. Yang, L. Zadjura, C. J. D'Arienzo, B. J. Eggers, K. Riccardi, P. Y. Shi, Y. F. Gong, M. R. Browning, Q. Gao, S. Hancel, K. Santone, P. F. Lin, N. A. Meanwell and J. K. Kadow, Bioorg. Med. Chem. Lett., 2013, 23, 198–202 CrossRef CAS PubMed.
- K. S. Yeung, Z. Qiu, Z. Yin, A. Trehan, H. Fang, B. Pearce, Z. Yang, L. Zadjura, C. J. D'Arienzo, K. Riccardi, P. Y. Shi, T. P. Spicer, Y. F. Gong, M. R. Browning, S. Hancel, K. Santone, J. Barker, T. Coulter, P. F. Lin, N. A. Meanwell and J. F. Kadow, Med. Chem. Lett., 2013, 23, 203–208 CrossRef CAS PubMed.
- K. S. Yeung, Z. Qiu, Z. Yang, L. Zadjura, C. J. D'Arienzo, M. R. Browning, S. Hansel, X. S. Huang, B. J. Eggers, K. Riccardi, P. F. Lin, N. A. Meanwell and J. F. Kadow, Bioorg. Med. Chem. Lett., 2013, 23, 209–212 CrossRef CAS PubMed.
- N. Zhang, X. Zhang, J. Zhu, A. Turpoff, G. Chen, C. Morrill, S. Huang, W. Lennox, R. Kakarla, R. Liu, C. Li, H. Ren, N. Almstead, S. Venkatraman, F. G. Njoroge, Z. Gu, V. Clausen, J. Graci, S. P. Jung, Y. Zheng, J. M. Colacino, F. Lahser, J. Sheedy, A. Mollin, M. Weetall, A. Nomeir and G. M. Karp, J. Med. Chem., 2014, 57, 2121–2136 CrossRef CAS PubMed.
- G. Cihan-Üstündağ, E. Gürsoy, L. Naesens, N. Ulusoy-Güzeldemirci and G. Çapan, Bioorg. Med. Chem., 2016, 24, 240–246 CrossRef PubMed.
- J. T. Randolph, C. A. Flentge, P. Donner, T. W. Rockway, S. V. Patel, L. Nelson, D. K. Hutchinson, R. Mondal, N. Mistry, T. Reisch and T. Dekhtyar, Bioorg. Med. Chem. Lett., 2016, 26, 5462–5467 CrossRef CAS PubMed.
- D. Dressen, A. W. Garofalo, J. Hawkinson, D. Hom, J. Jagodzinski, J. L. Marugg, M. L. Neitzel, M. A. Pleiss, B. Szoke, J. S. Tung and D. W. Wone, J. Med. Chem., 2017, 50, 5161–5167 CrossRef PubMed.
- C. Gibson, T. Tradler, K. Schnatbaum, J. Pfeifer, E. Locardi, D. Scharn, M. Paschke, U. Reimer, U. Richter, G. Hummel, U. Reineke, WO Pat., PCT WO 2008116620, 2008 Search PubMed.
- C. Gibson, K. Schnatbaum, T. Tradler, J. Pfeifer, D. Scharn, U. Reimer, U. Richter, G. Hummel, U. Reineke, E. Locardi, M. Paschke, WO Pat., PCT WO 2010031589, 2010 Search PubMed.
- C. Bolea, WO Pat., PCT WO 2010079239, 2010 Search PubMed.
- M. Sakagami, H. Hashizume, S. Tanaka, T. Okuno, H. Yari, K. Tonogaki, N. Kouyma, WO Pat., PCT WO 2009131096, 2009 Search PubMed.
- G. D. Glick, A. R. Hurd, C. B. Taylor, C. A. Vanhuis, WO Pat., PCT WO 2012078874, 2012 Search PubMed.
- W. Mingchun and L. Qingyi, CN Pat., CN 116655536A, 2023 Search PubMed.
- L. Rooney, A. Vidal, A. M. D'Souza, N. Devereux, B. Masick, V. Boissel, R. West, V. Head, R. Stringer, J. Lao and M. J. Petrus, J. Med. Chem., 2014, 57, 5129–5140 CrossRef CAS PubMed.
- J. Nan, J. Liu, G. Lin, S. Zhang, A. Xia, P. Zhou, Y. Zhou, J. Zhang, J. Zhao, S. Zhang, C. Huang, Y. Wang, Q. Hu, J. Chen, M. Xiang, X. Yang and S. Yang, J. Med. Chem., 2023, 66, 3460–3483 CrossRef CAS PubMed.
- L. Crocetti, I. A. Schepetkin, A. Cilibrizzi, A. Graziano, C. Vergelli, D. Giomi, A. I. Khlebnikov, M. T. Quinn and M. P. Giovannoni, J. Med. Chem., 2013, 56, 6259–6272 CrossRef CAS PubMed.
- H. Fujiwara, S. Mizumoto, Y. Kubo, H. Nakada, S. Hagiwara, Y. Baba, T. Tamura, H. Kuniyoshi, T. Masuko and M. Yamamoto, JP Pat., JP 2013136535, 2013 Search PubMed.
- F. Padilla, N. Bhagirath, S. Chen, E. Chiao, D. M. Goldstein, J. C. Hermann, J. Hsu, J. J. Kennedy-Smith, A. Kuglstatter, C. Liao, W. Liu, L. E. Lowrie, K. C. Luk, S. M. Lynch, J. Menke, L. Niu, T. D. Owens, C. O-Yang, A. Railkar, R. C. Schoenfeld, M. Slade, S. Steiner, Y. C. Tan, A. G. Villasenor, C. Wang, J. Wanner, W. Xie, D. Xu, X. Zhang, M. Zhou and M. C. Lucas, J. Med. Chem., 2013, 56, 1677–1692 CrossRef CAS PubMed.
- A. R. Hurd, D. J. Skalitzky, C. B. Taylor, P. L. Toogood and C. A. Van Huis, WO Pat., PCT WO 2013185045, 2013 Search PubMed.
- L. N. Casillas, S. J. Chakravorty, A. K. Charnley, P. Eidam, P. A. Haile, T. V. Hughes, J. U. Jeong, J. Kang, S. A. Lakdawala, L. K. Leister, R. W. Marquis, N. A. Miller, D. J. Price, C. L. Sehon, G. Z. Wang and D. Zhang, WO Pat., PCT WO 2011120025, 2011 Search PubMed.
- V. K. Patel and S. Swanson, WO Pat., PCT WO 2005073217, 2005 Search PubMed.
- K. B. Goodman, H. Cui, S. E. Dowdell, D. E. Gaitanopoulos, R. L. Ivy, C. A. Sehon, R. A. Stavenger, G. Z. Wang, A. Q. Viet, W. Xu, G. Ye, S. F. Semus, C. Evans, H. E. Fries, L. J. Jolivette, R. B. Kirkpatrick, E. Dul, S. Khandekar, T. Yi, D. K. Jung, L. L. Wright, G. K. Smith, D. J. Behm, C. P. Doe, R. Bentley, E. Hu and D. Lee, J. Med. Chem., 2007, 50, 6–9 CrossRef CAS PubMed.
- D. Lee and K. B. Goodman, WO Pat., PCT WO 2005082890, 2005 Search PubMed.
- C. A. Sehon, G. Z. Wang, A. Q. Viet, K. B. Goodman, S. E. Dowdell, P. A. Elkins, S. F. Semus, C. Evans, L. J. Jolivette, R. B. Kirkpatrick, E. Dul, S. S. Khandekar, T. Yi, L. L. Wright, G. K. Smith, D. J. Behm, R. Bentley, C. P. Doe, E. Hu and D. Lee, J. Med. Chem., 2008, 51, 6631–6634 CrossRef CAS PubMed.
- N. D. Smith, S. P. Govek, M. Kahraman, J. D. Julien, J. Y. Nagasawa, K. L. Douglas, C. Bonnefous and A. G. Lai, WO Pat., PCT WO 2013142266, 2013 Search PubMed.
- S. Usui, T. Takada and Y. Nishio, JP Pat., JP 2014166961, 2014 Search PubMed.
- M. Calderini, M. Wucherer-Plietker, U. Graedler and C. Esdar, WO Pat., PCT WO 2011006567, 2011 Search PubMed.
- B. S. Sathe, V. A. Jagtap, S. D. Deshmukh and B. V. Jain, Int. J. Pharm. Sci., 2011, 3, 220–222 CAS.
- B. D. Varpe and S. B. Jadhav, Int. J. Pharm. Invest., 2021, 11, 189–194 CrossRef CAS.
- A. J. Ayoub, G. A. El-Achkar, S. E. Ghayad, L. Hariss, R. H. Haidar, L. M. Antar, Z. I. Mallah, B. Badran, R. Grée, A. Hachem, E. Hamade and A. Habib, Int. J. Mol. Sci., 2023, 24, 10399 CrossRef CAS PubMed.
- T. Fuchigami, Phosphorus, Sulfur Relat. Elem., 1997, 120, 343–344 CrossRef.
- Z. Wang, C. Yi, K. Chen, T. Wang, K. Deng, C. Jin and G. Hao, Eur. J. Med. Chem., 2022, 228, 114025 CrossRef CAS PubMed.
- P. Limpachayaporn, B. Riemann, K. Kopka, O. Schober, M. Schäfers and G. Haufe, Eur. J. Med. Chem., 2013, 64, 562–578 CrossRef CAS PubMed.
- H. Wang, Y. Wang, S. Liu, Y. Mai, X. Zong, H. Gao, R. Hu, X. Jiang and Y. Y. Yeung, Adv. Synth. Catal., 2019, 361, 5334–5339 CrossRef CAS.
- S. Crosignani, S. Cauwenberghs, F. Deroose and G. Driessens, WO Pat., PCT WO2015067782, 2015 Search PubMed.
- M. Chochkova, B. Stoykova, G. Ivanova, A. Ranz, X. Guo, E. Lankmayr, T. Milkova and J. Fluor, Chem, 2013, 156, 203–208 CAS.
- Q. Meng, B. Zhao, Q. Xu, X. Xu, G. Deng, C. Li, L. Luan, F. Ren, H. Wang, H. Xu and Y. Xu, Bioorg. Med. Chem. Lett., 2012, 22, 2794–2797 CrossRef CAS PubMed.
- K. Sato, H. Sugimoto, K. Rikimaru, H. Imoto, M. Kamaura, N. Negoro, Y. Tsujihata, H. Miyashita, T. Odani and T. Murata, Bioorg. Med. Chem., 2014, 22, 1649–1666 CrossRef CAS PubMed.
- S. Nomura, Y. Yamamoto, Y. Matsumura, K. Ohba, S. Sakamaki, H. Kimata, K. Nakayama, C. Kuriyama, Y. Matsushita, K. Ueta and M. Tsuda-Tsukimoto, ACS Med. Chem. Lett., 2014, 5, 51–55 CrossRef CAS PubMed.
- X. Li, D. Bai, H. Dong, Y. Chen and F. He, WO Pat., PCT WO2023116862 A1, 2023 Search PubMed.
- A. Lantermann, E. L. P. Chekler, D. Blom, K. M. Couto, B. A. Lefker and D. B. Kitchen, WO Pat., PCT WO2023049438A1, 2023 Search PubMed.
- G. Semple, C. Averbuj, P. J. Skinner, T. Gharbaoui and Y. J. Shin, WO Pat., PCT WO 2004032928, 2004 Search PubMed.
- B. Pelcman, A. Sanin and P. Nilsson, WO Pat., PCT WO 2006032851, 2006 Search PubMed.
- B. Pelcman, A. Sanin, P. Nilsson, T. Boesen, S. B. Vogensen, H. Kromann and T. Groth, WO Pat., PCT WO 2007045868, 2007 Search PubMed.
- S. N. Haydar, H. Bothmann, C. Ghiron, L. Maccari, I. Micco, A. Nencini and R. Zanaletti, WO Pat., PCT WO 2010009290, 2010 Search PubMed.
- A. A. Trabanco-Suarez, H. J. M. Gijsen, M. L. M. Van Gool, J. A. Vega Ramiro and F. Delgado-Jimenez, WO Pat., PCT WO 2012117027, 2012 Search PubMed.
- C. Gege, O. Kinzel, C. Steeneck, G. Kleymann and T. Hoffmann, WO Pat., PCT WO 2014023367, 2014 Search PubMed.
- K. Ahn, M. Boehm, S. Cabral, P. A. Carpino, K. Futatsugi, D. Hepworth, D. W. Kung, S. Orr and J. Wang, WO Pat., PCT WO 2013150416, 2013 Search PubMed.
- C. Zhou, W. Zou, Y. Hua and Q. Dang, WO Pat., PCT WO 2013040790, 2013 Search PubMed.
- Q. Dang, C. Zhou, W. Zou and Y. Hua, WO Pat., PCT WO 2013043624, 2013 Search PubMed.
- S. K. Dodd, P. Furet, R. M. Grotzfeld, D. B. Jones, P. Manley, A. Marzinzik, X. F. A. Pelle, B. Salem and J. Schoepfer, WO Pat., PCT WO 2013171639, 2013 Search PubMed.
- P. Furet, R. M. Grotzfeld, D. B. Jones, P. Manley, A. Marzinzik, X. F. A. Pelle, B. Salem and J. Schoepfer, WO Pat., PCT WO 2013171642, 2013 Search PubMed.
- R. M. Claramunt, C. Lopez, C. Perez-Medina, M. Perez-Torralba, J. Elguero, G. Escames and D. Acuna-Castroviejo, Bioorg. Med. Chem., 2009, 17, 6180–6187 CrossRef CAS PubMed.
- T. M. Ballard, Z. K. Groebke, E. Pinard, T. Ryckmans and H. Schaffhauser, WO Pat., PCT WO 2015044072, 2015 Search PubMed.
- S. B. Hoyt, J. A. Taylor, C. London, Y. Xiong and A. J. Cooke, WO Pat., PCT WO 2014099833, 2014 Search PubMed.
- I. P. Buchler, M. J. Hayes, S. G. Hegde, S. L. Hockerman, D. E. Jones, S. W. Kortum, J. G. Rico, R. E. Tenbrink and K. K. Wu, WO Pat., PCT WO 2009106980, 2009 Search PubMed.
- S. Lin, L. Xu, E. Metzger, E. R. Parmee and S. D. Debenham, WO Pat., PCT WO 2010098994, 2010 Search PubMed.
- G. Henkel, M. Liu and T. Neuberger, WO Pat., PCT WO 2009145814, 2009 Search PubMed.
- R. M. Claramunt, C. López, A. López, C. Pérez-Medina, M. Pérez-Torralba, I. Alkorta, J. Elguero, G. Escames and D. Acuña-Castroviejo, Eur. J. Med. Chem., 2011, 46, 1439–1447 CrossRef CAS PubMed.
- A. Kousaxidis, A. Petrou, P. Rouvim, P. Bodo, M. Stefek, I. Nicolaou and A. Geronikaki, J. Mol. Struct., 2023, 1271, 134116 CrossRef CAS.
- A. Mallia, M. Nguyen, J. Sloop, N. Forlemu and J. Undergrad, Chem. Res., 2023, 22, 22 Search PubMed.
- K. M. Short, M. A. Estiarte, S. M. Pham, D. C. Williams, L. Igoudin, S. Dash, N. Sandoval, A. Datta, N. Pozzi, E. Di Cera and D. B. Kita, Eur. J. Med. Chem., 2023, 246, 114855 CrossRef CAS PubMed.
- J. O. Witt, A. L. McCollum, M. A. Hurtado, E. D. Huseman, D. E. Jeffries, K. J. Temple, H. C. Plumley, A. L. Blobaum, C. W. Lindsley and C. R. Hopkins, Bioorg. Med. Chem. Lett., 2016, 26, 2481–2488 CrossRef CAS PubMed.
- S. N. Mistry, J. Shonberg, C. J. Draper-Joyce, C. Klein Herenbrink, M. Michino, L. Shi, A. Christopoulos, B. Capuano, P. J. Scammells and J. R. Lane, J. Med. Chem., 2015, 58, 6819–6843 CrossRef CAS PubMed.
- M. Marcinkowska, M. Kołaczkowski, K. Kamiński, A. Bucki, M. Pawłowski, A. Siwek, T. Karcz, B. Mordyl, G. Starowicz, P. Kubowicz, E. Pękala, A. Wesołowska, J. Samochowiec, P. Mierzejewski and P. Bienkowski, Eur. J. Med. Chem., 2016, 124, 456–467 CrossRef CAS PubMed.
- T. E. Ali and R. M. Abdel-Rahman, J. Sulfur Chem., 2014, 35, 399–411 CrossRef CAS.
- J. F. Ge, Q. Q. Zhang, J. M. Lu, M. Kaiser, S. Wittlin, R. Brun and M. Ihara, MedChemComm, 2012, 3, 1435–1442 RSC.
- R. Aslanian, X. Zhu, H. A. Vaccaro, N. Y. Shih, J. J. Piwinski, S. M. Williams and R. E. West, Bioorg. Med. Chem. Lett., 2008, 18, 5032 CrossRef CAS PubMed.
- C. Ojeda-Gómez, H. Pessoa-Mahana, P. Iturriaga-Vásquez, C. D. Pessoa-Mahana, G. Recabarren-Gajardo and C. Méndez-Rojas, Arch. Pharm., 2014, 347, 174–184 CrossRef PubMed.
- Y. Xiong, J. S. K. Choi, B. M. Smith, S. Strah-Pleynet and B. Teegarden, WO Pat., PCT WO 2008054748, 2008 Search PubMed.
- H. M. Elokdah, A. A. Greenfield, K. Liu, R. E. McDevitt, G. R. McFarlane, C. Grosanu, J. R. Lo, Y. Li, A. J. Robichaud and R. C. Bernotas, US Pat., US 20070037802, 2007 Search PubMed.
- K. Mizuno, J. Ikeda, T. Nakamura, M. Iwata, H. Otakaand and N. Goto, JP Pat., JP 2014133739, 2014 Search PubMed.
- A. Pennell, J. Aggen, J. J. Wright, S. Sen, B. McMaster, D. Dairaghi, W. Chen and P. Zhang, US Pat., US 20050256130, 2005 Search PubMed.
- A. Pennell, J. Aggen, J. J. Wright, S. Sen, B. McMcmaster, D. Dairaghi, W. Chen and P. Zhang, US Pat., US 20060106218, 2006 Search PubMed.
- P. Zhang, A. M. K. Pennell, J. K. J. Wright, W. Chen, M. R. Leleti, Y. Li, L. Li, Y. Xu, M. M. Gleason, Y. Zeng and K. L. Greenman, US Pat., US 20080058341, 2008 Search PubMed.
- P. Zhang, A. M. K. Pennell, J. K. Wright, W. Chen, M. R. Leleti, Y. Li, L. Li, Y. Xu, M. M. Gleason, Y. Zeng, K. L. Greenman, WO Pat., PCT WO 2008147822 A1, 2008 Search PubMed.
- N. P. Barton, H. Hobbs, S. T. Hodgson, Y. M. L. Lacroix, P. A. Procopiou, CC.chemokine receptor 4 antagonists, WO Pat., WO 2012025473, 2012 Search PubMed.
- P. A. Procopiou, J. W. Barrett, N. P. Barton, M. Begg, D. Clapham, R. C. Copley, A. J. Ford, R. H. Graves, D. A. Hall, A. P. Hancock, A. P. Hill, H. Hobbs, S. T. Hodgson, C. Jumeaux, Y. M. L. Lacroix, A. H. Miah, K. M. L. Morriss, D. Needham, E. B. Sheriff, R. J. Slack, C. E. Smith, S. L. Sollis and H. Staton, J. Med. Chem., 2013, 56, 1946–1960 CrossRef CAS PubMed.
- K. Hata, M. Masuda, H. Nakai, D. Taniyama, H. Tobinaga, A. Hato and M. Fujo, WO Pat., PTC WO 2013061977, 2013 Search PubMed.
- K. Hata, M. Masuda, H. Nakai, D. Taniyama, H. Tobinaga, A. Hato and M. Fujo, JP Pat., JP 2014224104, 2014 Search PubMed.
- Y. K. Lee, D. J. Parks, T. Lu, T. V. Thieu, T. Markotan, W. Pan, D. F. McComsey, K. L. Milkiewicz, C. S. Crysler, N. Ninan, M. C. Abad, E. C. Giardino, B. E. Maryanoff, B. P. Damiano and M. R. Player, J. Med. Chem., 2008, 51, 282–297 CrossRef CAS PubMed.
- A. Katsifis, C. J. R. Fookes, T. Q. Pham, I. D. Greguric, M. F. P. S. Mattner, WO Pat., PCT WO 2008022396A1, 2008 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Thoraya A. Farghaly
a,
Thoraya A. Farghaly