
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic lignin as an efficient and sustainable homogenous catalyst for aqueous Knoevenagel condensation reactions†
Ahmed I. A. Soliman‡
 ab,
Ameena Bacchus‡a,
Rozita Zarea,
Shrikanta Sutradhara and
Pedram Fatehi*a
ab,
Ameena Bacchus‡a,
Rozita Zarea,
Shrikanta Sutradhara and
Pedram Fatehi*a
aGreen Processes Research Centre and Chemical Engineering Department, Lakehead University, 955 Oliver Road, Thunder Bay, P7B5E1, ON, Canada. E-mail: pfatehi@lakeheadu.ca; Tel: +1 807 343 8697
bChemistry Department, Faculty of Science, Assiut University, Assiut 71516, Egypt
First published on 18th September 2024
Abstract
Knoevenagel condensation is a chemical reaction between aldehydes and active methylene-containing compounds in the presence of heterogeneous, basic homogenous organic or inorganic catalysts and solvent or neat systems. Herein, we introduced a new strategy for this synthesis by using the aqueous solution of cationic kraft lignin (CKL) as a catalyst. The CKL was synthesized through the reaction of kraft lignin (KL) with glycidyltrimethylammonium chloride (GTMAC) in a basic medium. The optimal reaction conditions for the Knoevenagel reaction were 5% catalyst load (weight of catalyst to the weight of benzaldehyde), water as the solvent, and at room temperature, which generated the products with a yield of 97%, illustrating that the CKL was an effective homogenous and green catalyst. The results confirmed that the increase in CKL charge density improved the product yield. The water-insoluble products were easily separated by filtration, and the filtrate containing the catalysts was reused effectively for 5 cycles without a significant decrease in the production yield, which would confirm the advantages of this catalyst for this reaction system. The CKL catalyst exhibited biodegradability comparable to KL. This paper discusses a novel method for Knoevenagel condensation reactions for different aldehydes in a green system utilizing a sustainable, biodegradable catalyst at room temperature and in an aqueous system.
1 Introduction
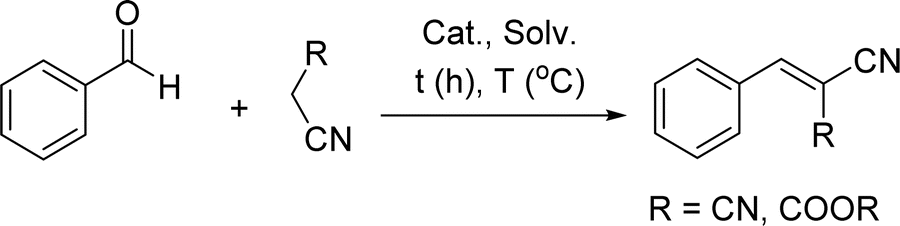
Knoevenagel condensation reaction is an abundant reaction in organic synthesis, where aldehydes react with active methylene-containing compounds to produce α,β-unsaturated carbonyl compounds, which are widely applied in pharmaceutical, flavor, and perfume industries.1–3 In the conventional method of this reaction, a homogenous organic nitrogen-containing base catalyst, such as pyridine, triethylamine, or piperidine, is used in solvent-based systems.2,4 However, the challenges associated with the organic solvent recovery and product purification, as well as the handling of residuals of the reaction, introduce operational and environmental challenges.4–6Another approach to catalyse the reaction is to use heterogeneous catalysts, such as hydrotalcite, zeolites, metal–organic frameworks (MOFs), metal oxide (MO), graphitic carbon nitride, ionic liquids (ILs), and metal-doped MO (M-MO), which would reduce the harmful impact of environmentally hazardous organic bases.4,7,8 Also, the use of these heterogenous catalysts has the advantage of catalyst recycling, which would reduce catalyst consumption.4,7,9 In addition, the reaction might occur in water or in a solvent-free system, which is considered a green approach that avoids the use of hazardous organic solvents.4,7 However, the development of these heterogenous catalysts requires multi-steps and harsh fabrication conditions, such as high pressure and high temperature.4,7,10,11 Also, the used catalysts that contain metals, such as hydrotalcite, zeolites, MO, M-MO, and MOFs, require special and careful disposal.4,7 Furthermore, when the reaction proceeds in organic solvents that dissolve both reactants and products; centrifugation, filtration, or other separation methods are required to isolate the catalyst for the recycling process, and product purification may be challenging.4,7 Another limitation in the use of heterogeneous catalysts in organic synthesis is the need for the reactivation of the recycled catalyst, which may require special treatment, such as catalyst washing and high-temperature drying, for long periods.4,7 Furthermore, if the heterogenous reaction occurred in a solvent-free or water environment, the catalyst recycling would be more tedious, as the products, which would be almost water-insoluble and mixed with the catalyst, should be dissolved for the catalyst separation. Such difficulties in catalyst development promote the use of green chemistry protocols to avoid complicated recycling and reactivation processes, which would be more suitable in the discipline of organic synthesis.
Lignin is considered one of the most abundant, natural, and sustainable polyphenolic polymers, which contains different functional groups, especially hydroxyl groups.12 The presence of these hydroxyl groups facilitates the derivatization reactions of lignin.12,13 One of these derivatization reactions is the amination through the reaction between lignin, e.g., kraft lignin (KL) and glycidyl trimethylammonium chloride (GTMAC), where quaternary-N cation is introduced to the lignin backbone. Previous reports showed that the cationic kraft lignin, CKL, produced from GTMAC,14,15 was water soluble and could be used as an antioxidant, flocculant, UV blocker, and oxygen barrier.16–19 The success in developing catalysts from lignin for catalyzing organic reactions would be advantageous due to the abundance, biodegradability, and easy derivatization of lignin.13,20–22 Different studies have reported the use of lignin-based heterogeneous catalysts in catalyzing different organic reactions,23 such as Cu@lignosulfonate,24,25 Fe3O4@lignin, amino-functionalized imidazolium-based ionic liquid immobilized on ammonium lignosulfonate,26 and Pd@lignin derived C as effective catalysts in different organic synthesis reactions such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogenation, Suzuki reactions, multicomponent reactions,26 Knoevenagel condensations,25 and Sonogashira–Hagihara reactions.23 However, the utilization of water-soluble lignin-based catalysts in organic synthesis would facilitate the environmentally friendly discarding of the catalyst and the product isolation without a complicated process.
C hydrogenation, Suzuki reactions, multicomponent reactions,26 Knoevenagel condensations,25 and Sonogashira–Hagihara reactions.23 However, the utilization of water-soluble lignin-based catalysts in organic synthesis would facilitate the environmentally friendly discarding of the catalyst and the product isolation without a complicated process.
In this paper, a water-soluble CKL was synthesized from the reaction between KL and GTMAC following the previously established procedure.14,16,17 The cationization reaction and properties of the CKL products were comprehensively assessed. Moreover, the aquatic biodegradation of CKL in an aerobic system was assessed, concluding the sustainability and environmental friendliness of CKL. After that, the synthesized CKL was used as a water-soluble catalyst to catalyze the Knoevenagel condensation without requiring any additives or mixing with metal, metal oxide, ionic liquids, etc. The optimal conditions of solvent, temperature, and catalyst load were investigated to generate the product with a maximum yield. The impact of charge density of CKL and the catalyst reuse were investigated, and the mechanism routes for the catalyzing process were postulated. Also, the efficiency of the reaction using different aldehydes was studied. The use of the aqueous CKL solution for Knoevenagel condensation would facilitate the development of an environmentally friendly aqueous-based reaction system and sustainable catalyst (i.e., lignin-based catalyst) with a high production yield and easy recycling. The primary novelty of this work was the use of a water-soluble and biodegradable catalyst (CKL) for the Knoevenagel condensation. Also, the impact of the charge density of CKL on the yield of the synthesized product was studied for the first time. Furthermore, the merits of easy product separation, no catalyst regeneration requirements, or complicated disposal strategies will be beneficial for the use of CKL in Knoevenagel condensation systems.
2 Experimental
2.1. Materials
Softwood kraft lignin (KL) was sourced from Hinton, Alberta. Glycidyltrimethylammonium chloride (GTMAC), H2SO4, NaOH, methanol (HPLC grade), glacial acetic acid (>99%), malononitrile (99%), benzaldehyde derivatives (>97%), Na2HPO4 (≥98%), KH2PO4 (≥99%), NH4Cl (≥99.5%), NaCl (≥99), CaCl2 (≥97), acetic acid (AcOH, 99.7), trimethylsilyl propanoic acid (99.7%, TMSP), and potassium polyvinyl sulfate (PVSK, 100–200 kg mol−1, 97.7 wt% esterified) were purchased from Sigma Aldrich.2.2. Synthesis of CKL
The synthesis of CKL was carried out as described in previous reports,14,15 whereby a 20 wt% solution of lignin in water was prepared, and the pH was adjusted to 11.5 using a 3 M NaOH solution. Then, 0.5, 1, and 1.5 molar ratios of lignin to GTMAC were prepared in 500 mL three-neck round bottom flasks to generate cationic lignin with three different charge densities. All the reactions were allowed to progress in a 70 °C water bath for 1 h with stirring at 150 rpm. Subsequently, the flasks were submerged in cold water for 15 minutes and neutralized using 0.1 M H2SO4 to pH 7. Finally, membrane dialysis (1000 Da cutoff) was used to remove any unreacted salts and to purify the product for 48 hours, and then the samples were freeze-dried.2.3. Biodegradability analysis
The assessment of CKL's biodegradability was carried out in a 500 mL bioreactor under an aerobic condition in a mineral salt aqueous environment by a respirometer instrument, the AER-800S respirometer system. Samples (0.2 g) were pulverized and dissolved in a nutrient solution at 22 °C. The nutrition culture medium contained Na2HPO4 (6 g L−1), KH2PO4 (3 g L−1), NH4Cl (1 g L−1), NaCl (0.5 g L−1), and CaCl2 (3 mg L−1).27 For the aquatic biodegradation test, microbial inoculum sourced from Polyseed, NX, was utilized, comprising a specialized blend of non-pathogenic Bacillus bacteria of soil origin. These bacteria, in a spore form, required rehydration with nutrient buffer water to initiate the sporulation process. To initiate the biodegradation process, 5 mL of microbial inoculum was introduced into each 500 mL sterilized mineral salt medium bioreactor. Subsequently, a KOH holder bottle containing 30% KOH solution, and a series of holes was employed to capture the CO2 released during biodegradation.28,29 The evaluation process involved monitoring the consumption of oxygen.Samples for the biodegradation test include CKL and KL (the mineral medium, microbial inoculum, and test samples), a blank control (a mineral with microbial inoculum), and starch as a reference sample.28,30 All test samples were analyzed in triplicate within their respective bioreactors to ensure consistency and reliability of the results. The biodegradability of the samples (denoted as R and presented as a percentage) was determined using eqn (1), where the QT and Qb represented the oxygen consumption values of the test substance and the blank, respectively, measured in mg L−1. CT denotes the concentration of the test substance in mg L−1, while ThODT stands for the theoretical oxygen demand of the test substance, expressed as mg of O2 per mg of the substance (mgO2 mg−1 substance).27,31
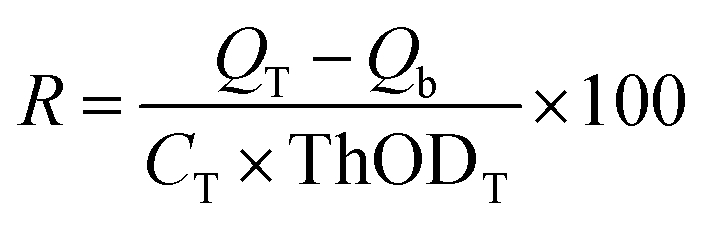 | (1) |
2.4. Knoevenagel condensation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) of methanol and aqueous AcOH (2.5%); the column temperature was fixed at 50 °C at a 0.9 mL s−1 flow rate. The reaction scope was investigated using other aldehydes under the optimized conditions determined for 3a.
:3) of methanol and aqueous AcOH (2.5%); the column temperature was fixed at 50 °C at a 0.9 mL s−1 flow rate. The reaction scope was investigated using other aldehydes under the optimized conditions determined for 3a.
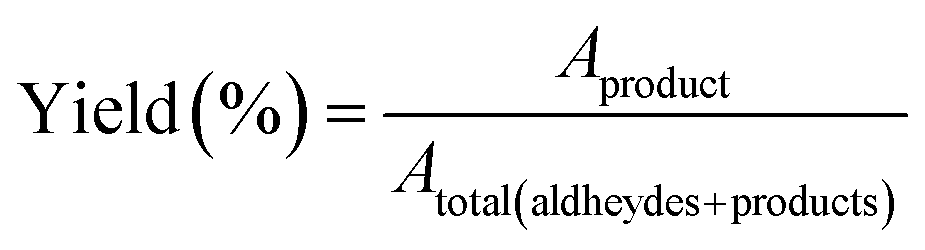 | (2) |
2.5. Characterization
The solubility of the samples was estimated by preparing 20 mL of 1 wt% solution of the CKL sample in Milli-Q water, which was allowed to mix for 1 h at 30 °C and 100 rpm in a shaker bath. Then, it was centrifuged at 1000 rpm to separate any potentially undissolved parts of the sample. The supernatant was used for charge density and solubility. The percentage of the solubilized sample in the supernatant was determined by comparing the weight of the solubilized samples after drying at 105 °C to the weight of the used sample before solubilization. The charge density was assessed using the Mütek Particle Charge Detector 04 titrator (BTG Instruments GmbH, Germany) via back titration against a solution of 0.005 M PVSK.To estimate the composition (i.e., carbon, hydrogen, oxygen, and nitrogen) of KL and CKL samples, the elemental analysis was performed using an elemental analyzer (Vario EL Cube, Elemental Analyzer, Germany), where a 2 mg of the sample was burned at 1200 °C in the instrument tube. The degree of substitution (DS) of CKL was calculated in the following eqn (3),14,16 where NCKL and NKL represent the nitrogen contents of CKL and KL samples, respectively. The values of 180, 151.5, and 14 g mol−1 represent the molecular weight (Mw) of C9 unit of lignin, Mw of GTMAC, and atomic weight (Aw) of nitrogen, respectively.
 | (3) |
1H NMR spectra of KL and CKL were collected using a Bruker Advance spectrometer (AVANCE Neo NMR-500 MHz instrument, Switzerland). DMSO-d6 was used as a solvent, and trimethylsilyl propanoic acid (0.01 wt%) was used as an internal standard. The samples of KL and CKL were prepared by dissolving and stirring 100 mg of each sample into 1 mL of DMSO-d6, along with 5 mg mL−1 TMSP. The spectra of these samples were registered using a Bruker Avance Neo NMR 500 MHz instrument with a 45° pulse and relaxation delay time of 2 seconds.14,15
The KL and CKL samples were characterized using HSQC NMR by dissolving 70 mg of the sample into 1 mL of DMSO-d6. The HSQC spectra were acquired using 13 ppm spectral width in the F2 (1H) dimension and 2048 data points (155 ms acquisition time), F1 (13C) dimension and 256 data points (6.2 millisecond acquisition time), a 2.5 s pulse delay and 16 scans at ambient temperature, using Bruker pulse program, hsqcetgpsisp2.3.
Quantitative 31P-NMR was used to quantify the aliphatic, phenolic, and carboxylate hydroxyl groups of KL and CKL samples with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP), a phosphorylating reagent. The samples were dissolved in a 1:1.6 (v/v) solution of CDCl3 and pyridine. After this, 50 μL of chromium(III) acetylacetonate as a relaxing agent, 100 μL of TMDP, and 70 μL of cyclohexanol-d12 as an internal standard were added to the samples. The 31P NMR spectra were acquired using a nuclear magnetic resonance spectrometer at room temperature with spectra parameters of a decoupling pulse sequence using a 90° pulse angle, 0.65 s acquisition time, and a 25 s pulse delay.15
The XPS analysis of CKL before and after the Knoevenagel condensation reaction was conducted and spectra were collected using a Kratos AXIS Supra XPS Axis Supra (Shimadzu Group Company, Japan) with a monochromatic Al Kα radiation (1486.7 eV). The obtained spectra were referenced to the CC components of C 1s at 284.5 eV, and CasaXPS software was used for deconvoluting and quantifying the components.32–34 The FTIR spectra were collected using a Bruker Tensor 37 equipped with ZnSe ATR accessory at the scan cycles of 1024 and at a 4 cm−1 resolution.35
3 Results and discussion
3.1. CKL synthesis
The cationic modification reaction of lignin occurs via the bimolecular nucleophilic substitution reaction mechanism, more commonly known as the SN2 mechanism.22 This reaction has been well documented in the literature for the preparation of cationic polysaccharides,36,37 and it was later adapted for lignin.14–18 Adjusting the pH of the lignin using sodium hydroxide serves two purposes: first, it solubilizes the lignin in water, improving the contact area for the reaction to occur, and second, it creates a strong nucleophile by deprotonating the lignin so that the reaction can progress. The nucleophile attacks the epoxy group of the GTMAC reagent via a ring-opening mechanism, effectively forming the new bond resulting in cationic lignin, as illustrated in Fig. 1a.14,15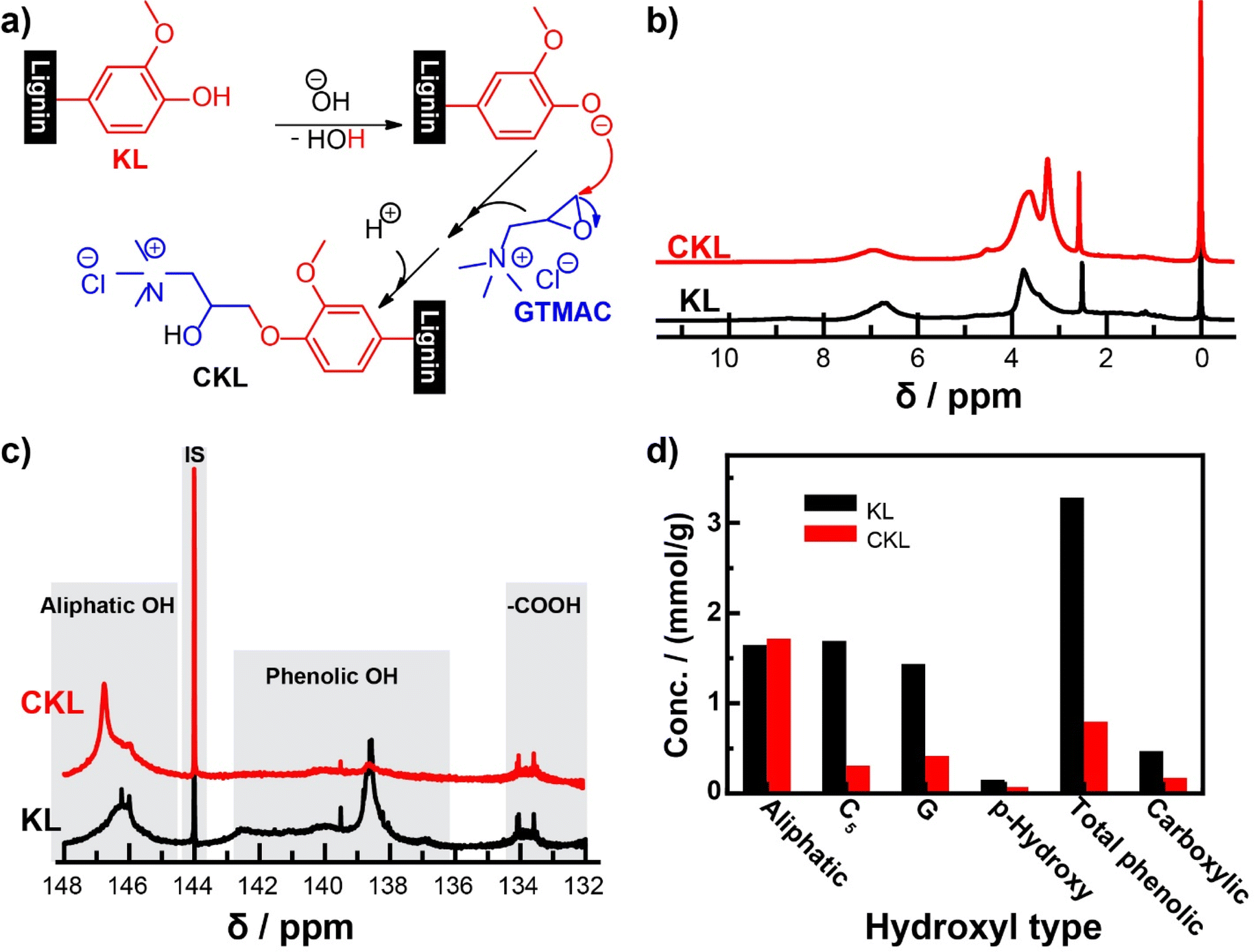 | ||
| Fig. 1 (a) Plausible reaction for CKL synthesis from KL, (b) 1H and (c) 31P NMR spectra of KL and CKL samples, and (d) quantitative comparison between the hydroxyl type obtained from 31P NMR spectra. | ||
Table 1 shows the properties of the CKL, where the charge density of the synthesized CKL was +1.1 meq g−1, with nitrogen content of 2.18% and water solubility of 10 g L−1. The degree of substitution was found to be 0.36. The Mw was 81679 g mol−1, and Mn was 70983 g mol−1. The charge density and nitrogen content found in this work were comparable to what was found by Kong et al.14 Guo et al.16 reported that the reaction of GTMAC with KL occurred at both aliphatic and phenolic positions of the KL structure when the reaction proceeded at a higher reaction temperature of 80 °C and a longer reaction time of 2 hours. Changing the charge density of KL from −1.1 to +1.1 could contribute to the presence of cationic ammonium moieties after cationization, which will be discussed later. Based on the applied reaction conditions, the reaction only took place at the phenolic position, as will be discussed later. Also, the molecular weight of CKL reported in this work is comparably higher than that reported in previous studies.16 Guo et al. reported Mn, Mw, and PDI of 9980 g mol−1, 32760 g mol−1, and 3.28, respectively.16 This can be explained by the use of freezing drying instead of oven drying at 105 °C. Freeze-drying, as used in this work, eliminates water by sublimation, while oven-drying evaporates water slowly, which may lead to a certain extent of polymer hydrolysis/degradation at high temperatures in the presence of water, which would result in a reduction in molecular weight reported in other papers.38,39 The increase in solubility and nitrogen content would be ascribed to the occurrence of cationization.
| KL | CKL | |
|---|---|---|
| Charge density (meq g−1) | −1.1 ± 0.30 | +1.1 ± 0.10 |
| Solubility (g L−1) in water | 7 ± 0.2 | 10 ± 0.6 |
| Nitrogen (%) | 0.04 ± 0.04 | 2.18 ± 0.05 |
| DS (mol mol−1) | — | 0.36 |
| Mw (g mol−1) | 17890 |
81679 |
| Mn (g mol−1) | 5150 | 70983 |
| Mw/Mn | 3.473 | 1.151 |
In 1H NMR (Fig. 1b), the broad signal in the range of 8–10 ppm is associated with unsubstituted phenolic protons; the signal between 6 and 7.7 ppm is associated with aromatic protons; the signal between 4.3 and 5.5 ppm is associated with oxygenated aliphatic protons; the signal between 3–4 ppm is associated with lignin's methoxy group; the signal at 2.5 ppm is association with the solvent, dimethyl sulfoxide-d6 (DMSO-d6); and the signal between 0.7 and 2.3 ppm is associated with the saturated aliphatic protons.14,40 The cationic modification reaction can be confirmed in two ways. First, there is a significant decrease of the broad signal between 8–10 ppm for the unsubstituted phenolic protons, which is where the cationic reagent attaches to the lignin structure. Second, the appearance of new peaks, such as 3.2 ppm and 3.7 ppm, is associated with the trimethyl ammonium group, which lends the cationic charge to the lignin and the methine.
31P NMR spectroscopy was used to ascertain the content of hydroxyl groups in the samples. The signals for phenolic OH components were significantly decreased after cationization, as illustrated in Fig. 1c. Still, the signals of both carboxylic and aliphatic OH components did not decrease significantly compared to those of phenolic OH content. The quantitative results provided in Fig. 1d indicate that the total phenolic content decreased significantly from 3.28 mmol g−1 in KL to 0.80 mmol g−1 in CKL. This was a result of the reduction in concentration of C5, G, and p-hydroxyphenyl substituted hydroxyl groups. No significant changes were observed in the aliphatic hydroxyl and carboxylic groups. Thus, it can be concluded that the reaction preferentially occurred at the phenolic position of KL. A covalent connection to the lignin structure is demonstrated by the fact that the reaction product was subjugated to thorough dialysis using membranes with a 1000 Da cut-off. This would largely preclude lower molecular weight compounds, such as any unreacted compounds.14,30 Previous literature has discussed the presence of side product formation due to hydroxide ions reacting with the reagent in a competing reaction to form an undesired, low-molecular-weight side product in the form of a glycol derivative.17,41 It has been shown that reaction conditions, such as excessive water content in the reaction medium, reagent ratio, temperature, and time, should be carefully considered when conducting alkaline synthesis of this type.41 Furthermore, longer reaction times and lower reagent ratio resulted in the increased formation of the undesired side product.17,41
HSQC NMR was conducted to ascertain more information on the structure of CKL. This technique showed the linkages within the KL structure as compared to the CKL structure. The HSQC spectra of KL and CKL are shown in Fig. 2a and b. The signals were assigned according to the literature.14,16,42 The β-O-4 aryl ether structure (A), resinol structure (B), and phenylcoumaran (C) were attributed to signals occurring at δC/δH 74.0/4.9 ppm (Aα), 86.0/4.3 ppm (Aβ), 63.0/3.4 ppm (Aγ), 85.1/4.7 ppm (Bα), 74.7/4.2 ppm (Bγ), 74.7/3.7 ppm (Bγ), 88.1/5.5 ppm (Cα) and 67.0/3.4 ppm (Cγ). When comparing the signals of CKL to KL in the HSQC spectra, certain signals were either absent or had a low resolution, particularly in the aromatic and aliphatic regions of the spectrum. This could be attributed to the low concentration of these structures,16 and or the shielding effect of the cationic nitrogen in the sample.43 It can also be seen that there are four new signals, which were attributed to structure D. The cross-peak at δC/δH 73.0/4.0 ppm was attributed to Ca–Ha. The cross-peak at 65.0/4.6 ppm was attributed to Cb–Hb. The cross-peak at 70.0/3.6 ppm was attributed to Cc–Hc. The cross-peak at 55/3.3 ppm was attributed to cationic methyl groups, Cd–Cd.16
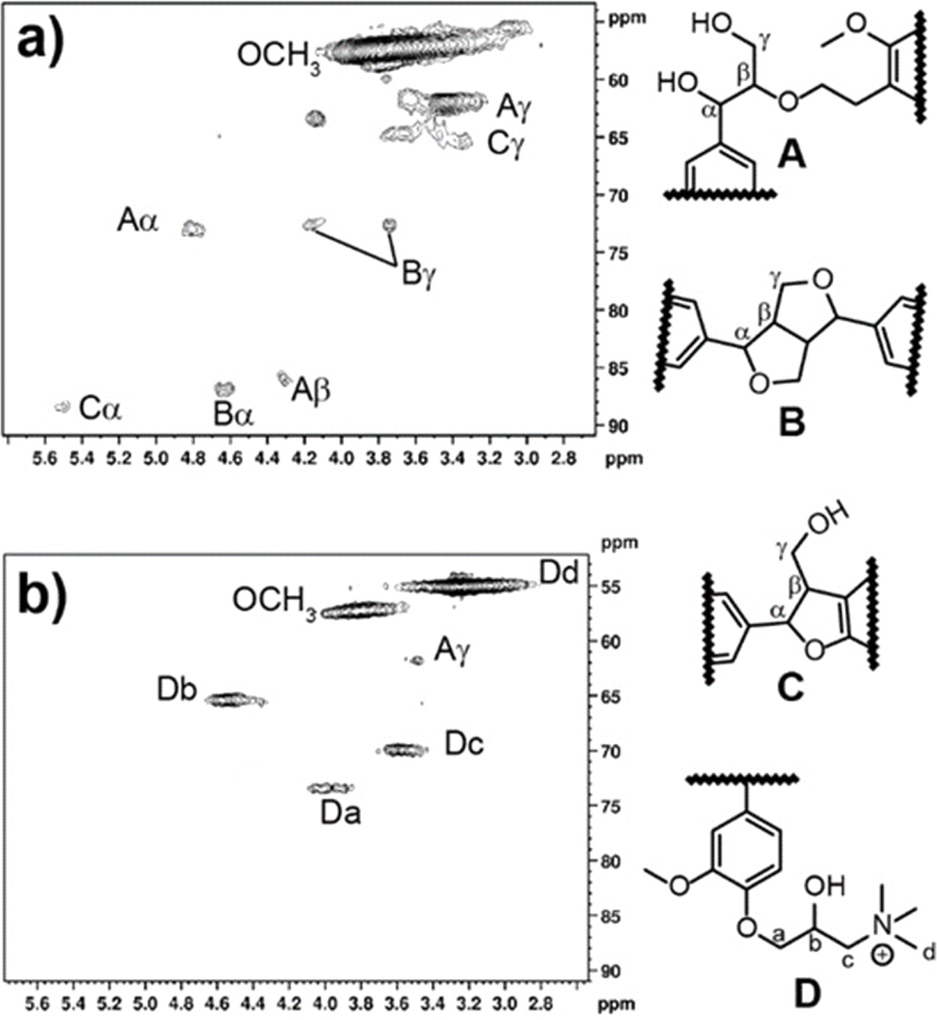 | ||
| Fig. 2 HSQC NMR spectra of (a) KL and (b) CKL with expecting the chemical structures of the observed signals. | ||
Fig. 3 shows the XPS analysis of KL and CKL conducted to confirm the cationic modification. The XPS C 1s spectra of KL and CKL were deconvoluted into 6 peaks at the binding energies (BE) of 284.3 eV, 285.1 eV, 286.0 eV, 287.1 eV, 288.6 eV, and 290.0 eV, which are assigned to the CC, C–C, C–O, –C–O, CO and COO components, respectively.32,33 After cationization, the abundance of C–C and –C–O components, which was originated from the GTMAC, increased. The O 1s spectra were deconvoluted into 531.3 eV, 532.5 eV, and 533.9 eV, and these peaks were assigned into CO, C–O, and O–H, respectively. The abundance of C–O–H components increased after the cationization resulting from GTMAC. The nitrogen content in both KL and CKL could be deconvoluted to two peaks at 399 eV and 401 eV, representing both N–C and cationic N–C, respectively. The atomic concentration of nitrogen in KL increased from 0.1% to 2.4% in CKL, which is also observed from elemental analysis, indicating the occurrence of cationization. The FT-IR spectrum (Fig. S2†) of KL showed bands at >3200 cm−1, 3035 cm−1, 2964–2839 cm−1, 1703 cm−1, 1600–1427 cm−1, 1370–1375 cm−1, 1215–1146 cm−1, and 854–813 cm−1, which were attributed to the vibration bands of ν(O–H), ν(C–H), ν(aliphatic C–H), ν(CO), ν(CC), ν(C–O) combined with δ(C–H), and δ(C–H), respectively.44 After cationization, bands at 1466 cm−1, 1222 cm−1, and 968 cm−1 were attributed to δ (CH2), ν(C–O), and δ (C–H) of GTMAC, respectively.14
 | ||
| Fig. 3 The XPS (a) C 1s, (b) O 1s, and (c) N 1s spectra of KL and CKL samples (d) the calculated atomic percentages of carbon, oxygen, and nitrogen in both KL and CKL samples. | ||
3.2. CKL biodegradability
Fig. 4 illustrates the biodegradation assessments of KL, CKL, and starch as a control sample. Wheat starch was selected as a control reference due to its favorable biodegradability, with an approximately 50% degradation observed within 12 days, which is in reasonable agreement with the starch degradation reported by Chen et al.,45 approaching 60% degradation after 30 days. Initially, the KL exhibited no discernible microbial degradation, which might be attributed to the microbial acclimatization process and the environmental conditions.28,46 Notably, KL, being more hydrophobic, potentially necessitated a longer duration for microbial action due to greater resistance to chain cleavage, unlike CKL, which is comparatively more hydrophilic (Table 1). After that, the KL graph depicted a slight increment from the CKL graph after 4 days, ultimately reaching a 20% biodegradation rate over a 14 days duration. In contrast, the CKL sample achieved a 15% biodegradation rate, and the biodegradable segments of the CKL sample were more readily accessible to the microorganisms. The slight suppression of the biodegradability after cationization might be ascribed to the antimicrobial activity of GTMAC induced by the cationic group, as reported by Beyler-Çigil et al.47 The degradation of lignin in an aqueous medium was reported to be in the range of 19% to 60% over 13 weeks to 2 years.30,48 Consequently, the observed biodegradation rates of 15–20% for KL and CKL within 14 days is reasonable and demonstrates the environmentally friendly characteristics of CKL promoting different applications for sustainable solutions.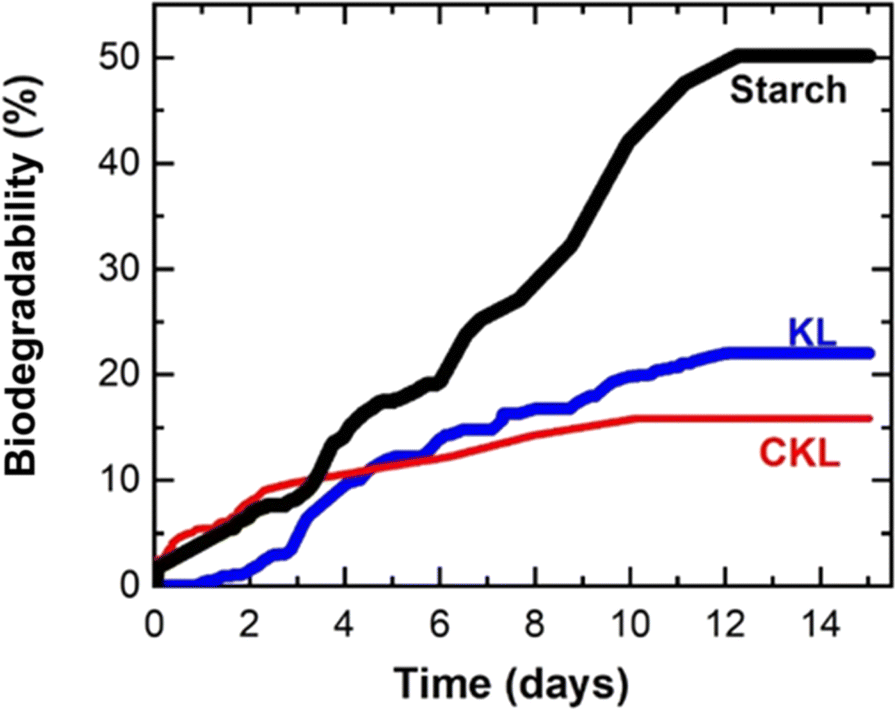 | ||
| Fig. 4 Biodegradability of KL, CKL, and starch. | ||
3.3. Knoevenagel condensation
 | ||
| Fig. 5 Reaction optimization for the synthesis of 3a in the presence of water as solvent (a) at different reaction times, different temperatures (RT and 50 °C), and in the presence of 5% CKL load (b) at RT and for 60 min and 120 min with different CKL loads of 1%, 5% and at 10%. | ||
The influence of different catalyst loads on the production yield is illustrated in Fig. 5b. The catalyst load of 1% accelerated the reaction and produced 3a with yields of 79% and 92% after 60 and 120 minutes reaction times, respectively. Increasing the catalyst load to 10% facilitated the reaction accomplishment with a yield of 97% and 98% after 60 and 120 min, respectively. In the absence of CKL, the 3a was not detectable, which would indicate that the catalyst was crucial for the reaction proceeding.
The influence of the charge density of CKL on the yield of Knoevenagel condensation is illustrated in Fig. 6, where the yields of the Knoevenagel condensation increased with increasing the charge density of the CKL, and the maximum yields occurred in the presence of CKL with the charge density of 1.1 meq g−1. The increase in the charge density would increase the polarization of the formyl group of 1a, as will be discussed below.
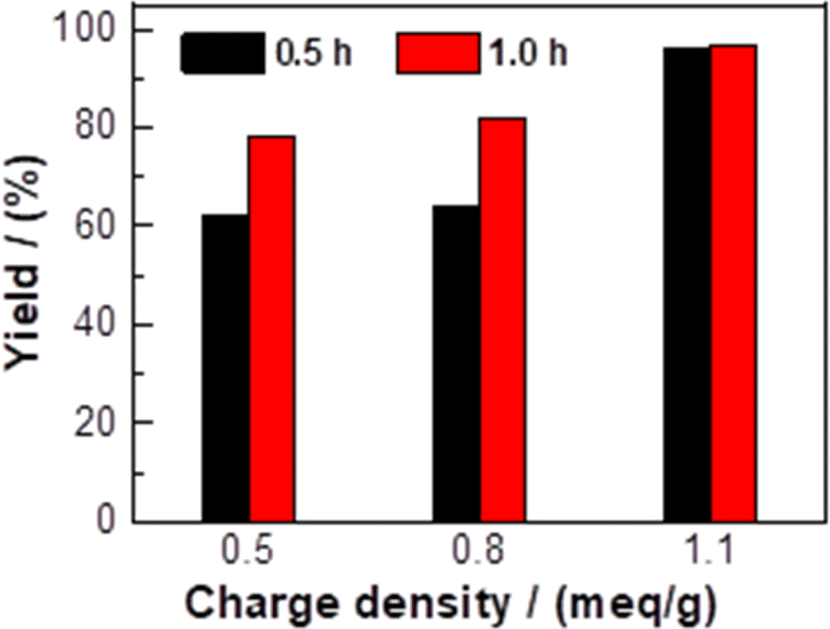 | ||
| Fig. 6 The influence of charge density of CKL on the yield of 3a. | ||
Based on these findings, the optimized procedures to perform the Knoevenagel condensation reaction were water as a reaction medium, 5% load of CKL with a charge density of 1.1 meq g−1, and 60 min as reaction time and at room temperature. Such procedures are considered a green and effective approach for performing Knoevenagel condensation without applying hazardous solvents, tiresome product isolation, applying high temperature, or special catalyst separation from the product.
O polarization. Also, the occurrence of steric hindrance negatively affected the reaction, causing a lowering in the product yield.52 However, the excellent yields undoubtedly illustrate that the CKL could be applied as a water-soluble, environmentally friendly, and sufficiently effective homogenous catalyst for Knoevenagel condensation reactions between different benzaldehydes containing electron withdrawing or donating groups with 2a.
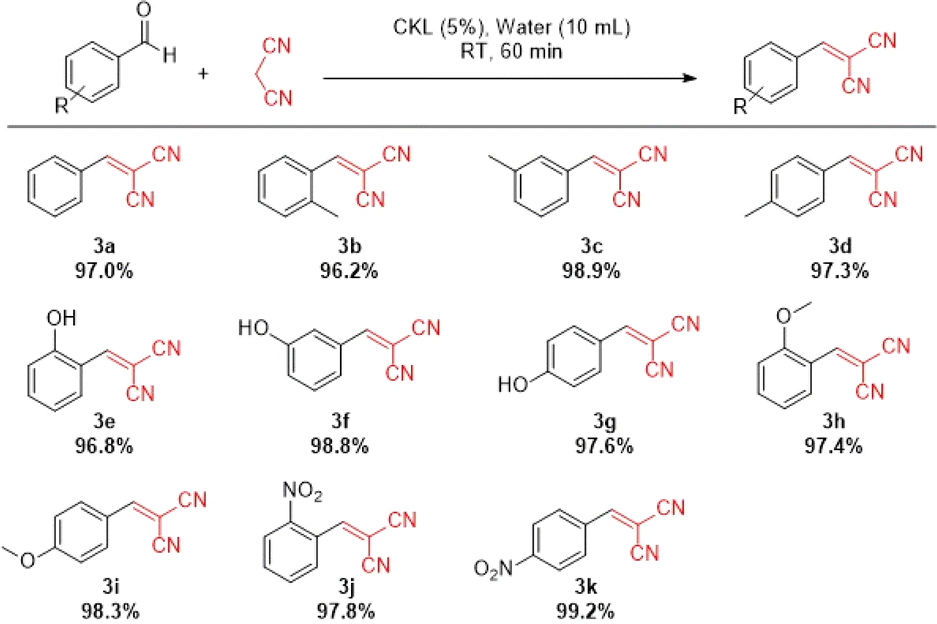 | ||
| Scheme 1 Scope of Knoevenagel condensation catalyzed by CKL. Yields are provided below the structures. | ||
 | ||
| Fig. 7 (a) Catalyst reusability for 3a synthesis under the optimum condition. XPS (b) C 1s and (c) O 1s spectra of the recycled CKL, (d) 1H NMR, and (e) HSQC spectra of the recycled CKL. | ||
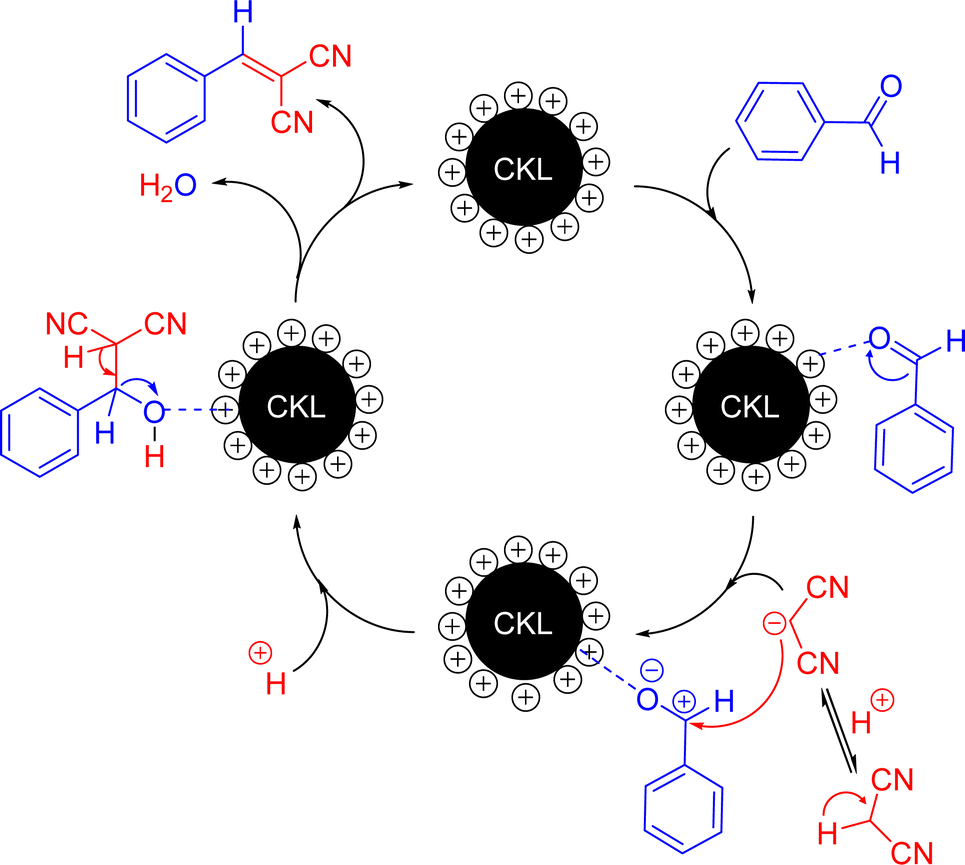 | ||
| Fig. 8 Postulated mechanism of Knoevenagel condensation using CKL. | ||
|
||||||
|---|---|---|---|---|---|---|
| Catalyst | Conditions | Yield (%) | Ref. | |||
| Solvent | t (h) | T (°C) | ||||
| Metal and metal oxide | ZnO | Neat | 24 | 100 | 74 | 54 |
| La2O2CO3@TiO2 | EtOH | 4 | 50 | 90 | 55 | |
| PdAlO(OH) | MeOH (65%) | 0.5 | RT | 99 | 56 | |
| Ag@TiO2 | ETOH | 1 | 65 | 95 | 49 | |
| g-C3N4 | FeWO4@g-C3N4 | CH3CN | 4 | RT | 75 | 57 |
| K-g-C3N4@r-GO | ETOH | 0.17–3 | RT | 99 | 58 | |
| Cellulose | CPILs | H2O | 3 | 80 | 84 | 53 |
| CMC-Na | Neat | 0.67 | RT | 94 | 59 | |
| Sulfonated Fe3O4@CNC | EtOH | 1 | RT | 95 | 35 | |
| MOF | Zn-MOF-NH2 | EtOH | 1 | 80 | 99 | 60 |
| [Cu2(μ-H3ddba)2(phen)2] | H2O | 1 | 25 | >99 | 61 | |
| Zeolite | Offretite zeolite | Neat | 1.5 | MV, 170 | 80 | 62 |
| Pomegranate-like zeolites | Toluene | 4 | 90 | 80 | 63 | |
| Lignin | CKL | H2O | 1 | RT | 97 | This study |
4 Conclusions
Herein, we introduced a novel water-soluble lignin-derived catalyst, CKL, with acceptable biodegradability for the Knoevenagel condensation reaction. The optimal procedure for catalysing the Knoevenagel reaction between benzaldehyde and malononitrile was to perform the reaction in an aqueous system at room temperature for 1 hour in the presence of 5% CKL, producing 3a with a yield of 97%. The recovery process of the catalyst involved filtration to remove the 3a residue and an aqueous system that contained aqueous CKL, implying that the recovery of CKL was easy without the need for catalyst activation. The reuse of the CKL aqueous solution was examined in 5 cycles without a significant decrease in the catalyst reactivity and product yield. The scope of the reaction showed that the catalyst was effective for different aryl aldehydes containing electron-withdraw or electron-donating groups, and the yields were almost >97%. The CKL, which is positively charged, is expected to polarize the CO group of aldehydes, facilitating the condensation with active methylene components to produce the α,β-unsaturated products. Hence, this study highlights a novel lignin-derived catalyst that can be applied to organic synthesis in water at room temperature, and the use of such an environmentally friendly and sustainable catalyst would be beneficial to both the economy and the environment.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†Author contributions
Ahmed I. A. Soliman; data acquisition, experimentation manuscript preparation, first draft, revision. Ameena Bacchus: data acquisition, experimentation Manuscript preparation. Rozita Zare: data acquisition, experimentation manuscript preparation. Shrikanta Sutradhar: experimentation. Pedram Fatehi: supervision, funding, manuscript preparation, revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AIAS acknowledges Assiut University and Zhejiang University for financial support. Also, financial support from NSERC, Canada, and Canada Research Chairs program is acknowledged.Notes and references
- M. M. Heravi, F. Janati and V. Zadsirjan, Monatsh. Chem., 2020, 151, 439–482 CrossRef CAS.
- K. van Beurden, S. de Koning, D. Molendijk and J. van Schijndel, Green Chem. Lett. Rev., 2020, 13, 85–100 Search PubMed.
- J. Kossmann, T. Heil, M. Antonietti and N. López-Salas, Chemsuschem, 2020, 13, 6643–6650 CrossRef CAS PubMed.
- S. Johari, M. R. Johan and N. G. Khaligh, Org. Biomol. Chem., 2022, 20, 2164–2186 RSC.
- Y. A. Sonawane, S. B. Phadtare, B. N. Borse, A. R. Jagtap and G. S. Shankarling, Org. Lett., 2010, 12, 1456–1459 CrossRef CAS.
- D. Koszelewski and R. Ostaszewski, Chem.–Eur. J., 2019, 25, 10156–10164 CrossRef CAS PubMed.
- J. N. Appaturi, R. Ratti, B. L. Phoon, S. M. Batagarawa, I. U. Din, M. Selvaraj and R. J. Ramalingam, Dalton Trans., 2021, 50, 4445–4469 RSC.
- Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, J. Mater. Chem., 2009, 19, 7263–7272 RSC.
- E. M. Schneider, M. Zeltner, N. Kränzlin, R. N. Grass and W. J. Stark, Chem. Commun., 2015, 51, 10695–10698 RSC.
- A. Rashidizadeh, H. R. Esmaili Zand, H. Ghafuri and Z. Rezazadeh, ACS Appl. Nano Mater., 2020, 3, 7057–7065 CrossRef CAS.
- F. Kalantari, S. Rezayati, A. Ramazani, H. Aghahosseini, K. Ślepokura and T. Lis, ACS Appl. Nano Mater., 2022, 5, 1783–1797 CrossRef CAS.
- R. Shorey, A. Salaghi, P. Fatehi and T. H. Mekonnen, RSC Sustainability, 2024, 2, 804–831 RSC.
- M. J. Suota, D. M. Kochepka, M. G. G. Moura, C. L. Pirich, M. Matos, W. L. E. Magalhaes and L. P. Ramos, Bioresources, 2021, 16, 6471–6511 CAS.
- F. Kong, K. Parhiala, S. Wang and P. Fatehi, Eur. Polym. J., 2015, 67, 335–345 CrossRef CAS.
- S. J. Wang, F. G. Kong, W. J. Gao and P. Fatehi, Ind. Eng. Chem. Res., 2018, 57, 6595–6608 CrossRef CAS.
- Y. Guo, W. Gao, F. Kong and P. Fatehi, Int. J. Biol. Macromol., 2019, 140, 429–440 CrossRef CAS.
- R. Wahlström, A. Kalliola, J. Heikkinen, H. Kyllönen and T. Tamminen, Ind. Crops Prod., 2017, 104, 188–194 CrossRef.
- S. Sabaghi and P. Fatehi, Chemsuschem, 2020, 13, 4722–4734 CrossRef CAS.
- Y. Matsushita, H. Endo, S. Takahashi, D. Aoki, K. Fukushima and T. Yamada, J. Wood Sci., 2018, 64, 683–689 CrossRef CAS.
- E. Melro, A. Filipe, D. Sousa, B. Medronho and A. Romano, New J. Chem., 2021, 45, 6986–7013 RSC.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2015, 116, 2275–2306 CrossRef PubMed.
- A. Eraghi Kazzaz, Z. Hosseinpour Feizi and P. Fatehi, Green Chem., 2019, 21, 5714–5752 RSC.
- C. D. G. Martin, J. I. H. Garcia, S. Bonardd and D. D. Diaz, Molecules, 2023, 28, 3513 CrossRef CAS PubMed.
- B. B. Lai, M. Ye, P. Liu, M. H. Li, R. X. Bai and Y. L. Gu, Beilstein J. Org. Chem., 2020, 16, 2888–2902 CrossRef CAS PubMed.
- S. H. Sun, R. X. Bai and Y. L. Gu, Chem.–Eur. J., 2014, 20, 549–558 CrossRef CAS.
- W. Chen, X. W. Peng, L. X. Zhong, Y. Li and R. C. Sun, ACS Sustainable Chem. Eng., 2015, 3, 1366–1373 CrossRef CAS.
- D. Šašinková, L. Serbruyns, M. Julinová, A. FayyazBakhsh, B. De Wilde and M. Koutný, Polym. Degrad. Stab., 2022, 203, 110085 CrossRef.
- S. Muniyasamy and A. Patnaik, J. Renewable Mater., 2021, 9, 1661–1671 CAS.
- S. Kwon, M. C. Zambrano, J. J. Pawlak and R. A. Venditti, Cellulose, 2021, 28, 2863–2877 CrossRef CAS.
- M. R. Martínez-Gallardo, M. J. Estrella-González, F. Suárez-Estrella, J. A. López-González, M. M. Jurado, A. J. Toribio and M. J. López, Agronomy, 2023, 13, 1638 CrossRef.
- L. J. Zhou, Z. Y. Rong, W. Gu, D. L. Fan, J. N. Liu, L. L. Shi, Y. H. Xu and Z. Y. Liu, Environ. Monit. Assess., 2020, 192, 278 CrossRef CAS PubMed.
- Q. Z. Wu, L. S. Gao, M. N. Huang, G. A. M. Mersal, M. M. Ibrahim, Z. M. El-Bahy, X. F. Shi and Q. L. Jiang, Adv. Compos. Hybrid Mater., 2022, 5, 1044–1053 CrossRef CAS.
- F. Mahmood, C. Zhang, Y. C. Xie, D. Stalla, J. Lin and C. X. Wan, RSC Adv., 2019, 9, 22713–22720 RSC.
- A. I. A. Soliman, A. M. A. Abdel-Wahab and H. N. Abdelhamid, RSC Adv., 2022, 12, 7075–7084 RSC.
- M. Sayed, A. A. Saddik, A. M. K. El-Dean, P. Fatehi and A. I. A. Soliman, RSC Adv., 2023, 13, 28051–28062 RSC.
- J. L. Ren, R. C. Sun, C. F. Liu, Z. Y. Chao and W. Luo, Polym. Degrad. Stab., 2006, 91, 2579–2587 CrossRef CAS.
- Z. Liu, Y. Ni, P. Fatehi and A. Saeed, Biomass Bioenergy, 2011, 35, 1789–1796 CrossRef CAS.
- R. D. Sudduth, Polym. Eng. Sci., 2004, 36, 2135–2141 CrossRef.
- N. Quiévy, N. Jacquet, M. Sclavons, C. Deroanne, M. Paquot and J. Devaux, Polym. Degrad. Stab., 2010, 95, 306–314 CrossRef.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- P. I. F. Pinto, S. Magina, E. Budjav, P. C. R. Pinto, F. Liebner and D. Evtuguin, Ind. Eng. Chem. Res., 2022, 61, 3503–3515 CrossRef CAS PubMed.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Hoboken, NJ, 7th edn, 2005 Search PubMed.
- C. G. Boeriu, D. Bravo, R. J. A. Gosselink and J. E. G. van Dam, Ind. Crops Prod., 2004, 20, 205–218 CrossRef CAS.
- H. B. Chen, B. S. Chiou, Y. Z. Wang and D. A. Schiraldi, ACS Appl. Mater. Interfaces, 2013, 5, 1715–1721 CrossRef CAS PubMed.
- F. Hussain, H. W. Yu, K. Chon, Y. G. Lee, H. Eom, K. J. Chae and S. E. Oh, J. Environ. Manage., 2021, 277, 111467 CrossRef CAS PubMed.
- A. Beyler-Çigil, H. Birtane, F. Sen and M. V. Kahraman, Mater. Today Commun., 2021, 27, 102463 CrossRef.
- F. Asina, I. Brzonova, K. Voeller, E. Kozliak, A. Kubátová, B. Yao and Y. Ji, Bioresour. Technol., 2016, 220, 414–424 CrossRef CAS.
- M. Sayed, Z. P. Shi, F. Gholami, P. Fatehi and A. I. A. Soliman, ACS Omega, 2022, 7, 32393–32400 CrossRef CAS.
- M. Sayed, A. Soliman and H. N. Abdelhamid, J. Solid State Chem., 2024, 332, 124352 CrossRef.
- P. A. Burate, B. R. Javle, P. H. Desale and A. K. Kinage, Catal. Lett., 2019, 149, 2368–2375 CrossRef CAS.
- Z. Wang, X. Yuan, Q. a. Cheng, T. Zhang and J. Luo, New J. Chem., 2018, 42, 11610–11615 RSC.
- Y. Q. Shen, C. P. Yuan, X. Y. Zhu, Q. Chen, S. J. Lu and H. B. Xie, Green Chem., 2021, 23, 9922–9934 RSC.
- M. R. Shakil, A. G. Meguerdichian, H. Tasnim, A. Shirazi-Amin, M. S. Seraji and S. L. Suib, Inorg. Chem., 2019, 58, 5703–5714 CrossRef CAS PubMed.
- J. N. Appaturi, T. Pulingam, J. R. Rajabathar, F. Khoerunnisa, T. C. Ling, S. H. Tan and E. P. Ng, Microporous Mesoporous Mater., 2021, 320, 111091 CrossRef CAS.
- H. Göksu and E. Gültekin, Chemistryselect, 2017, 2, 458–463 CrossRef.
- A. Rashidizadeh, H. Ghafuri, H. R. E. Zand and N. Goodarzi, ACS Omega, 2019, 4, 12544–12554 CrossRef CAS PubMed.
- T. Chhabra, A. Bahuguna, S. S. Dhankhar, C. M. Nagaraja and V. Krishnan, Green Chem., 2019, 21, 6012–6026 RSC.
- H. Ma, L. N. Zou, L. H. Mi, H. T. Pan, Y. H. Qiao, N. Li and J. J. Teng, Chemistryselect, 2018, 3, 11110–11117 CrossRef.
- G. Q. Huang, J. Chen, Y. L. Huang, K. Wu, D. Luo, J. K. Jin, J. Zheng, S. H. Xu and W. G. Lu, Inorg. Chem., 2022, 61, 8339–8348 CrossRef CAS PubMed.
- H. R. Zhang, J. Z. Gu, M. V. Kirillova and A. M. Kirillov, Inorg. Chem. Front., 2021, 8, 4209–4221 RSC.
- E. P. Ng, N. H. Ahmad, T. C. Ling, F. Khoerunnisa and T. J. Daou, Mater. Chem. Phys., 2021, 272, 125001 CrossRef CAS.
- H. K. Min, H. An, D. C. Kang, S. Kweon, S. H. Baek, M. B. Park and C. H. Shin, ACS Appl. Mater. Interfaces, 2020, 12, 57881–57887 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of the synthesized benzylidenemalononitrile derivatives. See DOI: https://doi.org/10.1039/d4ra05763e |
| ‡ These authors are equally contributed. |
| This journal is © The Royal Society of Chemistry 2024 |