
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid acids as cocatalysts in the chelation-assisted hydroacylation of alkenes and alkynes†
Blanca I. Vergara-Arenasa,
Eréndira García-Ríosb,
Rubén Gaviño*b,
Jorge Cárdenas b,
Alfredo Martinez-Garciac,
Erick A. Juarez-Arellanoc,
Adolfo López-Torresc and
José A. Morales-Serna*c
b,
Alfredo Martinez-Garciac,
Erick A. Juarez-Arellanoc,
Adolfo López-Torresc and
José A. Morales-Serna*c
aDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco No. 186, Ciudad de México, C. P. 09340, Mexico
bInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Ciudad de México, 04510, Mexico. E-mail: rgavino@unam.mx
cCentro de Investigaciones Científicas, Instituto de Química Aplicada, Universidad del Papaloapan, Tuxtepec, Oaxaca, 68301, Mexico. E-mail: joseantonio.moralesserna@gmail.com
First published on 7th October 2024
Abstract
The use of homogeneous Brønsted acid cocatalysts (such as benzoic acid) in hydroacylation reactions via imine intermediates has been extensively studied. However, the use of heterogeneous cocatalysts has been limited to montmorillonite K10. Thus, we can use other solid acids to increase the efficiency of the reaction. In this study, we describe the effects of sulfated zirconia, Al-MCM-41 or superacid modified montmorillonite on the hydroacylation of alkenes and alkynes with aldehydes via imine intermediates and in the presence of the Wilkinson complex. Furthermore, we addressed the dual role of montmorillonite, a redox reagent in the presence of TEMPO and an acid solid, allowing the direct use of benzyl alcohols as substrates to generate saturated or α,β-unsaturated ketones.
Introduction
The transformation of an aromatic or aliphatic aldehyde into a saturated or α,β-unsaturated ketone involves the hydroacylation of alkenes or alkynes.1 This strategy of synthesis considers the aldehydic C–H bond as a functional group that can be activated to form a C–C bond in the presence of transition metal catalysis.2 In the overall catalytic hydroacylation process, the principal drawback is the decarbonylation of aldehyde,3 which results in the formation of carbonyl-metal complexes and alkanes (Scheme 1a). The following strategies have been used to avoid this problem: (i) the use of a β-sulfide4 or β-OR5 group in the substrate2 (Scheme 1b) and (ii) the use of reversible imine formation with 2-aminopyridines6 (Scheme 1c). Both of these approaches involve the assistance of chelating groups to stabilise the intermediate that results in the formation of a ketone. In the last protocol, the in situ formation of imines from aldehydes is performed in the presence of aniline and benzoic acid as cocatalysts.7 This imine reacts with alkenes in the presence of Wilkinson's catalyst to generate ketimines, which are hydrolysed to saturated ketones (Scheme 1c).8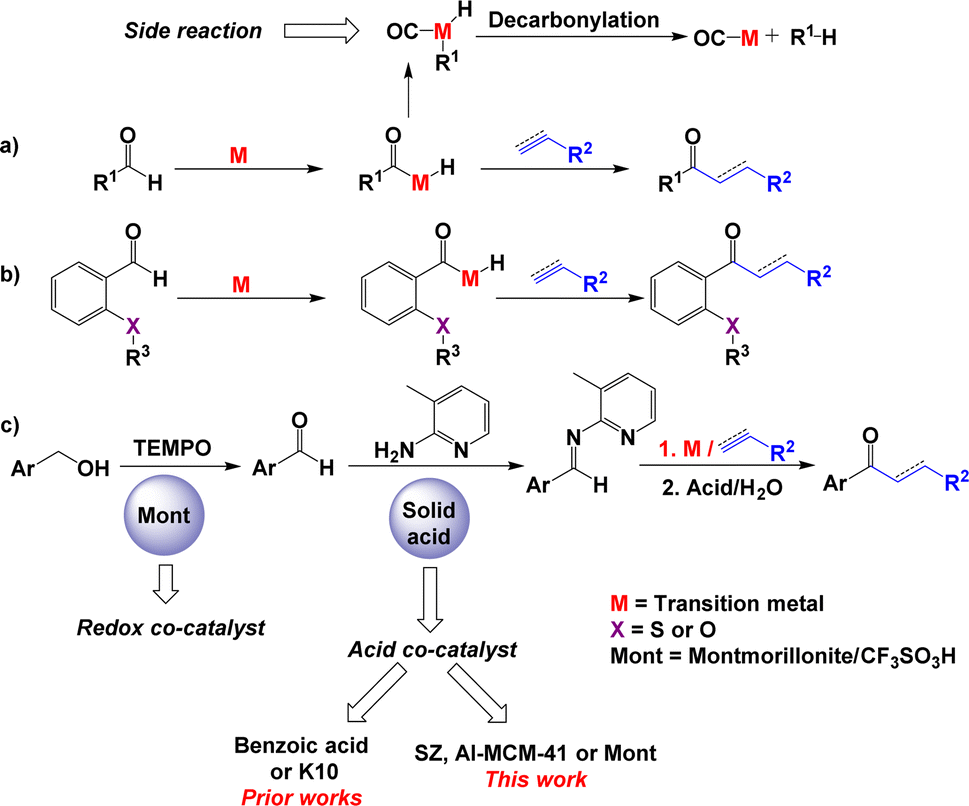 | ||
| Scheme 1 Hydroacylation reaction: (a) in the absence of chelating groups, (b) with the use of a β-sulfide or β-OR chelating group and (c) via an imine intermediate. | ||
Alternatively, the use of montmorillonite K10 (ref. 9) as an efficient acidic solid and reusable cocatalyst to generate imines has been described. However, the study of other solid acids with different properties and structural characteristics has not been explored, even though the use of solid acids as cocatalysts in organic reactions is attractive due to the recyclability of solid materials.10
Thus, in this work, we chose to use three structurally different solid acids, namely, sulfated zirconia (SZ),11 Al-MCM-41 (ref. 12) and montmorillonite13 modified with a superacid (CF3SO3H), to develop a cooperative system through which hydroiminoacylation of the corresponding aldimines generates ketones (Scheme 1c). Additionally, we considered extending this study to a process in which a simple alcohol is used as the aldehyde precursor in the presence of modified montmorillonite and TEMPO to generate the ketone. To the best of our knowledge, montmorillonite has not been used in similar dual processes (redox and acid catalysis).
Results and discussion
Materials
The SZ investigated in the present study was synthesised by the sol–gel technique and characterised by X-ray powder diffraction (XRD).14 Fig. 1a shows the diffractogram for SZ, in which the characteristic pattern of the crystalline tetragonal phase can be observed, given by the reflections at 2θ = 30.21°, 35.35°, 50.21°, 59.23°, 60.17°, 62.84°, 74.78° and 81.78°. These values are congruent with what is described in the literature14 for the crystalline tetragonal structure of a super acid material.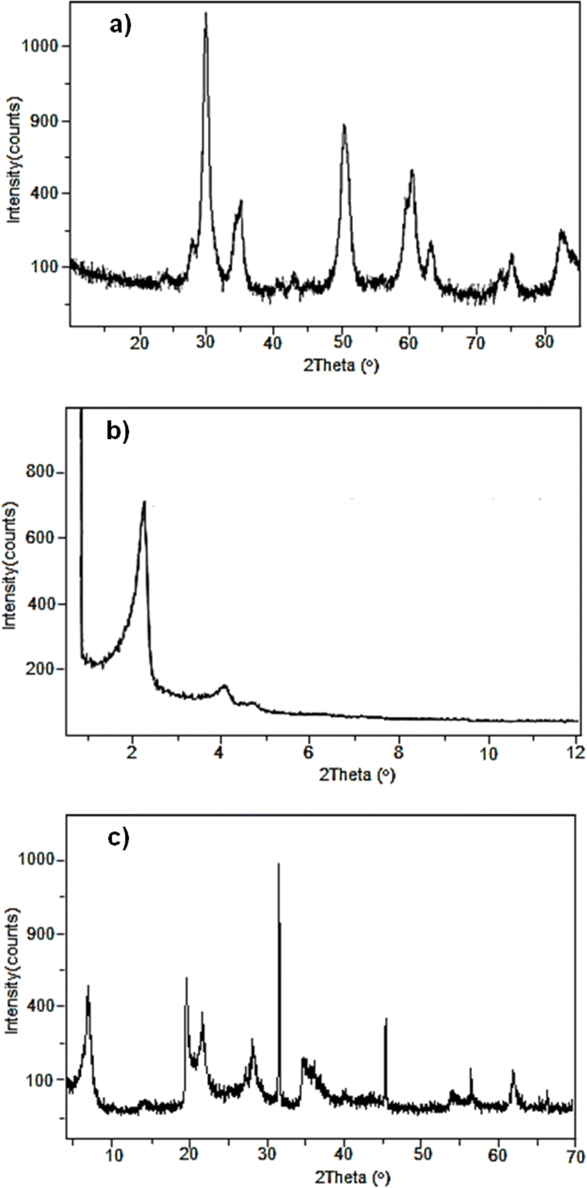 | ||
| Fig. 1 XRD patterns of (a) SZ, (b) Al-MCM-41 and (c) montmorillonite modified with CF3SO3H. | ||
The mesoporous material Al-MCM-41 was synthesised by an ultrasound-assisted technique.15 The structure of the mesoporous material was confirmed by XRD. Fig. 1b shows the diffractogram for Al-MCM-41, in which reflections can be observed at 2θ = 2.2° and 4.0°. These values are identical to those described in the literature for these kinds of materials.
On the other hand, natural montmorillonite was treated with concentrated CF3SO3H acid to generate the modified material. Fig. 1c shows the diffractogram with the characteristic reflections to a montmorillonite clay at 2θ = 7.0°, 20.0°, 35.0° and 61.8°.16
The textural properties of the synthesised materials were determined by nitrogen adsorption–desorption (BET) methods, allowing us to compare the surface area, pore volume and pore size of the catalysts. Compared with the SZ and Al-MCM-41 materials, the modified montmorillonite had a larger surface area and larger pore size (Table 1). This pore size makes this material go from being microporous to mesoporous according to IUPAC nomenclature.
| Materials | |||
|---|---|---|---|
| Properties | SZ | Al-MCM-41 | Modified montmorillonite |
| BET area (m2 g−1) | 90.35 | 1221.79 | 185.00 |
| Pore volume (cm3 g−1) | 0.12 | 0.85 | 0.60 |
| Pore size (Å) | 52.01 | 26.49 | 107.79 |
Initially, we optimised the typical reaction parameters, including the catalytic reagents, solvent, temperature, and time, for the hydroacylation of 1-butene with benzaldehyde, as shown in Scheme 1c and Table S10 in the ESI.† Subsequently, and based on the results previously obtained, we focused on studying the reaction trend with different substrates and the following different solid acids: modified montmorillonite, SZ or Al-MCM-41. As shown in Table 2, the hydroacylation of alkenes in the presence of 2-amino-3-picoline, aniline, Wilkinson's complex, and toluene as the solvent at 80 °C afforded saturated ketones 3a–3e in very good yields (80–92%). Similar yields were observed when the reaction was carried out with alkynes to generate α,β-unsaturated ketones 3f–3j. In contrast, the yields of the reaction decrease considerably when the aldehyde is aliphatic (acetaldehyde), as demonstrated by the yields obtained for ketones 3k and 3l (Table 2). It is important to note that, in this catalytic system, 2-amino-3-picoline and aniline were used simultaneously to enhance the formation of the imine, as described by Castillón et al.9
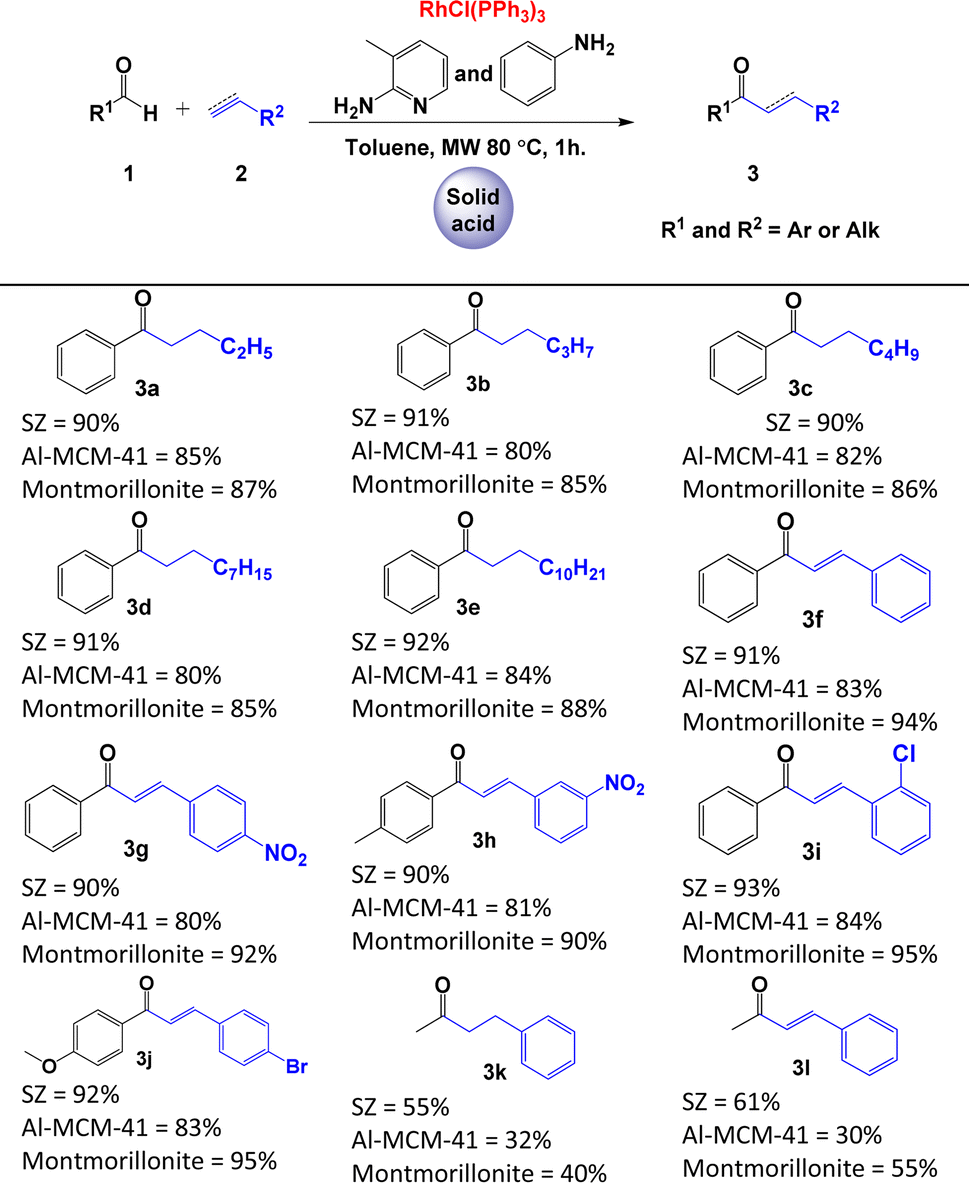
We think that the difference between the yields obtained with the three materials may be explained by their acidic properties and direct influence on the formation of picolyl imines. The SZ has H0 ≤ −14,17 Al-MCM-41 H0 ≤ 6.80 (ref. 18) and modified montmorillonite has H0 ≤ −12.75.19 Therefore, materials with major acidity properties catalyse the formation of imines more efficiently.
The efficiency of the SZ and modified montmorillonite catalysts are compared with previously reported for the synthesis of 1-phenylheptan-1-one (3c). As shown in Table 3, similar conversions were obtained when the reaction was carried out in presence of montmorillonite K10.9 However, the main differences lie in the excess moles of the 1-hexene used. We use an excess of 0.5 mmol, in contrast to the 5 mmol excess required when using montmorillonite K10.9
| Catalyst | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SZ | Mont/CF3SO3H | Mont/K10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Yield of the isolated product after chromatographic purification.b 80% Conversion and 98% yield, which were determined by GC. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Aldehyde mmol | 1 | 1 | 2.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Alkene mmol | 1.5 | 1.5 | 12.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Catalyst loading (mg) | 50 | 50 | 83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Temperature (°C) | 80 | 80 | 110 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Time (h) | 1 | 1 | 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Yield % | 90a | 86a | 98b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Reuse of solid acids
We studied the reuse of solid acids in the hydroacylation of 1-butene with benzaldehyde to form 3a by reactivation of the materials at 100 °C in an oven under an O2 atmosphere for 12 h. In the case of modified montmorillonite and SZ, the reaction yield did not decrease significantly after three reuses. After one run, Al-MCM-41 lost its catalytic activity (Fig. 2).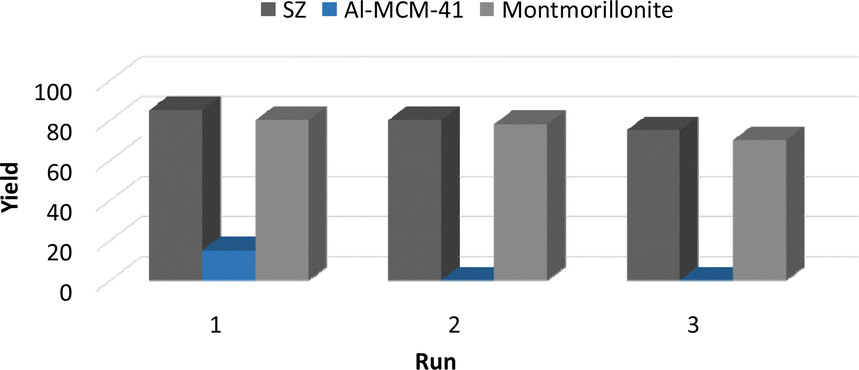 | ||
| Fig. 2 Reuse of materials in the synthesis of 3a. | ||
To confirm the homogeneity of catalysts in the reuse reactions, the structures of materials were verified by XRD before use. Fig. 3 shows the diffractograms of the three materials after drying at 100 °C and before being their reused. For SZ, the characteristic pattern of the crystalline tetragonal phase observed in the original material was still present (Fig. 3a). The plane reflections at 2θ = 30.21°, 35.35°, 50.21°, 59.23°, 60.17°, 62.84°, 74.78° and 81.78° were observed again. In the case of Al-MCM-41, the original structure was lost (Fig. 3b), which helps explain the significant decrease in reaction yield. Fig. 3c shows the plane reflections for montmorillonite at 2θ = 7.0°, 20.0°, 35.0° and 61.8°, which are the same as those of the original catalyst.
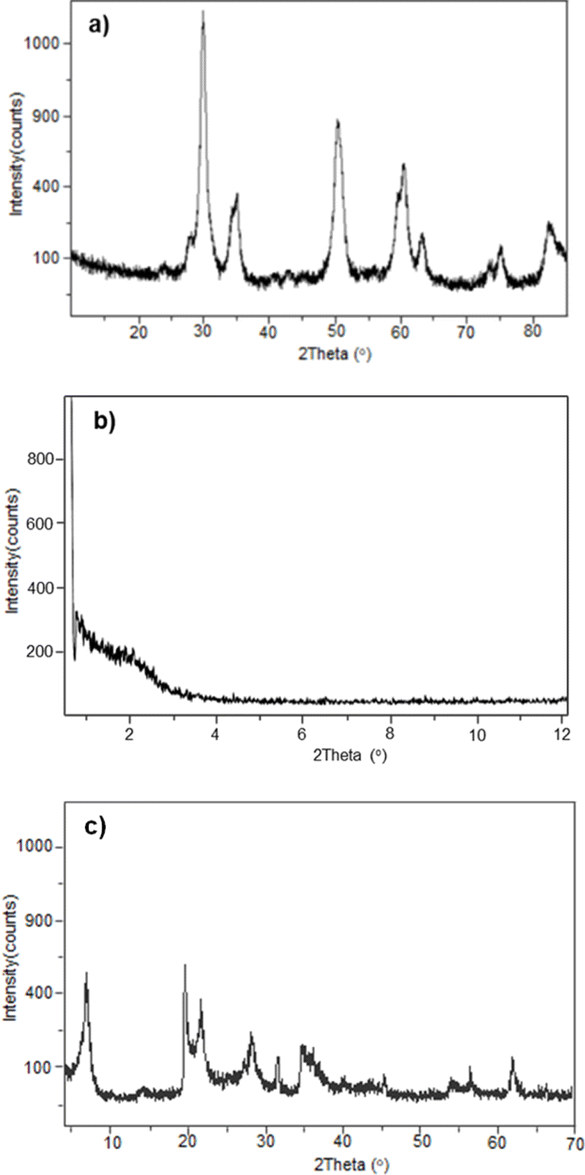 | ||
| Fig. 3 XRD patterns (a) SZ, (b) Al-MCM-41 and (c) montmorillonite modified before being reused. | ||
Oxidation of benzylic alcohols and hydroacylation
In previous studies, we observed that montmorillonite modified with CF3SO3H can participate as a Brønsted or Lewis acid catalyst in organic reactions20 or as a generator of free radical species in redox processes.21 From this double behaviour, we focus on studying the hydroacylation reaction of alkenes and alkynes with benzylic alcohols (Table 4).22 The process begins with the in situ oxidation of benzyl alcohol 4 to aldehyde 1 in the presence of modified montmorillonite and TEMPO23 to subsequently catalyse the hydroacylation of alkene or alkyne 2 and generate corresponding ketone 3 (Tables 4 and S11, ESI†). This strategy allowed us to obtain saturated (3a–3e) and α,β-unsaturated (3f–3j) ketones in good yields (55–70%). In the case of aliphatic alcohols, the formation of the final product was not observed; only the corresponding carboxylic acid was obtained. This suggests that, under our reaction conditions, the overoxidation of the aliphatic alcohol occurs significantly faster that the hydroacylation reaction.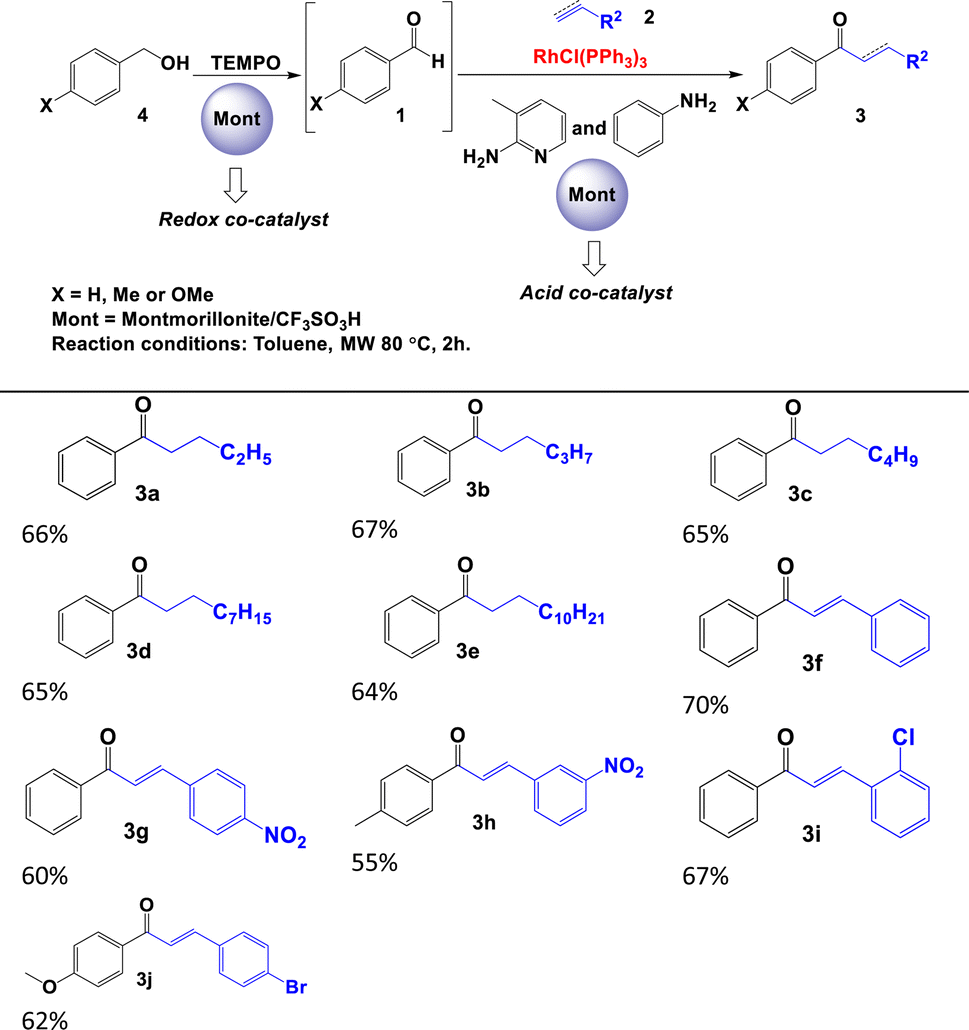
To propose a pathway for this direct hydroacylation reaction from benzyl alcohol 4a, a series of control experiments were carried out. Fe3+ was detected in a sample of modified montmorillonite by EPR (150 mT and g = 4.36, Scheme 2); thus, the treatment with CF3SO3H may dissolve structural iron in the montmorillonite, allowing these free cations to move to the interlayer of the montmorillonite where the oxidation of benzyl alcohol to aldehyde is catalysed. In the absence of montmorillonite or its replacement by another free-Fe3+ solid acid (SZ or Al-MCM-41), ketone 3a was only detected at 10%. When the reaction was carried out in N2 atmosphere, no final product formation was observed. In the absence of Wilkinson's complex and long reaction times (1 to 7 h), benzoic acid was generated in 65% yield. Moreover, in the absence of TEMPO, a complex mixture of benzyl alcohol oligomerization products was observed via NMR. Based on these results, a putative reaction mechanism is shown in Scheme 2. The Fe3+ present in the modified montmorillonite is responsible for the oxidation of TEMPO to generate TEMPO+, which oxidises alcohol 4a to aldehyde 1a.24 Air acts as an oxidant in the first step of the process, promoting the oxidation of Fe2+ to Fe3+ from montmorillonite. On the other hand, protons and aluminium ions present in solid acids (Brønsted or Lewis acid sites) are responsible for catalysing the formation of imine 5a. Then, Wilkinson's complex participates in the second part of the process,25 the hydroacylation of the imine to obtain 7a via the rhodium–aminoacyl complex 6a. Finally, the hydrolysis of 7a afforded saturated ketone 3a.
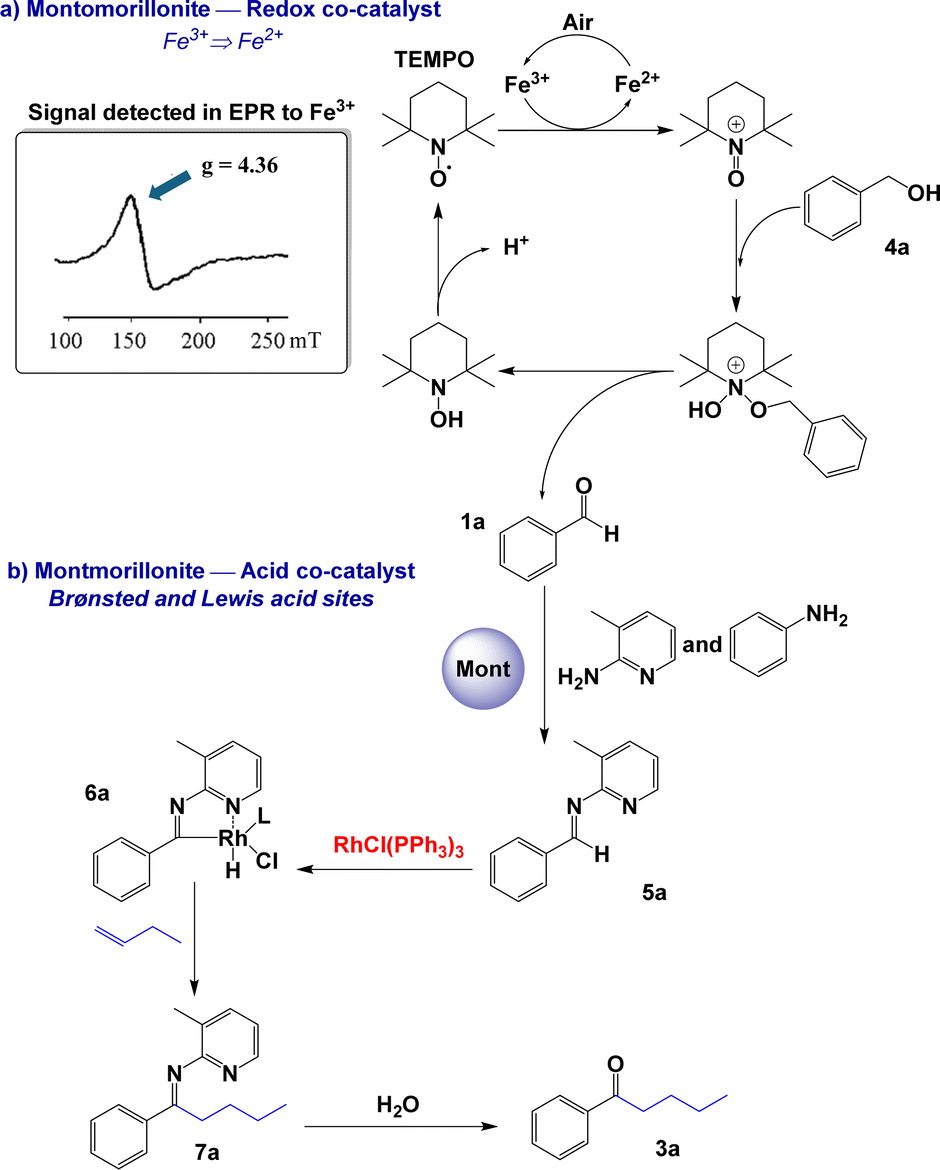 | ||
| Scheme 2 Proposed mechanism for the hydroacylation of benzyl alcohols. | ||
Conclusions
In summary, we demonstrated that different solid acids (sulfated zirconia, Al-MCM-41 or montmorillonite superacid) can be used as cocatalysts in the hydroacylation of alkenes and alkynes via an imine intermediate and in the presence of Wilkinson's complex. Due to the stability of SZ, Al-MCM-41 and modified montmorillonite under reaction conditions, the catalysts can be reused with similar yields. Additionally, the catalytic process was carried out from benzylic alcohols with in situ oxidation of alcohol in the presence of modified montmorillonite and TEMPO to afford the corresponding aldehyde. In the latter case, we demonstrate the usefulness of the dual character of montmorillonite by utilizing it first in a redox process and then as an acid catalyst.Experimental
General information
Trifluoromethanesulfonic acid, zirconium n-propoxide (70% n-propanol), 2-propanol, sulfuric acid, cetyltrimethylammonium bromide, triethylamine, tetramethylammonium hydroxide, TEOS, aluminium nitrate nonahydrate, benzaldehyde, chloridotris(triphenylphosphine)rhodium(I), 2-amino-3-picoline, aniline, 1-butene, 1-pentene, 1-hexene, 1-nonene, 1-dodecene, phenylacetylene, 1-ethynyl-4-nitrobenzene, 1-ethynyl-3-nitrobenzene, 1-chloro-2-ethynylbenzene, 1-bromo-4-ethynylbenzene, p-anisaldehyde, acetaldehyde, toluene, hexane, and ethyl acetate were purchased from Sigma-Aldrich. Microwave irradiation experiments to were performed using a Discover System (CEM Corporation) single-mode microwave with standard sealed microwave glass vials. The organic reactions were monitored by TLC carried out on 0.25 mm Merck silica gel plates. The developed TLC plates were visualised under a short-wave UV lamp or by heating after they were dipped in Ce(SO4)2. Flash column chromatography (FCC) was performed using silica gel (230–400) and employed a solvent polarity correlated with the TLC mobility. Yields refer to the chromatographically and spectroscopically (1H and 13C) homogeneous materials. NMR experiments were conducted on a Varian 300 or Bruker 500 MHz instruments in CDCl3 (99.9% D) and CD3OD (99.8% D) as solvents; the chemical shifts (δ) were referenced to CHCl3 (7.26 ppm 1H, 77.00 ppm 13C), CH3OH (4.87 ppm 1H, 49.00 ppm 13C), or TMS (0.00 ppm). The chemical shifts are reported in parts per million (ppm) and coupling constants J are given in hertz (Hz). The elemental analyses were carried out in an Elemental Analyzer Thermo Scientific/Flash 2000 equipment. The measurements of EPR were made with a Jeol JES-TE300 X band fashions spectrometer with a cylindrical cavity in the mode TE011. The external calibration of the magnetic field was carried out with a precision gaussmeter, Jeol ES-FC5. Powder X-ray diffraction (XRD) was performed using a Stoe Stadi-P Cu diffractometer with Cu Kα1 (using 40 kV and 30 mA). The nitrogen adsorption–desorption analysis of the materials was obtained at −196 °C on Micromeritics ASAP 2020 equipment. The chemical compositions of natural montmorillonite and modified montmorillonite were determined by energy-dispersive X-ray spectroscopy (EDXS), using an electronic spectrometer microprobe EPMA, JXA8900-R, JEOL. FT-IR spectra were recorded on a Nicolet Magna 750 spectrometer and data collection was performed using DRIFT.Synthesis of sulfated zirconia (SZ)
Zirconium n-propoxide (20 mL, 70% n-propanol) and 2-propanol (30 mL) were added to a 250 mL Erlenmeyer flask and stirred with a magnetic bar. Acid solution (1 mL 98% sulfuric acid in 3.2 mL distilled H2O) was added dropwise to hydrolyse the zirconium n-propoxide to obtain a gel. The solid was filtered and dried at 80 °C until complete alcohol evaporation, then calcined in air at 600 °C for 6 h.Synthesis of Al-MCM-41
Cetyltrimethylammonium bromide (7.3 g) was mixed with deionized H2O (200 mL) at 30 °C, then triethylamine (3.3 mL) was added. After stirring for 15 minutes, TMAOH (18 mL, 10 wt%), TEOS (22.4 mL) and Al(NO3)3·9H2O (18.75 g) were added, and the solution was stirred for 1 hour. The resulting gel, with molar composition 1TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2CTMABr:0.6EA:0.2TMAOH:0.05Al(NO3)3·9H2O:150H2O, was placed in glass bottles and sonicated for 4 h. The precipitated solid was recovered by filtration and washed with deionized water, dried at 80 °C overnight and calcinated at 540 °C for 6 h under air flow.
:0.2CTMABr:0.6EA:0.2TMAOH:0.05Al(NO3)3·9H2O:150H2O, was placed in glass bottles and sonicated for 4 h. The precipitated solid was recovered by filtration and washed with deionized water, dried at 80 °C overnight and calcinated at 540 °C for 6 h under air flow.
Modification of montmorillonite
50 g of natural montmorillonite was ground in a mortar and suspended in 1000 mL of deionized H2O. The mixture was stirred for 24 h and the suspended montmorillonite was separated by centrifugation (600 rpm for 15 min). This process was repeated three times and then the montmorillonite was dried at 100 °C under vacuum for 72 h to obtain a white solid. 10 g of this solid was suspended in 300 mL of deionized H2O and stirred for 72 h. Subsequently, a solution of CF3SO3H (100 mL, 0.18 M) was added, and the mixture was stirred for 24 h. The solid was recovered by vacuum filtration and washed with acetone (50 mL). Finally, the montmorillonite was dried at 100 °C under vacuum for 72 h to obtain a white solid.Standard reaction procedure to hydroacylation reaction from aldehydes
In a 10 mL tube of microwave with stir bar, 50 mg of solid acid, 1 mmol of aldehyde 1, 1.5 mmol of alkene or alkyne 2, 5% mmol of RhCl(PPh3)3, 20% mmol of 2-amino-3-picoline, 20% mmol of aniline and 3 mL of toluene were added. It was irradiated for 1 h in the microwave at 80 °C. The mixture was dried in a rotavapor and purified for chromatography column.Standard reaction procedure to oxidation of benzylic alcohols and hydroacylation
In a 10 mL tube of microwave with stir bar, 50 mg of solid acid, 1 mmol of alcohol 4, 1.5 mmol of alkene or alkyne 2, 10% TEMPO, 5% mmol of RhCl(PPh3)3, 20% mmol of 2-amino-3-picoline, 20% mmol of aniline and 3 mL of toluene were added. It was irradiated for 2 h in the microwave at 80 °C. The mixture was dried in a rotavapor and purified for chromatography column.Characterization data
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
J. A. M. S. and R. G. conceived the project and acquired the funds. B. I. V. A., E. G. R., R. G., J. C. and J. A. M. S designed the experiments. B. I. V. A., E. G. R., A. M. G., E. A. J. A. and A. L. T. conducted the experimental work. J. C., R. G. and J. A. M. S. coordinated the whole project. B. I. V. A., E. G. R., R. G., J. C. and J. A. M. S wrote the manuscript. All the authors contributed to the discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. A. M. S is grateful to the Royal Society (UK) for the International Alumni Grant (AL\180003 and AL\191055).Notes and references
- (a) J. Zhao, Y.-Y. Lu, R. Wang, P. Peters and H.-S. Lia, Rhodium(I) catalyzed hydroacylation reactions of alkenyl compounds, Adv. Synth. Catal., 2024, 366, 1 CrossRef; (b) A. Ghosh, K. F. Johnson, K. L. Vickerman, J. A. Walker and L. M. Stanley, Recent advances in transition metal-catalysed hydroacylation of alkenes and alkynes, Org. Chem. Front., 2016, 3, 639 RSC; (c) S. K. Murphy and V. M. Dong, Enantioselective hydroacylation of olefins with rhodium catalysts, Chem. Commun., 2014, 50, 13645 RSC; (d) J. C. Leung and M. J. Krische, Catalytic intermolecular hydroacylation of C–C π-bonds in the absence of chelation assistance, Chem. Sci., 2012, 3, 2202 RSC; (e) C.-H. Jun, E.-A. Jo and J. W. Park, Intermolecular hydroacylation by transition-metal complexes, Eur. J. Org Chem., 2007, 1869 CrossRef CAS.
- (a) H.-R. Wang and M.-C. Ye, Research advance on enantioselective transition metal-catalyzed hydroacylation reactions, Chin. J. Org. Chem., 2022, 42, 3152 CrossRef CAS; (b) M. C. Willis, Transition metal catalyzed alkene and alkyne hydroacylation, Chem. Rev., 2010, 110, 725 CrossRef CAS PubMed; (c) Y. J. Park, J.-W. Park and C.-H. Jun, Metal–organic cooperative catalysis in C–H and C–C bond activation and ats concurrent recovery, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed.
- B. Bosnich, Asymmetric catalysis. A somparative study of the mechanisms of intramolecular hydroacylation and hydrosilation, Acc. Chem. Res., 1998, 31, 667 CrossRef CAS.
- (a) Y.-N. Jiang, D.-C. Li, Y. Yang and Z.-P. Zhan, Porous organic polymers as heterogeneous ligands for highly selective hydroacylation, Org. Chem. Front., 2019, 6, 2964 RSC; (b) R. N. Straker, M. K. Majhail and M. C. Willis, Exploiting rhodium-catalysed ynamide hydroacylation as a platform for divergent heterocycle synthesis, Chem. Sci., 2017, 8, 7963 RSC; (c) J. F. Hooper, R. D. Young, A. S. Weller and M. C. Willis, Traceless chelation-controlled rhodium-catalyzed intermolecular alkene and alkyne hydroacylation, Chem.–Eur. J., 2013, 19, 312 CrossRef; (d) P. Lenden, D. A. Entwistle and M. C. Willis, An alkyne hydroacylation route to highly substituted furans, Angew. Chem., Int. Ed., 2011, 50, 10657 CrossRef CAS; (e) G. L. Moxham, H. E. Randell-Sly, S. K. Brayshaw, R. L. Woodward, A. S. Weller and M. C. Willis, A second-generation catalyst for intermolecular hydroacylation of alkenes and alkynes using β-S-substituted aldehydes: The role of a hemilabile P-O-P ligand, Angew. Chem., Int. Ed., 2006, 45, 7618 CrossRef CAS; (f) M. C. Willis, S. J. McNally and P. J. Beswick, Chelation controlled intermolecular hydroacylation: The direct addition of alkyl aldehydes to functionalised alkenes, Angew. Chem., Int. Ed., 2004, 43, 340 CrossRef CAS.
- (a) D. D. Thalakottukara and T. Gandhi, NHC-organocatalysed hydroacylation of unactivated or weakly activated C–C multiple bonds and ketones, Asian J. Org. Chem., 2022, 11, e202200080 CrossRef CAS; (b) G. You, Z.-X. Chang, J. Yan, C. Xia, F.-R. Lib and H.-S. Li, Rhodium-catalyzed sequential intermolecular hydroacylation and deconjugative isomerization toward diversified diketones, Org. Chem. Front., 2021, 8, 39 RSC; (c) J. Barwick-Silk, S. Hardy, M. C. Willis and A. S. Weller, Rh(dpephos)-catalyzed alkyne hydroacylation using β-carbonyl-substituted aldehydes: mechanistic insight leads to low catalyst loadings that enables selective catalysis on gram-scale, J. Am. Chem. Soc., 2018, 140, 734 CrossRef; (d) T. J. Coxon, M. Fernandez, J. Barwick-Silk, A. I. McKay, L. E. Britton, A. S. Weller and M. C. Willis, Exploiting carbonyl groups to control intermolecular rhodium-catalyzed alkene and alkyne hydroacylation, J. Am. Chem. Soc., 2017, 139, 10142 CrossRef CAS PubMed.
- (a) C.-H. Jun, C. W. Moon, H. Lee and D.-Y. Lee, Chelation-assisted carbon–carbon bond activation by Rh(I) catalysts, J. Mol. Catal. A: Chem., 2002, 189, 145 CrossRef CAS; (b) C.-H. Jun, C. W. Moon and D.-Y. Lee, Chelation-assisted carbon–hydrogen and carbon–carbon bond activation by transition metal catalysts, Chem.–Eur. J., 2002, 8, 2422 CrossRef CAS PubMed; (c) J. W. Suggs, Activation of aldehyde C-H bonds to oxidative addition via formation of 3-methyl-2-aminopyridyl aldimines and related compounds: Rhodium based catalytic hydroacylation, J. Am. Chem. Soc., 1979, 101, 489 CrossRef CAS.
- R. Ling, M. Yoshida and P. S. Mariano, Exploratory investigations probing a preparatively versatile, pyridinium salt photoelectrocyclization–solvolytic aziridine ring opening sequence, J. Org. Chem., 1996, 61, 4439 CrossRef CAS PubMed.
- Y. J. Park, J.-W. Park and C.-H. Jun, Metal–organic cooperative catalysis in C–H and C–C bond activation and its concurrent recovery, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS.
- (a) R. X. Yañez and S. Castillón, Rhodium-catalyzed intermolecular hydroacylation of 1-alkynes: Effect of phosphines and MK-10 on the reaction selectivity, J. Organomet. Chem., 2007, 692, 1628 CrossRef; (b) R. X. Yañez, C. Claver, S. Castillón and E. Fernández, Montmorillonite K10 as a suitable co-catalyst for atom economy in chelation-assisted intermolecular hydroacylation, Tetrahedron Lett., 2003, 44, 1631 CrossRef.
- (a) N. Mansir, Y. H. Taufiq-Yap, U. Rashid and I. M. Lokman, Investigation of heterogeneous solid acid catalyst performance on low grade feedstocks for biodiesel production: A review, Energy Convers. Manage., 2017, 141, 171 CrossRef CAS; (b) P. Gupta and S. Paul, Solid acids: Green alternatives for acid catalysis, Catal. Today, 2014, 236, 153 CrossRef CAS; (c) K. Nakajima and M. Hara, Amorphous carbon with SO3H groups as a solid brønsted acid catalyst, ACS Catal., 2012, 2, 1296 CrossRef CAS; (d) Y. Román-Leshkov and M. E. Davis, Activation of carbonyl-containing molecules with solid lewis acids in aqueous media, ACS Catal., 2011, 1, 1566 CrossRef; (e) F. Chen, X. Meng and F.-S. Xiao, Mesoporous solid acid catalysts, Catal. Surv. Asia, 2011, 15, 37 CrossRef CAS; (f) J. H. Clark, Solid acids for green chemistry, Acc. Chem. Res., 2002, 35, 791 CrossRef CAS.
- (a) P. Wang, Y. Yue, T. Wang and X. Bao, Alkane isomerization over sulfated zirconia solid acid system, Int. J. Energy Res., 2020, 44, 3270 CrossRef CAS; (b) G. X. Yan, A. Wang, I. E. Wachs and J. Baltrusaitis, Critical review on the active site structure of sulfated zirconia catalysts and prospects in fuel production, Appl. Catal., A, 2019, 572, 210 CrossRef CAS; (c) M. K. Patil, A. N. Prasad and B. M. Reddy, Zirconia-based solid acids: Green and heterogeneous catalysts for organic synthesis, Curr. Org. Chem., 2011, 15, 3961 CrossRef CAS; (d) K. Arata, Organic syntheses catalyzed by superacidic metal oxides: sulfated zirconia and related compounds, Green Chem., 2009, 11, 1719 RSC; (e) B. M. Reddy and M. K. Patil, Organic syntheses and transformations catalyzed by sulfated zirconia, Chem. Rev., 2009, 109, 2185 CrossRef CAS.
- (a) J. Szymczak and M. Kryjewski, Porphyrins and phthalocyanines on solid-state Mesoporous Matrices as catalysts in oxidation reactions, Materials, 2022, 15, 2532 CrossRef CAS; (b) C. T. Kresge and W. J. Roth, The discovery of mesoporous molecular sieves from the twenty year perspective, Chem. Soc. Rev., 2013, 42, 3663 RSC.
- (a) S. Meenakshy, M. Neetha and G. Anilkumar, Montmorillonite-catalysed coupling reactions: a green overview, Org. Biomol. Chem., 2024, 22, 1961 RSC; (b) T. Chellapandi and G. Madhumitha, Montmorillonite clay-based heterogenous catalyst for the synthesis of nitrogen heterocycle organic moieties: A review, Mol. Diversity, 2022, 26, 2311 CrossRef CAS; (c) G. Nagendrappa and R. R. Chowreddy, Organic reactions using clay and clay-supported catalysts: a survey of recent literature, Catal. Surv. Asia, 2021, 25, 231 CrossRef CAS; (d) M. Hechelski, A. Ghinet, B. Louvel, P. Dufrénoy, B. Rigo, A. Daïch and C. Waterlot, From conventional Lewis acids to heterogeneous montmorillonite K10: eco-friendly plant-based catalysts used as green Lewis acids, ChemSusChem, 2018, 11, 1249 CrossRef CAS PubMed; (e) B. S. Kumar, A. Dhakshinamoorthy and K. Pitchumani, K10 montmorillonite clays as environmentally benign catalysts for organic reactions, Catal. Sci. Technol., 2014, 4, 2378 RSC.
- R. González-Olvera, B. I. Vergara-Arenas, G. E. Negron-Silva, D. Angeles-Beltrán, L. Lomas-Romero, A. Gutiérrez-Carrillo, V. K. Lara and J. A. Morales-Serna, Synthesis of β-nitrostyrenes in the presence of sulfated zirconia and secondary amines, RSC Adv., 2015, 5, 99188 RSC.
- I. Vergara-Arenas, L. Lomas-Romero, D. Angeles-Beltrán, G. E. Negron-Silva, A. Gutiérrez-Carrillo, V. K. Lara and J. A. Morales-Serna, Multicomponent synthesis of 4-aryl-NH-1, 2, 3-triazoles in the presence of Al-MCM-41 and sulfated zirconia, Tetrahedron Lett., 2017, 58, 2690 CrossRef.
- (a) C. Mejri, W. Oueslati and A. Ben Haj Amara, Structure and reactivity assessment of dioctahedral montmorillonite during provoked variable sequential cation exchange process via XRD modelling approach, Appl. Surf. Sci. Adv., 2023, 15, 100403 CrossRef; (b) A. Ahmed, Y. Chaker, E. H. Belarbi, O. Abbas, J. N. Chotard, H. B. Abassi, A. N. Van Nhien, M. El Hadri and S. Bresson, XRD and ATR/FTIR investigations of various montmorillonite clays modified by monocationic and dicationic imidazolium ionic liquids, J. Mol. Struct., 2018, 1173, 653 CrossRef CAS.
- (a) C. Morterra, G. Cerrato, S. Ardizzone, C. L. Bianchi, M. Signorettob and F. Pinna, Surface features and catalytic activity of sulfated zirconia catalysts from hydrothermal precursors, Phys. Chem. Chem. Phys., 2002, 4, 3136 RSC; (b) F. R. Chen, G. Coudurier, J. F. Joly and J. C. Vedrine, Superacid and catalytic properties of sulfated zirconia, J. Catal., 1993, 143, 616 CrossRef CAS.
- (a) S. Weiming, L. Xing, J. Tao and D. Qigang, Characterization and catalytic properties of Al-MCM-41 mesoporous materials grafted with tributyltin chloride, Chin. J. Chem. Eng., 2012, 20, 900 CrossRef; (b) Q. Lili, J. Min, W. Xinkui, H. Min and C. Tianxi, AlCl3/MCM-41 as a Catalyst for Isomerization of Endo-tricyclodecane, Chin. J. Catal., 2010, 31, 383 CrossRef.
- G. Granados-Oliveros, V. Gómez-Vidales, A. Nieto-Camacho, J. A. Morales-Serna, J. Cárdenas and M. Salomón, Photoproduction of H2O2 and hydroxyl radicals catalysed by natural and super acid-modified montmorillonite and its oxidative role in the peroxidation of lipids, RSC Adv., 2013, 3, 937 RSC.
- J. A. Morales-Serna, B. N. Nguyen, E. García-Ríos, R. Gaviño, J. Cárdenas and G. García de la Mora, Glycosylation of stannyl ceramides promoted by modified montmorillonite in supercritical carbon dioxide, Synthesis, 2018, 50, 593 CrossRef CAS.
- J. A. Morales-Serna, B. A. Frontana-Uribe, R. Olguín, V. Gómez-Vidales, L. Lomas-Romero, E. García-Ríos, R. Gaviño and J. Cárdenas, Reaction control in heterogeneous catalysis using montmorillonite: switching between acid-catalysed and red-ox processes, RSC Adv., 2016, 6, 42613 RSC.
- (a) S.-Q. Zhanga and J.-B. Xia, Selective cross-coupling of α, β-unsaturated nitriles with aldehydes or alcohols by hydrogen transfer catalysis towards β-ketonitriles and glutaronitriles, Org. Chem. Front., 2024, 11, 2613 RSC; (b) C. H. Lee and C. H. Jun, Chelation-Assisted C–H and C–C Bond Activation of Allylic Alcohols by a Rh (I) Catalyst under Microwave Irradiation, Synlett, 2018, 29, 736 CrossRef CAS; (c) S. Hatanaka, Y. Obora and Y. Ishii, Iridium-catalyzed coupling reaction of primary alcohols with 2-alkynes leading to hydroacylation products, Chem.–Eur. J., 2010, 16, 1883 CrossRef CAS PubMed; (d) V. M. Williams, J. C. Leung, R. L. Patman and M. J. Krische, Hydroacylation of 2-butyne from the alcohol or aldehyde oxidation level via ruthenium catalyzed C–C bond forming transfer hydrogenation, Tetrahedron, 2009, 65, 5024 CrossRef CAS PubMed; (e) D.-W. Kim, S.-G. Lim and C.-H. Jun, Recyclable self-assembly-supported catalyst for chelation-assisted hydroacylation of an olefin with a primary alcohol, Org. Lett., 2006, 8, 2937 CrossRef CAS; (f) D.-H. Chang, D.-Y. Lee, B.-S. Hong, J.-H. Choi and C.-H. Jun, A new solvent system for recycling catalysts for chelation-assisted hydroacylation of olefins with primary alcohols, J. Am. Chem. Soc., 2004, 126, 424 CrossRef CAS PubMed; (g) C.-H. Jun, H.-S. Hong and C.-W. Huh, Direct conversion of benzyl alcohol to ketone by polymer-supported Rh catalyst, Tetrahedron Lett., 1999, 40, 8897 CrossRef CAS.
- R. Varala and V. Seema, Recent Applications of TEMPO in Organic Synthesis and Catalysis, SynOpen, 2023, 7, 408 CrossRef CAS.
-
(a) Z. Zhang, Y. Ma, S. Dai, L. Li, Y. Zhang and H. Li, Iron/TEMPO-catalyzed direct aerobic oxidative coupling of methyl-mubstituted N-heteroazaarenes with alcohols, Tetrahedron Lett., 2020, 61, 151885 CrossRef CAS;
(b) X. Jiang, J. Zhang and S. Ma, Iron catalysis for room-temperature aerobic oxidation of alcohols to carboxylic acids, J. Am. Chem. Soc., 2016, 138, 8344 CrossRef CAS;
(c) J. Yu, M. Shen and M. Lu, Aerobic oxidative synthesis of 2-arylbenzimidazoles, 2-arylbenzoxazoles, and 2-arylbenzothiazoles from arylmethanols or arylmethylamines catalyzed by Fe(III)/TEMPO under solvent-free conditions, J. Iran. Chem. Soc., 2015, 12, 771 CrossRef CAS;
(d) E. Zhang, H. Tian, S. Xu, X. Yu and Q. Xu, Iron-catalyzed direct synthesis of imines from amines or alcohols and amines via aerobic oxidative reactions under air, Org. Lett., 2013, 15, 2704 CrossRef CAS;
(e) J. Liu and S. Ma, Iron-Catalyzed Aerobic Oxidation of Allylic Alcohols: The Issue of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C Bond Isomerization, Org. Lett., 2013, 15, 5150 CrossRef CAS;
(f) X. Wang and X. Liang, Aerobic Oxidation of Alcohols to Carbonyl Compounds Catalyzed by Fe(NO3)3/4-OH-TEMPO under Mild Conditions, Chin. J. Catal., 2008, 29, 935 CrossRef CAS.
C Bond Isomerization, Org. Lett., 2013, 15, 5150 CrossRef CAS;
(f) X. Wang and X. Liang, Aerobic Oxidation of Alcohols to Carbonyl Compounds Catalyzed by Fe(NO3)3/4-OH-TEMPO under Mild Conditions, Chin. J. Catal., 2008, 29, 935 CrossRef CAS. - (a) M. Wang, X. Zhang, Z. Chen, Y. Tang and M. Lei, A theoretical study on the mechanisms of intermolecular hydroacylation of aldehyde catalyzed by neutral and cationic rhodium complexes, Sci. China: Chem., 2014, 57, 1264 CrossRef CAS; (b) D.-Y. Lee, C. W. Moon and C.-H. Jun, Synthesis of aliphatic ketones from allylic alcohols through consecutive isomerization and chelation-assisted hydroacylation by a rhodium catalyst, J. Org. Chem., 2002, 67, 3945 CrossRef CAS PubMed; (c) C.-H. Jun, K.-Y. Chung and J.-B. Hong, C–H and C–C bond activation of primary amines through dehydrogenation and transimination, Org. Lett., 2001, 3, 785 CrossRef CAS PubMed; (d) C.-H. Jun, C.-W. Huh and S.-J. Na, Direct Synthesis of Ketones from Primary Alcohols and 1-Alkenes, Angew. Chem., Int. Ed., 1998, 37, 145 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, full characterization of materials, analytical data of individual compounds and 1H, 13C NMR spectra of the synthesised compounds. See DOI: https://doi.org/10.1039/d4ra05791k |
| This journal is © The Royal Society of Chemistry 2024 |